

Summary
The understanding and prediction of the solubility of biomolecules, even of the simplest ones, reflect an open question and unmet need. Short aromatic tripeptides are among the most highly aggregative biomolecules. However, in marked contrast, Ala-Phe-Ala (AFA) was surprisingly found to be non-aggregative and could be solubilized at millimolar concentrations. Here, aiming to uncover the underlying molecular basis of its high solubility, we explore in detail the solubility, aggregation propensity, and atomic-level structure of the tripeptide. We demonstrate an unexpectedly high water solubility of AFA reaching 672 mM, two orders of magnitude higher than reported previously. The single crystal structure reveals an anti-parallel β sheet conformation devoid of any aromatic interactions. This study provides clear mechanistic insight into the structural basis of solubility and suggests a simple and feasible tool for its estimation, bearing implications for design of peptide drugs, peptides materials, and advancement of peptide nanotechnology.
Keywords: Aromatic peptide, aggregation, crystal packing-solubility relationship, mass spectrometry, NMR spectroscopy
Graphical abstract
Highlights
Balance between solubility versus aggregation of aromatic peptides is examined
Significant differences in solubility are observed for analogous tripeptides
Solid-state packing is found to be underlying molecular basis for high solubility
Revealing the importance of biology at the solid state in physiology and pathology
The solubility of biomolecules in aqueous solutions is a central issue with regard to their physiological and pathological activities. Bera et al. provide insight into the solid-state packing-solubility relationship of biomolecules. This work may further extend the importance of solid-state biology for basic and applied science.
Introduction
Although extremely important, the problem of insolubility of peptides and proteins in aqueous solution is still far from being completely understood.1, 2, 3 In nature, autonomous organization of disordered building blocks into ordered stable assemblies is a fundamental process.4 Many major degenerative disorders share the common etiology of aggregation into amyloid fibrils by proteins and peptides.5 Although, in more recent times, these nanostructures have found interesting applications as advanced materials for innovative medicine solutions, tissue engineering, renewable energy, environmental science, nanotechnology, and materials science,6,7 the high propensity to assemble into insoluble aggregates is very common but poses a serious limitation to the production and use of many peptides in a wide range of biotechnological and pharmaceutical applications.1,8 Increasing the aqueous solubility of poorly water-soluble drugs is the most frequent requirement in the pharmaceutical analysis and formulation fields. In principle, for a drug to exert its biological effect, it must be soluble in and transported by body fluids.9 Many peptide-based drugs with great therapeutic potential are rendered ineffective during preclinical or clinical trials1 simply because of the problems related to low water solubility or high propensity for irreversible self-assembly.
The self-assembly process is driven by a wide variety of weak non-covalent interactions,10, 11, 12 including hydrogen bonding, van der Waals forces, Coulombic interactions, and hydrophobic effects. The rate of aggregation of biomolecules has been shown to be altered significantly by the presence of metal ions.13, 14, 15, 16 Although aggregation does not occur under normal physiological conditions, it can be induced in the presence of certain metal ions, such as copper(II), zinc(II), and iron(II). However, among these interactions, π-π stacking between aromatic residues of peptide side chains has been postulated to be the major driving force for early recognition and acceleration of peptide self-assembly.17, 18, 19 A recent in silico analysis aimed to predict formation of nanostructures by de-novo-designed short peptides has suggested aromatic tripeptides to be the most aggregation-prone biomolecules.20 In accordance, the aromatic-rich small peptide segment 16KLVFFAE22 was designated as the nucleating core of the β-amyloid (Aβ) peptide because of high aggregation propensity in producing insoluble fibrillar deposits with cross-β architecture, the hallmark of the full-length sequence.21,22 In addition, the Phe residues play an important role in formation of key Aβ intermediates with a disordered 310 helical structure.23 The importance of aromatic moieties is also evident by the abundance of aromatic residues in amyloid-forming short peptide sequences as well as by the proposed design rule of amyloid inhibitors to include building units that specifically target the aromatic residues.24, 25, 26 Moreover, applying a reductionist approach, FF, FFF, and their derivatives have been shown to be strong promoters of peptide aggregation into a variety of nanoarchitechtures.27, 28, 29 Replacement of the aromatic residues with nonaromatic surrogates has been found to completely abrogate or attenuate aggregation, further signifying the role of aromatic moieties in peptide self-asssembly.30, 31, 32
The solubility of a building unit and the rate of its association process are inversely related to each other. Physiochemically, the degree of solubility depends on the power of peptide-solvent cross talk, the types and intensities of electrostatic and hydrogen bonding, and van der Waals interactions between the peptides and water molecules at the expense of peptide-peptide interactions. Protein and peptide aggregation is also strongly dependent on the concentration of the aggregating biomolecules because of their solubility limits. At the initial stage of the aggregation process, the biomolecular aggregates are completely soluble but gradually become insoluble while enhancing protein association as they exceed solubility limits, resulting in protein/peptide precipitation.33,34 However, significantly different solubility of analogous peptide sequences with similar polarities and numbers of probable H-bonding sites remains a mystery. In this context, uncovering the underlying features that determine the ultimate solubility of biomolecules is of the utmost importance for basic science as well as for design of efficient peptide drugs.
In contrast to the commonly observed self-assembly propensity of aromatic short peptides, manifested in fast and efficient aggregation into ordered nanostructures in aqueous solution, AFA has been found, surprisingly, to be non-aggregative and quite highly soluble, with a reported critical concentration of up to 10 mM.35, 36, 37 Primary study of its solution structure via solution NMR showed a combination of inverse γ turn and β strand conformations.36 Follow-up studies revealed that AFA exists as an ensemble of rapidly interconverting conformers in aqueous solution,37 but its unexpected solubility remains unexplained.
Here we extensively explore the solubility, aggregation propensity, and solid-state structure relationship of AFA and compare them with those of analogous peptides. We uncover that AFA can be solubilized up to a concentration of 672 mM at room temperature and 720 mM at 90°C. These values are unexpectedly high, considering the solubility of aromatic short peptides,2,38 and two orders of magnitude higher than the previously reported solubility of AFA. The solid-state structure reveals formation of an antiparallel β sheet organization surprisingly devoid of any aromatic π-π stacking between molecular units. Simple sequence shuffling results in decreased solubility by an order of magnitude for AAF (105 mM) and FAA (42 mM). The crystal structure of AAF demonstrates transformation of the molecular orientation into a helical structure with strong face-to-face π-π stacking interactions. In addition, screening of a focused peptide library at different length scales further confirms the aromatic stacking-dependent solubility of aromatic peptides. Our work clearly establishes that the solubility of analogous short peptides with similar polarity and H-bonding sites is determined by solid-state packing. These results could further advance our understanding of peptide solubility and pave the way for modular design of peptide drugs with high efficiency.
Results and discussion
Critical soluble concentration of tripeptide
We first established the threshold concentration of the AFA tripeptide that could be solubilized in pure water at room temperature and physiological pH of 6.8 (Figure 1A). Under these conditions, the maximum solubility of AFA was found to be 672 mM, unexpectedly high compared with other well-known short aromatic peptides.2,38 The ultraviolet-visible (UV-vis) spectra at different concentrations indicated no major shift of the peak position, emphasizing the appearance of a yellowish color because of the extremely high concentrated solution of peptides (Figure S1). Next, to understand the thermodynamics of solubility, we investigated the dependence of critical concentration on temperature in the range of 25°C–100°C with a minimum interval of 10°C (Figure 1A). The solubility increased gradually with temperature and showed a steep climb to 702 mM at 70°C. Near the boiling point of water, the solubility reached 710 mM. This variation of solubility with temperature indicated the existence of heat capacity, and the corresponding enthalpy and entropy change could be realized from a van’t Hoff relation.39 The favorable free energy could arise from various interactions, such as hydrogen bonding and the presence of salt bridges. The aromatic side chain could also be engaged in an extended π-stacked structure that would also cause favorable free energy.40 However, these types of interactions have only a moderate temperature dependence, and the temperature dependence of solubility or aggregation has been reported to most likely stem from the contribution of the hydrophobic effect of the aromatic side chains,41 suggesting that such an effect takes place in AFA. Next, the role of electrostatic interactions was explored by measuring the solubility at different pH values and at room temperature (Figure 1B). The minimal critical concentration was found around pH 6–7 (668 mM), which is the corresponding pH of AFA lyophilized powder in pure water. At this pH, the predominant species in solution is the globally neutral, zwitterionic AFA monomer. As the pH of the solution approached the pKa value of either terminus resulted rapidly increasing concentration of charged monomers; however, this increased the solubility only very slightly (670 mM and 672 mM). The relatively strong effect of temperature and weak effect of pH on the critical concentration of AFA can be rationalized on the basis of relatively weak absolute values of electrostatic driving forces, which are diminished by the strong hydrophobic effect of the aromatic side chains. At the same time, because the peptide molecules carry one positive and one negative charge, electrostatic interactions could also be involved in salt bridge formation.40 To verify the importance of these electrostatic interactions, the solubility was investigated at different ionic strengths, adjusted by increasing NaCl concentrations from 1 mM to 2 M (Figure 1C). The results showed that the solubility remained constant for a large range of NaCl concentrations and started to change only at a very high NaCl concentration of 500 mM or above. This observation clearly indicates that the effect of NaCl is related to the well-established “salting out” effect of hydrophobic molecules, further supporting no significant role of the electrostatic effect in solubility of AFA.42,43 Moreover, using different types of salts having altered cations or anions also had a similar effect, suggesting that simple electrostatic screening was not much responsible for the solubility of AFA (Figure S2). Therefore, the hydrophobic and aromatic interactions play the most striking role in maintaining the favorable energy landscape for the exceptional high solubility of AFA.
Figure 1.

Solubility versus self-assembly of tripeptides
(A and B) Water solubility of AFA relative to (A) temperature and (B) pH of the solution. The insets in (A) show images of the solution at three different temperatures. The data represent the mean ± SEM for 3 independent experiments.
(C) Solubility of AFA in the presence of different concentrations of NaCl. The data represent the mean ± SEM for 3 independent experiments.
(D and E) Comparison of the critical concentrations of AFA, AAF, and FAA at different (D) temperature and (E) pH values. The inset in (D) shows a magnified graph for AAF and FAA.
(F) Calculated Stokes hydrodynamic diameter distribution of three tripeptide clusters in solution at different concentrations at pH 6.8 and room temperature.
(G–N) Scanning electron microscopy images of the resultant architectures after 2 weeks of self-assembly of (G–J) AFA, (K and L) AAF, and (M and N) FAA. The top panels show the entire area of the solution drop, and the corresponding magnified areas marked by green squares are shown in the bottom panels. The insets in (L) and (N) show the probability distribution of fiber width.
(O–Q) FTIR spectra of (O) AFA, (P) AAF, and (Q) FAA.
To further understand the role of the hydrophobic effect, we shuffled the sequence of the native peptide and designed two other possible combinations, AAF and FAA. Surprisingly, although displaying similar hydrophobicity, the two modified tripeptides exhibited solubility of nearly one order of magnitude lower compared with the parent peptide (105 mM for AAF and 42 mM for FAA) under similar conditions (Figure 1D). The solubility of both modified peptides revealed a similar dependency on temperature, although, even at the highest measured temperature, the critical concertation was found to be nearly 6-fold and 15-fold lower for AAF and FAA, respectively. The critical soluble concentration was explored at different pH values to understand the effect of electrostatic interactions (Figure 1E). However, similar to AFA, the temperature dependence stemming from hydrophobic or aromatic effects was found to be more significant than the driving forces imparted by electrostatic interactions. To obtain the size of existing clusters in the solution, dynamic light scattering (DLS) experiments were performed with a range of sub- and supercritical concentrations in water (Figures 1F and S3). Below 40 mM, for all three peptides, the scattering signal was dominated by monomeric or small oligomeric species with a diffusion coefficient corresponding to an equivalent size of approximately 200–400 nm. Above 40 mM, the cluster size of AFA and AAF remained constant, whereas FAA showed a presence of larger insoluble subcritical structures approximately 2000 nm in size. These significantly larger clusters dominated the scattering intensity by the Rayleigh scattering relation.41 When the concentration reached 100 mM, AAF showed formation of insoluble large prenucleation clusters or micelles approximately 4,000 nm in size, indicating insolubility and an aggregation process. However, the size of clusters formed by AFA persisted, signifying the presence of non-aggregative monomeric species even at this high concentration or above.
Solubility and self-assembly/aggregation are opposite processes; the higher the solubility, the lower the aggregation. Support for this notion was obtained from the DLS results because AFA failed to form any higher-ordered structures even at very high concentrations. Aggregation was followed by investigating the nanostructures produced by the three peptides in solution. Scanning electron microscopy images showed AFA to mainly exist as monomers or small oligomers even after allowed to self-assemble for 2 weeks. Large-scale images capturing the entire area of solution drop clearly indicated the absence of any kind of ordered nanostructure (Figures 1G, 1H, and S4). During the drying process, the soluble AFA monomers were mainly deposited in a ring-like fashion along the original drop edge because of a coffee ring effect. Magnification of the edge of these drops showed deposition of unordered particulate materials resulting from coalescence of monomers. Upon increasing the concentration, larger crystalline clusters were found to deposit; however, no indication of formation of ordered assembled structures was observed (Figures 1I and 1J). On the other hand, AAF and FAA exhibited formation of well-ordered and closely entangled networks of fibers consisting of filaments that bundled into larger twisted fibrils because of self-assembly of small nucleating sites even at lower concentrations (Figures 1K–1N and S5). The average diameter of the filaments was approximately 2–3 μm with only a minor variation between the tripeptides, as calculated using a histogram. Therefore, the significant differences in solubility and aggregation of similar hydrophobic analogous peptides, along with an observed minimal effect of electrostatic interactions, most likely indicate that molecular structure, atomic-level packing, and aromatic stacking might play a pivotal role in solubility.
To characterize the secondary structure of the tripeptides, we used Fourier transform infrared (FTIR) and circular dichroism (CD) spectroscopy techniques. For AFA, the FTIR spectra at solid state manifested a sharp amide I peak at 1,631 cm−1 with a shoulder at 1,687 cm−1, indicating the predominant β sheet structure (Figure 1O).44 However, the amide I peak shifted to 1,636 cm−1 and 1,635 cm−1 for AAF and FAA, respectively, indicating structural transition toward a helical conformation (Figures 1P and 1Q). The CD spectrum of AFA at room temperature showed a clear positive ellipticity in the far-UV region with distinct double maxima at 197 and 217 nm, in accordance with previous studies (Figure S6A),36,37 and provide support for a dominant β strand conformation. The CD spectrum of AAF showed similar double maxima with positive peaks at the same positions but a significantly different intensity ratio compared with the CD spectra of AFA (Figure S6D). Apparently, the corresponding differences in intensity ratios for AFA and AAF indicate different dihedral angles.35 FAA exists preferably in a polyproline II (PPII)-type turn conformation because the CD spectra exhibited a negative maximum at 197 nm and a positive peak at 218 nm (Figure S6G).35 To gain knowledge regarding the effect of temperature and pH on molecular conformation, CD spectra were acquired at different pH values (2.4–10.5) and temperatures (10°C–90°C) (Figure S6). Identical spectra were obtained at all the studied pH values, indicating conformation rigidity in the studied pH range. Upon increasing the temperature, the intensity of the CD signal decreased very slightly, likely because of partial unfolding. Moreover, our earlier study showed that ROESY spectra acquired in the presence of 10 mM to 1 M NaCl and at pH 1.2 and 12.1 did not significantly alter the NOE pattern,36 further confirming the limited influence of the charged terminal groups on molecular conformation and solubility.
Solid-state packing of tripeptides
Next, to identify the most stable molecular conformation and preferred packing of the aromatic moieties, we grew diffraction-quality single crystals of AFA and AAF under similar conditions as used for other studies and analyzed their structure. Unfortunately, we were unable to produce diffractable single-crystal of FAA. The fundamental mechanisms underlying the oriented organization process into hierarchical self-assembly and macroscopic crystallization still remain unclear.12 Different kinds of nanostructures and morphologies are stabilized according to the balance between noncovalent acceptor (hydrogen-bond acceptor, HBA) ability, polarity, surface tension, and dielectric constant, which fuse or dissociate to produce macroscopic crystals. Thus, we speculate that a correlation of the differences in rate of aggregation at nanoscale within specific timescale and macroscopic crystallization between FAA, AFA, and AAF is not feasible. The single-crystal structure of AFA revealed that two peptide molecules crystallized with water, acetic acid, and trifluoroacetic acid, one molecule of each, in the asymmetric unit of the monoclinic P21 space group (Figure 2A).28 In the solid-state structure, all counterions were present with the peptide at a 1:1 ratio as part of their solid-phase synthesis and included in the crystals because of their role in packing; therefore, the solution should also contain equimolar concentrations of the tripeptide and its counterion. The two peptide molecules adopted a similar conformation with no intramolecular H-bond in the peptide molecules, which refutes the possibility of a γ turn structure. The torsion angles around the Phe2 residue appeared to play a pivotal role in determining the overall structural features. The allowed torsion angles of the Phe2 residue were constrained within the β sheet region of the Ramachandran plot,45 with φ2 and ψ2 values of −141.77° and 144.12° for molecule A and −140.72° and 130.43° for molecule B, respectively (Figures 2B–2D). The tripeptide molecules exhibited two conventional NH3+... –OOC head-to-tail hydrogen bonds, with the third amine hydrogen involving the acetic acid or trifluoroacetic acid moiety. In the crystallographic a direction, β strands were stacked in an antiparallel manner and interacted with the adjacent strands through intermolecular H-bonds incorporating terminal polar groups and internal amide groups, producing an antiparallel β sheet conformation (Figures 2D and 2E). The aromatic side of the Phe residue of one β strand resided in the opposite side of the next interacting strand, and, surprisingly, no aromatic π-π interactions were formed in the β sheet conformation (Figure 2F). Moreover, nearby β sheets were connected through head-to-tail H-bonds between polar head groups in the crystallographic b direction, generating a 1D layer where all aromatic residues were positioned on the same face of the layer (Figure S7). The individual layers stacked to afford a layer-by-layer structural organization of AFA stabilized through van der Waals interactions. However, π-π interactions between Phe residues were also not observed within cross-sheets along the b direction. The closest distance between two Phe residues was 9.5 Å (Figure S7), considerably higher than the defined distance for π-π interaction (4.5–7 Å).46 Therefore, the solid-state structure of AFA is a clear exception from the structures of other aromatic short peptides, which are mainly stabilized by strong aromatic π-π interactions.46 This special arrangement of the aromatic ring and the absence of π-π stacking could be the molecular basis of the exceptional solubility of AFA.
Figure 2.

Solid-state structures of AFA and AAF
(A–F) AFA (CCDC:1834550):28 shown are (A) the asymmetric unit; (B) the torsion angle of Phe2 for the two different molecules present in the asymmetric unit; (C) a Ramachandran plot of the allowed dihedral angles of Phe2 residues in red and orange, showing preference for the β sheet conformation; (D) the β sheet structure of AFA; (E) H-bonded antiparallel β sheet arrangement in higher-order packing; and (F) the absence of any π-π stacking interactions between two adjacent molecules.
(G–K) AAF: shown are (G) the asymmetric unit, (H) the allowed torsion angle of Ala2, (I) helical structure formation by head-to-tail H-bond connection and the helical dimer, (J) stacking of adjacent helices via H-bond and aromatic interactions, and (K) strong face-to-face π-π stacking in the structure.
The modified tripeptide AAF crystallized with one peptide molecule and one trifluoroacetic acid molecule in the asymmetric unit of the orthorhombic P212121 space group (Figures 2G–2K, Table S1). Shuffling the Phe moiety position from the central residue to the C terminus completely altered the molecular conformation of AAF compared with the native peptide. The allowed torsion angles of the Ala2 were constrained in the right handed α-helical region of the Ramachandran plot, with φ2 and ψ2 values of –86.28° and −29.49°, respectively (Figure 2H). In the crystallographic b direction, the AAF molecules were connected via head-to-tail H-bonds through trifluoroacetic acid molecules and produced a single helical structure (Figure 2I). Two nearby helices connected by H-bonds in an antiparallel fashion in the c direction fabricated the helical dimer (Figure 2I). Adjacent dimers were mated through a hydrophobic zipper-like structural interaction exerted by Phe residues and the trifluoroacetic acid side chain, creating a helical sheet-like organization (Figure S8).47,48 Most importantly, in the perpendicular direction (along the a axis), individual helices were stacked in parallel orientation to produce a single helical strand (Figure 2J). Together with H-bonds, strong aromatic face-to-face π-π interactions played a pivotal role to stabilize the helical strands (Figures 2K and S8). The centroid-to-centroid distance of Phe residues between two nearby AAF molecules was 5.32 Å in the lower region of the proposed π-π stacking distance.46 Therefore, in accordance with other reported aromatic short peptide structures, the presence of aromatic π-π interaction in AAF solid-state packing is probably the major influential factor that determines the lower solubility of AAF compared with AFA. Although we were unable to produce a diffraction-quality single crystal of FAA, the powder X-ray pattern of FAA exhibited peaks at similar 2θ value positions with AAF (Figure S9). This indicates a similar atomic-level orientation, equatorial and longitudinal distances between molecular units, and higher-order organization of FAA with structural arrangement of AAF, which also resulted in a similar lower solubility.
Effect of aromatic amyloid inhibitors on solubility
Polyphenols such as epigallocatechin-3-gallate (EGCG), tannic acid (TA), and rosmarinic acid (RA) have been well studied as protein aggregation modulators that inhibit formation of amyloid aggregations as well as dissociate preformed fibrils by preferentially and reversibly binding to the structures, breaking the initial ordered pattern of two polymers and greatly reducing their β sheet content and enlarging their conformational space.3,49, 50, 51, 52 The aromatic rings of these polyphenols appear to play a key role by interfering in amyloid aggregation and partially replacing the original amino acids in π-π stacking. To understand the effect of inhibitors in structural disruption-mediated modulation of tripeptide solubility, critical concentration was measured in the presence of different ratios of EGCG, TA, and RA. The presence of inhibitors at molar ratios ranging from 1:0.1–1:0.3 was unable to significantly alter the solubility of AFA, probably because of the inability of the inhibitors to disrupt interpeptide π-π stacking (Figure 3A). However, we observed a significant increase in critical soluble concentration of AAF and FAA in a dose-dependent manner in the presence of polyphenols (Figures 3B and 3C). The AAF solubility of 105 mM without inhibitor increased sharply to 130 mM and 210 mM in the presence of increasing amounts of EGCG (1:0.25–1: 0.5). The lower water solubility of EGCG hindered a further increase of its molar ratio. The effect of TA was found to be extremely potent because the solubility of AAF could be increased by 3-fold (340 mM), whereas the ratio reached 1:2 in favor of TA. Addition of RA was also caused substantial improvement of AAF solubility to 263 mM by the occurrence of a same ratio of RA. A similar sharp increase in critical soluble concentration of FAA was also observed by increasing the concentration of all three inhibitors interference with peptide stacking. The results clearly demonstrate the dependence of solubility of the low-soluble aromatic peptides, but not soluble AFA, on the π-π stacking interactions because increasing the stacking-disrupting agent enables a gradual increase in solubility by intercalating peptide aggregation and partial replacement of the peptide molecules in higher-order π-π stacking.
Figure 3.

Structure-dependent solubility of the tripeptides
(A–C) The effect of the polyphenols EGCG (A), TA (B), and RA (C) on modulation of solubility of the tripeptides because of interference with structural organization. Statistical distribution and p values of tripeptides solubility by the inhibitors compared with their absence is shown. The data represent the mean ± SEM for 3 independent experiments. N.S., not significant; ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.001.
(D–F) ESI-MS mass spectra of (D) AAF, (E) AFA, and (F) FAA (10 mM in water, pH 6.8, 25°C). Circles above the peaks denote the oligomer order. The number of circles represents the number of monomers in the aggregate. +1, +2, and +3 charge states are represented as cyan, pink, and green circles, respectively.
(G–I) ESI-IMS-MS 2D plot of the (G) AAF, (H) AFA, and (I) FAA monomers through to oligomers. ESI-IMS-MS 2D plots show the IMS drift time versus mass/charge (m/z) versus intensity (z, square-root scale). The different charge families are marked with white boxes. The numbers indicate the corresponding oligomeric states (for details, see Supplemental information).
(J) One-dimensional 1H NMR spectra of tripeptides (FAA, blue; AAF, red; AFA, green) dissolved in 90% H2O/10% 2H2O, with assignments labeled.
(K and L) 2D [1H-1H]-NOESY (blue) and [1H-1H]-TOCSY (red) NMR spectra of AFA (K) and AAF (L). For clarity, only the fingerprint region of TOCSY/NOESY spectra is shown, and the assignments are labeled.
Aggregation characterization using ESI-IMS-MS
In recent years, important advances in mass spectrometry (MS) have allowed the phenomenon of biological macromolecule aggregation to be uniquely characterized.53, 54, 55 Combination of MS with orthogonal techniques such as ion mobility spectrometry (IMS) manifests a new dimension that also yields insight into the gas-phase structural composition of the corresponding parent ions. When used in conjunction with electrospray ionization (ESI), ESI-IMS-MS has provided interesting structural information of building units in the gas phase.56, 57, 58, 59 To understand aggregation of higher-order oligomers and non-aggregative characteristics, the tripeptide samples were run on a homebuilt linear 4-m drift tube IMS paired with a time-of-flight (ToF) mass spectrometer (Figure S10). The ESI mass spectrum of a 10-mM water solution of AAF at pH 6.8 manifested a predominant presence of higher-order aggregates of different charge families, as shown in Figures 3D and S11. For the +1 charge state, monomeric and oligomeric species were the most abundant, followed by dimers through pentamers, with traces of hexamer ions. High-intensity peaks observed for +2 (heptamer through undecamer), +3 (decamer through hexadecamer), and +4 (heptadecamer and nonadecamer) charge states indicated strong aggregation. A typical 2D plot of the data (a nested drift-[flight] time distribution), recorded upon ESI of the AAF solution, is shown in Figure 3G and S12. As reported in earlier studies, for amino acids and peptides, peaks fall linearly into families according to their charge states.56,57 Under the experimental conditions employed here, AAF showed a multitude of higher-order species comprising five distributions of charge states: [n(AAF)+H]+ (n = 1–6, where n corresponds to the number of monomer units), [n(AAF)+2H]2+ (n = 6–13), [n(AAF)+3H]3+ (n = 10–21), [n(AAF)+4H]4+ (n = 17 to 24), and [n(AAF)+5H]5+ (Table S2). The extensive population of the +4 and higher charge families indicated a strong aggregation propensity to form higher-order oligomers that finally assembled into fibrillary structures, as observed in scanning electron microscopy images (Figure 1L). In sharp contrast, the ESI mass spectrum of AFA under similar experimental conditions exhibited a much lower affinity of monomeric conformers for aggregation, as shown in Figures 3E and S11. The spectrum showed a predominant presence of the +1 charge state, with a lesser intensity of dimers and trimers along with only a tiny amount of tetramers and pentamers. Although AFA was found to form clusters up to the +5 charge state, prominent intensity was observed only up to oligomers of +3: [n(AFA)+H]+ (n = 1–5), [n(AFA)+2H]2+ (n = 7–13), and [n(AFA)+3H]3+ (n = 12–15) (Figures 3H and S12; Table S3). The ESI-IMS-MS data presented clear differences between the spectral “fingerprints” of AAF undergoing strong aggregation into higher-order clusters, whereas AFA epitomized significantly lower aggregation propensity. This further clarified the behavior of AFA, which existed mainly as soluble monomeric conformers in solution and showed reluctance to form any ordered nanostructures (Figure 1G). Although FAA was expected to show a spectral pattern and ESI-IMS-MS plot similar to aggregated AAF, the traces of higher-order oligomers could not be detected clearly (Figures 3F, 3I, S11, and S12; Table S4). Under the applied experimental concentration, FAA converted into a gel-like, semi-solid matrix, which might affect the experimental results and the exact proportions of higher-order oligomeric structures compared with analogous tripeptides.
Solution structure and aggregation analysis by NMR spectroscopy
To probe the molecular conformation of the tripeptides in solution and ensure a similar molecular organization with the solid-state structure, comprehensive NMR spectroscopy studies were carried out in 9:1 H2O/2H2O solution at 10 mM concentration and pH ∼7. 1D 1H spectra showed a single set of resonances for all three tripeptides (Figure 3J). The NMR assignments were inferred from 2D [1H-1H]-TOCSY, 2D [1H-1H]-NOESY, [13C-1H]-HSQC, and [15N-1H]-HSQC spectra (Figures 3K, 3L, S13, and S14). The amide temperature coefficients were calculated from NMR spectra recorded at different temperatures ranging from 5°C–30°C (Figure S15). The amide temperature coefficients for Ala2 (FAA), Phe2 (AFA), Ala2 (AAF), and Phe3 (AAF) were less than 5 ppb/K (Table S5), suggesting their involvement in hydrogen bonding. The chemical shifts and 3JHNα values were measured from 1D and 2D NMR spectra to obtain evidence regarding the nature of the secondary structures (Table S5). For AAF, the measured 3JHNα values for Ala2 (5.8 Hz) and Phe3 (8 Hz) designate a helical-turn-like and an extended conformation for Ala2 and Phe3, respectively, showing the existence of a molecular organization in solution similar to that observed in the solid state. The 3JHNα values for the residues of AFA were between 6 and less than 8 Hz, indicating that the peptide has a disordered/extended conformation, which is in accordance with the solid-state structural arrangement. FAA shows 3JHNα values in a range similar to that of AFA. Additionally, for AAF, a large HN chemical shift dispersion was observed compared with FAA and AFA, supporting the conformations mentioned above for these peptides (Figures 3J–3L and S13). Further, the self-association of tripeptides under these conditions was monitored by measuring translational diffusion coefficient measurements using DOSY NMR experiments (Figure S16). However, the measured diffusion coefficients were found to be quite similar for all three peptides, with hydrodynamic radii between 1.18–1.28 nm (Table S6), suggesting the absence of higher-order aggregates in solution. DOSY spectra were also recorded for FAA and AAF at a higher concentration of 40 mM. Although AAF remained entirely in solution at this concentration, FAA produced larger subcritical aggregates that came out of the solution, leaving monomers and small oligomers in solution, resulting in similar diffusion coefficient values observed at a lower concentration. Also, identical chemical shifts were observed at different concentrations (10 and 40 mM) and at different time intervals (day 1 and day 28) (Figure S17), further showing the existence of mainly non-aggregative units in solution in an isotropic environment.
Molecular dynamics simulations
Molecular dynamics (MD) simulations are used widely to dissect the mechanisms of peptide aggregation and interactions.60, 61, 62 To understand the distinct solubility of AAF, FAA, and AFA at the molecular level, we performed all-atom MD simulations on systems consisting of 60 AAF, FAA, or AFA molecules in aqueous solutions (Figures 4, S18, and S19). All simulations started from randomly disordered states (Figures 4A–4C). Within 300 ns, AAF and FAA molecules aggregated into a large and relatively compact cluster with only a few molecules dispersed in solution and the compact aggregate maintained until the end of the simulation (Figures 4A and 4B). In contrast, AFA molecules first aggregated into a large but loosely packed cluster that split into two separate clusters at t = 500 ns (Figure 4C).
Figure 4.

Distinct solubility of AAF, FAA, and AFA probed by all-atom MD simulations
(A–C) Snapshots of (A) AAF, (B) FAA, and (C) AFA aggregates at three time points. Tripeptide molecules are shown in van der Waals (VDW) representation, with Ala in yellow and Phe in blue.
(D–F) Probability density function (PDF) of RgY/RgX and RgZ/RgX of the largest aggregates, where RgX, RgY, and RgZ are the radii of gyration (Rg) of a nanoassembly with respect to the three principle axes x, y, and z.
(G–I) PDF of the number of clusters and the size of the largest cluster, SASA of tripeptide molecules and Phe residues, and number of H-bonds formed between tripeptides (Np-p) and between tripeptide and water molecules (Np-w).
(J–L) The free energy landscape (FEL) of π-π stacking plotted as a function of the centroid distance and the angle between two aromatic rings.
(M–O) Probability of the parallel π-π stacking pattern within 0–200 ns and 200–500 ns.
To characterize the shape of the aggregates, we calculated the probability density function (PDF) of RgY/RgX and RgZ/RgX of the largest assemblies, where RgX, RgY, and RgZ are the radii of gyration (Rgs) of a nanoassembly with respect to the three principal axes x, y, and z. RgY and RgZ of AAF were almost the same but larger than RgX, which corresponds to a prolate aggregate, suggesting elongation potential of AAF during fibril formation (Figure 4D). As for FAA and AFA, RgY and RgZ of the aggregates were much closer to RgX, indicating formation of sphere-like nanoaggregates (Figures 4E and 4F). Comparison of the cluster number and the largest cluster size in the three systems showed that AAF and FAA formed much larger aggregates than AFA, indicating their stronger aggregation propensity and lower solubility (Figure 4G). The smaller solvent-accessible surface area (SASA) of AAF and FAA reveals that more AAF and FAA molecules assemble into aggregates, consistent with their stronger aggregation propensity compared with AFA. More importantly, the side chains of the Phe residues in AAF and FAA showed a smaller solvent-exposed surface than those in AFA, indicating that the aromatic rings of AAF and FAA play a major role in aggregate formation, in contrast to AFA (Figure 4H). As shown in Figure 4I, AAF and FAA form more inter-peptide H-bonds and fewer H-bonds with water molecules compared with AFA, revealing that AAF and FAA possess stronger aggregation propensity and lower solubility.
To reveal the contribution of each residue to the different aggregation propensities of the three peptides, we calculated the time evolution of their fraction of SASA (Figures S19A–S19C). The SASA fraction of the Phe side chains in AAF dropped rapidly from 0.38 to 0.29 within the first 100 ns and then fluctuated around this value until the end of the simulation, whereas the SASA fraction of the N-terminal Ala increased quickly to 0.22, indicating that the aromatic ring of AAF is mostly buried in the aggregates, whereas the N-terminal Ala is solvent exposed. As for the second Ala in the peptide sequence, its SASA fraction did not change significantly throughout the entire simulation (Figure S19A). Although FAA exhibited strong aggregation propensity similar to AAF, the SASA fraction of Phe side chains increased slightly to 0.41 before t = 129 ns and then decreased to 0.38 with fluctuation, revealing that the aromatic rings in FAA are not buried in the aggregates like those in AAF. Instead, the C-terminal Ala tended to be buried, as evidenced by the slight decrease of the SASA fraction (Figure S19B). In AFA systems, the SASA fraction of each residue exhibited no significant change throughout the entire simulation (Figure S19C), indicating that the aggregation propensity of AFA is very weak. The Phe and Ala residues make sequence-dependent contributions to the solvent exposure of the peptide, leading to different solubilities for the three peptides.
We also calculated the contact probability for each residue pair to uncover interactions crucial for the different self-assembly capabilities of AAF, FAA, and AFA (Figures S19D–S19F). The Phe-Phe pair in AAF and AFA showed the highest contact probability (0.732 and 0.449, respectively), indicating a dominant role of the Phe-Phe interaction in self-assembly of AAF and AFA. In contrast, the highest contact probability in FAA was detected between Phe and the C-terminal Ala, and the contact probability between Phe residues ranked second, with a value of 0.421. The contact probability between Phe residues in AAF is much higher than that in FAA and AFA, indicating that the Phe-Phe interaction is crucial for aggregate formation by AAF. The interaction strength among different residue pairs in FAA is quite similar, revealing strong interaction competition and lack of a dominant interaction, which may lead to imperfect molecular packing and explain the inability of FAA to grow good-quality single crystals.
To explore the role of aromatic stacking in self-assembly of the three tripeptides, we calculated the free energy landscape (FEL) of π-π stacking as a function of the centroid distance and the angle between two aromatic rings of AAF, FAA, and AFA (Figures 4J–4L). The stacking patterns are classified into three categories according to the angles: parallel (0°–30°), herringbone (30°–60°), and perpendicular (T-shaped, 60°–90°). The FEL shows that the aromatic stacking of AAF and FAA is much stronger than that of AFA. Perpendicular and herringbone stacking patterns, rather than the parallel pattern, are dominant, in contrast to the aromatic stacking pattern observed in tripeptide crystal structures. This is probably because of the fact that our MD simulations at a nanosecond timescale can only explore the early stage of tripeptide self-assembly, which is far from the timescale required for crystallization. The early-formed aggregates still undergo structural reorganization toward thermodynamically stable crystal structures. Furthermore, statistical analysis of the parallel stacking within 0–200 ns and 200–500 ns shows that the probability of the parallel pattern in AAF increases significantly over the simulation time (Figure 4M), showing a strong tendency toward the stacking pattern observed in the crystal structure. However, such a tendency is not observed in the FAA and AFA systems (Figures 4N and 4O).
To investigate the aggregation propensity of AAF, FAA, and AFA at a larger size scale, we performed 3-ms coarse-grained (CG) MD simulations on systems consisting of 720 AAF, FAA, or AFA molecules in aqueous solution. All three tripeptides self-assembled into large aggregates from the dispersed states (Figures 5A–5C). The cluster number of AAF and FAA dropped rapidly within the first 300 ns and reached a plateau of 12 and 5, respectively, at t = 500 ns, whereas the cluster number of AFA decreased slowly to a larger value of 86 (Figure 5D). The size of the largest cluster of FAA increased and reached a value of 706 at t = 770 ns, corresponding to formation of one large aggregate within 1 μs (purple line in Figure 5E and snapshot at t = 1 μs in Figure 5B). Interestingly, the size of the largest cluster of AAF was smaller than that of FAA after t = 770 ns, followed by an increase to a value only slightly smaller than that of FAA at t = 2.3 μs (blue line in Figure 5E). AAF self-assembled into two aggregates around t = 1 μs (snapshot at t = 1 μs in Figure 5A), which then started to fuse into a large prolate aggregate (snapshots at t = 2 μs and 3 μs in Figure 5A). AFA showed the smallest size of the largest cluster after t = 1 μs, and the largest cluster size only reached a value of 624 at t = 3 μs (orange line in Figure 5E), leaving almost 100 AFA molecules dispersed in solution (snapshot at t = 3 μs in Figure 5C). More importantly, the largest cluster size of AFA fluctuated the most, indicating that AFA molecules exchange dynamically into and out of the aggregates. Thus, AFA molecules display the highest mobility, in accordance with its highest solubility. The rapid decrease of the SASA also reveals that all three tripeptide molecules aggregate into large aggregates from dispersed states (Figure 5F). These data indicate that the aggregation propensities of the tripeptide molecules are in the order of FAA > AAF > AFA. Aggregates formed in CG MD simulations were indeed larger and more structurally ordered than those in all-atom MD simulations. We also calculated the FEL of π-π stacking in the largest cluster of AAF, FAA, and AFA as a function of the centroid distance and the angle between two aromatic rings (Figures 5G–5I). A minimum-energy basin located at 15° and 0.48 nm was observed in the free energy surface, corresponding to formation of the parallel stacking pattern in the aggregates of AAF, FAA, and AFA. These data reveal that the probability of parallel aromatic stacking patterns increases with system size and simulation timescale, but the perpendicular and herringbone stacking patterns are still dominant.
Figure 5.

Large size scale analysis of the different aggregation propensities of AAF, FAA, and AFA using microsecond-long CG MD simulations
(A–C) Snapshots of AAF, FAA, and AFA aggregates at four time points. The CG beads of the main chain and side chain are colored yellow and blue, respectively.
(D–F) Time evolution of (D) the number of clusters, (E) the size of the largest cluster, and (F) the SASA of the aggregates.
(G–I) FEL of intermolecular π-π stacking in the largest cluster, plotted as a function of the centroid distance and the angle between two aromatic rings. The basins of the parallel stacking pattern are marked by arrows.
Screening of peptide library
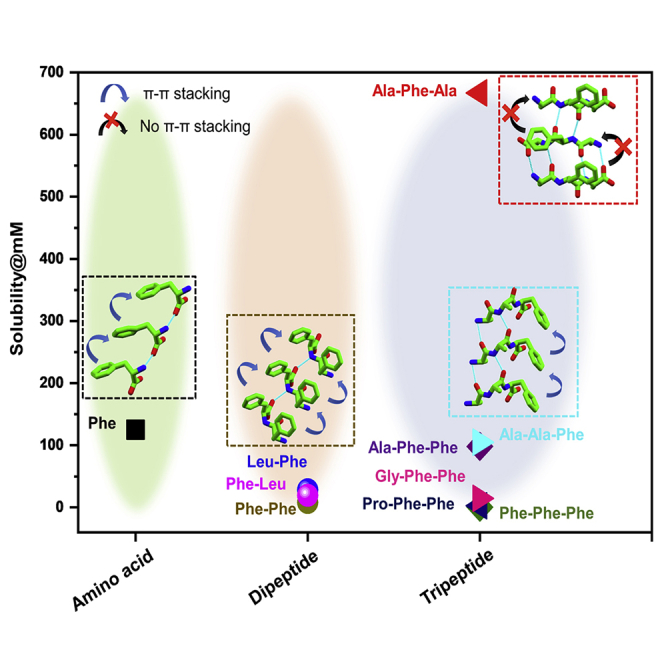
To further investigate whether the notion of solid-state packing-dependent solubility is applicable only to the studied tripeptide series or whether it is a generic phenomenon that is lawful for all peptides, we prepared a focused library of Phe-based peptides with similar compositions at different length scales (Figure 6; Table S7). For proof of principle, a number of building units staring from the single amino acid (F)63 through dipeptides (FF, LF, and FL)64 to tripeptides (FFF, PFF, GFF, and AFF),27,28,65 all having knowledge of structural information, were selected as queries to investigate water solubility at room temperature and pH 6.8 and compared with the three peptides studied here. The single amino acid F, with one π-π interaction per molecule, was soluble up to 125 mM. Upon increasing the chain length, the solubility of aromatic peptides decreased gradually. FF, comprising two types of π-π interactions per molecule, could be solubilized at a maximal concentration of 8.1 mM. However, replacing one F with an aliphatic amino acid raised the solubility to some extent because of the decrease in the number of aromatic-aromatic interactions. Thus, FL and LF with one aromatic stacking per molecule showed a higher solubility of 28.3 mM and 19.4 mM, respectively, than FF. The tripeptide FFF was found to exhibit significantly lower water solubility (0.5 mM) because of the increased number of π-π interaction (three types per molecule). However, lowering the number of aromatic side chains gradually increased the solubility; PFF and GFF, each comprising two types of aromatic-aromatic interactions per molecule, showed a threshold concentration of 2.6 mM and 14.5 mM, respectively. Surprisingly, despite the similar composition and hydrophobicity, AFF displayed a 6-fold higher solubility of 98.3 mM compared with GFF. The solid-state structure of AFF revealed absence of any π-π interaction in the packing because of antiparallel β-sheet structure formation (Figure S20). The observed lack of aromatic interactions in the molecular organization and the tremendous increase in AFF solubility is in accordance with the discovered 6-fold and 15-fold higher solubility of AFA without aromatic-aromatic stacking compared with the analogous peptides AAF and FAA, respectively, exerting strong aromatic interactions in the solid state. The solubility of a solid in water mainly depends on two factors: the stability of the solute crystal lattice and the interaction of the solute with water. For the molecule to dissolve in the aqueous solvent, it must be able to dissociate from its crystal lattice. Therefore, the general solubility equation (GSE) used to predict aqueous solubility indicates solubility at room temperature as a simple function of its melting temperature (Tm).66,67 However, many suggested structure-property associations in the literature are valid only for the immediate system described and lack full transferability to a wider context. To investigate the relation between solubility and melting point, the Tm of all of the above mentioned amino acids and peptides was measured at room temperature (Table S7). Correlation between solubility and melting point was found only within isostructural groups having a similar length of chains, but anomalies appear for different lengths of peptides, further indicating our motivation for extended elucidation of the process of solubility. On the other hand, screening of a peptide library and the observed solid-state packing-dependent water solubility perfectly validates our proposed principle of the molecular origin of peptide solubility based on solid-state structure.
Figure 6.

Solid-state packing underlies the solubility of Phe-containing peptides at different length scales
The solubility of single amino acids (NH3+-F-COO−), dipeptides (NH3+-FF-COO−.H2O, NH3+-FL-COO−.H2O, and NH3+-LF-COO−.H2O), and tripeptides (NH3+-FFF-COO−, NH2+-PFF-COO−, NH3+-GFF-COO−.H2O, NH3+-AFF-COO−.TFA.H2O, NH3+-AAF-COO−.TFA, and NH3+-AFA-COO−.TFA.AcOH.H2O) is represented by squares, circles, and triangles, respectively. The solid-state structure of the corresponding peptide is presented in a box of the same color as the symbol. Black and blue arrows indicate the presence of one and more than one type of aromatic-aromatic stacking, respectively, in the structure.
In summary, understanding and prediction of peptide solubility in water is of relevance in diverse areas such as basic chemistry, materials science, and molecular medicine. However, the puzzle still remains unresolved, and there is an extreme need for in-depth exploration. The experimental results presented here show that solid-state packing guides the water solubility of aromatic peptides. The analogous tripeptides AFA, AAF, and FAA, obtained only by sequence alteration, exhibit significantly different solubility stemming from their different molecular packing at the atomic level. Lacking any π-π interactions, AFA has 6-fold and 15-fold higher solubility than AAF and FAA, respectively, which comprise aromatic stacking in the solid-state structure. The extremely poor aggregation of AFA further supports its predominant presence as a soluble monomeric conformer in solution. Comprehensive NMR analysis reveals the molecular conformation in solution to be similar to the solid-state structure. All-atom and CG MD simulations demonstrate their differences in aggregation propensity and solubility and provide explanations at the molecular level for the unexpected solubility of AFA. Additionally, screening of a focused peptide library based on Phe-containing peptides at different length scales establishes this notion more firmly. Solubility and the number of aromatic side chains are inversely related as long as all aromatic side chains are involved in the π-π interaction. However, a major discrepancy of this general trend is found in peptides lacking π-π stacking in the solid-state structure, which exhibits a remarkably high critical soluble concentration. The results illustrate the molecular origin of high solubility of certain aromatic peptides and are significant steps forward in understanding peptide solubility, potentially paving the way for modular design of future peptide drugs based on highly soluble aromatic peptides.
Experimental procedures
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Ehud Gazit (ehudga@tauex.tau.ac.il).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The data supporting the findings of this study are available in the article and the Supplemental information. X-ray crystallography data for AAF (cif) have been deposited in the Cambridge Crystallographic Data Centre (CCDC). The accession number is CCDC:1942277. These data can be obtained free of charge from the CCDC. All other data are available from the lead contact upon reasonable request.
Preparation of peptide solution and concentration measurements
The peptides were synthesized by DGpeptides (Hangzhou, Zhejiang, China) and purified to more than 95% followed by MS confirmation of their identity (Figures S21–S23). All peptides were stored at −20°C. Solutions of peptides were prepared by suspension in distilled water, followed by vigorous vortexing for 2 min at room temperature. The samples were then centrifuged for 15 min at 6,000 rpm. The critical concentrations of the peptide solutions were determined by UV spectrophotometry using a Cary 100 spectrophotometer. Spectra were recorded between 300 and 200 nm with a reading every 0.5 nm and a scan rate of 400 nm/min. Glass cuvettes of 10-mm path length were used for the experiments. Concentration was calculated based on absorption at 257 nm. All other experimental details are described in the Supplemental experimental procedures.
Acknowledgments
S.B. thanks Tel Aviv University for a post-doctoral fellowship. This project received funding from ERC under the European Union Horizon 2020 Research and Innovation Program (grant agreement BISON-694426 to E.G and 5R01GM121751-03 to D.E.C). The authors thank Dr. Sigal Rencus-Lazar for help with scientific and language editing.
Author contributions
S.B. and E.G. designed the experiments. S.B. performed the experiments and crystallization of peptides. X.D., Y.T., and G.W. performed the all-atoms MD and CG MD analysis. B.K. and A.R. performed the NMR experiments. S.A.R., D.A.H., and D.E.C. performed the ESI-IMS-MS analysis. W.J. performed DLS of the peptides. L.J.W.S. solved the crystal structures. S.B. coordinated all of the work and analyzed the results. S.B., X.D., G.W., and E.G, wrote the paper. All authors commented on the manuscript.
Declarations of interest
The authors declare no competing interests.
Published: April 2, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrp.2021.100391.
Supplemental information
References
- 1.Malavolta L., Pinto M.R.S., Cuvero J.H., Nakaie C.R. Interpretation of the dissolution of insoluble peptide sequences based on the acid-base properties of the solvent. Protein Sci. 2006;15:1476–1488. doi: 10.1110/ps.051956206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perrett S., Buell A., Knowles T. Springer; 2019. Biological and Bio-inspired Nanomaterials. [Google Scholar]
- 3.Ji W., Xue B., Arnon Z.A., Yuan H., Bera S., Li Q., Zaguri D., Reynolds N.P., Li H., Chen Y. Rigid Tightly Packed Amino Acid Crystals as Functional Supramolecular Materials. ACS Nano. 2019;13:14477–14485. doi: 10.1021/acsnano.9b08217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tadepalli S., Slocik J.M., Gupta M.K., Naik R.R., Singamaneni S. Bio-Optics and Bio-Inspired Optical Materials. Chem. Rev. 2017;117:12705–12763. doi: 10.1021/acs.chemrev.7b00153. [DOI] [PubMed] [Google Scholar]
- 5.Knowles T.P.J., Buehler M.J. Nanomechanics of functional and pathological amyloid materials. Nat. Nanotechnol. 2011;6:469–479. doi: 10.1038/nnano.2011.102. [DOI] [PubMed] [Google Scholar]
- 6.Abdelrahman S., Alghrably M., Lachowicz J.I., Emwas A.-H., Hauser C.A.E., Jaremko M. “What Doesn’t Kill You Makes You Stronger”: Future Applications of Amyloid Aggregates in Biomedicine. Molecules. 2020;25:5245. doi: 10.3390/molecules25225245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei G., Su Z., Reynolds N.P., Arosio P., Hamley I.W., Gazit E., Mezzenga R. Self-assembling peptide and protein amyloids: from structure to tailored function in nanotechnology. Chem. Soc. Rev. 2017;46:4661–4708. doi: 10.1039/c6cs00542j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bak A., Leung D., Barrett S.E., Forster S., Minnihan E.C., Leithead A.W., Cunningham J., Toussaint N., Crocker L.S. Physicochemical and formulation developability assessment for therapeutic peptide delivery--a primer. AAPS J. 2015;17:144–155. doi: 10.1208/s12248-014-9688-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherje A.P., Desai K.J. Spectrophotometric Determination of Poorly Water Soluble Drug Rosiglitazone Using Hydrotropic Solubilization technique. Indian J. Pharm. Sci. 2011;73:579–582. doi: 10.4103/0250-474X.99021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang S. Fabrication of novel biomaterials through molecular self-assembly. Nat. Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 11.Whitesides G.M., Mathias J.P., Seto C.T. Molecular self-assembly and nanochemistry: a chemical strategy for the synthesis of nanostructures. Science. 1991;254:1312–1319. doi: 10.1126/science.1962191. [DOI] [PubMed] [Google Scholar]
- 12.Yuan C., Ji W., Xing R., Li J., Gazit E., Yan X. Hierarchically Oriented Organization in Supramolecular Peptide Crystals. Nat. Rev. Chem. 2019;3:567–588. [Google Scholar]
- 13.Alghrably M., Dudek D., Emwas A.-H., Jaremko Ł., Jaremko M., Rowińska-Żyrek M. Copper(II) and Amylin Analogues: A Complicated Relationship. Inorg. Chem. 2020;59:2527–2535. doi: 10.1021/acs.inorgchem.9b03498. [DOI] [PubMed] [Google Scholar]
- 14.Łoboda D., Rowińska-Żyrek M. Zn(II) - pramlintide: Stability, binding sites and unexpected aggregation. J. Inorg. Biochem. 2017;174:150–155. doi: 10.1016/j.jinorgbio.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 15.Alghrably M., Czaban I., Jaremko Ł., Jaremko M. Interaction of amylin species with transition metals and membranes. J. Inorg. Biochem. 2019;191:69–76. doi: 10.1016/j.jinorgbio.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Brender J.R., Hartman K., Nanga R.P.R., Popovych N., de la Salud Bea R., Vivekanandan S., Marsh E.N.G., Ramamoorthy A. Role of zinc in human islet amyloid polypeptide aggregation. J. Am. Chem. Soc. 2010;132:8973–8983. doi: 10.1021/ja1007867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fleming S., Ulijn R.V. Design of nanostructures based on aromatic peptide amphiphiles. Chem. Soc. Rev. 2014;43:8150–8177. doi: 10.1039/c4cs00247d. [DOI] [PubMed] [Google Scholar]
- 18.Gazit E. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002;16:77–83. doi: 10.1096/fj.01-0442hyp. [DOI] [PubMed] [Google Scholar]
- 19.Lampel A., McPhee S.A., Park H.-A., Scott G.G., Humagain S., Hekstra D.R., Yoo B., Frederix P.W.J.M., Li T.-D., Abzalimov R.R. Polymeric peptide pigments with sequence-encoded properties. Science. 2017;356:1064–1068. doi: 10.1126/science.aal5005. [DOI] [PubMed] [Google Scholar]
- 20.Frederix P.W.J.M., Scott G.G., Abul-Haija Y.M., Kalafatovic D., Pappas C.G., Javid N., Hunt N.T., Ulijn R.V., Tuttle T. Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels. Nat. Chem. 2015;7:30–37. doi: 10.1038/nchem.2122. [DOI] [PubMed] [Google Scholar]
- 21.Hsieh M.C., Liang C., Mehta A.K., Lynn D.G., Grover M.A. Multistep Conformation Selection in Amyloid Assembly. J. Am. Chem. Soc. 2017;139:17007–17010. doi: 10.1021/jacs.7b09362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Omosun T.O., Hsieh M.C., Childers W.S., Das D., Mehta A.K., Anthony N.R., Pan T., Grover M.A., Berland K.M., Lynn D.G. Catalytic diversity in self-propagating peptide assemblies. Nat. Chem. 2017;9:805–809. doi: 10.1038/nchem.2738. [DOI] [PubMed] [Google Scholar]
- 23.Vivekanandan S., Brender J.R., Lee S.Y., Ramamoorthy A. A partially folded structure of amyloid-beta(1-40) in an aqueous environment. Biochem. Biophys. Res. Commun. 2011;411:312–316. doi: 10.1016/j.bbrc.2011.06.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doran T.M., Kamens A.J., Byrnes N.K., Nilsson B.L. Role of amino acid hydrophobicity, aromaticity, and molecular volume on IAPP(20-29) amyloid self-assembly. Proteins. 2012;80:1053–1065. doi: 10.1002/prot.24007. [DOI] [PubMed] [Google Scholar]
- 25.Madhusudan Makwana K., Mahalakshmi R. Implications of aromatic-aromatic interactions: From protein structures to peptide models. Protein Sci. 2015;24:1920–1933. doi: 10.1002/pro.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma M., Kuang Y., Gao Y., Zhang Y., Gao P., Xu B. Aromatic-aromatic interactions induce the self-assembly of pentapeptidic derivatives in water to form nanofibers and supramolecular hydrogels. J. Am. Chem. Soc. 2010;132:2719–2728. doi: 10.1021/ja9088764. [DOI] [PubMed] [Google Scholar]
- 27.Reches M., Gazit E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science. 2003;300:625–627. doi: 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- 28.Bera S., Mondal S., Xue B., Shimon L.J.W., Cao Y., Gazit E. Rigid helical-like assemblies from a self-aggregating tripeptide. Nat. Mater. 2019;18:503–509. doi: 10.1038/s41563-019-0343-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayans E., Casanovas J., Gil A.M., Jiménez A.I., Cativiela C., Puiggalí J., Alemán C. Diversity and Hierarchy in Supramolecular Assemblies of Triphenylalanine: From Laminated Helical Ribbons to Toroids. Langmuir. 2017;33:4036–4048. doi: 10.1021/acs.langmuir.7b00622. [DOI] [PubMed] [Google Scholar]
- 30.Marshall K.E., Morris K.L., Charlton D., O’Reilly N., Lewis L., Walden H., Serpell L.C. Hydrophobic, aromatic, and electrostatic interactions play a central role in amyloid fibril formation and stability. Biochemistry. 2011;50:2061–2071. doi: 10.1021/bi101936c. [DOI] [PubMed] [Google Scholar]
- 31.Bowerman C.J., Liyanage W., Federation A.J., Nilsson B.L. Tuning β-sheet peptide self-assembly and hydrogelation behavior by modification of sequence hydrophobicity and aromaticity. Biomacromolecules. 2011;12:2735–2745. doi: 10.1021/bm200510k. [DOI] [PubMed] [Google Scholar]
- 32.Azriel R., Gazit E. Analysis of the minimal amyloid-forming fragment of the islet amyloid polypeptide. An experimental support for the key role of the phenylalanine residue in amyloid formation. J. Biol. Chem. 2001;276:34156–34161. doi: 10.1074/jbc.M102883200. [DOI] [PubMed] [Google Scholar]
- 33.Kodaka M. Interpretation of concentration-dependence in aggregation kinetics. Biophys. Chem. 2004;109:325–332. doi: 10.1016/j.bpc.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 34.Poulson B.G., Szczepski K., Lachowicz J.I., Jaremko L., Emwas A.-H., Jaremko M. Aggregation of Biologically Important Peptides and Proteins: Inhibition or Acceleration Depending on Protein and Metal Ion Concentrations. RSC Advances. 2020;10:215–227. doi: 10.1039/c9ra09350h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eker F., Griebenow K., Cao X., Nafie L.A., Schweitzer-Stenner R. Preferred peptide backbone conformations in the unfolded state revealed by the structure analysis of alanine-based (AXA) tripeptides in aqueous solution. Proc. Natl. Acad. Sci. USA. 2004;101:10054–10059. doi: 10.1073/pnas.0402623101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Motta A., Reches M., Pappalardo L., Andreotti G., Gazit E. The preferred conformation of the tripeptide Ala-Phe-Ala in water is an inverse γ-turn: implications for protein folding and drug design. Biochemistry. 2005;44:14170–14178. doi: 10.1021/bi050658v. [DOI] [PubMed] [Google Scholar]
- 37.Pizzanelli S., Forte C., Monti S., Zandomeneghi G., Hagarman A., Measey T.J., Schweitzer-Stenner R. Conformations of phenylalanine in the tripeptides AFA and GFG probed by combining MD simulations with NMR, FTIR, polarized Raman, and VCD spectroscopy. J. Phys. Chem. B. 2010;114:3965–3978. doi: 10.1021/jp907502n. [DOI] [PubMed] [Google Scholar]
- 38.Sarma R., Wong K.Y., Lynch G.C., Pettitt B.M. Peptide Solubility Limits: Backbone and Side-Chain Interactions. J. Phys. Chem. B. 2018;122:3528–3539. doi: 10.1021/acs.jpcb.7b10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mason T.O., Michaels T.C.T., Levin A., Dobson C.M., Gazit E., Knowles T.P.J., Buell A.K. Thermodynamics of Polypeptide Supramolecular Assembly in the Short-Chain Limit. J. Am. Chem. Soc. 2017;139:16134–16142. doi: 10.1021/jacs.7b00229. [DOI] [PubMed] [Google Scholar]
- 40.Mason T.O., Chirgadze D.Y., Levin A., Adler-Abramovich L., Gazit E., Knowles T.P.J., Buell A.K. Expanding the solvent chemical space for self-assembly of dipeptide nanostructures. ACS Nano. 2014;8:1243–1253. doi: 10.1021/nn404237f. [DOI] [PubMed] [Google Scholar]
- 41.Mason T.O., Michaels T.C.T., Levin A., Gazit E., Dobson C.M., Buell A.K., Knowles T.P.J. Synthesis of Nonequilibrium Supramolecular Peptide Polymers on a Microfluidic Platform. J. Am. Chem. Soc. 2016;138:9589–9596. doi: 10.1021/jacs.6b04136. [DOI] [PubMed] [Google Scholar]
- 42.Graziano G. On the Salting out of Benzene by Alkali Chlorides. J. Chem. Eng. Data. 2009;54:464–467. [Google Scholar]
- 43.Arakawa T., Timasheff S.N. Mechanism of protein salting in and salting out by divalent cation salts: balance between hydration and salt binding. Biochemistry. 1984;23:5912–5923. doi: 10.1021/bi00320a004. [DOI] [PubMed] [Google Scholar]
- 44.Kong J., Yu S. Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Biochim. Biophys. Sin. (Shanghai) 2007;39:549–559. doi: 10.1111/j.1745-7270.2007.00320.x. [DOI] [PubMed] [Google Scholar]
- 45.Ramachandran G.N., Ramakrishnan C., Sasisekharan V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963;7:95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- 46.Anjana R., Vaishnavi M.K., Sherlin D., Kumar S.P., Naveen K., Kanth P.S., Sekar K. Aromatic-aromatic interactions in structures of proteins and protein-DNA complexes: a study based on orientation and distance. Bioinformation. 2012;8:1220–1224. doi: 10.6026/97320630081220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tayeb-Fligelman E., Tabachnikov O., Moshe A., Goldshmidt-Tran O., Sawaya M.R., Coquelle N., Colletier J.P., Landau M. The cytotoxic Staphylococcus aureus PSMα3 reveals a cross-α amyloid-like fibril. Science. 2017;355:831–833. doi: 10.1126/science.aaf4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nelson R., Sawaya M.R., Balbirnie M., Madsen A.Ø., Riekel C., Grothe R., Eisenberg D. Structure of the cross-β spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maity S., Kumar R., Maity S.K., Jana P., Bera S., Haldar D. Synthesis and Study of 2-Acetyl Amino-3-[4-(2-Amino-5-Sulfo-Phenylazo)- Phenyl]-Propionic Acid: A New Class of Inhibitor for Hen Egg White Lysozyme Amyloidogenesis. MedChemComm. 2013;4:530–536. [Google Scholar]
- 50.Hora M., Carballo-Pacheco M., Weber B., Morris V.K., Wittkopf A., Buchner J., Strodel B., Reif B. Epigallocatechin-3-gallate preferentially induces aggregation of amyloidogenic immunoglobulin light chains. Sci. Rep. 2017;7:41515. doi: 10.1038/srep41515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lopez del Amo J.M., Fink U., Dasari M., Grelle G., Wanker E.E., Bieschke J., Reif B. Structural properties of EGCG-induced, nontoxic Alzheimer’s disease Aβ oligomers. J. Mol. Biol. 2012;421:517–524. doi: 10.1016/j.jmb.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 52.Wang Q., Guo J., Jiao P., Liu H., Yao X. Exploring the influence of EGCG on the β-sheet-rich oligomers of human islet amyloid polypeptide (hIAPP1-37) and identifying its possible binding sites from molecular dynamics simulation. PLoS ONE. 2014;9:e94796. doi: 10.1371/journal.pone.0094796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cukalevski R., Yang X., Meisl G., Weininger U., Bernfur K., Frohm B., Knowles T.P.J., Linse S. The Aβ40 and Aβ42 peptides self-assemble into separate homomolecular fibrils in binary mixtures but cross-react during primary nucleation. Chem. Sci. (Camb.) 2015;6:4215–4233. doi: 10.1039/c4sc02517b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bera S., Mondal S., Tang Y., Jacoby G., Arad E., Guterman T., Jelinek R., Beck R., Wei G., Gazit E. Deciphering the Rules for Amino Acid Co-Assembly Based on Interlayer Distances. ACS Nano. 2019;13:1703–1712. doi: 10.1021/acsnano.8b07775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Do T.D., de Almeida N.E.C., LaPointe N.E., Chamas A., Feinstein S.C., Bowers M.T. Amino Acid Metaclusters: Implications of Growth Trends on Peptide Self-Assembly and Structure. Anal. Chem. 2016;88:868–876. doi: 10.1021/acs.analchem.5b03454. [DOI] [PubMed] [Google Scholar]
- 56.Counterman A.E., Clemmer D.E. Magic Number Clusters of Serine in the Gas Phase. J. Phys. Chem. B. 2001;105:8092–8096. [Google Scholar]
- 57.Chen X., Raab S.A., Poe T., Clemmer D.E., Larriba-Andaluz C. Determination of Gas-Phase Ion Structures of Locally Polar Homopolymers Through High-Resolution Ion Mobility Spectrometry-Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2019;30:905–918. doi: 10.1007/s13361-019-02184-9. [DOI] [PubMed] [Google Scholar]
- 58.Young L.M., Saunders J.C., Mahood R.A., Revill C.H., Foster R.J., Tu L.H., Raleigh D.P., Radford S.E., Ashcroft A.E. Screening and classifying small-molecule inhibitors of amyloid formation using ion mobility spectrometry-mass spectrometry. Nat. Chem. 2015;7:73–81. doi: 10.1038/nchem.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trimpin S., Plasencia M., Isailovic D., Clemmer D.E. Resolving oligomers from fully grown polymers with IMS-MS. Anal. Chem. 2007;79:7965–7974. doi: 10.1021/ac071575i. [DOI] [PubMed] [Google Scholar]
- 60.Yuan C., Li S., Zou Q., Ren Y., Yan X. Multiscale simulations for understanding the evolution and mechanism of hierarchical peptide self-assembly. Phys. Chem. Chem. Phys. 2017;19:23614–23631. doi: 10.1039/c7cp01923h. [DOI] [PubMed] [Google Scholar]
- 61.Tang Y., Yao Y., Wei G. Expanding the structural diversity of peptide assemblies by coassembling dipeptides with diphenylalanine. Nanoscale. 2020;12:3038–3049. doi: 10.1039/c9nr09317f. [DOI] [PubMed] [Google Scholar]
- 62.Guo C., Luo Y., Zhou R., Wei G. Probing the self-assembly mechanism of diphenylalanine-based peptide nanovesicles and nanotubes. ACS Nano. 2012;6:3907–3918. doi: 10.1021/nn300015g. [DOI] [PubMed] [Google Scholar]
- 63.Ihlefeldt F.S., Pettersen F.B., von Bonin A., Zawadzka M., Görbitz C.H. The polymorphs of L-phenylalanine. Angew. Chem. Int. Ed. Engl. 2014;53:13600–13604. doi: 10.1002/anie.201406886. [DOI] [PubMed] [Google Scholar]
- 64.Görbitz C.H. Nanotube formation by hydrophobic dipeptides. Chemistry. 2001;7:5153–5159. doi: 10.1002/1521-3765(20011203)7:23<5153::aid-chem5153>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 65.Parthasarathy R., Chaturvedi S., Go K. Design of crystalline helices of short oligopeptides as a possible model for nucleation of α-helix: role of water molecules in stabilizing helices. Proc. Natl. Acad. Sci. USA. 1990;87:871–875. doi: 10.1073/pnas.87.3.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ran Y., Yalkowsky S.H. Prediction of drug solubility by the general solubility equation (GSE) J. Chem. Inf. Comput. Sci. 2001;41:354–357. doi: 10.1021/ci000338c. [DOI] [PubMed] [Google Scholar]
- 67.Bergström C.A.S., Larsson P. Computational prediction of drug solubility in water-based systems: Qualitative and quantitative approaches used in the current drug discovery and development setting. Int. J. Pharm. 2018;540:185–193. doi: 10.1016/j.ijpharm.2018.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available in the article and the Supplemental information. X-ray crystallography data for AAF (cif) have been deposited in the Cambridge Crystallographic Data Centre (CCDC). The accession number is CCDC:1942277. These data can be obtained free of charge from the CCDC. All other data are available from the lead contact upon reasonable request.