

Abstract
The loss of estrogen (E2) initiates a rapid phase of bone loss leading to osteoporosis in one-half of postmenopausal women, but the mechanism is not fully understood. Here, we show for the first time how loss of E2 activates low-grade inflammation to promote the acute phase of bone catabolic activity in ovariectomized (OVX) mice. E2 regulates the abundance of dendritic cells (DCs) that express IL-7 and IL-15 by inducing the Fas ligand (FasL) and apoptosis of the DC. In the absence of E2, DCs become long-lived, leading to increased IL-7 and IL-15. We find that IL-7 and IL-15 together, but not alone, induced antigen-independent production of IL-17A and TNFα in a subset of memory T cells (TMEM). OVX of mice with T-cell–specific ablation of IL15RA showed no IL-17A and TNFα expression, and no increase in bone resorption or bone loss, confirming the role of IL-15 in activating the TMEM and the need for inflammation. Our results provide a new mechanism by which E2 regulates the immune system, and how menopause leads to osteoporosis. The low-grade inflammation is likely to cause or contribute to other comorbidities observed postmenopause. © 2020 American Society for Bone and Mineral Research.
Introduction
Osteoporosis increases the risk of bone fracture after minimal trauma. Increase in circulatory system disease, malignant neoplasms, and dementia postfracture are a significant cause of mortality among the elderly.(1,2) It was recognized nearly eight decades ago that involutional osteoporosis in postmenopausal women is mediated by loss of estrogen (E2).(3) Since the studies by Reifenstein and Albright,(4) several hypotheses were proposed for how aging and E2 loss lead to reduced bone mass. Some reports suggested that decreased calcium absorption,(5,6) decline in renal function,(7) and impaired vitamin D metabolism,(8,9) along with aging and menopause, cause osteoporosis. In general, the early studies focused on the effect of E2 on nutrient absorption and metabolism as a cause of osteoporosis. At homeostasis, during remodeling the activity of bone resorbing cells, osteoclasts, and bone forming cells, osteoblasts, are coupled to maintain skeletal mass.(10) At menopause women undergo an early rapid phase, followed by a slower and extended period of bone loss. In the early phase, more cancellous bone is lost, whereas in the slower phase both cortical and cancellous bone are equally eroded.(11–13) Here we propose a new model based on our data for the events that lead to the early phase of bone loss.
The finding four decades ago, that sex steroid receptors are expressed in human, rat, and mouse osteoclasts, osteoblasts, and growth plate chondrocytes indicated that sex hormones directly regulate these cells to maintain bone mass.(14) In detail, experiments showed that E2 regulates the expression of the Fas ligand (FasL) in osteoclasts and osteoblasts inducing apoptosis in osteoclasts to limit bone resorption in premenopausal women.(15–18) At the same time E2 extends the lifespan of osteoblasts and osteocytes(15,19,20) to favor bone formation. Therefore, E2 loss leads to increased osteoclast numbers and concurrent decreased osteoblast numbers, in line with the notion that E2 regulates the bone remodeling unit (BRU) directly. This paradigm led to drug treatments for osteoporosis that have to date also focused on restoring the balance between bone resorption and formation by targeting cells in the BRU. For instance, antiresorptives such as bisphosphonates(21) and denosumab(22) (anti-RANKL antibody) suppress osteoclasts, whereas teriparatide(23) (PTH1–34) and romosozumab(24) (anti-sclerostin antibody) target osteoblasts.
Nearly two decades ago, the recognition of the effect of T-cell–produced cytokines on osteoclasts(25) led to coining of the term osteoimmunology.(26) Some cytokines, such as TNFα and IL-17A, lead to increased osteoclastogenesis.(27) Other cytokines, such as interferon-gamma (IFN-γ), suppress osteoclastogenesis.(25,28) Pioneering studies by Roggia and colleagues(29) showed that in mice, ovariectomy (OVX) leads to increased TNFα-producing T cells in the bone marrow. Furthermore, whereas postmenopausal women with normal bone density have undetectable levels of IL-17A, postmenopausal osteoporotic women have detectable levels of IL-17A in peripheral blood.(30) Together, these results suggest that low-grade inflammation promotes or is associated with bone loss in some postmenopausal women. Consistent with these findings, men and women with chronic inflammatory diseases such as rheumatoid arthritis (RA), Crohn’s disease, and some viral (ie, human immunodeficiency virus [HIV]) infections develop osteoporosis.(31–36) Despite these observations, questions as to the nature and the contribution of inflammation to postmenopausal osteoporosis have lingered. In some cases, how the immune system is activated is starting to be unraveled. For instance, self-reactive T cells play a central role in the pathogenesis and pathophysiology of RA.(37,38) Although controversial, antigen presentation by myeloid dendritic cells (mDCs) appears to play an important role in initiating joint damage.(39) OVX-induced oxidative stress has been suggested as an activator of DCs.(40) However, the mechanism(s) of how TNFα and IL-17A are induced in T cells postmenopause has not been identified to date.
It is accepted that E2 is an “anti-inflammatory” that works through a number of mechanisms to suppress inflammation.(41) The first Framingham study seven decades ago showed that premenopausal women were protected against osteoporosis and cardiovascular morbidity in comparison with age-matched men and postmenopausal women (or with oophorectomy).(42–44) Because osteoporosis often occurs in the sixth decade of life, age has been also suggested as the cause of skeletal frailty.(45) The molecular mechanisms by which E2 regulates the immune system, and conversely, how menopause effects the immune system, have not been elucidated. Here we describe a novel and coherent pathway by which E2 suppresses the immune system and, conversely, our studies show how E2 loss promotes a low-grade inflammation by T cells that leads to osteoporosis.
We previously discovered that osteoclasts are antigen-presenting cells that cross-present antigens to CD8 T cells. Osteoclasts induce FoxP3, CD25, and CTLA4, as well as interferon (IFN)-γ, IL-6, IL-10, and IL-2 in the CD8 T cells that are immunosuppressive. These osteoclast-induced regulatory T cells (TcREGs) also suppress bone resorption by osteoclasts to form a negative feedback loop. The mechanism by which TcREGs suppress osteoclasts has been detailed by our laboratory, but the mechanism by which they suppress the immune system in OVX mice is not well understood. We recently initiated studies to reveal the immune cells that TcREGs target. In the process of discovering the targets of TcREGs we determined how the loss of E2 by OVX leads to activation of memory T cells (TMEM) to secrete TNFα and IL-17A, leading to a low-grade persistent inflammation that is the focus of this work.
Materials and Methods
Mice
C57BL/6J (model 000664), IL15RA-floxed (model 022365),(46) and Lck-Cre Tg540-I (model 006889)(47) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and bred in house. Breeding trio of IL-7CFP reporter mice(48) were generously provided by Dr. Scott Durum (NIH-NCI, Bethesda, MD, USA).
OVX
Mice were randomly assigned to sham surgery or OVX groups. Each cage had at least one sham or one OVX mouse to minimize alterations of the microbiome between groups. Bilateral OVX was performed on 12-week-old mice. Mice were anesthetized using 2.5% isoflurane to initiate anesthesia, and 1% for maintenance. The ovaries were accessed through a single incision in the skin and exteriorized through the muscle wall on each side. Each ovary was clamped using a hemostat and removed by a single cut. Skin staples (3M) were used to close the skin incision. To minimize discomfort postsurgery, three doses of 0.1 mg/kg buprenorphine HCl over2 days or a single dose of 1 mg/kg buprenorphine SR was administered subcutaneously. No adverse responses or effects were observed in any of the mice used in these experiments.
Animal use statement
All animals were maintained in the Department of Comparative Medicine, Saint Louis University School of Medicine in accordance with institutional and Public Health Service Guidelines. The mice were housed in microisolator caging, tested and found to be specific pathogen free (SPF). The mice used in experiments described in this study were maintained on rodent chow (Lab Diets cat# 5LOB). Saint Louis University School of Medicine Institutional Animal Care and Use Committee (IACUC) approved all procedures performed on mice (protocol numbers 2072 and 2184).
Antibodies and FACS
Anti-mouse antibodies for FACS were as follows: Brilliant violent (BV) 711-conjugated anti-mouse CD45 (clone 30-F11; BD Biosciences, San Jose, CA, USA); AF700-conjugated anti-mouse CD44 (IM7; BD Pharmingen, San Jose, CA, USA); BV605-conjugated CD62L (MEL-14; BD Biosciences); PE-Cy7-cojugated anti-CD3e (500A2; BioLegend, San Diego, CA, USA); R700-conjugated anti-CD8a (5H10; Caltag); anti-CD4 (RM4–5; BD Pharmingen), PE-conjugated TNFα (MP6-XT22; BD Biosciences); and V450-cojugated IL-17A (TC11–18H10; BD Biosciences). For FACS, cells were blocked with anti-mouse FcgRIII/IIR (Fc-block; BD Pharmingen) for 10 min and then stained for 45 min on ice with fluorophore-conjugated antibody. Stained cells were washed, fixed with 3% paraformaldehyde, and analyzed on LSRII instrument with CellQuest (BD Biosciences) software. Gates were determined using combination of single-color and fluor-minus-one controls. For live cell sorting, cells for isolation TMEM or BMDCs (Fig. 2 through Fig. 4), bone marrow cells (BMCs) were first incubated with Fc-block for 5 min, then stained with fluor-conjugated antibodies against cell-surface proteins (see figure legends or text for details) for 30 min in PBS + 1% FBS (PBS + 1%), washed and resuspended in PBS + 1% for sorting on a FACS-DIVA (BD Biosciences) The sorted cells were collected directly in 15-mL tubes containing 4 mL RPMI +10% FBS to improve viability. Data analyses were performed with FlowJo software (version 8.73; FlowJo, LLC, Ashland, OR, USA).
Fig. 2.
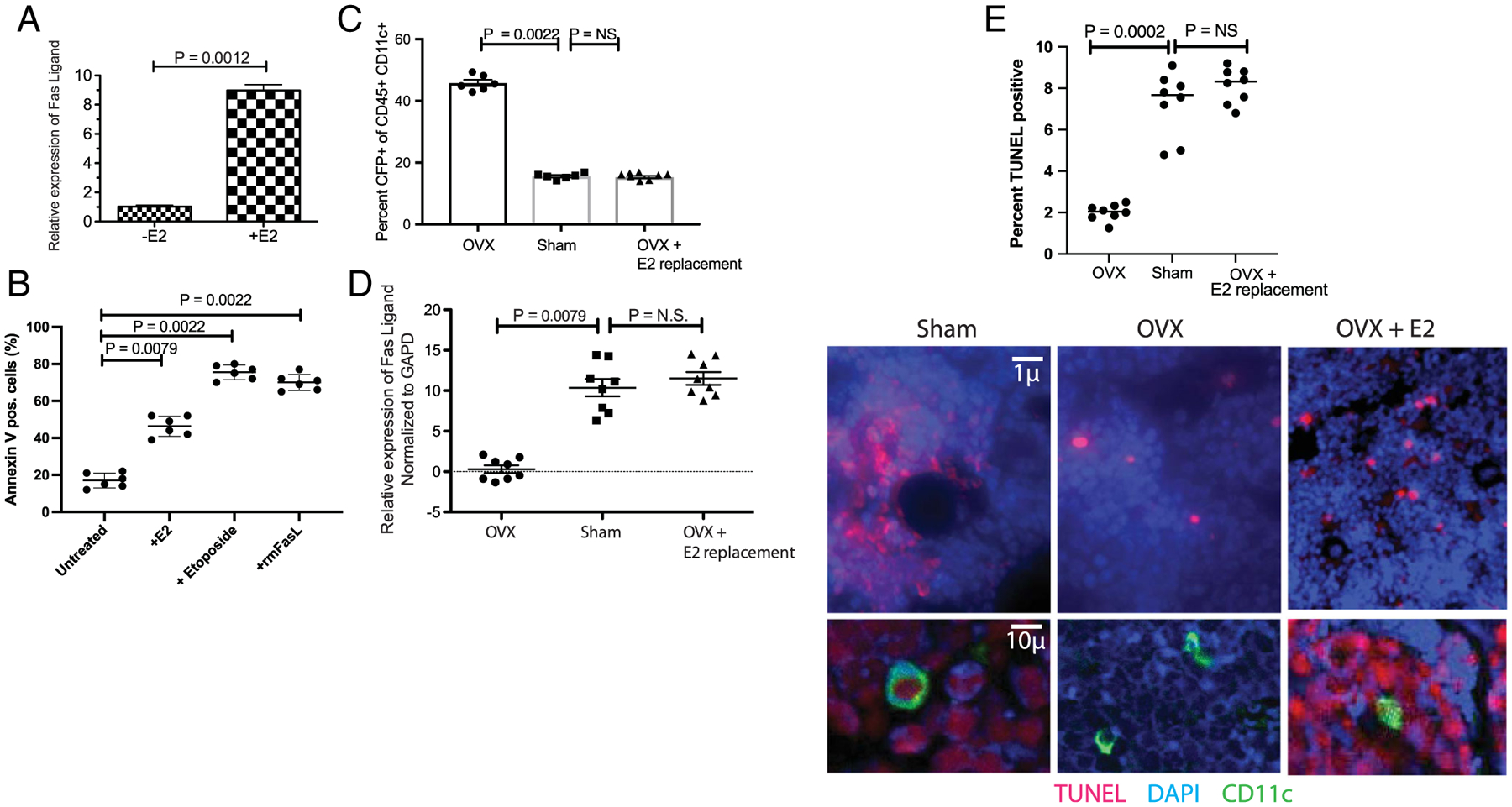
E2 regulates dendritic cell lifespan by inducing apoptosis via FasL. (A) Estrogen induced FasL in BMDCs: BMDC (CD45+ CD11c+ CFP+) cells isolated by FACS were cultured for 4 hours with 10nM 17β-estradiol. FasL mRNA was quantified by qPCR relative to GAPDH message (average of six independent experiments [BMC preparations] are shown). (B) FasL induced apoptosis in BMDC: apoptosis was assessed by Annexin V staining in cells used in A after 24 hours. Etoposide (10−8M) was used as a positive control and recombinant murine FasL (10 ng/mL) was used to assess expression of Fas (receptor). (C) E2 replacement in OVX mice restores CFP+ BMDC: Increased number of CD45 CD11c and CFP positive cells are observed in OVX mice (cf. Fig. 1B) but decrease with E2 replacement (0.1 mg/pellet 21-day release). The decrease is observed in BMDC when 17β-estradiol pellets are implanted 1 or 3 weeks post-OVX. Data from 8 mice/group. (D) E2 regulates FasL in vivo: RNA was isolated 3 weeks postsurgery from bone marrow cells of OVX, sham-operated, and OVX with 17β-estradiol pellets implanted mice. FasL expression was assessed by qRT-PCR. (Same mice from C). (E) E2 loss limits apoptosis: foci of apoptotic cells are observed in E2-replete mice (sham-surgery), but fourfold to fivefold fewer foci are observed in OVX mice (also see Supplementary Fig. 3). As the ovary produces multiple factors/hormones, we performed E2 replacement in OVX mice, and show that 17β-estradiol restored the levels of apoptotic foci. BMDC = bone marrow dendritic cell; E2 = estrogen; FasL = Fas ligand.
Fig. 4.
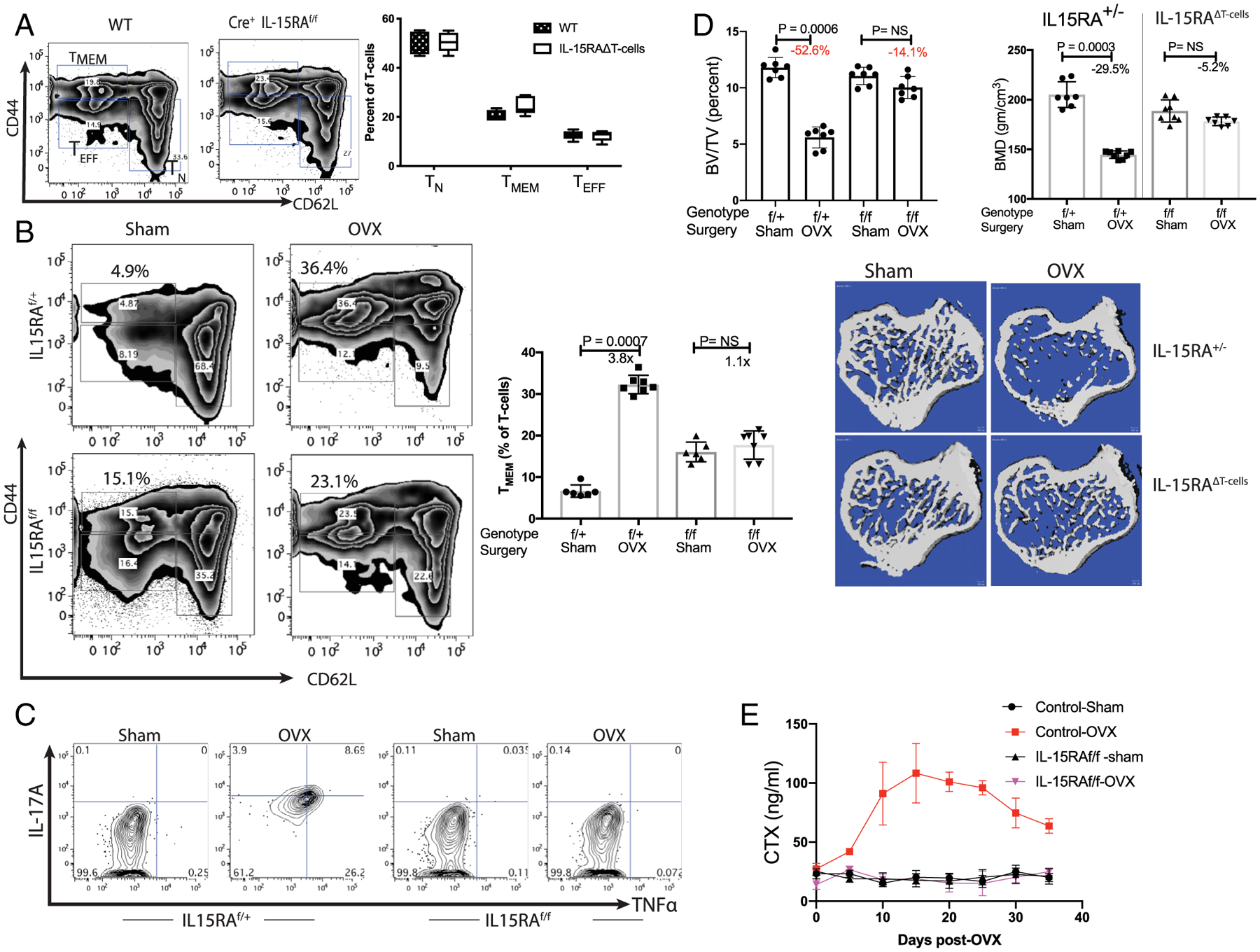
Ablation of IL-15 receptor α chain in T-cell prevents bone loss post-ovariectomy: IL15RAf/f mice were mated with Lck-Cre mice in two rounds of crosses to obtain Cre-positive IL15RAf/f, Cre+ IL15RAf/+ and Cre+ IL15RA+/+ littermates. The control (IL15RA sufficient) mice included both Cre+ homozygote (IL15RA+/+) and heterozygotes (IL15RAf/+) as they were phenotypically similar, if not identical, in assays used here (i.e. T-cell populations, IL-17A, TNFα induction and bone parameters). IL15RAf/f Cre+ mice are labeled as IL15RAΔT-cells in some panels. (A) No significant change in T-cell populations in IL15RAΔT-cells mice: Percent of T-cell subsets in the bone marrow of WT C57BL/6J and Cre+ IL15RAf/f in 10-week-old female mice were compared. No significant difference is observed in in bone marrow T-cells indicating that all T-cell subsets develop normally in the absence of IL15RA. Data representative of six mice/group is shown. (B) No change in TMEM subset is observed in IL15RA-deficient mice post-OVX: 12-week-old Cre-positive flox/flox (IL15RAf/f) or 1L15RA-sufficient mice were OVX or sham-operated. Mice were euthanized 3 weeks postsurgery and bone marrow T cells analyzed by flow-cytometry. Two-factor ANOVA (genotype and surgery) indicates that genotype (baseline values) account for 1.29% of the variance (p < .03) and surgery accounts for 55.6% of the variance (p < .0001); the interaction term accounts for 37.8% (p < .0001) indicating that OVX had a larger effect in WT but not in mutant mice. These results are consistent with the model in Fig. 3D, where an increase in T (CD45+ CD3+ CD44high CD62Llow) is observed in the control mice, the TMEM in IL15RAΔT-cells mice do not change. (C) T-cells with IL15RA deficiency do not express IL-17A and TNFα: While Cre+IL15RAf/+ mice express IL-17A and TNFα, these cytokines are not observed in OVX Cre+IL15RAf/f mice. (D) IL15RAΔT-cell mice maintain bone mass post-OVX: 12-week-old female Cre+IL15RAf/f and control liter-mates (Cre+IL15RAf/+) were sham-operated or OVX. Mice were sacrificed 3.5 weeks post-OVX and were evaluated by measuring bone-volume over total-volume (% BV/TV) and BMD. For BV/TV a two-factor ANOVA indicates that genotype contributed 13.15% (p < .0001) of the variance, surgery accounted for 50.3% (p < .0001) of the variance and the interaction term accounted for 25.9% (p < .0001). While control mice lost bone mass, mice with T-cell deficient in IL15RA maintained bone mass. Trabecular parameters by μCT of tibias is shown in Table 1. (E) Time course of CTX post-OVX in IL15RA sufficient and T-cell specific 1L15RA-deficient mice: Although control IL15RA-sufficient mice lost bone, no bone loss is observed in IL15RA-deficient mice, demonstrating the role of inflammation in promoting bone loss.
Magnetic bead–aided cell sorting
CD45-positive bone cells were isolated using positive selection; the flow through was considered to comprise CD45-negative cells. Anti-CD45 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany; Cat # 130-052-301) were used to isolate cells from single-cell suspension of BMCs. Cells were lysed and RNA was isolated (Agilent Technologies, Santa Clara, CA, USA).
Culturing of BMDC and memory T cells
After FAC sorting, both T-cells and BMDC were pelleted and resuspended in complete T-cell growth media (RPMI +10% heat-inactivated FBS + Penicillin + Streptomycin +2mM glutamine +1mM sodium pyruvate that was supplemented with non-essential amino acids and 55μM β-mercaptoethanol; all from Sigma-Aldrich, St. Louis, MO, USA) and cocultured for 2 to 5 days, as indicated in the figure legends.
Immunofluorescence staining
Tibias were fixed in 10% neutral buffered formalin for 24 hours. The bones were washed briefly with water, placed in 70% ethanol, and submitted to Washington University Musculoskeletal Histology and Morphometry Core. Tibias were decalcified for 2 weeks in EDTA and embedded in paraffin, then sectioned (5 μm), mounted, and stored. Prior to immunofluorescence (IF) staining, slides were baked at 55°C for 2 hours, washed with Xylene, and decreasing concentrations of ethanol (100% to 30%) in Coplin jars. Citrate buffer was used for antigen retrieval and 30% BSA in PBS was used for block. The following primary antibodies were used: rabbit anti-mouse IL-7 (Abcam, Cambridge, MA, USA; cat# ab9732); rat anti-mouse IL-15 (eBioscience, Santa Clara, CA, USA; cat# MAB477), and Armenian hamster anti-mouse CD11c (Abcam; cat# ab33483). Binding of primary antibody was visualized using fluor-conjugated secondary antibodies: goat anti-rabbit-AF594; donkey anti-Armenian hamster-AF555; and chicken anti-rat-AF488 (for triple labeling) or Texas Red (in Fig. 1D). The slides were imaged, and cell counts were obtained using Image J/Fiji(49) (NIH, Bethesda, MD, USA; https://imagej.nih.gov/ij/) by an operator blinded to treatments. No region of interest was selected, instead a total of 96 fields, four randomly selected fields (using 10× objective) from each slide, and eight mice/group in two groups (sham or OVX) were used for quantitation.
Fig. 1.
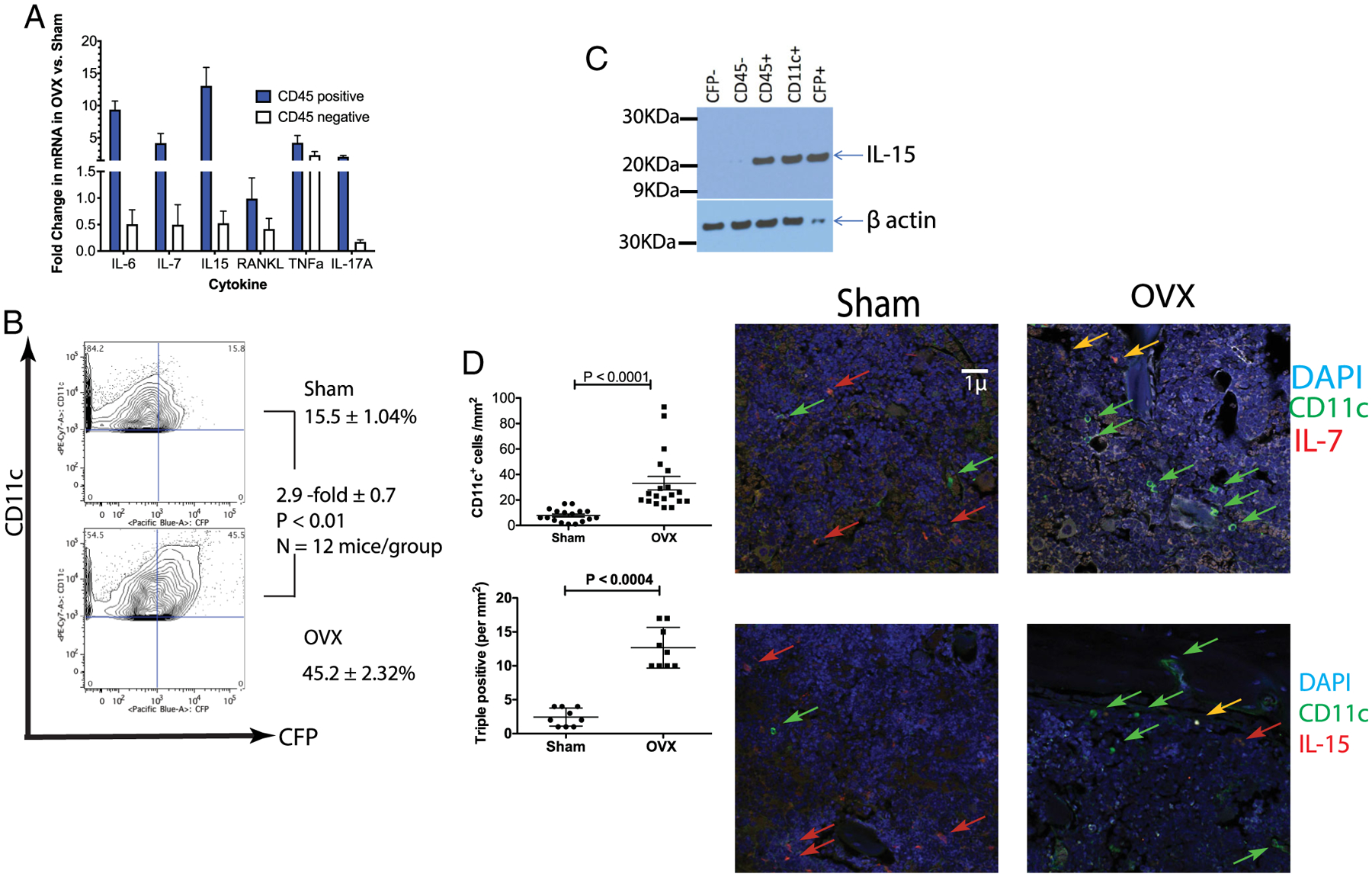
Ovariectomy increases IL-7 and IL-15 levels that are produced by bone marrow CD11c+ cells. (A) Increased IL-7 and IL-15 mRNA expression in OVX mice: BMCs from sham-operated and OVX mice were separated by magnetic beads into CD45-positive and CD45-negative populations using magnetic beads. RNA was isolated from each subset and expression of cytokines analyzed by qRT-PCR (SYBR green; ABI). The data is expressed as the fold difference between OVX and sham-surgery, after normalizing to GAPDH expression (data is from 5 mice/group with 3 technical replicates/mouse). (B) Identification of IL-7–expressing cells: BMCs from OVX or sham-operated from IL-7eCFP reporter mice were analyzed by flow cytometry. Although two cell populations were found to express CFP in the bone marrow, only the CD45+ CD11b + CD11c + population changed in ovariectomized mice. (C) IL-7–expressing cells co-express IL-15: expression of IL-15 was examined using Western blots in sorted BMC CD45 negative (lane 2), hematopoietic (CD45+), CD45+ CD11c+, CD45+CFP+, and CD45+CFP− (lane 1) cells. Representative Western blot of four experiments. (D) OVX leads to increased IL-7+ IL-15+ and CD11c+ cells: increased CD11c+ cells are observed post-OVX (top left panel) in bone sections, as well as triple-positive CD11c+ IL-7+ and IL-15+ (bottom-left) cells, but not all CD11c+ cells express IL-7 and IL-15 concurrently as shown in representative photomicrographs (right panels; yellow arrows are DAPI, CD11c, and cytokine positive). BMC = bone marrow cell; CFP = cyan-fluorescent protein; OVX = ovariectomized.
TUNEL assay
Formalin-fixed, paraffin-embedded bone sections were TUNEL stained (In situ cell death detection kit TMR red) per the manufacturer’s instructions (Roche Diagnostics, Mannheim, Germany). Quantitation was performed as described for IF.
Cytokines
Recombinant murine IL-7 and IL-15 (PeproTech, Rocky Hill, NJ, USA; cat # 217–17 and 210–15, respectively) were used at 20 and 50 ng/mL, respectively. 17-β estradiol (Sigma-Aldrich; cat # E8875) powder was dissolved in DMSO and used in culture assays. 17β-estradiol pellets (Innovative Research of America, Sarasota, FL, USA; 0.1 mg/pellet 21 day-release; cat #SE-121) or placebo pellets (Innovative Research of America; cat# C-111) were implanted 3 days post-OVX subcutaneously using a 10G trocar.
CTX assay
Mice were fasted overnight prior to blood collection. Blood (100 to 200 μL), obtained via submandibular vein, was allowed to clot for 1 hour at room temperature and serum was collected by spinning down the cell pellet (1000g for 10 min). Serum was flash frozen and stored at −80°C. Serum C-terminal telopeptide of type 1 collagen (serum CTX) was measured using ELISA according to the manufacturer’s instructions (Immunodiagnostic Systems, Gaithersburg, MD, USA; cat # AC-06F1)
Micro–CT data collection and analysis
Scanning settings and thresholding used for analysis were determined empirically to account for known age differences in bone microarchitecture. The bones, left proximal tibia, were scanned in μCT40 (Scanco Medical) at 55 kV, 72 μA, 4 W, at a resolution of 10 μm. Gauss sigma of 0.8, Gauss support of 1, lower threshold of 220 permilles for cancellous bone and 240 permilles for cortical bone, and upper threshold of 1000 permilles were used for all analyses. These thresholds correspond to 468.8, 592.2, and 3000 mg hydroxyapatite (HA)/cm3. Regions of interest were selected 50 slices below the growth plate of the proximal tibia to evaluate the trabecular compartment. Bone mineral density was obtained by quantitative micro–CT (μCT) using Phantoms for calibration.(50) All μCT data was collected and analyzed by LC, who was blinded to the treatment performed.
Statistical analysis
Statistical significance (p values) was assessed in all cases using unpaired two-tailed Mann-Whitney U test in GraphPad Prism8.2 (GraphPad Software, Inc., La Jolla, CA, USA) because no assumption of normal distribution is assumed. ANOVA was also performed in Prism 8.2.
Results
Increased IL-7 and IL-15 by DCs in the bone marrow of OVX mice
To characterize the bone marrow environment, we harvested bone marrow cells from sham-operated and OVX mice and sorted cells into CD45-positive (hematopoietic origin) and CD45-negative populations. We analyzed the cytokine expression in each cell subset by quantitative PCR (qPCR). As shown in Fig. 1A, the number of cytokines were found to be upregulated in OVX mice relative to sham-operated mice, including IL-6, IL-7, IL-15, RANKL, and TNFα. These cytokines were upregulated in the Cytokine Super Array (Qiagen, Valencia, CA, USA). With the exception of TNFα, all of the cytokines are differentially increased in CD45+ cells. Although these five cytokines have been previously observed to increase in bone marrow cells of OVX animals and in serum from postmenopausal women,(51–56) the regulation of these cytokines by ovarian hormones has not been established. Furthermore, IL-7 and IL-15 play an important role in T-cell homeostasis and in the formation and reactivation of TMEM responses. Data in Fig. 1A suggest that TMEM may play an important role following loss of ovarian function, so we focused our attention on the source of IL-7 and IL-15.
To identify the source of IL-7, IL-7eCFP reporter mice (cyanofluorescent protein [CFP])(48) were OVX (ovariectomized) or sham-operated and euthanized 10 days postsurgery. Bone marrow cells were examined by flow cytometry to assess the change in CFP-positive cells between mice with sham surgery and OVX. Consistent with the observations of Mazzucchelli and colleagues,(48) two populations of CFP-expressing cells were observed: one minor population that were CD45-negative (most likely stromal cells), and a second CD45+ CD11b+ CD11c+ population (Supplementary Fig. 1). Of the two CFP+ populations in the bone marrow, only the CD11c+ population increased post-OVX (Fig. 1B), confirming the qPCR data. The CFP-positive (ie, IL-7–expressing) cells also produce IL-15, as shown in Western blots (Fig. 1C). Because the CFP+ cells are CD11b+ and CD11c+ we refer to them here as bone marrow myeloid dendritic cells (BMDCs). Further characterization (Supplementary Fig. 1) of the CFP+ BMDCs showed that in addition to CD11b these BMDCs express IRF4 and IRF8 and both CD8 and CD4. Based on these markers, these myeloid DCs do not fit into the “classical pDC/DC1/DC2” paradigm.(57–59)
We confirmed the results of FACS analysis by immunofluorescence (IF) of bone sections from OVX and sham-operated mice. The IF results (Fig. 1D; Supplementary Fig. 2) show that the number of (CD11c+) DCs increased by 2.5-fold/mm2 in OVX mice, in agreement with FACS data (Fig. 1B). Also, consistent with the FACS (Fig. 1B) the IF data shows that 40 to 50% of DC express IL-7 or IL-15 (Fig. 1D; see also Supplementary Fig. 2 for representative images and Materials and Methods for details of quantification). The quantitation of triple-positive cells (CD11c, IL-7, and IL-15) in bone sections shows a 2.5-fold/mm2 increase in OVX relative to sham surgery, with one-half of the CD11c+ also IL-7–postivite and IL-15–positive (Fig. 1D). In summary, data in Fig. 1 show increased levels of DCs that express IL-7 and IL-15 post-OVX relative to sham-operated mice.
Estrogen regulates the lifespan of the DCs via inducing FasL
E2 regulates osteoclast lifespan by autocrine FasL-induced apoptosis.(15,16,60) As both osteoclasts and BMDC are derived from myeloid lineage, we hypothesized that increased BMDC numbers in the absence of E2 is due to their increased lifespan. To test this hypothesis, first we isolated BMDC and cultured them with vehicle (DMSO) or 10nM 17-β estradiol for 4 hours. We quantitated expression of FasL by PCR (qPCR) using BMDCs (Fig. 2A) and found increased expression of FasL, which led to increased apoptosis in the presence of E2 relative to vehicle (Fig. 2B). Increased apoptosis, as assessed by annexin V, was also observed with recombinant FasL, confirming that DCs express the receptor (Fas for FasL; Fig. 2B). Etoposide, a topoisomerase inhibitor that induces apoptosis by creating DNA breaks, was used as a positive control (Fig. 2B). Second, to test the hypothesis that E2 regulates the lifespan of the BMDCs in vivo, IL-7eCFP reporter mice were sham-operated or OVX. Three-weeks post-OVX, 17β-estradiol pellets were implanted subcutaneously in one-half of the OVX mice. Mice were euthanized 10 days post-pellet implantation and the number of CD11c+CFP+ cells assessed by flow cytometry of freshly isolated bone marrow cells in all three groups. CFP-expressing BMDCs increased in OVX (Fig. 2C), relative to sham-operated mice; additionally, E2 replacement restored the percent of CFP+CD11c+ cells to levels in sham-operated mice (Fig. 2C). FasL mRNA expression by qPCR in the three groups was also assessed in freshly isolated BMDCs. OVX mice had nearly undetectable levels of FasL, but significantly higher levels of FasL were detected in both sham-operated and 17β-estradiol–treated OVX mice. Together, these results show that increase in CFP+ BMDC and decrease in FasL expression was attributable to E2 loss (Fig. 2D).
Finally, to measure E2-induced apoptosis in vivo, we used TUNEL staining of histological sections of long bones collected from sham-operated and OVX mice. As shown in Fig. 2E, a number of TUNEL-positive foci (per mm2) are observed in sham-operated mice, including BMDCs (Fig. 2E lower panel). Examination of the micrographs indicates that cells proximal to DCs also stain TUNEL-positive (Fig. 2E; Supplementary Fig. 3), suggesting that either E2 induces apoptosis in cells other than DCs or FasL produced by DCs induces paracrine apoptosis. In contrast, very few foci are observed in OVX mice (Fig. 2E center panel). Furthermore, no TUNEL staining is observed in OVX mice in the proximity of the BMDCs. In summary, these experiments show that in estrogen-replete animals, DC lifespan is shortened by induction of FasL. Next, we examined the effect of IL-7 and/or IL-15, as well as BMDCs in the presence or absence of E2, on bone marrow T cells.
Together IL-7 and IL-15 induced IL-17A and TNFα in a subset of bone marrow T cells
Previous reports have shown that IL-7 and IL-15 together, but not individually, lead to the proliferation of human TMEM.(61,62) Here, to assess cytokine production, bone marrow CD4 and CD8 were cultured with IL-7, IL-15, or both for 48 hours. TNFα and IL-17A are found in abundance in postmenopausal women with osteoporosis, and TNFα and IL17A null mice do lose bone post-OVX.(29,63,64) Therefore, these two cytokines were assessed in the T cells. The data show that TMEM (CD44high CD62Lint) not only proliferate (as assessed by Ki-67) but also produce TNFα and IL-17A when treated with both IL-7 and IL-15 for 48 hours, but not when treated with each cytokine individually (Fig. 3A; Supplementary Fig. 4). No IFN-γ or IL-4 expression was detected in the TMEM (not shown). Although prior studies have shown that these cytokines lead to proliferation of TMEM,(61,62) the induction of proinflammatory cytokines has not been previously shown and was unexpected. To assess whether IL-7 and IL-15 produced by BMDCs also activate TMEM to induce TNFα and IL-17A, we cocultured the two cell populations in the presence and absence of 10nM 17β-estradiol for 48 hours. The DCs and TMEM were isolated by FACS: BMDC were sorted using CD45, CD11c and CFP fluorescence and thus are the only source of IL-7 and IL-15 in the well; the T cells were isolated using CD45, CD44 and CD62L signals. Use of anti-CD3 was avoided because CD3 crosslinking can activate T cells.(65) In the absence of E2, a fourfold expansion of TMEM and a twofold decrease in effector T cells (Fig. 3B) was observed in cocultures with BMDC that was not observed in the presence of E2. The increase in TMEM is consistent with the effects of IL-7 and IL-15 produced by the BMDCs (Fig. 1B,C). We also observed induction of TNFα, IL-17A, or both in TMEM cocultured with BMDCs in the absence of E2 but not in the presence of E2 (Fig. 3B lower panel). Although the effect of BMDCs in inducing TNFα or IL-17A (Fig. 3B) reproduce the results with recombinant IL-7 and IL-15 (Fig. 3A), to determine whether IL-7 and IL-15 from the BMDCs was responsible for this effect we titrated several anti-mouse IL-7 or IL-15 antibodies to neutralize these cytokines. At low (<25 μg/mL) concentrations we observed no effect. At higher concentrations of either IL-7 or IL-15 antibody (≥50 μg/mL), with two different antibody clones for each cytokine, we observed reduced numbers of BMDCs, suggesting that these cytokines may also have an autocrine or paracrine function for the BMDC.
Fig. 3.
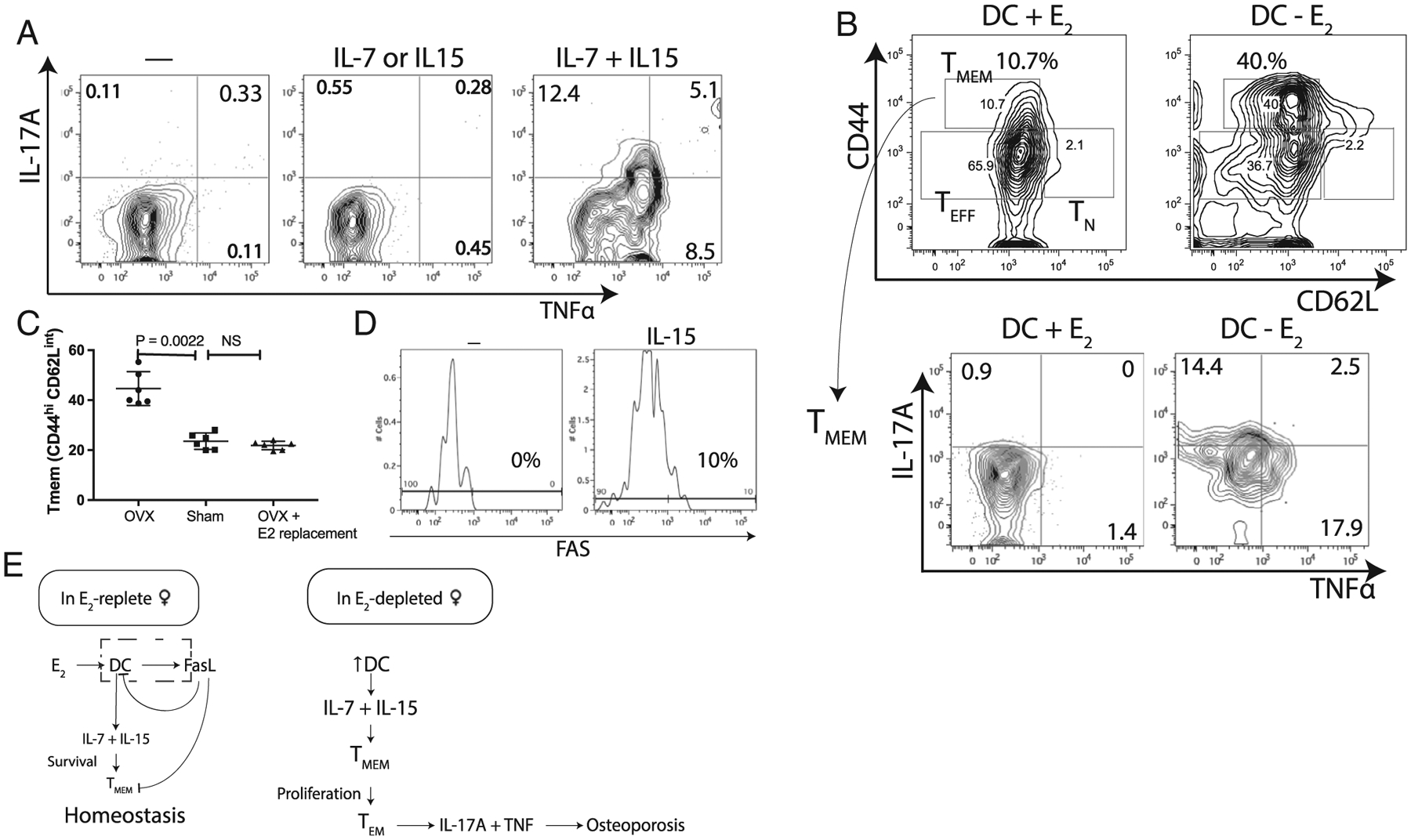
IL-7 and IL-15 together lead to induction of IL-17A, TNFα or both in memory T-cells. (A) Cytokine-dependent conversion of TMEM to TEM: culturing memory T-cells (TMEM: CD3 CD44high CD62Llow) with IL-7 and IL-15 together induces TNFα and IL-17 in the absence of antigen stimulation. Representative flow contour plot of six experiments is shown. (B) Co-culturing TMEM with BMDCs induced TNFα and IL-17A in TMEM: culturing T with CFP+ BMDC in the absence of E2 results in a fourfold increase in TMEM and twofold decrease in TEFF levels relative to DC in the presence of 10nM E2; BMDC induced IL-17A and TNFα in TMEM in the absence of E2 mimicking OVX. Representative flow contour plot from six experiments is shown. (C) E2 replacement in OVX mice restores TMEM levels to that of sham-operated mice. Data from six mice/group. (D) IL-15 induces FAS in TMEM: culturing BM-TMEM with IL-15 induced FAS in this subset. (E) A model summarizing the data from Fig. 1 through Fig. 3D.
To verify the role of E2 on TMEM in vivo we compared the TMEM population in sham, OVX, and OVX mice with 17β-estradiol replacement. The TMEM population increased in OVX mice relative to sham-operated mice and 17β-estradiol replacement restored TMEM to sham-surgery levels (Fig. 3C). Based on the observation that BMDCs express FasL in the presence of E2, we checked if T-cell subsets express Fas (CD95). Culturing bone marrow T cells with IL-15 (Fig. 3D) but not IL-7 (not shown), induced Fas in TMEM (Fig. 3D). These results indicate that in E2-replete females, BMDCs regulate memory T cells via IL-7 and IL-15, but also by inducing Fas via IL-15 to prevent expansion of TMEM. In the absence of E2, BMDCs use IL-7 and IL-15 to activate TMEM to proliferate and secrete proinflammatory cytokines.
Summarizing, our data show IL-7 and IL-15 together lead to production of IL-17A, TNFα, or both in TMEM. Recombinant IL-7 and IL-15 activated a subset of TMEM, leading to expression of TNFα, IL-17A, or both. The induction TNFα and IL-17A was recapitulated using CFP+BMDCs (ie, IL-7–expressing and IL-15–expressing DCs, as shown in Fig. 1B,C) and TMEM cocultures (Fig. 3B). E2 induces FasL and apoptosis in the BMDCs (Fig. 2A,D), leading to fewer BMDCs, low levels of IL-7 + IL-15, and low-level proliferation of TMEM; IL-15–induced FAS in TMEM thus maintains TMEM homeostasis (Fig. 3D). In the absence of E2, BMDCs have longer lifespans because they do not produce FasL, leading to higher levels of IL-7 and IL-15, which induces TNFα, IL-17A, or both in a subset of TMEM. We hypothesized that these proinflammatory cytokines contribute to bone loss (Fig. 3E).
Mice with a T-cell–specific deletion of IL15RA fail to activate TMEM post-OVX
To test our hypothesis and validate the model shown in Fig. 3E in vivo we developed a new mouse model. As our results showed that IL-7 and IL-15 produced by DCs (Fig. 1), whose abundance is controlled by E2 (Fig. 2), are primarily responsible for activation of TMEM in the bone marrow (Fig. 3), one possible approach was to delete either of these cytokines. IL-7 is required for T cell development, and for generation of TMEM,(66) deleting IL-7 would lead to depletion of T cells and TMEM. We then considered deleting IL-15 because the role of IL-15 in reactivating TMEM in the context of menopause has not been defined. A global knockout or antibody-mediated depletion of IL-15 was rejected because this cytokine plays an important role in NK cell homeostasis that would make interpretation of the results questionable and because anti-IL-15 antibodies have been shown to produce adverse neurological effects.(67–69) Further, IL-15 has been shown to directly regulate osteoclastogenesis(70) and mineralization by osteoblasts.(71) Because we do not know the developmental lineage of the CFP+DC cell-specific deletion, IL-15-blockade was deemed to not be a feasible approach. Therefore, we chose to delete the IL-15 receptor-α (IL15RA) gene in T cells by crossing Lck-Cre with IL15RA-floxed (IL15RAf/f) mice. Lck, a kinase, is constitutively expressed primarily in CD4 and CD8 T cells.(47) High expression of Lck-Cre has been reported to have off-target effects(72); therefore, we used a mouse strain with low-expression of Cre (see Materials and Methods). We first compared levels of bone marrow T cells between C57BL/6J (WT) and mice with T-cell–specific deletion of the IL15RA gene (IL15RAΔT-cells) to assess changes due to IL15RA deletion. Our results show that in 8-week-old mice, there are modestly higher levels of memory (TMEM), but no significant change in naïve (TN) or effector (TEFF) populations of T cells in the bone marrow (Fig. 4A). The moderate increase in TMEM in these E2-replete mice may be attributed to the observation that IL-15 induces Fas in TMEM (Fig. 3D). The absence of the IL-15 receptor leads most likely to low levels of Fas expression in these cells and allowing the cells to live longer even with FasL expression in E2-replete mice.
Next, to determine the effect of E2 deficiency on the immune and skeletal systems, 12-week-old female IL15RAf/f Cre+ or control IL-15+/− (Cre+ IL15RAf/+) littermates were OVX or sham-operated. A significant increase in TMEM was observed in (control) IL15RAf/+ mice, but not in IL15RAΔT-cells (Fig. 4B). The levels of TN decrease in both the IL15RAf/f and control mice to similar levels (Fig. 4B). We also observed TNFα and IL-17A expression in control littermates but not in mice with IL15RA-deficient T cells (Fig. 4C). Consistent with expression of IL-17A and TNFα in the control littermates, these mice also lost bone volume and trabecular bone and had decreased bone mineral density as assessed by μCT measurements of the tibias in the four groups (Fig. 4D; Table 1). While the number of osteoclasts increased in control mice, a modest increase in osteoclasts was observed in OVX IL15RAf/f mice post-OVX (Table 1). We also noted a slight increase in osteoclasts at baseline between the control and IL15RAf/f mice. We examined bone formation in both control and IL15RAΔT-cells and, as expected, the control mice had increased bone formation but no change in mineral apposition rate (MAR) or bone formation rate per unit of bone surface (BFR/BS) was observed in the IL15RAf/f mice (Table 1). Finally, we studied the kinetics of bone resorption post-OVX in both groups of mice. Our results show (Fig. 4E) that no increase in CTX is observed in the absence of TNFα and IL-17A in the IL15RAf/f Cre+ mice. These results also confirm that IL-7 and IL-15 together are needed to induce TNFα and IL-17A in T cells, which then promote bone loss.
Table 1.
Summary of Trabecular and Histomorphometry Data in IL15RAΔT-cells and Control Mice
| Control (IL15RAf/+ Cre+) | IL15RAΔT-cells(IL15RAf/f Cre+) | ||||
|---|---|---|---|---|---|
| Experiment | Parameter | Sham (n = 10) | OVX (n = 10) | Sham1 (n = 10) | OVX (n = 12) |
| Cancellous μCT (proximal tibia) | Tb.N | 4.338 ± 0.1194 | 3.561 ± 0.3040*** | 4.489 ± 0.2939 NS | 4.298 ± 0.1022 NS |
| Tb.Th (mm) | 0.04895 ± 0.000169 | 0.04896 ± 0.000119 NS | 0.04930 | 0.04595 | |
| 0.0002268 NS | 0.004272 NS | ||||
| Tb.Sp (mm) | 0.2521 ± 0.004573 | 0.2423 ± 0.005556** | 0.2646 ± 0.003119*** | 0.2615 ± 0.004100 NS | |
| Conn.D (mm−3) | 17.53 ± 0.2804 | 13.30 ± 0.5949*** | 18.44 ± 1.113 NS | 17.85 ± 0.4840 NS | |
| Histomorphometry (proximal tibia) | N.Oc/BS (mm−1) | 3.335 ± 1.153 | 4.360 ± 0.4840** | 3.568 ± 1.025 NS | 3.955 ± 0.3771 NS |
| Oc.S/BS (%) | 11.80 ± 0.6541 | 24.84 ± 0.9230*** | 12.28 ± 0.7114 NS | 14.22 ± 0.7162*** | |
| Dynamic histomorphometry (proximal tibia) | MAR (μm/day) | 1.60 ± 0.2418 | 2.092 ± 0.3687** | 1.628 ± 0.3005 NS | 1.5890 ± 0.2630 NS |
| BFR/BS (μm3/μm2/day) | 0.546 ± 0.112 | 0.773 ± 0.0540**** | 0.525 ± 0.077 NS | 0.565 ± 0.091 NS | |
Mean ± SD is shown for all parameters for each genotype. All measurements are from 3 weeks post-OVX. Unpaired two-tailed Mann-Whitney p values were computed.
Conn.D = connectivity density; NS = nonsignificant; Tb.N = trabecular number; Tb.Sp = trabecular spacing; Tb.Th = trabecular thickness.
Values of p were computed using values between control and IL15RAf/f Cre+ to assess the effect of genotype.
p < .01.
p < 1 × 10−3.
< 1E-4.
Summarizing, the results show that TMEM develop normally (at baseline) in absence of the IL-15 receptor. However, no TNFα and IL-17 is induced by OVX in the TMEM, relative to littermate controls that express the IL-15 receptor. Furthermore, in the absence of TNFα, IL-17A, or both, we observed no bone resorption, decrease in bone mass, change in trabecular parameters, or increase in osteoclast numbers (Fig. 4D,E; Table 1). These studies confirm or validate the model proposed in Fig. 3E and show that inflammation is needed for bone loss leading to osteoporosis.
Discussion
Involutional osteoporosis is mediated by E2 loss at menopause. Osteoporosis leads to effete bone that is prone to fracture with minimal trauma. Although the effect of E2 was recognized decades ago, a number of causes of osteoporosis have been proposed. Early studies by Avioli and others suggested that aging and metabolic changes associated with menopause lead to osteoporosis.(73–76) Later, molecular studies suggested the direct effect of E2 on maintaining balance in the cells of BRUs.(20,60,77,78) The prevailing view has been that E2 loss leads to uncoupled increase in bone resorption by osteoclasts and deficit in osteoblasts.(79,80) Most strategies to treat osteoporosis have focused on rebalancing the activities of cells in the BRU.
Elegant studies from Pacifici, Weitzmann, Takayanagi, and colleagues have highlighted the role of proinflammatory cytokines produced by T cells as a cause for promoting bone loss. These studies laid the foundations of osteoimmunology.(26,29,81–84) A number of studies have shown both in humans and mice, that loss of E2 leads to T-cell–dependent inflammation, but the underlying mechanism is undetermined. Here, we have identified for the first time a definitive mechanism by which E2 suppresses the activation of T cells. Conversely, we also show how loss of E2 leads to activation of TMEM, unexpectedly converting them to TNFα-secreting and IL-17–secreting TMEM that lead to bone loss. The activation of TMEM post-OVX is mediated by IL-7 and IL-15 produced by increased numbers of bone marrow DCs (Fig. 1). To the best of our knowledge, no previous study has identified the critical role of BMDCs in induction of TNFα, IL-17A, or both by T cells in the context of postmenopausal osteoporosis. Although a previous study showed that OVX causes the maturation of BMDCs (CD11c) to express costimulatory signal CD80,(40) the authors did not demonstrate that the expression of proinflammatory cytokines in the T cells was due to antigen-dependent activation by BMDCs. The steady state levels of BMDCs is maintained by E2 by inducing apoptosis via FasL in the DCs (Fig. 2). With declining E2 levels postmenopause, the levels of FasL also decline, leading to increased lifespan and levels of DCs and hence to higher levels of IL-7 and IL-15 (Figs. 1 and 2).
DCs are a heterogenous population of professional antigen-presenting cells that arise from different hematopoietic lineages.(85,86) The DCs that produce IL-7 and IL-15 in the bone marrow are closest in lineage to plasmacytoid DCs (pDCs) because these express the transcription factor IRF4 and IRF8, but they also express CD11b and CD11c, which are typically expressed by myeloid DCs. In addition, these DCs expressed both CD4 and CD8 markers, which is unusual for tissue-resident DCs (Supplementary Fig. 1). Because these DCs express novel marker combinations that have not been previously described, additional studies are needed because they are important for maintaining TMEM that reside in the bone marrow and because the role of this DC population in inducing proinflammatory T cells in postmenopausal osteoporosis has not been previously described.
IL-7 is required for T cell homeostasis, because loss of functional IL-7 leads to thymic hypoplasia, decreased numbers of B cells and T cells.(87) Importantly, IL-7 is needed for development of T cell memory.(88,89) Similarly, IL-15 is also important for the development of NK cells and memory CD8 T cells.(90) Resting memory CD4 and CD8 T cells are dependent on signals from IL-7–producing and IL-15–producing cells for their survival and intermittent homeostatic proliferation.(91) For this reason, IL-7 and IL-15 are considered to be redundant in their functions. Furthermore, Geginat and colleagues(61,92) have shown that IL-7 and IL-15 together, but not alone, lead to antigen-independent proliferation of human TMEM. Therefore, increased levels of IL-7 and IL-15 in OVX mice, which lead to TMEM proliferation, could be anticipated. Indeed, we observed 12-fold expansion due to proliferation (assayed by Ki-67 staining) of TMEM in culture in presence of IL-7 and IL-15 (Supplementary Fig. 4). However, we were not successful in measuring proliferation in vivo by Ki-67 or BrdU pulsing (at 1 and 3 days postsurgery). The lack of success may be due to several factors: first, it is possible that the TMEM proliferated very rapidly (<24 hours) after surgery, prior to pulsing with BrdU or isolation of BMC for Ki-67 staining. We selected days 1 and 3 post-OVX because we observed increased serum CTX 5 days post-OVX (Fig. 4E). Alternatively, the levels of E2 post-OVX decrease progressively over days, leading to buildup of IL-7–producing and IL-15–producing DCs over this time. This is the only population of IL-7–producing and IL-15–producing cells that reacted to E2 loss and replacement (Figs. 1B and 2C). The buildup of IL-7 and IL-15 over days leads to induction and accumulation of TNFα-expressing and IL-17A–expressing TMEM. If initial levels of TMEM that respond to IL-7 and IL-15 represent a small subset that divide nonsynchronously over days, BrdU or Ki-67 will not be sufficiently sensitive to detect proliferation. To increase sensitivity additional tools are needed to identify the IL-7 and IL-15 responsive subset of TMEM. Nonetheless, we observed a 3.8-fold expansion of TMEM in vivo (Fig. 4B) post-OVX over 3 weeks. Importantly and not previously shown, we found that together these two cytokines also induce IL-17A, TNFα, or both, in a subset of TMEM (Fig. 3A; see also Supplementary Fig. 4). Because these TNFα+ or IL-17A+ cells do not alter the expression of CD44 and CD62L (Fig. 3B), we refer to them as effector memory T cells (TEM). IL-15 has also been shown to directly regulate osteoclasts and osteoblasts, either alone or in combination with RANKL.(70,71,93–96) Together with the CTX time course (Fig. 4E), our results are most consistent with the notion that the acute (early) phase of bone loss stems from the accumulation of BMDCs, IL-7 and IL15 expressing, and TNFα and IL-17–expressing TMEM over ~3 weeks of time. In support of our data, polymorphisms in IL-15 are associated with bone mineral density in postmenopausal women.(97) Additionally, Djaafar and colleagues(70) have shown that IL-15−/− mice have a high bone mass; furthermore, OVX of the IL-15 null mice did not increase osteoclast numbers in distal femur relative to control mice. In our study, IL-15 also induced the Fas receptor in the TMEM, so in the presence of E2, which induces FasL, these T cells undergo apoptosis (Fig. 3C). Notably, both DCs and proliferating TMEM would undergo apoptosis consistent with the foci of the TUNEL stain (Fig. 2E). The dying cells display annexin V (Fig. 2B), an “eat-me” signal that would allow safe removal of apoptotic cells by phagocytes.(98) We note that although the current paradigm of TMEM activation requires antigen exposure, E2 loss leads to a cytokine-dependent but antigen-independent activation of TMEM by exposure to high levels of IL-7 and IL-15.
Due to the pleiotropic nature of IL-15, the effect on NK cells, on osteoclasts, endothelial cells, epithelial cells, and myocytes,(99) to assess the role of IL-15 in TEM activation in vivo, we ablated IL-15Rα specifically in T cells by crossing mice engineered with loxP sites flanking exons 2 and 3 of the IL15RA gene with Lck-Cre (a pan αβ T-cell expressed gene). Fortunately, no effect on the development of TMEM was observed in E2-replete Cre+IL15RAf/f female mice (Fig. 4A). However, although an increase in TMEM levels was observed in control (IL15RAf/+Cre+) mice post-OVX, no such increase in TMEM was observed in IL15RAΔT-cells mice, confirming that TMEM expansion is dependent on IL-15 (with IL-7; Fig. 4B). Consistent with lack of expansion, no TNFα or IL-17A expression was observed in TMEM (Fig. 4C) in the IL15RAΔT-cells. In summary, we show that OVX leads to the expansion of BMDC which produce IL-7 and IL-15 (we note that only CFP+ BMDC express IL-15; Fig. 1) because E2 directly induces apoptosis of the CFP+ BMDC in vivo (Fig. 2). We show that coculturing CFP+ BMDC with BM T cells leads to the expansion of TMEM and because the CFP+BMDCs are the only source of IL-7 and IL-15, this result confirms that these cytokines also induce TNFα, IL-17, or both in the TMEM in absence of E2 (Fig. 3B). We observed similar expansion of BM TMEM (Fig. 4B) and induction of TNFα, IL-17A, or both in TMEM post-OVX (Fig. 4B,C). One limitation of the current study is that we cannot directly show in vivo that IL-7 and IL-15 produced by the BMDCs are the cause of TNFα and IL-17A induction in TMEM. As discussed, blockade of IL-7 and IL-15 using antibodies is not feasible and genetic ablation of these cytokines will require identification of cell-specific promoters for Cre expression. Nonetheless, the increase in TMEM post-OVX is consistent with the increase in IL-7 and IL-15 and expression of CD127 (IL-7 receptor) on the TMEM.(100,101) Our data with T-cell–specific deletion of the IL-15Rα gene (IL15RAΔT-cell) shows that IL-15 is required for expansion and induction of TNFα, IL-17A by TMEM.
Because IL15RAΔT-cell mice do not express the proinflammatory cytokines post-OVX, this mouse model allows us to assess the contribution of inflammation and the direct effect of E2 loss on the BRU and on bone loss. Our results show that although OVX of control mice leads to induction of TNFα and IL-17 produced by TEMs and to significant trabecular bone loss by activating bone resorption, the IL15RAΔT-cell mice did not lose bone volume and no increase in serum CTX or loss of trabecular bone was observed (Fig. 4; Table 1). We observed a modest but statistically significant increase in osteoclast surface/bone surface (Oc.S/BS) (a modest increase in number of osteoclasts/bone surface [N.Oc/BS] is also observed but did not achieve statistical significance) post-OVX showing that E2 loss does lead to increased osteoclasts, even in the absence of inflammation, but interestingly this increase does not translate to a significant decrease in bone mass (Fig. 4D) or bone resorption as measured by CTX (Fig. 4E) in the absence of TNFα and IL-17A. Further, we note based on our previous experience, that exposure to IL-7 and IL-15 activation of TMEM leads to low levels of proinflammatory cytokines, relative to infection (antigen-dependent activation). Both IL-17A and TNFα promote osteoclastogenesis and increased bone resorption.(27) Neutralization of IL-17A(102) and TNFα(103) have been shown to decrease osteoporosis in response to OVX in mice. Additionally, no bone loss is observed in OVX in TNFα-deficient mice,(104) or in transgenic mice expressing soluble TNF receptor.(105) In humans, polymorphisms that effect TNFα(106–108) or IL-17A(109) function are also modulators of osteoporosis. Therefore, our results place inflammatory signals upstream or epistatic to the direct effect of E2 on the BRU via FasL and/or other mechanism. Taken together, our results show that production of these proinflammatory cytokines is required for acute phase bone loss leading to osteoporosis.
The finding that inflammation is due to activation of TMEM may provide a population-level explanation. Because TMEM encode the lifetime of exposure to commensal and pathogenic microbes, the levels of TMEM that can be induced to produce TNFα and IL-17A are likely to vary in a population. Nearly one-half of postmenopausal women develop osteoporosis, but the remaining one-half do not. Based on current knowledge, T cells that produce TNFα, IL-17A, or both in the initial activation by antigen and that develop into the memory repertoire are reactivated by IL-7 + IL-15 to produce TNFα, IL-17A, or both.(110–112) The levels of TMEM in individuals in a population that have the potential to produce TNFα and IL-17A will vary because each individual’s exposure to these microbes vary. We hypothesize that postmenopausal women having healthy bone mass have low levels of TMEM capable of producing TNFα and IL-17A or both. In contrast, there are more TNFα, IL-17A–producing TMEM in postmenopausal women with osteoporosis. Thus, our pathway (Fig. 3E) may explain a population-level phenomenon. In this context, we note that many studies have implicated the gut microbiome as a factor in maintaining bone health in postmenopausal women.(113,114) Our studies implicate the role of T cells in the crosstalk between the immune system maintaining the gut microbiota and the gut microbiota affecting the immune system.(115,116) The gut microbiota is likely to influence bone marrow–resident TMEM populations.(112,117) In mice, recent studies have shown that segmented filamentous bacteria (SFB) increase TH17 response in the gut and that this response protects against infection by pathogenic Citrobacter rodentium.(118) Although, it is not clear if SFB colonization leads to TMEM development, OVX of SFB-colonized mice may produce increased bone loss relative to mice lacking SFB. Additional studies are needed to test this hypothesis. We have tested for, and find no evidence for, presence of SFB in our colony of mice (originally purchased from The Jackson Laboratory, Bar Harbor, ME, USA).(118)
Although aspects of these findings such as the increase in IL-7,(82) IL-15,(51,97) and IL-17A(30,119) have been noted to increase post-OVX in mice and in postmenopausal women with osteoporosis, our study connects the cellular and signaling components into a coherent description. The role of DCs and FasL have also been previously noted.(120) However, the connection between these individual cells and molecules into a pathway that is activated at menopause has not been described previously. The model described in Fig. 3E is entirely consistent with all of the previous studies including genetic engineering, cellular, epidemiological, and population genetic studies in both rodents and humans. A number of features are novel, including the antigen-independent activation of TMEM, the role of IL-7 and IL-15–producing DCs, and the requirement for T-cell–dependent inflammation for bone loss.
The physiological (and evolutionary) basis for activation of this low-grade inflammation associated with loss of E2 may be reconciled by considering the link between pregnancy, immune system, and bone as proposed by Pacifici.(81) During pregnancy calcium is stored in the bones because of increased weight and high levels of placenta-produced E2, which promotes osteoblast activity while blocking osteoclast activity. High E2 levels during pregnancy also decreases the immune response to protect the fetus, because it is partially self. E2 levels drop when the placenta is delivered, leading to an acute increase in osteoclast numbers, bone resorption, and to release of stored calcium in bone. The calcium is supplied to the rapidly growing skeleton of the neonate via lactation. In contrast to postpartum, where these events are limited in duration (months in humans and weeks in mice), in postmenopausal women inflammation persists (for decades), because the ovaries restart E2 production postlactation but not postmenopause. Unpublished data show that the same panel of cytokines shown in Fig. 1A are upregulated 2 days postpartum, relative to age-matched nulliparous females in the bone marrow.
Nearly one-half of postmenopausal women develop osteoporosis, but menopause also increases incidence of other comorbidities including coronary artery disease, certain cancers, chronic obstructive pulmonary disease, stroke, and dementia.(121–124) It is tempting to speculate that the proinflammatory T cells produced by E2 loss also contribute to the pathogenesis of these conditions because all these conditions are known to be caused by both chronic inflammation and metabolic syndrome.(125–129) In conclusion, our studies establish a new regulatory circuit between E2, the immune system, and the skeletal system. Our data shows IL-7 and IL-15 together activate TMEM to induce TNFα, IL-17A, or both (referred to as T-effector memory cells or TEM for short). In mice that have a T-cell–specific deletion of IL-15 receptor, TMEM do not produce TNFα and IL-17A and no bone loss is observed. These results show that the uncoupled activity of the BRU in osteoporosis is due to T-cell–dependent inflammation and not due to the direct effect of E2 on the BRU. Because bone is a mechanosensing organ, a store for calcium and phosphate, and the bone marrow a site for hematopoiesis and a nest for immune memory cells, the BRU is regulated by a number of different feedback loops. Knowledge of this pathway will, most likely, lead to additional discoveries on how bone homeostasis is maintained and how these feedback loops may intersect in health and pathological states.
Supplementary Material
Acknowledgments
We dedicate this work to Prof. Roberto Pacifici for his longstanding contributions to osteoimmunology and mechanisms that promote involutional osteoporosis. This work was partially supported by the NIH National Institute of Arthritis and Musculoskeletal and Skin Disease of the NIH under Award Number AR064821 and AR068438; the Washington University Musculoskeletal Research Core (AR057235) also partially supported this study. We acknowledge Sheri Koehm and Joy Eslick in the flow cytometry core facility at St. Louis University School of Medicine. Crystal Idleburg (Washington University in St. Louis Musculoskeletal Histology core) is acknowledged for her support preparing slides and bone sections for histomorphometry and immunofluorescence. RA thanks Sara McBride-Gagyi for helpful discussions through the course of these studies and also help from Mariah Hastert is acknowledged in the early stages of these studies.
Footnotes
Additional Supporting Information may be found in the online version of this article.
Disclosures
The authors confirm they have no conflicts of interest.
References
- 1.Bolland MJ, Grey AB, Gamble GD, Reid IR. Effect of osteoporosis treatment on mortality: a meta-analysis. J Clin Endocrinol Metab. 2010;95(3):1174–81. [DOI] [PubMed] [Google Scholar]
- 2.Panula J, Pihlajamaki H, Mattila VM, et al. Mortality and cause of death in hip fracture patients aged 65 or older: a population-based study. BMC Musculoskelet Disord. 2011;12:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Albright F, Smith PH, Richardson AM. Postmenopausal osteoporosis: its clinical feature. JAMA. 1941;116(22):2465–74. [Google Scholar]
- 4.Reifenstein EC Jr, Albright F. The metabolic effects of steroid hormones in osteoporosis. J Clin Invest. 1947;26(1):24–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujita T Calcium intake, calcium absorption, and osteoporosis. Calcif Tissue Int. 1996;58(4):215. [DOI] [PubMed] [Google Scholar]
- 6.Avioli LV, McDonald JE, Lee SW. The influence of age on the intestinal absorption of 47-Ca absorption in post-menopausal osteoporosis. J Clin Invest. 1965;44(12):1960–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ledger GA, Burritt MF, Kao PC, O’Fallon WM, Riggs BL, Khosla S. Role of parathyroid hormone in mediating nocturnal and age-related increases in bone resorption. J Clin Endocrinol Metab. 1995;80(11):3304–10. [DOI] [PubMed] [Google Scholar]
- 8.Eastell R, Yergey AL, Vieira NE, Cedel SL, Kumar R, Riggs BL. Interrelationship among vitamin D metabolism, true calcium absorption, parathyroid function, and age in women: evidence of an age-related intestinal resistance to 1,25-dihydroxyvitamin D action. J Bone Miner Res. 1991;6(2):125–32. [DOI] [PubMed] [Google Scholar]
- 9.Francis RM, Peacock M, Taylor GA, Storer JH, Nordin BE. Calcium malabsorption in elderly women with vertebral fractures: evidence for resistance to the action of vitamin D metabolites on the bowel. Clin Sci (Lond). 1984;66(1):103–7. [DOI] [PubMed] [Google Scholar]
- 10.Martin RB. Targeted bone remodeling involves BMU steering as well as activation. Bone. 2007;40(6):1574–80. [DOI] [PubMed] [Google Scholar]
- 11.Slemenda C, Longcope C, Peacock M, Hui S, Johnston CC. A prospective study of pre-, peri-, and postmenopausal women. J Clin Invest. 1996;97(1):14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garnero P, Sornay-Rendu E, Chapuy MC, Delmas PD. Increased bone turnover in late postmenopausal women is a major determinant of osteoporosis. J Bone Miner Res. 1996;11(3):337–49. [DOI] [PubMed] [Google Scholar]
- 13.Riggs BL, Melton LJ 3rd. Involutional osteoporosis. N Engl J Med. 1986;314(26):1676–86. [DOI] [PubMed] [Google Scholar]
- 14.Vanderschueren D, Vandenput L, Boonen S, Lindberg MK, Bouillon R, Ohlsson C. Androgens and bone. Endocr Rev. 2004;25(3):389–425. [DOI] [PubMed] [Google Scholar]
- 15.Krum SA, Miranda-Carboni GA, Hauschka PV, et al. Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J. 2008;27(3):535–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura T, Imai Y, Matsumoto T, et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130(5):811–23. [DOI] [PubMed] [Google Scholar]
- 17.Kovacic N, Grcevic D, Katavic V, et al. Fas receptor is required for estrogen deficiency-induced bone loss in mice. Lab Invest. 2010;90(3):402–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawakami A, Eguchi K, Matsuoka N, et al. Fas and Fas ligand interaction is necessary for human osteoblast apoptosis. J Bone Miner Res. 1997;12(10):1637–46. [DOI] [PubMed] [Google Scholar]
- 19.Bradford PG, Gerace KV, Roland RL, Chrzan BG. Estrogen regulation of apoptosis in osteoblasts. Physiol Behav. 2010;99(2):181–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jilka RL, Takahashi K, Munshi M, Williams DC, Roberson PK, Manolagas SC. Loss of estrogen upregulates osteoblastogenesis in the murine bone marrow. Evidence for autonomy from factors released during bone resorption. J Clin Invest. 1998;101(9):1942–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83(9):1032–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diedhiou D, Cuny T, Sarr A, Norou Diop S, Klein M, Weryha G. Efficacy and safety of denosumab for the treatment of osteoporosis: a systematic review. Ann Endocrinol (Paris). 2015;76(6):650–7. [DOI] [PubMed] [Google Scholar]
- 23.Harvey NC, Kanis JA, Oden A, et al. FRAX and the effect of teriparatide on vertebral and non-vertebral fracture. Osteoporos Int. 2015;26(11):2677–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bandeira L, Lewiecki EM, Bilezikian JP. Romosozumab for the treatment of osteoporosis. Expert Opin Biol Ther. 2017;17(2):255–63. [DOI] [PubMed] [Google Scholar]
- 25.Takayanagi H, Ogasawara K, Hida S, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. 2000;408(6812):600–5. [DOI] [PubMed] [Google Scholar]
- 26.Arron JR, Choi Y. Bone versus immune system. Nature. 2000;408(6812):535–6. [DOI] [PubMed] [Google Scholar]
- 27.Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203(12):2673–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takayanagi H, Sato K, Takaoka A, Taniguchi T. Interplay between interferon and other cytokine systems in bone metabolism. Immunol Rev. 2005;208:181–93. [DOI] [PubMed] [Google Scholar]
- 29.Roggia C, Tamone C, Cenci S, Pacifici R, Isaia GC. Role of TNF-alpha producing T-cells in bone loss induced by estrogen deficiency. Minerva Med. 2004;95(2):125–32. [PubMed] [Google Scholar]
- 30.Zhao R, Wang X, Feng F. Upregulated cellular expression of IL-17 by CD4+ T-cells in osteoporotic postmenopausal women. Ann Nutr Metab. 2016;68(2):113–8. [DOI] [PubMed] [Google Scholar]
- 31.Blaschke M, Koepp R, Cortis J, et al. IL-6, IL-1beta, and TNF-alpha only in combination influence the osteoporotic phenotype in Crohn’s patients via bone formation and bone resorption. Adv Clin Exp Med. 2018;27(1):45–56. [DOI] [PubMed] [Google Scholar]
- 32.Sapir-Koren R, Livshits G. Postmenopausal osteoporosis in rheumatoid arthritis: the estrogen deficiency-immune mechanisms link. Bone. 2017;103:102–15. [DOI] [PubMed] [Google Scholar]
- 33.Klingberg E, Geijer M, Gothlin J, et al. Vertebral fractures in ankylosing spondylitis are associated with lower bone mineral density in both central and peripheral skeleton. J Rheumatol. 2012;39(10):1987–95. [DOI] [PubMed] [Google Scholar]
- 34.Shaiykova A, Pasquet A, Goujard C, et al. Reduced bone mineral density among HIV-infected, virologically controlled young men: prevalence and associated factors. AIDS. 2018;32(18):2689–96. [DOI] [PubMed] [Google Scholar]
- 35.Moran CA, Weitzmann MN, Ofotokun I. Bone loss in HIV infection. Curr Treat Options Infect Dis. 2017;9(1):52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piodi LP, Poloni A, Ulivieri FM. Managing osteoporosis in ulcerative colitis: something new? World J Gastroenterol. 2014;20(39):14087–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lubberts E Role of T lymphocytes in the development of rheumatoid arthritis: implications for treatment. Curr Pharm Des. 2015;21(2):142–6. [DOI] [PubMed] [Google Scholar]
- 38.Cope AP, Schulze-Koops H, Aringer M. The central role of T cells in rheumatoid arthritis. Clin Exp Rheumatol. 2007;25(Suppl 46):S4–11. [PubMed] [Google Scholar]
- 39.Yu MB, Langridge WHR. The function of myeloid dendritic cells in rheumatoid arthritis. Rheumatol Int. 2017;37(7):1043–51. [DOI] [PubMed] [Google Scholar]
- 40.Grassi F, Tell G, Robbie-Ryan M, et al. Oxidative stress causes bone loss in estrogen-deficient mice through enhanced bone marrow dendritic cell activation. Proc Natl Acad Sci U S A. 2007;104(38):15087–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chakrabarti S, Lekontseva O, Davidge ST. Estrogen is a modulator of vascular inflammation. IUBMB Life. 2008;60(6):376–82. [DOI] [PubMed] [Google Scholar]
- 42.Felson DT, Zhang Y, Hannan MT, Anderson JJ. Effects of weight and body mass index on bone mineral density in men and women: the Framingham study. J Bone Miner Res. 1993;8(5):567–73. [DOI] [PubMed] [Google Scholar]
- 43.Hannan MT, Felson DT, Anderson JJ. Bone mineral density in elderly men and women: results from the Framingham osteoporosis study. J Bone Miner Res. 1992;7(5):547–53. [DOI] [PubMed] [Google Scholar]
- 44.Gordon T, Kannel WB, Hjortland MC, McNamara PM. Menopause and coronary heart disease: the Framingham study. Ann Intern Med. 1978;89(2):157–61. [DOI] [PubMed] [Google Scholar]
- 45.Aspray TJ, Hill TR. Osteoporosis and the ageing skeleton. Subcell Biochem. 2019;91:453–76. [DOI] [PubMed] [Google Scholar]
- 46.Mortier E, Advincula R, Kim L, et al. Macrophage- and dendritic-cell-derived interleukin-15 receptor alpha supports homeostasis of distinct CD8+ T cell subsets. Immunity. 2009;31(5):811–22. [DOI] [PubMed] [Google Scholar]
- 47.Hennet T, Hagen FK, Tabak LA, Marth JD. T-cell-specific deletion of a polypeptide N-acetylgalactosaminyl-transferase gene by site-directed recombination. Proc Natl Acad Sci U S A. 1995;92(26):12070–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mazzucchelli RI, Warming S, Lawrence SM, et al. Visualization and identification of IL-7 producing cells in reporter mice. PLoS One. 2009;4(11):e7637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nazarian A, Snyder BD, Zurakowski D, Muller R. Quantitative microcomputed tomography: a non-invasive method to assess equivalent bone mineral density. Bone. 2008;43(2):302–11. [DOI] [PubMed] [Google Scholar]
- 51.Arsenovic-Ranin N, Kosec D, Nacka-Aleksic M, et al. Ovarian hormone level alterations during rat post-reproductive life-span influence CD8 + T-cell homeostasis. Exp Biol Med (Maywood). 2015;240(10):1319–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robbie-Ryan M, Pacifici R, Weitzmann MN. IL-7 drives T cell-mediated bone loss following ovariectomy. Ann N Y Acad Sci. 2006;1068:348–51. [DOI] [PubMed] [Google Scholar]
- 53.Kimble RB, Matayoshi AB, Vannice JL, Kung VT, Williams C, Pacifici R. Simultaneous block of interleukin-1 and tumor necrosis factor is required to completely prevent bone loss in the early postovariectomy period. Endocrinology. 1995;136(7):3054–61. [DOI] [PubMed] [Google Scholar]
- 54.Lin SC, Yamate T, Taguchi Y, et al. Regulation of the gp80 and gp130 subunits of the IL-6 receptor by sex steroids in the murine bone marrow. J Clin Invest. 1997;100(8):1980–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kimble RB, Bain S, Pacifici R. The functional block of TNF but not of IL-6 prevents bone loss in ovariectomized mice. J Bone Miner Res. 1997;12(6):935–41. [DOI] [PubMed] [Google Scholar]
- 56.Papadopoulos NG, Georganas K, Skoutellas V, Konstantellos E, Lyritis GP. Correlation of interleukin-6 serum levels with bone density in postmenopausal women. Clin Rheumatol. 1997;16(2):162–5. [DOI] [PubMed] [Google Scholar]
- 57.Schlitzer A, Zhang W, Song M, Ma X. Recent advances in understanding dendritic cell development, classification, and phenotype. F1000Res. 2018;7 F1000 Faculty Rev-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. 2018;154(1):3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takeuchi S, Furue M. Dendritic cells: ontogeny. Allergol Int. 2007;56(3):215–23. [DOI] [PubMed] [Google Scholar]
- 60.Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nat Med. 1996;2(10):1132–6. [DOI] [PubMed] [Google Scholar]
- 61.Geginat J, Lanzavecchia A, Sallusto F. Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood. 2003;101(11):4260–6. [DOI] [PubMed] [Google Scholar]
- 62.Geginat J, Sallusto F, Lanzavecchia A. Cytokine-driven proliferation and differentiation of human naive, central memory, and effector memory CD4(+) T cells. J Exp Med. 2001;194(12):1711–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu S, He H, Gao C, et al. Ovariectomy-induced bone loss in TNF-alpha and IL6 gene knockout mice is regulated by different mechanisms. J Mol Endocrinol. 2018;60(3):185–98. [DOI] [PubMed] [Google Scholar]
- 64.Goswami J, Hernandez-Santos N, Zuniga LA, Gaffen SL. A bone-protective role for IL-17 receptor signaling in ovariectomy-induced bone loss. Eur J Immunol. 2009;39(10):2831–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsoukas CD, Landgraf B, Bentin J, et al. Activation of resting T lymphocytes by anti-CD3 (T3) antibodies in the absence of monocytes. J Immunol. 1985;135(3):1719–23. [PubMed] [Google Scholar]
- 66.Fry TJ, Mackall CL. The many faces of IL-7: from lymphopoiesis to peripheral T cell maintenance. J Immunol. 2005;174(11):6571–6. [DOI] [PubMed] [Google Scholar]
- 67.Kwon KW, Kim SJ, Kim H, et al. IL-15 generates IFN-gamma-producing cells reciprocally expressing lymphoid-myeloid markers during dendritic cell differentiation. Int J Biol Sci. 2019;15(2):464–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.White EJ, Trigatti BL, Igdoura SA. Suppression of NK and CD8(+) T cells reduces astrogliosis but accelerates cerebellar dysfunction and shortens life span in a mouse model of Sandhoff disease. J Neuroimmunol. 2017;306:55–67. [DOI] [PubMed] [Google Scholar]
- 69.DeGottardi MQ, Okoye AA, Vaidya M, et al. Effect of Anti-IL-15 Administration on T Cell and NK Cell Homeostasis in Rhesus Macaques. J Immunol. 2016;197(4):1183–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Djaafar S, Pierroz DD, Chicheportiche R, Zheng XX, Ferrari SL, Ferrari-Lacraz S. Inhibition of T cell-dependent and RANKL-dependent osteoclastogenic processes associated with high levels of bone mass in interleukin-15 receptor-deficient mice. Arthritis Rheum. 2010;62(11):3300–10. [DOI] [PubMed] [Google Scholar]
- 71.Loro E, Ramaswamy G, Chandra A, et al. IL15RA is required for osteoblast function and bone mineralization. Bone. 2017;103:20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shi J, Petrie HT. Activation kinetics and off-target effects of thymus-initiated cre transgenes. PLoS One. 2012;7(10):e46590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Avioli LV. Impact of the menopause on skeletal metabolism and osteoporotic syndromes. Exp Gerontol. 1994;29(3–4):391–415. [DOI] [PubMed] [Google Scholar]
- 74.Pacifici R, Droke D, Avioli LV. Intestinal lactase activity and calcium absorption in the aging female with osteoporosis. Calcif Tissue Int. 1985;37(1):101–2. [DOI] [PubMed] [Google Scholar]
- 75.Avioli LV. Postmenopausal osteoporosis: prevention versus cure. Fed Proc. 1981;40(9):2418–22. [PubMed] [Google Scholar]
- 76.Heaney RP. Nutritional factors in osteoporosis. Annu Rev Nutr. 1993;13:287–316. [DOI] [PubMed] [Google Scholar]
- 77.Oursler MJ. Estrogen regulation of gene expression in osteoblasts and osteoclasts. Crit Rev Eukaryot Gene Expr. 1998;8(2):125–40. [DOI] [PubMed] [Google Scholar]
- 78.Oursler MJ, Pederson L, Pyfferoen J, Osdoby P, Fitzpatrick L, Spelsberg TC. Estrogen modulation of avian osteoclast lysosomal gene expression. Endocrinology. 1993;132(3):1373–80. [DOI] [PubMed] [Google Scholar]
- 79.Cosman F. Anabolic and antiresorptive therapy for osteoporosis: combination and sequential approaches. Curr Osteoporos Rep. 2014;12(4):385–95. [DOI] [PubMed] [Google Scholar]
- 80.Khosla S, Oursler MJ, Monroe DG. Estrogen and the skeleton. Trends Endocrinol Metab. 2012;23(11):576–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pacifici R Role of T cells in ovariectomy induced bone loss-revisited. J Bone Miner Res. 2012;27(2):231–9. [DOI] [PubMed] [Google Scholar]
- 82.Weitzmann MN, Roggia C, Toraldo G, Weitzmann L, Pacifici R. Increased production of IL-7 uncouples bone formation from bone resorption during estrogen deficiency. J Clin Invest. 2002;110(11):1643–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7(4):292–304. [DOI] [PubMed] [Google Scholar]
- 84.Takayanagi H. Osteoimmunology and the effects of the immune system on bone. Nat Rev Rheumatol. 2009;5(12):667–76. [DOI] [PubMed] [Google Scholar]
- 85.Wu L, Liu YJ. Development of dendritic-cell lineages. Immunity. 2007;26(6):741–50. [DOI] [PubMed] [Google Scholar]
- 86.Murphy TL, Grajales-Reyes GE, Wu X, et al. Transcriptional control of dendritic cell development. Annu Rev Immunol. 2016;34:93–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miller CN, Hartigan-O’Connor DJ, Lee MS, et al. IL-7 production in murine lymphatic endothelial cells and induction in the setting of peripheral lymphopenia. Int Immunol. 2013;25(8):471–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Abdelsamed HA, Zebley CC, Youngblood B. Epigenetic maintenance of acquired gene expression programs during memory CD8 T cell homeostasis. Front Immunol. 2018;9:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sprent J, Surh CD. Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol. 2011;12(6):478–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kennedy MK, Glaccum M, Brown SN, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med. 2000;191(5):771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.van Leeuwen EM, Sprent J, Surh CD. Generation and maintenance of memory CD4(+) T cells. Curr Opin Immunol. 2009;21(2):167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Geginat J, Campagnaro S, Sallusto F, Lanzavecchia A. TCR-independent proliferation and differentiation of human CD4+ T cell subsets induced by cytokines. Adv Exp Med Biol. 2002;512:107–12. [DOI] [PubMed] [Google Scholar]
- 93.Okabe I, Kikuchi T, Mogi M, et al. IL-15 and RANKL play a synergistically important role in osteoclastogenesis. J Cell Biochem. 2017;118(4):739–47. [DOI] [PubMed] [Google Scholar]
- 94.Takeda H, Kikuchi T, Soboku K, et al. Effect of IL-15 and natural killer cells on osteoclasts and osteoblasts in a mouse coculture. Inflammation. 2014;37(3):657–69. [DOI] [PubMed] [Google Scholar]
- 95.Miranda-Carus ME, Benito-Miguel M, Balsa A, et al. Peripheral blood T lymphocytes from patients with early rheumatoid arthritis express RANKL and interleukin-15 on the cell surface and promote osteoclastogenesis in autologous monocytes. Arthritis Rheum. 2006;54(4):1151–64. [DOI] [PubMed] [Google Scholar]
- 96.Ogata Y, Kukita A, Kukita T, et al. A novel role of IL-15 in the development of osteoclasts: inability to replace its activity with IL-2. J Immunol. 1999;162(5):2754–60. [PubMed] [Google Scholar]
- 97.Koh JM, Oh B, Ha MH, et al. Association of IL-15 polymorphisms with bone mineral density in postmenopausal Korean women. Calcif Tissue Int. 2009;85(5):369–78. [DOI] [PubMed] [Google Scholar]
- 98.Munoz LE, Frey B, Pausch F, et al. The role of annexin A5 in the modulation of the immune response against dying and dead cells. Curr Med Chem. 2007;14(3):271–7. [DOI] [PubMed] [Google Scholar]
- 99.Budagian V, Bulanova E, Paus R, Bulfone-Paus S. IL-15/IL-15 receptor biology: a guided tour through an expanding universe. Cytokine Growth Factor Rev. 2006;17(4):259–80. [DOI] [PubMed] [Google Scholar]
- 100.Racape M, Vanhove B, Soulillou JP, Brouard S. Interleukin 7 receptor alpha as a potential therapeutic target in transplantation. Arch Immunol Ther Exp (Warsz). 2009;57(4):253–61. [DOI] [PubMed] [Google Scholar]
- 101.Surh CD, Boyman O, Purton JF, Sprent J. Homeostasis of memory T cells. Immunol Rev. 2006;211:154–63. [DOI] [PubMed] [Google Scholar]
- 102.Deselm CJ, Takahata Y, Warren J, et al. IL-17 mediates estrogen-deficient osteoporosis in an Act1-dependent manner. J Cell Biochem. 2012;113(9):2895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ozmen B, Kirmaz C, Aydin K, Kafesciler SO, Guclu F, Hekimsoy Z. Influence of the selective oestrogen receptor modulator (raloxifene hydrochloride) on IL-6, TNF-alpha, TGF-beta1 and bone turnover markers in the treatment of postmenopausal osteoporosis. Eur Cytokine Netw. 2007;18(3):148–53. [DOI] [PubMed] [Google Scholar]
- 104.Roggia C, Gao Y, Cenci S, et al. Up-regulation of TNF-producing T cells in the bone marrow: a key mechanism by which estrogen deficiency induces bone loss in vivo. Proc Natl Acad Sci U S A. 2001;98(24):13960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ammann P, Rizzoli R, Bonjour JP, et al. Transgenic mice expressing soluble tumor necrosis factor-receptor are protected against bone loss caused by estrogen deficiency. J Clin Invest. 1997;99(7):1699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kotrych D, Dziedziejko V, Safranow K, et al. TNF-alpha and IL10 gene polymorphisms in women with postmenopausal osteoporosis. Eur J Obstet Gynecol Reprod Biol. 2016;199:92–5. [DOI] [PubMed] [Google Scholar]
- 107.Kim H, Chun S, Ku SY, Suh CS, Choi YM, Kim JG. Association between polymorphisms in tumor necrosis factor (TNF) and TNF receptor genes and circulating TNF, soluble TNF receptor levels, and bone mineral density in postmenopausal Korean women. Menopause. 2009;16(5):1014–20. [DOI] [PubMed] [Google Scholar]
- 108.Furuta I, Kobayashi N, Fujino T, et al. Bone mineral density of the lumbar spine is associated with TNF gene polymorphisms in early postmenopausal Japanese women. Calcif Tissue Int. 2004;74(6):509–15. [DOI] [PubMed] [Google Scholar]
- 109.Boron D, Agnieszka SM, Daniel K, Anna B, Adam K. Polymorphism of interleukin-17 and its relation to mineral density of bones in perimenopausal women. Eur J Med Res. 2014;19:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jaigirdar SA, MacLeod MK. Development and function of protective and pathologic memory CD4 T cells. Front Immunol. 2015;6:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lauvau G, Soudja SM. Mechanisms of memory T cell activation and effective immunity. Adv Exp Med Biol. 2015;850:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dunn J, McCuaig R, Tu WJ, Hardy K, Rao S. Multi-layered epigenetic mechanisms contribute to transcriptional memory in T lymphocytes. BMC Immunol. 2015:16(1):27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Quach D, Britton RA. Gut microbiota and bone health. Adv Exp MedBiol. 2017;1033:47–58. [DOI] [PubMed] [Google Scholar]
- 114.Chen YC, Greenbaum J, Shen H, Deng HW. Association between gut microbiota and bone health: potential mechanisms and prospective. J Clin Endocrinol Metab. 2017;102(10):3635–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cassady-Cain RL, Hope JC, Stevens MP. Direct manipulation of T lymphocytes by proteins of gastrointestinal bacterial pathogens. Infect Immun. 2018;86(5):e00683–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hegazy AN, West NR, Stubbington MJT, et al. Circulating and tissue-resident CD4(+) T cells with reactivity to intestinal microbiota are abundant in healthy individuals and function is altered during inflammation. Gastroenterology. 2017;153(5):1320–37.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Teng F, Felix KM, Bradley CP, et al. The impact of age and gut microbiota on Th17 and Tfh cells in K/BxN autoimmune arthritis. Arthritis Res Ther. 2017;19(1):188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: a new candidate in the pathogenesis of osteoporosis. PLoS One. 2012;7(9):e44552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Di Rosa F T-lymphocyte interaction with stromal, bone and hematopoietic cells in the bone marrow. Immunol Cell Biol. 2009;87(1):20–9. [DOI] [PubMed] [Google Scholar]
- 121.Bae JM, Yoon BK. The role of menopausal hormone therapy in reducing all-cause mortality in postmenopausal women younger than 60 years: an adaptive meta-analysis. J Menopausal Med. 2018;24(3):139–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mikkola TS, Tuomikoski P, Lyytinen H, et al. Increased cardiovascular mortality risk in women discontinuing postmenopausal hormone therapy. J Clin Endocrinol Metab. 2015;100(12):4588–94. [DOI] [PubMed] [Google Scholar]
- 123.Cheung AM. Review: hormone therapy reduces mortality in younger postmenopausal women. Ann Intern Med. 2010;152(8):JC4–9. [DOI] [PubMed] [Google Scholar]
- 124.Kannel WB, Hjortland MC, McNamara PM, Gordon T. Menopause and risk of cardiovascular disease: the Framingham study. Ann Intern Med. 1976;85(4):447–52. [DOI] [PubMed] [Google Scholar]
- 125.Robison LS, Gannon OJ, Salinero AE, Zuloaga KL. Contributions of sex to cerebrovascular function and pathology. Brain Res. 2019;1710:43–60. [DOI] [PubMed] [Google Scholar]
- 126.Wang X, Ping FF, Bakht S, Ling J, Hassan W. Immunometabolism features of metabolic deregulation and cancer. J Cell Mol Med. 2019;23(2):694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chornenkyy Y, Wang WX, Wei A, Nelson PT. Alzheimer’s disease and type 2 diabetes mellitus are distinct diseases with potential overlapping metabolic dysfunction upstream of observed cognitive decline. Brain Pathol. 2019;29(1):3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Machado FVC, Pitta F, Hernandes NA, Bertolini GL. Physiopathological relationship between chronic obstructive pulmonary disease and insulin resistance. Endocrine. 2018;61(1):17–22. [DOI] [PubMed] [Google Scholar]
- 129.Varghese JF, Patel R, Yadav UCS. Novel insights in the metabolic syndrome-induced oxidative stress and inflammation-mediated atherosclerosis. Curr Cardiol Rev. 2018;14(1):4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.