

Abstract
Protein phosphorylation is one of the most prevalent posttranslational modifications found in eukaryotic systems. It serves as a key molecular mechanism that regulates protein function in response to environmental stimuli. The Mut9-like kinases (MLKs) are a plant-specific family of Ser/Thr kinases linked to light, circadian, and abiotic stress signaling. Here we use quantitative phosphoproteomics in conjunction with global proteomic analysis to explore the role of the MLKs in daily protein dynamics. Proteins involved in light, circadian, and hormone signaling, as well as several chromatin-modifying enzymes and DNA damage response factors, were found to have altered phosphorylation profiles in the absence of MLK family kinases. In addition to altered phosphorylation levels, mlk mutant seedlings have an increase in glucosinolate metabolism enzymes. Subsequently, we show that a functional consequence of the changes to the proteome and phosphoproteome in mlk mutant plants is elevated glucosinolate accumulation and increased sensitivity to DNA damaging agents. Combined with previous reports, this work supports the involvement of MLKs in a diverse set of stress responses and developmental processes, suggesting that the MLKs serve as key regulators linking environmental inputs to developmental outputs.
Keywords: quantitative phosphoproteomics, kinase, arabidopsis, circadian, DNA damage, quantitative proteomics, plants
Abbreviations: ABA, abscisic acid; BRM, Brahma; CCA1, circadian clock associated; DDR, DNA damage repair; FDR, false discover rate; GLS, glucosinolate; GO, gene ontology; HY5, elongated hypocotyl 5; MLK, Mut9-like kinase; phyB, phytochrome B; RVE8, Reveille 8; SYD, SPLAYED; TMT, tandem mass tag; ZT12, Zeitgeber 12; ZT14, Zeitgeber 14
Graphical abstract
Highlights
-
•
MUT9-LIKE KINASE mutant quantitative proteome and phosphoproteome measured.
-
•
Changes to proteome and phosphoproteome are specific to genotype and environment.
-
•
Loss of MLKs alters glucosinolate enzyme abundance and metabolism.
-
•
Loss of MLKs increases plant sensitivity to UV radiation and DNA damage agents.
In Brief
The MUT9-like kinases are a family of plant-specific nuclear-localized kinases with roles in diverse signaling pathways, including light sensing, phytohormone perception, and the circadian clock. The proteome and phosphoproteome of compound mlk mutant seedlings have been determined under light and dark conditions. These experiments identify new roles for these kinases regulating secondary plant metabolism and stress responses, tested through metabolite analysis and assaying seedling sensitivity to DNA damaging agents.
Protein phosphorylation is a dynamic posttranslational modification that is key in the regulation of protein function and turnover, making it an integral part of complex signaling networks. Rapid and reversible posttranslational regulation is advantageous to plants, as they are often required to adapt to changing environments quickly. Moreover, protein phosphorylation is at the core of various biological processes, including stress response, light signaling, circadian regulation, and hormone perception and transduction. In Arabidopsis, nearly 4% of protein-encoding genes are kinases (1), which is a testament to the importance of phosphorylation-based protein regulation (2). Despite the upswing of large-scale phosphoproteomic studies in plant species (2, 3), a recent study suggests that the identification of Arabidopsis phosphoproteins and phosphosites is far from comprehensive (4).
The four-member family of Ser/Thr protein kinases known as the MUT9-like kinase (MLK)/Photoregulatory Protein Kinases/Arabidopsis EL1-like kinases, herein referred to as the MLKs, are involved in the phosphoregulation of several key signaling proteins (5, 6, 7, 8). The MLKs are a plant and green algae-specific family of kinases related to casein kinase I. The MLKs show significant divergence from casein kinase I, with similarities restricted to their catalytic domains (9). MLK family kinases are capable of phosphorylating histones H3 and H2A in Arabidopsis (10, 11) as well as the green algae Chlamydomonas (9). The Chlamydomonas MLK ortholog, MUT9, is also required for transgene silencing and response to DNA damaging agents (9, 12). In addition to phosphorylating histones, MLK family kinases phosphorylate proteins involved in multiple signaling pathways. Early studies of a rice MLK ortholog, early flowering1, have linked this kinase family to hormone signaling and the regulation of flowering time (13), a role which is at least in part conserved in Arabidopsis (8, 11, 14, 15, 16). MLKs also interact with core components of the morning (7, 14) and evening (15) loops of the Arabidopsis circadian clock. The association of the MLKs with the evening complex components, early flowering 3 and 4, is dependent on the presence of the red light receptor phytochrome B (15). Additionally, the MLKs phosphorylate the blue light receptor cryptochrome2 and the red light-regulated transcription factor phytochrome interacting factor 3 (5, 6). In sum, these studies suggest that the MLKs provide a link between light and circadian signaling, which in turn regulates plant growth and development.
In this study, we used quantitative phosphoproteomic techniques to expand our understanding of the various signaling pathways and cellular protein networks regulated by the MLK family of kinases. We combined high pH reversed-phase prefractionation and TiO2-based phosphopeptide enrichment with isobaric labeling to achieve an in-depth quantitative phosphoproteomic analysis of WT and mlk mutant seedlings at two different time points, one at the end of the day [Zeitgeber 12 (ZT12)] and the other several hours into the night [Zeitgeber 14 (ZT14)]. We identified over 20,000 phosphosites mapping to nearly 5000 protein groups. Notably, MLK mutant seedlings have altered abundance of glucosinolate (GLS) metabolism enzymes and differential phosphorylation of proteins involved in a diverse set of biological processes, including RNA processing, chromatin organization, and stress responses. The confluence of stress and chromatin factors suggested that MLKs may also regulate DNA-damage responses in Arabidopsis thaliana, which was tested by assessing the sensitivity of mlk mutants to DNA-damaging agents.
Experimental Procedures
Plant Material
The mlk1 (SALK_002211; AT5G18190), mlk2 (SALK_064333; AT3G03940), and mlk3 (SALK_017102; AT2G25760) mutant lines were obtained from the ABRC (Ohio State University). The mlk4 (GABI_756G08; AT3G13670) mutant line was obtained from the Nottingham Arabidopsis Stock Centre. All are in the Colombia (Col-0) background and were isolated as previously described (15).
Experimental Design and Statistical Rationale
For each of the two time points (ZT12 and ZT14), three biological replicate samples from WT (control), mlk123 mutant, and mlk134 mutant lines were collected and analyzed for a total of nine samples per time point. A 10th reference pooled sample composed of equal amounts of material from all samples was also generated to link tandem mass tag (TMT) experiments. Peptide identification was searched against a TAIR10 database with false discover rate (FDR) set to 1%, and peptide abundance changes were set with a statistical significance that was determined by Student’s t test (p-value ≤ 0.05).
Tissue Collection for Mass Spectrometry
Arabidopsis WT (Col-0) and mutant seedlings were grown on sterilized qualitative filter paper (Whatman) overlaid on ½ x MS (Murashige and Skoog) plates containing 1% sucrose and 0.8% agar at 22 °C. Seedlings were entrained under 12 h white light (100–110 μmol/m2/s)/12 h dark cycle. Tissue was collected on the 10th day of growth immediately before lights off (ZT12) or after 2 h of dark (ZT14).
Protein Isolation and Digestion
The seedlings were transferred into a liquid N2 chilled 35 ml ball mill and disrupted in a reciprocal mixer mill [30 Hz, 45 s, repeated three times (Retsch USA)] under liquid nitrogen. Ground tissue was gently resuspended in 1 ml (approximately 1 packed tissue volume) of SII buffer (100 mM sodium phosphate, pH 8.0, 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 0.1% Triton X-100, 1 mM PMSF, 1x protease inhibitor cocktail [Roche], 1x phosphatase inhibitors II & III [Sigma], and 50 μM Mg-132 [Peptides International]) and sonicated twice at 40% power, 1 s on/off cycles for 20 s total on ice (Fisher Scientific model FB505, with microtip probe). Extracts were clarified by centrifugation twice at 4 °C for 10 min at ≥20,000g, then subjected to cold acetone precipitation to remove buffer contaminants and lipophilic metabolite contaminants. Protein concentrations were determined by BCA protein assay (Thermo Fisher Scientific). Protein samples were reduced with 10 mM tris(2-carboxyethyl)phosphine and alkylated with 25 mM iodoacetamide before trypsin digestion in 1/40 enzyme/protein ratio at 37 °C overnight. Finally, samples were desalted with a C18 column before dry down.
Phosphopeptide Enrichment
Phosphopeptide enrichment was performed using the High-Select TiO2 Phosphopeptide Enrichment kit (Thermo Scientific PN32993) following the vendor’s protocol. Briefly, dried peptides were reconstituted in 150 μl of binding/equilibration buffer provided and applied to the TiO2 spin that was previously equilibrated with binding buffer/equilibration. After reapplying sample once, the tip was sequentially washed twice with 20 μl of binding buffer and wash buffer and once with 20 μl of LC-MS grade water. Bound peptides were eluted by two applications of 50 μl of elution buffer (also provided). Eluates containing the enriched phosphopeptides were dried down and subsequently resuspended with 50 μl 0.1% formic acid for peptide concentration measurement using the Pierce Quantitative Colorimetric Assay kit (Thermo Scientific PN23275).
Tandem Mass Tag Labeling
Hundred microgram of each digested sample (both whole proteome or after phosphopeptide enrichment) was added to 100 μl of 100 mM Hepes pH 8.5 buffer. A reference pooled sample composed of equal amounts of material from all samples was also generated to link TMT experiments. Isobaric labeling of the samples was performed using 10-plex TMT reagents (Thermo Fisher Scientific). All individual and pooled samples were labeled according to the TMT 10-plex reagent kit instructions. Briefly, TMT reagents were brought to room temperature and dissolved in anhydrous acetonitrile. Peptides were labeled by the addition of each label to its respective digested sample. Labeling reactions were incubated for 1 h at room temperature. Reactions were terminated with the addition of hydroxylamine.
High pH Reverse Phase Fractionation
High pH reverse phase fractionation was performed using the Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo Scientific PN84868) according to the manufacturer’s instructions. Briefly, peptide samples were dissolved in 300 μl of 0.1% TFA solution in LC-MS grade water and subsequently loaded onto reversed-phase fractionation spin columns also equilibrated with 0.1% TFA. Samples were then washed with 300 μl of 5% acetonitrile/0.1% TFA to remove unreacted TMT reagent. Peptides were eluted into eight peptide fractions with an acetonitrile step gradient (i.e., 10%, 12.5%, 15%, 17.5%, 20%, 22.5%, 25%, and 50%). Samples were acidified and dried down before LC-MS.
LC-MS/MS Analysis
Two microliters (1 μg) of each sample was injected onto a 0.075 × 500 mm EASY-Spray Pepmap C18 column equipped with a 0.100 × 5 mm EASY-Spray Pepmap C18 trap column (Thermo Fisher Scientific) attached to an EASY-nLC 1000 (Thermo Fisher Scientific). The peptides were separated using water (A) and acetonitrile (B) containing 0.1% formic acid as solvents at a flow rate of 300 nl per minute with a 3-h gradient. Data were acquired in positive ion data-dependent mode on an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) with a resolution of 120,000 (at m/z 200) and a scan range from m/z 380 to 1500. Precursor isolation was performed using the quadrupole before either collision-induced dissociation activation in the ion trap and detection in the Orbitrap at a resolution of 30,000 or higher-energy collisional dissociation activation with detection in the Orbitrap at a resolution of 60,000.
Data Analysis
All MS/MS data were analyzed using Proteome Discoverer 2.1 (Thermo Fisher Scientific). The search algorithm used in the study was Byonic v2.11 as part of the Proteome Discoverer software platform. Precursor mass tolerance was set to 10 ppm, fragment ion tolerance was 20 ppm while assuming the digestion enzyme trypsin and allowing up to two missed cleavages. Carbamidomethylation (+57.021 Da) on cysteine and TMT tag (+229.163 Da) on peptide N termini as well as lysine residue were set as static modifications. Dynamic modifications included acetylation (+42.011 Da) on protein N termini, oxidation (+15.995 Da) on methionine and phosphorylation (+79.966 Da) on serine, threonine, and tyrosine. Data were searched against the TAIR10 database (20101214, 35,386 entries) with FDR set to 1%. Note that the FDR in a list of identifications (either proteins or peptide sequences) is the number of incorrect identifications divided by the total number of identifications.
For quantitation, reporter ion intensity integration tolerance was set to 20 ppm. Reporter ion abundances were corrected for isotopic impurities based on manufacturer’s specifications. For each peptide, a minimal average reporter signal to noise threshold of 2 and a co-isolation threshold of 100% are required. The signal to noise values for all peptides were summed within each TMT channel, and each channel was scaled according to the reference channel. Both unique and razor peptides were used for quantification.
Peptides of altered abundance were identified from the Byonic output list generated from higher-energy collisional dissociation MS2 analysis using Microsoft Excel. Abundance ratios for mutant/WT pairwise comparisons were calculated from the average peptide abundance of mutant and WT biological replicates. Only peptides identified in at least two biological replicates were considered for further analysis. Statistical significance was determined by Student’s t test (p-value ≤ 0.05).
Bioinformatic Analysis
The Motif-X algorithm (17) was used to extract significantly enriched phosphorylation motifs from mlk1/2/3 and mlk1/3/4 phosphopeptide datasets. Only phosphopeptides with high confidence phosphorylation sites were used in the analysis. The peptides were aligned and extended to a width of 15 amino acids using the online utility PEPTIDEXTENDER ver.0.2.2 alpha (schwartzlab.uconn.edu/pepextend/). The aligned peptides were used to extract motifs. The probability threshold was set to p-value ≤10−5; the occurrence threshold was set to 10. The default IPI Arabidopsis Proteome dataset was used as the background dataset.
Enrichment analysis of gene ontology (GO) categories was performed with g:Profiler (18). AGI accession numbers for Arabidopsis were uploaded to the g:Profiler web server (http://biit.cs.ut.ee/gprofiler/), and GO enrichment was determined using default settings (significance level 0.05). Enriched terms were summarized, and redundancy removed using the online tool REVIGO (19). Semantic similarity threshold (dispensability) was set to 0.5 (default) for all global proteome analysis and cellular component category of the phosphoproteome analysis. Dispensability was increased to 0.7 for all other analyses.
Glucosinolate Extraction and Analysis by HPLC and LC-MS/MS
Arabidopsis seeds (Col-0, mlk 1/2/3, and mlk 1/3/4) were sown on ½ x MS (Murashige and Skoog) plates containing 1% sucrose and 0.8% agar and grown under 12 h white light (100–110 μmol/m2/s)/12 h dark cycle at 22 °C for 10 days before harvesting at ZT12. GLSs were extracted from approximately 350 mg of whole seedlings and desulfonated (in quadruplicate) as previously described (20) using sinigrin as an internal standard. Desulfo-GLS extracts were analyzed by HPLC (Waters) equipped with a photodiode array detector and separated using a Gemini C-18 column (150 × 2.00 mm, 5 μm; Phenomenex) with a flow rate of 0.5 ml per minute and the following solvents and binary gradient: solvent A-water and solvent B-acetonitrile; where solvent B was held at 1.5% for 1 min, then 1 to 6 min 1.5 to 5% B, 6 to 8 min 5 to 7% B, 8 to 18 min 7 to 21% B, 18 to 23 min 21 to 29% B, 23 to 30 min 29 to 43% B, 30 to 33 min 43 to 100% B, 33 to 37 100% B, 37 to 38 min 100 to 1.5% B, and held at 1.5% B for an additional 7 min. GLS peaks were identified using previously published UV spectra in addition to reported relative retention times and quantitated using peak areas of desulfo-GLS and internal standard along with published response factors (21, 22). GLS identities were confirmed by LC-MS/MS (SCIEX 6500 QTRAP) using enhanced product ion (ion trap MS/MS) scans to verify the presence of previously published fragment ions (20) from each GLS ion. Mass spectrometric data were collected in positive ion mode using the same gradient/solvents/column as for HPLC-UV analysis with the following source conditions: curtain gas, 20; ion-spray voltage, 5500 V; temperature, 500 °C; gas 1, 40; gas 2, 45; declustering potential, 80 V; entrance potential, 10 V; collision energy 20 eV.
Methyl Methanesulfonate Treatment
Arabidopsis WT (Col-0) and mutant seed was surface-sterilized and sown on ½ x MS (Murashige and Skoog) plates containing 1% sucrose and 0.8% agar with or without methyl methanesulfonate (MMS, Sigma). After stratification for 2 days at 4 °C, seedlings were grown under 12 h white light (100–110 μmol/m2/s)/12 h dark cycle at 22 °C. For growth sensitivity assays, seedlings were germinated on control media, and after 5 days of growth, they were transferred to MMS treatment media. Fresh weight was measured after 15 days of growth in the presence of MMS. For postgermination developmental assessment, the seed was germinated on ½ x MS plates containing 1% sucrose, 0.8% agar, and 150 ppm MMS. Seedlings were imaged and scored for arrest at 12 days after germination.
UV-C Tolerance Assay
Whole-plant sensitivity to UV-C (254 nm) was evaluated as described in Castells et al. (23) with the following modifications. Eight-day-old seedlings were irradiated with 2000 or 4000 J m−2 of UV-C twice during a 48 h time period using a Stratalinker UV Crosslinker 1800. Following each treatment, plants were returned to growth conditions of 12 h white light (100–110 μmol/m2/s)/12 h dark cycle at 22 °C. Seedlings were imaged after 5 days of recovery, and their phenotypes were measured.
Results
Proteomic Analysis Reveals Changes in Stress Response Pathways in mlk Mutant Seedlings
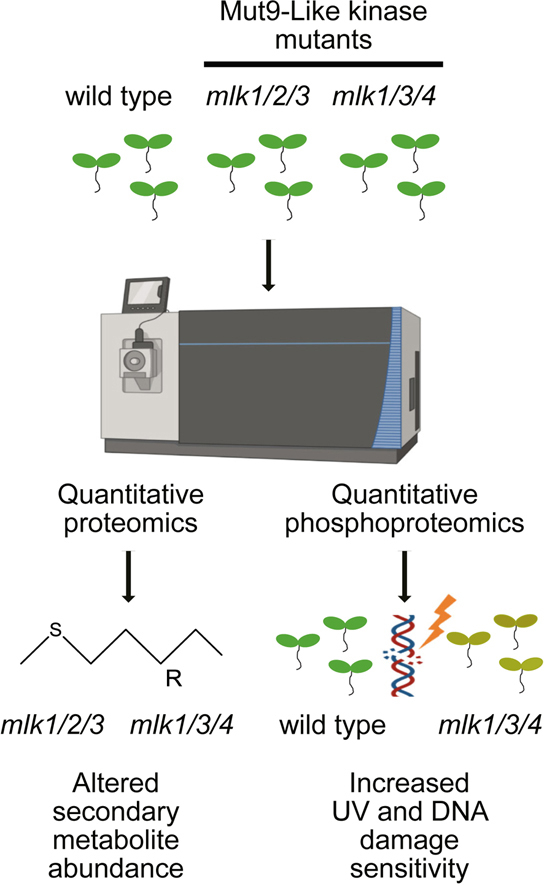
We measured the regulatory effects of the MLKs on proteome dynamics by TMT labeling combined with high pH reversed-phase fractionation and tandem mass spectrometry (Fig. 1). WT and mlk mutant Arabidopsis seedlings were entrained under 12 h light and 12 h dark conditions. We compared mlk1/2/3 and mlk1/3/4 mutant seedlings, as we were unable to isolate viable mlk1/2/3/4 mutant seed (6, 15). This mutant combination will allow us to assess potential redundancy within the MLK family and facilitate the identification of mlk2- or mlk4-specific changes. As the MLKs are associated with circadian and light-signaling pathways (15), we collected tissue near the dusk transition when the co-precipitating proteins from the evening complex, early flowering 3 and early flowering 4, peak. To capture changes associated with light-signaling during the light/dark transition at dusk, we sampled both immediately before lights off (ZT12) and after 2 h of dark (ZT14) to allow for photoreceptors to revert to their inactive state (24, 25). We identified nearly 50,000 peptides combined, mapping to over 7500 protein groups at both ZT12 and ZT14. Pairwise comparisons between mlk mutants and WT identified peptides showing altered abundance in the mlk mutant backgrounds at both the ZT12 and ZT14 time-points (supplemental Dataset S1). Peptides were classified as altered in abundance if both the log2 FC was at least ±1 (2 fold-change), and the p-value was <0.05. Only 13 unique proteins met our altered abundance criteria in the mlk1/2/3 mutant when compared with WT at both ZT12 and ZT14 (Fig. 2 and supplemental Table S1). In the mlk1/3/4 mutant background, more than 225 peptides mapping to over 110 unique proteins met our altered abundance threshold at ZT12 and ZT14 when compared with WT (Fig. 2 and supplemental Table S1). These results suggest that the mlk1/3/4 mutant combination has a greater impact on the global proteome than the mlk1/2/3 mutant combination at the light-to-dark transition.
Fig. 1.

Schematic representation of the quantitative proteomics workflow. Tissue samples were collected at ZT12 and ZT14 from WT and mutant seedlings entrained with a 12L:12D light/dark cycle (1). Total protein was extracted and digested (2). Following TMT10plex isobaric labeling (3), samples were subjected to high pH reversed phase prefractionation (5). Phosphopeptides were enriched using a TiO2-based method (4). Both phosphopeptide-enriched and global samples were analyzed by LC-MS/MS (6). ZT 12, Zeitgeber 12; ZT 14, Zeitgeber 14.
Fig. 2.

Global proteomic analysis of mlk mutant seedlings. Volcano plots of peptides identified in mutant and WT seedlings at ZT12 and ZT14. The x-axis specifies the log2 fold-change (FC) of mutant/WT, and the y-axis specifies the negative logarithm to the base 10 of the t test p-values. Open circles represent individual peptides, with blue circles specifying those considered statistically significant. Black vertical and horizontal lines reflect the filtering criteria (log2 FC = ±1 and p-value = 0.05) for significance. ZT 12, Zeitgeber 12; ZT 14, Zeitgeber 14.
In silico classification using gene ontology analysis (https://biit.cs.ut.ee/gprofiler/) revealed that proteins exhibiting altered abundance were associated with biotic and abiotic stresses (Fig. 3). To simplify the enriched GO term lists and focus on the most relevant terms, we performed additional analysis using REVIGO (default settings, dispensability threshold = 0.7) to remove functionally redundant terms (19). We found increased abundance of proteins involved in GLS biosynthesis (GO:0019761) and related processes (GO:1901659, GO:0016143, and GO:0044272) in mlk1/3/4 mutant seedlings at ZT12 and in both mlk1/2/3 and mlk1/3/4 mutant seedlings at ZT14 (Fig. 3). Nearly 75% of the peptides with an increased abundance of 3-fold or greater in the mlk1/3/4 mutant seedlings at ZT14 mapped to proteins directly involved in GLS biosynthesis (supplemental Dataset S1). These proteins include enzymes responsible for the early reactions leading to methionine-derived GLSs (branched-chain aminotransferase 4 and methylthioalkymalate synthase 1) as well as, desulfo-glucosinolate sulfotransferase 17 and 18, which are involved in the final step of GLS core structure biosynthesis (26). Other proteins involved in GLS biosynthesis that showed increased abundance in mlk1/3/4 mutants compared with WT include isopropylmalate dehydrogenase 1, isopropylmalate isomerase 2, 2-isopropylmalate synthase 2, flavin-monooxygenase glucosinolate S-oxygenase 1, and the cytochrome P450 proteins CYP83A1 and CYP79F1 (supplemental Dataset S1). Proteins involved in GLS catabolism, such as glucoside glucohydrolase 2, nitrile specifier protein 1, and beta glucosidase 34 and 35, were decreased in abundance in mlk1/3/4 mutant seedlings at ZT12 compared with WT.
Fig. 3.

Gene ontology (GO) enrichment analysis. GO enrichment analysis of proteins identified as having increased (A) or decreased (B) abundance in mlk mutant seedlings at indicated ZTs when compared with WT. Cluster representative GO terms identified with REVIGO (semantic similarity threshold <0.7) in the category of Biological Process are shown. ZT, Zeitgeber.
To begin testing the hypothesis that MLKs are involved in the regulation of GLS metabolism, we quantified GLS levels at the end of day (ZT12) when GLS levels peak (27). Total GLSs were extracted from whole seedlings and analyzed by HPLC. Peaks corresponding to individual GLSs were identified by comparison with published UV absorbance spectra and expected retention times, and the identities were further validated using LC-MS/MS. These analyses revealed an increase in aliphatic GLSs (Met-derived) in both mlk1/2/3 and mlk1/3/4 mutant seedlings compared with WT. In contrast, the levels of indolic GLSs (Trp-derived) remained unchanged in the mutant backgrounds (Fig. 4). Interestingly, the first seven GLSs originating from the earliest steps in the Met-derived GLS biosynthetic pathway were increased 2- to 4-fold over WT (Fig. 4), a pattern which correlates with the increased abundance of GLS-associated biosynthetic enzymes (branched-chain aminotransferase 4, methylthioalkymalate synthase 1, desulfo-glucosinolate sulfotransferase 17/18, etc.) in the mlk mutants. Together these findings support a role for the MLKs in early stages of aliphatic GLS biosynthesis and overall GLS metabolism.
Fig. 4.

mlk mutant seedlings contain elevated levels of Met-derived glucosinolates. Glucosinolate (GLS) content of 10-day-old mutant and WT seedlings at ZT12 was quantified using HPLC. GLS identity determined using UV spectra and confirmed by LC-MS/MS. The average of four biological replicates is presented. Error bars indicate standard deviation. ∗p < 0.01, compared with WT seedlings (Student’s t test). ZT 12, Zeitgeber 12.
In addition to GLS biosynthetic enzymes, several proteins involved in hormone signaling and diverse stress responses changed in abundance in mlk1/3/4 mutant seedlings compared with WT at either ZT12 or ZT14, including BRI1-EMS-suppressor 1, super sensitive to abscisic acid (ABA) and DROUGHT2, coronatine induced 1, pathogenesis-related gene 5, lipoxygenase 2, thylakoidal ascorbate peroxidase, and cold regulated 15a and b. Peptides mapping to the blue light receptor cryptochrome 2 also increased 2-fold in the mlk1/3/4 mutant when compared with WT at ZT12 (supplemental Dataset S1). These observations are in agreement with the role of MLKs in hormone signaling, stress response, and light signaling (5, 6, 8, 9, 13).
Quantitative Phosphoproteomic Comparisons of mlk Mutants
The MLKs physically interact with and phosphorylate important regulatory proteins (5, 6, 8, 13). Thus, we characterized the phosphoproteome of WT, mlk1/2/3, and mlk1/3/4 mutant seedlings in the light (ZT12) and after transition to dark (ZT14) to gain insight into the role of the MLKs in global phosphorylation. We applied a TiO2-based phosphopeptide enrichment technique to the TMT 10-plex labeled samples described above to achieve an in-depth phosphoproteomic analysis using the Thermo Scientific Orbitrap Fusion Lumos Tribrid mass spectrometer (Fig. 1). Byonic software, run as a node within the Proteome Discover V2.1 platform identified and derived the relative quantitation of phosphoproteins. Using this strategy, we identified a combined total of 23,386 phosphosites on 15,222 unique peptides mapping to 4854 protein groups at ZT12 and slightly fewer at ZT14 (19,947 phosphosites on 12,818 unique peptides mapping to 4467 protein groups). At ZT12, over 80% of the identified phosphosites were serine residues, approximately 15% were threonine, and less than 2% were tyrosine (Fig. 5A), which is consistent with phosphosite distributions previously reported for Arabidopsis (28, 29). The phosphosite residue distributions were similar at ZT14 (Fig. 5A).
Fig. 5.

Analysis of quantitative phosphoproteomics of mlk mutant seedlings. A, the distribution of threonine (T), serine (S), and tyrosine (Y) phosphorylation sites identified at ZT12 and ZT14. B, volcano plot of phosphopeptides identified in mutant and WT seedlings at ZT12 and ZT14. The x-axis specifies the log2 fold-change (FC) of mutant/WT, and the y-axis specifies the negative logarithm to the base 10 of the t test p-values. Open circles represent individual peptides, with blue circles specifying those considered statistically significant. Black vertical and horizontal lines reflect the filtering criteria (log2 FC = ±0.585 and p-value = 0.05) for significance. C and D, size-proportional Venn diagrams of differentially regulated phosphoproteins in mlk1/2/3 and mlk1/3/4 mutants at ZT12 (C) and ZT14 (D). Numbers indicate unique phosphoproteins. ZT 12, Zeitgeber 12; ZT 14, Zeitgeber 14.
We performed mlk-to-WT pairwise comparisons for each time point (supplemental Table S1 and supplemental Dataset S2) to identify peptides that showed altered phosphorylation in the absence of the MLKs at either ZT12 or ZT14. We considered peptides differentially phosphorylated if they had a minimum log2 FC of ±0.585 (1.5 fold-change) and a p-value < 0.05. We identified 113 phosphopeptides corresponding to 93 unique protein groups that met the cutoff in the mlk1/2/3 ZT12 set. In the mlk1/3/4 ZT12 set, 429 phosphopeptides corresponding to 284 unique proteins were differentially phosphorylated, a 300% increase (Fig. 5B). One hundred seventy and 274 phosphopeptides, corresponding to 149 and 215 unique protein groups, have altered phosphorylation in the mlk1/2/3 ZT14 and mlk1/3/4 ZT14 analysis, respectively (Fig. 5B and supplemental Table S1). The phosphosite residue distribution was similar in all datasets analyzed (Fig. 5A and supplemental Fig. S1). These data show that the mlk1/3/4 mutant combination has a larger impact on the phosphoproteome than the mlk1/2/3 mutant combination at each time point, particularly at ZT12. This observation supports a role for MLK4 in regulating the phosphoproteome in a light-dependent manner, likely through MLK4 phosphorylating blue light photoreceptors and red-light signaling pathway transcription factors (5, 6).
Over 50% (48 out of 93) of the differentially phosphorylated proteins identified in the mlk1/2/3 ZT12 set were present in the mlk1/3/4 ZT12 set. In contrast, over 80% of those identified in the mlk1/3/4 ZT12 set were specific to the mlk1/3/4 mutant (Fig. 5C). When comparing the ZT14 sets, 42 phosphoproteins were shared, accounting for 28% and 19.5% of the proteins identified in the mlk1/2/3 ZT14 and mlk1/3/4 ZT14 sets, respectively (Fig. 5D). Many of the proteins shared between mlk1/2/3 and mlk1/3/4 protein sets are involved in gene silencing and chromatin organization (supplemental Dataset S2), supporting a conserved role for the MLK family kinases in these processes (9, 12). Some of these proteins, including SUO and SERRATE (SE)—which are involved in microRNA biogenesis pathways, exhibit altered phosphopeptide abundance at both ZT12 and ZT14. However, proteins involved in chromatin organization, such as increased in bonsai methylation 1 and SPLAYED (SYD), only showed altered phosphorylation in both mlk1/2/3 and mlk1/3/4 mutant backgrounds at ZT12 (supplemental Dataset S2). These results suggest that the MLKs are involved in regulating gene expression, possibly through modulating light-dependent chromatin organization.
MLKs Influence Diverse Kinase Signaling Networks
The proteins identified as differentially phosphorylated in the mlk1/3/4 mutant background at both ZT12 and ZT14 are associated with a diverse set of biological processes, suggesting a possible disruption of multiple protein kinase networks. Motif-X (http://motif-x.med.harvard.edu/motif-x.html) (17, 30) was used to isolate overrepresented sequence motifs present in the phosphopeptide sets that are associated with known kinase families. Following extension of differentially phosphorylated high-confidence, unambiguous peptides using PEPTIDEXTENDER ver.0.2.2 alpha (http://schwartzlab.uconn.edu/pepextend), the resulting 15-mers were submitted for motif analysis using a significance threshold of p < 10−6 and a minimum occurrence requirement of 20. Peptides exhibiting increased or decreased abundance when compared with WT were analyzed separately. Three serine phosphorylation (Sp) motifs (S-x-x-K, R-x-x-S, and K-x-x-S) were overrepresented in peptides that were decreased in abundance at ZT12 in the mlk1/3/4 mutant seedling background (Fig. 6A). The K-x-x-S motif, along with an acidic S-type motif (S-x-x-x-x-x-E), was also overrepresented at ZT14 in mlk1/3/4 mutants (Fig. 6B). The CDPK-SnRK superfamily of protein kinases is known to recognize R/K-x-x-S/T basic motifs. The R-x-x-S motif has also been associated with the AGC family kinases, PKA and PKC (31, 32), which are involved in mid-to late-day rhythmic phosphorylation (33). The kinase(s) responsible for phosphorylation at the S-x-x-K site in plants is unknown. However, the highly conserved eukaryotic cyclin B-dependent protein kinase Cdk1 that recognizes several non-S/T-P motifs, including the S/T-x-x-R/K motif, is a candidate (34). The classical minimal motif required for recognition by proline-directed kinases families (mitogen-activated protein kinase, cyclin-dependent kinase, and glycogen synthase kinase 3), S-P, was found to be overrepresented in peptides that were increased in abundance in mlk1/3/4 mutants at ZT12 and ZT14. While the S-x-S motif, which is associated with the receptor-like protein kinase family (35), was only found to be overrepresented at ZT12 (Fig. 6). Using these parameters, there were no overrepresented motifs identified from the sites that decreased in abundance in either of the mlk1/2/3 datasets. However, if we reduced the minimum occurrence to 10, then the R-x-x-S and S-P motifs were overrepresented in peptides with decreased or increased abundance at ZT12 in mlk1/2/3 mutants (supplemental Fig. S2). The diversity of identified overrepresented kinases motifs suggests that MLK family kinases influence numerous biological processes through systemic regulation of multiple kinase signaling networks.
Fig. 6.

Motif analysis of differentially phosphorylated peptides. Phosphopeptides with altered abundance in mlk1/3/4 mutants at ZT12 (A) and ZT14 (B) where extended (http://schwartzlab.uconn.edu/pepextend) and centered. Motif-X analysis was then preformed with the probability threshold set to p-value ≤ 10−6, the occurrence threshold was set to 20, and the default IPI Arabidopsis Proteome dataset was used as the background dataset. ZT 12, Zeitgeber 12; ZT 14, Zeitgeber 14.
MLKs Influence the Phosphorylation Status of Nuclear Localized Proteins
Using g:Profiler (https://biit.cs.ut.ee/gprofiler/), 149 GO terms were enriched in the differentially phosphorylated proteins from the mlk1/3/4 ZT12 set; 107 of these were classified as biological process, 34 as cellular component, and 5 as molecular function. Forty-nine terms were enriched in the mlk1/3/4 ZT14 set, 28 in biological process, and 21 in cellular component. Fewer terms were found to be enriched in the mlk1/2/3 phosphoprotein sets, with only eight enriched terms at ZT12 and 18 at ZT14. A complete list of enriched GO terms can be found in supplemental Dataset S2. Functionally redundant terms were removed using REVIGO [default settings, dispensability threshold = 0.7 (cellular component) or 0.5 (biological process); supplemental Tables S2 and S3; (19)]. Under the cellular component category, there was strong enrichment for terms associated with the nucleus: “nucleus” (GO:0005634), “nuclear part” (GO:0044428), and “nucleoplasm” (GO:0005654). Terms including “chromosome” (GO:0005694), “chromosomal part” (GO:0044427), and “chromatin” (GO:0000785) were also found to be enriched in at least one of the differentially phosphorylated protein lists (Fig. 7A). These results are in agreement with the known nuclear localization of the MLKs and their role in modifying chromatin (7, 10, 15).
Fig. 7.

Gene ontology (GO) enrichment analysis of differentially phosphorylated proteins. A, heat map showing the p-value significance of enriched cellular component GO categories of proteins with altered abundance in mlk mutant seedlings at ZT12 and ZT14. B and C, treemap representation of biological process GO categories enriched in mlk1/3/4 mutant seedlings at ZT12 (B) and ZT14 (C). The box size correlates to the −log10 p-value of the GO-term enrichment. Boxes with the same color indicate related GO-terms and correspond to the representative GO-term which is found in the upper-left of each box. REVIGO was used to eliminate redundant GO-terms with a dispensability value ≥0.7 (A) or ≥0.5 (B and C). ZT 12, Zeitgeber 12; ZT 14, Zeitgeber 14.
Circadian-Associated Proteins Exhibit Altered Phosphorylation in mlk1/3/4 Mutant Seedlings
The mlk1/3/4 ZT12 differentially phosphorylated protein set was enriched in proteins associated with rhythmic processes (GO:0048511) and/or circadian rhythms (GO:0007623). These observations agree with previous reports linking the MLKs to light signaling and circadian regulation (5, 6, 7, 14, 15). The core circadian clock proteins pseudo-response regulator 7, time for coffee, and Reveille 8 (RVE8) were all differentially phosphorylated in the mlk1/3/4 mutant background at ZT12 (Table 1 and supplemental Table S3). We observed decreased phosphorylation of time for coffee at S324 and pseudo-response regulator 7 at S355 and S275, while RVE8 showed increased phosphorylation of the C-terminal half (Table 1). The red-light photoreceptor phytochrome B (phyB) and the transcriptional master regulator elongated hypocotyl 5 (HY5) also showed reduced phosphorylation at Threonine 42 and T64, respectively (Table 1); to the best of our knowledge, these phosphosites are previously unreported. Table 1 lists additional circadian-associated proteins with altered phospho-abundance in the mlk1/3/4 mutant at ZT12.
Table 1.
Phosphosites identified in circadian-associated proteins
| Protein group accession | Protein names | Protein description | Peptide sequence | Log2 FC | p-value |
|---|---|---|---|---|---|
| AT2G18790 | PHYB | Phytochrome B | GGEQAQSSGT42K | −1.34 | 5.95E-06 |
| AT4G30200 | VEL1 | Vernalization5/VIN3-like protein | KPS506SKNEDNNSPSVDESAAK | −0.90 | 3.00E-02 |
| AT5G11260 | HY5 | Basic-leucine zipper (bZIP) transcription factor family protein | ESGSAT64GQER | −0.80 | 1.46E-02 |
| AT3G22380 | TIC | Time for coffee | MPS324TSKQEAAGNDLTEAAK | −0.78 | 1.98E-04 |
| AT5G02810 | PRR7 | Pseudo-response regulator 7 | QDNS355FEK | −0.73 | 1.04E-05 |
| AT2G17840 | ERD7 | Senescence/dehydration-associated protein-like protein | SAAS488QKK | −0.68 | 3.95E-02 |
| AT5G02810 | PRR7 | Pseudo-response regulator 7 | AVS275LWDR | −0.61 | 1.93E-03 |
| AT4G30200 | VEL1 | Vernalization5/VIN3-like protein | LCSSALESLETIAT330TPPDVAALPS340PR | 0.61 | 1.53E-02 |
| AT4G30200 | VEL1 | Vernalization5/VIN3-like protein | NEDNNSPS516VDESAAK | 0.62 | 1.01E-04 |
| AT5G52310 | LTI78 | Low-temperature-responsive protein 78 (LTI78)/desiccation-responsive protein 29A (RD29A) | NEYSPES387DGGLGAPLGGNFPVR | 0.64 | 6.53E-03 |
| AT3G09600 | RVE8 | Homeodomain-like superfamily protein | GLLNVSSPSTSGMGSSSR | 0.84 | 1.72E-03 |
| AT3G46780 | PTAC16 | Plastid transcriptionally active 16 | ADAVGVT410VDGLFNK | 0.95 | 9.29E-03 |
Gene Ontology Analysis Reveals Enrichment of Proteins Involved in Chromatin Organization
Owing to the large number of enriched GO terms identified from both the mlk1/3/4 ZT12 and mlk1/3/4 ZT14 list, the dispensability threshold was reduced to 0.5 for REVIGO analysis. This resulted in the identification of 34 and 15 representative and nonredundant-enriched biological process terms in mlk1/3/4 ZT12 and mlk1/3/4 ZT14 altered phosphoprotein lists, respectively. Enriched GO terms and their underlying gene identifiers shared between the mlk1/3/4 ZT12 and ZT14 sets included “organic cyclic compound metabolism”, GO:1901360, “nitrogen compound metabolism”, GO:0006807, “chromosome organization”, GO:0051276, and “negative regulation of gene expression” GO:0006807 (Fig. 7, B and C and supplemental Table S3). The chromatin modifying proteins Brahma (BRM), SIN3-like3, vernalization5/VIN3-like, and high mobility ground B1 were shared among these GO terms. Additional proteins associated with chromatin modifications were present in the mlk1/3/4 ZT12 list including the histone methyltransferase early flowering in short days/set domain group 8 (SDG8), the histone acetyltransferase TBP-associated factor 1, as well as increased in bonsai methylation 1, actin-related protein 4, alfin-like 7, GLIOMAS 41 (GAS41/YAF9a), and stress-induced histone H2A protein 9. Few biological process GO-terms were found to be enriched in the mlk1/2/3 datasets. Nevertheless, those that were [“regulation of gene expression”, “epigenetic” (ZT12) and “chromosome organization”, “chromatin organization”, and “mitotic sister chromatid cohesion” (ZT14)] were also enriched in the mlk1/3/4 protein list (supplemental Dataset S2). These results support a role for the MLKs in regulating chromatin organization and gene expression at the assessed time points.
mlk1/3/4 Mutants Show Altered Phosphorylation of Proteins Involved in Nuclear Organization and DNA Damage Response
Analysis of differentially phosphorylated peptides has shown that the loss of mlk1, mlk3, and mlk4 at ZT12 has the greatest impact on both the global and phosphoproteome. Therefore, we chose to expand on our analysis exclusively for the mlk1/3/4 ZT12 dataset. To elucidate further the biological processes influenced by the MLKs at the end of the day (ZT12), we independently analyzed the phosphoproteins that were increased or decreased in abundance. Of the 429 phosphopeptides found to have altered abundance in the mlk1/3/4 mutant background at ZT12 (supplemental Dataset S2), 133 were increased, and 296 were decreased in abundance and mapping to 103 increased and 190 decreased unique proteins. Interestingly, nine gene identifiers were shared between the increased and decreased groups, including several that are involved in various aspects of nuclear organization such as LITTLE NUCLEI 1/CROWED NUCLEI 1 (LINC1/CRWN1), vernalization5/VIN3-like, and high mobility group B1 (Fig. 8A and supplemental Dataset S2). Proteins that were increased in abundance were associated with the representative GO-terms “response to organic substance” (GO:0010033) and “response to stimulus” (GO:0050896) (Fig. 8B). However, we found that the majority of GO terms enriched in the inclusive set (both increased and decreased peptides) such as “‘RNA processing” (GO:0006396), “chromosome organization” (GO:0051276), and “developmental processes” (GO:0032592) are associated with decreased phosphorylation (Fig. 8C). The GO term “cellular response to DNA damage stimulus” (GO:0006974) was also enriched in proteins exhibiting decreased phosphorylation. Proteins associated with this term include a catalytic subunit of DNA polymerase alpha INCARVATA2, as well as X-ray cross complementation group 4 and MUTM homolog-1 both of which are directly involved in DNA repair (36, 37, 38).
Fig. 8.

Analysis of differentially phosphorylated peptides in mlk1/3/4 mutants at ZT12. A, size-proportional Venn diagram of proteins which show increased, decreased, or both increased and decreased abundance of identified phosphosites in mlk1/3/4 mutant seedlings at ZT12. Numbers indicate unique phosphoproteins. Analysis of phosphoproteins that show increased (B) or decreased (C) in abundance in the mlk1/3/4 mutant seedlings background at ZT12. Treemap representations of biological process GO category enrichment are shown. The box size correlates to the −log10 p-value of the GO-term enrichment. Boxes with the same color indicate related GO-terms and correspond to the representative GO-term which is found in the upper-left of each box. REVIGO was used to eliminate redundant GO-terms with a dispensability value ≥0.5. ZT 12, Zeitgeber 12; ZT 14, Zeitgeber 14.
mlk Mutants Show Increased Sensitivity to DNA-Damaging Agents
Nuclear organization and chromatin dynamics strongly influence DNA damage repair efficacy (Reviewed in (39, 40)). Additionally, the Chlamydomonas MLK ortholog, Mut9, is required for survival when grown in the presence of genotoxic agents (12). Since many of the proteins exhibiting changes in phosphorylation abundance in the mlk1/3/4 mutants at ZT12 are proteins associated with nuclear organization (LINC/CRWN family members, SAD1/UNC-84 domain protein 2, BRM, and SYD) and DNA damage, we further explored what role the Arabidopsis MLKs might play in DNA damage response. To do so, we evaluated the sensitivity of mutant and WT seedlings to the genotoxic agent MMS and UV-C. MMS is a monofunctional DNA alkylating agent that induces replication fork stalling and subsequent double strand breaks (41). In addition to the mlk1/2/3 and mlk1/3/4 mutants, the mlk4 single and mlk quadruple amiRNA line (amiR4k) (6) were included in our analysis.
Mutant and WT seedlings were imaged and weighed after 14 days of exposure to titrations of MMS. All genotypes showed reduced aerial mass with increasing levels of MMS, with the mlk mutants showing increased sensitivity compared with WT (Fig. 9, A and B). In WT seedlings, growth was reduced by less than 10% (fresh weight) in the presence of 50 ppm MMS compared with the mock-treated seedlings. The mlk4 single mutant seedlings showed more than 20% reduction of fresh weight, the mlk1/3/4 seedlings a 39%, and amiR4K lines a 44% reduction, compared with the mock-treated seedlings (Fig. 9B). The mlk1/3/4 and amiR4k mutants continued to decline in fresh weight at higher concentrations of MMS. At 100 ppm MMS, mlk1/2/3 seedlings showed approximately a 10% greater reduction in fresh weight than what was observed for WT (Fig. 9B). In addition to stunted growth, chlorotic tissue was observed in the mlk1/3/4 mutants growing on 50 ppm MMS and in the amiR4K mutants at 75 PPM MMS (Fig. 9A). 150 ppm MMS caused severe growth reduction and lethality in all assessed genotypes; thus, seedlings were imaged but not weighed (Fig. 9A). Next, we assessed seedling germination and growth in the continuous presence of 150 ppm MMS. Postgermination growth was severely impaired in the mlk1/3/4 mutant, with more than 65% of seedlings exhibiting complete developmental arrest compared with approximately 10% of WT seedlings (Fig. 9, C and D). In contrast, the amiR4k mutant seedlings developed similar to WT seedlings when germinated in the presence of 150 ppm MMS (Fig. 9, C and D), which could be a result of near endogenous expression levels of MLK2 and MLK3 (5).
Fig. 9.

mlk1/3/4 mutants have increased sensitivity to MMS treatment. A, representative images of mutant and WT seedlings 15 days after transfer to solid media containing the indicated concentration of MMS. Chlorotic tissue is denoted with a blue arrowhead. Scale bar = 5 mm. B, fresh weight of 21-day-old WT and mutant seedlings grown in the presence of MMS relative to mock treated samples. The average of three biological replicates of ≥10 seedlings each is presented. C, percent of seedlings exhibiting postgermination developmental arrest after 12 days of growth in the presence of 150 ppm MMS. The average of three biological replicates of ≥30 seedlings is presented. B and C, error bars indicate standard deviation. ∗p < 0.05, ∗∗p < 0.01 compared with WT seedlings (Student’s t test). D, representative images of mutant and WT seedlings germinated in the presence of 150 ppm MMS. E, representative images of mutant and WT seedlings irradiated with indicated levels of UV-C. Scale bar = 2 mm. MMS, methyl methane sulfonate.
We also tested the mlk mutants for sensitivity to UV-induced DNA damage through periodic exposure to multiple doses of UV-C irradiation. The phenotypic impact of chronic irradiation with either 2000 or 4000 J m−2 on seedlings was evaluated 5 days after a recovery period. All genotypes had cotyledon cell death and reduced growth after exposure to both doses of UV-C. However, only mlk1/3/4 mutant seedlings showed tissue chlorosis after exposure to both 2000 and 4000 J m−2 UV-C. amiR4k mutants had minimal chlorosis after irradiation with 4000 J m−2 (Fig. 9E). Taken together, these data suggest that mlk mutants have increased sensitivity to DNA damage.
Discussion
MLK Protein Kinases Alter Protein Phosphorylation in Developmental and Stress Responsive Pathways
The current repertoire of MLK substrates is composed of a photoreceptor, multiple transcription factors, hormone receptors, and histones (5, 6, 7, 8, 9, 10, 11, 13). MLK mutants have defects in circadian period length, hypocotyl elongation, flowering time, osmotic stress responses, seed set, chromatin organization, and hormone sensitivity (6, 7, 8, 9, 11, 14, 15). These observations support a model where MLK family members function as central regulators of numerous interconnected signaling pathways. Our quantitative analysis of the global- and phosphoproteomes of mlk triple mutant seedlings supports a diverse and complex role for the MLKs in the regulation of cellular signaling and response. Interestingly, these kinases seem to share a balance of functional redundancy and substrate specificity, which, for example, results in opposing circadian period and hypocotyl elongation phenotypes (6, 15). We found the mlk1/3/4 mutant displays a much more severe proteomic phenotype relative to the mlk1/2/3 mutant, with over 10-fold more proteins showing altered abundance in the mlk1/3/4 mutant (Fig. 2). This increase holds for the phosphoproteome as well. However, the difference was greatest in tissue sampled in the light. An explanation for the light dependence could be the result of MLK4 acquiring substrate specificity or MLK4 having a greater tendency than other MLKs for interacting with light-signaling proteins in planta. MLK4 has a higher affinity for PIF3 when compared with other MLKs (5), and both phyB and HY5 have altered phosphorylation only at ZT12 in the mlk1/3/4 mutants (Table 1). While some proteins with altered abundance were unique to the mlk1/3/4 or mlk1/2/3 mutants, shared targets include GLS biosynthesis (global proteome; Fig. 3) and chromosome organization (phosphoproteome; Fig. 7, B and C and supplemental Dataset S2). Further work is needed to determine if any of the proteins showing altered phosphorylation are direct substrates of the MLKs or whether changes in phosphorylation status is occurring indirectly through additional kinases.
MLKs Regulate Hormone Signaling and Stress Responses
Several proteins responsible for GLS metabolism showed altered abundance in mlk mutant seedlings before and after dark transition (Fig. 3 and supplemental Dataset S1). GLSs are nitrogen- and sulfur-containing secondary metabolites known for their role in plant defense (42, 43, 44, 45) and anticarcinogenic properties (46). Accumulation of GLSs in A. thaliana is rhythmic, controlled in part by circadian clock regulated jasmonate accumulation and the activity of the basic leucine zipper transcription factor, HY5 (27, 47). HY5 is a positive regulator of light signaling and functions as a central regulator of light-dependent growth and development by integrating various environmental signals (48). Peak GLS levels occur during the day, possibly to protect against rhythmic herbivory. Two GLS biosynthesis genes that showed increased protein abundance in mlk mutants, CYP79F1 and SOT18, are expressed at lower levels in the hy5 mutant background (27). The phosphorylation of HY5 is associated with its activity and stability, with the nonphosphorylated form being more active (49). Thus, it is possible that increased HY5 activity, resulting from decreased phosphorylation in the mlk mutant background, could be influencing GLS metabolism. Additionally, ABA induces GLS accumulation in plants (50, 51). MLK3 regulates ABA signaling through the phosphorylation of the PYR/PYL ABA receptor family of proteins (8). In agreement with altered ABA signaling, increased phosphorylation of proteins associated with ABA responses and the SnRK consensus motif, R/K-x-x-S/T, was found to overrepresented in the mlk1/3/4 mutant background. Thus, the MLKs may be involved in the regulation of defense responses through multiple converging signaling pathways.
The Phosphorylation Status of Key Circadian and Light Signaling Components Are Altered in the Absence of MLK Family Kinases
Several differentially phosphorylated proteins that are involved in chromatin organization also function as core circadian clock components (RVE8) or are central regulators of clock input pathways such as temperature and light signaling (phyB and HY5). RVE8 is a MYB-like transcription factor that regulates the expression of the clock gene timing of CAB expression1 by promoting histone 3 (H3) acetylation of its promoter (52). RVE8 shares structural similarity to the core clock transcription factors circadian clock associated (CCA1) and late elongated hypocotyl. Phosphorylation of CCA1 by the Ser/Thr protein kinase CK2 antagonistically regulates CCA1 transcriptional activity by reducing its ability to bind to the promoters of clock gene targets, which in turn alters circadian period (53). Further exploration of the impact of RVE8 phosphorylation could reveal a new avenue of posttranslational regulation of the circadian clock.
Temperature and light signaling are critical circadian inputs that allow plants to coordinate growth and development (e.g., germination and photoperiodic flowering) with their environment. The phyB photoreceptor is central to both temperature and light signaling pathways (54). PhyB activity is regulated in part by phosphorylation of its N terminus (55, 56). Altered phospho-status of phyB Ser86 and Y104 influences phyB rate dark-reversion rates, hypocotyl elongation, and flowering time in Arabidopsis (55, 56, 57). Here, we report decreased phosphorylation of a previously unidentified phyB N-terminal phosphosite, T42, in the mlk mutant background at ZT12 (Table 1). MLKs are known to associate with phyB, phosphorylate the phytochrome–interacting factor, PIF3, and display a variety of red light–dependent growth phenotypes (5, 15). In addition to the well-established light-induced phyB-PIF signaling cascade, there is an ample amount of evidence supporting the role of phyB in large-scale chromatin organization (58, 59). Thus, the MLK-phyB interaction may contribute to light-dependent chromatin re-organization in addition to regulating PIF3 turnover. We also found decreased phosphorylation of another key light signaling component, HY5 at T64. Whether the phosphorylation of HY5T64 and/or phyBT42 is directly or indirectly influenced by the MLKs and how those phosphosites fit into the existing light signaling paradigm will be an exciting line of future research.
The Role of MLK Family Kinases in Modulating Nuclear Architecture
The role of histone modifications in the regulation of developmental processes and stress response has been well-established, yet our understanding of the responsible modifiers, modification crosstalk, and targeted genes is incomplete (60, 61). Early observations have implicated the MLK family in the regulation of environmentally stimulated chromatin organization. MLK1, like its Chlamydomonas homolog MUT9, has been shown to phosphorylate histone H3 on threonine 3 (H3T3p) and to function redundantly with MLK2 to promote H3T3p in response to salt stress (9, 10). Accordingly, the mlk1mlk2 double mutant has abnormal chromatin organization and increased sensitivity to osmotic stress (10). Comparisons have been drawn between the defects in chromosomal organization observed in the mlk1mlk2 mutants and those occurring in plants harboring mutations in members of the little nuclei/crowded nuclei (LINC/CRWN) gene family, which are involved in controlling nuclear size and heterochromatin organization (62, 63). Our analysis of the mlk mutant phosphoproteomes found that peptides mapping to multiple members of the LINC/CRWN family were altered in abundance in mlk1/2/3 and mlk1/3/4 mutants, suggesting that MLKs may influence nuclear organization in part through regulation of the LINC proteins.
MLKs Are Involved in DNA Damage Repair Through Multiple Pathways
Plants are exposed to DNA damage from their external environment (e.g., ultraviolet light, ionizing radiation, heat stress, and bacterial and fungal toxins) as well as endogenous sources such as DNA-alkylating metabolic byproducts. Maintenance of genomic integrity requires an efficient DNA damage repair (DDR) system that can identify, access, remove, and reassemble damaged genomic regions within the context of chromatin. Mutations in genes involved in chromatin organization and remodeling often exhibit defects in DDR and enhanced susceptibility to DNA damaging reagents (40). The increased sensitivity of mlk mutants to DNA damaging agents could result from the dysregulation of proteins involved in chromatin remodeling, such as GAS41/YAFa, actin-related protein 4, BRM, and SYD (Fig. 9). The mlk1/3/4 mutant also shows altered phosphorylation of several proteins directly involved in DDR, such as MUTM homolog-1 and X-ray cross complementation group 4 (64, 65). Additionally, there is accumulating evidence supporting a role for small regulatory RNAs in DDR (66); proteins associated with small RNA metabolism are enriched in mlk1/3/4 at ZT12 (Fig. 7). There is no question that full elucidation and validation of the mechanisms linking MLKs and DDR will require further exploration. However, taken together, our data suggests the MLKs play an important role in mitigating DNA damage through the regulation of multiple response pathways.
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier Project accession: PXD020762 and Project DOI: 10.6019/PXD020762.
Supplemental data
This article contains supplemental data.
Conflict of interest
The authors declare no competing interests.
Acknowledgments
This material is based upon work supported by the National Science Foundation under Grant No. DBI-1827534 for acquisition of the Orbitrap Fusion Lumos LC-MS/MS. The authors thank Maria L. Sorkin, He Huang, and Rebecca Bindbeutel for helpful comments in the preparation of the manuscript.
Author contributions
M. E. W., M. M. A., X. J., J. H. C., J. C. R., B. S. E., T. M. K., and D. A. N. designed the experiments, M. E. W., S.-C. T., M. M. A., X. J., and J. H. C. performed the experiments, M. E. W., S.-C. T., M. M. A., M. M., X. J., J. H. C., B. S. E., and D. A. N. analyzed the data, M. E. W., S.-C. T., M. M. A., J. H. C., B. S. E., and D. A. N. wrote the manuscript.
Supplemental Data
References
- 1.Wang Y., Liu Z., Cheng H., Gao T., Pan Z., Yang Q., Guo A., Xue Y. EKPD: A hierarchical database of eukaryotic protein kinases and protein phosphatases. Nucleic Acids Res. 2014;42:D496–D502. doi: 10.1093/nar/gkt1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mergner J., Frejno M., List M., Papacek M., Chen X., Chaudhary A., Samaras P., Richter S., Shikata H., Messerer M., Lang D., Altmann S., Cyprys P., Zolg D.P., Mathieson T. Mass-spectrometry-based draft of the Arabidopsis proteome. Nature. 2020;579:409–414. doi: 10.1038/s41586-020-2094-2. [DOI] [PubMed] [Google Scholar]
- 3.Silva-Sanchez C., Li H., Chen S. Recent advances and challenges in plant phosphoproteomics. Proteomics. 2015;15:1127–1141. doi: 10.1002/pmic.201400410. [DOI] [PubMed] [Google Scholar]
- 4.Vlastaridis P., Kyriakidou P., Chaliotis A., Van de Peer Y., Oliver S.G., Amoutzias G.D. Estimating the total number of phosphoproteins and phosphorylation sites in eukaryotic proteomes. Gigascience. 2017;6:1–11. doi: 10.1093/gigascience/giw015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ni W., Xu S.L., González-Grandío E., Chalkley R.J., Huhmer A.F.R., Burlingame A.L., Wang Z.Y., Quail P.H. PPKs mediate direct signal transfer from phytochrome photoreceptors to transcription factor PIF3. Nat. Commun. 2017;8:15236. doi: 10.1038/ncomms15236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Q., Wang Q., Deng W., Wang X., Piao M., Cai D., Li Y., Barshop W.D., Yu X., Zhou T., Liu B., Oka Y., Wohlschlegel J., Zuo Z., Lin C. Molecular basis for blue light-dependent phosphorylation of Arabidopsis cryptochrome 2. Nat. Commun. 2017;8:15234. doi: 10.1038/ncomms15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su Y., Wang S., Zhang F., Zheng H., Liu Y., Huang T., Ding Y. Phosphorylation of histone H2A at serine 95: A plant-specific mark involved in flowering time regulation and H2A.Z deposition. Plant Cell. 2017;29:2197–2213. doi: 10.1105/tpc.17.00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen H.H., Qu L., Xu Z.H., Zhu J.K., Xue H.W. EL1-like casein kinases suppress ABA signaling and responses by phosphorylating and destabilizing the ABA receptors PYR/PYLs in arabidopsis. Mol. Plant. 2018;11:706–719. doi: 10.1016/j.molp.2018.02.012. [DOI] [PubMed] [Google Scholar]
- 9.Casas-Mollano J.A., Jeong B.R., Xu J., Moriyama H., Cerutti H. The MUT9p kinase phosphorylates histone H3 threonine 3 and is necessary for heritable epigenetic silencing in Chlamydomonas. Proc. Natl. Acad. Sci. U. S. A. 2008;105:6486–6491. doi: 10.1073/pnas.0711310105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z., Casas-Mollano J.A., Xu J., Riethoven J.J., Zhang C., Cerutti H. Osmotic stress induces phosphorylation of histone H3 at threonine 3 in pericentromeric regions of Arabidopsis thaliana. Proc. Natl. Acad. Sci. U. S. A. 2015;112:8487–8492. doi: 10.1073/pnas.1423325112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang J., Cui H., Jia S., Liu W., Yu R., Wu Z., Wang Z. Arabidopsis thaliana MLK3, a plant-specific casein kinase 1, negatively regulates flowering and phosphorylates histone H3 in vitro. Genes (Basel) 2020;11:345. doi: 10.3390/genes11030345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeong B.R., Wu-Scharf D., Zhang C., Cerutti H. Suppressors of transcriptional transgenic silencing in Chlamydomonas are sensitive to DNA-damaging agents and reactivate transposable elements. Proc. Natl. Acad. Sci. U. S. A. 2002;99:1076–1081. doi: 10.1073/pnas.022392999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai C., Xue H.W. Rice early flowering1, a CKI, phosphorylates della protein SLR1 to negatively regulate gibberellin signalling. EMBO J. 2010;29:1916–1927. doi: 10.1038/emboj.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng H., Zhang F., Wang S., Su Y., Ji X., Jiang P., Chen R., Hou S., Ding Y. MLK1 and MLK2 coordinate RGA and CCA1 activity to regulate hypocotyl elongation in Arabidopsis thaliana. Plant Cell. 2017;30:67–82. doi: 10.1105/tpc.17.00830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang H., Alvarez S., Bindbeutel R., Shen Z., Naldrett M.J., Evans B.S., Briggs S.P., Hicks L.M., Kay S.A., Nusinow D.A. Identification of evening complex associated proteins in arabidopsis by affinity purification and mass spectrometry. Mol. Cell. Proteomics. 2016;15:201–217. doi: 10.1074/mcp.M115.054064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun K., Xue X., Liu N., Zhu Z., Li H. A point-to-point protein–protein interaction assay reveals the signaling interplays among plant hormones and environmental cues. Plant Direct. 2020;4 doi: 10.1002/pld3.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou M.F., Schwartz D. Biological sequence motif discovery using motif-x. Curr. Protoc. Bioinformatics. 2011 doi: 10.1002/0471250953.bi1315s35. Chapter 13:Unit 13.15-24. [DOI] [PubMed] [Google Scholar]
- 18.Reimand J., Arak T., Adler P., Kolberg L., Reisberg S., Peterson H., Vilo J. g:Profiler-a web server for functional interpretation of gene lists (2016 update) Nucleic Acids Res. 2016;44:W83–W89. doi: 10.1093/nar/gkw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Supek F., Bošnjak M., Škunca N., Šmuc T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6 doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crocoll C., Halkier B.A., Burow M. Analysis and quantification of glucosinolates. Curr. Protoc. Plant Biol. 2016;1:385–409. doi: 10.1002/cppb.20027. [DOI] [PubMed] [Google Scholar]
- 21.Brown P.D., Tokuhisa J.G., Reichelt M., Gershenzon J. Variation of glucosinolate accumulation among different organs and developmental stages of Arabidopsis thaliana. Phytochemistry. 2003;62:471–481. doi: 10.1016/s0031-9422(02)00549-6. [DOI] [PubMed] [Google Scholar]
- 22.Grosser K., van Dam N.M. A Straightforward method for glucosinolate extraction and analysis with high-pressure liquid chromatography (HPLC) J. Vis. Exp. 2017:55425. doi: 10.3791/55425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castells E., Molinier J., Drevensek S., Genschik P., Barneche F., Bowler C. det1-1-induced UV-C hyposensitivity through UVR3 and PHR1 photolyase gene over-expression. Plant J. 2010;63:392–404. doi: 10.1111/j.1365-313X.2010.04249.x. [DOI] [PubMed] [Google Scholar]
- 24.Herbel V., Orth C., Wenzel R., Ahmad M., Bittl R., Batschauer A. Lifetimes of Arabidopsis cryptochrome signaling states in vivo. Plant J. 2013;74:583–592. doi: 10.1111/tpj.12144. [DOI] [PubMed] [Google Scholar]
- 25.Rausenberger J., Hussong A., Kircher S., Kirchenbauer D., Timmer J., Nagy F., Schäfer E., Fleck C. An integrative model for phytochrome B mediated photomorphogenesis: From protein dynamics to physiology. PLoS One. 2010;5 doi: 10.1371/journal.pone.0010721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sønderby I.E., Geu-Flores F., Halkier B.A. Biosynthesis of glucosinolates--gene discovery and beyond. Trends Plant Sci. 2010;15:283–290. doi: 10.1016/j.tplants.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Huseby S., Koprivova A., Lee B.R., Saha S., Mithen R., Wold A.B., Bengtsson G.B., Kopriva S. Diurnal and light regulation of sulphur assimilation and glucosinolate biosynthesis in Arabidopsis. J. Exp. Bot. 2013;64:1039–1048. doi: 10.1093/jxb/ers378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Champion A., Kreis M., Mockaitis K., Picaud A., Henry Y. Arabidopsis kinome: After the casting. Funct. Integr. Genomics. 2004;4:163–187. doi: 10.1007/s10142-003-0096-4. [DOI] [PubMed] [Google Scholar]
- 29.Sugiyama N., Nakagami H., Mochida K., Daudi A., Tomita M., Shirasu K., Ishihama Y. Large-scale phosphorylation mapping reveals the extent of tyrosine phosphorylation in Arabidopsis. Mol. Syst. Biol. 2008;4:193. doi: 10.1038/msb.2008.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwartz D., Gygi S.P. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 2005;23:1391–1398. doi: 10.1038/nbt1146. [DOI] [PubMed] [Google Scholar]
- 31.Rademacher E.H., Offringa R. Evolutionary adaptations of plant AGC kinases: From light signaling to cell polarity regulation. Front. Plant Sci. 2012;3:250. doi: 10.3389/fpls.2012.00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marondedze C., Groen A.J., Thomas L., Lilley K.S., Gehring C. A quantitative phosphoproteome analysis of cGMP-dependent cellular responses in Arabidopsis thaliana. Mol. Plant. 2016;9:621–623. doi: 10.1016/j.molp.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 33.Choudhary M.K., Nomura Y., Wang L., Nakagami H., Somers D.E. Quantitative circadian phosphoproteomic analysis of arabidopsis reveals extensive clock control of key components in physiological, metabolic, and signaling pathways. Mol. Cell. Proteomics. 2015;14:2243–2260. doi: 10.1074/mcp.M114.047183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki K., Sako K., Akiyama K., Isoda M., Senoo C., Nakajo N., Sagata N. Identification of non-Ser/Thr-Pro consensus motifs for Cdk1 and their roles in mitotic regulation of C2H2 zinc finger proteins and Ect2. Sci. Rep. 2015;5:7929. doi: 10.1038/srep07929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Wijk K.J., Friso G., Walther D., Schulze W.X. Meta-analysis of Arabidopsis thaliana phospho-proteomics data reveals compartmentalization of phosphorylation motifs. Plant Cell. 2014;26:2367–2389. doi: 10.1105/tpc.114.125815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.West C.E., Waterworth W.M., Jiang Q., Bray C.M. Arabidopsis DNA ligase IV is induced by γ-irradiation and interacts with an Arabidopsis homologue of the double strand break repair protein XRCC4. Plant J. 2000;24:67–78. doi: 10.1046/j.1365-313x.2000.00856.x. [DOI] [PubMed] [Google Scholar]
- 37.Ohtsubo T., Matsuda O., Iba K., Terashima I., Sekiguchi M., Nakabeppu Y. Molecular cloning of AtMMH, an Arabidopsis thaliana ortholog of the Escherichia coli mutM gene, and analysis of functional domains of its product. Mol. Gen. Genet. 1998;259:577–590. doi: 10.1007/s004380050851. [DOI] [PubMed] [Google Scholar]
- 38.Barrero J.M., González-Bayón R., del Pozo J.C., Ponce M.R., Micol J.L. INCURVATA2 encodes the catalytic subunit of DNA polymerase and interacts with genes involved in chromatin-mediated cellular memory in Arabidopsis thaliana. Plant Cell. 2007;19:2822–2838. doi: 10.1105/tpc.107.054130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vergara Z., Gutierrez C. Emerging roles of chromatin in the maintenance of genome organization and function in plants. Genome Biol. 2017;18:96. doi: 10.1186/s13059-017-1236-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donà M., Mittelsten Scheid O. DNA damage repair in the context of plant chromatin. Plant Physiol. 2015;168:1206–1218. doi: 10.1104/pp.15.00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ensminger M., Iloff L., Ebel C., Nikolova T., Kaina B., Lӧbrich M. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 2014;206:29–43. doi: 10.1083/jcb.201312078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim J.H., Lee B.W., Schroeder F.C., Jander G. Identification of indole glucosinolate breakdown products with antifeedant effects on Myzus persicae (green peach aphid) Plant J. 2008;54:1015–1026. doi: 10.1111/j.1365-313X.2008.03476.x. [DOI] [PubMed] [Google Scholar]
- 43.Kos M., Houshyani B., Achhami B.B., Wietsma R., Gols R., Weldegergis B.T., Kabouw P., Bouwmeester H.J., Vet L.E., Dicke M., van Loon J.J. Herbivore-mediated effects of glucosinolates on different natural enemies of a specialist aphid. J. Chem. Ecol. 2012;38:100–115. doi: 10.1007/s10886-012-0065-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bednarek P., Pislewska-Bednarek M., Svatos A., Schneider B., Doubsky J., Mansurova M., Humphry M., Consonni C., Panstruga R., Sanchez-Vallet A., Molina A., Schulze-Lefert P. A glucosinolate metabolism pathway in living plant cells mediates broad-spectrum antifungal defense. Science. 2009;323:101–106. doi: 10.1126/science.1163732. [DOI] [PubMed] [Google Scholar]
- 45.Clay N.K., Adio A.M., Denoux C., Jander G., Ausubel F.M. Glucosinolate metabolites required for an Arabidopsis innate immune response. Science. 2009;323:95–101. doi: 10.1126/science.1164627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Higdon J.V., Delage B., Williams D.E., Dashwood R.H. Cruciferous vegetables and human cancer risk: Epidemiologic evidence and mechanistic basis. Pharmacol. Res. 2007;55:224–236. doi: 10.1016/j.phrs.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goodspeed D., Chehab E.W., Min-Venditti A., Braam J., Covington M.F. Arabidopsis synchronizes jasmonate-mediated defense with insect circadian behavior. Proc. Natl. Acad. Sci. U. S. A. 2012;109:4674–4677. doi: 10.1073/pnas.1116368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gangappa S.N., Botto J.F. The multifaceted roles of HY5 in plant growth and development. Mol. Plant. 2016;9:1353–1365. doi: 10.1016/j.molp.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 49.Hardtke C.S., Gohda K., Osterlund M.T., Oyama T., Okada K., Deng X.W. HY5 stability and activity in Arabidopsis is regulated by phosphorylation in its COP1 binding domain. EMBO J. 2000;19:4997–5006. doi: 10.1093/emboj/19.18.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Z., Yang R., Guo L., Fang M., Zhou Y., Gu Z. Effects of abscisic acid on glucosinolate content, isothiocyanate formation and myrosinase activity in cabbage sprouts. Int. J. Food Sci. Technol. 2015;50:1839. [Google Scholar]
- 51.Zhu M., Assmann S.M. Metabolic signatures in response to abscisic acid (ABA) treatment in Brassica napus guard cells revealed by metabolomics. Sci. Rep. 2017;7:12875. doi: 10.1038/s41598-017-13166-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Farinas B., Mas P. Functional implication of the MYB transcription factor RVE8/LCL5 in the circadian control of histone acetylation. Plant J. 2011;66:318–329. doi: 10.1111/j.1365-313X.2011.04484.x. [DOI] [PubMed] [Google Scholar]
- 53.Portolés S., Más P. The functional interplay between protein kinase CK2 and cca1 transcriptional activity is essential for clock temperature compensation in Arabidopsis. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Legris M., Klose C., Burgie E.S., Rojas C.C., Neme M., Hiltbrunner A., Wigge P.A., Schäfer E., Vierstra R.D., Casal J.J. Phytochrome B integrates light and temperature signals in Arabidopsis. Science. 2016;354:897–900. doi: 10.1126/science.aaf5656. [DOI] [PubMed] [Google Scholar]
- 55.Medzihradszky M., Bindics J., Ádám É., Viczián A., Klement É., Lorrain S., Gyula P., Mérai Z., Fankhauser C., Medzihradszky K.F., Kunkel T., Schäfer E., Nagy F. Phosphorylation of phytochrome B inhibits light-induced signaling via accelerated dark reversion in arabidopsis. Plant Cell. 2013;25:535–544. doi: 10.1105/tpc.112.106898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nito K., Wong C.C., Yates J.R., Chory J. Tyrosine phosphorylation regulates the activity of phytochrome photoreceptors. Cell Rep. 2013;3:1970–1979. doi: 10.1016/j.celrep.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hajdu A., Ádám É., Sheerin D.J., Dobos O., Bernula P., Hiltbrunner A., Kozma-Bognár L., Nagy F. High-level expression and phosphorylation of phytochrome B modulates flowering time in Arabidopsis. Plant J. 2015;83:794–805. doi: 10.1111/tpj.12926. [DOI] [PubMed] [Google Scholar]
- 58.van Zanten M., Tessadori F., McLoughlin F., Smith R., Millenaar F.F., van Driel R., Voesenek L.A.C.J., Peeters A.J.M., Fransz P. Photoreceptors CRYTOCHROME2 and phytochrome B control chromatin compaction in arabidopsis. Plant Physiol. 2010;154:1686–1696. doi: 10.1104/pp.110.164616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tessadori F., van Zanten M., Pavlova P., Clifton R., Pontvianne F., Snoek L.B., Millenaar F.F., Schulkes R.K., van Driel R., Voesenek L.A., Spillane C., Pikaard C.S., Fransz P., Peeters A.J. PHYTOCHROME B and HISTONE DEACETYLASE 6 control light-induced chromatin compaction in Arabidopsis thaliana. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Probst A.V., Mittelsten Scheid O. Stress-induced structural changes in plant chromatin. Curr. Opin. Plant Biol. 2015;27:8–16. doi: 10.1016/j.pbi.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 61.Rosa S., Shaw P. Insights into chromatin structure and dynamics in plants. Biology (Basel) 2013;2:1378–1410. doi: 10.3390/biology2041378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang H., Dittmer T.A., Richards E.J. Arabidopsis CROWDED NUCLEI (CRWN) proteins are required for nuclear size control and heterochromatin organization. BMC Plant Biol. 2013;13:200. doi: 10.1186/1471-2229-13-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sakamoto Y., Takagi S. LITTLE NUCLEI 1 and 4 regulate nuclear morphology in arabidopsis thaliana. Plant Cell Physiol. 2013;54:622–633. doi: 10.1093/pcp/pct031. [DOI] [PubMed] [Google Scholar]
- 64.Yuan D., Lai J., Xu P., Zhang S., Zhang J., Li C., Wang Y., Du J., Liu Y., Yang C. AtMMS21 regulates DNA damage response and homologous recombination repair in Arabidopsis. DNA Repair (Amst.) 2014;21:140–147. doi: 10.1016/j.dnarep.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 65.Roy S., Choudhury S.R., Sengupta D.N., Das K.P. Involvement of AtPol in the repair of high salt- and DNA cross-linking agent-induced double strand breaks in arabidopsis. Plant Physiol. 2013;162:1195–1210. doi: 10.1104/pp.113.219022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hawley B.R., Lu W.T., Wilczynska A., Bushell M. The emerging role of RNAs in DNA damage repair. Cell Death Differ. 2017;24:580–587. doi: 10.1038/cdd.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier Project accession: PXD020762 and Project DOI: 10.6019/PXD020762.