

Abstract
Directed evolution experiments are typically carried out using in vitro systems, bacteria, or yeast—even when the goal is to probe or modulate mammalian biology. Performing directed evolution in systems that do not match the intended mammalian environment severely constrains the scope and functionality of targets that can be evolved. We review new platforms that are now making it possible to use the mammalian cell itself as the setting for directed evolution, and present an overview of frontier challenges and high-impact targets for this approach.
Editor’s Summary
This Perspective describes advances that have enabled robust directed evolution in mammalian cells. These approaches are poised to improve the development of new generations of tools to probe or modulate mammalian biology.
Directed evolution is a powerful methodology for creating biomolecules with new and improved properties. In a typical directed evolution experiment, a library of genetic variants is generated through mutagenesis of an initial sequence. The expressed biomolecules are then selected or screened for a desired activity. Iterative cycles of this process produce increasingly optimized biomolecules. This approach has yielded molecules useful to industry and research, numerous critical biotechnologies, and was the subject of the 2018 Nobel Prize in Chemistry.1 Aside from the many practical applications, valuable fundamental information about biomolecular structure, evolutionary biology, and organismal fitness can often be obtained using directed evolution.
Directed evolution can be used to create biomolecular tools to perturb or interrogate many mammalian systems, with both fundamental research and therapeutic applications. Directed evolution could even optimize for function in a specific cell type or genetic background, providing a platform for developing targeted or personalized medicine. From a basic science perspective, researchers could gain new insights into the structure and function of understudied mammalian proteins or those with unknown structures, as well as explore principles of mammalian evolutionary biology.
Yet, despite all this potential, the promised impact of directed evolution on mammalian biology and medicine has not yet been fully realized. Instead, many impactful targets and pathways have never been pursued using directed evolution, and many attempted directed evolution campaigns have yielded products that fail to function properly in mammalian cells. Published examples of the latter include monobodies,2 fluorescent proteins,3 inteins,4, 5 aminoacyl-tRNA synthetases,6 and membrane proteins.7
What is responsible for these failures? Directed evolution experiments are most commonly performed in vitro, in bacteria, or in yeast (Figure 1a), even when the product is intended to be used in mammalian cells or when the goal is to study mammalian biology.8 Unfortunately, most mammalian proteins do not retain their function when expressed in lower organisms, including in single-celled eukaryotes like yeast,9 commonly owing to misfolding or aggregation, the absence of complex signaling pathways unique to mammalian cells, or other differences (Figure 1b). Even when these obstacles can be overcome, the lower organism is still a highly artificial selection system. As a consequence, the desired functions of evolved products can be derailed in mammalian cells by unintended intermolecular interactions, poor folding, unexpected modifications or cellular localization, and many other serious problems (Figure 1c). Importantly, this limitation applies not only to mammalian proteins themselves but also to those derived from other sources, including bacteria and yeast, that could be evolved to perturb or probe mammalian biology.
Figure 1.
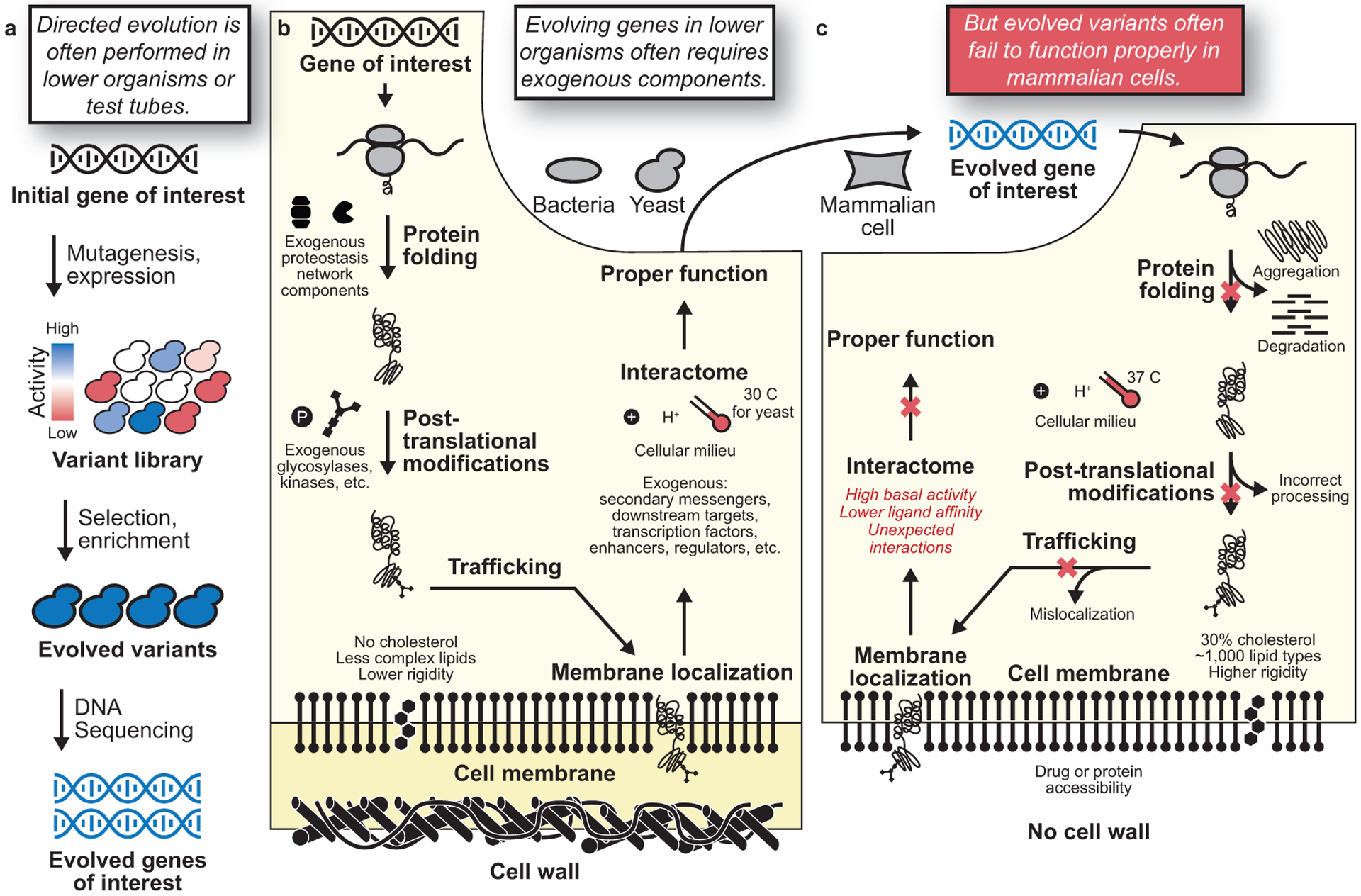
Directed evolution for function in mammalian cells. (a) In most directed evolution experiments, protein variants are expressed in test tubes, bacteria, or yeast. Genes of interest are mutagenized and expressed, creating a mutant library with varying activities. Successful variants (blue) are selected and enriched in the population more than unsuccessful variants (red). Evolved variants are then identified through DNA sequencing. (b–c) Illustration of some of the challenges associated with performing directed evolution in lower organisms—in this example, directed evolution of a mammalian membrane protein. (b) To evolve mammalian proteins in lower organisms, exogenous components from mammalian cells frequently need to be introduced for proper folding, modification, trafficking, and more. Membrane proteins also often require extensive interactions with other downstream exogenous components for proper function, requiring additional exogenous components. (c) Proteins evolved in vitro, in bacteria, or in yeast often fail to function properly once expressed in mammalian cells. Function can be derailed in mammalian cells because the evolved protein may aggregate, be degraded, be incorrectly processed or modified, mislocalize, interact with inappropriate partners, or lack catalytic or other activity for any number of additional reasons.
The apparent solution to all these issues is to use the mammalian cell itself as the design, engineering, and quality control setting for directed evolution. Although simple on its face, this solution immediately runs up against numerous obstacles. Many practical reasons, both technical (e.g., slow growth rate of mammalian cells, engineering challenges) and economic (e.g., labor-intensive mammalian cell culture, expensive media), have deterred researchers from using mammalian cells in directed evolution campaigns. These challenges, while real, have led the directed evolution field down what can prove to be an unproductive path—attempting to evolve function in poorly matched model systems.
Technological advances have yielded a comprehensive reshaping of the mammalian cell directed evolution landscape. New mammalian cell-specific platforms, including bespoke strategies for library creation and variant selection, are democratizing mammalian cell-based directed evolution. In this review, we present these emerging methods and some frontier challenges in the field. After first discussing key elements and challenges of mammalian cell-based directed evolution campaigns, we focus attention on ex mammalia and in mammalia mutagenesis methods. Next, we highlight virus-aided continuous evolution methods that integrate in mammalia mutagenesis with efficient expression and selection. Finally, we present a litany of high-impact directed evolution targets that were once too formidable to pursue but are now within reach.
General principles and challenges of mammalian cell-based directed evolution
Individual cycles of a directed evolution experiment have three main components: mutagenesis, expression, and screening or selection (Figure 2). For each component, mammalian cells present unique challenges.
Figure 2.

Directed evolution in mammalian cells. (a–b) First, genes of interest are mutagenized. (a) Ex mammalia mutagenesis strategies, such as error-prone PCR10 or mutator strains of Escherichia coli like XL-1 Red11 and bacteria transformed with the mutagenic MP6 plasmid,12 mutate genes of interest outside of the mammalian cell. (b) In mammalia mutagenesis strategies, such as targeted mutagenesis or viral mutagenesis, mutate genes directly in the mammalian cell. (c) Once genes of interest are mutagenized, the variant library must be expressed in mammalian cells, commonly through chemical transfection, electroporation, or viral transduction. (d–e) Cells must then be assessed for appropriate activity, either by screening or selection, and genes encoding active variants enriched in the population. (d) High-throughput screening methods include flow cytometry, cellular imaging followed by robotic cell picking,30 and cellular binding to affinity beads. (e) Selection methods may include cytotoxic drug treatment and viral selection.
Mutagenesis.
The first step in each cycle of a directed evolution experiment is mutagenesis, in which a large library of genetic variants of a gene of interest is created. Global mutagenesis approaches introduce off-target mutations throughout the genome. Considering the immense size and complexity of the mammalian genome, there is a high probability that such off-target mutations can lead to the creation of ‘cheaters’ that bypass screening or selection in unproductive ways, completely derailing directed evolution experiments.
Mammalian directed evolution strategies therefore rely upon targeted mutagenesis, meaning mutagenesis directed to a specific genetic region. Targeted mutagenesis methods can be categorized as either ex mammalia or in mammalia. Ex mammalia mutagenesis, or mutagenesis that occurs outside of mammalian cells, includes random techniques, such as error-prone PCR10 and mutator strains of bacteria,11, 12 as well as non-random techniques, such as site-directed mutagenesis and synthetic library generation (Figure 2a). Non-random mutagenesis provides precise control over which genetic variants are created, but these libraries are typically much smaller in size than those produced using random methods.13 Therefore, most ex mammalia mutagenesis strategies rely on random mutagenesis.
In mammalia mutagenesis, or mutagenesis that occurs inside mammalian cells, currently relies on somatic hypermutation,14 CRISPR-based DNA targeting,15 highly processive RNA polymerases,16, 17 or viral replication (Figure 2b). As opposed to ex mammalia mutagenesis, in mammalia mutagenesis enables multiple rounds of mutagenesis without needing to excise genetic libraries from the mammalian cell population to allow for further rounds of mutagenesis. Many of these strategies currently have a limited mutational spectrum, owing to their reliance on DNA-damaging enzymes to drive mutagenesis. Non-random in mammalia mutagenesis methods, such as Cas9-induced homology-directed repair using designed single-stranded DNA repair templates, can promote the introduction of a greater spectrum of point mutations18, 19 but generate smaller libraries.
Expression.
Once a mutagenized genetic library is generated, the protein variants must be expressed either transiently or stably within mammalian cells. In transient expression methods, including chemical transfection and electroporation, cells express protein variants without genomically encoding or replicating the variant’s genetic information (Figure 2c). Challenges associated with transient expression include transfection efficiency, inconsistent expression levels, expression of multiple variants in a single cell, and compatibility with a user-desired cell line. Further, if the genes encoding transiently expressed biomolecule variants are not somehow replicated in mammalia over multiple generations, they cannot be mutagenized in mammalia, hampering iterability. If, however, viruses are used to encode, propagate, and transiently express genetic variants, then in mammalia mutagenesis can be used alongside transient expression.
Stable expression, including retroviral transduction, can address many of the problems inherent to transient expression. Stably encoded genes can be mutated in mammalia, and protein expression levels may be more consistent. Nonetheless, cells can be infected by multiple retroviruses, and some commonly used retroviruses such as lentivirus are packaged with two genomes.
Selection or screening.
Once a library of protein variants is expressed in mammalian cells, the expressed variants must somehow be assessed for appropriate activity and then enriched in the population. This step can be categorized as either screening or selection, and the details depend entirely on the particular biomolecular activity being evolved. Screening involves physically separating cells on the basis of a given phenotype, commonly fluorescence-based flow cytometry or cellular display (Figure 2d). Screening-based methods can be quite labor-intensive since cells usually must be manually manipulated before, during, and after each round. Another challenge is determining appropriate activity cutoffs for each round, which must be modified to become more stringent over the course of a directed evolution experiment.
Selection-based strategies usually rely on the survival and replication of cells or viruses to enrich genetic variants (Figure 2e). Commonly, selection involves treating cells that encode a diverse library of genetic variants with a cytotoxic drug. Only those cells encoding genetic variants that provide resistance to the cytotoxic drug can survive selection to replicate and enrich in the population. Selection is in many ways much preferable to screening, as it requires minimal researcher intervention, is easy to scale and iterate, and requires little specialized equipment. Nonetheless, the slow growth rate of mammalian cells, the relatively narrow scope of biomolecules that negate the impact of cytotoxic perturbations, and the tendency for cells to spontaneously attain resistance to many such perturbations via undesired ‘cheating’ pathways, mean that selection-based approaches have historically been mostly limited to studies on oncoproteins.20–24 Recently developed virus-based selection methods25, 26, described below, overcome many of the limitations of these past cell-based methods, leading to a vastly expanded scope of biomolecules evolvable through selection in mammalian cells.
Mammalian cell-based directed evolution enabled by ex mammalia mutagenesis
Ex mammalia mutagenesis combined with cellular screening is historically the most common approach for directed evolution in mammalian cells (Figure 3). Ex mammalia mutagenesis techniques were originally designed to support directed evolution in test tubes or lower organisms,11, 12 and typically do not account for the unique challenges associated with mutagenesis within mammalian cells. Nonetheless, despite the constraints that ex mammalia techniques place on library size and throughput, these strategies can produce valuable evolved biomolecules that interact with and depend on the unique mammalian cellular environment.13, 20, 21, 27–31
Figure 3.
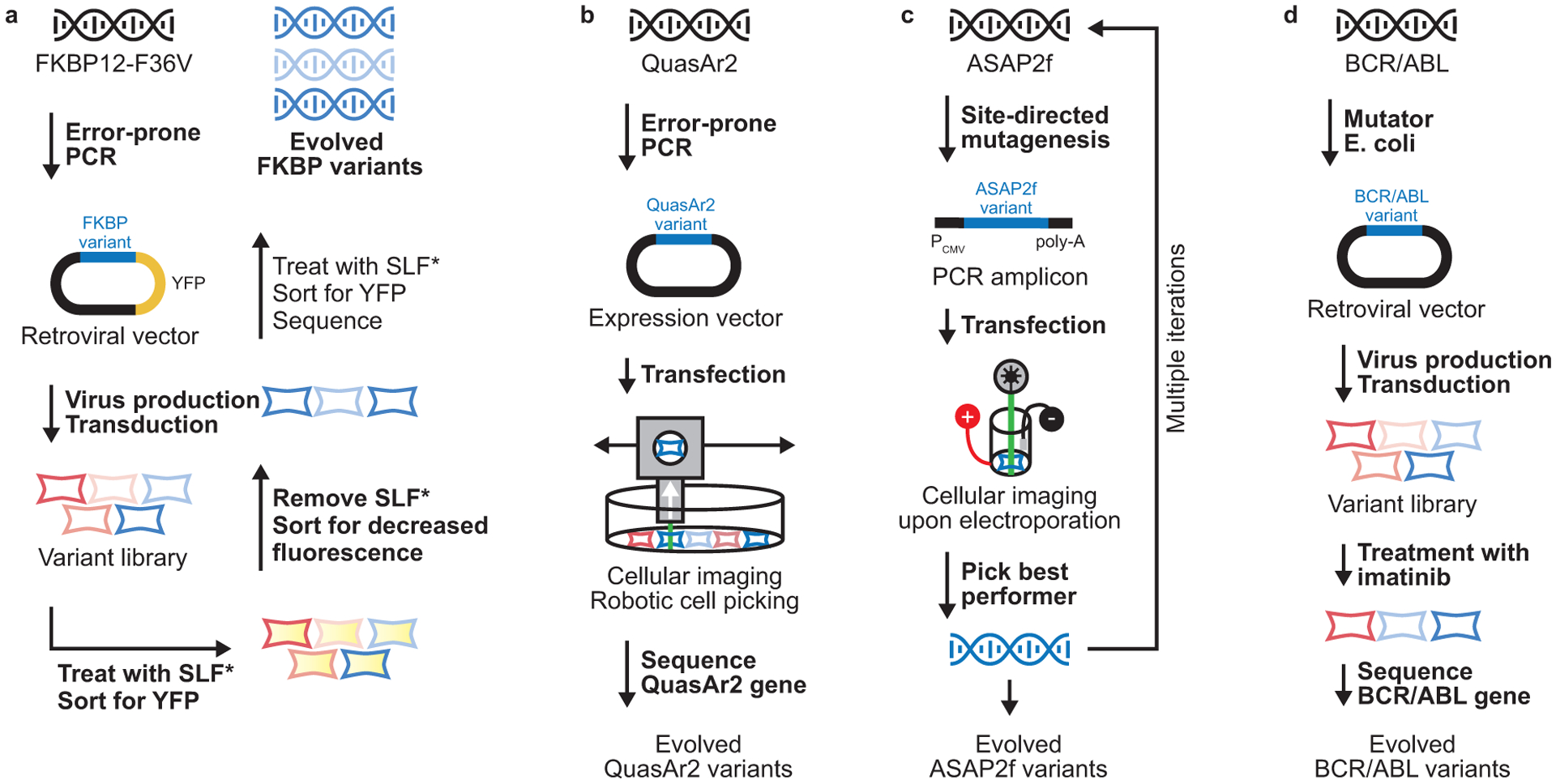
Selected examples of directed evolution experiments that use ex mammalia mutagenesis. (a) Directed evolution of a small-molecule regulated destabilizing domain by Wandless and co-workers.27 (b) Robotic cell sreening-based directed evolution of a voltage reporter by Boyden and co-workers.30 (c) Iterative directed evolution of a voltage reporter by Dieudonné, Lin, and co-workers.13 (d) Directed evolution of BCR/ABL by Daley and co-workers.21
An informative example of ex mammalia mutagenesis followed by cellular screening is the development of small molecule-controlled destabilizing domains by Wandless and co-workers.27 Destabilizing domains are proteins that exist largely in an unfolded state. Proteins fused to destabilizing domains are rapidly degraded by the proteasome. However, in the presence of a stabilizing small molecule, the folded state is more prevalent. The result is small molecule-mediated control of the level of any destabilizing domain-fused protein in cells and animals. It is critical that destabilizing domains intended for use in mammalian systems are optimized directly in mammalian cells, as lower organisms can have vastly different proteostasis networks and degradation pathways. To evolve a destabilizing domain based on the human protein FKBP12, Wandless and co-workers used error-prone PCR and retroviral transduction to create and then stably express a library of ~30,000 FKBP12–YFP fusion protein variants in 3T3 mouse cells (Figure 3a).27 From this library, 10,000 cells expressing FKBP12–YFP variants were treated with the FKBP12-stabilizing ligand SLF* and screened for YFP fluorescence using flow cytometry. Selected cells were then cultured in the absence of SLF* and subsequently screened again by flow cytometry for a decrease in YFP fluorescence. A final round of flow cytometry was performed upon addition of SLF* and, from the population expressing fluorescent YFP, 72 FKBP12 clones were sequenced, revealing several frequently occurring mutations and resulting in small molecule-regulated destabilizing domains for use in mammalian cells. A similar strategy has since been used to identify small molecule-regulated destabilizing domain versions of other proteins.28, 29 These destabilizing domains are now widely used to confer small molecule control of the levels of proteins ranging from Cas932 to membrane proteins28, 33 in mammalian cells.
Another instructive example is the directed evolution of membrane-spanning ion channels with valuable new functions. Boyden and co-workers used ex mammalia mutagenesis, transient transfection, and an innovative robotic screening approach to evolve the membrane-bound voltage reporter QuasAr234 for improved properties such as brightness and localization.30 A library of Arch variants was generated through error-prone PCR (Figure 3b) and subsequently transfected into HEK293 cells. Detailed images of ~10,000 cells were computationally evaluated for desired biomolecular properties, such as membrane localization, brightness, and more. Cells expressing QuasAr2 variants with the desired properties were then physically detached from the dish using a robotic micropipette and sequenced. Two of the identified variants, Archon1 and Archon2, displayed improved brightness, photostability, and cellular localization, and allow detailed imaging of neuronal activity from many neurons simultaneously.
A second approach to ion channel directed evolution in mammalian cells was demonstrated by Dieudonné, Lin, and co-workers.13 ASAP family voltage sensors are created via fusion of a circularly permuted GFP variant within the transmembrane region of a voltage-sensing phosphatase.35 First-generation ASAP voltage sensors suffer from relatively slow kinetics and are, therefore, unable to temporally resolve rapid action potentials. Dieudonné, Lin, and co-workers developed a streamlined platform for directed evolution of ASAP voltage sensors by transfecting PCR-amplified, mutated constructs directly into mammalian cells (Figure 3c), avoiding the time-consuming and inefficient step of plasmid library generation. They then screened ~400 variants per round of evolution by inducing an electrical potential and observing, through imaging, cells with improved voltage sensing. After six iterative rounds of evolution, a sextuple-mutant of ASAP2f was identified, termed ASAP3, that showed more rapid activation kinetics similar to Archon1 and with improved molar brightness.
While less common than screening-based methods, selection-based methods combined with ex mammalia mutagenesis have also been used, especially in efforts to illuminate how oncoproteins evolve drug resistance during cancer treatment. The small molecule imatinib inhibits BCR-ABL, an oncogene encoding an aberrant tyrosine kinase that can cause chronic myeloid leukemia and other cancers.36 Within just two years of FDA approval, 16 imatinib-resistant genetic mutations were observed in patients, creating a need to more comprehensively understand and predict the possible mutations that may permit imatinib resistance. Daley and co-workers used a single-round directed evolution technique to profile drug-resistant BCR-ABL variants in murine B cells.21 They employed a mutator strain of bacteria to create a BCR-ABL variant library (Figure 3d), introduced the library into a retroviral vector, transduced ~500,000 murine B cells with the library, and then treated the cells with imatinib for 10 days. Upon picking surviving cell colonies and sequencing the BCR-ABL variants, they identified 13 of the 16 known drug-resistance mutations. More significantly, they also discovered 100 novel drug-resistant variants, some of which were later observed in imatinib-treated cancer patients.37 Similar approaches have since been deployed to illuminate drug resistance pathways in other oncoproteins, including farnesyl transferase31 and mitogen-activated protein kinase.20
Summary.
Proteins such as destabilizing domains, ion channels, and oncoproteins intimately rely on the unique mammalian cellular environment and complex mammalian pathways for proper function. These features cannot feasibly be moved into lower organisms or a test tube and, therefore, the success of these directed evolution campaigns depended on optimizing function within mammalian cells.
Unfortunately, while ex mammalia mutagenesis techniques are well-established and straightforward to employ, they have significant drawbacks. Most significantly, the labor-intensive nature of variant library expression in and extraction from mammalian cells mean that in most cases only a single round of evolution is performed on what is typically quite a small variant library. In cases where multiple rounds of evolution are performed, sometimes fewer than 103 variants are tested in each round. More complex directed evolution targets typically require multiple rounds of evolution to be performed iteratively on variant libraries that are orders of magnitude larger than in these studies.
Mammalian cell-based directed evolution enabled by in mammalia targeted mutagenesis
A key bottleneck in most early examples of mammalian cell-based directed evolution experiments is that only one round of mutagenesis is performed. Breakthrough techniques to enable in mammalia targeted mutagenesis (Figures 2b and 4a) facilitate directed evolution experiments that require multiple rounds of evolution without the inefficient and cumbersome steps of library extraction, ex mammalia mutagenesis, and re-expression.
Figure 4.
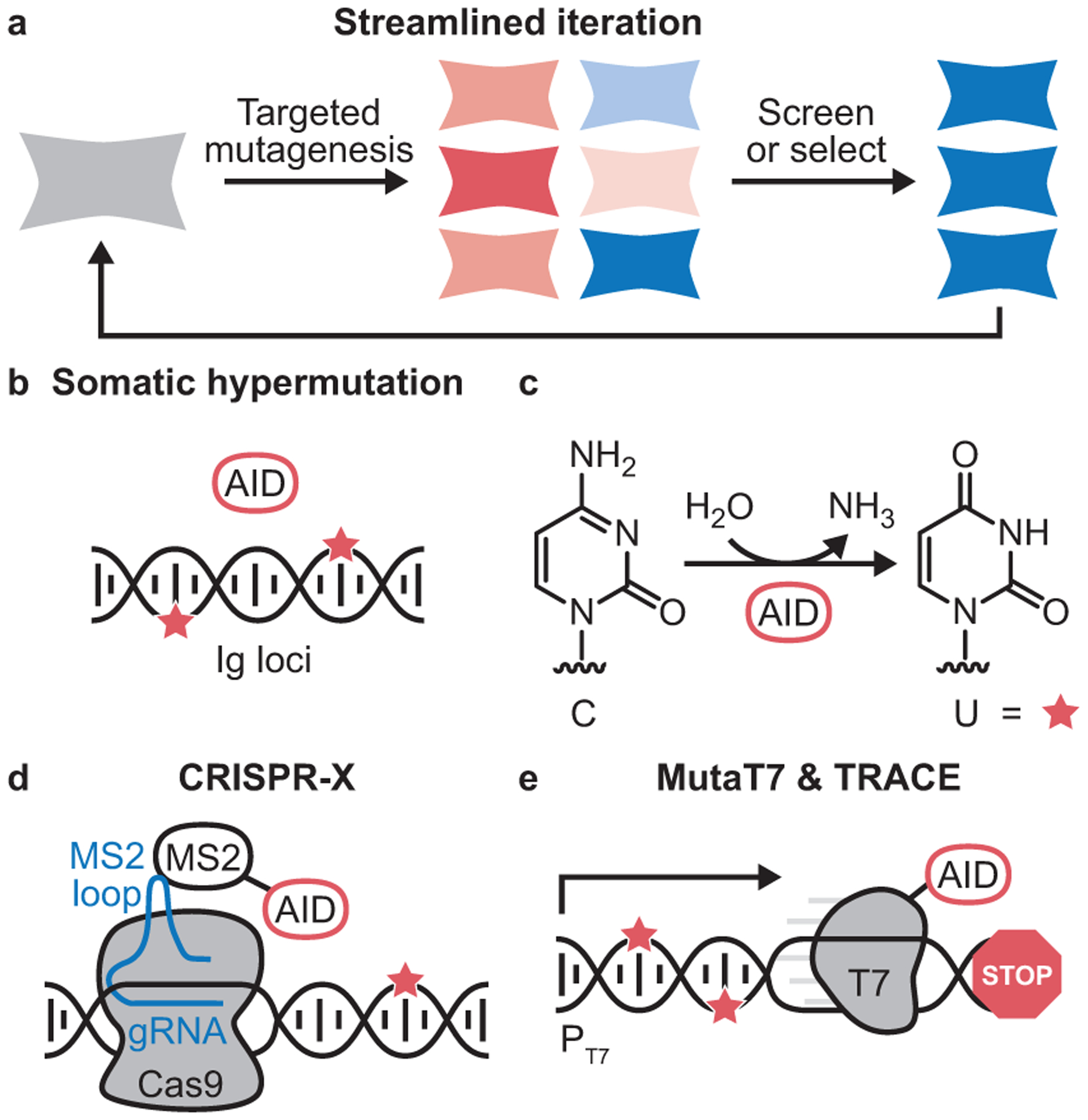
Targeted in mammalia mutagenesis. (a) Mutating genes of interest directly in mammalian cells streamlines iteration and increases the number of rounds of evolution that can be performed, but targeted mutagenesis of only the gene of interest for evolution is essential to avoid cheaters that escape selection pressure or subvert screening strategies. (b) Activation-induced cytidine deaminase (AID) mutagenizes hypervariable regions of immunoglobulin (Ig) loci,14 although evidence suggests that significant off-target mutagenesis occurs.44,45 (c) AID induces point mutations by deaminatingcytosine (C), creating uracil (U) bases that can be unfaithfully repaired or may base-pair with adenine upon replication, resulting in the introduction of C➔T mutations. (d) CRISPR-X targets mutagenesis to user-defined DNA sequences through a targeted guide RNA (gRNA) with a MS2 binding loop that recruits an MS2-AID fusion.50 This fusion protein mutagenizes DNA within a relatively narrow window of DNA (10–50 bp), but with much higher target specificity than AID alone. (e) Processive mutagenesis methods like TRACE17 leverage the MutaT716 concept, in which AID is fused to a highly processive T7 polymerase. MutaT7 targets genes preceded by the T7 promoter (PT7) and delimited by a terminator sequence.
Perhaps the most significant obstacle to diversifying genes in mammalia is the immense size of the mammalian genome, providing many opportunities for off-target mutations that can cheat screening or selection strategies. Unfortunately, many of the highly specific genomic editing strategies or commonly used genetic tools that function well in lower organisms do not exist for mammalian cells. For example, mammalian plasmids (e.g., latent viruses such as SV4038) and mammalian artificial chromosomes,39 both of which facilitate in vivo mutagenesis in bacteria or yeast (e.g., in the OrthoRep platform for directed evolution in yeast40), do not nearly approach the utility of the corresponding systems in lower organisms. Despite these obstacles, the last five years have witnessed the development of a number of targeted mutagenesis strategies that use bespoke technologies to address problems presented by the large mammalian genome and other aspects of mammalian cell biology.
Somatic hypermutation.
In mammalia targeted mutagenesis occurs naturally in B cells during the somatic hypermutation of antibody genes, mediated in part by the mutagenic activity of activation-induced cytidine deaminase (AID, Figure 4b).41 Cytidine deaminases are a class of DNA-damaging enzymes that convert cytosine into uracil (Figure 4c). The resulting uracil bases may be unfaithfully repaired or may base pair with adenine instead of guanine upon replication, thereby introducing C➔T mutations throughout targeted gene regions.
Several groups have taken advantage of AID-mediated mutagenesis to perform screening-based directed evolution of antibodies42 and fluorescent proteins43 in mammalian cells. Often, B cells that naturally express AID are used as a host cell to which genes of interest are introduced and continuously mutagenized. Unfortunately, although originally thought to be targeted to specific genetic loci, it is now clear that AID induces mutations genome-wide in cultured cells, especially at highly transcribed regions.44, 45 Supporting this observation, AID expression is cytotoxic when highly expressed, requiring researchers to artificially moderate expression levels during directed evolution experiments.44 Thus, the need for truly targeted DNA-damaging enzymes has emerged.
Deactivated RNA-guided endonucleases paired with DNA-damaging enzymes.
CRISPR enzymes such as Cas9 are bacteria-derived endonucleases that can be targeted to any genomic sequence containing an appropriate protospacer adjacent motif (PAM) using a single guide RNA (sgRNA). At an sgRNA-targeted genomic site, the endonuclease induces double-strand breaks that are commonly repaired by either non-homologous end joining or homology-directed repair.15
In principle, the resulting DNA damage can be used to diversify genes of interest for directed evolution experiments. Indeed, Cas9-induced in-frame insertions and deletions in targeted oncogenes have been used to generate and enrich drug-resistant protein variants.46, 47 However, non-homologous end-joining is much less likely to generate functional variants than homology-directed repair, owing to the high potential for insertions and deletions to induce frameshifts and potentially cause early termination. A Cas9-based method that specifically promotes homology-directed repair has been used to evolve a red fluorescent protein to function in mammalian lysosomes.19 Griesbeck and co-workers inserted a single copy of a mRuby-based fluorescent protein into mammalian genomes using the Flp-recombinase/FRT system. Single-stranded oligonucleotides encoding point mutants were then used as donor templates to promote the desired homology-directed repair at five amino acid codons with ~20% efficiency. Using this approach, a new variant of mRuby was identified, termed mCRISPRed, that showed stability at low pH and proved functional even in mammalian lysosomes.
Researchers have also sought to develop CRISPR-based mutagenesis methods that specifically promote point mutations. Inspired by base editors pioneered by Liu and co-workers,48 two technologies were developed that combine the DNA-targeting ability of Cas9 with the point mutation-inducing activity of wild-type or engineered variants of cytidine deaminases such as AID and APOBEC (Figure 4d). In an approach termed targeted AID-mediated mutagenesis (TAM), Xing and co-workers fused AID to deactivated Cas9 variants (dCas9) that bind, but do not cut, specific DNA sequences.49 In a proof-of-principle directed evolution experiment, this dCas9-AID fusion was then used to target point mutations to BCR-ABL in K562 cells. Upon passaging cells in the presence of imatinib, cells encoding drug-resistant BCR-ABL variants were readily enriched. In a related approach, termed CRISPR-X, a cytidine deaminase was fused to the RNA aptamer-binding protein MS2. The MS2-AID fusion can be recruited to a desired genetic locus by interaction with a dCas9-binding sgRNA that also contains an MS2-binding RNA aptamer domain.50 CRISPR-X was used to target mutations to a proteasome subunit in K562 cells to generate and identify variants resistant to the proteasome inhibitor bortezomib. These methods have also been applied to evolve other endogenous genes, including antibodies.51 Excitingly, the advent of additional DNA-damaging enzymes, such as adenosine deaminases,52, 53 may expand the accessible mutational spectrum using this type of approach for targeted mutagenesis.
Emerging CRISPR-based tools constitute a major improvement over natural somatic hypermutation as a strategy to target point mutations to DNA in mammalian cells. Nonetheless, there are still some significant limitations. TAM and CRISPR-X target mutations only to very small (~10–50 bp) windows of DNA that surround designed sgRNAs.49, 50 These small targeting windows mean that many different sgRNAs must be tiled along a gene to mutagenize the entire sequence. Critically, some regions may not be accessible owing to the absence of Cas9-specific PAMs.15 Further, since repeated rounds of directed evolution result in mutation accumulation, new sgRNAs are required as the guide recognition sequences mutate.
Processive targeted mutagenesis.
Bearing in mind the limitations of CRISPR-based technologies, a new type of strategy for targeted in mammalia mutagenesis was more recently developed. Like Cas9, RNA polymerases can be precisely targeted to specific regions of DNA. Unlike Cas9, RNA polymerases can be extremely processive. For example, the bacteriophage-derived T7 RNA polymerase is able to traverse >10,000 bp of DNA,54 directed by the highly-specific T7 promoter. In the Escherichia coli-based MutaT7 system, a T7 polymerase was fused to a cytidine deaminase to create a processive, highly targeted mutagenic protein that introduces point mutations in vivo in any stretch of DNA downstream of a T7 promoter (Figure 4e).16, 55 The TRACE platform built on this MutaT7 concept to mutagenize specific genes of interest directly in mammalian cells.17 TRACE has been used to screen for GFP variants with blue-shifted emission spectra in HEK293T cells and to select for MEK variants resistant to small molecule inhibitors in A375 cells.
The high processivity of RNA polymerases permits the mutation of kilobases of DNA without significant sequence limitations, such as the availability of PAM sequences or the need to synthesize and introduce multiple, custom sgRNAs for each gene of interest during each round of evolution. Multiple strategies to terminate T7 polymerase can be used to prevent unwanted mutagenesis downstream of the targeted region,16, 56 and installation of a second T7 promoter facing the opposite direction of the protein coding sequence allows for the introduction of both C➔T and G➔A mutations.16 Moreover, fusion to other DNA-damaging enzymes, such as improved variants of the adenosine deaminases52, 53 and a related approach to induce C➔G transversions57 are beginning to allow expansion of the mutational spectrum of these technologies,56 as was also the case in the CRISPR systems. A key limitation of MutaT7-based systems, such as TRACE, is that they require installation of a T7 promoter upstream of the gene of interest. The significance of this limitation depends largely on whether the user seeks to evolve an endogenous gene encoded in its unmodified genomic context.
Summary.
By simplifying the process of mutant library generation and expression, the capacity of targeted in mammalia mutagenesis techniques to facilitate multiple rounds of directed evolution is potentially transformative. In theory, it is now possible to easily mutagenize any gene targeted by an RNA-guided endonuclease or by a T7 polymerase directly in mammalian cells. Ultimately, improvements in targeted in mammalia mutagenesis focus only on mutagenesis but not on further steps. Directed evolution experiments using these advanced genetic diversification techniques still rely on traditional methods of selection or screening to enrich cells encoding desired variants.
Virus-based continuous directed evolution in mammalian cells
Despite improvements with in mammalia mutagenesis, a number of issues with typical cell-based platforms continue to restrict broad application. First, enrichment of functional variants can be limited by the slow rate of replication of mammalian cells, often requiring individual rounds of evolution to take a week or longer. Second, assessment techniques limit the types of biomolecules that can be evolved. Selection-based methods can only evolve functions that impact cell viability, while screening methods require an observable phenotype that can be screened. Many valuable targets of directed evolution do not fit easily in either category.
New implementations of virus-based continuous directed evolution in mammalian cells are primed to address these obstacles and have potential to revolutionize the field. The concept of virus-based directed evolution is simple: Introduce the gene of interest into the genome of a mammalian virus that replicates with low fidelity. Then, implement a selection couple that ties the ability of the virus to propagate in mammalian cells to the function of the directed evolution target. From there, simply propagating the virus results in the generation and enrichment of desired gene variants. In the field of bacteria-based directed evolution, PACE (phage-assisted continuous evolution)58 has enabled the rapid and efficient evolution of many diverse protein functions by simply coupling the desired protein function to the ability of M13 phage to replicate in E. coli. Analogous mammalian-cell based systems are poised to do the same.
A number of general principles must be considered when designing a broadly useful virus-based mammalian directed evolution platform. First, the rate of viral growth is important. Some mammalian viruses replicate faster than others or replicate with a larger burst size. However, viral replication must not outpace the rate of expression, maturation, and function of the protein of interest. Second, platforms should be able to evolve as many genes of interest in as many different mammalian cell types as possible. Viruses with large packaging capacities and broad tropisms are preferable.59 Third, platforms should be designed with practicability in mind. Virus generation, packaging efficiency, and other design features such as visual aids to monitor viral propagation should be considered. Lastly, the most important consideration is safety. Many laboratory viral strains have built-in safety strategies to prevent their replication in wild-type hosts. These strategies include deleting genes non-essential to cell culture,60 using extensive trans-complementation,61 or developing laboratory-adapted strains that are incapable of producing a replicative infection in organisms. These types of safety features should always be included and verified in any mammalian virus-based directed evolution platform.
While there exist some intriguing early reports of viral evolution in mammalian cells,62–64 early methods are ill-equipped for a broad range of substrates and often have serious safety liabilities. The landscape has changed with the reports of two adaptable virus-based directed evolution platforms for mammalian cells from our laboratory25 and from Roth, English, and co-workers.26 Both of these mammalian PACE (mPACE)-style platforms rely on custom engineering of a virus and a host cell to enable mutagenesis, selection, and amplification to occur simultaneously and continuously throughout an evolution experiment. First, essential viral genes are deleted from the viral genome (Figure 5a). Second, the gene of interest for evolution is introduced into the viral genome. Finally, the desired function of the gene of interest is then coupled to transcription, translation, or activation of the missing viral proteins in the host cell (Figure 5b). In this way, viruses that contain active genes of interest are rescued by trans-complementation and rapidly enrich in the population (Figure 5c). Viruses that do not contain active genes of interest are not rescued and cannot replicate. Critically, in an mPACE-style system, the mammalian cells are replaced after each round of evolution, avoiding the problem off-target mutation accumulation. Creativity is required for designing an appropriate selection couple, but the success of bacterial PACE highlights the tremendous breadth of functions that can be evolved using this type of system, ranging from protein stability to diverse enzymatic activities.58, 65–68
Figure 5.
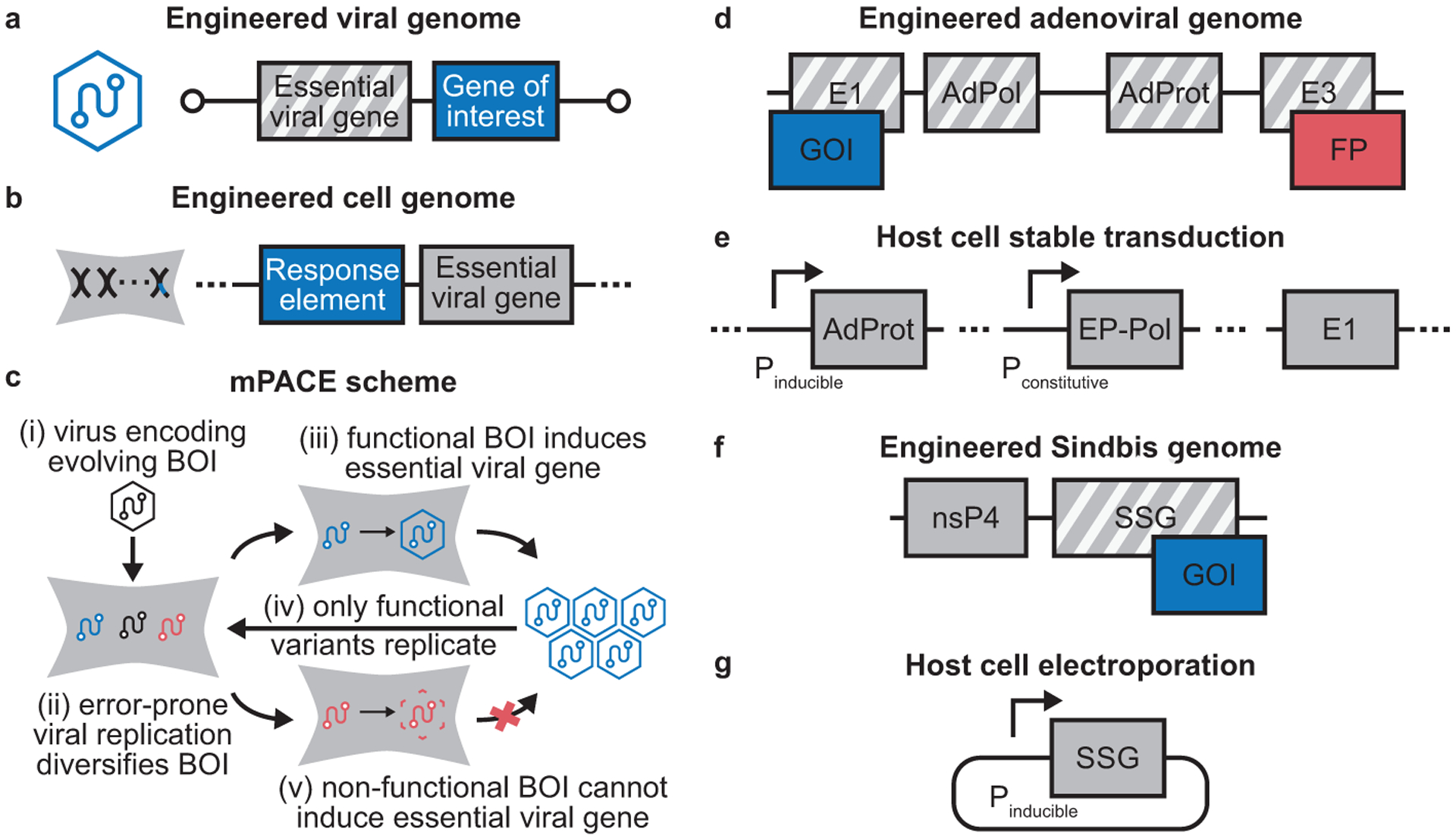
mPACE-style directed evolution platforms. (a–b) Mammalian PACE, or mPACE, utilizes two engineered components, a virus and a host cell. (a) The engineered virus lacks essential viral genes required for replication, but contains an evolving gene of interest. (b) The engineered mammalian cell encodes the deleted essential viral genes. Transcription, translation, or the activity of the conditionally trans-complemented viral genes is coupled to the desired activity of the evolving gene of interest. (c) These components combine to create a directed evolution platform, where the activity of a biomolecule of interest (BOI) drives viral replication. Over time, viruses encoding active genes of interest (GOIs) enrich in the population while viruses with inactive genes of interest do not replicate and are depleted. (d–e) Overview of the adenovirus-based mPACE directed evolution platform.25 (d) For safety and to clear space for insertion of a gene of interest (GOI) to be evolved, the engineered adenoviral genome has the E1 genes, E3 genes, the adenoviral polymerase (AdPol), and the adenoviral protease (AdProt) deleted from the wild-type genome. A gene encoding a fluorescent protein (FP) is also included to allow for easy visualization of directed evolution campaigns and viral titering. (e) E1, AdPol, and AdProt are stably trans-complemented by the host cell. The E3 region, which is dispensable in cell culture, is not trans-complemented at all to provide an additional safety feature. Mutagenesis of an encoded gene of interest is conferred by a constitutively expressed, highly error-prone variant of AdPol (EP-Pol). AdProt activity is inducibly coupled to the desired function of the GOI. (f–g) Overview of the Sindbis virus-based mPACE directed evolution platform (VEGAS).26 (f) Multiple genes are deleted from the wild-type Sindbis genome, including the capsid protein, E1, E2, 6K, and E3, known collectively as the Sindbis structural genome (SSG). (g) The missing SSG genes are inducibly expressed in response to the desired activity of the GOI via a gene cassette introduced by electroporation before each round of evolution. Mutagenesis of an encoded gene of interest is conferred by the wild-type Sindbis replicase (nsP4; or nonstructural protein 4).
The mPACE-style system introduced by our group employs the double-stranded DNA virus adenovirus to achieve continuous directed evolution in mammalian cells (Figures 5d and 5e), with mutations introduced during viral replication by a highly error-prone, engineered version of the adenoviral DNA polymerase.25 The second mPACE-style system, introduced by Roth, English, and co-workers and termed VEGAS (viral evolution of genetically actuating sequences), operates on the same principles but uses the single-stranded RNA alphavirus Sindbis (Figures 5f and 5g) with mutations introduced by the endogenous Sindbis replicase.26 Both systems rely on conditional expression of essential viral genes to drive selection, either the adenoviral protease (AdProt) or the Sindbis structural genome (SSG), respectively. Proof-of-principle experiments from these mPACE-style platforms demonstrate their potential. Both systems were employed to rapidly evolve inhibitor-insensitive, highly active versions of the tetracycline transactivating transcription factor (tTA) by placing AdProt (in HEK293A cells) or SSG expression (in BHK21 cells) under control of a tTA-dependent promoter. Further, VEGAS was used to evolve a constitutively active version of the G-protein coupled receptor MRGPRX2 by placing SSG expression under control of the serum response element. By introducing an ex mammalia-diversified nanobody library as a starting point for selection, Roth, English, and co-workers were also able to obtain a nanobody that stabilized the active state of the serotonin 2A GPCR.
Both platforms present distinct advantages. The adenovirus-based system has a number of features that enhance ease-of-use, including a fluorescent marker to visualize viral infection and compatibility with Gateway™ cloning to introduce genes of interest for evolution. The approach is compatible with a small molecule inhibitor69 of the selection marker, AdProt, which can be applied to tune the stringency of selection pressure over the course of a directed evolution campaign.25 Because adenovirus contains a double-stranded DNA genome, this system is also compatible with the targeted mutagenesis techniques discussed earlier. The system includes multiple distinct safety features to prevent production of a replication-competent virus. Three separate genetic regions are trans-complemented, decreasing the chance that a viable virus can be created. In addition, the E3 region essential for replication in living organisms is deleted and not trans-complemented at all. Lastly, adenovirus has an unusually broad tropism, allowing application in a vast array of different mammalian cell types.70
VEGAS also has unique advantages. Sindbis virus can be generated more quickly than adenovirus and is readily compatible with ex mammalia-diversified libraries. Sindbis has a faster replication cycle than adenovirus, ~4 hours for Sindbis versus ~24 hours for adenovirus, accelerating iterative rounds of evolution. Roth and co-workers estimate a mutation rate of 4 × 10–4 mutations per base per replication, whereas the adenoviral system has a lower estimated rate of 4 × 10–5 mutations per base per replication. It is important to balance useful mutation rates with mutational error catastrophe and so it is difficult to know the best mutation rate a priori—a single mutation is sufficient to render tTA fully functional yet inhibitor-insensitive25 whereas the consensus tTA variant from VEGAS had 47 mutations—but a higher mutation rate is often valuable. More significant than the mutation rate, Roth, English, and co-workers report a largely unbiased mutational spectrum for the Sindbis replicase. It is noteworthy, in this regard, that evolved sequences produced by VEGAS show a high percentage of A➔G transitions, including 46 of 47 reported mutations in the consensus sequence from the tTA evolution experiment.26
Summary.
Amongst all mammalian cell-based directed evolution platforms, virus-based continuous directed evolution platforms provide the largest library sizes, fastest experimental timescales, and allow, in principle, for the broadest range of biomolecules to be evolved. These mPACE-style techniques would still greatly benefit from additional features to improve scope and efficiency. First, current mPACE selection schemes only allow for positive selection, which may lead to poly-specific, promiscuous, or even constitutive activity. Developing negative selection schemes would allow the evolution of specific protein activity or environment-dependent protein activity, such as in the presence of a small molecule, in a particular cell type, or in a particular genetic background. Second, implementing fully continuous mPACE systems may increase library size, accelerate experimental timescales, and reduce researcher effort. Third, mutations are currently driven entirely by error-prone viral polymerases. The result is both some bias in mutational spectra as well as continual mutagenesis of the viral genome, which may eventually cause error catastrophe. Finally, published mPACE directed evolution campaigns all rely on direct transcriptional couples. Demonstration of adaptability to other types of couples that evolve non-transcription-related activities is essential.
Although there is more work to be done, virus-based directed evolution in mammalian cells presents a quantum leap over previous in mammalia evolution methods, providing simultaneous mutagenesis, selection, and enrichment on rapid timescales. As has proven readily achievable in bacterial PACE, mPACE-style approaches are likely to enable the evolution of targets previously unattainable by other screening or selection-based methods. The two platforms presented here therefore lay the groundwork for new discoveries and innovative applications in mammalian cells..
Overview of targets for mammalian cell-based directed evolution
With the development of broadly useful and efficient platforms for mammalian cell-based directed evolution, new targets for directed evolution are now within reach. Below, we highlight examples of targets for which mammalian cell-based directed evolution should prove particularly impactful, categorized into broadly conserved cellular processes, metazoan-specific processes, or mammalian-specific processes (Figure 6). Within each of these categories, targets include biomolecules that natively perform relevant biological functions and non-native proteins that can be evolved to perturb those same native functions.
Figure 6.
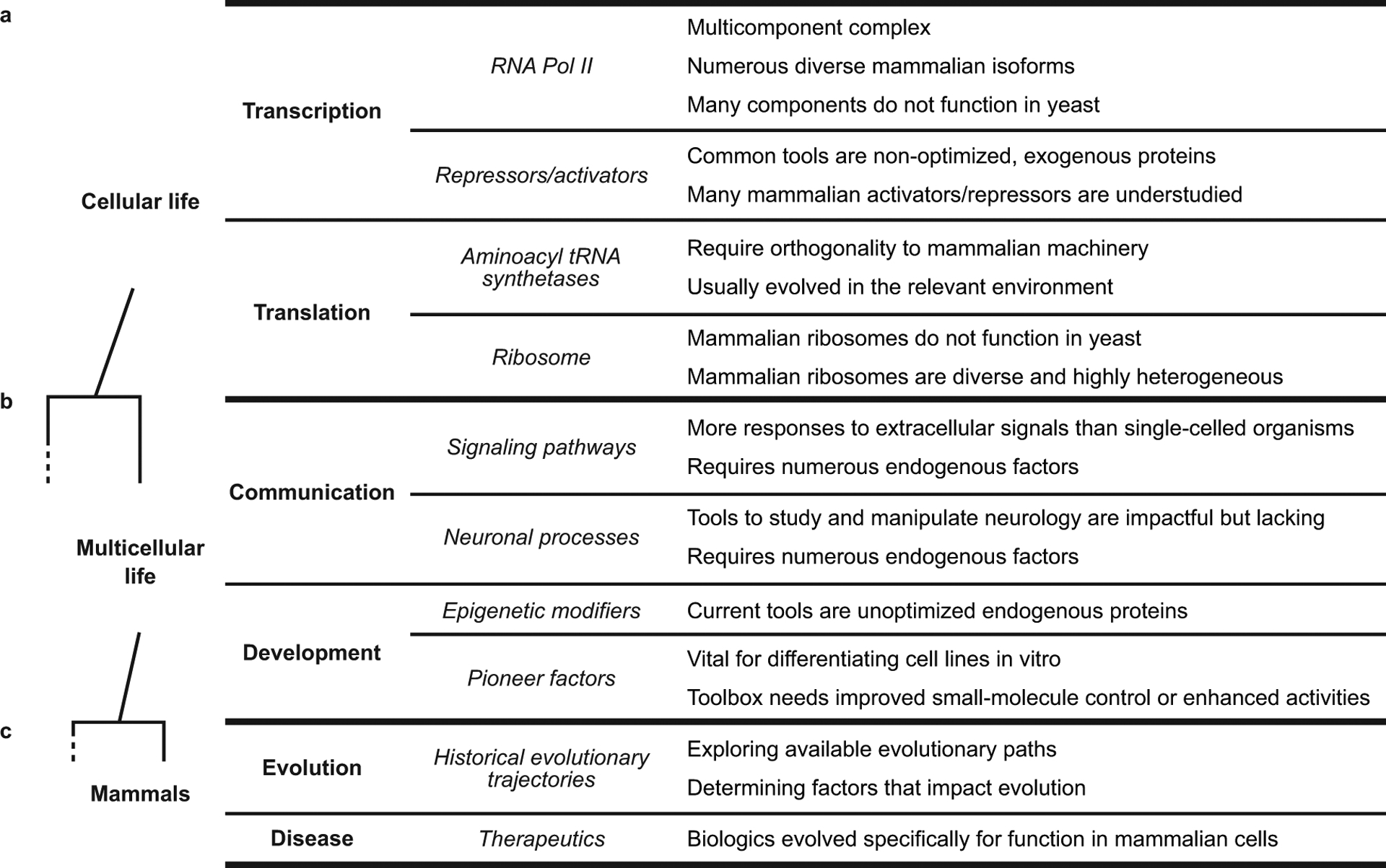
Selected examples of high-impact targets for mammalian cell-based directed evolution. Targets for mammalian cell-based directed evolution encompass targets across the phylogenetic tree of life, from (a) processes conserved in all kingdoms of cellular life to (b) processes specific to multicellular life to (c) processes specific to mammals.
Genes unique to mammals provide perhaps the most obvious examples of targets that would benefit from mammalian cell-based directed evolution. Even when a given conserved process is shared between kingdoms, it is often the case that directed evolution experiments to understand or perturb the mammalian version of that process still need to be performed in mammalian cells. As discussed above, protein variants evolved and optimized in lower organisms often fail to function properly once they are reintroduced into mammalian cells. Further, even for well-conserved biological processes, mammalian protein families often contain numerous paralogs that have highly differentiated functions. Likewise, there are many distinctive mammalian cell types with unique features that can greatly influence protein folding, modification, or activity.71 Finally, even successful instances of directed evolution experiments involving conserved targets in lower organisms should not necessarily preclude similar experiments being performed in mammalian cells. On the contrary, they facilitate such experiments by providing a roadmap for researchers interested in evolving the same or improved protein activities in a vastly different environment.
Cellular processes conserved across all kingdoms.
Although all cells share some common processes, the specific mechanisms by which these processes are carried out are typically not readily transferable between organisms. Some illustrative examples (Figure 6a) of general cellular processes that present compelling targets for mammalian cell-based directed evolution are discussed below.
Transcription and translation.
Tools to regulate and control transcription and translation are crucially important for molecular biology and are active areas of research for the treatment of diseases such as cancer.72–74 While RNA polymerase II is highly conserved from yeast to mammals,75, 76 many mammalian transcription factors, activators, and repressors do not function appropriately in yeast.77 Moreover, mammalian cells have many more RNA polymerase II subunits than do yeast—subunits with important and diverse functions.75, 76 Studying these subunits, and the complex transcriptional machinery altogether, requires numerous known and unknown components already present in mammalian cells but absent in lower organisms.
Tools to control and enhance translation are also invaluable, both for fundamental and medical research. For example, genetic code expansion through unnatural amino acid incorporation has profoundly impacted our understanding of translation and spurred the growth of the genetically encoded biological toolbox.78 Although each organism requires tools that are orthogonal to endogenous machinery, the current standard practice to create engineered amino acyl tRNA synthetases (aaRSs) for use in mammalian cells is to transport functional aaRSs from yeast and hope they retain orthogonality.79 As a result, the toolkit for unnatural amino acid incorporation in mammalian cells is limited as compared to yeast or bacterial systems, although efforts of Chatterjee and co-workers highlight the potential of new strategies for generation of such tools.6, 80 Likewise, efforts to engineer the ribosome have seen exciting progress, but are almost exclusively focused on ribosomes from lower organisms like E. coli.81 Successfully and reliably evolving new mammalian cell-compatible tools for enhancing both transcription and translation will often require mammalian cell-based directed evolution.
Post-translational engagement.
Almost as soon as translation initiates, nascent proteins begin to interact with many cellular components that aid in attaining proper structure and function. These include interactions with the proteostasis network, functional partners, and post-translational protein modification machineries. The differences in these systems between mammals and lower organisms can dramatically impact protein function.
Many high-impact targets for directed evolution require the mammalian proteostasis machinery to assist their folding and maturation.82 This challenge is particularly acute for complex and large proteins, such as membrane proteins83 or protein trafficking machineries. Beyond just the clients of mammalian proteostasis networks, the directed evolution of chaperones84 or chaperone modulators in mammalian cells could lead to a much deeper understanding of the mammalian proteostasis network, as well as to strategies for enhanced production of well-folded mammalian proteins. Mammalian proteins also show a much larger diversity of post-translational modifications than yeast or bacteria, but the tools to genetically encode and control post-translational modification in mammalian cells are primitive at best.85 Although directed evolution schemes have been developed in simple systems to evolve kinases,86 phosphatases,87 lectins,88 glycosyltransferases,89 and proteases,52 these targets are very rarely evolved in mammalian cells. Developing specialized tools to control and enhance mammalian-specific proteostasis machinery and post-translational modifications, particularly through mammalian cell-based directed evolution, would have enormous fundamental and biomedical research impacts.
Metazoan-specific processes.
Metazoans have evolved many biological functions that do not exist in single-celled organisms. Pathways involved in these functions will provide fertile ground for many important mammalian cell-based directed evolution experiments, both for natively functioning proteins as well as for non-native proteins that perturb these processes (Figure 6b).
Differentiation and development.
All metazoan organisms develop from a single cell, the descendants of which create a vast diversity of cell types. The underlying regulatory pathways that drive differentiation and development cannot be easily replicated in single-celled organisms such as yeast. Efforts to study or control differentiation and development, for example by employing epigenetic modifiers90 and pioneer factors,91 would benefit greatly from mammalian cell-based directed evolution.
Efforts to control epigenetic modification include orthogonal write-read epigenetic markers92 and CRISPR-Cas9-based epigenome modifiers,93 and the application of naturally occurring pioneer factors allows researchers to generate differentiated cell lines from pluripotent stem cells. Directed evolution of modified versions of these tools with improved small molecule control, more or less promiscuous activity, and expanded scope of activity would dramatically improve our ability to study and control cellular development and differentiation.
Communication.
Unlike single-celled organisms, metazoans must implement higher-order communication to interpret environmental and endogenous cues. Engineering biomolecules in these pathways can create fundamental insights into the neurological pathways that drive behavior and may even allow precision engineering of organismal behavior. G-protein coupled receptors, involved in the sensing of light, smell, and aspects of taste,94 are well-suited for mammalian cell-based directed evolution.26 The study of ion channels, involved in the sensing of heat, touch, and aspects of taste,95 has benefited tremendously from engineering efforts but has yet to be significantly impacted by directed evolution. Conveniently, successfully engineering biomolecules in the complex senses will directly impact organism behavior, allowing researchers to easily employ these engineered biomolecules in model mammalian organisms.
Along similar lines, neuroscientists often rely on electrophysiology and imaging techniques to identify biochemical underpinnings of neurological activity. Engineered proteins that allow precise control through small molecules or light can help elucidate many neurological pathways. Indeed, evolved neurological receptors integral to neurological pathways have been important for advances in molecular neuroscience.30 Further development of these proteins and others associated with neural function will undoubtedly provide even greater levels of control and expand the molecular neuroscientist’s toolbox even further.
Mammalian-specific processes and needs.
Some processes are important to study simply because we, as mammals, find them to be particularly important (Figure 6c).
Historical evolution.
Directed evolution is an accelerated laboratory mimic of natural evolution. Thus, it can be used to study factors that impact natural evolutionary history. Protein fitness landscapes can be probed by directed evolution, yielding evolutionary insights and detailed information about protein function and stability.96 Such studies are rarely performed in mammalian cells but, when they are, they often identify important cellular factors that may have impacted historical evolution.97, 98 Studying some of the fastest evolving genes in the human genome may help characterize the traits that evolved during the recent speciation of Homo sapiens. By evolving mammalian genes in the mammalian cell setting, but on a laboratory-timescale, we may be able to glimpse into our own history.
Treatments for human diseases.
Directed evolution has already transformed the way we treat disease. One class of proteins, antibodies, demonstrates the tremendous therapeutic potential of evolving mammalian proteins.99 The clever application of directed evolution of antibodies has also spawned the development of chimeric antigen receptors, where immune cell receptors can be engineered to target specific antigens.100 But opportunities in this space go far beyond just antibodies. Many other mammalian proteins can have therapeutic impacts through other modalities. By using directed evolution, we may even be able to generate new proteins to solve problems for which natural evolution did not provide any answer. A compelling modern example is CRISPR enzymes that may be valuable for disease treatment. Optimization of gene editing and other key functions directly in mammalian cells may prove critical for successful development of CRISPR-based therapeutics.
Concluding remarks
The development of biotechnologies purpose-built for application in mammalian cells have produced remarkable advances in mammalian cell-based directed evolution platforms. With numerous established techniques and a wide range of useful targets to evolve, there is ample opportunity for growth in this field. Impactful and potentially transformative targets abound in the mammalian genome and beyond, many of which have never been evolved or engineered before. Looking forward, improvements in library size and library complexity could be achieved by employing large synthetic DNA libraries or less biased random mutagenesis techniques. Furthermore, improved strategies for negative selection, more efficient methods for cellular screening, and strategies for the routine implementation of effective selection couples should expand the scope of activities that can be efficiently and reliably evolved in mammalian cells. We expect that the methods described in this Perspective, and their descendants, will serve to democratize mammalian cell-based directed evolution, such that researchers may soon routinely use directed evolution to augment their own specific fields of study without needing to invent or develop the techniques. If so, we will more rapidly enter a new age of mammalian biology—one not only of discovery, but of invention.
Acknowledgements
S.J.H. acknowledges support from the National Science Foundation Graduate Research Fellowship Program under Grant No. 1745302. M.D.S. acknowledges support from the National Institutes of Health, National Institute of General Medical Sciences (1R35GM136354).
Footnotes
Ethics Declaration
S.J.H. and M.D.S. are co-inventors on a patent application filed by MIT related to the use of adenoviruses for mammalian cell-based directed evolution. M.D.S. is a co-inventor on a patent application filed by MIT related to the MutaT7 system.
References
- 1.Drummond DA & Bloom JD A Nobel Prize for evolution. Evolution 73, 630–631 (2019). [Google Scholar]
- 2.Visintin M, Tse E, Axelson H, Rabbitts TH & Cattaneo A Selection of antibodies for intracellular function using a two-hybrid in vivo system. Proc. Natl. Acad. Sci. U. S. A 96, 11723–11728 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herwig L et al. Directed evolution of a bright near-infrared fluorescent rhodopsin using a synthetic chromophore. Cell Chem. Biol 24, 415–425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buskirk AR, Ong YC, Gartner ZJ & Liu DR Directed evolution of ligand dependence: small-molecule-activated protein splicing. Proc. Natl. Acad. Sci. U. S. A 101, 10505–10510 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peck SH, Chen I & Liu DR Directed evolution of a small-molecule-triggered intein with improved splicing properties in mammalian cells. Chem. Biol 18, 619–630 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Italia JS et al. Genetically encoded protein sulfation in mammalian cells. Nat. Chem. Biol 16, 379–382 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armbruster BN, Li X, Pausch MH, Herlitze S & Roth BL Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. U. S. A 104, 5163–5168 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pourmir A & Johannes TW Directed evolution: selection of the host organism. Comput. Struct. Biotechnol. J 2, e201209012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kachroo AH et al. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 348, 921–925 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cirino PC, Mayer KM & Umeno D Generating mutant libraries using error-prone PCR. Methods Mol. Biol 231, 3–9 (200). [DOI] [PubMed] [Google Scholar]
- 11.Muteeb G & Sen R Random mutagenesis using a mutator strain. Methods Mol. Biol 634, 411–419 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Badran AH & Liu DR Development of potent in vivo mutagenesis plasmids with broad mutational spectra. Nat. Commun 6, 8425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villette V et al. Ultrafast two-photon imaging of a high-gain voltage indicator in awake behaving mice. Cell 179, 1590–1608.e1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Odegard VH & Schatz DG Targeting of somatic hypermutation. Nat. Rev. Immunol 6, 573–583 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Pickar-Oliver A & Gersbach CA The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol 20, 490–507 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore CL, Papa LJ 3rd & Shoulders MD A processive protein chimera introduces mutations across defined DNA regions in vivo. J. Am. Chem. Soc 140, 11560–11564 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H et al. Efficient, continuous mutagenesis in human cells using a pseudo-random DNA editor. Nat. Biotechnol 38, 165–168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sansbury BM, Hewes AM & Kmiec EB Understanding the diversity of genetic outcomes from CRISPR-Cas generated homology-directed repair. Commun. Biol 2, 458 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erdogan M, Fabritius A, Basquin J & Griesbeck O Targeted in situ protein diversification and intra-organelle validation in mammalian cells. Cell Chem. Biol 27, 610–621 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Emery CM et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. U. S. A 106, 20411–20416 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Azam M, Latek RR & Daley GQ Mechanisms of autoinhibition and STI-571/Imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 112, 831–843 (2003). [DOI] [PubMed] [Google Scholar]
- 22.McDermott M et al. In vitro development of chemotherapy and targeted therapy drug-resistant cancer cell lines: a practical guide with case studies. Front. Oncol 4, 40 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Booth L et al. The afatinib resistance of in vivo generated H1975 lung cancer cell clones is mediated by SRC/ERBB3/c-KIT/c-MET compensatory survival signaling. Oncotarget 7, 19620–19630 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forment JV et al. Genome-wide genetic screening with chemically mutagenized haploid embryonic stem cells. Nat. Chem. Biol 13, 12–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berman CM et al. An adaptable platform for directed evolution in human cells. J. Am. Chem. Soc 140, 18093–18103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.English JG et al. VEGAS as a platform for facile directed evolution in mammalian cells. Cell 178, 748–761 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG & Wandless TJ A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iwamoto M, Bjorklund T, Lundberg C, Kirik D & Wandless TJ A general chemical method to regulate protein stability in the mammalian central nervous system. Chem. Biol 17, 981–988 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyazaki Y, Imoto H, Chen LC & Wandless TJ Destabilizing domains derived from the human estrogen receptor. J. Am. Chem. Soc 134, 3942–3945 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piatkevich KD et al. A robotic multidimensional directed evolution approach applied to fluorescent voltage reporters. Nat. Chem. Biol 14, 352–360 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raz T, Nardi V, Azam M, Cortes J & Daley GQ Farnesyl transferase inhibitor resistance probed by target mutagenesis. Blood 110, 2102–2109 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maji B et al. Multidimensional chemical control of CRISPR-Cas9. Nat. Chem. Biol 13, 9–11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dietz O et al. Characterization of the small exported Plasmodium falciparum membrane protein SEMP1. PLoS One 9, e103272 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hochbaum DR et al. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat. Methods 11, 825–833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.St-Pierre F et al. High-fidelity optical reporting of neuronal electrical activity with an ultrafast fluorescent voltage sensor. Nat. Neurosci 17, 884–889 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Capdeville R, Buchdunger E, Zimmermann J & Matter A Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov 1, 493–502 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Soverini S et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood 118, 1208–1215 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Mahon MJ Vectors bicistronically linking a gene of interest to the SV40 large T antigen in combination with the SV40 origin of replication enhance transient protein expression and luciferase reporter activity. Biotechniques 51, 119–128 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown DM & Glass JI Technology used to build and transfer mammalian chromosomes. Exp. Cell Res 388, 111851 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Ravikumar A, Arzumanyan GA, Obadi MKA, Javanpour AA & Liu CC Scalable, continuous evolution of genes at mutation rates above genomic error thresholds. Cell 175, 1946–1957 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pavri R & Nussenzweig MC AID targeting in antibody diversity. Adv. Immunol 110, 1–26 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Bowers PM et al. Coupling mammalian cell surface display with somatic hypermutation for the discovery and maturation of human antibodies. Proc. Natl. Acad. Sci. U. S. A 108, 20455–20460 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, Jackson WC, Steinbach PA & Tsien RY Evolution of new nonantibody proteins via iterative somatic hypermutation. Proc. Natl. Acad. Sci. U. S. A 101, 16745–16749 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Al-Qaisi TS, Su YC & Roffler SR Transient AID expression for in situ mutagenesis with improved cellular fitness. Sci. Rep 8, 9413 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang CL, Harper RA & Wabl M Genome-wide somatic hypermutation. Proc. Natl. Acad. Sci. U. S. A 101, 7352–7356 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donovan KF et al. Creation of novel protein variants with CRISPR/Cas9-mediated mutagenesis: turning a screening by-product into a discovery tool. PLoS One 12, e0170445 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ipsaro JJ et al. Rapid generation of drug-resistance alleles at endogenous loci using CRISPR-Cas9 indel mutagenesis. PLoS One 12, e0172177 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma Y et al. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat. Methods 13, 1029–1035 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Hess GT et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods 13, 1036–1042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Devilder MC et al. Ex vivo evolution of human antibodies by CRISPR-X: from a naive B cell repertoire to affinity matured antibodies. BMC Biotechnol. 19, 14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaudelli NM et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaudelli NM et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol (2020). [DOI] [PubMed] [Google Scholar]
- 54.Thiel V, Herold J, Schelle B & Siddell SG Infectious RNA transcribed in vitro from a cDNA copy of the human coronavirus genome cloned in vaccinia virus. The Journal of general virology 82, 1273–1281 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Park H & Kim S Gene-specific mutagenesis enables rapid continuous evolution of enzymes in vivo. Nucleic Acids Res. (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alvarez B, Mencia M, de Lorenzo V & Fernandez LA In vivo diversification of target genomic sites using processive base deaminase fusions blocked by dCas9. Nat. Commun 11, e6436 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kurt IC et al. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol 39, 41–46 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Esvelt KM, Carlson JC & Liu DR A system for the continuous directed evolution of biomolecules. Nature 472, 499–503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dai X, Zhang X, Ostrikov K & Abrahamyan L Host receptors: the key to establishing cells with broad viral tropism for vaccine production. Crit. Rev. Microbiol 46, 147–168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Danthinne X & Imperiale MJ Production of first generation adenovirus vectors: a review. Gene Ther. 7, 1707–1714 (2000). [DOI] [PubMed] [Google Scholar]
- 61.Chira S et al. Progresses towards safe and efficient gene therapy vectors. Oncotarget 6, 30675–30703 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mullick A et al. The cumate gene-switch: a system for regulated expression in mammalian cells. BMC Biotechnol. 6, 43 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uil TG et al. Directed adenovirus evolution using engineered mutator viral polymerases. Nucleic Acids Res. 39, e30 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Das AT et al. Viral evolution as a tool to improve the tetracycline-regulated gene expression system. J. Biol. Chem 279, 18776–18782 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Richter MF et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol 38, 883–891(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang T, Badran A, Huang T & Liu D Continuous directed evolution of proteins with improved soluble expression. Nat. Chem. Biol 14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dickinson BC, Packer MS, Badran AH & Liu DR A system for the continuous directed evolution of proteases rapidly reveals drug-resistance mutations. Nat. Commun 5, 5352 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miller SM et al. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat. Biotechnol 38, 471–481 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mac Sweeney A et al. Discovery and structure-based optimization of adenain inhibitors. ACS Med. Chem. Lett 5, 937–941 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Havenga MJ et al. Exploiting the natural diversity in adenovirus tropism for therapy and prevention of disease. J. Virol 76, 4612–4620 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ellgaard L, Molinari M & Helenius A Setting the standards: quality control in the secretory pathway. Science 286, 1882–1888 (1999). [DOI] [PubMed] [Google Scholar]
- 72.Warren L & Lin C mRNA-based genetic reprogramming. Mol. Ther 27, 729–734 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trepotec Z, Lichtenegger E, Plank C, Aneja MK & Rudolph C Delivery of mRNA therapeutics for the treatment of hepatic diseases. Mol. Ther 27, 794–802 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen A & Koehler AN Transcription factor inhibition: lessons learned and emerging targets. Trends Mol. Med 26, 508–518 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Conaway RC & Conaway JW Origins and activity of the Mediator complex. Semin. Cell Dev. Biol 22, 729–734 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Conaway JW et al. The mammalian Mediator complex. FEBS Lett. 579, 904–908 (2005). [DOI] [PubMed] [Google Scholar]
- 77.Kennedy BK Mammalian transcription factors in yeast: strangers in a familiar land. Nat. Rev. Mol. Cell Biol 3, 41–49 (2002). [DOI] [PubMed] [Google Scholar]
- 78.Wang L, Xie J & Schultz PG Expanding the genetic code. Annu. Rev. Biophys. Biomol. Struct 35, 225–249 (2006). [DOI] [PubMed] [Google Scholar]
- 79.Chin JW Expanding and reprogramming the genetic code of cells and animals. Annu. Rev. Biochem 83, 379–408 (2014). [DOI] [PubMed] [Google Scholar]
- 80.Italia JS et al. Expanding the genetic code of mammalian cells. Biochem. Soc. Trans 45, 555–562 (2017). [DOI] [PubMed] [Google Scholar]
- 81.Hammerling MJ et al. In vitro ribosome synthesis and evolution through ribosome display. Nat. Commun 11, 1108 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sebastian RM & Shoulders MD Chemical biology framework to illuminate proteostasis. Annu. Rev. Biochem 89, 529–555 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marinko JT et al. Folding and misfolding of human membrane proteins in health and disease: from single molecules to cellular proteostasis. Chem. Rev 119, 5537–5606 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sachsenhauser V & Bardwell JC Directed evolution to improve protein folding in vivo. Curr. Opin. Struct. Biol 48, 117–123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chuh KN, Batt AR & Pratt MR Chemical methods for encoding and decoding of posttranslational modifications. Cell Chem. Biol 23, 86–107 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Christians FC, Scapozza L, Crameri A, Folkers G & Stemmer WP Directed evolution of thymidine kinase for AZT phosphorylation using DNA family shuffling. Nat. Biotechnol 17, 259–264 (1999). [DOI] [PubMed] [Google Scholar]
- 87.Xu H. f., Zhang X. e., Zhang Z. p., Zhang Y. m. & Cass AEG Directed evolution of E. coli alkaline phosphatase towards higher catalytic activity. Biocatal. Biotransfor 21, 41–47 (2009). [Google Scholar]
- 88.Hu D, Tateno H, Kuno A, Yabe R & Hirabayashi J Directed evolution of lectins with sugar-binding specificity for 6-sulfo-galactose. J. Biol. Chem 287, 20313–20320 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang G et al. Fluorescence activated cell sorting as a general ultra-high-throughput screening method for directed evolution of glycosyltransferases. J. Am. Chem. Soc 132, 10570–10577 (2010). [DOI] [PubMed] [Google Scholar]
- 90.Atlasi Y & Stunnenberg HG The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet 18, 643–658 (2017). [DOI] [PubMed] [Google Scholar]
- 91.Iwafuchi-Doi M & Zaret KS Pioneer transcription factors in cell reprogramming. Genes Dev. 28, 2679–2692 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Park M, Patel N, Keung AJ & Khalil AS Engineering epigenetic regulation using synthetic read-write modules. Cell 176, 227–238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pulecio J, Verma N, Mejia-Ramirez E, Huangfu D & Raya A CRISPR/Cas9-Based engineering of the epigenome. Cell Stem Cell 21, 431–447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Julius D & Nathans J Signaling by sensory receptors. Cold Spring Harb. Perspect. Biol 4, a005991 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Subramanyam P & Colecraft HM Ion channel engineering: perspectives and strategies. J. Mol. Biol 427, 190–204 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Romero PA & Arnold FH Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol 10, 866–876 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Geller R, Pechmann S, Acevedo A, Andino R & Frydman J Hsp90 shapes protein and RNA evolution to balance trade-offs between protein stability and aggregation. Nat. Commun 9, 1781 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Phillips AM et al. Host proteostasis modulates influenza evolution. eLife 6, e28652 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu RM et al. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci 27, 1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guedan S, Calderon H, Posey AD Jr. & Maus MV Engineering and design of chimeric antigen receptors. Mol. Ther. Methods Clin. Dev 12, 145–156 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]