

Abstract
SUMMARY
N6-methyladenosine (m6A) is a conserved ribonucleoside modification that regulates many facets of RNA metabolism. Using quantitative mass spectrometry, we find that the universally conserved tandem adenosines at the 3′ end of 18S rRNA, thought to be constitutively di-methylated (m62A), are also mono-methylated (m6A). Although present at substoichiometric amounts, m6A at these positions increases significantly in response to sulfur starvation in yeast cells and mammalian cell lines. Combining yeast genetics and ribosome profiling, we provide evidence to suggest that m6A-bearing ribosomes carry out translation distinctly from m62A-bearing ribosomes, featuring a striking specificity for sulfur metabolism genes. Our work thus reveals methylation multiplicity as a mechanism to regulate translation.
In brief
Ribosome heterogeneity has become increasingly evident. Liu et al. report an example in the form of rRNA methylation. They show two conserved adenosines in the 18S rRNA are modified with varying numbers of methyl groups. Differentially methylated ribosomes translate differently, suggesting methylation multiplicity as a mechanism to regulate translation.
Graphical abstract
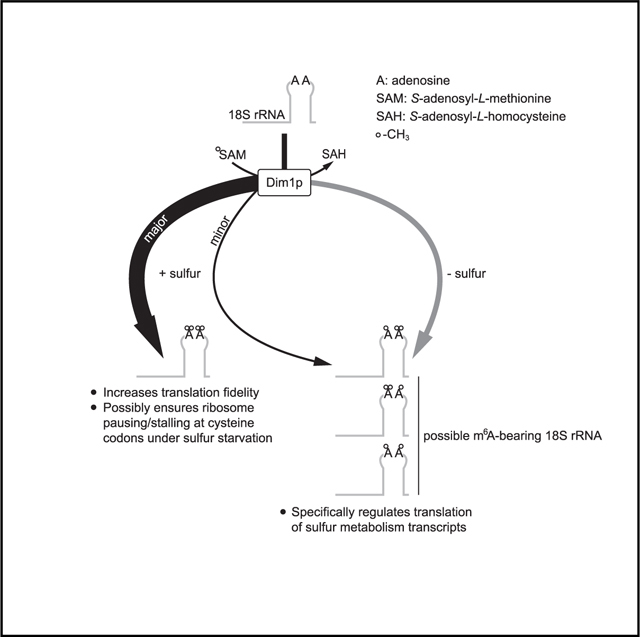
INTRODUCTION
RNA, despite its simple composition, is ornamented with more than 150 distinct modifications (Boccaletto et al., 2018). Curiously, ~40% of them involve methylation (https://iimcb.genesilico.pl/modomics/), an alkylation reaction that covalently adds a methyl group to a ribonucleoside. Methylation is an energetically expensive process, requiring an energy input equivalent to the hydrolysis of ~13 ATP molecules (Atkinson, 1977). From an evolutionary perspective, this implies that methylation likely confers functional importance. However, except for a few well-characterized examples, many RNA methylation events remain functionally enigmatic (Motorin and Helm, 2011).
Of all methylated ribonucleosides, only a handful have been conserved across all three domains of life (Motorin and Helm, 2011), two prime examples of which are mono-methylated N6-methyladenosine (m6A) and di-methylated N6-methyladenosine (m62A). Despite their structural resemblance (one versus two methyl groups at the N6 position of the adenine ring), m6A and m62A are distinct modifications with respect to their spatial distribution and synthesis. The m6A modification was initially discovered in mammalian mRNA (Desrosiers et al., 1974) and subsequently in other RNA species, including rRNA, tRNA, and small nuclear RNA (snRNA) (Yue et al., 2015). In accordance with its promiscuous residencies, several m6A methyltransferases targeting different RNA substrates have been identified and characterized (Bokar et al., 1994, 1997; Clancy et al., 2002; Liu et al., 2014; Ma et al., 2019; Pendleton et al., 2017; van Tran et al., 2019), which enabled the functional interrogation of m6A. At the molecular level, m6A is known to regulate many facets of RNA metabolism, such as mRNA stability and translation efficiency (TE) (Wang et al., 2014, 2015; Zhou et al., 2015), microRNA (miRNA) processing and maturation (Alarcón et al., 2015a, 2015b), RNA-protein interaction (Liu et al., 2015), and phase separation (Ries et al., 2019). At the cellular level, m6A has been implicated in pluripotency (Geula et al., 2015), heat shock response (Zhou et al., 2015), viral infection (Gokhale et al., 2016; Kennedy et al., 2016), and development (Clancy et al., 2002; Zhao et al., 2017). These findings reinforce the notion that addition of a simple methyl group can profoundly affect RNA metabolism and cellular physiology.
In contrast to m6A, m62A has an extremely confined distribution. With a few exceptions, it is found universally at two adjacent adenosines (A1781 and A1782 in Saccharomyces cerevisiae) near the 3′ end of the small subunit (SSU) rRNA (Rife, 2009; Van Knippenberg et al., 1984). This region forms a highly conserved hairpin loop (helix 45), and the tandem m62A modifications reside at the apex of this loop, which situates them close to the ribosome decoding site (DCS) (Sharma and Lafontaine, 2015). Despite their remarkable conservation and occupation of a structurally important position within the ribosome, the functional importance of these tandem m62A modifications remains incompletely understood.
Here, we report our serendipitous discovery that the m62A methyltransferase Dim1p is capable of modifying the conserved tandem adenosines with a single methyl group (i.e., m6A). Although present at low stoichiometry, m6A increases significantly during sulfur starvation in yeast cells and mammalian cell lines. Ribosome profiling experiments further indicate that m6A-bearing ribosomes carry out translation distinctly from m62A-bearing ribosomes, featuring a striking specificity for sulfur metabolism genes. Our work thus suggests that methylation multiplicity of these tandem adenosines functions as a mechanism to regulate translation.
RESULTS
Identification of m6A as a bona fide modification at A1781/A1782 in yeast 18S rRNA
Contrary to its prevalence in mammalian cells (Yue et al., 2015), m6A in budding yeast cells is present only in mRNA from sporulating diploid cells (Agarwala et al., 2012; Bodi et al., 2010; Clancy et al., 2002). Accordingly, one might expect little m6A in the sporulation-deficient haploid cells. Surprisingly, using quantitative liquid chromatography coupled to tandem mass spectrometry, we could readily detect m6A in total RNA isolated from haploid cells grown in a synthetic defined (SD) medium (Figures 1A and S1A). Consistent with previous studies (Agarwala et al., 2012; Bodi et al., 2010; Clancy et al., 2002), poly(A)+ RNA was essentially devoid of m6A (Figure S1B), except in sporulating diploid cells (Figure S1C). Subsequent RNA fractionation revealed the presence of m6A in 18S rRNA (Figures 1B and S1D), 25S rRNA (Figure S1E), and small RNA (Figure S1F). However, m6A from the latter two sources is likely derived from N1-methyl-adenosine (m1A) via Dimroth rearrangement (Engel, 1975; Macon and Wolfenden, 1968), because loss of the corresponding m1A methyltransferase or methyltransferases eliminated both m1A and m6A (Figures S1E and S1F). Therefore, only m6A detected in 18S rRNA appears to be a bona fide modification. m6A is a substoichiometric modification, with ~4% of 18S rRNA on average harboring one m6A in haploid yeast cells grown in SD medium.
Figure 1. m6A is a bona fide modification located at A1781 and/or A1782 of 18S rRNA.

(A) Detection of m6A in total RNA from vegetatively growing haploid S. cerevisiae.
(B) m6A is detected in 18S rRNA of vegetatively growing haploid S. cerevisiae (strain: CEN.PK).
(C) m6A is located in the last 22 nucleotides of 18S rRNA. ac4C, N4-acetylcytidine; m62A, N6, N6-dimethyladenosine. See Data S1 for other regions of 18S rRNA surveyed using the MBN protection assay.
(D) m6A is located at A1781 and/or A1782 of 18S rRNA. The peak area of m6A was first normalized to that of m62A, and the m6A/m62A ratio was then normalized to the fragment protected by oKL204. Mean ± SD (n = 7 biological replicates). The p value was calculated using unpaired two-tailed Student’s t test, assuming equal variances.
(E) m62A methyltransferase Dim1p is responsible for the m6A modification in yeast 18S rRNA. p > 0.05 (n.s.).
CPS, counts per second.
See also Figures S1 and S2 and Data S1.
To precisely map m6A in 18S rRNA, we performed a mung bean nuclease (MBN) protection assay (Figure S2A). As validation, we isolated a fragment of yeast 18S rRNA corresponding to the region from 601 to 660 and detected the expected 2′-O-methyladenosine (Am) (Figure S2B). We next scanned the entire 18S rRNA and found that m6A was located within the last 22 nucleotides (nt) (Figure 1C; Data S1). Using a DNA oligo that only partially protects this 22-nt region (Figure 1C), we pinpointed m6A to A1781 and/or A1782 of 18S rRNA (Figure 1D). However, due to technical difficulties, we could not further distinguish between these two positions. Nevertheless, an 15N-tracing experiment showed that m6A in 18S rRNA was derived from cultured yeast, not from contamination during sample preparation (Figure S2C). Altogether, these results indicate that m6A is a bona fide modification that maps to A1781 and/or A1782 of 18S rRNA, a site also known to accommodate the conserved tandem m62A modifications.
The co-occupation of m6A and m62A prompted us to speculate that the m62A methyltransferase Dim1p (Lafontaine et al., 1994) might also be responsible for installing m6A. To test this hypothesis, we created a mutant Dim1p by changing the glutamic acid at 85 (E85) to alanine, which reportedly abolishes its methyltransferase activity (Pulicherla et al., 2009). As expected, we found little m62A in 18S rRNA from the E85A mutant, but surprisingly, we could still detect m6A, albeit at slightly reduced amounts (Figure S2D). In the structure of the Dim1 homolog from Methanocaldococcus jannaschii, the glutamic acid at 59 (equivalent to E85 in S. cerevisiae Dim1p) is within hydrogen-bonding distance to the substrate S-adenosyl-L-methionine (SAM) (Figure S2E) (O’Farrell et al., 2010). Substitution of glutamic acid with alanine might destabilize, but might not eliminate, SAM binding. By contrast, swapping glutamic acid with a bulkier residue might be more effective at disrupting SAM binding through physical hindrance. Indeed, changing the glutamic acid to tryptophan resulted in complete loss of m62A and m6A (Figure 1E), confirming that Dim1p is responsible for both modifications. Furthermore, a highly conservative change of glutamic acid to glutamine inactivated Dim1p, because no m6A or m62A was detected in 18S rRNA of the E85Q mutant (Figure 1E).
m6A levels increase specifically and significantly in response to sulfur starvation
The unexpected presence of m6A in yeast 18S rRNA raises the question of its biological significance. To this end, we first tested whether m6A levels might change according to growth conditions. Deprivation of carbon, nitrogen, or phosphate had little impact on m6A levels (Figure 2A). By contrast, sulfate starvation caused a significant increase of m6A (Figure 2A), without eliciting apparent changes in amounts of other methylated nucleosides in 18S rRNA (Figure S3A). These observations indicate that m6A levels respond specifically to sulfate availability, a notion that is reinforced by the periodic changes of m6A in response to sulfate fluctuations (Figure 2B). A stable isotope-tracing experiment further demonstrated that under sulfate starvation, most m6A was derived from de novo synthesis (Figure 2C), whereas only a minority of m62A was newly synthesized (Figure S3B). Moreover, this starvation response is not specific for sulfate, because deprivation of methionine or SAM, two reduced sulfur sources, also increased m6A levels (Figures 2D and 2E). Supplying cells with S-adenosyl-L-homocysteine (SAH), a product formed following transfer of the methyl group from SAM, was sufficient to increase m6A levels even in the presence of sulfate (Figure 2F). Without a sulfur source, the impact of SAH on m6A levels was even more pronounced (Figure 2G). Lastly, using the MBN protection assay, we found that HeLa, HEK293T, and 3T3 cells cultured with methionine contained very low levels of m6A, only slightly above our detection limit, compared with yeast cells (Figure S3C). By contrast, methionine starvation led to a significant increase of m6A in all three cell lines (Figure 2H), suggesting that both yeast and mammalian cells sense sulfur starvation to increase m6A in their 18S rRNA. These observations also imply that m6A might be functionally important under sulfur starvation.
Figure 2. Sulfur starvation increases m6A levels in 18S rRNA in yeast cells and mammalian cell lines.

(A) m6A levels in 18S rRNA increase specifically under sulfate starvation. com, complete medium; −C, carbon starvation; −N, nitrogen starvation; −P, phosphate starvation; −S, sulfate starvation. Mean ± SD (n = 3 biological replicates).
(B) Changes in m6A levels in response to sulfate availability. Mean ± SD (n = 2–3 biological replicates).
(C) Increased m6A under sulfate starvation is synthesized de novo. Cells were fully labeled in [15N] SD and starved in [14N] sulfur-free medium + 50 mg L−1 adenine ([14N] S + A) for 2 h. Peak areas of [U-14N] and [U-15N] ac4C were combined to normalize the differentially labeled m6A. Normalized abundance was further divided by that of preswitch samples. Mean ± SD (n = 3 biological replicates).
(D and E) Starvation of methionine (D) and SAM (E) increases m6A levels in 18S rRNA. Mean ± SD (n = 3 biological replicates). Methionine (D) and SAM (E) were supplemented at 1 and 0.5 mM, respectively. Data for −S + SAM in (E) were also used in (G).
(F) SAH increases m6A levels in 18S rRNA. Mean ± SD (n = 2 biological replicates).
(G) SAH enhances the impact of SAM starvation on increasing m6A levels in 18S rRNA. SAM and SAH were used at 0.5 mM. Mean ± SD (n = 3 biological replicates).
(H) Methionine starvation increases m6A levels at the 3′ end of mammalian 18S rRNA. MBN protection assay was performed to specifically examine m6A in the last 37 nucleotides of mammalian 18S rRNA. Top panels are representative chromatograms, and bottom panels are quantification results. Mean ± SD (n = 3–7 biological replicates). Chromatograms were normalized to the peak area of ac4C to allow comparison between samples. The peak area of m6A was first normalized to that of ac4C and to samples with methionine.
Ordinary one-way analysis of variance (ANOVA) and Dunnett’s multiple comparison test with a single pooled variance were performed to calculate the p values for (A) and (B), and unpaired two-tailed Student’s t test, assuming equal variances, was used for (D)–(H). p > 0.05 (n.s.), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
See also Figure S3.
m6A and m62A in 18S rRNA are not functionally equivalent
Investigating the functional role of m6A necessitates a mutant Dim1p that ideally installs only m62A, but not m6A. Sequence alignment from 20 phylogenetically diverse species revealed two universally conserved residues, E85 and D87, in Dim1 homologs, as well as the GAA triplet in SSU rDNA (18S rDNA or 16S rDNA), in which the two adenosines are modified as m6A or m62A (Figure 3A). Inspired by the E85A mutant (Figure S2D), we sought to systematically mutate the glutamic acid to encompass all possible changes at the 85 position. Substitution with small amino acids, e.g., glycine and serine, eliminated m62A but largely spared m6A (Figure 3B). By contrast, replacement with bulky amino acids inactivated Dim1p (Figure 3B). Surprisingly, a conservative change of glutamic acid to aspartic acid led to an approximately nine-fold increase of m6A and a commensurate decrease of m62A (Figures 3B–3D). A reciprocal change at the 87 position (D87E) resulted in even more pronounced elevation of m6A (Figures 3C and 3D). Simultaneous introduction of the E85D and D87E substitutions appeared to convert Dim1p into a mono-methyltransferase (Figures 3C and 3D). None of these mutants were able to boost m6A levels in response to sulfate starvation (Figure S3D). Collectively, these observations suggest that the E85 and D87 residues of Dim1p are critical for determining methylation multiplicity at A1781/A1782 of 18S rRNA and for relaying a deficiency in sulfur availability to an increase in m6A levels.
Figure 3. m6A and m62A in 18S rRNA are not functionally equivalent.

(A) Partial sequence alignment of Dim1 homologs and 18S (16S) rDNA. Highlighted are E85 and D87 of S. cerevisiae Dim1p and the two adenosines modified as m6A or m62A.
(B) E85 is a key determinant of the catalytic activity and methylation multiplicity of Dim1p. Data were acquired from the MBN protection assay using oKL204. Peak areas were normalized to that of ac4C. Chromatograms of E (WT), W (E85W), Q (E85Q), and A (E85A) were also presented in Figures 1E and S2D.
(C) E85D and D87E mutations alter the methylation multiplicity of Dim1p. Chromatograms from the MBN protection assay using oKL204 were normalized to the peak area of ac4C to allow comparison between samples.
(D) Quantification of m6A and m62A in 18S rRNA from dim1 mutants. Mean ± SD (n = 3–7 biological replicates). The p values were calculated using ordinary one-way ANOVA and Dunnett’s multiple comparison test with a single pooled variance. Data were also used for plotting Figure S3D (prestarvation).
(E) Dim1p E85D, D87E, and E85Q mutants have lower fitness than WT. Mean ± SD (n = 4–6 biological replicates). The p values were calculated using one-sample Student’s t test.
(F) m6A-bearing ribosomes participate in active translation. The peak area of m6A was normalized to that of ac4C. Mean ± SD (n = 5–6 biological replicates). **p < 0.01, ***p < 0.001, ****p < 0.0001.
See also Figures S3, S4, and S8.
Although our search for an m62A-only Dim1p was unsuccessful, we found the E85D and D87E mutants useful for inferring the functions of m6A at A1781/A1782. The remarkable conservation of E85 and D87 in Dim1 homologs suggests that these two residues are so crucial that few changes were tolerated during evolution. Consistent with this idea, mutating these two residues, even in the form of conservative changes such as E85D and D87E, invariably reduced cellular fitness (Figure 3E). Because the abundance of Dim1p was not obviously affected by these mutations (Figure S3E), one likely explanation for the preservation of E85 and D87 is to maintain high stoichiometry of m62A and to relay sulfur availability to m6A levels. This interpretation would further imply that m6A and m62A at A1781/A1782 may not be functionally equivalent.
Regulation of translation by m6A in 18S rRNA
Given the proximity of m6A and m62A to the ribosome peptidyl site (P site) (Figure S4A) (Hussain et al., 2014; Tesina et al., 2019) and the DCS (Sharma and Lafontaine, 2015), we speculated that the number of methyl groups might affect translation differentially. To test this hypothesis, we first verified that m6A-bearing ribosomes are translation competent (Figures 3F and S4B). Next, we performed polysome profiling to qualitatively examine translation under methionine-replete and methionine-starvation conditions. Three strains were compared: wild type (WT) (~3% m6A with methionine and ~10% m6A without methionine), the D87E mutant (~80% m6A irrespective of methionine availability), and the E85Q mutant (no detectable m6A or m62A under either condition). With methionine, the D87E mutant was highly similar to WT, whereas the E85Q mutant accumulated higher levels of the large subunit (LSU) (Figure S5A), a manifestation of defective SSU biogenesis. Consistently, the E85Q mutant accumulated slightly less SSU (Figure S5B) and showed mild defects in rRNA processing (Figures S6A and S6B). Dim1p is reportedly required for early pre-rRNA processing at A1 and A2, and its depletion reduces 20S and 27SA pre-rRNA but increases 33/32S and 22S pre-rRNA (Lafontaine et al., 1995). Although the E85Q mutant bore resemblance to Dim1p depletion in rRNA processing, it accumulated rather than decreased 20S pre-rRNA (Figure S6B). This observation would argue against the notion that the E85Q mutation impairs cleavage at A1 and/or A2, because inhibition at either site or both is expected to severely reduce the 20S pre-rRNA. Moreover, it illustrates the challenges in uncoupling the methyltransferase activity of Dim1p from its rRNA processing functions (see Discussion). Nevertheless, without methionine, all three strains exhibited reduced polysomes and concomitant increase of monosomes (Figure S5A), indicative of global repression of translation under methionine starvation. Lastly, both yeast and mammalian cells appeared to restrict ribosome biogenesis when methionine is deficient (Figures S6C and S6D), consistent with a previous report (Wejksnora and Haber, 1974).
We next performed ribosome profiling (Ingolia et al., 2009) to quantitatively examine translation in the three strains under methionine-replete and methionine-starvation conditions (Figure S7A). Because of its known artifacts (Gerashchenko and Gladyshev, 2014; Hussmann et al., 2015; Santos et al., 2019), the translation inhibitor cycloheximide was excluded during sample harvest. Nevertheless, ribosome footprints were enriched in 28-to 29-mers as anticipated (Figure S7B), and excellent reproducibility was observed across ribosome profiling and RNA sequencing (RNA-seq) samples (Figure S7C). At the transcript level, the D87E mutant and WT were indistinguishable, irrespective of methionine availability (Figure S7D; Table S1). By contrast, the transcriptome of the E85Q mutant showed clear differences compared with WT in a methionine-dependent fashion (Figure S7D; Table S1). With methionine, 42 genes were significantly altered in the E85Q mutant (9 upregulated genes and 33 downregulated genes). The downregulated group includes several amino acid metabolism genes (e.g., MET13, MET17, STR3, and ARG1), as well as two SSU genes RPS9A and RPS22B (Figures S7D–S7F; Table S1). Although the rps9AΔ and rps22BΔ mutants resemble the E85Q mutant in polysome profiles (Figures S7G and S7H), loss of either gene did not lead to the rRNA processing defects observed in the E85Q mutant (Figure S7I). This observation would argue against their low expression as the reason for the defective rRNA processing in the E85Q mutant, although it may still contribute to the under-accumulation of SSU. Nevertheless, without methionine, the transcriptome-wide differences were more prominent between the E85Q mutant and the WT, with 402 genes showing significant changes (Figure S7D). Interestingly, ~54% (120/224) of the upregulated genes in the E85Q mutant encode the ribosome SSU and LSU (Table S1).
We next calculated TE (see STAR methods) to quantitatively examine translation in the three yeast strains. With methionine, 10 genes showed significantly altered TE in the E85Q mutant compared with WT (higher TE: CST9, FIG2, STR3, and YBR191W-A; lower TE: CHA1, FIT2, FIT3, KDX1, MAL31, and YER186C) (Figure 4A; Table S2). In the D87E mutant, we identified 16 significantly changed genes (lower TE: AGP3, FIT2, FIT3, GRX8, JLP1, MET2, MET3, MET28, MET32, MMP1, OPT1, PDC6, SOA1, SUL1, SUL2, and YCT1) (Figure 4A; Table S2). Remarkably, 12 genes (underlined) are involved in sulfur metabolism, and none of them were significantly changed in the E85Q mutant (Figures 4A–4C; Table S2). This observation suggests that m6A at A1781/A1782 of 18S rRNA functions differently from m62A, with a striking specificity for sulfur metabolism genes. It also argues strongly against the notion that the presence of m6A at these two conserved adenosines is a fortuitous phenomenon.
Figure 4. Translational regulation of sulfur metabolism genes via methylation multiplicity.

(A) Change of translation efficiency (TE) under methionine-replete and methionine-starvation conditions. A 10% false discovery rate (FDR) (−log10(Padj) ≥1) and 2-fold change of TE (log2(TE fold change) ≥1 or log2(TE fold change) ≤ −1) are considered significant, and genes with significantly changed TE are highlighted in black.
(B) Representative tracks of ribosome footprint (RFP) and mRNA for JLP1, YCT1, MET3, and RPN10. Two biological replicates for each genotype are shown, and tracks are comparable only within each RFP or RNA group.
(C) Simplified schematic of yeast sulfur metabolism. Highlighted are proteins whose transcripts are translated with significantly lower TE in the D87E mutant under methionine-replete conditions.
(D) Impact of methionine starvation on TE and mRNA levels of sulfur metabolism genes listed in (C).
The p values were calculated using two-sided Mann-Whitney test. ****p < 0.0001.
See also Figures S5–S7 and Tables S1, S2, and S3.
These translational differences between the D87E mutant and the WT disappeared under methionine starvation (Figures 4A and 4B; Table S2). Although methionine deprivation generally decreased TE of these sulfur metabolism genes in WT and the E85Q mutant, most of them were translated with higher TE in the D87E mutant (Figure 4D). The only significantly changed gene in the D87E mutant under methionine starvation is JIP5, which encodes an essential LSU biogenesis factor (Li et al., 2009) (Figure 4A; Table S2). Methionine starvation drastically reduced TE of JIP5 in all three strains (Table S3), but the reduction was significantly more pronounced in the D87E mutant (Figure 4A; Table S2). However, methionine starvation led to significantly more TE changes in the E85Q mutant compared with WT, with 114 genes showing altered TE (55 with higher TE and 59 with lower TE) (Figure 4A; Table S2). Of the genes with lower TE, approximately half (29/59) encode the ribosome SSU and LSU (Table S2).
The E85Q mutant that lacks methylation at A1781/A1782 fails to pause/stall at cysteine codons under methionine starvation
Lastly, we observed that methionine starvation led to strong pausing/stalling at cysteine codons within the ribosome aminoacyl site (A site) in WT and the D87E mutant, which strikingly was absent from the E85Q mutant that lacks both m6A and m62A (Figures 5A and 5B). Surprisingly, no pausing at the methionine codon was observed in any of the three strains. Methionine starvation is expected to lower many sulfurous metabolites, including cysteine, which may result in lower cysteinyl-tRNA amounts to cause ribosome pausing/stalling at cysteine codons. Indeed, many sulfurous metabolites plummeted under methionine starvation, but surprisingly, the E85Q mutant was able to maintain higher levels of cysteine, homocysteine, cystathionine, reduced glutathione (GSH), and oxidized glutathione (GSSG) (Figure 5C). Other amino acids were not necessarily increased in the E85Q mutant, suggesting that the higher levels of sulfurous metabolites are not simply due to its slower growth rate (Figures 3E, S8A, and S8B). Nevertheless, although the slightly increased cysteine amounts (~60%) might increase cysteinyl-tRNA to alleviate pausing/stalling, the E85Q mutant still exhibited a substantial reduction of cysteine under methionine starvation (Figure 5C), which led us to consider additional explanations for its lack of pausing/stalling at cysteine codons.
Figure 5. The E85Q mutation abolishes pausing/stalling at cysteine codons under methionine starvation.

(A) Relative enrichment of ribosomes at each codon under methionine-replete and methionine-starvation conditions. Reads were mapped to the ribosome aminoacyl site (A site). Two biological replicates for each genotype are shown (two circles per genotype), with their average being represented by a solid line. The p values were calculated using unpaired two-tailed Student’s t test, assuming equal variances for comparison between WT and the E85Q mutant.
(B) Representative tracks showing enrichment of ribosomes at two cysteine codons of the MET30 transcript following methionine starvation.
(C) Changes in sulfurous metabolites under methionine-replete and methionine-starvation conditions. Mean ± SD (n = 2 biological replicates).
(D) Schematic of the dual luciferase reporters.
(E) E85Q mutant has higher decoding errors. Mean ± SD (n = 6 biological replicates).
(F) Loss of RPS22B/snR44 or RPS9A does not affect decoding fidelity. Mean ± SD (n = 5–11 and n = 7–11 biological replicates for the control and the H245R reporter, respectively).
(G) rRNA processing defects due to loss of TSR3 do not increase decoding errors. Mean ± SD (n = 3 biological replicates).
The p values were calculated using ordinary one-way ANOVA and Dunnett’s multiple comparison test with a single pooled variance for (C) and (E)–(G). p > 0.05 (n.s.), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Given that the tandem m62A modifications reside close to the DCS, we speculated that loss of methylation might render ribosomes less sticky at cysteine codons despite limited cysteinyl-tRNA. Perhaps the unmethylated ribosomes are intrinsically prone to errors and could decode cysteine codons using their near-cognate aminoacyl-tRNAs, whose availability is unlikely to be limited by methionine starvation (Figure S8B). To test this hypothesis, we used a luciferase reporter (Figure 5D) (Salas-Marco and Bedwell, 2005) in which a histidine codon (CAC) critical for Firefly luciferase activity is mutated to an arginine encoded by its near-cognate codon CGC. This change severely reduces Firefly luciferase activity, which can be restored if the CGC codon is decoded by histidinyl-tRNAGUG. With this reporter, we found that the E85Q mutant exhibited significantly higher decoding errors than WT and the D87E mutant (Figure 5E), similar to the previously reported E85A mutant (Ghalei et al., 2017). As an important control, we found that disruption of RPS22B or RPS9A did not increase decoding errors (Figure 5F). Moreover, we deleted the TSR3 gene, which encodes the aminocarboxypropyl transferase for the N1-methyl-N3-aminocarboxypropyl-pseudouridine (m1acp3ψ) modification in yeast 18S rRNA (Meyer et al., 2016). Its deletion causes rRNA processing defects (Li et al., 2009) reminiscent of those observed in the E85Q mutant (Figure S8C). The tsr3Δ mutant showed similar decoding fidelity compared with WT cells (Figure 5G). Collectively, these results suggest that the increased decoding errors in the E85Q mutant likely stem from the absence of methylation at A1781/A1782, rather than rRNA processing defects. Loss of methylation at these tandem adenosines may facilitate decoding of cysteine codons at the cost of translation fidelity to alleviate pausing/stalling under methionine starvation. This interpretation might also explain the attenuated pausing/stalling at CCG (proline) and CGA (arginine) codons in the E85Q mutant (Figure 5A), because both are rare codons without their cognate tRNAs (Tuller et al., 2010).
DISCUSSION
Methylation multiplicity as a mechanism to diversify ribosomes to regulate translation
Ribosomes have long been perceived as a homogeneous population. However, it has become evident that they may exist as a group of heterogeneous entities, with respect to not only their protein subunit composition but also modifications of these subunits and rRNA (Byrgazov et al., 2013; Dinman, 2016; Genuth and Barna, 2018). Here, we present another example of ribosome heterogeneity in the form of rRNA methylation via methylation multiplicity. The presence of m6A at the conserved tandem adenosines in 18S rRNA, together with m62A, increases the complexity of the SSU. By conducting a comprehensive mutagenesis analysis, we were able to increase m6A levels in bulk by introducing an E85D or D87E mutation to Dim1p (Figures 3B–3D). Such conservative changes by a single methylene group (-CH2-) minimize perturbations that could be inadvertently introduced to the cell. Analyses of polysome and ribosome subunit profiles (Figures S5A and S5B), rRNA processing (Figure S6B), the transcriptome (Figure S7D), intracellular metabolites (Figures 5C and S8B), and 18S rRNA modifications (Figure S8D) indicate that the D87E mutant is virtually indistinguishable from WT. Still, this mutant bearing more m6A in its ribosomes carries out translation distinctly compared with WT cells, featuring a striking specificity for sulfur metabolism genes and a peculiar dependency on sulfur availability (Figures 4A–4C). It is unclear how this specificity is determined, although all of these genes are heavily induced at the transcriptional level by methionine starvation (Figure 4D; Table S3). Perhaps a cis-regulatory element is embedded in their transcripts to confer the specificity, as reported previously (Xue et al., 2015). However, ongoing bioinformatic investigation has yet to identify promising candidates. In addition, trans-acting factors (e.g., RNA-binding proteins) might assist in determining the specificity (Leppek et al., 2018).
With respect to the sulfur dependency, two outstanding questions remain. The first concerns how sulfur metabolism transcripts are translated with higher TE in the D87E mutant under methionine starvation (Figure 4D), when global translation is repressed (Figure S5A). One possible explanation is that m6A-bearing SSU might function more efficiently using non-canonical translation pathways, e.g., internal ribosome entry site (IRES)-mediated translation, which is known to operate under stress conditions (Gilbert et al., 2007; Holcik and Sonenberg, 2005; Spriggs et al., 2008). Under methionine-replete conditions, m6A-bearing SSU might be inefficient at translating sulfur metabolism transcripts using the canonical cap-dependent pathway, perhaps because of the presence of IRES in their 5′ untranslated regions. Under methionine starvation, cap-dependent translation may be inhibited because of methionine scarcity. m6A-bearing SSU, perhaps with assistance from IRES trans-acting factors(King et al., 2010; Komar and Hatzoglou, 2011), might be able to efficiently recognize IRES within these sulfur metabolism transcripts to support their translation during methionine starvation.
The second question concerns the dependency of the TE differences between the WT and the D87E mutant on methionine availability (Figure 4A). Because translation of these sulfur metabolism transcripts is seemingly recalcitrant to methionine deprivation in the D87E mutant (Figure 4D), we speculate that they might be translated predominantly by m6A-bearing ribosomes in WT under methionine starvation. A single yeast cell is estimated to contain ~200,000 ribosomes (von der Haar, 2008; Warner, 1999), and a stoichiometry of ~10% would equal ~20,000 m6A-bearing ribosomes in methionine-starved WT cells. If the yeast transcriptome comprises ~60,000 mRNA molecules (Zenklusen et al., 2008), there should be sufficient m6A-bearing ribosomes for these sulfur metabolism transcripts.
Functional importance of the tandem m62A modifications
Modified ribonucleosides are prevalent in rRNAs. Because many of them reside in structurally important positions within the ribosome (Sloan et al., 2017), it is perhaps not surprising that rRNA modifications have been shown to play key roles in maintaining translation efficiency and translation accuracy (Baudin-Baillieu et al., 2009; Jack et al., 2011; King et al., 2003; Lafontaine et al., 1998; Liang et al., 2009; Ma et al., 2019; Schosserer et al., 2015). rRNA modifications can also be selectively impactful, because some appear to regulate translation of only a subset of mRNAs, such as rRNA pseudouridylation (Bellodi et al., 2010a, 2010b; Yoon et al., 2006) and 2′-O-methylation (Basu et al., 2011; Erales et al., 2017; Marcel et al., 2013) in IRES-mediated translation. Moreover, a recent study suggests that m6A in C. elegans 18S rRNA is important for translating an mRNA involved in lipid oxidation (Liberman et al., 2020).
Among all known rRNA modifications, the tandem m62A modifications are remarkably conserved; they are found almost universally at the 3′ end of SSU rRNA (Rife, 2009; Van Knippenberg et al., 1984), which situates them close to the ribosome P site (Figure S4A) and the DCS (Sharma and Lafontaine, 2015). Despite their conservation and occupation of a structurally important location, the functional importance of the tandem m62A modifications remains incompletely understood. One obstacle is that Dim1p (and its homologs) is a dual-function protein required for both m62A methylation and rRNA processing (Connolly et al., 2008; Lafontaine et al., 1995; Zorbas et al., 2015), and a dim1 mutant that completely uncouples these two functions has hitherto been elusive (see Limitations of study). A previous study showed that cellular extract from a dim1–2 mutant was incompetent at translating reporter genes (Lafontaine et al., 1998). Although some defects may result from the loss of m62A, effects of impaired rRNA processing in the dim1–2 mutant have not been excluded. In our work, some changes in the E85Q mutant in the ribosome profiling and RNA-seq experiments (Figures 4A and S7D), as well as the growth assays (Figures 3E and S8A), may stem from rRNA processing defects. However, we have performed important controls to rule out defects in SSU biogenesis and rRNA processing as an explanation for the increased decoding errors in the E85Q mutant (Figures 5F and 5G). Therefore, minimally, we conclude that the tandem m62A modifications are important for maintaining translation fidelity and possibly for pausing/stalling at cysteine codons under methionine starvation.
Limitations of study
Challenges in uncoupling the methyltransferase activity of Dim1p from its rRNA processing functions
Functional interrogation of the tandem m62A modifications necessitates a dim1 mutant that uncouples its methyltransferase activity from its rRNA processing functions. Despite significant efforts in previous studies (Connolly et al., 2008; Lafontaine et al., 1998) and our work, such a mutant has yet to be found. An early study in yeast constructed a temperature-sensitive dim1–2 mutant that is reportedly defective in methylation (Lafontaine et al., 1998). However, this dim1–2 mutant, containing six substitutions, still exhibits rRNA processing defects and retains some methyltransferase activity for m62A and perhaps for m6A as well (primer extension may not effectively distinguish between m6A and A). Similar efforts in E. coli were also unsuccessful (Connolly et al., 2008): the tested KsgAE66A (equivalent to Dim1pE85A) might still be partially active based on our findings with Dim1pE85A (Figures 3B and S2D), and importantly it leads to defects in SSU biogenesis and rRNA processing (Connolly et al., 2008). Here, we introduced a highly conservative E85Q mutation to eliminate the methyltransferase activity of Dim1p (Figure 1E), but unfortunately, this E85Q mutant is still defective in SSU biogenesis (Figures S5A and S5B) and rRNA processing (Figure S6B). Recent structural work reveals that the E85A mutation has little impact on the overall conformation of human DIMT1 (Shen et al., 2020), and conceivably, the E85Q mutation would be expected to be even less disruptive. Still, even such a conservative change leads to rRNA processing defects. With the aforementioned early findings, this led us to speculate that the methyltransferase activity of Dim1p might be involved in rRNA processing, as proposed previously (Connolly et al., 2008).
Therefore, the difficulty in uncoupling the dual functions of Dim1p may limit our interpretation of the ribosome profiling and RNA-seq experiments (Figures 4A and S7D) and of the growth assays (Figures 3E and S8A), because some changes may not stem directly from the loss of methylation. However, we have included important controls to rule out defects in SSU biogenesis and rRNA processing as an explanation for the increased decoding errors in the E85Q mutant (Figures 5F and 5G). In closing, the tandem m62A modifications are important for ensuring translational fidelity and possibly for pausing/stalling at cysteine codons under methionine starvation. Nonetheless, these and other phenotypes of the E85Q mutant, such as changes in transcript levels or translational efficiency, could be secondary and compensatory because of defects in rRNA processing and/or SSU biogenesis.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Benjamin Tu (benjamin.tu@utsouthwestern.edu).
Materials availability
Reagents are available upon request from the Lead Contact.
Data and code availability
All sequencing data have been deposited in Gene Expression Omnibus with the accession number GEO: GSE142528.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains and growth conditions
Prototrophic CEN.PK Saccharomyces cerevisiae (van Dijken et al., 2000) was used for strain construction (Table S4), using the lithium acetate based transformation protocol (Longtine et al., 1998). Unless otherwise stated, yeast strains were grown in SD medium containing 20 g L−1 glucose and 6.7 g L−1 yeast nitrogen base without amino acids (BD Difco) at 30 °C and 300 rpm. For nutrient starvation experiments, medium formulas are listed in Table S5. Plasmids were constructed using the Gibson assembly protocol (Gibson et al., 2009) and site directed mutagenesis was performed using Phusion HF polymerase (NEB) with primers bearing the desired mutations (Table S4), followed by DpnI (NEB) digestion and subsequent transformation into E. coli DH5α. All the plasmids were verified by DNA sequencing.
Mammalian cell lines and growth conditions
All cell lines (HeLa, HEK293T, and 3T3, see Key resources table for details) were cultured in a Heracell humidified incubator (Thermo-fisher, HERAcell 150i) at 37 °C with 5% CO2. HeLa cells were maintained in RPMI-1640 (GIBCO A14517–01), and 3T3 and HEK293T cells in DMEM (GIBCO 21013–024). Both media were supplemented with the required amino acids and 5% fetal bovine serum (Sigma F6178). When confluency reached ~80%, cells were washed with PBS twice and subsequently cultured in either RPMI-1640 (GIBCO A14517–01) or DMEM (GIBCO 21013–024) with or without methionine. After six hours, cells were harvested and total RNA was isolated using the TRIzol reagent (Invitrogen, Thermo Fisher), as described below.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-FLAG antibody | Sigma | Cat#F11804 |
| Rabbit polyclonal anti-G6pdh antibody | Sigma | Cat#A9521 |
| Rabbit polyclonal anti-Rpn10 antibody | Abcam | Cat#ab98843 |
| Chemicals, peptides, and recombinant proteins | ||
| Nuclease P1 from Penicillium citrinum | Sigma | Cat#N8630 |
| Phusion® High-Fidelity DNA Polymerase | New England Biolabs | Cat#M0530S |
| DpnI | New England Biolabs | Cat#R0176S |
| RNase A | Lucigen | Cat#MRNA092 |
| Phosphodiesterase I from Crotalus atrox | Sigma | Cat#P4506 |
| Alkaline phosphatase calf intestinal | New England Biolabs | Cat#M0290 |
| Mung bean nuclease | New England Biolabs | Cat#M0250L |
| Critical commercial assays | ||
| RPMI-1640 | GIBCO | Cat#A14517–01 |
| DMEM | GIBCO | Cat#21013–024 |
| Fetal bovine serum | Sigma | Cat#F6178 |
| Yeast nitrogen base without amino acids | BD Difco | Cat#BD233520 |
| complete EDTA-free protease inhibitor cocktail tablets | Roche | Cat#11873580001 |
| Chemiluminescent nucleic acid detection module kit | Thermo Fisher | Cat#89880 |
| D-Tube Dialyzer Mini (MWCO 6–8 kDa) | EMD Millipore | Cat#71–504-3 |
| Dual-Luciferase® Reporter assay system | Promega | Cat#E1910 |
| NucleoSpin® gel and PCR clean-up kit | Takara | Cat#740609 |
| PureLink miRNA isolation kit | Invitrogen | Cat#K157001 |
| NuPAGE 4–12% polyacrylamide Bis-Tris Gels | Invitrogen | Cat#WG1403BX10 |
| Ribo-Zero Gold for yeast | Illumina | Cat#MRZY1306 |
| Dynabeads mRNA purification kit | Invitrogen | Cat#61006 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE142528 |
| Experimental models: cell lines | ||
| HeLa | ATCC | Cat#CCL-2 |
| Lenti-X 293T | Takara | Cat#632180 |
| 3T3 | ATCC | Cat#CRL-1658 |
| Experimental models: organisms/strains | ||
| CEN.PK MATa | van Dijken et al., 2000 | NA |
| CEN.PK MATa/α | van Dijken et al., 2000 | NA |
| S288C MATα | ATCC | Cat#204508 |
| W303 MATa | Korolev et al., 2012 | NA |
| A full list of yeast mutant strains is provided in Table S4 | NA | NA |
| Oligonucleotides | ||
| A full list of oligos is provided in Table S4 | NA | NA |
| Recombinant DNA | ||
| A full list of plasmids is provided in Table S4 | NA | NA |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Software and algorithms | ||
| Analyst software package | AB SCIEX | Versions 1.6.2 or 1.6.3 |
| Image Lab | Bio-Rad | Version 6.0 |
| ImageJ | National Institutes of Health | Version 1.50e |
| Integrative Genomics Viewer | Broad Institute | 2.8.0 |
| GraphPad Prism | GraphPad Software | Versions 6, 7, 8, and 9 |
METHOD DETAILS
Total RNA isolation
Yeast cell pellet stored at −80 °C was thawed on ice and washed with ice-cold sterile water once. TES (10 mM Tris-HCl pH 7.5, 10 mM EDTA pH 8.0, and 0.5% SDS) was added to resuspend the cell pellet followed by addition of an equal volume of acidic phenol (pH 4.3). Cells were lysed using acid-washed glass beads on a bead beater (two cycles of one-minute beating followed by one-minute cooling on ice). Cell debris and glass beads were removed by centrifugation. The aqueous phase was transferred into a clean centrifuge tube and extracted with an equal volume of acidic phenol, followed by a third extraction with chloroform to remove the residual phenol. RNA was ethanol precipitated, washed with 70% ethanol, and dried before finally being dissolved in nuclease-free water. Purified RNA was examined electrophoretically and quantified spectrophotomically.
Mammalian RNA was isolated using the TRIzol reagent (Invitrogen, Thermo Fisher). Briefly, 2 mL TRIzol was dispensed into 15-cm dish and cell suspension was collected by pipetting. Cell pellet was either stored at −80 °C or processed immediately. To 1 mL TRIzol cell suspension, 250 μL chloroform was added. The mixture was vortexed vigorously and centrifuged. The aqueous phase was transferred to a clean centrifuge tube with 600 μL isopropanol and precipitated on ice. RNA was pelleted by centrifugation and washed with 70% ethanol once to remove salts. RNA was then resuspended in nuclease-free water and digested with DNase I to remove DNA. DNase I was subsequently removed using phenol (pH 4.3):chloroform (1:1) extraction and RNA was recovered by ethanol precipitated. RNA was washed once with 70% ethanol and resuspended in nuclease-free water.
RNA fractionation
Small RNA was purified using the PureLink miRNA isolation kit (Invitrogen) and polyA+ RNA was isolated using the Dynabeads mRNA purification kit (Invitrogen) following the manufacturers’ instructions. To isolate 18S and 25S rRNA, we mixed total RNA with an equal volume of 2 × RNA loading solution (95% formamide, 0.02% SDS, 0.02% bromophenol blue, and 1 mM EDTA pH 8.0) and denatured RNA samples at 75 °C for five minutes followed by rapid chilling on ice. Denatured RNA was loaded onto a 1.3% TAE low melting agarose gel. The 18S and 25S rRNA were visualized by ethidium bromide staining, excised using a clean scalpel, and purified using the NuceloSpin Gel and PCR clean-up kit (Takara) following the manufacturer’s instructions.
Estimation of stoichiometry of m6A in yeast 18S rRNA
We used HeLa 18S rRNA as the standard, since it is modified with a single m6A at A1832 to ~98% (Liu et al., 2013). Yeast and HeLa 18S rRNA were digested and ribonucleosides were quantified as described below. The abundance of m6A was normalized to each of the four ribonucleosides (A, G, C, and U). These ratios were further corrected to account for the differences in the abundance of each ribonucleoside between yeast and HeLa cells. The stoichiometry of m6A in yeast 18S rRNA was then estimated by comparing the normalized m6A (e.g., m6A/A) between yeast and HeLa cells. Normalization by each of the four ribonucleosides gave similar results, averaging between 4%–5% of m6A per 18S rRNA.
Mung bean nuclease (MBN) protection assay
This assay was performed as described previously (Andersen et al., 2004; Peifer et al., 2013; Sharma et al., 2013) with some modifications. A total of ~1000 pmole DNA oligo was mixed with 200 μg total RNA. After ethanol precipitation and wash, the nucleic acid mixture was resuspended in 40 μL 1 × hybridization buffer (40 mM PIPES pH 6.4, 400 mM NaCl, 1 mM EDTA, and 20% formamide) and heated at 85 °C for ten minutes followed by incubation at 35 °C for three hours. The hybridization mixture was then mixed with 497 μL nuclease-free water, 60 μL 10 × RNA digestion buffer (100 mM Tris-HCl pH 7.5, 3 M sodium acetate pH 5.2, 50 mM EDTA pH 8.0), and 3 μL 5 mg ml−1 RNase A (Epicenter), and incubated at 37 °C for one hour. The digestion mixture was then extracted with an equal volume of acidic phenol (pH 4.3):chloroform (1:1) and ethanol precipitated. The nucleic acid pellet was washed, resuspended in 100 μL 1 × MBN buffer (30 mM NaCl, 50 mM sodium acetate, 1 mM ZnSO4, pH 5.0) supplemented with 4.5 μL 10 U μl−1 MBN (NEB), and incubated at 30 °C for one hour. The digestion mixture was then extracted with an equal volume of acidic phenol (pH 4.3): chloroform (1:1) and ethanol precipitated. The precipitated nucleic acid was loaded onto a 15% polyacrylamide gel and the RNA:DNA hybrid was visualized by ethidium bromide staining and excised using a clean scalpel. The gel slice was transferred to a clean microcentrifuge tube, crushed, and soaked in 200 μL elution buffer containing 300 mM sodium acetate (pH 5.3), 1 mM EDTA (pH 8.0), and 0.1% SDS. The eluted RNA:DNA fragment was ethanol precipitated, washed, and resuspended in nuclease-free water. Alternatively, the RNA:DNA hybrid was extracted using D-Tube Dialyzer Mini (MWCO 6–8 kDa) (EMD Millipore) by electro-elution following the manufacturer’s instructions.
LC-MS/MS detection of nucleosides
RNA digestion and nucleoside detection were performed essentially as described by Laxman et al. (2013). Briefly, RNA was resuspended in 102 μL of ultrapure water and 7 μL of acidic buffer (0.1 M sodium acetate, 20 mM ZnCl2, pH 6.8) was added, followed by the addition of 5 μg RNase A (Epicenter) and 1.5 U nuclease P1 (Sigma). Digestion was carried out at 37 °C for four hours before 7 μL of basic buffer (0.3 M sodium acetate, pH 7.8) was added. The digestion mixture was further treated with 10 U alkaline phosphatase (calf intestinal, NEB) and 5 μL 8 mg ml−1 snake venom phosphodiesterase I (Sigma) overnight to maximize digestion and dephosphorylation efficiency.
The digested samples were separated on a Synergi Fusion-RP column (4 mm particle size, 80 Å pore size, 150 mm × 2 mm, Phenomenex) using a Shimadzu high performance liquid chromatography (HPLC) machine and simultaneously detected under positive mode by a triple quadrupole mass spectrometer (3200 QTRAP, ABSCIEX). The total run time was 25 minutes at a flow rate of 0.5 mL min−1, with 5 mM ammonium acetate (pH 5.5) in water as solvent A and 5 mM ammonium acetate in methanol as solvent B. The following gradient elution was performed: 0.01 min, 0% B, 4 min, 0% B, 5 min, 0.2% B, 6 min, 1% B, 7 min, 3% B, 8 min, 5% B, 14 min, 25% B, 16 min, 50% B, 18 min, 100% B, 22 min, 100% B, 23 min, 0% B, 25 min, 0% B. Ribonucleosides were quantified using the Analyst software package 1.6.2 or 1.6.3 by calculating the total peak area. For each experiment, authentic standards were injected and analyzed alongside samples.
LC-MS/MS detection of metabolites
The extraction protocol comprised two sequential steps: quenching and extraction, as described in Castrillo et al. (2003) and Gonzalez et al. (1997), respectively. To quench cells, one volume of cell culture was mixed with three volumes of methanol-water solution (60% v/v, buffered with 10 mM Tricine to pH 7.4) kept at −40 °C. Quenched cells were centrifuged and resuspended in extraction buffer containing ethanol-water solution (75% v/v, 0.1% formic acid to minimize oxidation of thiols) and heated at 80 °C for three minutes. Cell extraction was immediately chilled on ice and subsequently centrifuged at maximum speed at 0 °C to remove cell debris. The supernatant was vacuum dried and stored at −80 °C until analysis.
Samples were analyzed using reversed-phase HPLC coupled to tandem mass spectrometry as described previously (Tu et al., 2007). Metabolites were separated on a Synergi Fusion-RP column (4 μm particle size, 80 Å pore size, 150 mm × 2 mm, Phenomenex) using a Shimadzu HPLC machine and simultaneously detected by a triple quadrupole mass spectrometer (3200 QTRAP, AB SCIEX). The total run time was 22 minutes at a flow rate of 0.5 mL min−1, with 0.1% (v/v) formic acid in water as solvent A and 0.1% (v/v) formic acid in methanol as solvent B. The following gradient elution was performed: 0.01 min, 0% B, 4 min, 0% B, 11 min, 50% B, 13 min, 100% B, 17 min, 100% B, 18 min, 0% B, 22 min, 0% B. Metabolites were detected by multiple reaction monitoring (MRM) with transitions listed in Table S6. Metabolites were quantified using the Analyst software package 1.6.2 or 1.6.3 by calculating total peak area.
Competition assay
Competition between various strains was performed as described previously (Sankar et al., 2016), with some modifications. To distinguish between dim1 and WT, the E. coli lacZ gene controlled by the TEF1 promoter and CYC1 terminator was inserted into an intergenic region between NCA3 and ASF1. This site was selected according to Mikkelsen et al. (2012), who reported that a similar integration in this region supports robust lacZ expression and does not noticeably impact growth. To control for the impact of lacZ expression on fitness, the lacZ cassette was integrated into both dim1 mutants and WT. For example, for competition between WT and the E85Q mutant, two experiments (a and b) were performed in parallel with the following combinations: WT-lacZ versus E85Q and WT versus E85Q-lacZ. Cells were first acclimated in complete medium and then grown in fresh complete medium to log phase. To start the competition, two competitors were mixed at a ratio of 1:1, with each having an initial OD600 ~0.01. Cells were grown to saturation (~7–8 doublings) and were diluted into fresh complete medium in 1:200 after approximately 24 hours. This was then repeated for a minimum of four times and the first round of competition was typically excluded for fitness calculation as cells were just beginning to adapt to the new environment. Nevertheless, cells were plated onto synthetic defined agar plates (6.7 g L−1 yeast nitrogen base without amino acids (BD Difco), 0.79 g L−1 CSM (Sunrise Science), 20 g L−1 glucose, and 20 g L−1 agar), supplemented with 80 μg mL−1 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) and BU salts (26.1 mM Na2HPO4 and 25 mM NaH2PO4, pH 7.0). Blue (competitor expressing lacZ) and white colonies were counted in duplicate to minimize counting errors. Fitness was calculated relative to WT using the following equations:
,where are the relative fitness of dim1-lacZ to WT and dim1 to WT-lacZ in the ith round of competition, respectively; is the number of doublings of a particular strain during a 24-hour competition and i is the ith round of competition; are the arithmetic mean fitness of dim1-lacZ to WT and dim1 to WT-lacZ, respectively. is the geometric mean fitness of dim1 relative to WT, which was reported in Figure 3E.
Dual-luciferase assay
The dual-luciferase assay was performed as described previously (Ghalei et al., 2017; Salas-Marco and Bedwell, 2005), using the Dual Luciferase Reporter Assay System (Promega) with some modifications. When cells reached log phase, one mL of culture was collected by centrifugation and cell pellets were snap frozen in liquid nitrogen and stored at −80 °C until analysis. Cell pellets were first thawed on ice and washed once with ice-cold water to remove the residual medium. Cells were then lysed in 100 μL passive lysis buffer at room temperature for one minute and 10 μL was added to 30 μL Luciferase Assay Reagent II to measure Firefly luciferase activity, followed by the addition of 30 μL Stop & Glow Reagent to measure Renilla luciferase activity. Luminescence was recorded for ten seconds in a 96-well flat-bottom black polystyrene plate (COSTAR) on a Synergy 2 plate reader (BioTek) at room temperature and reported in relative luminescence units (RLU). Background-subtracted Firefly luciferase activity was subsequently normalized to background-subtracted Renilla luciferase activity.
Polysome profiling
Samples for polysome profiling were prepared as described with some modifications (Mašek et al., 2011). When cells were ready, cycloheximide (dissolved in 100% ethanol) was added to a final concentration of 0.1 mg mL−1 and frozen ice at −20 °C (2 g per 10 mL culture) was added to rapidly chill cells. After further incubation on ice for five minutes, cells were centrifuged at 4 °C, snap frozen in liquid nitrogen, and stored at −80 °C until processing. Frozen cell pellets were thawed on ice and washed twice in polysome extraction buffer (PEB) (20 mM Tris-HCl pH 7.5, 140 mM KCl, 5 mM MgCl2, 0.1 mg mL−1 cycloheximide, 1% Triton X-100, and 0.5 mM DTT). Cells were subsequently lysed in PEB with glass beads on a bead beater following three rounds of beating (30 s beating and two-minute cooling on ice). Cell debris was removed by centrifugation at 8000 × g for five minutes at 4 °C and supernatant was collected and measured spectrophotometrically at 260 nm. A 10%–50% (w/v) sucrose gradient was prepared using BIOCOMP Gradient Station ip in 20 mM Tris-HCl pH 7.5, 140 mM KCl, 5 mM MgCl2, 0.1 mg mL−1 cycloheximide, and 0.5 mM DTT. Approximately one A260 unit of cell lysates was carefully loaded onto the top of the sucrose gradient and centrifuged at 41,000 × g for two hours at 4 °C. Polysome profiles were recorded using BIOCOMP Gradient Station ip by measuring absorbance at 260 nm.
Western blot
Samples for western blot were quenched in 10% trichloroacetic acid (TCA) for ten minutes on ice and then stored at −80 °C until analysis. Cell pellets were washed once in cold acetone to remove the residual TCA, before bead-beating in urea lysis buffer containing 6 M urea, 1% SDS, 50 mM Tris-HCl (pH 7.5), 5 mM EDTA, 1 mM DTT, 1 mM PMSF, 10 μM leupeptin, 5 mM pepstatin A, and 1 × protease inhibitor cocktail (Roche). Lysates were heated for five minutes at 75 °C and then centrifuged at maximum speed for five minutes. Protein concentration was estimated using the bicinchoninic acid assay (Thermo Fisher Scientific) and equal amounts of proteins were separated by electrophoresis using 4%–12% NuPAGE gels. Proteins were then transferred to a nitrocellulose membrane and blotted with the corresponding antibodies. Blocking was performed in 5% dry milk/TBST, while antibody incubation was in 1% dry milk/TBST. Antibodies were used at the following dilutions: α-FLAG 1:3,000 (Sigma F1804), α-Rpn10p 1:40,000 (Abcam ab98843), α-G6pdhp 1:20,000 (Sigma A9521).
Northern blot
Northern blot was performed as described previously (Josefsen and Nielsen, 2011; Tafforeau et al., 2013), with minor modifications. Briefly, total RNA of equal amounts was separated on a 1.2% denaturing formaldehyde agarose gel and transferred to a nylon membrane (Hybond-N+, GE healthcare). RNA was crosslinked to the membrane using a UV-cross linker (Stratagene) and stained with methylene blue (0.02% in 0.3 M sodium acetate, pH 5.0). Membrane was washed in nuclease-free water a few times to remove the dye and images were taken using the ChemiDoc MP Imaging System (Bio-Rad). Membrane was pre-hybridized in hybridization solution containing 50% deionized formamide, 5× SSPE, 5× Denhardt’s solution, and 1% SDS at 65 °C for one hour. The hybridization solution was discarded and fresh solution with 10 pM biotinylated DNA probe was added. Membrane was incubated at 65 °C for another hour and then at 37 °C overnight. Membrane-bound biotinylated DNA probes were detected using the Chemiluminescent Nucleic Acid Detection Module (Thermo Fisher) following the manufacturer’s recommendations. Images were taken using the ChemiDoc MP Imaging System and processed using Image Lab 6.0 (Bio-Rad). Biotinylated DNA probes were stripped from the membrane in 50% deionized formamide and 2× SSPE at 65 C for one hour to allow for subsequent hybridization.
Ribosome profiling
Yeast cells were grown in sulfur free (see formula in Table S5) + 1 mM methionine (SFM) to saturation and diluted into 20 mL SFM with a starting OD600 ~0.1. Cells were grown to log phase and diluted into 320 mL SFM with a starting OD600 ~0.005. When OD600 reached ~0.5–0.6, cells were harvested according to Santos et al. (2019). Briefly, 200 mL pre-starvation culture was transferred into a pre-warmed (30 °C) vacuum filtration apparatus and cells were collected onto a 0.45 μm cellulose nitrate membrane filter (Whatman). Before the medium was completely drained, cell pellet was rapidly scraped using a clean metal spatula and transferred into liquid nitrogen. The remaining culture was spun down at 4000 × g for one minute and washed once with an equal volume pre-warmed (30 °C) SF medium once. Washed cells were resuspended in an equal volume pre-warmed (30 °C) SF. After two hours, cells were collected exactly as described above and cell pellets were stored at −80 °C until analysis.
Frozen cell pellets were cryogenically pulverized on a SPEX 6870 Freezer/Mill for one minute at 15 cycles per minute and frozen droplets of lysis buffer (20 mM Tris-HCl pH 8.0, 140 mM KCl, 5 mM MgCl2, 1 mM dithiothreitol, 100 μg mL−1 cycloheximide, 1% Triton X-100, and 0.025 U μL−1 Turbo DNase) were added. The cell lysate was thawed, and cell debris was removed by two sequential centrifugation steps at 4 °C: first at 3000 × g for five minutes and then at 20,000 × g for ten minutes.
Libraries for ribosome profiling and RNA-seq were constructed essentially as described by McGlincy and Ingolia (2017). Briefly, ribosome-protected RNA fragments ranging from ~15–34 nt were isolated after RNase I digestion and denaturing PAGE separation. Cloning linkers with 3′ barcode sequences were ligated to RNA footprints and samples of unique barcodes were pooled together post-ligation whenever possible. rRNA was depleted sequentially using Ribo-Zero Gold for Yeast (Illumina) and biotinylated antisense oligos against rRNA species that co-migrate with ribosome footprints as described by Brar et al. (2012). For RNA-seq, RNA was extracted from the clarified lysates using TRIzol (Invitrogen) and rRNA was depleted using Ribo-Zero Gold for Yeast (Illumina). The processed RNA was then used to generate TruSeq Stranded libraries (Illumina) following the manufacturer’s recommendations. Libraries of ribosome profiling and RNA-seq were sequenced on an Illumina HiSeq 4000 in single read 50-base mode. Each set of matched ribosome profiling and RNA-seq data are derived from a single biological sample (two biological replicates in total for each strain under each condition).
Sequencing data analysis
FASTX-clipper and -barcode splitter (http://hannonlab.cshl.edu/fastx_toolkit/) were used to remove the linker sequences and de-multiplex ribosome profiling data, respectively. Unique molecular identifiers and sample barcodes were subsequently removed using a custom Python script. Reads corresponding to rRNAs and tRNAs were excluded using Bowtie v1.1.2 (http://bowtie-bio.sourceforge.net/) and the remaining reads were aligned to Saccharomyces cerevisiae genome using tophat v2.1.1 (https://ccb.jhu.edu/software/tophat/). We then used the plastid cs program (Dunn and Weissman, 2016) to calculate counts per gene and normalized counts per gene (in reads per kilobase per million mapped reads, or RPKM), with counts assigned to the ribosome P-site determined by the plastid psite program. Genome regions that could not be uniquely mapped from a 26-base read with two mismatches were identified by the plastid crossmap program, which, together with the first 30 and last five codons of each coding sequence (CDS), were excluded from count assignments and RPKM calculations. RNA-seq data were analyzed similarly. However, because of the TruSeq Stranded chemistry, the reads had to be reverse-complemented prior to plastid analysis, and counts were assigned to the 5′-most aligned base. We used the plastid make wiggle program to generate Wiggle files from genome alignments for subsequent data visualization in the IGV browser (http://software.broadinstitute.org/software/igv/). For visualization purposes, Wiggle counts were assigned to the ribosome P-site for ribosome profiling, or equally apportioned across reads for RNA-seq. Dubious ORFs listed in the Saccharomyces Genome Database (SGD, https://www.yeastgenome.org/) were not considered for analysis.
Translation efficiency (TE) is defined as the ratio of normalized ribosome footprint counts to normalized mRNA counts. TE and mRNA fold changes and adjusted p values were calculated from raw counts with DESeq2 (Love et al., 2014). TE changes were calculated using the design formula ~sample + sample:assay, where the sample interaction term denotes the growth condition and genotype (e.g., SFM_E85Q) and the assay interaction term specifies whether counts are derived from RNA-seq or ribosome profiling. mRNA changes were calculated from RNA-seq counts using the design formula ~sample.
Metacodon analysis
Ribosome profiling data were processed to produce mean relative enrichment values for each codon as described in Hussmann et al. (2015). Briefly, footprint sequencing reads were trimmed of adaptor sequence, aligned to the yeast genome and spliced transcriptome with TopHat2 (Kim et al., 2013), and assigned to the codon positioned in the A-site of the footprint as in Ingolia et al. (2009). For each gene, the raw counts of uniquely mapped footprints with their A-site over each codon were normalized by dividing by the average count for all codons in that gene to produce a relative enrichment value for each codon. The mean relative enrichment for each codon type was then calculated by averaging the relative enrichment value at every occurrence of that codon type located at least 90 codons away from the start or stop codon of its gene. To reduce noise, genes with less than 0.1 mean footprints per codon were excluded from averaging.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed in GraphPad Prism (versions 6, 7, 8, and 9), with details provided in the corresponding figure legends, such as statistical tests employed, values and definition of n, and definition of center and dispersion. No methods were used to determine whether the data met assumptions of the statistical approach.
Supplementary Material
Highlights.
Two conserved adenosines in the 18S rRNA can be modified as either m6A or m62A
m6A levels increase under sulfur starvation in yeast and mammalian cell lines
m6A-bearing ribosomes translate distinctly from m62A-bearing ribosomes
Loss of methylation impairs translation fidelity and ribosome pausing/stalling
ACKNOWLEDGMENTS
We thank Dr. David Bedwell from University of Alabama at Birmingham for the generous gifts of pDB722 and pDB868. We also thank Dr. Juan Manuel Povedano for critically reading the manuscript and Venkat Malladi at the Bioinformatics Core Facility of UT Southwestern Medical Center for assistance with sequencing data analysis. J.S.W. is supported by the Howard Hughes Medical Institute. D.A.S. is supported by the National Science Foundation (1650113) and Moritz-Heyman Discovery Fellowship. J.A.H. is the Rebecca Ridley Kry Fellow of the Damon Runyon Cancer Research Foundation (DRG-2262-16). B.P.T. is supported by the Welch Foundation (I-1797) and NIH (grants R35GM136370 and R01NS115546).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following references appear in the supplemental information: Calvo et al. (1999); Gueldener et al. (2002); Keeling et al. (2004); Miller et al. (2013); Taoka et al. (2016); Voth et al. (2001).
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2021.108825.
REFERENCES
- Agarwala SD, Blitzblau HG, Hochwagen A, and Fink GR (2012). RNA methylation by the MIS complex regulates a cell fate decision in yeast. PLoS Genet. 8, e1002732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, and Tavazoie SF (2015a). HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell 162, 1299–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón CR, Lee H, Goodarzi H, Halberg N, and Tavazoie SF (2015b). N6-methyladenosine marks primary microRNAs for processing. Nature 519, 482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen TE, Porse BT, and Kirpekar F (2004). A novel partial modification at C2501 in Escherichia coli 23S ribosomal RNA. RNA 10, 907–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson D (1977). Cellular energy metabolism and its regulation (Academic Press; ), p. 75. [Google Scholar]
- Basu A, Das P, Chaudhuri S, Bevilacqua E, Andrews J, Barik S, Hatzoglou M, Komar AA, and Mazumder B (2011). Requirement of rRNA methylation for 80S ribosome assembly on a cohort of cellular internal ribosome entry sites. Mol. Cell. Biol. 31, 4482–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin-Baillieu A, Fabret C, Liang XH, Piekna-Przybylska D, Fournier MJ, and Rousset JP (2009). Nucleotide modifications in three functionally important regions of the Saccharomyces cerevisiae ribosome affect translation accuracy. Nucleic Acids Res. 37, 7665–7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellodi C, Kopmar N, and Ruggero D (2010a). Deregulation of oncogene-induced senescence and p53 translational control in X-linked dyskeratosis congenita. EMBO J. 29, 1865–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellodi C, Krasnykh O, Haynes N, Theodoropoulou M, Peng G, Montanaro L, and Ruggero D (2010b). Loss of function of the tumor suppressor DKC1 perturbs p27 translation control and contributes to pituitary tumorigenesis. Cancer Res. 70, 6026–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, de Crécy-Lagard V, Ross R, Limbach PA, Kotter A, et al. (2018). MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46 (D1), D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodi Z, Button JD, Grierson D, and Fray RG (2010). Yeast targets for mRNA methylation. Nucleic Acids Res. 38, 5327–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, and Rottman F (1994). Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J. Biol. Chem. 269, 17697–17704. [PubMed] [Google Scholar]
- Bokar JA, Shambaugh ME, Polayes D, Matera AG, and Rottman FM (1997). Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 3, 1233–1247. [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, and Weissman JS (2012). High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335, 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrgazov K, Vesper O, and Moll I (2013). Ribosome heterogeneity: another level of complexity in bacterial translation regulation. Curr. Opin. Microbiol. 16, 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo O, Cuesta R, Anderson J, Gutiérrez N, García-Barrio MT, Hinnebusch AG, and Tamame M (1999). GCD14p, a repressor of GCN4 translation, cooperates with Gcd10p and Lhp1p in the maturation of initiator methionyl-tRNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 19, 4167–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrillo JI, Hayes A, Mohammed S, Gaskell SJ, and Oliver SG (2003). An optimized protocol for metabolome analysis in yeast using direct infusion electrospray mass spectrometry. Phytochemistry 62, 929–937. [DOI] [PubMed] [Google Scholar]
- Clancy MJ, Shambaugh ME, Timpte CS, and Bokar JA (2002). Induction of sporulation in Saccharomyces cerevisiae leads to the formation of N6-methyladenosine in mRNA: a potential mechanism for the activity of the IME4 gene. Nucleic Acids Res. 30, 4509–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly K, Rife JP, and Culver G (2008). Mechanistic insight into the ribosome biogenesis functions of the ancient protein KsgA. Mol. Microbiol. 70, 1062–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrosiers R, Friderici K, and Rottman F (1974). Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 71, 3971–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinman JD (2016). Pathways to specialized ribosomes: the Brussels lecture. J. Mol. Biol. 428 (10 Pt B), 2186–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JG, and Weissman JS (2016). Plastid: nucleotide-resolution analysis of next-generation sequencing and genomics data. BMC Genomics 17, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel JD (1975). Mechanism of the Dimroth rearrangement in adenosine. Biochem. Biophys. Res. Commun. 64, 581–586. [DOI] [PubMed] [Google Scholar]
- Erales J, Marchand V, Panthu B, Gillot S, Belin S, Ghayad SE, Garcia M, Laforêts F, Marcel V, Baudin-Baillieu A, et al. (2017). Evidence for rRNA 2′-O-methylation plasticity: Control of intrinsic translational capabilities of human ribosomes. Proc. Natl. Acad. Sci. USA 114, 12934–12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genuth NR, and Barna M (2018). The discovery of ribosome heterogeneity and its implications for gene regulation and organismal life. Mol. Cell 71, 364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerashchenko MV, and Gladyshev VN (2014). Translation inhibitors cause abnormalities in ribosome profiling experiments. Nucleic Acids Res. 42, e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. (2015). m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 347, 1002–1006. [DOI] [PubMed] [Google Scholar]
- Ghalei H, Trepreau J, Collins JC, Bhaskaran H, Strunk BS, and Karbstein K (2017). The ATPase Fap7 tests the ability to carry out translocation-like conformational changes and releases Dim1 during 40S ribosome maturation. Mol. Cell 67, 990–1000.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Gilbert WV, Zhou K, Butler TK, and Doudna JA (2007). Cap-independent translation is required for starvation-induced differentiation in yeast. Science 317, 1224–1227. [DOI] [PubMed] [Google Scholar]
- Gokhale NS, McIntyre ABR, McFadden MJ, Roder AE, Kennedy EM, Gandara JA, Hopcraft SE, Quicke KM, Vazquez C, Willer J, et al. (2016). N6-methyladenosine in Flaviviridae viral RNA genomes regulates infection. Cell Host Microbe 20, 654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez B, François J, and Renaud M (1997). A rapid and reliable method for metabolite extraction in yeast using boiling buffered ethanol. Yeast 13, 1347–1355. [DOI] [PubMed] [Google Scholar]
- Gueldener U, Heinisch J, Koehler GJ, Voss D, and Hegemann JH (2002). A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 30, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcik M, and Sonenberg N (2005). Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 6, 318–327. [DOI] [PubMed] [Google Scholar]
- Hussain T, Llácer JL, Fernández IS, Munoz A, Martin-Marcos P, Savva CG, Lorsch JR, Hinnebusch AG, and Ramakrishnan V (2014). Structural changes enable start codon recognition by the eukaryotic translation initiation complex. Cell 159, 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussmann JA, Patchett S, Johnson A, Sawyer S, and Press WH (2015). Understanding biases in ribosome profiling experiments reveals signatures of translation dynamics in yeast. PLoS Genet. 11, e1005732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack K, Bellodi C, Landry DM, Niederer RO, Meskauskas A, Musalgaonkar S, Kopmar N, Krasnykh O, Dean AM, Thompson SR, et al. (2011). rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol. Cell 44, 660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefsen K, and Nielsen H (2011). Northern blotting analysis. In RNA: Methods and Protocols, Nielsen H, ed. (Humana Press; ), pp. 87–105. [DOI] [PubMed] [Google Scholar]
- Keeling KM, Lanier J, Du M, Salas-Marco J, Gao L, Kaenjak-Angeletti A, and Bedwell DM (2004). Leaky termination at premature stop codons antagonizes nonsense-mediated mRNA decay in S. cerevisiae. RNA 10, 691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy EM, Bogerd HP, Kornepati AV, Kang D, Ghoshal D, Marshall JB, Poling BC, Tsai K, Gokhale NS, Horner SM, and Cullen BR (2016). Posttranscriptional m6A editing of HIV-1 mRNAs enhances viral gene expression. Cell Host Microbe 19, 675–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King TH, Liu B, McCully RR, and Fournier MJ (2003). Ribosome structure and activity are altered in cells lacking snoRNPs that form pseudouridines in the peptidyl transferase center. Mol. Cell 11, 425–435. [DOI] [PubMed] [Google Scholar]
- King HA, Cobbold LC, and Willis AE (2010). The role of IRES trans-acting factors in regulating translation initiation. Biochem. Soc. Trans. 38, 1581–1586. [DOI] [PubMed] [Google Scholar]
- Komar AA, and Hatzoglou M (2011). Cellular IRES-mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle 10, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolev KS, Müller MJ, Karahan N, Murray AW, Hallatschek O, and Nelson DR (2012). Selective sweeps in growing microbial colonies. Phys. Biol. 9, 026008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine D, Delcour J, Glasser AL, Desgrès J, and Vandenhaute J (1994). The DIM1 gene responsible for the conserved m62Am62A dimethylation in the 3′-terminal loop of 18 S rRNA is essential in yeast. J. Mol. Biol. 241, 492–497. [DOI] [PubMed] [Google Scholar]
- Lafontaine D, Vandenhaute J, and Tollervey D (1995). The 18S rRNA dimethyl-lase Dim1p is required for pre-ribosomal RNA processing in yeast. Genes Dev. 9, 2470–2481. [DOI] [PubMed] [Google Scholar]
- Lafontaine DL, Preiss T, and Tollervey D (1998). Yeast 18S rRNA dimethy-lase Dim1p: a quality control mechanism in ribosome synthesis? Mol. Cell. Biol. 18, 2360–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laxman S, Sutter BM, Wu X, Kumar S, Guo X, Trudgian DC, Mirzaei H, and Tu BP (2013). Sulfur amino acids regulate translational capacity and metabolic homeostasis through modulation of tRNA thiolation. Cell 154, 416–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppek K, Das R, and Barna M (2018). Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 19, 158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Lee I, Moradi E, Hung NJ, Johnson AW, and Marcotte EM (2009). Rational extension of the ribosome biogenesis pathway using network-guided genetics. PLoS Biol. 7, e1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Liu Q, and Fournier MJ (2009). Loss of rRNA modifications in the decoding center of the ribosome impairs translation and strongly delays pre-rRNA processing. RNA 15, 1716–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman N, O’Brown ZK, Earl AS, Boulias K, Gerashchenko MV, Wang SY, Fritsche C, Fady PE, Dong A, Gladyshev VN, and Greer EL (2020). N6-adenosine methylation of ribosomal RNA affects lipid oxidation and stress resistance. Sci. Adv. 6, eaaz4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Parisien M, Dai Q, Zheng G, He C, and Pan T (2013). Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 19, 1848–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. (2014). A METTL3-METTL14 °Complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Dai Q, Zheng G, He C, Parisien M, and Pan T (2015). N6-methyl-adenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, and Pringle JR (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R, Chen K, Lu Z, Chen H, Shi YG, et al. (2019). N6-Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat. Chem. Biol. 15, 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macon JB, and Wolfenden R (1968). 1-Methyladenosine. Dimroth rearrangement and reversible reduction. Biochemistry 7, 3453–3458. [DOI] [PubMed] [Google Scholar]
- Marcel V, Ghayad SE, Belin S, Therizols G, Morel AP, Solano-Gonzàlez E, Vendrell JA, Hacot S, Mertani HC, Albaret MA, et al. (2013). p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell 24, 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mašek T, Valášek L, and Pospíšek M (2011). Polysome analysis and RNA purification from sucrose gradients. Methods Mol. Biol. 703, 293–309. [DOI] [PubMed] [Google Scholar]
- McGlincy NJ, and Ingolia NT (2017). Transcriptome-wide measurement of translation by ribosome profiling. Methods 126, 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B, Wurm JP, Sharma S, Immer C, Pogoryelov D, Kötter P, Lafontaine DL, Wöhnert J, and Entian KD (2016). Ribosome biogenesis factor Tsr3 is the aminocarboxypropyl transferase responsible for 18S rRNA hypermodification in yeast and humans. Nucleic Acids Res. 44, 4304–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen MD, Buron LD, Salomonsen B, Olsen CE, Hansen BG, Mortensen UH, and Halkier BA (2012). Microbial production of indolylglu-cosinolate through engineering of a multi-gene pathway in a versatile yeast expression platform. Metab. Eng. 14, 104–111. [DOI] [PubMed] [Google Scholar]
- Miller AW, Befort C, Kerr EO, and Dunham MJ (2013). Design and use of multiplexed chemostat arrays. J. Vis. Exp. 72, e50262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y, and Helm M (2011). RNA nucleotide methylation. Wiley Interdiscip. Rev. RNA 2, 611–631. [DOI] [PubMed] [Google Scholar]
- O’Farrell HC, Musayev FN, Scarsdale JN, and Rife JP (2010). Binding of adenosine-based ligands to the MjDim1 rRNA methyltransferase: implications for reaction mechanism and drug design. Biochemistry 49, 2697–2704. [DOI] [PubMed] [Google Scholar]
- Peifer C, Sharma S, Watzinger P, Lamberth S, Kötter P, and Entian KD (2013). Yeast Rrp8p, a novel methyltransferase responsible for m1A 645 base modification of 25S rRNA. Nucleic Acids Res. 41, 1151–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, and Conrad NK (2017). The U6 snRNA m6A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell 169, 824–835.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulicherla N, Pogorzala LA, Xu Z, O Farrell HC, Musayev FN, Scarsdale JN, Sia EA, Culver GM, and Rife JP (2009). Structural and functional divergence within the Dim1/KsgA family of rRNA methyltransferases. J. Mol. Biol. 391, 884–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries RJ, Zaccara S, Klein P, Olarerin-George A, Namkoong S, Pickering BF, Patil DP, Kwak H, Lee JH, and Jaffrey SR (2019). m6A enhances the phase separation potential of mRNA. Nature 571, 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rife JP (2009). Roles of the ultra-conserved ribosomal RNA methyltransferase KsgA in ribosome biogenesis. In DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution, Grosjean H, ed. (Landes Bioscience; ), pp. 512–526. [Google Scholar]
- Salas-Marco J, and Bedwell DM (2005). Discrimination between defects in elongation fidelity and termination efficiency provides mechanistic insights into translational readthrough. J. Mol. Biol. 348, 801–815. [DOI] [PubMed] [Google Scholar]
- Sankar TS, Wastuwidyaningtyas BD, Dong Y, Lewis SA, and Wang JD (2016). The nature of mutations induced by replication–transcription collisions. Nature 535, 178–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos DA, Shi L, Tu BP, and Weissman JS (2019). Cycloheximide can distort measurements of mRNA levels and translation efficiency. Nucleic Acids Res. 47, 4974–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schosserer M, Minois N, Angerer TB, Amring M, Dellago H, Harreither E, Calle-Perez A, Pircher A, Gerstl MP, Pfeifenberger S, et al. (2015). Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat. Commun. 6, 6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, and Lafontaine DLJ (2015). ‘View from a bridge’: A new perspective on eukaryotic rRNA base modification. Trends Biochem. Sci. 40, 560–575. [DOI] [PubMed] [Google Scholar]
- Sharma S, Watzinger P, Kötter P, and Entian KD (2013). Identification of a novel methyltransferase, Bmt2, responsible for the N-1-methyl-adenosine base modification of 25S rRNA in Saccharomyces cerevisiae. Nucleic Acids Res. 41, 5428–5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Stoute J, and Liu KF (2020). Structural and catalytic roles of the human 18S rRNA methyltransferases DIMT1 in ribosome assembly and translation. J. Biol. Chem. 295, 12058–12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan KE, Warda AS, Sharma S, Entian KD, Lafontaine DLJ, and Bohnsack MT (2017). Tuning the ribosome: The influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 14, 1138–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spriggs KA, Stoneley M, Bushell M, and Willis AE (2008). Re-programming of translation following cell stress allows IRES-mediated translation to predominate. Biol. Cell 100, 27–38. [DOI] [PubMed] [Google Scholar]
- Tafforeau L, Zorbas C, Langhendries JL, Mullineux ST, Stamatopoulou V, Mullier R, Wacheul L, and Lafontaine DL (2013). The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of Pre-rRNA processing factors. Mol. Cell 51, 539–551. [DOI] [PubMed] [Google Scholar]
- Taoka M, Nobe Y, Yamaki Y, Yamauchi Y, Ishikawa H, Takahashi N, Nakayama H, and Isobe T (2016). The complete chemical structure of Saccharomyces cerevisiae rRNA: partial pseudouridylation of U2345 in 25S rRNA by snoRNA snR9. Nucleic Acids Res. 44, 8951–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesina P, Heckel E, Cheng J, Fromont-Racine M, Buschauer R, Kater L, Beatrix B, Berninghausen O, Jacquier A, Becker T, and Beckmann R (2019). Structure of the 80S ribosome-Xrn1 nuclease complex. Nat. Struct. Mol. Biol. 26, 275–280. [DOI] [PubMed] [Google Scholar]
- Tu BP, Mohler RE, Liu JC, Dombek KM, Young ET, Synovec RE, and McKnight SL (2007). Cyclic changes in metabolic state during the life of a yeast cell. Proc. Natl. Acad. Sci. USA 104, 16886–16891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, and Pilpel Y (2010). An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141, 344–354. [DOI] [PubMed] [Google Scholar]
- van Dijken JP, Bauer J, Brambilla L, Duboc P, Francois JM, Gancedo C, Giuseppin ML, Heijnen JJ, Hoare M, Lange HC, et al. (2000). An interlaboratory comparison of physiological and genetic properties of four Saccharomyces cerevisiae strains. Enzyme Microb. Technol. 26, 706–714. [DOI] [PubMed] [Google Scholar]
- Van Knippenberg PH, Van Kimmenade JM, and Heus HA (1984). Phylogeny of the conserved 30 terminal structure of the RNA of small ribosomal subunits. Nucleic Acids Res. 12, 2595–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tran N, Ernst FGM, Hawley BR, Zorbas C, Ulryck N, Hackert P, Bohnsack KE, Bohnsack MT, Jaffrey SR, Graille M, and Lafontaine DLJ (2019). The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 47, 7719–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Haar T (2008). A quantitative estimation of the global translational activity in logarithmically growing yeast cells. BMC Syst. Biol. 2, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth WP, Richards JD, Shaw JM, and Stillman DJ (2001). Yeast vectors for integration at the HO locus. Nucleic Acids Res. 29, E59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, and He C (2015). N6-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner JR (1999). The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 24, 437–440. [DOI] [PubMed] [Google Scholar]
- Wejksnora PJ, and Haber JE (1974). Methionine-dependent synthesis of ribosomal ribonucleic acid during sporulation and vegetative growth of Saccharomyces cerevisiae. J. Bacteriol. 120, 1344–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S, Tian S, Fujii K, Kladwang W, Das R, and Barna M (2015). RNA regulons in Hox 5′ UTRs confer ribosome specificity to gene regulation. Nature 517, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon A, Peng G, Brandenburger Y, Zollo O, Xu W, Rego E, and Ruggero D (2006). Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 312, 902–906. [DOI] [PubMed] [Google Scholar]
- Yue Y, Liu J, and He C (2015). RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 29, 1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenklusen D, Larson DR, and Singer RH (2008). Single-RNA counting reveals alternative modes of gene expression in yeast. Nat. Struct. Mol. Biol. 15, 1263–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A, Ho RK, and He C (2017). m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542, 475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, and Qian SB (2015). Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 526, 591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorbas C, Nicolas E, Wacheul L, Huvelle E, Heurgué-Hamard V, and Lafontaine DL (2015). The human 18S rRNA base methyltransferases DIMT1L and WBSCR22-TRMT112 but not rRNA modification are required for ribosome biogenesis. Mol. Biol. Cell 26, 2080–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data have been deposited in Gene Expression Omnibus with the accession number GEO: GSE142528.