

Abstract
Statins are a family of drugs that are used for treating hyperlipidaemia with a recognized capacity to prevent cardiovascular disease events. They inhibit β‐hydroxy β‐methylglutaryl‐coenzyme A reductase, i.e. the rate‐limiting enzyme in mevalonate pathway, reduce endogenous cholesterol synthesis, and increase low‐density lipoprotein clearance by promoting low‐density lipoprotein receptor expression mainly in the hepatocytes. Statins have pleiotropic effects including stabilization of atherosclerotic plaques, immunomodulation, anti‐inflammatory properties, improvement of endothelial function, antioxidant, and anti‐thrombotic action. Despite all beneficial effects, statins may elicit adverse reactions such as myopathy. Studies have shown that mitochondria play an important role in statin‐induced myopathies. In this review, we aim to report the mechanisms of action of statins on mitochondrial function. Results have shown that statins have several effects on mitochondria including reduction of coenzyme Q10 level, inhibition of respiratory chain complexes, induction of mitochondrial apoptosis, dysregulation of Ca2+ metabolism, and carnitine palmitoyltransferase‐2 expression. The use of statins has been associated with the onset of additional pathological conditions like diabetes and dementia as a result of interference with mitochondrial pathways by various mechanisms, such as reduction in mitochondrial oxidative phosphorylation, increase in oxidative stress, decrease in uncoupling protein 3 concentration, and interference in amyloid‐β metabolism.
Overall, data reported in this review suggest that statins may have major effects on mitochondrial function, and some of their adverse effects might be mediated through mitochondrial pathways.
Keywords: Statin, Myopathy, SAMS, Mitochondria, Apoptosis, Coenzyme Q10, Respiratory chain, Diabetes, Cognitive impairment
Introduction
Cardiovascular diseases (CVDs), such as myocardial infarction, angina, heart failure, and cerebrovascular accidents, contribute significantly to global mortality. World Health Organization estimated 17.9 million deaths in 2017 due to CVDs, representing 31% of the whole global mortality. 1 The systematic analysis for the Global Burden of Disease Study estimates that CVD‐associated mortality will raise to 23 million deaths per year by 2030. 2 Among the major risk factors, CVDs are associated with high plasma total cholesterol, low‐density lipoprotein (LDL) cholesterol, and reduced high‐density lipoprotein cholesterol. CVD risk is reduced by ~22% per each 1 mmol/L LDL cholesterol decrease. 3 There is overwhelming evidence showing an increasing clinical benefit when greater and earlier LDL cholesterol reductions are achieved. 4 Apo B‐containing lipoproteins (like LDLs), which are the key sources of cholesterol, are responsible for initiation and progression of atherogenesis. 5 Therefore, one of the main goals of CVD prevention is LDL cholesterol level reduction.
β‐Hydroxy β‐methylglutaryl‐coenzyme A (HMG‐CoA) reductase inhibitors, better known as statins, are among the most effective lipid‐lowering agents. 6 Besides lifestyle approaches like diet and exercise interventions, 7 additional cholesterol‐lowering therapies include nutraceuticals, 8 , 9 , 10 ezetimibe, proprotein convertase subtilisin/kexin type 9 inhibitors, lomitapide, and mipomersen. 11 , 12
In 1976, Akira Endo isolated from Penicillium citrinum, a molecule (mevastatin) able to inhibit HMG‐CoA reductase, a crucial enzyme in endogenous cholesterol biosynthesis. 13 Statins are reversible and competitive HMG‐CoA reductase inhibitors and determine a reduction in serum and tissue levels of total cholesterol, LDL cholesterol, apo B, and triglycerides. Statins are prescribed in order to reduce plasma cholesterol levels both in primary and secondary prevention of CVD.
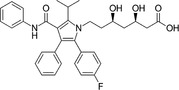
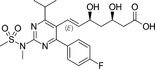
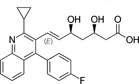
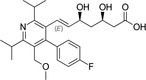
Because HMG‐CoA is a key enzyme in the mevalonate pathway, its inhibition results in a reduced bioavailability of farnesyl pyrophosphate, geranylgeranyl pyrophosphate, heme A, coenzyme Q10 (CoQ10; also known as ubiquinone), and other metabolites that play essential roles in cellular physiology. 14 , 15 Moreover, cholesterol is not only a final product of this biochemical pathway but also itself an intermediate for many other compounds, such as corticosteroids, bile acids, and vitamin D. 15 This explains why statins pharmacodynamically exert pleiotropic effects, 16 , 17 , 18 , 19 , 20 , 21 which are responsible for both beneficial and adverse consequences during treatment. Apart from their cholesterol‐lowering effect, statins are known to contribute to stabilization of atherosclerotic plaques, 22 improvement of endothelial dysfunction, 23 and immunomodulation and show anti‐inflammatory, 24 antioxidant, and anti‐thrombotic effects. 25 Lipid‐dependent and lipid‐independent effects of statins may vary according to the potency, 6 lipophilicity (Table 1 ), and pharmacokinetic properties 14 , 26 , 27 , 28 of different agents.
Table 1.
Statins and their structures and classification
| Statin | Generation | Structure | Hydrophilicity/lipophilicity |
|---|---|---|---|
| Lovastatin | I |
|
Hydrophilic |
| Pravastatin | I |
|
Lipophilic |
| Fluvastatin | I |
|
Hydrophilic |
| Simvastatin | II |
|
Hydrophilic |
| Atorvastatin | II |
|
Hydrophilic |
| Rosuvastatin | III |
|
Lipophilic |
| Pitavastatin | III |
|
Hydrophilic |
| Cerivastatin | Withdrawn |
|
Lipophilic |
Statins have been proved to be safe drugs. However, they may elicit adverse reactions (muscle, metabolic, liver, and neurological side effects), being muscle adverse effects the most frequently reported. 29 These adverse reactions are commonly named as statin‐associated symptoms (SAS). 29 It is important to underline that the causative relationship between statins and SAS is not always clearly demonstrable: some patients complaining of SAS can tolerate a low statin dose, and some non‐specific side effects have been reported by patients treated with placebo. 29 Most of SAS appear to be dose dependent; half of the patients on high statin dosages stop the drug within 1 year because of SAS. 30 , 31 , 32 The risk of drug–drug interaction increases alongside with age and number of administered drugs. 33 Other risk factors for statin intolerance are frailty, surgery, infection, female sex, exertion, hypothyroidism, chronic kidney disease, and genetic predisposition. 15 , 34
Statins may interfere with mitochondrial activity both via direct mechanisms, i.e. impairment of electron transport chain (ETC) complexes, and via indirect ones, i.e. as a consequence of mevalonate pathway metabolites depletion, e.g. CoQ10 and isoprenoids. Growing evidence is being provided showing that SAS are mostly due not to their direct cholesterol‐lowering effect but rather to the induced impairment on mitochondrial function, as supported by histopathological, laboratory, and molecular findings in cell lines, rat models, and humans. 34 , 35 A better understanding of the pathogenic background of SAS will be fundamental for quickly identifying patients who are at major risk and acquire best results of treatment, so learning about their mechanisms, progression, and treatment is essential for better counteracting adverse reactions of statins responsible for therapy discontinuation.
Aims and methods
The aim of this review was to provide an overview of achieved knowledge about mechanisms that underlie SAS, focusing on muscle effects, statin‐induced diabetes, and cognitive impairment. In particular, we focused on statin‐driven molecular mechanisms interfering both directly and indirectly with mitochondrial function.
This review is based on evidence gathered performing a PubMed query by using ‘statin AND mitochondria AND adverse reaction’ as search terms. The search strategy was implemented by hand searching the references reported by the most relevant studies on this topic.
Statin‐associated muscle symptoms
Among all SAS, muscle side effects are the most frequently reported. 29 , 36 , 37 About 7–29% of patients complain of statin‐associated muscle symptoms (SAMS). 38 Symptoms usually occur after 4–6 weeks of treatment initiation but sometimes after many years. 39 Weakness, pain, and muscle fatigue are responsible for 30–62% statin treatment discontinuation. 40 SAMS present as a wide range of clinical manifestations, such as myalgia, weakness, tendon pain, night muscle cramping, and rhabdomyolysis in most severe cases. 41 , 42 Weakness and pain usually involve large back muscles and proximal muscle groups symmetrically. 43 Underlying these manifestations is a statin‐induced myopathy (SIM), whose pathophysiology has not been fully understood yet. 44 Further, the lack of reliable laboratory biomarkers for diagnosing myopathy complicates its clinical identification. The most used laboratory test is creatine kinase (CK) blood concentration, which however suffers from low sensibility and specificity. 45
Up to now, there is no universally established terminology used to describe SIM. The National Lipid Association describes myopathy as muscle pain, soreness, weakness, and cramps together with an at least 10‐fold increase in CK levels above the upper limit of normal (ULN). 46 European Atherosclerosis Society (EAS) defines myopathy as the presence of muscle symptoms accompanied by CK over 10‐fold the ULN. 38 According to the American College of Cardiology/American Heart Association/National Heart, Lung, and Blood Institute, myopathy can be identified by either myalgia (without CK elevation), myositis (CK elevation within 10‐fold the ULN), or blunt rhabdomyolysis (CK elevation over 10‐fold the ULN, increase in creatininemia, and reddish or brown urine). 47 On the contrary, rhabdomyolysis is defined by the Food and Drug Administration (FDA) as a condition characterized by an increase in CK blood concentration over 50‐fold the ULN associated with myoglobin‐induce acute renal injury. 48 Conversely, according to EAS, CK over 40‐fold the ULN associated with renal impairment and/or myoglobinuria configures the presence of rhabdomyolysis. 38 Importantly, albeit rhabdomyolysis is the most severe statin‐associated side effect, it is a rare complication occurring in about 0.01% of treated patients. 49 Myoglobinuria, deriving from muscle necrosis and the release of myoglobin in the blood circulation, can cause mechanical obstruction and acute necrosis of the renal tubules and subsequently acute renal failure, a potentially life‐threatening complication. 50
Statin‐associated muscle symptom definition refers to any muscle symptom related to statin treatment. In this frame, the EAS Consensus suggested three criteria for the diagnosis of SAMS: correspondence between CK levels increase and onset of statin treatment; decline of symptoms at treatment withdrawal; and symptoms recurrence at statin treatment rechallenging. 38 However, it must be underlined that SAMS might occur in patients who do not show increased CK blood concentrations. 29
Mitochondria
Mitochondria are organelles often referred to as the power station of eukaryotic cells, because several phases of cellular respiration take place there, such as the Krebs cycle, the oxidative phosphorylation, and fatty acid β‐oxidation. 51 Oxidative phosphorylation is a key biochemical pathway in cellular respiration and is carried out by several protein complexes and molecules that are located at the inner mitochondrial membrane (IMM). It consists of two subsequent steps, the ETC and ATP synthesis. ATP synthesis consists of phosphorylation of ADP to ATP catalysed by ATP synthase, thanks to the proton motive force following the electrochemical gradient across the IMM. This electrochemical gradient is sustained by the proton translocation from mitochondrial matrix to intermembrane space (IMS) mediated by complexes I, III, and IV of the ETC. 52
Complex I (NADH : ubiquinone oxidoreductase) is responsible for reduction of NADH yielded during glycolysis and Krebs cycle. While transferring electrons from NADH, thereby reducing ubiquinone to ubiquinol, it mediates transport of four protons (H+) from mitochondrial matrix to the IMS. Complex II (succinate : ubiquinone oxidoreductase) is part of the Krebs cycle. It catalyses succinate oxidation to fumarate while reducing ubiquinone to ubiquinol. Different to other complexes, complex II does not transport protons across the membrane. Complex III (cytochrome bc 1) catalyses reoxidation of ubiquinol (moving freely along the IMM and yielded by complex I and II enzymatic activities) to ubiquinone, transferring electrons to the membrane‐attached cytochrome c and four protons from matrix to IMS. Eventually, cytochrome c is again oxidized by complex IV (cytochrome c oxidase) that yields H2O from transferring electrons to O2 and transfers four protons against gradient across the IMS. 52 The electron transport system (ETS) is thought to be an important source of reactive oxygen species (ROS). As a consequence of many redox reactions, mitochondria yield an important amount of ROS, which can act either as cell signalling molecules or as oxidative stress factors 52 , 53 , 54 (Figure 1 ).
Figure 1.
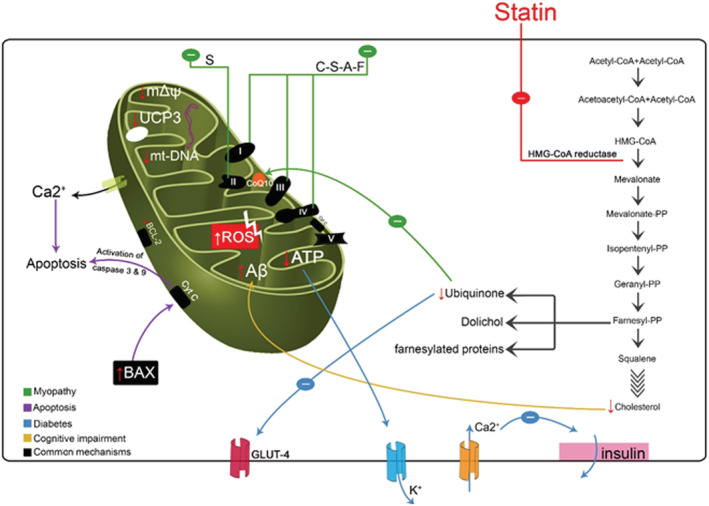
Effects of statins on mitochondrial function: (i) reduction in mitochondrial membrane potential; (ii) reduction in coenzyme Q10 (CoQ10) and GLUT‐4 expression; (iii) increased reactive oxygen species (ROS) level and induction of intrinsic apoptosis; (iv) deregulation of Ca2+ metabolism; (v) mitochondrial depletion; (vi) decrease in uncoupling protein 3 (UCP3) expression and reduction in β‐oxidation efficiency; (vii) increase in amyloid‐β (Aβ) concentration in mitochondria; (viii) direct inhibition in respiratory chain. A, atorvastatin; C, cerivastatin; F, fluvastatin; HMG‐CoA, β‐hydroxy β‐methylglutaryl‐coenzyme A; mtDNA, mitochondrial DNA; PP, pyrophosphate; S, simvastatin.
Statin‐associated muscle symptoms and mitochondrial dysfunction
Mitochondrial dysfunction is believed to play a pivotal role in the pathogenesis of SAMS, because skeletal muscles are avid energy consumers and closely depend on mitochondrial activity. 55 This is supported by histopathological findings, evidencing how SIM is characterized by lipid aggregation in mitochondria and ragged red fibres accumulating in subsarcolemmal region visible with Gömöri staining. 56 , 57 Abnormal high blood lactate/pyruvate ratio 58 and increased mean metabolic recovery time as assessed by 31P magnetic resonance spectroscopy 59 in statin‐treated patients when compared with untreated hypercholesterolaemic and healthy controls add evidence to this hypothesis. However, neither the LIFESTAT study nor 8 week treatment with simvastatin 80 mg/day on healthy subjects proved any difference in mitochondrial function after statin treatment. 61 , 62 According to the LIFESTAT study, 20 healthy middle‐aged men were treated with 80 mg/day simvastatin (n = 10) or 40 mg/day pravastatin (n = 10). After 14 days, 50% of participants presented a different range of side effects from soreness, myalgia, to stomach ache, but no changes in mitochondrial function with respect to respiratory chain capacity, mitochondrial density, or Q10 content, except a reduction in complex IV activity in both groups. 61 Hou et al. found the presence of mitochondrial dysfunction and abnormal respiratory chain enzyme activity in 24% and 10%, respectively, among 279 biopsies from patients with SIM. Hence, whether mitochondrial dysfunction may be considered a frequent pathogenic hallmark of SAMS is still debated. 63
In the following sections, the proposed pathogenetic mechanisms of SAMS involving mitochondrial dysfunction are described (Table 2 ).
Table 2.
Overview on statin mechanisms of mitochondrial toxicity
| Type of statin | Mechanism | Adverse effect | Reference |
|---|---|---|---|
| Atorvastatin | Inhibition of complexes I, III, and IV | Myopathy | Salviati et al. 68 |
| Dysregulation of calcium homoeostasis | Myopathy | Liantonio et al. 60 | |
| Reduction in CoQ10 concentration | Myopathy | Saha and Whayne 69 | |
| mtDNA depletion | Myopathy | Saha and Whayne 69 | |
| PGC‐1/Akt pathway disruption | Myopathy | Mullen et al. 83 | |
| ROS accumulation and oxidative stress | Induced apoptosis | Dirks and Jones 104 | |
| Akt pathway disruption | Induced apoptosis | Mikus et al. 81 | |
| Impaired insulin secretion | Diabetes | Shepherd et al., Sattar et al., and Sadighara et al. 130 , 132 , 133 | |
| Unknown | Cognitive impairment | Ott et al. and Swiger et al. 153 , 154 | |
| Fluvastatin | Inhibition of complexes I, III, and IV | Myopathy | Salviati et al. 68 |
| Dysregulation of calcium homoeostasis | Myopathy | Liantonio 60 | |
| Lovastatin | CPT‐2 inhibition | Myopathy | Ghatak et al. 123 |
| Reduced activity of complex IV | Myopathy | Sirvent et al. 91 | |
| Impaired insulin secretion | Diabetes | Shepherd et al. 130 | |
| Pravastatin | Inhibition of complex IV | Myopathy | Phillips et al. 57 |
| Rosuvastatin | Akt pathway disruption | Induced apoptosis | Mikus et al. 81 |
| Simvastatin | Impaired ETC function | Myopathy | Salviati et al., Saha and Whayne, Vaughan et al., Kwak et al., and Sirvent et al. 68 , 69 , 85 , 88 , 91 |
| Reduction in CoQ10 muscle concentration | Myopathy | Saha and Whayne and Sirvent et al. 69 , 91 | |
| mtDNA depletion or reduced citrate synthase activity | Myopathy | Saha and Whayne, Lamperti et al., Banach et al., Qu et al., and Schick et al. 69 , 76 , 77 , 78 , 79 | |
| PGC‐1/Akt pathway disruption | Myopathy | Åberg et al., Stringer et al., and Singla et al. 67 , 80 , 82 | |
| Dysregulation of calcium homoeostasis | Myopathy | De Pinieux et al., Murlasists and Radák, and Herminghaus et al. 58 , 95 , 96 | |
| Inhibition of complex IV | Myopathy | Phillips et al. 57 | |
| Calpain activation | Induced apoptosis | Hermann et al. 103 | |
| ROS accumulation and oxidative stress | Induced apoptosis | Vaughan et al. 85 | |
| Isoprenoid depletion | Induced apoptosis | Novelli and D'Apice and Guijarro et al. 109 , 110 | |
| Akt pathway disruption | Induced apoptosis | Stringer et al. and Mikus et al. 80 , 81 | |
| Peripheral insulin resistance | Diabetes | Sadighara et al. and Moro et al. 136 , 140 | |
| Unknown | Cognitive impairment | Ott et al. 153 | |
| Cerivastatin | Inhibition of complexes I, III, and IV | Myopathy | Salviati et al. 68 |
| Reduced activity of complex IV | Myopathy | Brookes 53 | |
| Isoprenoid depletion | Induced apoptosis | Hosseinzadeh et al. 108 |
CoQ10, coenzyme Q10; CPT‐2, carnitine palmitoyltransferase‐2; ETC, electron transport chain; mtDNA, mitochondrial DNA; PGC‐1, peroxisome proliferator‐activated receptor gamma coactivator 1; ROS, reactive oxygen species.
Depletion of coenzyme Q10
Coenzyme Q10 (alias ubiquinone) is a hexameric quinone ring containing a 10‐isoprenyl unit side chain; it is structurally similar to vitamin K and can be found in the hydrophobic layer of all cell membranes in mammalians. Ubiquinone is an end product of the mevalonate pathway but can be supplied by diet being mostly present in some meats like beef, pork, and chicken 64 , 65 and by dietary supplements/nutraceuticals. 66 Because of its chemical structure, CoQ10 presents a high antioxidant activity. The highest tissue concentrations of CoQ10 can be found in the kidney, liver, and heart. 67 As previously described, CoQ10 is an essential component of the ETC and therefore plays a pivotal role in cellular energy yield. Primary CoQ10 deficiency has been detected in several diseases comprising heart failure, nephrotic syndrome, and muscular and neurological disorders. 68 , 69 In addition, secondary CoQ10 deficiency may occur. In this regard, the inhibitory effect of statins on the mevalonate pathway (Figure 1 ) may reduce CoQ10 levels by 16–54% 64 and cause mitochondrial dysfunction, revealed by inhibition of mitochondrial ETC complexes, 70 , 71 disruption of mitochondrial membrane potential, decrease in mitochondrial DNA (mtDNA) copy number, interference with oxidative phosphorylation, mitochondrial swelling, and release of cytochrome c. 71 , 72 However, decrease in plasma CoQ10 level is mostly due to the reduction of circulating lipoproteins, because around 74% of CoQ10 is carried in the blood by apo B‐containing lipoproteins. 64 Accordingly, no significant difference in CoQ10 to total cholesterol ratio was found before and after statin treatment.
Scarce and contradictory data are available about the impact of statin treatment on skeletal muscle CoQ10 concentration. 73 In a randomized controlled trial, 8 weeks of treatment with simvastatin 80 mg/day but not the treatment with atorvastatin 40 mg/day or placebo was associated with a significant decrease in muscle ubiquinone concentration. 72 Analyses from a subgroup of patients showed that those with markedly reduced muscle CoQ10 had concurrent reduction in respiratory chain enzyme and citrate synthase activities. 72 However, conflicting data showing either increase or no change in muscle ubiquinone concentrations after both short‐term and long‐term statin treatments have been provided. 74 , 75 , 76
Recent meta‐analyses on the topic have led to controversial results about the effectiveness of CoQ10 supplementation in statin‐treated patients in improving SIM and SAMS. 77 , 78
Mitochondrial depletion and oxidative stress
A significative reduction in mtDNA/nuclear DNA ratio was observed after 8 weeks of 80 mg/day simvastatin but not after 8 weeks of 40 mg/day atorvastatin. 79 The findings of this randomized controlled trial suggested a major impact of simvastatin on mitochondrial depletion due to its lipophilicity. Notably, changes in mtDNA/nuclear DNA ratio were irrespective of muscle symptoms but strongly associated with muscle CoQ10 concentration. 79 These results were replicated in a small cohort of patients experiencing SIM. 80
Exercise training can enhance mitochondrial function by increasing oxygen uptake by ~10% and citrate synthase activity, a marker of skeletal muscle mitochondrial content, by 13% in overweight or obese patients after 12 week exercise training. 81 The addition of 40 mg/day simvastatin led to a lower increment of oxygen uptake during exercise training (1.5%) and a 4.5% reduction in citrate synthase activity. 81 Similar results (3.6% reduction in citrate synthase activity and cardiorespiratory fitness impairment) have been reported in a cohort of patients with type 2 diabetes mellitus (T2DM) treated with 40 mg/day simvastatin for 12 weeks. 82 Intriguingly, vitamin D supplementation resulted effective in preventing statin‐related decrease in mitochondrial content and impairment in cardiorespiratory fitness. 82
Statin‐induced toxicity could be due to the reduced phosphorylation of protein kinase B (Akt) and subsequent disruption of its pathway, which plays an important role in the mitochondrial health. 83 , 84 The Akt pathway disruption is probably caused by decreased transcription and translation rates of peroxisome proliferator‐activated receptor gamma coactivator (PGC)‐1α and PGC‐1β. In fact, these transcriptional co‐factors act as activators of the Akt pathway and are considered master inductors of mitochondrial biogenesis. 85 Different cell lines both in vitro and in vivo have shown to be differentially susceptible to the disruption of the Akt pathway. Primary mouse skeletal muscle myocytes, C2C12 myotubes, 83 human rhabdomyosarcoma cells, 85 and skeletal muscle cells from deltoid biopsies 70 were susceptible to statin treatment in terms of mitochondrial dysfunction, as assessed by several parameters, including PGC‐1α mRNA transcription and mitochondrial oxidative capacity. Interestingly, mitochondrial dysfunction could be rescued either by administration of ubiquinol, 85 exogenous antioxidant agents, or by PGC‐1α overexpression. 70 In similar studies done on various models such as murine liver HepG2 cells, 83 oxidative muscle fibres, 86 and human cardiac muscle fibres, 70 statins showed mitochondrial dysfunctions. The production of ROS and oxidative stress could be crucial hallmarks of statin‐induced toxicity and have a role in regulating mitochondrial biogenesis. 87 , 88 In cells with high‐efficient antioxidant systems, as cardiac myofibres, statin‐induced ROS accumulation is limited and stimulates PGC‐1α activity, thus promoting mitochondrial biogenesis and improving mitochondrial function. On the other hand, in cells with lower antioxidant capacity, such as fast glycolytic muscle fibres, a greater ROS increase could be responsible for the Akt pathway disruption. 70 , 86 The differential response in terms of mitochondrial biogenesis according to ROS production is known as mitochondrial hormesis and can explain why some tissues and cells, in particular fast muscle fibres, are more susceptible to statin toxicity. 70 It is notable that PGC‐1β knockout mice showed mitochondrial dysfunction, increased ROS production, and induced apoptosis after oral administration of 5 mg/kg/day atorvastatin per 2 weeks both in glycolytic and oxidative muscle fibres. 86
Inhibition of electron transport chain complexes
Direct inhibition of ETC complexes has been proposed as a responsible mechanism for SIM. 63 , 88 , 89 , 90 In vitro, functional impairment of complexes I, III, and IV occurred in L6 rat skeletal muscle cells after a 24 hour exposure to 100 μmol/L cerivastatin, simvastatin, fluvastatin, or atorvastatin. On the contrary, no functional impairment was reported after exposure up to 1 mmol/l pravastatin. 71 These findings were confirmed in human myocytes, onto which simvastatin acts mostly as an inhibitor of complex I. 91 However, assays on fresh and frozen muscle samples from rats treated with different statin regimens failed to identify abnormalities in ETC complexes activity. 92 , 93 A randomized controlled trial showed significant reduction in all ETC complexes activity in patients treated with simvastatin 80 mg/day for 8 weeks but not in those treated with atorvastatin 40 mg/day or placebo. 72 However, a systematic review of several studies on cell cultures, animal models, and humans showed highly contradicting results on this topic. 34
In two patients affected by statin‐induced rhabdomyolysis, skeletal muscle biopsies showed reductions in ubiquinone concentration and in complex IV activity. 94 A significant reduction by 60% of complex IV activity was identified in a patient presenting myoglobinuria after treatment with cerivastatin and gemfibrozil. 56
Statins are known to interfere with beneficial exercise training adaptations, which mostly rely on improvements of the mitochondrial function, 81 , 82 , 95 and could be due to ETC complexes activity impairment. 81 Finally, it has been suggested that impaired ETC function could be found out in other tissues as well, though data on these aspects are still very scarce. 96
Dysregulation of Ca2+ metabolism
As a consequence of mitochondrial dysfunction, studies on both animal and human models have identified an increased Ca2+ efflux from the sarcoplasmic reticulum, leading to abnormalities in excitation–contraction coupling in skeletal muscle cells. 97 Sirvent et al. hypothesized that ETC disruption (and, above all, complex I inhibition) due to statin treatment could be responsible both in vitro and in vivo for calcium metabolism deregulation. 98 Alterations in spontaneous Ca2+ sparks (e.g. frequency and amplitude) in muscle cells indicate impaired Ca2+ homoeostasis. 99 Decreased frequency of spontaneous Ca2+ sparks was found in patients treated with statin and complaining muscle symptoms but not in asymptomatic statin‐treated patients when compared with healthy controls. 98 Increased Ca2+ spark amplitude was found in all statin‐treated patients when compared with controls 98 and in individuals treated with 80 mg/day simvastatin for 8 weeks displaying increased serum CK concentrations. 62 Statins have been demonstrated both in vivo and in vitro in animal and human models to raise cytosolic and sarcoplasmic Ca2+ concentration: this increase does not seem to be attributable to abnormal sarcolemma permeability but rather to a primary Ca2+ efflux from mitochondria mediated by mitochondrial permeability transition pore and Na+–Ca2+ exchanger. 99 The latter secondarily leads to Ca2+ release from the sarcoplasmic reticulum mediated by ryanodine receptors. 60 , 70 , 96 The abnormal opening of mitochondrial permeability transition pore and Na+–Ca2+ exchanger may be caused by the decrease of mitochondrial membrane potential and depolarization of the IMM as a consequence of disrupted ETC activity. 99
Lactone toxicity
Statins are available in two different forms: hydroxy acid (active molecule) and the lactone (inactive prodrug). Interconversion through these two forms occurs spontaneously according to environmental pH or can be catalysed by enzymes, such as glucuronosyltransferases. 100 Lactone forms are more lipophilic and therefore more capable of crossing cell membranes and exerting their pharmacological activity. 100 Lactone forms have been described as more myotoxic both in rat and human cells in vitro. 101 , 102 Individuals affected by SIM showed higher circulating levels of lactone metabolites than controls. 103 Acute administration of lactone forms of atorvastatin, cerivastatin, and pitavastatin in C2C12 myoblasts elicited a fast decrease of oxygen consumption rate, indicating a net inhibitory effect on mitochondrial respiratory function. 90 In particular, lactone forms showed a higher capacity to impair ETC complex III activity. 90 Therefore, the conditions altering the hydroxy acid/lactone forms ratio, such as pH disturbances and pharmacokinetic interactions, might play a role in amplifying the muscle damage of statin treatment.
Apoptosis
Statin‐induced mitochondrial dysfunction has been correlated in vitro with an abnormal activation of the intrinsic apoptosis pathway, as a consequence of a decrease in the Bcl‐2/Bax ratio. The statin‐induced apoptosis occurs in different tissues and might be responsible for both beneficial pleiotropic and detrimental side effects of statin treatment. 88 , 104
Several mechanisms have been proposed to explain the abnormal apoptosis in cells exposed to statins. In vitro studies on human primary skeletal muscle cells identified calpain, a calcium‐dependent protease implicated in a variety of cellular processes such as signal transduction, cell proliferation, cell cycle progression, differentiation, and apoptosis, 105 as a plausible upstream effector for the statin‐induced initiation of the intrinsic apoptotic pathway, which is only partially dependent on mevalonate depletion. 106 In fact, statin‐induced calpain activation could be subsequent to increased Ca2+ sarcoplasmic concentration, as discussed previously, and is responsible for Bax translocation to the outer mitochondrial membrane (OMM), leading to cytochrome c release into the cytosol and eventually to apoptosome assembly and activation of the caspases cascade. 106
Accumulation of ROS as a consequence of ETC impairment could contribute to inducing mitochondria‐mediated apoptosis. 88 , 107 Treatment with 10 mg/kg/day atorvastatin for 2 weeks suggests that statin‐induced muscle cell apoptosis could be strictly dependent on the muscle fibre's metabolic phenotype. However, administration of quercetin, an antioxidant agent, resulted in apoptosis inhibition in fast glycolytic skeletal muscle fibres. 107 On the other hand, pretreatment with atorvastatin was able to protect human chondrocytes from high‐glucose oxidative stress, by inducing expression of antioxidant enzymes, reducing ROS levels, and down‐regulating the Bax/Bcl‐2 ratio and caspase‐3 activation. 108 Altogether, these findings imply that the differential cellular response to oxidative stress is an important determinant of tissue susceptibility to statins not only in terms of mitochondrial function but also in terms of apoptosis induction, according with the aforementioned concept of mitochondrial hormesis. 70
Impairment of mitochondrial energy production affects isoprenoid biosynthesis. Statin treatment leads to depletion of isoprenoids compounds, such as farnesyl pyrophosphate and geranylgeranyl pyrophosphate, as a consequence of the blockade on the mevalonate pathway. 14 Protein farnesylation and geranylgeranylation are essential reactions for maintaining cellular homoeostasis. 109 Depletion of isoprenoids may have a role in statin‐related apoptosis induction, because HMG‐CoA—but not squalene synthase inhibition—resulted in myotoxicity. 104 , 110 , 111 Isoprenoid depletion is likely to cause abnormal activation of γ‐phospholipase C and PI3K signalling pathways, 89 , 112 , 113 thus leading to alteration in calcium metabolism with the related detrimental consequences. 104 Finally, statins might alter the phosphorylation state of Akt by interfering with pathways involved in cell survival. 83 , 84 Despite a large amount of in vitro experiments, very few in vivo studies are currently available about the apoptotic effects of statins.
Genetic background influences susceptibility to statin‐associated symptoms
Polymorphisms on genes encoding for proteins involved in mitochondrial processes can modify the cell susceptibility to statin mitochondrial adverse effects. In fact, subclinical genetic mutations or polymorphisms have been associated with higher risk of SAS and SAMS. 114 Polymorphisms on gene COQ2, encoding for an enzyme involved in endogenous ubiquinone biosynthesis, have been associated to SAS and SAMS. 115 Similar findings have been reported for ATP2B1, encoding for a calcium transporting ATPase, and DMPK, encoding for a protein kinase with pathogenic implications in myotonic dystrophy. 116
GATM gene
The GATM gene encodes for the mitochondrial enzyme glycine amidinotransferase, which is the rate‐limiting enzyme in creatine biosynthesis. 117 In a study carried out on lymphoblastoid cells of 480 statin‐treated individuals, acute in vitro administration of simvastatin was associated with reduced gene expression levels and with reduced creatine synthesis in carriers of the rs9806699 single nucleotide polymorphism. 118 Moreover, the minor GATM allele, which is responsible for a reduced creatine yield, was associated with lower SIM rates but not with CK concentration in two different populations treated with 40 mg/day simvastatin for 6 weeks. 118 Although no causal mechanism can be inferred, it was speculated that reduced phosphocreatine storage impairs adenosine monophosphate‐activated protein kinase signalling, in a way that could be protective against glucose and/or cholesterol deprivation. 118 , 119 However, these data were not replicated in subsequent studies. 120 , 121 Areas of uncertainty still exist on this topic.
Mitochondrial encephalopathy, lactic acidosis, and stroke‐like episodes syndrome
Mitochondrial encephalopathy, lactic acidosis, and stroke‐like episodes syndrome is an archetypical model of mitochondrial genome inheritable disease. Because all mtDNA genes encode for mitochondrial proteins, these pathologies are characterized by dysfunctions in organs and tissues with high‐energy requirements. 122 Statin treatment has been shown to disclose several cases of previously asymptomatic mitochondrial encephalopathy, lactic acidosis, and stroke‐like episodes syndrome, possibly as a consequence of reduction in mitochondrial number and volume 79 , 123 or as a consequence of CoQ10 depletion, 123 , 124 as discussed previously.
Carnitine palmitoyltransferase‐2 deficiency
Carnitine palmitoyltransferase‐2 (CPT‐2) is an IMM enzyme that allows acyl‐CoA to undergo β‐oxidation in mitochondrial matrix. CPT‐2 deficiency is the most common disorder of lipid metabolism and shows up with recurrent myalgia, rhabdomyolysis, and myoglobinuria that is precipitated by physical exercise, cold, infections, emotional stress, or fasting, usually during adulthood. Laboratory tests display high plasma acylcarnitine levels and acylcarnitine/carnitine ratio. 125 Such laboratory findings were found in a rabbit model treated with 30 mg/day lovastatin for 16 weeks, whereas the liver, heart, and skeletal muscle carnitine concentrations decreased after treatment. Interestingly, CPT‐2 activity was significantly increased in the heart and liver but not in skeletal muscle. 126 In a case–control study, the frequency of the CPT‐2 heterozygous mutation was 13‐fold higher over the expected general population rate in patients affected by SIM. Statin treatment in those patients could be responsible for an impairment in CPT‐2 activity, leading to a critical reduction of the enzyme overall activity and eliciting homozygous‐like clinical manifestations. 114 These results corroborate the hypothesis that statin‐induced mitochondrial dysfunction could also occur as impaired fatty acid oxidation, as suggested by histopathological findings. 57 The abnormally increased respiratory exchange ratio in patients who had experienced myositis 127 but not in those without SAMS 128 provides further evidence. Moreover, in high‐fat‐fed apoE knockout mice experiencing impaired exercise training adaptation after statin treatment, the administration of trimetazidine, a β‐oxidation inhibitor, was successful in reversing statin‐induced muscle injury, 129 suggesting that lipid metabolism disruption could be one of the several pathological mechanisms for SAMS.
Statin‐induced diabetes and mitochondrial function
In the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER), the use of pravastatin (40 mg/day) in old patients was associated with an increased incidence of T2DM. 130 Similarly, in the Justification for the Use of Statins in Primary Prevention: Intervention Trial Evaluating Rosuvastatin (JUPITER) trial, a significantly higher incidence of new‐onset diabetes occurred in the rosuvastatin group when compared with the placebo group during the 2 years of statin administration. 131 A large meta‐analysis of 13 trials including 91 140 patients reported a significant 9% increase in the risk of T2DM among patients treated with statins. 132 Because of these findings, FDA imposed to report the risk of new‐onset diabetes on statin safety labels.
In an experimental setting, rats treated with 80 mg/kg/day lovastatin or 20 mg/kg/day atorvastatin for 2 weeks displayed increased glucose levels and decreased insulin secretion compared with controls. 133 Statin‐treated rats also showed increased pancreatic cells ROS production, disrupted membrane potential, mitochondrial swelling, and reduced complex IV activity. 133 Administration of CoQ10 and l‐carnitine separately or together was able to partially but significantly blunt these statin‐induced abnormalities. 133
We herein discuss the mitochondria‐related mechanisms potentially explaining the increased risk of T2DM in statin‐treated patients.
Impaired insulin secretion
ATP levels are crucial in beta cells because they regulate the KATP channel closure that leads to cell depolarization and to insulin secretion. Mitochondrial dysfunction in beta cells could impair insulin secretion. 134 In a study on human pancreatic islets and in rat insulinoma INS‐1 cells, atorvastatin—but not pravastatin—impaired both basal and glucose‐induced insulin secretion. 135 This phenomenon is likely to be attributed to a significant decrease in ATP basal levels and ATP production rate, which is subsequent to a time and dose‐dependent reduction in complexes I, III, and IV, and ATP synthase expression in atorvastatin‐exposed cells. 135 Both mevalonate administration and N‐acetylcysteine, an antioxidant agent, were able to rescue the impaired insulin secretion in statin‐exposed cells. 135 In rat isolated pancreatic mitochondria, exposure to >75 μM atorvastatin significantly impaired complex II activity and reduced ATP levels. 136
Nevertheless, experiments with patch clamping suggested that statins might impair insulin secretion by inhibiting L‐type voltage‐dependent Ca2+ channel as well, preventing the Ca2+ cytosolic concentration increase that is fundamental for insulin secretion. 137 The contrasting results obtained with atorvastatin and pravastatin account for their different diabetogenic potential. 138
Atorvastatin exposure determined a significant increase in ROS levels both in human pancreatic islets and in INS‐1 rat insulinoma cells 135 and in rat isolated pancreatic mitochondria 136 in a time and dose‐dependent manner. Because pancreatic beta cells have weak antioxidant systems, oxidative stress may be crucial for beta‐cell dysfunction, thus increasing the risk of new‐onset diabetes during statin treatment. 136
Finally, CoQ10 concentration was reduced by atorvastatin administration as well, leading to ETC activity impairment. 135 As a consequence of ETC activity disruption, atorvastatin caused a decline in mitochondrial membrane potential, mitochondrial swelling, and cytochrome c release, 136 thus leading to intrinsic apoptosis induction in the pancreas.
Peripheral insulin resistance
Statins could also induce peripheral insulin resistance via different pathways. Simvastatin determined a reduction in insulin‐dependent glucose transporter GLUT4 expression in 3T3‐L1 adipocytes, and this phenomenon could be rescued by CoQ10 administration. 139 The accumulation of non‐esterified fatty acids in skeletal muscle cells is responsible for insulin resistance. 140 Mitochondrial uncoupling protein (UCP)‐3 is a protein located in the IMM that acts as a fatty acid carrier, favouring their β‐oxidation. 141 Patients affected by T2DM show lower expression of UCP3. 142 A case–control study pointed out a significantly reduced UCP3 expression in patients treated with simvastatin. 143 The complex relationship among statins, UCP3, and insulin resistance has not been fully clarified yet. Though no causative mechanisms can be inferred, it can be hypothesized that statins could play a role in determining skeletal muscle insulin resistance by down‐regulating UCP3 expression.
Cognitive impairment and dementia
Many studies have been published in the last two decades about the relationship between statin treatment and cognitive function. Like skeletal muscle, brain tissue has a high metabolic demand and hence suffers from mitochondrial vulnerability. 144
A large retrospective study showed an increased risk for both Alzheimer's disease and vascular dementia in hypercholesterolaemic patients after a three‐decade follow‐up. 145
Intriguingly, several case reports 146 , 147 and two randomized controlled trials 148 , 149 suggested a potential association between statin treatment and cognitive impairment, leading FDA to include in the warning section of all statin labels a statement about the risk for reversible memory loss or impairment. Contrariwise, large trials, 130 , 150 a Cochrane database systematic review, 151 a consensus statement by the Statin Cognitive Safety Task Force, 152 and more recent meta‐analyses 153 , 154 failed to demonstrate any effect of statin treatment on cognitive function, while a large meta‐analysis even showed a great risk reduction for dementia associated with statin treatment. 155 An extensive and punctual review about the state of the art on this topic has been recently published. 156
It is plausible that these contradicting results derive from different mechanisms that might be responsible for both cognitive protective effect of statins and treatment‐related adverse cognitive effects. 156
In terms of mitochondrial function, the pathological basis for these contradictory results is completely to be elucidated. It is conceivable that the mechanisms implicated in SAMS, such as CoQ10 depletion, ROS accumulation, and mitochondrial membrane depolarization, could account for cognitive impairment too. Accordingly, genetic disorders, dysthyroidisms, or the metabolic syndrome, which all underlie mitochondrial disfunction, are well‐known risk factors for statin‐related cognitive adverse effects. 15 , 157 However, it must be underlined that simvastatin was able to protect a neuroblastoma cell line (i.e. SH‐SY5Y) from amyloid‐β neurotoxicity by improving several aspects of mitochondrial function (e.g. cytochrome c release, ROS production, Bcl‐2/Bax ratio, and membrane potential). 158 In addition, in a cellular model of mitochondrial dysfunction (i.e. cybrid), low‐dose (1 μM) simvastatin reduced hypoxia‐inducible factor 1α and β‐site amyloid precursor protein cleaving enzyme, the latter molecules promoting amyloid‐β production. However, higher dose of simvastatin (10 μM) increased hypoxia‐inducible factor 1α and β‐site amyloid precursor protein cleaving enzyme expression both in the cybrid and control cell models, 159 thus suggesting that statin dose might differentially affect neuronal function irrespective of mitochondrial activity.
Conclusions
Besides the well‐known beneficial effects in primary and secondary prevention of CVD, statins may induce adverse effects, like myopathy. Mitochondrial dysfunction is likely to play an important role in the pathogenesis of these adverse reactions due to CoQ10 depletion, inhibition of ETC complexes, depletion of mevalonate pathway end products, membrane depolarization and induction of intrinsic apoptosis, dysregulation of calcium metabolism, and fatty acid oxidation. Chronic statin treatment has been associated with increased risk for T2DM and cognitive impairment. The beneficial effects of statin treatment are not questionable; however, the widespread usage of these drugs and the considerable prevalence of side effects require the underlying pathological mechanisms to be carefully studied.
Conflict of interest
Dr M.B. has served on the speakers bureau of Abbott/Mylan, Abbott Vascular, Actavis, Akcea, Amgen, Biofarm, KRKA, MSD, Sanofi‐Aventis, Servier, and Valeant and has served as a consultant to Abbott Vascular, Akcea, Amgen, Daichii Sankyo, Esperion, Lilly, MSD, Resverlogix, and Sanofi‐Aventis; he also received grants from Sanofi and Valeant. S.v.H. has been a paid consultant for and/or received honoraria payments from Bayer, Boehringer Ingelheim, BRAHMS, Chugai, Grünenthal, Helsinn, Hexal, ovartis, Respicardia, Roche, Sorin, and Vifor; owns shares in Actimed. SvH; and reports research support from IMI and the German Center for Cardiovascular Research (DZHK).
Other authors have no competing interests to declare.
Funding
None.
Acknowledgements
The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle. 160 The authors also thank Dr Andy Gitsham for his help in editing the manuscript.
Mollazadeh H., Tavana E., Fanni G., Bo S., Banach M., Pirro M., von Haehling S., Jamialahmadi T., and Sahebkar A. (2021) Effects of statins on mitochondrial pathways, Journal of Cachexia, Sarcopenia and Muscle, 12, 237–251, 10.1002/jcsm.12654
References
- 1. Mensah GA, Roth GA, Fuster V. The global burden of cardiovascular diseases and risk factors. J Am Coll Cardiol 2019;74:2529–2532. [DOI] [PubMed] [Google Scholar]
- 2. Forouzanfar MH, Afshin A, Alexander LT, Anderson HR, Bhutta ZA, Biryukov S, et al. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016;388:1659–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sampson UK, Fazio S, Linton MF. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Curr Atheroscler Rep 2012;14:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, et al. Low‐density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2017;38:2459–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ference BA, Majeed F, Penumetcha R, Flack JM, Brook RD. Effect of naturally random allocation to lower low‐density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both. J Am Coll Cardiol 2015;65:1552–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kapur NK, Musunuru K. Clinical efficacy and safety of statins in managing cardiovascular risk. Vasc Health Risk Manag 2008;4:341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Franklin BA, Durstine JL, Roberts CK, Barnard RJ. Impact of diet and exercise on lipid management in the modern era. Best Pract Res Clin Endocrinol Metab 2014;28:405–421. [DOI] [PubMed] [Google Scholar]
- 8. Pirro M, Vetrani C, Bianchi C, Mannarino MRR, Bernini F, Rivellese AAA. Joint position statement on “Nutraceuticals for the treatment of hypercholesterolemia” of the Italian Society of Diabetology (SID) and of the Italian Society for the Study of Arteriosclerosis (SISA). Nutr Metab Cardiovasc Dis 2017;27:2–17. [DOI] [PubMed] [Google Scholar]
- 9. Bianconi V, Mannarino MR, Sahebkar A, Cosentino T, Pirro M. Cholesterol‐lowering nutraceuticals affecting vascular function and cardiovascular disease risk. Curr Cardiol Rep 2018;20:53. [DOI] [PubMed] [Google Scholar]
- 10. Pirro M, Mannarino MR, Bianconi V, Simental‐Mendía LE, Bagaglia F, Mannarino E, et al. The effects of a nutraceutical combination on plasma lipids and glucose: a systematic review and meta‐analysis of randomized controlled trials. Pharmacol Res 2016;110:76–88. [DOI] [PubMed] [Google Scholar]
- 11. Johnston TP, Korolenko TA, Pirro M, Sahebkar A. Preventing cardiovascular heart disease: promising nutraceutical and non‐nutraceutical treatments for cholesterol management. Pharmacol Res 2017;120:219–225. [DOI] [PubMed] [Google Scholar]
- 12. Sahebkar A, Simental‐Mendía LE, Reiner Ž, Kovanen PT, Simental‐Mendía M, Bianconi V, et al. Effect of orlistat on plasma lipids and body weight: a systematic review and meta‐analysis of 33 randomized controlled trials. Pharmacol Res 2017;122:53–65. [DOI] [PubMed] [Google Scholar]
- 13. Endo A, Kuroda M, Tsujita Y. ML‐236A, ML‐236B, and ML‐236C, new inhibitors of cholesterogensis produced by penicillium citrinum. J Antibiot (Tokyo) 1976;29:1346–1348. [DOI] [PubMed] [Google Scholar]
- 14. Stancu C, Sima A. Statins: mechanism of action and effects. J Cell Mol Med 2001;5:378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Golomb BA, Evans MA. Statin adverse effects. Am J Cardiovasc Drugs 2008;8:373–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sahebkar A, Serban C, Ursoniu S, Mikhailidis DP, Undas A, Lip GYH, et al. The impact of statin therapy on plasma levels of von Willebrand factor antigen. J Thromb Haemost 2016;115:03:520–532. 10.1160/th15-08-0620 [DOI] [PubMed] [Google Scholar]
- 17. Afshari AR, Mollazadeh H, Henney NC, Jamialahmad T, Sahebkar A. Effects of statins on brain tumors: a review. Sem Cancer Biol 2020. 10.1016/j.semcancer.2020.08.002 [DOI] [PubMed] [Google Scholar]
- 18. Bagheri H, Ghasemi F, Barreto GE, Sathyapalan T, Jamialahmadi T, Sahebkar A. The effects of statins on microglial cells to protect against neurodegenerative disorders: A mechanistic review. BioFactors 2020;46:3:309–325. 10.1002/biof.1597 [DOI] [PubMed] [Google Scholar]
- 19. Parizadeh SMR, Azarpazhooh MR, Moohebati M, Nematy M, Ghayour‐Mobarhan M, Tavallaie S, et al. Simvastatin therapy reduces prooxidant‐antioxidant balance: results of a placebo‐controlled cross‐over trial. Lipids 2011;46:333–340. [DOI] [PubMed] [Google Scholar]
- 20. Chruściel P, Sahebkar A, Rembek‐Wieliczko M, Serban M‐C, Ursoniu S, Mikhailidis DP, et al. Impact of statin therapy on plasma adiponectin concentrations: a systematic review and meta‐analysis of 43 randomized controlled trial arms. Atherosclerosis 2016;253:194–208. [DOI] [PubMed] [Google Scholar]
- 21. Mohammadzadeh N, Montecucco F, Carbone F, Xu S, Al‐Rasadi K, Sahebkar A. Statins: Epidrugs with effects on endothelial health? Eur J Clin Invest 2020;50:12. 10.1111/eci.13388 [DOI] [PubMed] [Google Scholar]
- 22. Calabrò P, Yeh ETH. The pleiotropic effects of statins. Curr Opin Cardiol 2005;20:541–546. [DOI] [PubMed] [Google Scholar]
- 23. Ii M, Losordo DW. Statins and the endothelium. Vascul Pharmacol 2007;46:1–9. [DOI] [PubMed] [Google Scholar]
- 24. Blanco‐Colio LM, Tuñón J, Martín‐Ventura JL, Egido J. Anti‐inflammatory and immunomodulatory effects of statins. Kidney Int 2003;63:12–23. [DOI] [PubMed] [Google Scholar]
- 25. Stoll LL, McCormick ML, Denning GM, Weintraub NL. Antioxidant effects of statins. Drugs of Today 2004;40:975–990. [DOI] [PubMed] [Google Scholar]
- 26. Mahley RW, Berson TP. Drug therapy for hypercholesterolemia and dyslipidemia. Goodman Gilman's Pharmacol Basis Ther 2006;933–966. [Google Scholar]
- 27. Gazzerro P, Proto MC, Gangemi G, Malfitano AM, Ciaglia E, Pisanti S, et al. Pharmacological actions of statins: a critical appraisal in the management of cancer. Pharmacology 2012;64:102–146. [DOI] [PubMed] [Google Scholar]
- 28. McTaggart F. Comparative pharmacology of rosuvastatin. Atheroscler Suppl 2003;4:9–14. [DOI] [PubMed] [Google Scholar]
- 29. Thompson PD, Panza G, Zaleski A, Taylor B. Statin‐associated side effects. J Am Coll Cardiol 2016;67:2395–2410. [DOI] [PubMed] [Google Scholar]
- 30. Lemstra M, Blackburn D, Crawley A, Fung R. Proportion and risk indicators of nonadherence to statin therapy: a meta‐analysis. Can J Cardiol 2012;28:574–580. [DOI] [PubMed] [Google Scholar]
- 31. Mann DM, Woodward M, Muntner P, Falzon L, Kronish I. Predictors of nonadherence to statins: a systematic review and meta‐analysis. Ann Pharmacother 2010;44:1410–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maningat P, Gordon BR, Breslow JL. How do we improve patient compliance and adherence to long‐term statin therapy? Curr Atheroscler Rep 2013;15:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Venturini CD, Engroff P, Ely LS, de Araújo Zago LF, Schroeter G, Gomes I, et al. Gender differences, polypharmacy, and potential pharmacological interactions in the elderly. Clinics 2011;66:1867–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Apostolopoulou M, Corsini A, Roden M. The role of mitochondria in statin‐induced myopathy. Eur J Clin Invest 2015;45:745–754. [DOI] [PubMed] [Google Scholar]
- 35. Auer J, Sinzinger H, Franklin BARRY, Berent R. Muscle‐ and skeletal‐related side‐effects of statins: tip of the iceberg? Eur J Prev Cardiol 2016;23:88–110. [DOI] [PubMed] [Google Scholar]
- 36. Scott RS, Lintott CJ, Wilson MJ. Simvastatin and side effects. N Z Med J 1991;104:493–495. [PubMed] [Google Scholar]
- 37. Wierzbicki AS, Lumb PJ, Semra Y, Chik G, Christ ER, Crook MA. Atorvastatin compared with simvastatin‐based therapies in the management of severe familial hyperlipidaemias. QJM 1999;92:387–394. [DOI] [PubMed] [Google Scholar]
- 38. Stroes ES, Thompson PD, Corsini A, Vladutiu GD, Raal FJ, Ray KK, et al. Statin‐associated muscle symptoms: impact on statin therapy—European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur Heart J 2015;36:1012–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thompson PD. In Thompson PD, Taylor BA, eds. The Clinical Presentation of Statin‐Associated Muscle Symptoms (SAMS). Cham: Springer; 2020. p 21–26. [Google Scholar]
- 40. Zaleski AL, Taylor BA, Thompson PD. Coenzyme Q10 as treatment for statin‐associated muscle symptoms—a good idea, but …. Adv Nutr 2018;9:519S–523S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Parker BA, Capizzi JA, Grimaldi AS, Clarkson PM, Cole SM, Keadle J, et al. Effect of statins on skeletal muscle function. Circulation 2013;127:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krishnan GM, Thompson PD. The effects of statins on skeletal muscle strength and exercise performance. Curr Opin Lipidol 2010;21:324–328. [DOI] [PubMed] [Google Scholar]
- 43. Thompson PD, Clarkson P, Karas RH. Statin‐associated myopathy. JAMA 2003;289:1681–1690. [DOI] [PubMed] [Google Scholar]
- 44. Toth PP, Harper CR, Jacobson TA. Clinical characterization and molecular mechanisms of statin myopathy. Expert Rev Cardiovasc Ther 2008;6:955–969. [DOI] [PubMed] [Google Scholar]
- 45. Magni P, Macchi C, Morlotti B, Sirtori CR, Ruscica M. Risk identification and possible countermeasures for muscle adverse effects during statin therapy. Eur J Intern Med 2015;26:82–88. [DOI] [PubMed] [Google Scholar]
- 46. McKenney JM, Davidson MH, Jacobson TA, Guyton JR. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol 2006;97:S89–S94. [DOI] [PubMed] [Google Scholar]
- 47. Pasternak RC, Smith SC, Bairey‐Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Circulation 2002;106:1024–1028. [DOI] [PubMed] [Google Scholar]
- 48. Maji D, Shaikh S, Solanki D, Gaurav K. Safety of statins. Indian J Endocrinol Metab Med 2013;17:636. 10.4103/2230-8210.113754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Newman CB, Tobert JA. Statin‐related myopathy and rhabdomyolysis. Endocr Metab Med 2018;760–774. [Google Scholar]
- 50. Chavez LO, Leon M, Einav S, Varon J. Beyond muscle destruction: a systematic review of rhabdomyolysis for clinical practice. Crit Care 2016;20:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bindoff L. Mitochondria and the heart. Eur Heart J 2003;24:221–224. [DOI] [PubMed] [Google Scholar]
- 52. Rich PR, Maréchal A. The mitochondrial respiratory chain. Essays Biochem 2010;47:1–23. [DOI] [PubMed] [Google Scholar]
- 53. Brookes PS. Mitochondrial H+ leak and ROS generation: an odd couple. Free Radic Biol Med 2005;38:12–23. [DOI] [PubMed] [Google Scholar]
- 54. Raha S, Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem Sci 2000;25:502–508. [DOI] [PubMed] [Google Scholar]
- 55. Chan K, Truong D, Shangari N, O'Brien PJ. Drug‐induced mitochondrial toxicity. Expert Opin Drug Metab Toxicol 2005;1:655–669. [DOI] [PubMed] [Google Scholar]
- 56. Arenas J, Fernández‐Moreno MA, Molina JA, Fernández V, Del Hoyo P, Campos Y, et al. Myoglobinuria and COX deficiency in a patient taking cerivastatin and gemfibrozil. Neurology 2003;60:124–126. [DOI] [PubMed] [Google Scholar]
- 57. Phillips PS, Haas RH, Bannykh S, Hathaway S, Gray NL, Kimura BJ, et al. Statin‐associated myopathy with normal creatine kinase levels. Ann Intern Med 2002;137:581–585. [DOI] [PubMed] [Google Scholar]
- 58. De Pinieux G, Chariot P, Ammi‐Saïd M, Louarn F, Lejonc JL, Astier A, et al. Lipid‐lowering drugs and mitochondrial function: effects of HMG‐CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol 2003;42:333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wu JS, Buettner C, Smithline H, Ngo LH, Greenman RL. Evaluation of skeletal muscle during calf exercise by 31‐phosphorus magnetic resonance spectroscopy in patients on statin medications. Muscle Nerve 2011;43:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liantonio A, Giannuzzi V, Cippone V, Camerino GM, Pierno S, Camerino DC. Fluvastatin and atorvastatin affect calcium homeostasis of rat skeletal muscle fibers in vivo and in vitro by impairing the sarcoplasmic reticulum/mitochondria Ca2+‐release system. J Pharmacol Exp Ther 2007;321:626–634. [DOI] [PubMed] [Google Scholar]
- 61. Asping M, Stride N, Søgaard D, Dohlmann TL, Helge JW, Dela F, et al. The effects of 2 weeks of statin treatment on mitochondrial respiratory capacity in middle‐aged males: the LIFESTAT study. Eur J Clin Pharmacol 2017;73:679–687. [DOI] [PubMed] [Google Scholar]
- 62. Galtier F, Mura T, Raynaud de Mauverger E, Chevassus H, Farret A, Gagnol J‐P, et al. Effect of a high dose of simvastatin on muscle mitochondrial metabolism and calcium signaling in healthy volunteers. Toxicol Appl Pharmacol 2012;263:281–286. [DOI] [PubMed] [Google Scholar]
- 63. Hou T, Li Y, Chen W, Heffner RR, Vladutiu GD. Histopathologic and biochemical evidence for mitochondrial disease among 279 patients with severe statin myopathy. J Neuromuscul Dis 2017;4:77–87. [DOI] [PubMed] [Google Scholar]
- 64. Marcoff L, Thompson PD. The role of coenzyme Q10 in statin‐associated myopathy. J Am Coll Cardiol 2007;49:2231–2237. [DOI] [PubMed] [Google Scholar]
- 65. Garrido‐Maraver J, Cordero MD, Oropesa‐Ávila M, Fernández Vega A, de la Mata M, Delgado Pavón A, et al. Coenzyme Q10 therapy. Mol Syndromol 2014;5:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sahebkar A, Simental‐Mendía LE, Stefanutti C, Pirro M. Supplementation with coenzyme Q10 reduces plasma lipoprotein(a) concentrations but not other lipid indices: a systematic review and meta‐analysis. Pharmacol Res 2016;105:198–209. [DOI] [PubMed] [Google Scholar]
- 67. Åberg F, Appelkvist EL, Dallner G, Ernster L. Distribution and redox state of ubiquinones in rat and human tissues. Arch Biochem Biophys 1992;295:230–234. [DOI] [PubMed] [Google Scholar]
- 68. Salviati L, Trevisson E, Doimo M, Navas P. Primary Coenzyme Q10 Deficiency. In Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., eds. GeneReviews®. Seattle, WA: University of Washington; 2017. [Google Scholar]
- 69. Saha SP, Whayne TF. Coenzyme Q10 in human health: supporting evidence? South Med J 2016;109:17–21. [DOI] [PubMed] [Google Scholar]
- 70. Bouitbir J, Charles A‐L, Echaniz‐Laguna A, Kindo M, Daussin F, Auwerx J, et al. Opposite effects of statins on mitochondria of cardiac and skeletal muscles: a ‘mitohormesis’ mechanism involving reactive oxygen species and PGC‐1. Eur Heart J 2012;33:1397–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kaufmann P, Török M, Zahno A, Waldhauser KM, Brecht K, Krähenbühl S. Toxicity of statins on rat skeletal muscle mitochondria. Cell Mol Life Sci 2006;63:2415–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Päivä H, Thelen KM, Van Coster R, Smet J, De Paepe B, Mattila KM, et al. High‐dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther 2005;78:60–68. [DOI] [PubMed] [Google Scholar]
- 73. du Souich P, Roederer G, Dufour R. Myotoxicity of statins: mechanism of action. Pharmacol Ther 2017;175:1–16. [DOI] [PubMed] [Google Scholar]
- 74. Laaksonen R, Jokelainen K, Laakso J, Sahi T, Härkönen M, Tikkanen MJ, et al. The effect of simvastatin treatment on natural antioxidants in low‐density lipoproteins and high‐energy phosphates and ubiquinone in skeletal muscle. Am J Cardiol 1996;77:851–854. [DOI] [PubMed] [Google Scholar]
- 75. Laaksonen R, Jokelainen K, Sahi T, Tikkanen MJ, Himberg JJ. Decreases in serum ubiquinone concentrations do not result in reduced levels in muscle tissue during short‐term simvastatin treatment in humans. Clin Pharmacol Ther 1995;57:62–66. [DOI] [PubMed] [Google Scholar]
- 76. Lamperti C, Naini AB, Lucchini V, Prelle A, Bresolin N, Moggio M, et al. Muscle coenzyme Q10 level in statin‐related myopathy. Arch Neurol 2005;62:1709–1712. [DOI] [PubMed] [Google Scholar]
- 77. Banach M, Serban C, Sahebkar A, Ursoniu S, Rysz J, Muntner P, et al. Effects of coenzyme Q10 on statin‐induced myopathy: a meta‐analysis of randomized controlled trials. Mayo Clin Proc 2015;90:24–34. [DOI] [PubMed] [Google Scholar]
- 78. Qu H, Guo M, Chai H, Wang W, Gao Z, Shi D. Effects of coenzyme Q10 on statin‐induced myopathy: an updated meta‐analysis of randomized controlled trials. J Am Heart Assoc 2018;7:e009835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schick BA, Laaksonen R, Frohlich JJ, Päivä H, Lehtimäki T, Humphries KH, et al. Decreased skeletal muscle mitochondrial DNA in patients treated with high‐dose simvastatin. Clin Pharmacol Ther 2007;81:650–653. [DOI] [PubMed] [Google Scholar]
- 80. Stringer HAJ, Sohi GK, Maguire JA, Côté HCF. Decreased skeletal muscle mitochondrial DNA in patients with statin‐induced myopathy. J Neurol Sci 2013;325:142–147. [DOI] [PubMed] [Google Scholar]
- 81. Mikus CR, Boyle LJ, Borengasser SJ, Oberlin DJ, Naples SP, Fletcher J, et al. Simvastatin impairs exercise training adaptations. J Am Coll Cardiol 2013;62:709–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Singla M, Rastogi A, Aggarwal AN, Bhat OM, Badal D, Bhansali A. Vitamin D supplementation improves simvastatin‐mediated decline in exercise performance: a randomized double‐blind placebo‐controlled study. J Diabetes 2017;9:1100–1106. [DOI] [PubMed] [Google Scholar]
- 83. Mullen PJ, Zahno A, Lindinger P, Maseneni S, Felser A, Krähenbühl S, et al. Susceptibility to simvastatin‐induced toxicity is partly determined by mitochondrial respiration and phosphorylation state of Akt. Biochim Biophys Acta – Mol Cell Res 2011;1813:2079–2087. [DOI] [PubMed] [Google Scholar]
- 84. Bonifacio A, Sanvee GM, Bouitbir J, Krähenbühl S. The AKT/mTOR signaling pathway plays a key role in statin‐induced myotoxicity. Biochim Biophys Acta – Mol Cell Res 2015;1853:1841–1849. [DOI] [PubMed] [Google Scholar]
- 85. Vaughan RA, Garcia‐Smith R, Bisoffi M, Conn CA, Trujillo KA. Ubiquinol rescues simvastatin‐suppression of mitochondrial content, function and metabolism: implications for statin‐induced rhabdomyolysis. Eur J Pharmacol 2013;711:1–9. [DOI] [PubMed] [Google Scholar]
- 86. Singh F, Zoll J, Duthaler U, Charles A‐L, Panajatovic MV, Laverny G, et al. PGC‐1β modulates statin‐associated myotoxicity in mice. Arch Toxicol 2019;93:487–504. [DOI] [PubMed] [Google Scholar]
- 87. Liu A, Wu Q, Guo J, Ares I, Rodríguez J‐L, Martínez‐Larrañaga M‐R, et al. Statins: adverse reactions, oxidative stress and metabolic interactions. Pharmacol Ther 2019;195:54–84. [DOI] [PubMed] [Google Scholar]
- 88. Kwak H‐B, Thalacker‐Mercer A, Anderson EJ, Lin C‐T, Kane DA, Lee N‐S, et al. Simvastatin impairs ADP‐stimulated respiration and increases mitochondrial oxidative stress in primary human skeletal myotubes. Free Radic Biol Med 2012;52:198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sirvent P, Mercier J, Lacampagne A. New insights into mechanisms of statin‐associated myotoxicity. Curr Opin Pharmacol 2008;8:333–338. [DOI] [PubMed] [Google Scholar]
- 90. Schirris TJJ, Renkema GH, Ritschel T, Voermans NC, Bilos A, van Engelen BGM, et al. Statin‐induced myopathy is associated with mitochondrial complex III inhibition. Cell Metab 2015;22:399–407. [DOI] [PubMed] [Google Scholar]
- 91. Sirvent P, Bordenave S, Vermaelen M, Roels B, Vassort G, Mercier J, et al. Simvastatin induces impairment in skeletal muscle while heart is protected. Biochem Biophys Res Commun 2005;338:1426–1434. [DOI] [PubMed] [Google Scholar]
- 92. Nakahara K, Kuriyama M, Sonoda Y, Yoshidome H, Nakagawa H, Fujiyama J, et al. Myopathy induced by HMG‐CoA reductase inhibitors in rabbits: a pathological, electrophysiological, and biochemical study. Toxicol Appl Pharmacol 1998;152:99–106. [DOI] [PubMed] [Google Scholar]
- 93. Schaefer WH, Lawrence JW, Loughlin AF, Stoffregen DA, Mixson LA, Dean DC, et al. Evaluation of ubiquinone concentration and mitochondrial function relative to cerivastatin‐induced skeletal myopathy in rats. Toxicol Appl Pharmacol 2004;194:10–23. [DOI] [PubMed] [Google Scholar]
- 94. Duncan AJ, Hargreaves IP, Damian MS, Land JM, Heales SJR. Decreased ubiquinone availability and impaired mitochondrial cytochrome oxidase activity associated with statin treatment. Toxicol Mech Methods 2009;19:44–50. [DOI] [PubMed] [Google Scholar]
- 95. Murlasits Z, Radák Z. The effects of statin medications on aerobic exercise capacity and training adaptations. Sports Med 2014;44:1519–1530. [DOI] [PubMed] [Google Scholar]
- 96. Herminghaus A, Laser E, Schulz J, Truse R, Vollmer C, Bauer I, et al. Pravastatin and gemfibrozil modulate differently hepatic and colonic mitochondrial respiration in tissue homogenates from healthy rats. Cell 2019;8:983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ramachandran R, Wierzbicki A. Statins, muscle disease and mitochondria. J Clin Med 2017;6:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sirvent P, Fabre O, Bordenave S, Hillaire‐Buys D, Raynaud De Mauverger E, Lacampagne A, et al. Muscle mitochondrial metabolism and calcium signaling impairment in patients treated with statins. Toxicol Appl Pharmacol 2012;259:263–268. [DOI] [PubMed] [Google Scholar]
- 99. Sirvent P, Mercier J, Vassort G, Lacampagne A. Simvastatin triggers mitochondria‐induced Ca2+ signaling alteration in skeletal muscle. Biochem Biophys Res Commun 2005;329:1067–1075. [DOI] [PubMed] [Google Scholar]
- 100. Taha DA, De Moor CH, Barrett DA, Lee JB, Gandhi RD, Hoo CW, et al. The role of acid‐base imbalance in statin‐induced myotoxicity. Transl Res 2016;174:140–160.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Skottheim IB, Gedde‐Dahl A, Hejazifar S, Hoel K, Åsberg A. Statin induced myotoxicity: the lactone forms are more potent than the acid forms in human skeletal muscle cells in vitro. Eur J Pharm Sci 2008;33:317–325. [DOI] [PubMed] [Google Scholar]
- 102. Nakahara K, Yada T, Kuriyama M, Osame M. Cytosolic Ca2+ increase and cell damage in L6 rat myoblasts by HMG‐CoA reductase inhibitors. Biochem Biophys Res Commun 1994;202:1579–1585. [DOI] [PubMed] [Google Scholar]
- 103. Hermann M, Bogsrud MP, Molden E, Åsberg A, Mohebi BU, Ose L, et al. Exposure of atorvastatin is unchanged but lactone and acid metabolites are increased several‐fold in patients with atorvastatin‐induced myopathy. Clin Pharmacol Ther 2006;79:532–539. [DOI] [PubMed] [Google Scholar]
- 104. Dirks AJ, Jones KM. Statin‐induced apoptosis and skeletal myopathy. Am J Physiol Physiol 2006;291:C1208–C1212. [DOI] [PubMed] [Google Scholar]
- 105. Suzuki K, Hata S, Kawabata Y, Sorimachi H. Structure, activation, and biology of calpain. Diabetes 2004;53:S12–S18. [DOI] [PubMed] [Google Scholar]
- 106. Sacher J, Weigl L, Werner M, Szegedi C, Hohenegger M. Delineation of myotoxicity induced by 3‐hydroxy‐3‐methylglutaryl CoA reductase inhibitors in human skeletal muscle cells. J Pharmacol Exp Ther 2005;314:1032–1041. [DOI] [PubMed] [Google Scholar]
- 107. Bouitbir J, Singh F, Charles A‐L, Schlagowski A‐I, Bonifacio A, Echaniz‐Laguna A, et al. Statins trigger mitochondrial reactive oxygen species‐induced apoptosis in glycolytic skeletal muscle. Antioxid Redox Signal 2016;24:84–98. [DOI] [PubMed] [Google Scholar]
- 108. Hosseinzadeh A, Bahrampour Juybari K, Kamarul T, Sharifi AM. Protective effects of atorvastatin on high glucose‐induced oxidative stress and mitochondrial apoptotic signaling pathways in cultured chondrocytes. J Physiol Biochem 2019;75:153–162. [DOI] [PubMed] [Google Scholar]
- 109. Novelli G, D'Apice MR. Protein farnesylation and disease. J Inherit Metab Dis 2012;35:917–926. [DOI] [PubMed] [Google Scholar]
- 110. Guijarro C, Blanco‐Colio LM, Ortego M, Alonso C, Ortiz A, Plaza JJ, et al. 3‐Hydroxy‐3‐methylglutaryl coenzyme A reductase and isoprenylation inhibitors induce apoptosis of vascular smooth muscle cells in culture. Circ Res 1998;83:490–500. [DOI] [PubMed] [Google Scholar]
- 111. Johnson TE, Zhang X, Bleicher KB, Dysart G, Loughlin AF, Schaefer WH, et al. Statins induce apoptosis in rat and human myotube cultures by inhibiting protein geranylgeranylation but not ubiquinone. Toxicol Appl Pharmacol 2004;200:237–250. [DOI] [PubMed] [Google Scholar]
- 112. Mutoh T, Kumano T, Nakagawa H, Kuriyama M. Involvement of tyrosine phosphorylation in HMG‐CoA reductase inhibitor‐induced cell death in L6 myoblasts. FEBS Lett 1999;444:85–89. [DOI] [PubMed] [Google Scholar]
- 113. Mutoh T, Kumano T, Nakagawa H, Kuriyama M. Role of tyrosine phosphorylation of phospholipase C γ1 in the signaling pathway of HMG‐CoA reductase inhibitor‐induced cell death of L6 myoblasts. FEBS Lett 1999;446:91–94. [DOI] [PubMed] [Google Scholar]
- 114. Vladutiu GD, Simmons Z, Isackson PJ, Tarnopolsky M, Peltier WL, Barboi AC, et al. Genetic risk factors associated with lipid‐lowering drug‐induced myopathies. Muscle Nerve 2006;34:153–162. [DOI] [PubMed] [Google Scholar]
- 115. Oh J, Ban MR, Miskie BA, Pollex RL, Hegele RA. Genetic determinants of statin intolerance. Lipids Health Dis 2007;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ruaño G, Windemuth A, Wu AHB, Kane JP, Malloy MJ, Pullinger CR, et al. Mechanisms of statin‐induced myalgia assessed by physiogenomic associations. Atherosclerosis 2011;218:451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Humm A, Pritsche E, Steinbacher S. Structure and reaction mechanism of l‐arginine:glycine amidinotransferase. Biol Chem 1997;193–197. [PubMed] [Google Scholar]
- 118. Mangravite LM, Engelhardt BE, Medina MW, Smith JD, Brown CD, Chasman DI, et al. A statin‐dependent QTL for GATM expression is associated with statin‐induced myopathy. Nature 2013;502:377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ballard KD, Thompson PD. Does reduced creatine synthesis protect against statin myopathy? Cell Metab 2013;18:773–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Floyd JS, Bis JC, Brody JA, Heckbert SR, Rice K, Psaty BM. GATM locus does not replicate in rhabdomyolysis study. Nature 2014;513:E1–E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Carr DF, Alfirevic A, Johnson R, Chinoy H, van Staa T, Pirmohamed M. GATM gene variants and statin myopathy risk. Nature 2014;513:E1–E1. [DOI] [PubMed] [Google Scholar]
- 122. Schon EA, Bonilla E, DiMauro S. Mitochondrial DNA mutations and pathogenesis. J Bioenerg Biomembr 1997;29:131–149. [DOI] [PubMed] [Google Scholar]
- 123. Ghatak A, Faheem O, Thompson PD. The genetics of statin‐induced myopathy. Atherosclerosis 2010;210:337–343. [DOI] [PubMed] [Google Scholar]
- 124. Chariot P, Abadia R, Agnus D, Danan C, Charpentier C, Gherasdi RK. Simvastatin‐induced rhabdomyolysis followed by a MELAS syndrome. Am J Med 1993;94:109–110. [DOI] [PubMed] [Google Scholar]
- 125. Bonnefont JP, Djouadi F, Prip‐Buus C, Gobin S, Munnich A, Bastin J. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med 2004;25:495–520. [DOI] [PubMed] [Google Scholar]
- 126. Bhuiyan J, Seccombe DW. The effects of 3‐hydroxy‐3‐methylglutaryl‐CoA reductase inhibition on tissue levels of carnitine and carnitine acyltransferase activity in the rabbit. Lipids 1996;31:867–870. [DOI] [PubMed] [Google Scholar]
- 127. Phillips PS, Phillips CT, Sullivan MJ, Naviaux RK, Haas RH. Statin myotoxicity is associated with changes in the cardiopulmonary function. Atherosclerosis 2004;177:183–188. [DOI] [PubMed] [Google Scholar]
- 128. Traustadóttir T, Stock AA, Harman SM. High‐dose statin use does not impair aerobic capacity or skeletal muscle function in older adults. Age (Omaha) 2008;30:283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Song M, Chen F, Li Y, Zhang L, Wang F, Qin R, et al. Trimetazidine restores the positive adaptation to exercise training by mitigating statin‐induced skeletal muscle injury. J Cachexia Sarcopenia Muscle 2018;9:106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Shepherd J, Blauw GJ, Murphy MB, Bollen ELEM, Buckley BM, Cobbe SM, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002;360:1623–1630. [DOI] [PubMed] [Google Scholar]
- 131. Ridker PM, Danielson E, Fonseca FAH, Genest J, Gotto AM, Kastelein JJP, et al. Rosuvastatin to prevent vascular events in men and women with elevated C‐reactive protein. N Engl J Med 2008;359:2195–2207. [DOI] [PubMed] [Google Scholar]
- 132. Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJ, et al. Statins and risk of incident diabetes: a collaborative meta‐analysis of randomised statin trials. Lancet 2010;375:735–742. [DOI] [PubMed] [Google Scholar]
- 133. Sadighara M, Joktaji J, Hajhashemi V, Minaiyan M. Protective effects of coenzyme Q 10 and L‐carnitine against statin‐induced pancreatic mitochondrial toxicity in rats. Res Pharm Sci 2017;12:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Supale S, Li N, Brun T, Maechler P. Mitochondrial dysfunction in pancreatic β cells. Trends Endocrinol Metab 2012;23:477–487. [DOI] [PubMed] [Google Scholar]
- 135. Urbano F, Bugliani M, Filippello A, Scamporrino A, Di Mauro S, Di Pino A, et al. Atorvastatin but not pravastatin impairs mitochondrial function in human pancreatic islets and rat β‐cells. Direct effect of oxidative stress. Sci Rep 2017;7:11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Sadighara M, Amirsheardost Z, Minaiyan M, Hajhashemi V, Naserzadeh P, Salimi A, et al. Toxicity of atorvastatin on pancreas mitochondria: a justification for increased risk of diabetes mellitus. Basic Clin Pharmacol Toxicol 2017;120:131–137. [DOI] [PubMed] [Google Scholar]
- 137. Yada T, Nakata M, Shiraishi T, Kakei M. Inhibition by simvastatin, but not pravastatin, of glucose‐induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L‐type Ca2+ channels in rat islet β‐cells. Br J Pharmacol 1999;126:1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Cederberg H, Stančáková A, Yaluri N, Modi S, Kuusisto J, Laakso M. Increased risk of diabetes with statin treatment is associated with impaired insulin sensitivity and insulin secretion: a 6 year follow‐up study of the METSIM cohort. Diabetologia 2015;58:1109–1117. [DOI] [PubMed] [Google Scholar]
- 139. Ganesan S, Ito MK. Coenzyme Q10 ameliorates the reduction in GLUT4 transporter expression induced by simvastatin in 3T3‐L1 adipocytes. Metab Syndr Relat Disord 2013;11:251–255. [DOI] [PubMed] [Google Scholar]
- 140. Moro C, Bajpeyi S, Smith SR. Determinants of intramyocellular triglyceride turnover: implications for insulin sensitivity. Am J Physiol Metab 2008;294:E203–E213. [DOI] [PubMed] [Google Scholar]
- 141. Schrauwen P, Hesselink MKC, Blaak EE, Borghouts LB, Schaart G, Saris WHM, et al. Uncoupling protein 3 content is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 2001;50:2870–2873. [DOI] [PubMed] [Google Scholar]
- 142. Liu J, Li J, Li W‐J, Wang C‐M. The role of uncoupling proteins in diabetes mellitus. J Diabetes Res 2013;2013:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Larsen S, Stride N, Hey‐Mogensen M, Hansen CN, Bang LE, Bundgaard H, et al. Simvastatin effects on skeletal muscle. J Am Coll Cardiol 2013;61:44–53. [DOI] [PubMed] [Google Scholar]
- 144. Sastre J. The role of mitochondrial oxidative stress in aging. Free Radic Biol Med 2003;35:1–8. [DOI] [PubMed] [Google Scholar]
- 145. Solomon A, Kivipelto M, Wolozin B, Zhou J, Whitmer RA. Midlife serum cholesterol and increased risk of Alzheimer's and vascular dementia three decades later. Dement Geriatr Cogn Disord 2009;28:75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wagstaff LR, Mitton MW, Arvik BM, Doraiswamy PM. Statin‐associated memory loss: analysis of 60 case reports and review of the literature. Pharmacotherapy 2003;23:871–880. [DOI] [PubMed] [Google Scholar]
- 147. Rojas‐Fernandez CH, Cameron J‐CF. Is statin‐associated cognitive impairment clinically relevant? A narrative review and clinical recommendations. Ann Pharmacother 2012;46:549–557. [DOI] [PubMed] [Google Scholar]
- 148. Muldoon MF, Ryan CM, Sereika SM, Flory JD, Manuck SB. Randomized trial of the effects of simvastatin on cognitive functioning in hypercholesterolemic adults. Am J Med 2004;117:823–829. [DOI] [PubMed] [Google Scholar]
- 149. Muldoon MF, Barger SD, Ryan CM, Flory JD, Lehoczky JP, Matthews KA, et al. Effects of lovastatin on cognitive function and psychological well‐being. Am J Med 2000;108:538–546. [DOI] [PubMed] [Google Scholar]
- 150. Collins R, Armitage J, Parish S, Sleight P, Peto R. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20 536 high‐risk individuals: a randomised placebocontrolled trial. Lancet 2002;360:7–22. [DOI] [PubMed] [Google Scholar]
- 151. McGuinness B, Craig D, Bullock R, Passmore P. Statins for the prevention of dementia. Cochrane Database Syst Rev 2016;2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Rojas‐Fernandez CH, Goldstein LB, Levey AI, Taylor BA, Bittner V. An assessment by the Statin Cognitive Safety Task Force: 2014 update. J Clin Lipidol 2014;8:S5–S16. [DOI] [PubMed] [Google Scholar]
- 153. Ott BR, Daiello LA, Dahabreh IJ, Springate BA, Bixby K, Murali M, et al. Do statins impair cognition? A systematic review and meta‐analysis of randomized controlled trials. J Gen Intern Med 2015;30:348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Swiger KJ, Manalac RJ, Blumenthal RS, Blaha MJ, Martin SS. Statins and cognition: a systematic review and meta‐analysis of short‐ and long‐term cognitive effects. Mayo Clin Proc 2013;88:1213–1221. [DOI] [PubMed] [Google Scholar]
- 155. Song Y, Nie H, Xu Y, Zhang L, Wu Y. Association of statin use with risk of dementia: a meta‐analysis of prospective cohort studies. Geriatr Gerontol Int 2013;13:817–824. [DOI] [PubMed] [Google Scholar]
- 156. Schultz BG, Patten DK, Berlau DJ. The role of statins in both cognitive impairment and protection against dementia: a tale of two mechanisms. Transl Neurodegener 2018;7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Müller WE, Eckert A, Kurz C, Eckert GP, Leuner K. Mitochondrial dysfunction: common final pathway in brain aging and alzheimer's disease—therapeutic aspects. Mol Neurobiol 2010;41:159–171. [DOI] [PubMed] [Google Scholar]
- 158. Li Y, Liu Q, Sun J, Wang J, Liu X, Gao J. Mitochondrial protective mechanism of simvastatin protects against amyloid β peptide‐induced injury in SH‐SY5Y cells. Int J Mol Med 2018;41:2997–3005. [DOI] [PubMed] [Google Scholar]
- 159. Jeong J‐H, Yum KS, Chang JY, Kim M, Ahn J, Kim S, et al. Dose‐specific effect of simvastatin on hypoxia‐induced HIF‐1α and BACE expression in Alzheimer's disease cybrid cells. BMC Neurol 2015;15:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]