

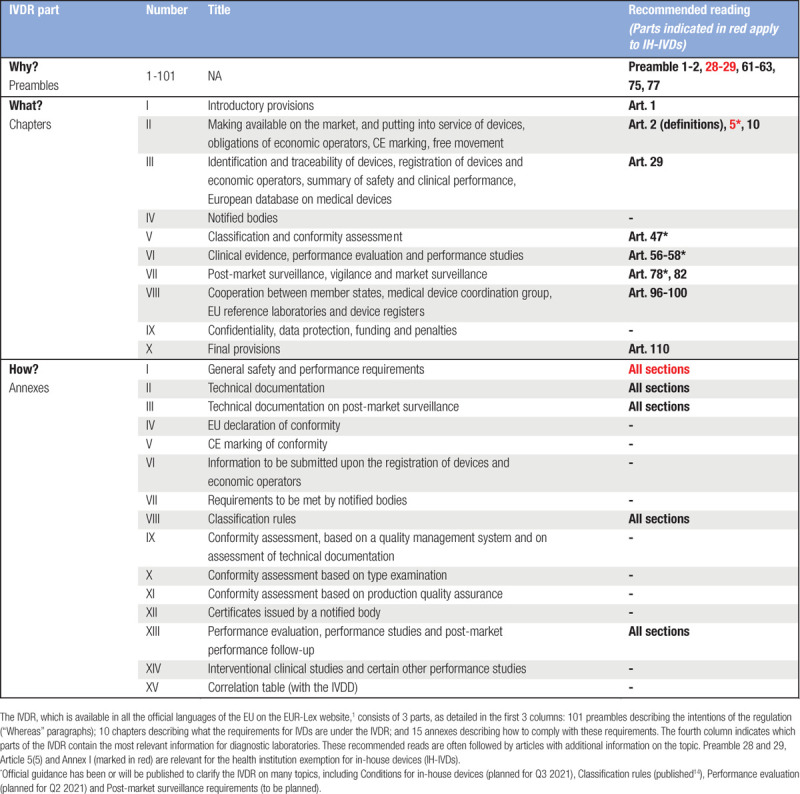
On May 26, 2022, after a transitional period of 5 years, the new Regulation (EU) 2017/746 on in vitro diagnostic medical devices1 (IVDR; Table 1) will fully replace Directive 98/79/EC on in vitro diagnostic medical devices2 (IVDD). The aim of the IVDR is to further establish a well-regulated and smoothly functioning market for in vitro diagnostic medical devices (IVDs; Box 1) within the European Union (EU) that is better aligned with international guidelines. Moreover, following vast technical and medical developments during the previous 2 decades—for example, in the area of genetic testing and companion diagnostics—it was deemed critical to secure protection of safety and health by setting high standards for safety and performance of IVDs.
Table 1.
Structure of the IVDR and Recommended Reading for Diagnostic Laboratories
Box 1. Definition of in vitro diagnostic medical device (IVDR Article 2(2)).1.
“‘in vitro diagnostic medical device’ means any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information on one or more of the following:
(a) concerning a physiological or pathological process or state;
(b) concerning congenital physical or mental impairments;
(c) concerning the predisposition to a medical condition or a disease;
(d) to determine the safety and compatibility with potential recipients;
(e) to predict treatment response or reactions;
(f) to define or monitoring therapeutic measures.
Specimen receptacles shall also be deemed to be in vitro diagnostic medical devices.”
Both the IVDD and IVDR dictate that, depending on their intended use/purpose (and associated risk), IVDs either have to be assessed and certified by an independent third party (a notified body) or can be self-certified by their manufacturer. After certification, manufacturers are allowed to label products with the “Conformité Européenne” (CE) mark, which is required to distribute and sell “CE-IVD” products on the EU market. Even though both legislations share the general concept of IVD certification and CE marking, a much wider range of IVDs will have to be certified by notified bodies and more (clinical) performance data and documentation will be required to legitimately commercialize IVDs under the IVDR. However, the replacement of the IVDD by the IVDR will not only have major consequences for manufacturers of IVDs but also for all diagnostic laboratories.
This report aims to help diagnostic laboratories understand the scope of the IVDR and its consequences for commercially available CE-IVDs and, even more importantly, for in-house devices (IH-IVDs). Although many aspects of the IVDR still have to be clarified, laboratories are urged to take a number of actions to prepare for this new regulation.
Action 1: Appoint a dedicated team and stay informed
It is worthwhile to appoint a small dedicated team with the responsibility for regulatory compliance with the IVDR as early as possible. Looking into the relevant articles and annexes of the IVDR (see Table 1 for recommended reading) is strongly advised for those working in diagnostic laboratories and involved in regulatory compliance. Regularly consulting other information sources, such as guidance documents, publications and white papers, as well as participation in (inter)national conferences and information/consultation sessions on the subject, will help to stay informed about the implementation of the IVDR and the consequences and requirements for laboratories.
The IVDR will affect assay portfolios of diagnostic laboratories
The IVDR primarily regulates CE-IVDs, but also addresses IH-IVDs that are manufactured and used by health institutions (referring to reagents, control materials, software, etc; see also Box 1). The requirements of the IVDR do explicitly not apply to the latter devices—with the exception of the relevant general safety and performance requirements (GSPR) in Annex I—as long as a number of conditions are met (see section “The IVDR dictates requirements for use of IH-IVDs” for details). Since the IVDR has major consequences for both categories of IVDs, an understanding of what the IVDR means for the availability and use of CE-IVDs and IH-IVDs is crucial for diagnostic laboratories.
More demanding requirements for manufacturers will affect the availability of CE-IVD tests
The IVDR requires that all existing and new IVDs are (re)classified on the basis of a risk-based classification system. The class of an IVD depends on its intended purpose and the level of associated risks to patients and the public. Table 2 lists the different classes described in the IVDR (see also IVDR Annex VIII). The highest risk class, class D, includes tests for infectious/transmissible agents that cause life-threatening diseases (eg, HIV, hepatitis B, and SARS-CoV-2) and the most critical blood grouping tests. Many other tests used in the hematology/hemato-oncology field, such as cancer tests and genetic tests, fall within class C.
Table 2.
IVD Classes Under the IVDR
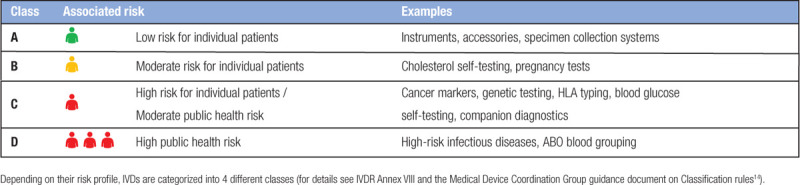
The IVD class determines the exact requirements and assessment route for a given IVD, as well as the depth of documentation required to demonstrate that all requirements have been fulfilled. Non-sterile class A devices can be self-certified by the manufacturer after reaching compliance with the IVDR. All other devices need to undergo conformity assessment by a notified body. In addition, expert panels and EU reference laboratories are also involved in the assessment procedure of class D devices. Following certification, manufacturers are required to update documentation whenever relevant, with a minimum of once per year for class C and D devices.
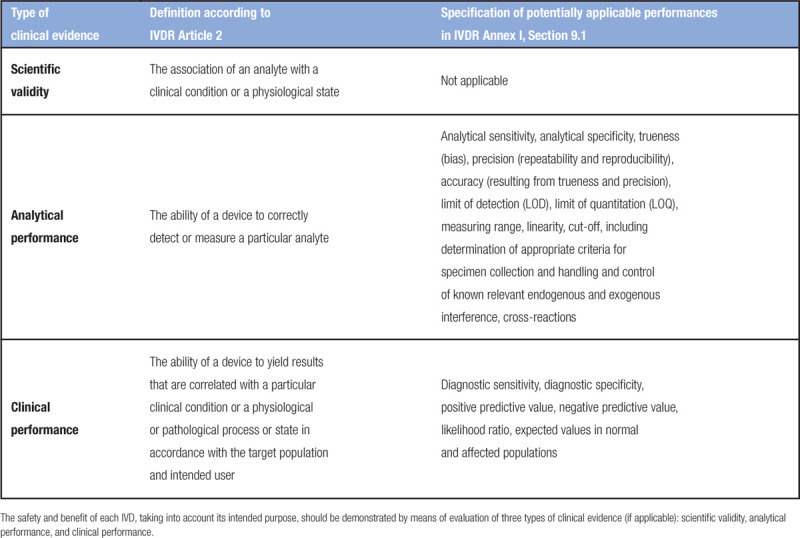
Importantly, a direct result of the new classification rules is that many products that are currently self-certified according to the IVDD will have to be certified by a notified body under the IVDR. The percentage of IVDs that requires notified body certification is estimated to increase from 15% (or even less) under the IVDD to 70%–90% under the IVDR.3 Moreover, the requirements for clinical evidence (with a major emphasis on clinical performance; Table 3) and post-market surveillance (systematic evaluation of experience with IVDs as a basis for necessary corrective and preventive action) will become more stringent under the IVDR.4
Table 3.
Specification of Clinical Evidence Requirements According to the IVDR
For IVD manufacturers, these changes translate to a need for additional well-trained staff, increased time investment and higher costs. In addition, the fact that many IVDs cannot be self-certified anymore means a huge workload for notified bodies. As only 4 notified bodies have been designated for the IVDR so far,5 the total notified body capacity might not be sufficient to assess all IVDs by May 2022. Finally, other vital infrastructure (eg, EU reference laboratories) and official guidance documents are still lacking. This means that companies might fail to engage with a notified body on time or might otherwise come to a halt in the conformity assessment process, and as a result do not manage to have all their IVDs certified before full application of the IVDR.
This makes clear that, even though the overall quality of CE-IVDs is expected to increase under the IVDR (consistent with its objectives), it is likely that there will be consequences for the availability of CE-IVDs. Manufacturers might need to prioritize, for example, their most important and profitable products will be submitted for CE marking, while other products might be temporarily unavailable or even discontinued.
Use of IH-IVDs will be restricted under the IVDR
IH-IVDs are described in the IVDR as in vitro diagnostic medical devices (IVDs; see Box 1)1 that are manufactured and used by a health institution. They are often referred to as laboratory-developed tests (LDTs), but it must be stated that the term “LDT” is not used in the IVDR. Importantly, the IVDR sets out the (quality) requirements for IVDs, but not for the complete diagnostic testing process chain as covered by ISO 15189 (Figure 1). For example, protocols for medical laboratory examinations are not themselves considered to be medical devices in the context of the IVDR.6 Moreover, the scope of the term IH-IVD, and to what extent it covers modified/off-label CE-IVDs and research use only (RUO) kits, remains to be clarified. Therefore, in the context of the IVDR, “LDT” can be regarded as a less accurate term to refer to IH-IVDs.
Figure 1.

Relation between the ISO 15189 standard and the IVDR. Given the important overlap between ISO 15189 and the IVDR, in particular regarding equipment, reagents and other in vitro diagnostic medical devices, ISO 15189 is an important basis for compliance to the IVDR for diagnostic laboratories. At the same time, ISO 15189 covers a much broader range of quality aspects, including management, personnel, and reporting. IVDR = Regulation (EU) 2017/746 on in vitro diagnostic medical devices.
Depending on the diagnostic field and level of specialization of a laboratory, IH-IVDs can form a significant part of its assay portfolio. An inventory among laboratories and hospitals in the Netherlands showed that in approximately 1 in 4 laboratories, at least half of the implemented tests are IH-IVDs (Figure 2).7 Whereas clinical chemistry and general hematology laboratories predominantly use CE-IVDs, fields such as special hematology, immunology, and medical microbiology are expected to use IH-IVDs much more frequently.
Figure 2.
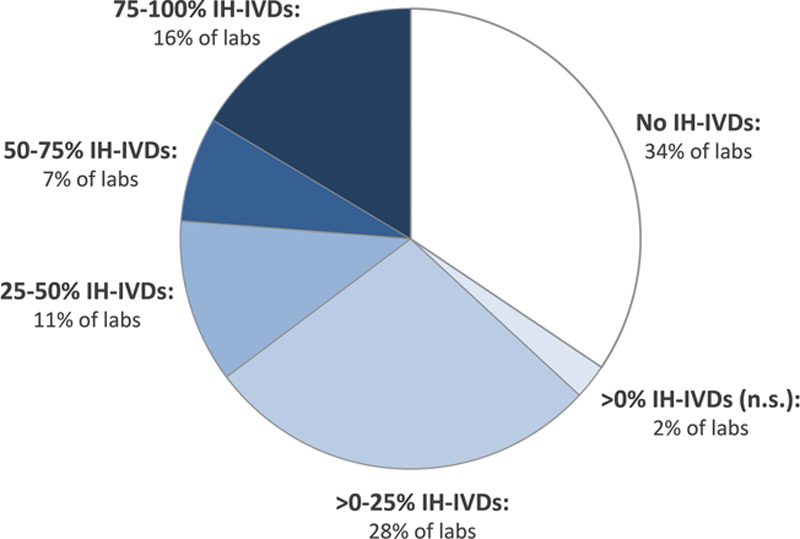
Use of IH-IVDs by hospitals and laboratories in the Netherlands.7 Pie chart shows the share of institutions that use no in-house devices (IH-IVDs; ie, 100% CE-IVDs), >0%–25%, 25%–50%, 50%–75%, 75%–100%, or >0% (percentage not specified) IH-IVDs. Approximately 2 of 3 laboratories have implemented IH-IVDs (all blue slices combined) and in almost 1 of 4 laboratories more than half of the implemented IVDs are IH-IVDs (2 darkest slices combined). The graph was made based on data from the Dutch National Institute for Public Health and the Environment (RIVM), 2015.7 CE-IVDs = CE marked in vitro diagnostic medical devices; IH-IVDs = in-house in vitro diagnostic medical devices.
This is consistent with a recent study that analyzed diagnostic tests performed at University Hospitals Leuven,8 showing that 42% of tests implemented in the hospital’s laboratories were CE-IVDs, 47% were LDTs, 11% were modified/off-label CE-IVDs, and 0.3% were RUO tests. The latter 3 categories were relatively often used in the immunology, special chemistry, and molecular microbiology groups. Finally, data collected at the initiative of the European Hematology Association (EHA) via a questionnaire shared with special hematology laboratories indicated that on average 80% of the tests implemented in these specialized laboratories are LDTs. LDTs/IH-IVDs tend to be more complex and/or less frequently used, and alternative CE-IVDs are frequently not available (eg, the Leuven study reports that for almost 3 of 4 LDTs, no CE-IVD is currently available on the market8).
It can be concluded that IH-IVDs are commonly used and essential for high-quality healthcare in many diagnostic fields. The IVDD did not regulate tests that are produced and used within health institution laboratories in a noncommercial context. In the current situation, IH-IVDs are generally implemented in line with applicable national regulations and the Quality Management System (QMS) used by the laboratory. Hence, current rules and practice can differ significantly from one member state to another. In contrast to the IVDD, the IVDR does specify conditions and requirements for IH-IVDs that need to be fulfilled by laboratories—requiring significant expertise, manpower, and time. Furthermore, the IVDR limits IH-IVD use to applications for which no appropriate CE-IVD is available (see below for a more detailed discussion of the conditions and requirements for IH-IVDs). Even though the exact interpretation of the requirements for IH-IVDs remain to be determined, this means that use of IH-IVDs will be more restricted under the IVDR.
Action 2: Make an assay inventory
As described above, the IVDR will affect the availability of CE-IVDs as well as a laboratory’s freedom and ability to implement IH-IVDs. To plan their assay portfolio under the IVDR, an elementary step for diagnostic laboratories is to make an assay inventory (Figure 3). This can be done according to the following questions:
Figure 3.
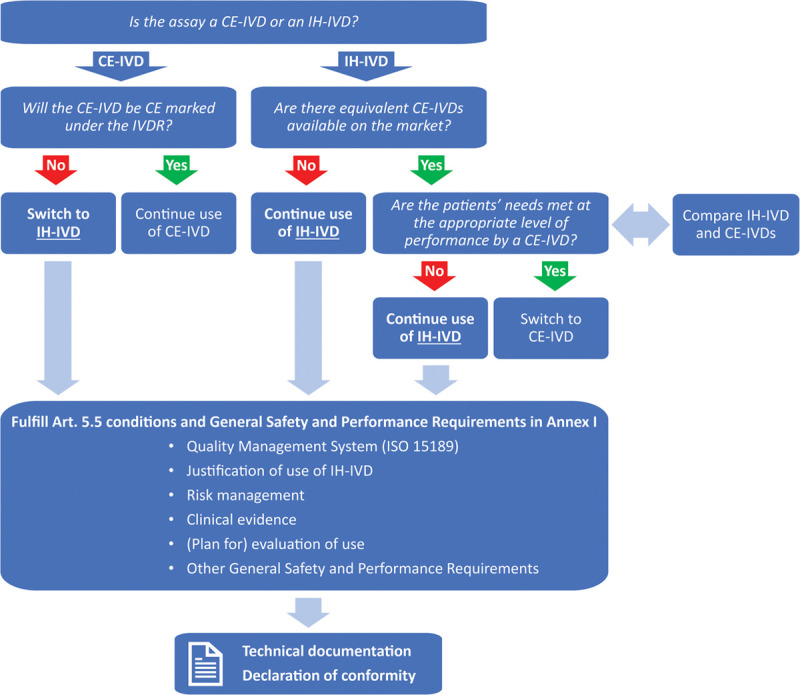
Assay inventory and IVDR compliance workflow. The assay inventory decision tree allows laboratories to plan their assay portfolio under the IVDR based on the tests that are currently implemented. Key questions are whether current CE-IVD tests will be CE marked under the IVDR (for the same intended purpose) and whether appropriate alternatives for in-house devices (IH-IVDs) are available (see also the discussion on IVDR Art. 5(5)d,e). Subsequently, for all IH-IVDs that are used after May 2022, the conditions and requirements for IH-IVDs as dictated by the IVDR should be fulfilled. The most important requirements are listed above and are discussed in more detail in the main text. Finally, fulfillment of the requirements should be documented in the technical documentation and publicly stated in the declaration of conformity. CE-IVDs = CE marked in vitro diagnostic medical devices; IH-IVDs = in-house in vitro diagnostic medical devices; IVDR = Regulation (EU) 2017/746 on in vitro diagnostic medical devices.
Which CE-IVDs and IH-IVDs are currently implemented? Laboratories should be aware that CE-IVDs should be used in line with their intended purpose and instructions for use; it is not yet clear to what extent modifications will be allowed.8,9
Will all CE-IVDs stay on the market, that is, will the manufacturers CE mark the tests under the IVDR on time? For most CE-IVDs, this means that they will have to be certified by a notified body by May 2022. Even though end users might be notified in time in the case of discontinuation of CE-IVDs, laboratories are advised to contact manufacturers or distributors in the case of doubt. In this respect it is also relevant to mention that all IVDD CE-IVDs placed on the market before the date of application, for example, at a distributor, can still be sold until May 2025 (the “sell-off provision”), and that CE-IVDs with a valid IVDD certificate issued by a notified body can be sold until May 2024 (the “grace period”; note that this only concerns certain high-risk tests; see IVDR Art. 110(2)-110(4)).
Should new IH-IVDs be developed/implemented to replace CE-IVDs that will be (temporarily) discontinued/unavailable?
Are CE-IVDs available that are equivalent to currently used IH-IVDs? If so, will these be certified according to the IVDR? If such CE-IVDs have an appropriate level of performance, the corresponding IH-IVDs cannot be used anymore after May 2022.
Whereas it will be possible to already answer these questions for some CE-IVD tests, for others it might be necessary to make an informed guess based on available information and to wait until a manufacturer can provide more accurate information. For IH-IVDs, decisive information on equivalent CE-IVDs might only become available at a late stage, when many manufacturers have certified their tests under the IVDR and the European Databank on Medical Devices (EUDAMED)10 has been launched (manufacturers will be obliged to submit information about all of their medical devices that are available in the EU to this database). In addition, a more detailed specification of the IVDR requirements for IH-IVDs is needed to answer the questions above for some IH-IVDs. When deciding whether or not to keep their IH-IVDs implemented under the IVDR, diagnostic laboratories are advised to critically assess whether the use of IH-IVDs can be justified (see discussion on IVDR Art. 5(5)d,e below) based on currently available information, making sure not to risk disruptions in diagnostic services around the date of application of the IVDR. This implies that they should start preparations to reach regulatory compliance with the IVDR now, and reassess their assay portfolio at regular intervals after the transition period ends.
The IVDR dictates requirements for use of IH-IVDs
As described in Preamble 28 and 29, the IVDR also intends to ensure the highest level of health protection by clarifying and strengthening the rules that govern in-house devices. However, not all the requirements for CE-IVDs need to be fulfilled for IH-IVDs (ie, only the GSPR in Annex I apply), as according to the preambles “the aims of the IVDR would still be met in a proportionate manner.” Importantly, this exemption for EU health institutions (organizations with the primary purpose to care for or treat patients, or to promote the public health, for example, hospitals, laboratories, and public health institutes) for using IH-IVDs only applies in case a number of specific conditions are met.
This entails that from the date of application of the IVDR onward, all diagnostic laboratories in the EU that implement IH-IVDs for diagnostic patient care are obliged to be in compliance with the health institution exemption of the IVDR. The conditions and requirements applicable to IH-IVDs are briefly discussed below.
Conditions for health institutions that use IH-IVDs
A number of conditions that uniquely apply to laboratories that make use of IH-IVDs are listed in Article 5(5) of the IVDR (Box 2).1
Box 2. Conditions for health institutions that use IH-IVDs (IVDR Article 5(5)).1.
“With the exception of the relevant general safety and performance requirements set out in Annex I, the requirements of this Regulation shall not apply to devices manufactured and used only within health institutions established in the Union, provided that all of the following conditions are met:
(a) the devices are not transferred to another legal entity;
(b) manufacture and use of the devices occur under appropriate quality management systems;
(c) the laboratory of the health institution is compliant with standard EN ISO 15189 or where applicable national provisions, including national provisions regarding accreditation;
(d) the health institution justifies in its documentation that the target patient group’s specific needs cannot be met, or cannot be met at the appropriate level of performance by an equivalent device available on the market;
(e) the health institution provides information upon request on the use of such devices to its competent authority, which shall include a justification of their manufacturing, modification and use;
(f) the health institution draws up a declaration which it shall make publicly available, including:
(i) the name and address of the manufacturing health institution,
(ii) the details necessary to identify the devices,
(iii) a declaration that the devices meet the general safety and performance requirements set out in Annex I to this Regulation and, where applicable, information on which requirements are not fully met with a reasoned justification therefore;
(g) as regards class D devices in accordance with the rules set out in Annex VIII, the health institution draws up documentation that makes it possible to have an understanding of the manufacturing facility, the manufacturing process, the design and performance data of the devices, including the intended purpose, and that is sufficiently detailed to enable the competent authority to ascertain that the general safety and performance requirements set out in Annex I to this Regulation are met. Member States may apply this provision also to class A, B, or C devices in accordance with the rules set out in Annex VIII;
(h) the health institution takes all necessary measures to ensure that all devices are manufactured in accordance with the documentation referred to in point (g); and
(i) the health institution reviews experience gained from clinical use of the devices and takes all necessary corrective actions.
Member States may require that such health institutions submit to the competent authority any further relevant information about such devices which have been manufactured and used on their territory. Member States shall retain the right to restrict the manufacture and use of any specific type of such devices and shall be permitted access to inspect the activities of the health institutions.
This paragraph shall not apply to devices that are manufactured on an industrial scale.”
Art. 5(5)a: No transfer of devices
It is not allowed to distribute devices such as reagents and control materials to other legal entities. Distribution of materials for external quality assessment rounds are an exception to this (see IVDR Art. 1(3)). It is also allowed to distribute documents such as protocols/standard operating procedures (SOPs), as these are not devices. Moreover, there is no constraint on testing samples from external sources. Hence, reference hospitals can continue analyzing samples from, for example, hospitals that are not able to perform the test in question, as long as this is not done on an industrial scale (see below).
Article 5(5)b,c: QMS/ISO 15189
Diagnostic laboratories using IH-IVDs are required to comply with the EN ISO 15189 standard, which specifies requirements for quality and competence in medical laboratories, or with applicable national provisions. Unless specifically required by your member state, accreditation is not strictly necessary, but it is good to be aware that external audits are a sound basis for improving a QMS. A QMS that is setup under ISO 15189 is appropriate for manufacture and use of IH-IVDs. It will be important to determine the optimal way to comply with ISO 15189 and the IVDR requirements in Annex I in parallel, as there is significant overlap (a proposal has been published by the Dutch Task Force IVDR9).
Article 5(5)d,e: Justification of use
Under the IVDR, use of CE-IVDs is the default option. Only when no equivalent CE-IVD is available, or a target patient group’s specific needs cannot be met at the appropriate level of performance by an equivalent CE-IVD, use of an IH-IVDs is allowed (Figure 3). This implies that IH-IVDs can be used when they perform better, that is, their use ultimately benefits the safety and health of patients. A written statement justifying the manufacture, modification, and use of IH-IVDs should be available for review upon request by the national competent authority, which is in charge of enforcing the IVDR and judging the validity of the justification.
Justification of the use of IH-IVDs, in particular when equivalent CE-IVDs are available, is one of the most urgent topics that still requires clarification. Which arguments count as valid justifications will depend on what exactly is meant by “equivalent” and by “the target patient group’s specific needs cannot be met at the appropriate level of performance.” A better analytical performance and/or a better clinical performance (eg, less false negatives/a higher sensitivity or less false positives/a higher specificity), broader applicability, and faster results/turnaround time (when this is relevant) appear to be straightforward justifications (see also Bank et al9). The European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) has developed a toolbox to substantiate the justification of the need for IH-IVDs in a standardized and rational manner11:
Identification of unmet clinical needs according to a structured checklist;
Definition of the target population; and
Description of the specific clinical pathway, including a detailed specification of the IH-IVD.
Justification of use is a continuous responsibility of laboratories that have implemented IH-IVDs, so they need to regularly monitor and evaluate new CE-IVDs for the lifetime of the IH-IVD. Publication of the results from comparisons between CE-IVDs and IH-IVDs will be worthwhile in this process. However, what exactly is expected from laboratories in terms of CE-IVD monitoring, comparison of performance, and speed of implementation in case an appropriate CE-IVD becomes available remains to be defined. Based on this information, laboratories will need to set up a strategy to fulfill the requirements to monitor CE-IVD equivalents, namely to define where, how, and how often to search for such alternatives. Importantly, transparency regarding clinical evidence documented by manufacturers will be essential for making well-informed decisions and maintaining an optimal assay portfolio.12,13
Article 5(5)f: Public declaration/GSPR
A publicly available declaration should specify the name and address of the manufacturing health institution and details that allow identification of the IH-IVDs. In addition, the declaration should state that the IH-IVDs meet the GSPR of Annex I, or alternatively, give a reasoned justification why specific requirements are not fully met. Important requirements of Annex I are, for example, provision of information on the design, safety, and performance of the device and setting up a risk management system (see below).
Article 5(5)g,h: Extra requirements for class D devices
For class D tests, the requirements are stricter than for tests in class A-C (as determined according to the same classification rules as applicable to CE-IVDs14). In particular, additional information on manufacturing, design, and performance of the IH-IVDs is required. This means that the GSPR for class D tests should be met and documented with an amount of detail that more closely resembles that of CE-IVDs, that is, according to the requirements for technical documentation in Annex II. Health institutions are required to ensure that all IH-IVDs are manufactured in accordance with their corresponding documentation.
Article 5(5)i: Evaluation of use
Experience gained from clinical use should be reviewed, and corrective action should be taken if necessary. A strategy for evaluation of use of IH-IVDs can be based on relevant parts of Article 78–79 and Annex III (on post-market surveillance for CE-IVDs).
Other Article 5(5) requirements
The exemption for health institutions does not apply to devices that are manufactured on an industrial scale (an expression that remains to be defined). Finally, the national competent authority should be permitted access to health institutions to inspect their activities. They can choose to demand that the inspected health institutions submit any additional relevant information, as well as restrict the manufacture and use of any specific type of IH-IVD.
General safety and performance requirements for IH-IVDs
In addition to meeting the conditions described above, the relevant GSPR in Annex I of the IVDR need to be met for IH-IVDs. Two important requirements concern risk management and performance evaluation.
Risk management
A risk management system needs to be set up for all IVDs (see Annex I, Section 3). Risk management is a component of ISO 15189 that relates to the evaluation and reduction of risks that might affect patient safety, so ISO 15189 compliance assures you a basic risk management. However, there are some open questions regarding compliance with the IVDR requirements, for example, how much is needed on top of the ISO 15189 requirements, and how often should the risk management be updated. For more detailed guidance on this topic, it is advised to look into ISO 14971: Application of risk management to medical devices, or ISO 22367: Application of risk management to medical laboratories, although ISO 14971 or ISO 22367 compliance is not an IVDR requirement.
Performance evaluation
Analytical and clinical performance (Table 3) of all IVDs should be demonstrated. A description of what the requirements for manufacturers are for these types of clinical evidence can be found in Article 56 and Annex XIII, but detailed guidance from the European Commission on performance evaluation for CE-IVDs or IH-IVDs is not yet available. Health institutions can get a head start by looking into the medical devices guideline MEDDEV 2.7/1 revision 4 on clinical evaluation.15 In addition, a new standard for good study practice concerning execution of clinical performance studies to assess IVDs for regulatory purposes has recently been published, which has been harmonized with the IVDR (ISO 20916: In vitro diagnostic medical devices—Clinical performance studies using specimens from human subjects—Good study practice).
An outline of how documentation for IH-IVDs might be organized can be found in a guidance on the health institution exemption of the MHRA (the competent authority of the United Kingdom).16
Concerted action can reduce the regulatory burden for IH-IVDs
The reality is that some IH-IVDs are developed and used by a single health institution, but others by (inter)national networks of health institutions. This is for example the case in EU-funded research consortia consisting of diagnostic laboratories. Such networks can share some of the efforts required by the IVDR to reach regulatory compliance for IH-IVDs. For example, performance data are often collected together in multicenter studies, and it is possible to generate some of the required documentation and to evaluate use collectively. By combining their datasets, institutes can jointly analyze large patient groups, even for rare diseases. Benefit from group efforts might also arise when networks, medical societies, or other groups support their members, for example by giving advice, publishing (opinion) papers, and developing guidance and/or templates.
Action 3: Obtain regulatory compliance for IH-IVDs
When a laboratory’s assay portfolio under the IVDR is established, the next step is to identify the gaps between the information that is already available, and the information that will be required to comply with IVDR Article 5(5) and Annex I (GSPR) for all IH-IVDs. Based on these gaps, the steps required to collect the missing information and data can be planned (Figure 3). In addition to establishing or maintaining compliance with ISO 15189 (and if necessary, adaptation of procedures based on the GSPR, for example, verification and validation procedures, or risk management), generation of performance data and documentation can be a major effort. Especially collection of clinical performance data can be a lengthy process, so a gap analysis for this requirement should be a priority.
Unfortunately, exact instructions on how to fulfill the requirements for IH-IVDs, and how to draft the corresponding documentation, are still lacking. As clinical evidence and evaluation of use/post-market surveillance are critical requirements for IH-IVDs as well as for CE-IVDs, the requirements for manufacturers can give an indication of what might be expected for IH-IVDs. Therefore, it will be informative to consult guidance documents that are being drafted for manufacturers of CE-IVDs on these and other topics, and to look at the way manufacturers comply with the requirements of the IVDR. However, considering the innovative nature of health institutions, it might not be feasible (or desirable) to reach compliance to the same extent as is required for CE-IVDs. Ultimately, an official guidance on Conditions for in-house devices is expected to clarify the conditions and requirements for IH-IVDs, but drafting of this document has only recently been initiated by the European Commission (Box 3).
Box 3. Summary of IVDR implementation.
The IVDR entered into force in May 2017. When entering into force, EU regulations are immediately applicable and enforceable by law in all EU member states. On top of the definitive text of the regulation that has been available since 2017, many topics require additional clarification in order for manufacturers, notified bodies, competent authorities, health institutions and other actors to understand in more detail how the requirements formulated for them should be interpreted. Such clarifications are published by the European Commission in the format of guidance documents.14
The MDCG, consisting of representatives of the European Commission and all EU member states, assists the European Commission with the implementation of the IVDR and the MDR (Regulation (EU) 2017/745 on medical devices17). One of the tasks of the MDCG is to coordinate task forces that draft guidance documents for the IVDR and MDR. For each guidance document, a task force is created that principally consists of representatives from a number of member states. However, if relevant, stakeholders (such as representatives of manufacturers, notified bodies or medical societies) are invited at a certain stage to provide input on the draft. A large number of medical societies is represented by the BioMed Alliance,20 which is recognized as a relevant stakeholder by the European Commission.
The date of full application of the IVDR is approaching fast. Many guidance documents are still pending, such as the documents on Performance evaluation and on Conditions for in-house devices, the latter one being highly relevant for diagnostic laboratories. In the meantime, many independent (inter)national IVDR working groups and task forces have been formed to provide advice on appropriate interpretation of the IVDR to their national governments or the European Commission, and to inform their followers about the IVDR.9,21
Finally, collective development of IH-IVDs can greatly reduce the regulatory burden for individual laboratories. At the same time, such collaborations have the potential to improve quality and comparability of results through standardization. Nevertheless, health institutions will always be held individually responsible by the competent authority for all of its IH-IVDs. They need to make sure to work under an appropriate QMS, to justify the use of each IH-IVD, to justify the corresponding documentation that has been generated, and to make a public declaration.
Discussion
The IVDR has major implications for diagnostic laboratories and the CE-IVDs and/or IH-IVDs in their assay portfolios. This report highlights that, despite the unknowns and missing guidance, laboratories can already undertake substantial steps to prepare for the day of full application of the IVDR (May 26, 2022):
Appoint a small dedicated team for regulatory compliance and stay informed about the IVDR implementation.
Make an assay inventory to plan their assay portfolio under the IVDR.
Work toward IVDR compliance (including a written justification based on the intended purpose) for their IH-IVDs—this can partly be a collective effort of networks of laboratories.
At least some competent authorities appear supportive of collective development and implementation of IH-IVDs (provided that each laboratory generates some validation data). However, whether it regards collective action or any other topic, it is important to realize that enforcement of the IVDR is the responsibility of individual EU member states (national competent authorities, typically part of the ministry of health, are assigned this role). As a consequence, the exact interpretation of the regulation might differ between member states. This underlines an urgent need for EU-wide harmonization of interpretation and enforcement of the health institution exemption, so that internationally coordinated efforts to collect and share information and to develop robust, multicenter validated IH-IVDs are not hampered. It can be wise for diagnostic laboratories to establish a dialog with their national competent authority to learn about their interpretation of the IVDR and their expectations, as well as to voice any questions and concerns.
With the introduction of the IVDR, laboratories have to comply to EU legislation for the first time. Especially for laboratories that use many different IH-IVDs, the workload to fulfill the IVDR requirements can be substantial. Instructive guidance on in-house devices is critical for efficient preparation for the IVDR, but, as mentioned repeatedly in this report, guidance documents on this topic and on various other topics are still pending.
The drafting of these documents is an ongoing process coordinated by the European Commission’s Medical Device Coordination Group (MDCG; Box 3).14 However, the European Commission has limited resources available for implementation of the IVDR and the new regulation on medical devices17 (MDR; Box 3). These resources have so far predominantly been allotted to the MDR, even more so because of the coronavirus pandemic and the subsequent delay of the date of application of the MDR from May 2020 to May 2021. Because of the limited progress with the IVDR implementation and the lack of guidance, the diagnostic health sector (eg, the BioMed Alliance18 and the German AWMF19), as well as other IVDR stakeholders, have raised serious concerns about the feasibility of the IVDR transition timeline. However, no intention to adjust the application date of the IVDR (or to partially implement the IVDR initially) has yet been expressed by the European Commission.
Implementation and enforcement of a law is an iterative process; final interpretation will only become clear while the law is being applied. In the absence of complete instructions, regulatory professionals generally advice to embrace the grey: get started without delay and do what appears reasonable. This will probably turn out to be a bit too little or too much, but that should not be a problem as long as choices are justified and documentation is adapted according to new information and feedback (it should also be kept in mind that laboratories are experts on their own tests and know what contributes to quality and what does not). Furthermore, laboratories are advised to give priority to IH-IVDs that are used most frequently and/or are associated with the highest risk, in line with the risk-based approach that will guide inspections by the competent authorities.
Finally, since implementation of the MDR precedes that of the IVDR, and given that the MDR is built on the same principles as the IVDR, it is also possible to learn from the MDR implementation process. In particular, it will be interesting to see how the MDR requirements are interpreted, what happens to the availability of CE marked medical devices at the end of the MDR transition period, and the experiences with the exemption for in-house devices.
In conclusion, the IVDR will bring about a significant change for diagnostic laboratories. Even though the exact requirements of the IVDR are not set in stone yet, it is advised to start preparations to reach regulatory compliance in a timely manner and to take along new information when it becomes available.
Disclosures
DG reports honoraria for participation in advisory boards, consultancy and/or speakers’ bureau from AstraZeneca, Roche, Novartis, Incyte, Lilly, Illumina, and Janssen, and grant funding from Roche, AstraZeneca and Novartis. DG is co-founder of Univ8 Genomics Ltd. MB reports personal fees from Incyte (advisory board), financial support for reference diagnostics from Affimed and Regeneron, grants and personal fees from Amgen (advisory board, speakers bureau, travel support), personal fees from Janssen (speakers bureau). JJMD reports being inventor on several EuroClonality and EuroFlow patents, which are licensed to industry and provide royalties to the EuroClonality and EuroFlow consortia. JJMD reports to be chairman of the EuroFlow scientific foundation, which receives royalties from licensed patents, which are collectively owned by the participants of the EuroFlow Foundation. In addition, JJMD reports an Educational Services Agreement with BD Biosciences and a Scientific Advisorship with Cytognos, with honorarium income for Leiden University Medical Center. The other authors have no conflicts of interest to disclose.
References
- 1.Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU. 2017. Available at: https://eur-lex.europa.eu/eli/reg/2017/746. Accessed April 8, 2021.
- 2.Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in vitro diagnostic medical devices. 1998. Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:31998L0079. Accessed April 8, 2021.
- 3.van Drongelen A, de Bruijn A, Pennings J, van der Maaden T. The impact of the new European IVD classification rules on the notified body involvement; a study on the IVDs registered in the Netherlands. RIVM Letter Report. 2018;2018-0082:1–32. [Google Scholar]
- 4.MedTech Europe. Clinical Evidence Requirements for CE certification under the in-vitro Diagnostic Regulation in the European Union. 2020. Available at: https://www.medtecheurope.org/resource-library/clinical-evidence-requirements-for-ce-certification-under-the-in-vitro-diagnostic-regulation-in-the-european-union/. Accessed March 12, 2021.
- 5.European Commission. Nando (New Approach Notified and Designated Organisations) Information System: Notified bodies designated for the IVDR. Available at: https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=35. Accessed March 12, 2021.
- 6.Vogeser M, Brüggemann M. Complex analytical procedures in diagnostic laboratories and the IVDR. Clin Chem Lab Med. 2020;59:457–458. [DOI] [PubMed] [Google Scholar]
- 7.de Bruijn ACP, Roszek BR. In-huis ontwikkelde IVD testen; Gebruik en kwaliteitsborging [Use of In-House IVDs]. RIVM Letter Report. 2015;2015-0152:1–70. [Google Scholar]
- 8.Vermeersch P, Van Aelst T, Dequeker EMC. The new IVD Regulation 2017/746: a case study at a large university hospital laboratory in Belgium demonstrates the need for clarification on the degrees of freedom laboratories have to use lab-developed tests to improve patient care. Clin Chem Lab Med. 2020;59:101–106. [DOI] [PubMed] [Google Scholar]
- 9.Bank PCD, Jacobs LHJ, van den Berg SAA, et al. The end of the laboratory developed test as we know it? Recommendations from a national multidisciplinary taskforce of laboratory specialists on the interpretation of the IVDR and its complications. Clin Chem Lab Med. 2021;59:491–497. [DOI] [PubMed] [Google Scholar]
- 10.European Commission. EUDAMED - European Database on Medical Devices. Available at: https://ec.europa.eu/tools/eudamed/#/screen/home. Accessed March 12, 2021.
- 11.European Federation of Clinical Chemistry and Laboratory Medicine (EFLM). Interactive unmet needs checklist. Available at: https://www.eflm.eu/site/page/a/1203. Accessed March 12, 2021.
- 12.Fraser AG, Butchart EG, Szymański P, et al. The need for transparency of clinical evidence for medical devices in Europe. Lancet. 2018;392:521–530. [DOI] [PubMed] [Google Scholar]
- 13.Horvath AR, Lord SJ, StJohn A, et al. ; Test Evaluation Working Group of the European Federation of Clinical Chemistry Laboratory Medicine. From biomarkers to medical tests: the changing landscape of test evaluation. Clin Chim Acta. 2014;427:49–57. [DOI] [PubMed] [Google Scholar]
- 14.European Commission. Guidance - MDCG endorsed documents and other guidance. Available at: https://ec.europa.eu/health/md_sector/new_regulations/guidance_en. Accessed March 12, 2021.
- 15.European Commission. MEDDEV 2.7/1 revision 4. Clinical evaluation: a guide for manufacturers and notified bodies under directives 93/42/EEC and 90/385/EEC. Available at: https://ec.europa.eu/docsroom/documents/17522/attachments/1/translations/en/renditions/native. Accessed March 12, 2021.
- 16.Medicines and Healthcare products Regulatory Agency (Gov.UK). MHRA guidance on the health institution exemption (HIE) – IVDR and MDR (Northern Ireland). 2021. Available at: https://www.gov.uk/government/publications/mhra-guidance-on-the-health-institution-exemption-hie-ivdr-and-mdr-northern-ireland. Accessed March 12, 2021.
- 17.Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC. 2017. Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32017R0745. Accessed April 8, 2021. [Google Scholar]
- 18.Biomedical Alliance in Europe. BioMed Alliance calls for rapid progress in IVDR implementation. 2020. Available at: https://www.biomedeurope.org/news/2020/226-biomed-alliance-calls-for-rapid-progress-in-ivdr-implementation.html. Accessed March 12, 2021.
- 19.AWMF Online. Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (Association of the Scientific Medical Societies). Available at: https://www.awmf.org/en/awmf-online-portal-for-scientific-medicine/awmf-news.html. Accessed March 12, 2021. [DOI] [PubMed]
- 20.BioMed Alliance. Biomedical alliance in Europe. Available at: https://www.biomedeurope.org/. Accessed March 21, 2021.
- 21.Macintyre E, Gribben J, Döhner K. EU-wide access to high-quality, affordable precision diagnostics: an EHA position paper. Hemasphere. 2020;4:e412. [DOI] [PMC free article] [PubMed] [Google Scholar]