

Summary
The characterization of cancer genomes has provided insight into somatically altered genes across tumors, transformed our understanding of cancer biology, and enabled tailoring of therapeutic strategies. However, the function of most cancer alleles remains mysterious, and many cancer features transcend their genomes. Consequently, tumor genomic characterization does not influence therapy for most patients. Approaches to understand the function and circuitry of cancer genes provide complementary approaches to elucidate both oncogene and non-oncogene dependencies. Emerging work indicates that the diversity of therapeutic targets engendered by non-oncogene dependencies is much larger than the list of recurrently mutated genes. Here we describe a framework for this expanded list of cancer targets, providing novel opportunities for clinical translation.
The majority of recently approved molecularly targeted cancer drugs are specific for oncoproteins encoded by somatically mutated genes. These agents include proteins essential for the survival of, expressed in specific cell lineages (e.g. estrogen receptor and androgen receptor), or proteins that inhibit immune responses such as PD-1. The strong association of these genes with specific tumor subtypes or with immunological phenotypes led to the discovery of small molecules and antibodies specific for these targets in particular cancers. Although large scale efforts to sequence cancer genomes facilitated the identification of recurrent somatically altered cancer genes, deciphering the deluge of information from these efforts created new challenges for cancer target discovery. In prior work, the Cancer Target Discovery and Development (CTD2) Network defined criteria to assess the strength of evidence supporting a specific target (Cancer Target and Development, 2016), allowing the prioritization of drivers that play a direct role in tumor initiation, maintenance or metastasis. Work from the network using this framework has facilitated the translation of discoveries in experimental models to clinical trials designed to test whether modulating the activity of specific targets leads to clinical responses (Table 1).
Table 1.
List of clinical trials initiated based on the CTD2 Network research findings (two different CTD2 centers are based at UCSF)
| CTD2 center |
ClinicalTrials.gov identifier |
Cancer type | Intervention/treatment | |
|---|---|---|---|---|
| 1 | Columbia University | NCT04028245 | Clear cell renal cell carcinoma | SPARC1 (Spartalizumab and Canakinumab) |
| 2 | Columbia University | NCT03211988 | Gastroenteropancreatic neuroendocrine tumors | Entinostat (deacetylase inhibitor) |
| 3 | Columbia University | NCT02066532 | HR−/HER2+ breast cancer | Ruxolitinib (JAK inhibitor) + trastuzumab (HER2 receptor antagonist) |
| 4 | Columbia University | NCT02632071 | Inflammatory breast cancer | Ricolinostat (HDAC6 inhibitor) + nab-paclitaxel (mitotic inhibitor) |
| 5 | Columbia University | NCT03951831 | Metastatic prostate cancer | PRIME-CUT (REGN2810 + Degarelix + Leuprolide Acetate + Docetaxel) |
| 6 | Columbia University | NCT04301414 | Prostate cancer | NEO-RED-P (BMS-986218 and Degarelix + Degarelix) |
| 7 | DFCI | NCT03654716 | Rhabdoid tumors | ALRN-6924 (MDM2/MDMX inhibitor) |
| 8 | Emory University | NCT04348292 | Lung non-small cell carcinoma | (Durvalumab + Sirolimus) |
| 9 | FHCRC | NCT02508246 | Head and neck squamous cell carcinoma | MK-1775 (WEE1 inhibitor) + docetaxel (antimicrotubule agent) + cisplatin (alkylating agent) |
| 10 | OHSU | NCT03557970 | Acute myeloid leukemia | JNJ-40346527 (inhibitor of CSF1R) |
| 11 | OHSU | NCT03874052 | Acute myeloid leukemia | Ruxolitinib (JAK inhibitor) + venetoclax (Bcl2 inhibitor) |
| 12 | UCSD | NCT03841110 | Solid tumors | FT500 (iPSC-derived natural killer cells) |
| 13 | UCSF (1) | NCT01402284 | Multiple myeloma | Carfilzomib (proteasome inhibitor) + lenalidomide (angiogenesis inhibitor) + dexamethasone (antiinflammatory) |
| 14 | UCSF (1) | NCT04085315 | Non-small cell lung cancer— EGFR mutant | Osimertinib (EGFR inhibitor) + alisertib (aurora kinase inhibitor) |
| 15 | UCSF (2) | NCT02947165 | Squamous cell carcinomas | NIS 793 (anti-TGF beta antibody) + PDR 001 (anti-PD-1 antibody) |
In addition, systematic functional studies performed by CTD2 and others have identified new categories of cancer targets beyond those inhibited by approved cancer drugs. Indeed, the number and diversity of these putative cancer targets far exceed oncogenes identified to date. This expanded repertoire of targets can be classified as tumor cell autonomous (intrinsic) or microenvironment-mediated (extrinsic) (Figure 1). Tumor intrinsic factors include mutated oncogenes, epigenetic changes, transcriptional and signal transduction dysregulation, aberrant pathway and metabolic activity, and changes in DNA damage responses. Together the effects of oncogenes as well as cell state changes result in stress responses that require mitigation strategies for the tumor to survive (Luo et al., 2009), often eliciting oncogene dependency (Weinstein, 2002). In particular, tumors that harbor these oncogenes often exhibit cell death when signaling induced by these oncogenes is extinguished, a phenomenon referred to as oncogene addiction. These cell state changes may introduce therapeutic vulnerabilities that can be exploited through synthetic lethal strategies as well as emergent therapeutic opportunities due to regulatory, signaling and metabolic network rewiring. Tumor extrinsic factors include changes in the tumor microenvironment and alterations in the differentiation state and composition of tumor-infiltrating stromal cells, such as immune cells and fibroblasts. At the organismal level, aberrant endocrine signals and the microbiome impact cancer pathogenesis and response to therapy. Intrinsic and extrinsic factors can affect cancer cell plasticity and their persistence during therapy. In this perspective, we describe an growing repertoire of intrinsic and extrinsic classes of targets as well as therapeutic opportunities.
Figure 1. Categories of cancer targets.
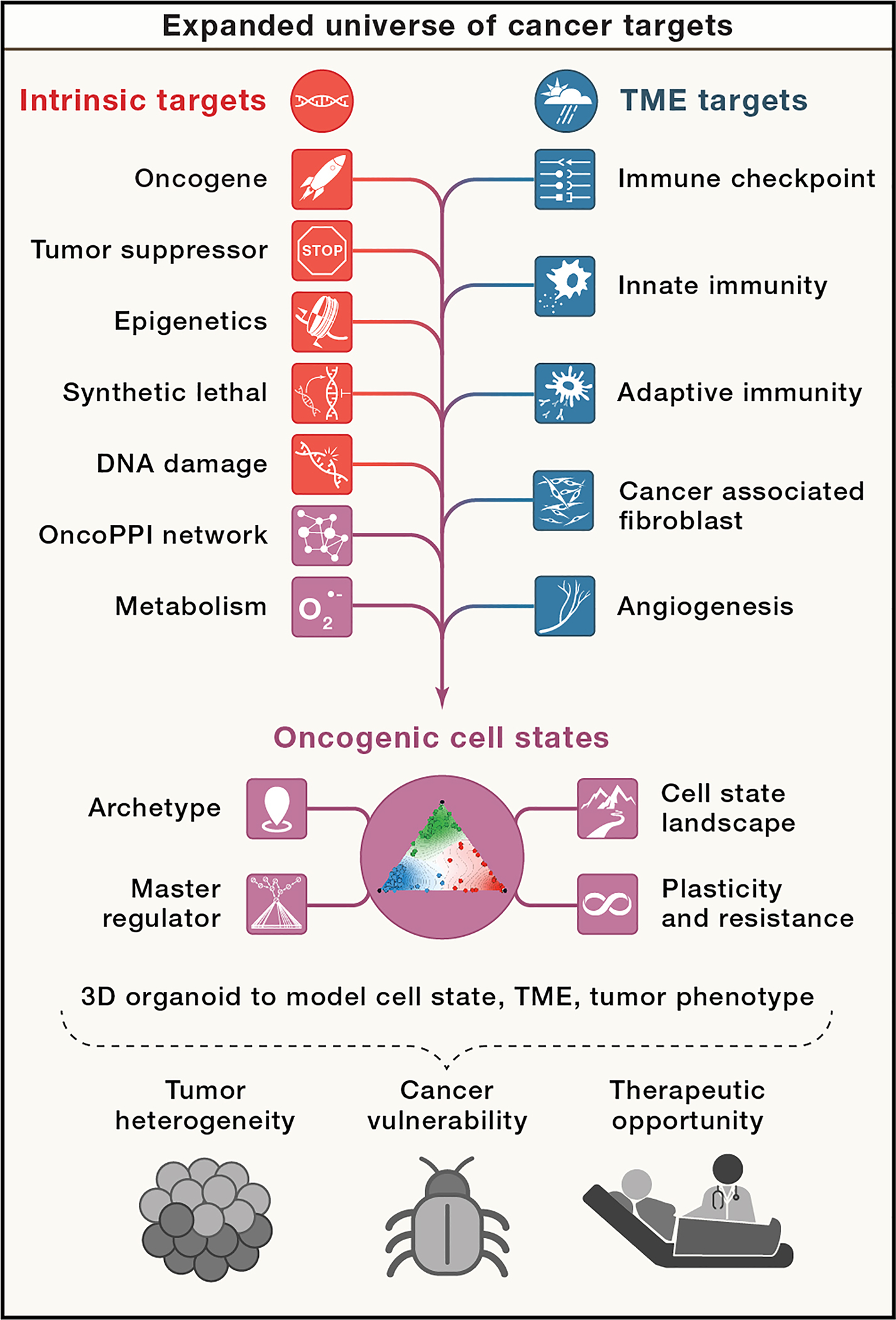
Recent studies have identified an expanded universe of cancer targets that include both tumor intrinsic targets and extrinsic targets including components of the tumor microenvironment. Among growing number of emerging targets (in green), the concept of oncogenic cell states captures the multi-dimensional complexity of a tumor with dynamic transition states, underlying regulatory mechanisms, and subpopulation-dependent plasticity that defines cellular phenotype and therapeutic response. These new targets are the consequence of tumor heterogeneity and represent non-oncogene-dependent cancer vulnerabilities, which inform new types of therapeutic opportunities.
I. Tumor intrinsic targets
Oncogene Addiction
The initial drugs targeting oncogenic tyrosine kinases showed remarkable efficacy. For example, Imatinib (Gleevec) transformed the treatment of chronic myeloid leukemia (CML), due to the robustness of BCR-ABL as a driver oncogene in CML (Braun et al., 2020). Other success stories include epidermal growth factor receptor (EGFR)-, ALK- or ROS1-driven lung cancers (Arbour and Riely, 2019), KIT-mutant gastrointestinal stromal tumors (Bannon et al., 2017), and ERBB2 in HER2+-breast cancer (Waks and Winer, 2019). However, oncogenic signaling in solid tumors is genetically complex and empirical experience suggests limited activity for a majority of drugs blocking any given individual oncogene aberrantly expressed in solid tumors.
As examples of genetic complexity driving oncogenic signaling in solid tumors, aberrant tumorigenic protein function arises from deleterious events in coding genes, such as point mutations, fusions, translocations, copy number changes, methylation, and transcriptional dysregulation. Gene copy-number alterations, among the most prevalent somatic mutational events in human cancers (Beroukhim et al., 2010), (Stratton et al., 2009), are fundamental drivers for tumor initiation and evolution. Chromosomal rearrangements produce deleterious events including creation of fusion genes with novel functions, altered gene dosage to levels that drive regulatory pathway activity towards cancer progression (Tsherniak et al., 2017) or translocations that change physiologic regulation of gene expression. Aneuploidy is also frequent, yet it recapitulates similar issues as large-scale paralog formation, making it difficult to unravel bystander passenger events acquired during cancer evolution from bona fide driver events that cause cancer or result in more aggressive tumors.
Dysregulated signaling represents an important conserved oncogenic mechanism. For example, RAS is mutated in ~19% of all human cancers (Prior et al., 2020). In addition, RAS is activated by mutation of tyrosine kinases and RAS regulatory proteins (NF1, SOS1, PTPN11). This pathway is also activated by mutation of downstream mediators (BRAF, RAF1). Therapeutic inhibition of RAS signaling has proven difficult, due to the challenging biochemical properties of RAS itself, and redundancies in downstream signaling. However, recent work has renewed enthusiasm for direct RAS inhibition, including approaches that target RAS dimerization or drugs that covalently bind G12C mutant RAS, locking it in a GDP-bound inactive state (Moore et al., 2020). In contrast, targeting MEK and other downstream proteins in the RAS signaling cascade has shown limited efficacy in RAS-driven malignancies, likely owing to dose-limiting toxicity and loss of auto-inhibitory feedback. Inhibitors of BRAF have shown marked, albeit temporary, efficacy in BRAF-mutant melanoma. Both tyrosine kinase- and RAS-pathway mutations also result in activation of other downstream, druggable signaling pathways, such as phosphoinositide-3 kinase (PI3K). PI3K can also be activated by direct mutations of the PIK3CA gene, and inhibitors of PI3K or downstream mediators AKT or mTOR have shown variable efficacy against cancers with PIK3CA or upstream mutations (Cox et al., 2014), (Gysin et al., 2011).
Oncogene amplification has proven challenging to target. The transcription factors MYC and MYCN, for instance, are recurrently amplified or translocated in a subset of cancers and the MYC network is dysregulated in a significant percentage of human cancers (Dang, 2012; Schaub et al., 2018). Therapeutic targeting of MYC has been challenging due to the lack of enzymatic activity or suitable binding sites for drugs. Indirect targeting of MYC via BRD4 inhibition, regulators of MYC stability, or inhibition of downstream interacting kinases are important areas of investigation. Other approaches to reverse effects of amplified genes, such as targeting proteins for degradation or use of CRISPR, siRNA and antisense therapeutics, remain of clinical and conceptual interest.
Epigenetic regulators are also mutated recurrently in cancer. While challenging to target as a class, an increasing number are becoming tractable. For example, two DNA methyltransferase (DNMT) inhibitors, azacytidine and decitabine, are approved for the treatment of myelodysplastic syndrome. Numerous drug-development efforts are focused on other recurrently mutated epigenetic regulators, either via direct inhibition of enzymatic activity (i.e. EZH2) or immediate downstream dependencies (MLL-Menin or MLL-DOT1L). However, the broad actions of epigenetic regulators on many genes raises questions regarding the therapeutic index for these inhibitors.
Tumor Suppressor Rescue
Loss of tumor suppressor genes occurs commonly in cancer and several approaches to target these loss of function events are now in development. For example, loss-of-function mutations in the ubiquitin ligase CBL result in increased signaling through a number of tyrosine kinases. Similarly, mutations in PTEN, which inactivate its phosphatase activity, increase PI3K signaling. In these cases, pharmacological targeting of the activated pathway, rather than the aberrant protein itself, holds promise. Targeting mutant TP53 with drugs that stabilize its 3D-structure and restore its normal function has also emerged as a promising approach (Levine, 2019). More generally, a variant-directed protein–protein interaction inducer screen identified small molecules that restored interactions lost as a result of tumor suppressor mutations (Tang et al., 2020). This study identified a bisindolylmaleimide derivative that restored interaction between mutated SMAD4 and SMAD3 and reactivated the cell growth suppression function of the SMAD4/SMAD3 complex. Collectively, these approaches offer versatile strategies to target many tumor suppressor mutations.
Synthetic Lethal Targets
Experimental approaches from yeast genetics (Hartwell et al., 1997) led to screens that identify proteins whose activities are essential for cancers harboring specific oncogenic mutations. Synthetic lethality allows targeting of cancers that harbor mutations in undruggable proteins with potential to improve therapeutic index. The success of poly (ADP-ribose) polymerase (PARP) inhibition in the setting of BRCA1 and BRCA2 deficiency for instance, provided an important proof of principle (Lord and Ashworth, 2017). Inhibition of PARP capitalized on defects in DNA repair induced by loss of BRCA1/2 and other homologous recombination DNA repair mediators, leading to mitotic catastrophe. One mechanism of resistance to PARP inhibition is the reversion of BRCA1 to produce a wild-type protein, confirming a direct relationship between BRCA1 mutation and PARP inhibitor sensitivity. Recent genome-scale genetic and small-molecule screens identified several new synthetic lethal combinations, including the finding that the helicase WRN necessary for the survival of tumors exhibiting microsatellite instability (Chan et al., 2019) and BET, SRC and BCL2 family inhibitors synergize with PARP inhibitors (Lui et al., 2020).
Recent studies have defined new classes of synthetic lethal interactions. As predicted by Elledge and coworkers (Luo et al., 2009), some of these synthetic lethal genes are required to suppress cell-stress mechanisms induced by particular oncogenes. For example, CSNK1E is required for the survival of Myc-amplified tumors (Toyoshima et al., 2012). Additional synthetic lethal opportunities are created when essential genes are located near tumor suppressors. The CYCLOPS (Nijhawan et al., 2012) (Copy number alterations Yielding Cancer Liabilities Owing to Partial losS) genes are never deleted homozygously and gene expression closely matches copy number. Inhibition of CYCLOPS genes induces cell death only in cells that lack the corresponding tumor suppressor gene due to copy number loss. For example, PRMT5 is required for survival of tumors that lose MTAP, which is frequently lost due to its proximity to the commonly deleted tumor suppressor gene, CDKN2A (Mavrakis et al., 2016) (Kryukov et al., 2016). CYCLOPS genes are currently the most common copy-number associated gene dependency (Tsherniak et al.) and likely represent a general paradigm in cancer synthetic dependencies.
Finally, synthetic essentiality also occurs when deletion of one paralog or family member renders the cell dependent on the remaining one. For example, tumors that harbor deletions of ENO1 require expression of ENO2 (Muller et al., 2012). A recent interrogation of cancer genomes identified 87 instances for which functional loss of one paralog was associated with a dependency on another paralog (Tsherniak et al.). Identifying and characterizing paralog dependencies may reveal new genes that are sensitive to expression dosage, uncovering new targetable dependencies. Similarly, in cancer cell lines, mutation of ARID1A confers sensitivity to knockdown of the paralog ARID1B, while superimposed loss of ARID1B in ARID1A mutant cells increases radiosensitivity (Helming et al., 2014; Niedermaier et al., 2019).
DNA Damage Response
Unrepaired cellular DNA damage results in mutations or altered chromatin structure, events that drive the initiation and progression of tumors. Deleterious events in genes that mediate DNA damage response (DDR) are frequent across multiple tumor lineages. Thus, understanding DDR and its clinical impact is critical to improving cancer outcomes. Defects in DDR cause genomic instability that promotes oncogenesis, so accurate annotation contributes to early detection and cancer prevention. DDR deficiencies can also sensitize cells to specific therapeutic cancer regimens and are associated with responses to conventional chemo- and radiation treatments (Brown et al., 2017). As noted above, mutations in the BRCA1/2 genes result in synthetic lethality for PARP inhibitors.
Despite systematic identification of genes critical for DDR function and drug sensitivity, many observed mutations have not been assessed for functional, therapeutic, and clinical relevance. It is thus difficult to predict whether a mutation highlights a critical dependency or lacks therapeutic relevance. Saturation mutagenesis, an approach to address this challenge, identified BRCA1 and PARP1 mutations as inducing protein function loss (Findlay et al., 2018) or PARP inhibitor resistance (Pettitt et al., 2018), respectively. Identifying downstream effects caused by DDR remains another promising approach.
II. Emerging Cancer Targets
Protein-protein Interactions
Oncogenic missense mutations can alter protein–protein interactions and drive cancer progression. Thus, identifying and characterizing critical nodes and hubs in cancer-related protein–protein interaction networks, which control the output of oncogenic programs, may reveal unique opportunities for therapeutic intervention. In particular, the systematic interrogation of new protein-protein interactions driven by mutant oncoproteins may reveal new types of cancer targets that are cancer specific.
For example, the cancer-associated protein-protein interaction network (OncoPPi) focuses on experimentally generated interactions among proteins with known or likely involvement in cancer (Li et al., 2017b). In contrast to interactomes established by proteomics approaches, such as immunoprecipitation-mass spectrometry, OncoPPi presents binary interactions of cancer-related proteins derived from a proximity-based biosensor to reflect direct interactions and, coupled with genomics, clinical and pharmacological information, facilitates therapeutic targeting. OncoPPi reveals prominent protein-interaction hubs with new partners and suggest novel mechanisms of action for major oncogene drivers, such as MYC with NSD3. Importantly, OncoPPi uncovers interactions for non-enzymatic proteins, offering potential intervention strategies by perturbing their interactions to target those once “undruggable” classes of proteins. Similarly, efforts to comprehensively map protein–protein interactions in human cells (Huttlin et al., 2017) or in specific oncogenic contexts provide new ways to identify protein-protein interactions and complexes involved in cancer phenotypes. For structure-based prediction of protein-protein interactions, the PrePPI algorithm and database (Zhang et al., 2012) offers a valuable resource for the scientific community.
Metabolic Vulnerabilities
The rapid proliferation of cancer cells requires extensive escalation of cellular catabolic and anabolic metabolism to meet energy and structural needs (Li et al., 2019). For example, it has long been recognized that expanded nucleotide synthesis leads to sensitivity to nucleoside analogs including 5-fluorouracil (5-FU), gemcitabine and cytarabine (Wagner et al., 2007), while elevated glucose uptake (part of the Warburg effect) enables tumor imaging using fluorodeoxyglucose positron emission tomography (FDG-PET) (Zhu et al., 2011). Recently, significant improvements in metabolomics and isotope tracing (Jang et al., 2018) propelled the discovery of many other metabolic alterations in cancer. While most metabolic changes are neutral, or only slightly modify cancer cell fitness under stress (Vander Heiden and DeBerardinis, 2017), certain pathways are essential for the progression of selected cancers and can be exploited therapeutically.
Despite the general increase in glucose uptake and consumption in tumors, direct inhibition of aerobic glycolysis using glucose mimetics or inhibitors of pyruvate kinase has seen limited preclinical and clinical success (Cortes-Cros et al., 2013) (Raez et al., 2013). In addition to their dependence on glucose metabolism, cancer cells are also dependent on the uptake or de novo biosynthesis of a subset of amino acids, including glutamine (DeBerardinis et al., 2007), glycine (Jain et al., 2012), serine (Kim et al., 2015; Piskounova et al., 2015), and aspartate (Sullivan et al., 2015; Sullivan et al., 2018) in a context-, lineage-, and oncogene-dependent manner. These amino acids not only contribute to protein synthesis, but also to the biosynthesis of other essential metabolites, such as pyrimidines, purines, phospholipids, glutathione and regeneration of NADPH, as approaches to limit toxicity from reactive oxygen species (ROS) (Luengo et al., 2017).
Certain tumors contain high levels of polyunsaturated lipids Due to limited de novo fatty acid biosynthesis in normal tissues other than the liver, adipose tissue, and lactating breast (Menendez and Lupu, 2006), lipid metabolism in tumors is thus an attractive target for therapeutic intervention, by modulating fatty acid uptake, synthesis, unsaturation, and incorporation into structural lipids (Viswanathan et al., 2017). For example, the tumorigenic potential of ovarian cancer stem cells is dependent on SCD1 (Li et al., 2017a), whereas metastasis-initiating cells are reliant on fatty acid receptor CD36-mediated fatty acid uptake (Pascual et al., 2017). While lipids are often available through regular diet, pharmacological inhibition of fatty acid synthesis may need to be combined with dietary interventions to generate a therapeutic window. One of the major challenges in targeting lipid metabolism is our limited understanding in the plasticity of cancer lipidome.
More recently, mutations in isocitrate dehydrogenase 1 and 2 (IDH1, IDH2), which are frequent in myeloid malignancies, glioma, chondrosarcoma, cholangiosarcoma, and hepatocellular carcinoma (Tommasini-Ghelfi et al., 2019), direct production of the oncometabolite 2-hydroxyglutarate (2-HG), which inhibits the demethylation of DNA, leading to gene silencing. This neomorphic enzymatic activity can be blocked by drugs that inhibit 2-HG production.
In addition, it has long been appreciated that some tumors aberrantly express specific metabolites, such as serine. In some tumors, increased expression of the serine synthesis enzyme phosphoglycerate dehydrogenase (PHGDH) results from copy number gain of a region on chromosome 1p (Beroukhim et al., 2010(Locasale, 2011 #3083(Possemato, 2011 #3084), while other tumors exhibit increased serine levels as a consequence of oncogenic signaling, including NRF2 and ATF4 signaling (DeNicola et al., 2015) or hypoxia responses (Samanta et al., 2016). Suppressing serine metabolism in these tumor cells leads to tumor regression.
A class of synthetic lethal genes acts by targeting aberrant metabolism necessary for cancer-cell survival. For example, ODGH is required for the survival of tumors that harbor mutations in PIK3CA due to increased reliance on aspartate signaling (Ilic et al., 2017). Although targeting tumor metabolism requires a therapeutic index between tumor and normal tissue, this class of cancer targets remains promising.
Cell States
Cell states, archetypes, and tumor checkpoints:
The enabling technologies that represent the pillars of Systems Biology, such as molecular interaction networks and multivariate analysis, have led naturally to the concept of “cell states.” Cell states represent high-dimensional vectors of variables that uniquely determine a cell’s phenotype. For instance, a tumor may comprise multiple subcellular populations with distinct drug sensitivity, metabolism, or stemness characteristics. In that case, the cell state would be a vector that uniquely identifies the cells in each subtype. Thus, this paradigm is especially useful to account for the heterogeneity of primary tumors as composites that incorporate multiple, often highly plastic cellular and micro-environment states (Figure 2).
Figure 2: Oncogenic targets and cell states.

This schematic represents the coexistence of distinct, yet isogenic malignant cell states within a tumor, each one presenting an equally distinct repertoire of non-oncogene dependencies. To avoid cluttering, tumor microenvironment-related cell states are not shown, even though they are involved in critical paracrine interactions with tumor cells. (A) Schematic representation of isogenic tumor cells presenting with distinct transcriptional states and epigenetic profiles. These include: (i) a low-proliferative (i.e., quiescent) meta-stable stem-like progenitor cell state capable of self-renewal and asymmetric differentiation (ii) two stable, differentiated cell states, persisting for long time periods, associated with either an epithelial (proliferative) or a mesenchymal (quiescent) cell phenotype, and (iii) an additional neuroendocrine (quiescent) stable state that can only be achieved by drug-induced transdifferentiation. The size of the arrows illustrates the likelihood of transition from one cell state to another. Stem-like progenitors can differentiate into either epithelial or mesenchymal cells, which can plastically reprogram between these states, albeit at different rates, for instance as a result of epithelial mesenchymal transformation processes. The neuroendocrine state is not pathophysiologically accessible but can be reached via drug-mediated transdifferentiation from the epithelial cell state. (B) Schematic representation of the canalization entropy landscape that underlies the possible cell states. Cells tend to move from a higher to a lower entropy state with a probability that is inversely proportional to the entropic barrier that separates them DE1 (i.e., height of the peak separating two adjacent valleys) and directly proportional to their differential entropy DE2 (i.e. differential depth of two adjacent valleys). For instance, an epithelial state cell can reprogram to a mesenchymal state cell because the entropic barrier between the two is low (plasticity) but the forward direction is more likely than the reverse one because the entropy of the mesenchymal state is lower. (C) Schematic representation of the tumor composition changes following drug treatment or spontaneous progression. For simplicity, only four transitions are illustrated, including: (i) Metastatic progression, associated with an increased ratio of mesenchymal to epithelial cells but no change in the fraction of stem-like progenitors, (ii) Chemotherapy treatment, resulting in ablation of the proliferative epithelial state and increase of the stem-like progenitor and mesenchymal quiescent states, (iii) Targeted Therapy A, inducing reduction of the stem-like progenitor and mesenchymal quiescent states and increase of the epithelial proliferative state, and (iv) Targeted Therapy B, resulting in the emergence of a novel neuroendocrine state resulting from drug-induced transdifferentiation.
One paradigm to identify oncogenic cell states is to model them as reproducible configurations of pathway activation or repression, defined by transcriptional, dependency or sensitivity profiles. The most common and representative configurations provide operational “archetypes” or “tumor checkpoint modules” (Califano and Alvarez, 2017) that help categorize cancers into functional taxonomies. While archetypes define the cell state itself, tumor checkpoints define regulatory protein modules comprising Master Regulator proteins responsible for mechanistically implementing and maintaining cell state. For example, the mesenchymal cell state of glioblastoma, which co-exists with isogenic proneural states at the single cell level (Neftel et al., 2019), was shown to be mechanistically induced by aberrant activation of a tumor checkpoint comprising three synergistic Master Regulator proteins, CEBPβ, CEBPδ, and STAT3 (Carro et al., 2010), whose aberrant activation is induced mechanistically by specific mutations in upstream pathways, including focal STAT3 amplifications, and homozygous KLHL9 deletions (Chen et al., 2014).
Cell states can be stable, meta-stable, or transient. Stable states represent regions of the state-space where cells are “trapped” for days – such as at the end stages of lineage development (Stadhouders et al., 2019) (Arendt et al., 2016). In contrast, meta-stable states correspond to regions where cells are trapped for several hours, rather than days, on their way to a more stable state or following a transient perturbation. Finally, transient states represent the most sparsely populated regions of state-space, populated by cells rapidly transitioning between states. This concept has increased relevance for cancer biology and therapeutics, because of the advent of methods allowing the direct observation of tumorigenesis, tumor plasticity, and adaptive resistance at the single-cell level. However, these advances are currently limited by the reliance on measuring gene expression (Grosse-Wilde et al., 2015) (Tsoi et al., 2018). It is also important to provide a framework to interpret the results from studies of transitions within a complex “landscape” of states (Janes, 2016) (MacLean et al., 2018) (Neftel et al., 2019).
Both tumor progression (e.g., to metastasis) and sensitivity to therapeutic agents largely depend on the presence of specific stable and meta-stable cell states – both tumor- and microenvironment-related – rather than on tumor histology, genetics and natural history of the tumor evolution (Kim et al., 2017). For instance, poor outcomes in breast cancer patients have been linked to a mixed epithelial–mesenchymal chimeric cell state (MacLean et al., 2018). Further, novel states were identified in acute myeloid leukemia (AML) that did not recapitulate traditional cell-surface markers, but whose gene-expression signature was predictive of patient survival. Neuroendocrine tumors cell states were also predictive of response to therapy (Alvarez et al., 2018). Finally, melanoma cell lines and tumors displayed a treatment-induced differentiation trajectory with four phases, and an inverse relationship between differentiation progress and sensitivity to iron-dependent compound-induced oxidative stress (Viswanathan et al., 2017).
Cell states and master regulators:
Master regulator (MR) proteins represent the mechanistic regulators of transcriptional cell-state homeostasis. As such, MR activity analysis permits identification of both stable and meta-stable cancer-related cell states and cell-state transitions (Califano and Alvarez, 2017), as well as of proteins that can reprogram cell state when ectopically expressed. For example, ectopic co-expression of three computationally-inferred prostate lineage MRs, AR, FOXA1, and NKX3.1, was sufficient to reprogram fibroblast to a competent prostate epithelium tissue (Talos et al., 2017) while shRNA-mediated co-silencing of computationally-inferred mesenchymal GBM MRs, CEBPβ and STAT3, was sufficient to reprogram mesenchymal cells to a proneural state and abrogate tumorigenesis in vivo (Carro et al., 2010). Candidate MR proteins can effectively be identified by network-based analysis of mRNA profiles, e.g., via the VIPER algorithm, which—akin to a multiplexed gene-reporter assay—measures enrichment of particular protein transcriptional targets in genes that are differentially expressed in a specific cell state. MR-based studies have identified novel, experimentally validated drivers and mechanisms in malignancies ranging from lymphoma, neuroblastoma, and glioma to prostate, breast, and gastroenteropancreatic neuroendocrine tumors (Califano and Alvarez, 2017).
Elucidation of the cellular logic responsible for the implementation and maintenance of cancer cell states has identified tightly autoregulated modular structures (Tumor Checkpoints), which comprise a small number of MR proteins working together to maintain cancer cell-state homeostasis by integrating the effect of genetic and epigenetic alterations in their upstream pathways (Paul et al., 2019). Indeed, genetic or pharmacological inhibition of Tumor Checkpoint MRs leads to their collapse and loss of cancer viability in vitro and in vivo (Alvarez et al., 2018), providing a means to abrogate the effects of multiple genetic alterations.
Cell-state plasticity and resistance:
Tumor-cell plasticity and adaptive responses to therapeutic stress play key roles in acquired drug resistance. Drug-tolerant cancer persister cells are a prime example. Found within a wide range of tumor types, persister cells are proposed to arise as a result of stochastic processes (Kupiec, 1997), introducing transient phenotypic heterogeneity into tumor-cell populations (Raj and van Oudenaarden, 2008). For example, non-genetic transcriptional variability within cell populations may identify cells likely to resist drug treatment in melanoma (Shaffer et al., 2017). In general, persister cells share common features with the stress mitigation pathways of myofibroblastic non-cancer cells under stress conditions other than those induced by cancer treatments (see cancer-associated fibroblasts (CAFs) discussed later). They possess: 1) a distinct chromatin state, 2) increased stemness, and 3) a switch in lipid content from highly saturated and monounsaturated to highly polyunsaturated lipids and phosphoplipids. Persister cells often implement a more quiescent state to escape chemotherapeutic stress and can in some cases, become resensitized to treatment upon drug withdrawal, pointing toward a non-genetic mechanism of drug tolerance. They can maintain this pro-survival state for weeks to months of drug treatment. However, in some patients, a subset of persister cells can re-enter the cell cycle and regrow tumors that are often irreversibly drug-resistant due to selection of genetic or epigenetic alterations. Therefore, targeting persister cells holds great promise to prevent or delay acquired drug resistance and tumor relapse (Viswanathan et al., 2017).
Persister cells present specific vulnerabilities. Many have increased levels of polyunsaturated phospholipids, making them exquisitely sensitive to chemical inducers of ferroptosis. Ferroptosis is a caspase-independent non-apoptotic cell death due to excessive polyunsaturated lipid peroxidation within cellular membranes. The antioxidant enzyme glutathione peroxidase 4 (GPX4) catalyzes the conversion of the resulting and otherwise lethal hydroperoxides into their corresponding alcohols (many of which regulate hematopoietic cells in the cancer microenvironment), protecting them from ferroptotic death. Chemical inhibition or genetic ablation of GPX4 selectively and potently induces persister cell ferroptosis in a wide array of solid tumor types (Viswanathan et al., 2017) and prevents melanoma relapse following targeted therapy in mouse xenografts (Hangauer et al., 2017). GPX4 is a vulnerability in epithelial tumors that undergo EMT, melanoma cells that transdifferentiate from a high-MITF to a high AXL-cell state in response to targeted therapy and immunotherapy (Tsoi et al., 2018), prostate cancer cells that transdifferentiate into an androgen-indifferent state, and cancers derived from tissues with an intrinsic high-polyunsaturated lipid state such as sarcomas (Viswanathan et al., 2017). Therefore, GPX4 and targets related to this ferroptotic cell circuitry represent promising non-oncogene therapeutic targets for a spectrum of high polyunsaturated lipid-based tumors.
III. Tumor Microenvironment
The tumor microenvironment (TME) is a rich milieu in which diverse non-neoplastic cell types and extracellular matrix proteins interact to regulate the biology of cancer cells (Binnewies et al., 2018). Deeper mechanistic insights into these dynamic molecular exchanges have enabled therapeutic strategies directly targeting aspects of the TME that are essential for tumor function (Figure 3). Proof-of-principle for TME-directed cancer therapy was achieved by anti-angiogenic treatments targeting Vascular Endothelial Growth Factor (VEGF), inhibiting the tumor’s recruitment of new blood vessels and exploiting the relative lack of neovascularization in healthy organs (Apte et al., 2019). The intricate cellular complexity of the TME spans immune cells, fibroblasts, ECM (Wong et al., 2017) and even neuronal elements (Monje, 2020; Venkatesh et al., 2019), creating a multitude of potential targets.
Figure 3. Tumor microenvironment targets.
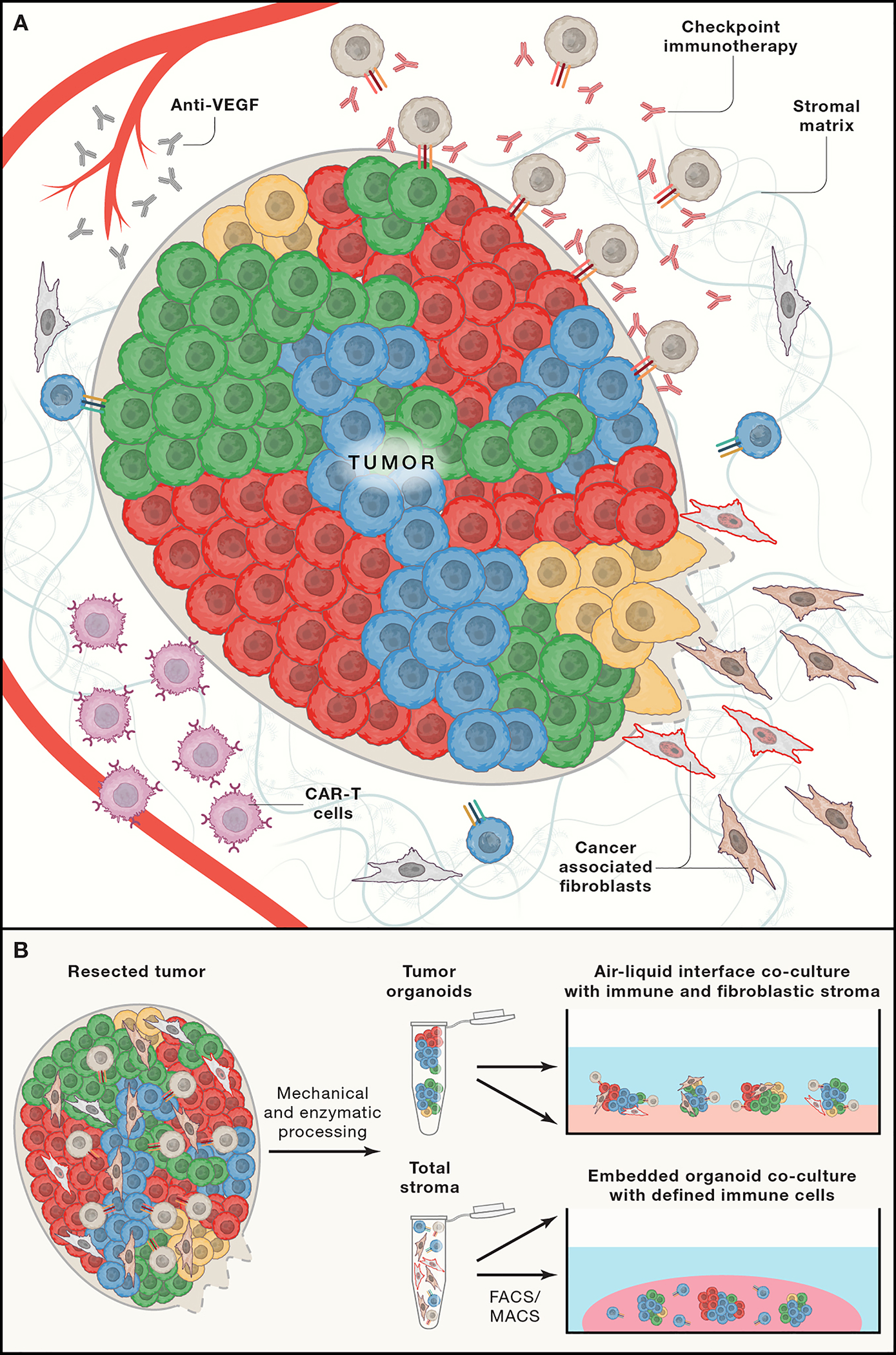
(A) Tumors are composed of a complex mixture of cancer cells in genetically and phenotypically distinct cell states surrounded by a fibrillar extracellular matrix and a diverse fibroblastic and immune stroma. Current therapies target VEGF to inhibit angiogenesis, disrupt T-cell immune checkpoints with antibody based immunotherapy, disrupt survival signaling pathways that are mediated by cancer associated fibroblasts, and deliver engineered immune cells to eliminate cancer cells. Our understanding of the molecular basis of interactions among cell types remains incomplete and therefore new methods are required to understand the individual and collaborative functions of these various cell populations. (B) Recent technical advances enable primary and metastatic tumors to be deconstructed into their constituent parts and then reassembled in culture with either total immune and fibroblastic stroma or through selective incorporation of molecularly defined stromal cell populations. These assays from a platform to identify new cancer targets and to test candidate therapeutic compounds in a systematic fashion.
Adaptive and innate immune cells
Recent TME-targeting strategies have focused on immune cells, spanning both adaptive and innate immunity, including lymphocytes, macrophages, natural killer (NK) cells, myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs) and others. The immune system is regulated by autocrine and paracrine cell-cell and cytokine interactions and exhibits a dynamic equilibrium between tumor-infiltrating and peripheral pools (Gajewski et al., 2013; Palucka and Coussens, 2016; Quail and Joyce, 2013). Modulation of immune function represents a high priority for cancer target identification and drug development, with strategies either exploiting the immunogenicity of tumor cells, spurring the host immune response, or both. Advances in immunotherapy have transformed the cancer treatment landscape in a number of cancers, embodied by antibody-based inhibitors of immune checkpoints such as PD-1/PD-L1 and CTLA-4, with a progression towards combinations of checkpoint inhibitors with themselves or with targeted therapies, chemotherapy and radiotherapy (Larkin et al., 2019; Pardoll, 2012; Socinski et al., 2018) as well as new immune populations such as macrophages (Kaneda et al., 2017). In parallel, “adoptive” cellular T-cell therapies involve infusion of chimeric antigen receptor T cells (CAR-T) effective for hematologic malignancies or T-cell receptor (TCR)-T cells directed against tumor antigen(s), the former having substantial efficacy in hematologic malignancies, or bulk infusion of ex vivo-expanded tumor-infiltrating lymphocytes (Hekman et al.) or NK cells (Hammerl et al., 2018; Rosenberg and Restifo, 2015; Tran et al., 2017).
New techniques link high-throughput CRISPR screening to tumor immunology, both in vitro or in vivo. Systematic CRISPR perturbation of cancer cells have revealed PTPN2, ADAR1, SWI/SNF complex members and other essential modulators of tumor immunotherapeutic action (Ishizuka et al., 2019; Lane-Reticker et al., 2018; Manguso et al., 2017; Pan et al., 2018; Patel et al., 2017). Conversely, CRISPR screens in T cells or dendritic cells have identified potential novel immunoregulatory molecules with relevenace to cancer immunotherapy (Dong et al., 2019; Parnas et al., 2015; Shifrut et al., 2018; Ye et al., 2019). Future tumor immunology screens may incorporate organoid approaches that preserve tumor cells along with infiltrating immune components, including T, B, NK cells and macrophages without reconstitution (Neal et al., 2018) or microfluidic approaches (Jenkins et al., 2018). Alternatively, tumor cell lines or organoids can be reconstituted with peripheral blood lymphocyte populations to identify both tumor cell intrinsic and extrinsic immune response modifiers (Dijkstra et al., 2018; Mo et al., 2019; Yuki et al., 2020).
Similar methods also enable co-culture with natural killer (NK) cells, a population known to be early responders to disseminated cancer cells and which can be directly modulated or used as cell-based therapies. Notably, in one set of studies, cancer cells were found to “co-opt” and convert NK cells to an alternative molecular state that promotes metastatic outgrowth (Chan et al., 2020). Analyses of receptor-ligand pairings between cancer cells and NK cells identified multiple cell surface receptors (e.g. Klgr1 and TIGIT) that can be inhibited with antibodies to block metastatic colonization and may have clinical relevance. Additionally, ligands for NK cell activating receptors are often upregulated on tumor cells or during infection (Miller and Lanier, 2019) and the loss of these ligands reduces NK cell recognition and killing. Notably, the NK cell activating ligands MHC Class I Polypeptide-Related Sequence A/B (MICA/B) are often down regulated by diverse tumors including leukemia, prostate cancer, melanoma, breast, lung, ovarian and colon carcinomas leading to decreased engagement of the NK cell activating receptor NKG2D and decreased anti-tumor activity (Lanier, 2019). Additionally, NKG2D ligands can be secreted by tumor cells leading to impaired NK cell function (Ashiru et al., 2010; Clayton et al., 2008; Hedlund et al., 2011; Lundholm et al., 2014). Therapeutically, a novel antibody that blocks this shedding of NKG2D ligands was found to improve NK cell-mediated anti-tumor activity (Ferrari de Andrade et al., 2018). Expression of NKG2D ligands by non-tumor cells such as endothelial cells can also lead to desensitization of NK cells and reduced anti-tumor activity (Thompson et al., 2017).
Cancer Associated Fibroblasts (CAFs)
Fibroblasts are an abundant cell type in normal and malignant tissues and accumulate along with fibrillar collagens in cancers characterized by a fibrotic response in pancreatic cancer. Genetically wild-type CAFs may be intrinsically less able to acquire resistance to targeted agents, compared to genomically unstable cancer cells. The CAF cell state changes during malignant progression, typically to a contractile myofibroblast or secretory phenotype, and therefore has similarities to the high-polyunsaturated lipid state described above, associated with therapy-resistant persister cancer cells. Single-cell RNA sequencing (scRNAseq) has identified additional phenotypic diversity in this population, including those with contractile vs. secretory behaviors. Mouse models of pancreatic cancer revealed context-specific tumor-promoting or tumor-inhibitory CAF functions (Biffi and Tuveson, 2020; Elyada et al., 2019; Hosein et al., 2020; LeBleu and Kalluri, 2018; Sahai et al., 2020). This complexity highlights the importance of robust physiologically relevant model systems that can define the precise function of a specific fibroblastic phenotype, enabling development of biomarkers to inform the selection of patients for targeted treatments, leading to the direct therapeutic targeting of CAFs for therapeutic purposes. These studies have greatly benefitted from co-cultures of CAFs with cancer cells or organoids which can then be exposed to candidate therapies (Ohlund et al., 2017). Alternatively, air-liquid interface tumor organoid cultures incorporate endogenous CAFs without requiring reconstitution (Neal and Kuo, 2016).
IV. Future opportunities
Identification of robust therapeutic targets across cancer types is a high priority. Tremendous efforts have led to the successful targeting of somatically altered oncoproteins. However, many cancers fail to express kinase oncogenes, and resistance to single agent therapy often rapidly occurs. As such, developing new therapeutic agents targeting the emerging classes of cancer targets described in this perspective may provide the means to use targeted approaches for more patients but also provide complementary agents to create combination regimens. The large and diversity of new types of cancer targets provides hope that all patients will benefit in the future from precision medicine-guided therapies.
In parallel, future efforts must consider cell-state heterogeneity in both the cancer cells themselves and the cells of the tumor microenvironment. The development of complex organoid co-cultures that incorporate fibroblastic, immune, and vascular compartments by reconstitution or by intrinsic preservation should further enable modeling of tumor micro-environmental influences on cell state (Neal et al., 2018; Ohlund et al., 2017; Yuki et al., 2020). For example, three dimensional (3D) cultures of primary tumor tissue maintain some of the cellular heterogeneity and cell state transitions observed in vivo (Roerink et al., 2018). 3D cultures also allow analysis of tumor-level phenotypes, like collective invasion, immune surveillance, and organ colonization, which are difficult to model in 2D culture (Lo et al., 2020). These types of approaches may also permit the development of more accurate animal models of human cancer.
In addition, single-cell RNA-seq analyses have revealed multiple distinct cell states with unequal contributions to tumor relapse. The selective depletion of stem-like progenitor populations or of a subset of persister cells, which may seed tumor relapse, represents a more tractable goal than completely eliminating all residual tumor cells. Furthermore, deeper understanding of the processes by which persister cells gain the ability to survive therapeutic stress and regrow may suggest new therapeutic approaches.
To improve patient outcomes, it is essential to target cancer not only in isolation, but also as part of a larger ecosystem. While deconvolution of the intricate composition and activities of the TME is in its infancy, notable functional demonstrations have included the clinical efficacy of immune- and blood vessel-targeted therapies, raising the potential for further target identification. The ability to effectively disrupt the tumor ecosystem may help address a major unmet clinical challenge of having to target multiple heterogeneous cell populations within tumors and the microenvironment. Further, as TME components fulfill important functions in other contexts, such as in healthy organs, the potential toxicity of therapeutic stromal targeting will require thorough evaluation. The cell state concept could significantly assist this process, by collapsing a daunting, essentially infinite universe of possible mutational patterns and TME to a very finite repertoire of distinct tumor cell states.
Although our initial framework to define criteria to assess the strength of evidence supporting a specific target was focused on oncogene targets, the same principles apply to these new categories of cancer drivers. Specifically, experimental evidence in multiple experimental systems from several different laboratories both in vitro and in vivo provides a clear path to prioritizing targets for clinical translation. Since in some cases only a few experimental models exist to study specific categories of the cancer targets described here, a concerted effort to create and characterize more experimental models must remain a priority.
Finally, the discovery and credentialing of non-oncogene and TME targets will be accompanied by a need to define markers or features that permit the stratification of patients who are most likely to benefit from such new classes of cancer therapies (Chari et al., 2019). For some targets, immunohistochemical methods or molecular profiling or sequencing will be sufficient to stratify patients likely to respond to treatment. However, for other targets, defining tumors that are likely or not to respond to particular tumor intrinsic or extrinsic focused therapies will require new tools that can be applied to clinical samples, such as the ability to assess multi-parameter gene expression in tumor tissue. Although a small number of diagnostic tests are based on gene expression, expansion of these efforts into clinical practice will require substantial changes to sample acquisition and testing.
Despite these challenges, defining and credentialing an expanded universe of cancer targets is now possible. In addition, future work will be needed to develop robust methods to test potential combinations, particularly if the one is targeting both the tumor and TME. These approaches will require both scale as well as novel endpoints that go beyond cell fitness. Combining approaches to target both tumor intrinsic and extrinsic factors will lead to rational combinations, which promise to provide robust and durable outcomes.
Acknowledgements
Data generated by the CTD2 Network can be found in the Data Portal and Dashboard More information about the Centers can be found on the Network web site.
Declaration of interests
G.B.M. is a SAB/Consultant with AstraZeneca, Chrysallis Biotechnology, GSK, ImmunoMET, Ionis, Lilly, PDX Pharmaceuticals, Signalchem Lifesciences, Symphogen, Tarveda, Turbine, Zentalis Pharmaceuticals; has Stock/Options/Financial engagement with Catena Pharmaceuticals, ImmunoMet, SignalChem, Tarveda; has Licensed Technology: HRD assay to Myriad Genetics, DSP patents with Nanostring; has sponsored research: Nanostring Center of Excellence, Ionis (Provision of tool compounds) and Clinical trials support (funding or in kind) with AstraZeneca, Genentech, GSK, Lilly.
S.L.S. is a shareholder and serves on the Board of Directors of Jnana Therapeutics; is a shareholder of Forma Therapeutics and Decibel Therapeutics; is a shareholder and advises Kojin Therapeutics, Kisbee Therapeutics and Eikonizo Therapeutics; serves on the Scientific Advisory Boards of Eisai Co., Ltd., Ono Pharma Foundation, Exo Therapeutics, and F-Prime Capital Partners; and the Board of Advisers of the Genomics Institute of the Novartis Research Foundation; and is a Novartis Faculty Scholar.
W.C.H. is a consultant for ThermoFisher, Solasta, MPM Capital, iTeos, RAPPTA Therapeutics, Jubilant Therapeutics and Paraxel and is a Scientific Founder and serves on the Scientific Advisory Board (SAB) for KSQ Therapeutics.
J.S.B. is a founder and director of Neochromosome, Inc., and holds an ownership equity interest in the company.
A.C. is the founder and an equity holder in DarwinHealth Inc., a company that has licensed algorithms for the analysis of regulatory networks and Master Regulator proteins from Columbia University. Columbia University is also an equity holder in DarwinHealth”
C.K. is a founder and an equity holder in SEngine Precision Medicine, a company that harnesses 3D organoid technology and AI for more effective treatment options and accelerated drug development”
W.K. and P.T. receive research support from Pfizer Oncology.
W.W. is co-founder of StemSynergy Therapeutics.
B.J.D.: SAB: Aileron Therapeutics, Therapy Architects (ALLCRON), Cepheid, Vivid Biosciences, Celgene, RUNX1 Research Program, Novartis, Gilead Sciences (inactive), Monojul (inactive); SAB & Stock: Aptose Biosciences, Blueprint Medicines, EnLiven Therapeutics, Iterion Therapeutics, Third Coast Therapeutics, GRAIL (SAB inactive); Scientific Founder: MolecularMD (inactive, acquired by ICON); Board of Directors & Stock: Amgen; Board of Directors: Burroughs Wellcome Fund, CureOne; Joint Steering Committee: Beat AML LLS; Founder: VB Therapeutics; Sponsored Research Agreement: EnLiven Therapeutics; Clinical Trial Funding: Novartis, Bristol-Myers Squibb, Pfizer; Royalties from Patent 6958335 (Novartis) and OHSU and Merck and one CytoImage, Inc. exclusive license).
Literature Cited
- Alvarez MJ, Subramaniam PS, Tang LH, Grunn A, Aburi M, Rieckhof G, Komissarova EV, Hagan EA, Bodei L, Clemons PA, et al. (2018). A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat Genet 50, 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte RS, Chen DS, and Ferrara N (2019). VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 176, 1248–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbour KC, and Riely GJ (2019). Systemic Therapy for Locally Advanced and Metastatic Non-Small Cell Lung Cancer: A Review. JAMA 322, 764–774. [DOI] [PubMed] [Google Scholar]
- Arendt D, Musser JM, Baker CVH, Bergman A, Cepko C, Erwin DH, Pavlicev M, Schlosser G, Widder S, Laubichler MD, et al. (2016). The origin and evolution of cell types. Nat Rev Genet 17, 744–757. [DOI] [PubMed] [Google Scholar]
- Ashiru O, Boutet P, Fernandez-Messina L, Aguera-Gonzalez S, Skepper JN, Vales-Gomez M, and Reyburn HT (2010). Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res 70, 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon AE, Klug LR, Corless CL, and Heinrich MC (2017). Using molecular diagnostic testing to personalize the treatment of patients with gastrointestinal stromal tumors. Expert Rev Mol Diagn 17, 445–457. [DOI] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G, and Tuveson DA (2020). DIVERSITY AND BIOLOGY OF CANCER-ASSOCIATED FIBROBLASTS. Physiol Rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun TP, Eide CA, and Druker BJ (2020). Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 37, 530–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JS, O’Carrigan B, Jackson SP, and Yap TA (2017). Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov 7, 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Califano A, and Alvarez MJ (2017). The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat Rev Cancer 17, 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Target D, and Development N (2016). Transforming Big Data into Cancer-Relevant Insight: An Initial, Multi-Tier Approach to Assess Reproducibility and Relevance. Mol Cancer Res 14, 675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, et al. (2010). The transcriptional network for mesenchymal transformation of brain tumours. Nature 463, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EM, Shibue T, McFarland JM, Gaeta B, Ghandi M, Dumont N, Gonzalez A, McPartlan JS, Li T, Zhang Y, et al. (2019). WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 568, 551–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan IS, Knútsdóttir H, Ramakrishnan G, Padmanaban V, Warrier M, Ramirez JC, Dunworth M, Zhang H, Jaffee EM, Bader JS, et al. (2020). Cancer cells educate natural killer cells to a metastasis-promoting cell state. J Cell Biol 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari A, Vogl DT, Gavriatopoulou M, Nooka AK, Yee AJ, Huff CA, Moreau P, Dingli D, Cole C, Lonial S, et al. (2019). Oral Selinexor-Dexamethasone for Triple-Class Refractory Multiple Myeloma. The New England journal of medicine 381, 727–738. [DOI] [PubMed] [Google Scholar]
- Chen JC, Alvarez MJ, Talos F, Dhruv H, Rieckhof GE, Iyer A, Diefes KL, Aldape K, Berens M, Shen MM, et al. (2014). Identification of causal genetic drivers of human disease through systems-level analysis of regulatory networks. Cell 159, 402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, and Tabi Z (2008). Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol 180, 7249–7258. [DOI] [PubMed] [Google Scholar]
- Cortes-Cros M, Hemmerlin C, Ferretti S, Zhang J, Gounarides JS, Yin H, Muller A, Haberkorn A, Chene P, Sellers WR, et al. (2013). M2 isoform of pyruvate kinase is dispensable for tumor maintenance and growth. Proc Natl Acad Sci U S A 110, 489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AD, Fesik SW, Kimmelman AC, Luo J, and Der CJ (2014). Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 13, 828–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, and Thompson CB (2007). Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 104, 19345–19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al. (2015). NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 47, 1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, Slagter M, van der Velden DL, Kaing S, Kelderman S, et al. (2018). Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 174, 1586–1598 e1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong MB, Wang G, Chow RD, Ye L, Zhu L, Dai X, Park JJ, Kim HR, Errami Y, Guzman CD, et al. (2019). Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell 178, 1189–1204 e1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. (2019). Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 9, 1102–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari de Andrade L, Tay RE, Pan D, Luoma AM, Ito Y, Badrinath S, Tsoucas D, Franz B, May KF Jr., Harvey CJ, et al. (2018). Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 359, 1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM, and Shendure J (2018). Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Schreiber H, and Fu YX (2013). Innate and adaptive immune cells in the tumor microenvironment. Nature immunology 14, 1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse-Wilde A, Fouquier d’Herouel A, McIntosh E, Ertaylan G, Skupin A, Kuestner RE, del Sol A, Walters KA, and Huang S (2015). Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS One 10, e0126522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gysin S, Salt M, Young A, and McCormick F (2011). Therapeutic strategies for targeting ras proteins. Genes Cancer 2, 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerl D, Smid M, Timmermans AM, Sleijfer S, Martens JWM, and Debets R (2018). Breast cancer genomics and immuno-oncological markers to guide immune therapies. Semin Cancer Biol 52, 178–188. [DOI] [PubMed] [Google Scholar]
- Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Szankasi P, Roberts CJ, Murray AW, and Friend SH (1997). Integrating genetic approaches into the discovery of anticancer drugs. Science 278, 1064–1068. [DOI] [PubMed] [Google Scholar]
- Hedlund M, Nagaeva O, Kargl D, Baranov V, and Mincheva-Nilsson L (2011). Thermal- and oxidative stress causes enhanced release of NKG2D ligand-bearing immunosuppressive exosomes in leukemia/lymphoma T and B cells. PLoS One 6, e16899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekman RM, Hume AJ, Goel RK, Abo KM, Huang J, Blum BC, Werder RB, Suder EL, Paul I, Phanse S, et al. (2020). Actionable Cytopathogenic Host Responses of Human Alveolar Type 2 Cells to SARS-CoV-2. Mol Cell 80, 1104–1122 e1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, Kim Y, Kryukov GV, Ghandi M, Aguirre AJ, et al. (2014). ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat Med 20, 251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosein AN, Brekken RA, and Maitra A (2020). Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic N, Birsoy K, Aguirre AJ, Kory N, Pacold ME, Singh S, Moody SE, DeAngelo JD, Spardy NA, Freinkman E, et al. (2017). PIK3CA mutant tumors depend on oxoglutarate dehydrogenase. Proc Natl Acad Sci U S A 114, E3434–E3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, Miller BC, Du PP, Yates KB, Dubrot J, et al. (2019). Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, and Mootha VK (2012). Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336, 1040–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes KA (2016). Single-cell states versus single-cell atlases - two classes of heterogeneity that differ in meaning and method. Curr Opin Biotechnol 39, 120–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Chen L, and Rabinowitz JD (2018). Metabolomics and Isotope Tracing. Cell 173, 822–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, Bowden M, Deng J, Liu H, Miao D, et al. (2018). Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov 8, 196–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, et al. (2017). Corrigendum: PI3Kgamma is a molecular switch that controls immune suppression. Nature 542, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Fiske BP, Birsoy K, Freinkman E, Kami K, Possemato RL, Chudnovsky Y, Pacold ME, Chen WW, Cantor JR, et al. (2015). SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 520, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Abudayyeh OO, Yeerna H, Yeang CH, Stewart M, Jenkins RW, Kitajima S, Konieczkowski DJ, Medetgul-Ernar K, Cavazos T, et al. (2017). Decomposing Oncogenic Transcriptional Signatures to Generate Maps of Divergent Cellular States. Cell Syst 5, 105–118 e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, Vazquez F, Weir BA, Fitzgerald ME, Tanaka M, et al. (2016). MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351, 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupiec JJ (1997). A Darwinian theory for the origin of cellular differentiation. Mol Gen Genet 255, 201–208. [DOI] [PubMed] [Google Scholar]
- Lane-Reticker SK, Manguso RT, and Haining WN (2018). Pooled in vivo screens for cancer immunotherapy target discovery. Immunotherapy 10, 167–170. [DOI] [PubMed] [Google Scholar]
- Lanier LL (2019). Plastic fantastic innate lymphoid cells. J Exp Med 216, 1726–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J, Dummer R, et al. (2019). Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. The New England journal of medicine 381, 1535–1546. [DOI] [PubMed] [Google Scholar]
- LeBleu VS, and Kalluri R (2018). A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis Model Mech 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ (2019). The many faces of p53: something for everyone. J Mol Cell Biol 11, 524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ning S, Ghandi M, Kryukov GV, Gopal S, Deik A, Souza A, Pierce K, Keskula P, Hernandez D, et al. (2019). The landscape of cancer cell line metabolism. Nat Med 25, 850–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD, Matei D, and Cheng JX (2017a). Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 20, 303–314 e305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Ivanov AA, Su R, Gonzalez-Pecchi V, Qi Q, Liu S, Webber P, McMillan E, Rusnak L, Pham C, et al. (2017b). The OncoPPi network of cancer-focused protein-protein interactions to inform biological insights and therapeutic strategies. Nat Commun 8, 14356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, and Ashworth A (2017). PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luengo A, Gui DY, and Vander Heiden MG (2017). Targeting Metabolism for Cancer Therapy. Cell Chem Biol 24, 1161–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui GYL, Shaw R, Schaub FX, Stork IN, Gurley KE, Bridgwater C, Diaz RL, Rosati R, Swan HA, Ince TA, et al. (2020). BET, SRC, and BCL2 family inhibitors are synergistic drug combinations with PARP inhibitors in ovarian cancer. EBioMedicine 60, 102988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundholm M, Schroder M, Nagaeva O, Baranov V, Widmark A, Mincheva-Nilsson L, and Wikstrom P (2014). Prostate tumor-derived exosomes down-regulate NKG2D expression on natural killer cells and CD8+ T cells: mechanism of immune evasion. PLoS One 9, e108925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Solimini NL, and Elledge SJ (2009). Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean AL, Hong T, and Nie Q (2018). Exploring intermediate cell states through the lens of single cells. Curr Opin Syst Biol 9, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. (2017). In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis KJ, McDonald ER 3rd, Schlabach MR, Billy E, Hoffman GR, deWeck A, Ruddy DA, Venkatesan K, Yu J, McAllister G, et al. (2016). Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 351, 1208–1213. [DOI] [PubMed] [Google Scholar]
- Menendez JA, and Lupu R (2006). Oncogenic properties of the endogenous fatty acid metabolism: molecular pathology of fatty acid synthase in cancer cells. Curr Opin Clin Nutr Metab Care 9, 346–357. [DOI] [PubMed] [Google Scholar]
- Mo X, Tang C, Niu Q, Ma T, Du Y, and Fu H (2019). HTiP: High-Throughput Immunomodulator Phenotypic Screening Platform to Reveal IAP Antagonists as Anti-cancer Immune Enhancers. Cell Chem Biol 26, 331–339 e333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje M (2020). Synaptic communication in brain cancer. Cancer Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AR, Rosenberg SC, McCormick F, and Malek S (2020). RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov 19, 533–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, Eisenson D, Narurkar R, Deng P, Nezi L, et al. (2012). Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 488, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal JT, and Kuo CJ (2016). Organoids as Models for Neoplastic Transformation. Annu Rev Pathol 11, 199–220. [DOI] [PubMed] [Google Scholar]
- Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, Liu IH, Chiou SH, Salahudeen AA, Smith AR, et al. (2018). Organoid Modeling of the Tumor Immune Microenvironment. Cell 175, 1972–1988 e1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM, et al. (2019). An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178, 835–849 e821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedermaier B, Sak A, Zernickel E, Xu S, Groneberg M, and Stuschke M (2019). Targeting ARID1A-mutant colorectal cancer: depletion of ARID1B increases radiosensitivity and modulates DNA damage response. Sci Rep 9, 18207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhawan D, Zack TI, Ren Y, Strickland MR, Lamothe R, Schumacher SE, Tsherniak A, Besche HC, Rosenbluh J, Shehata S, et al. (2012). Cancer vulnerabilities unveiled by genomic loss. Cell 150, 842–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ, et al. (2017). Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palucka AK, and Coussens LM (2016). The Basis of Oncoimmunology. Cell 164, 1233–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, et al. (2018). A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM (2012). The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnas O, Jovanovic M, Eisenhaure TM, Herbst RH, Dixit A, Ye CJ, Przybylski D, Platt RJ, Tirosh I, Sanjana NE, et al. (2015). A Genome-wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks. Cell 162, 675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA, et al. (2017). Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541, 41–45. [DOI] [PubMed] [Google Scholar]
- Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM, Yamamoto TN, et al. (2017). Identification of essential genes for cancer immunotherapy. Nature 548, 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul P, Malakar AK, and Chakraborty S (2019). The significance of gene mutations across eight major cancer types. Mutat Res 781, 88–99. [DOI] [PubMed] [Google Scholar]
- Pettitt SJ, Krastev DB, Brandsma I, Drean A, Song F, Aleksandrov R, Harrell MI, Menon M, Brough R, Campbell J, et al. (2018). Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun 9, 1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, and Morrison SJ (2015). Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, Hood FE, and Hartley JL (2020). The Frequency of Ras Mutations in Cancer. Cancer Res 80, 2969–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, and Joyce JA (2013). Microenvironmental regulation of tumor progression and metastasis. Nat Med 19, 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, Rocha Lima CM, Schlesselman JJ, Tolba K, Langmuir VK, et al. (2013). A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol 71, 523–530. [DOI] [PubMed] [Google Scholar]
- Raj A, and van Oudenaarden A (2008). Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 135, 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roerink SF, Sasaki N, Lee-Six H, Young MD, Alexandrov LB, Behjati S, Mitchell TJ, Grossmann S, Lightfoot H, Egan DA, et al. (2018). Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 556, 457–462. [DOI] [PubMed] [Google Scholar]
- Rosenberg SA, and Restifo NP (2015). Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, et al. (2020). A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 20, 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta D, Park Y, Andrabi SA, Shelton LM, Gilkes DM, and Semenza GL (2016). PHGDH Expression Is Required for Mitochondrial Redox Homeostasis, Breast Cancer Stem Cell Maintenance, and Lung Metastasis. Cancer Res 76, 4430–4442. [DOI] [PubMed] [Google Scholar]
- Schaub FX, Dhankani V, Berger AC, Trivedi M, Richardson AB, Shaw R, Zhao W, Zhang X, Ventura A, Liu Y, et al. (2018). Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst 6, 282–300 e282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, Beqiri M, Sproesser K, Brafford PA, Xiao M, et al. (2017). Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, Li PJ, Diolaiti ME, Ashworth A, and Marson A (2018). Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 175, 1958–1971 e1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, Rodriguez-Abreu D, Moro-Sibilot D, Thomas CA, Barlesi F, et al. (2018). Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. The New England journal of medicine 378, 2288–2301. [DOI] [PubMed] [Google Scholar]
- Stadhouders R, Filion GJ, and Graf T (2019). Transcription factors and 3D genome conformation in cell-fate decisions. Nature 569, 345–354. [DOI] [PubMed] [Google Scholar]
- Stratton MR, Campbell PJ, and Futreal PA (2009). The cancer genome. Nature 458, 719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, and Vander Heiden MG (2015). Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Luengo A, Danai LV, Bush LN, Diehl FF, Hosios AM, Lau AN, Elmiligy S, Malstrom S, Lewis CA, et al. (2018). Aspartate is an endogenous metabolic limitation for tumour growth. Nat Cell Biol 20, 782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talos F, Mitrofanova A, Bergren SK, Califano A, and Shen MM (2017). A computational systems approach identifies synergistic specification genes that facilitate lineage conversion to prostate tissue. Nat Commun 8, 14662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson TW, Kim AB, Li PJ, Wang J, Jackson BT, Huang KTH, Zhang L, and Raulet DH (2017). Endothelial cells express NKG2D ligands and desensitize antitumor NK responses. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tommasini-Ghelfi S, Murnan K, Kouri FM, Mahajan AS, May JL, and Stegh AH (2019). Cancer-associated mutation and beyond: The emerging biology of isocitrate dehydrogenases in human disease. Sci Adv 5, eaaw4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima M, Howie HL, Imakura M, Walsh RM, Annis JE, Chang AN, Frazier J, Chau BN, Loboda A, Linsley PS, et al. (2012). Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc Natl Acad Sci U S A 109, 9545–9550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HC, Wan Z, Sheard MA, Sun J, Jackson JR, Malvar J, Xu Y, Wang L, Sposto R, Kim ES, et al. (2017). TGFbetaR1 Blockade with Galunisertib (LY2157299) Enhances Anti-Neuroblastoma Activity of the Anti-GD2 Antibody Dinutuximab (ch14.18) with Natural Killer Cells. Clin Cancer Res 23, 804–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 33, 890–904 e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, and DeBerardinis RJ (2017). Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, Arzt M, Tam LT, Espenel C, Ponnuswami A, Ni L, et al. (2019). Electrical and synaptic integration of glioma into neural circuits. Nature 573, 539–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AD, Buechner-Steudel P, Wein A, Schmalenberg H, Lindig U, Moehler M, Behrens R, Kleber G, Kuss O, and Fleig WE (2007). Gemcitabine, oxaliplatin and weekly high-dose 5-FU as 24-h infusion in chemonaive patients with advanced or metastatic pancreatic adenocarcinoma: a multicenter phase II trial of the Arbeitsgemeinschaft Internistische Onkologie (AIO). Ann Oncol 18, 82–87. [DOI] [PubMed] [Google Scholar]
- Waks AG, and Winer EP (2019). Breast Cancer Treatment: A Review. JAMA 321, 288–300. [DOI] [PubMed] [Google Scholar]
- Weinstein IB (2002). Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science 297, 63–64. [DOI] [PubMed] [Google Scholar]
- Wong KM, Horton KJ, Coveler AL, Hingorani SR, and Harris WP (2017). Targeting the Tumor Stroma: the Biology and Clinical Development of Pegylated Recombinant Human Hyaluronidase (PEGPH20). Curr Oncol Rep 19, 47. [DOI] [PubMed] [Google Scholar]
- Ye L, Park JJ, Dong MB, Yang Q, Chow RD, Peng L, Du Y, Guo J, Dai X, Wang G, et al. (2019). In vivo CRISPR screening in CD8 T cells with AAV-Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nat Biotechnol 37, 1302–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuki K, Cheng N, Nakano M, and Kuo CJ (2020). Organoid Models of Tumor Immunology. Trends Immunol 41, 652–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, Bisikirska B, Lefebvre C, Accili D, Hunter T, et al. (2012). Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 490, 556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Li H, Zhang Y, Li Y, Liang S, and Liu J (2011). Concomitant pulmonary and thyroid tumors identified by FDG PET/CT and immunohistochemical techniques. World J Surg Oncol 9, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]