

Abstract
Blood vessel epicardial substance (BVES, otherwise known as POPDC1) is an integral membrane protein known to regulate tight junction formation and epithelial–mesenchymal transition. BVES is underexpressed in a number of malignancies, including colorectal cancer. BVES loss leads to activation of the Wnt pathway, suggesting that decreased BVES expression functionally contributes to tumorigenesis. However, the mechanism by which BVES modulates Wnt signaling is unknown. Here, we confirm that BVES loss increases β-catenin protein levels, leads to Wnt pathway activation in a ligand-independent fashion and coordinates with Wnt ligand to further increase Wnt signaling. We show that BVES loss increases levels and activation of the Wnt co-receptor, LRP6, in cell lines, murine adenoma tumoroids and human-derived colonoids. We also demonstrate that BVES interacts with LRP6. Finally, murine tumor modeling using a Wnt-driven genetic model and a chemically induced model of colorectal carcinogenesis demonstrate that BVES loss increases tumor multiplicity and dysplasia. Together, these results implicate BVES as an inhibitor of Wnt signaling, provide one of the first examples of a tight junction-associated protein regulating Wnt receptor levels, and expand the number of putative molecular targets for therapeutic intervention in colorectal cancer.
Alterations in Wnt signaling are fundamental drivers of cancer. We identified an unexpected interaction between BVES and the Wnt co-receptor LRP6 and show that loss of BVES increases receptor activation, Wnt signaling and intestinal tumorigenesis.
Introduction
Colorectal cancer (CRC) is the third most common cancer in men and the second most common cancer in women worldwide. In the United States, CRC is the second leading cause of cancer-related deaths (1,2). Over 90% of CRC tumors exhibit disruptions in the Wnt signaling pathway with inactivating mutations in the tumor suppressor adenomatous polyposis coli (APC) or activating mutations in β-catenin present in about 80% of cases (3). β-catenin is the key effector of canonical Wnt signaling, and cytoplasmic β-catenin is maintained at low levels via a destruction complex composed of the scaffolding proteins AXIN and APC and the protein kinases glycogen synthase kinase 3 (GSK3) and casein kinase 1 alpha (CK1α) (4). CK1α phosphorylates β-catenin, priming it for subsequent phosphorylation by GSK3, which allows for recognition by the β-transducin repeats-containing protein (β-TrCP) ubiquitin ligase and subsequent proteasomal degradation (5,6).
LRP6 is a Wnt co-receptor that, in the presence of ligand, complexes with the seven-transmembrane-domain receptor Frizzled (FZ) and the scaffold protein Dishevelled (DVL) (7,8). LRP6 is ultimately phosphorylated by GSK3β and CK1α at multiple sites including Thr1479, Ser1490 and Thr1493 on stimulation, which effectively recruits AXIN to the membrane, preventing the activity of the β-catenin destruction complex and allowing β-catenin to translocate to the nucleus to mediate TCF/β-catenin target gene transcription (9). AXIN2 is one such Wnt target gene and functions in a negative feedback loop to degrade β-catenin and attenuate Wnt signaling (10). Given the dependence of CRC on hyperactive Wnt signaling, identifying novel regulators of this pathway will be essential to the development of targeted therapeutic strategies.
Blood vessel epicardial substance (BVES or POPDC1) is a tight junction-associated adhesion molecule that was isolated in a complementary DNA (cDNA) screen of the developing heart (11). It is proposed that BVES promotes epithelial junctional competence and can set epithelial–mesenchymal states via sensing of cell–cell contacts. The BVES promoter is frequently hypermethylated in malignancy leading to its reduced expression (12). Accordingly, BVES loss is observed in all stages of CRC, including in premalignant adenomas with APC loss, suggesting that its suppression may contribute to tumor initiation (12). Restoring BVES in CRC cell lines results in attenuated growth, migration and invasion (12) and BVES interacts with a protein phosphatase 2A (PP2A) regulatory subunit to reduce c-Myc protein levels (13). Furthermore, in the inflammation-driven azoxymethane/dextran sodium sulfate (AOM) carcinogenesis model, tumors from Bves–/– mice displayed increased cytoplasmic and nuclear β-catenin staining (13). RNA-seq analysis of these tumors also indicated hyperactive Wnt signaling networks (13). Small intestinal crypts isolated from Bves–/– mice and grown in 3D-Matrigel™ cultures as enteroids displayed increases in plating efficiency and in the ratio of stem spheroids, a morphology associated with hyperactive Wnt signaling (14). Importantly, Bves–/– enteroids are also hyperresponsive to Wnt activation on Wnt3a stimulation (14). Taken together, these data suggest that BVES may tune cellular responses to Wnt and growth factor signaling; however, the mechanisms by which BVES influences Wnt signaling are unknown.
Given the baseline and Wnt-dependent phenotypes associated with BVES loss, we hypothesized that BVES may regulate Wnt signaling in a ligand-independent fashion that is enhanced upon ligand stimulation. We observed that BVES knockdown using multiple approaches indeed increases Wnt pathway activation and β-catenin levels at baseline and coordinated with Wnt3a stimulation to further enhance Wnt signaling. Furthermore, BVES interacts with the Wnt co-receptor LRP6, and BVES loss increases LRP6 receptor levels and activation. Adenoma tumoroids isolated from a Wnt-driven genetic mouse model and human samples derived from normal colon also demonstrated increased LRP6 receptor levels and receptor activation, suggesting that BVES loss may aid in the transition from premalignant to malignant lesions by augmenting Wnt signaling. Finally, loss of BVES increased tumor multiplicity and dysplasia in two separate mouse models of colon tumorigenesis. Together, these results implicate BVES in the regulation of Wnt signaling through modulating LRP6 receptor levels and confirm its role as a tumor suppressor by inhibiting Wnt ligand-independent signaling cascades.
Materials and methods
Animal care
Animals were provided water and standard chow diet from Harlan Laboratories ad libitum and kept on a 12-h light/12-h dark cycle. All mice were bred and housed in the same facility throughout the duration of the experiments. BvesWT, Bves+/– and Bves–/– mice were maintained in the same room for a year before beginning the experiment to ensure a controlled microenvironment. Bves–/– mice were previously characterized (13,15). All in vivo procedures were carried out in accordance with protocols approved by the Vanderbilt Institutional Animal Care and Use Committee.
AOM and Lrig1-CreER;Apc tumorigenesis protocols
For the AOM tumorigenesis study, 8-week-old C57BL/6 wild-type (WT; n = 11) or Bves–/– (n = 10) mice were injected with 5 weekly injections of AOM at 12.5 mg/kg (Sigma–Aldrich) intraperitoneally as described previously (16) and harvested at 30 weeks.
For the Lrig1-CreER;Apcfl/+ experiments, Lrig1-CreER;Apcfl/fl;Bves+/– mice were crossed with Bves+/– mice to generate both WT and Bves–/– mice with appropriate age-matched littermate controls. Between 8 and 12 weeks of age, mice were injected intraperitoneally on 3 sequential days with 2 mg of Tamoxifen (Sigma–Aldrich). Briefly, a 10 mg/ml Tamoxifen solution was prepared by dissolving 10 mg of Tamoxifen in 100 μl of 100% ethanol. This solution was vortexed for 15 min at room temperature and pre-warmed corn oil was then added to bring the total volume up to 1 ml. 200 μl was injected intraperitoneally per mouse. Mice were harvested 100 days after tamoxifen injection.
All mice were killed using isoflurane followed by cervical dislocation and intestines resected. Intestines were irrigated with phosphate-buffered saline (PBS) then opened longitudinally and tumor numbers/size were quantified visually. The sections were then rolled from distal to proximal and the tissues were fixed in 10% formalin overnight before paraffin-embedding. Five micrometer sections were cut and stained with hematoxylin and eosin by the Vanderbilt Translational Pathology Shared Resource (TPSR) Core.
Cell lines and culture
HEK293 SuperTopFlash (293STF) cells were a kind gift from Dr Ethan Lee, Vanderbilt University and J. Nathans, Johns Hopkins University (17,18) and 293DVL TKO were a kind gift from Dr Ethan Lee, Vanderbilt University and S. Angers, University of Toronto (19). 293STF cells were not authenticated in our laboratory but demonstrate expected TopFlash activity in response to Wnt pathway activation and expected G418-resistance conferred during generation of the cell line (18). 293DVL TKO cells were confirmed to be DVL knockout by western blot. L-cells, L-Wnt3a, HEK293T and HCT116 cells were purchased from ATCC, which characterizes cell lines using short tandem repeat DNA profiles. HCT116 cells were maintained in McCoy’s 5A and all other cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM), all supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in 5% CO2. All cells used were passaged in our laboratory for less than 3 months after resurrection. Cells were tested using the polymerase chain reaction (PCR) Mycoplasma Detection Kit (ABM).
Transfections
For small interfering RNA (siRNA) knockdown experiments, Control siRNA-A (SantaCruz, sc-37007) and individual as well as pooled BVES siRNAs (sc-60295) were transfected into 293STF and HCT116 cells using Lipofectamine RNAimax (Life Technologies) as per manufacturer’s instructions. The overexpression plasmid for chick-BVES has been described previously (20), LRP6-green fluorescent protein (GFP) was a gift from Dr Ethan Lee and Randall Moon (21), and Myc-LRP6ICD was a gift from Dr Ethan Lee. pcDNA3.2/V5/GW-CAT (Thermo Scientific) was used as filler to maintain equivalent DNA concentrations.
Lentivirus generation
Lentivirus was packaged using pMD2.G (a gift from Didier Trono, Addgene plasmid # 12259) and psPAX2 (a gift from Didier Trono, Addgene plasmid # 12260) vectors and the pLKO.1 non-target short hairpin RNA (shRNA) (Sigma–Aldrich) or pLKO.1 shBVES (Mission shRNA—Sigma, TRCN0000153094). HEK293T at 50% confluency in 10 cm plates were transfected with 1 μg each of pMD2.G and psPAX2 and 2 μg of shRNA using polyethylenimine. Media was changed the following morning then viral supernatant was harvested 48 h later, centrifuged, and filtered through a 0.45 μM filter. For transient shRNA knockdown, polybrene was added to 5 μg/ml and target cells were transduced overnight then assayed 48–60 h later. Lentiviral transduction of human colonoids and human tumoroids was performed as described previously (17).
TopFlash assays
293STF cells were seeded into 12-well plates and knockdown was performed when cells were at about 50% confluency. Forty-eight hours after knockdown, cells were stimulated with 50% L-cell-conditioned media or 50% Wnt3a-conditioned media overnight. Cells were then lysed in 250 μl of 1× Glo lysis buffer (Promega).Thereafter, 30 μl of lysate was mixed 1:1 with the Steady-Glo Luciferase Assay solution (Promega) in 96-well polystyrene white opaque plates (Thermo Scientific) and luminescence measured using a GloMax Discover plate reader (Promega). Assays were performed in triplicate and repeated at least 4×. TopFlash experiments in HCT116 were performed by co-transfection of 1 μg of M50 Super8x TopFlash (gift from Randal Moon, Addgene plasmid #12456) or M51 Super 8× FOPFlash (gift from Randal Moon, Addgene plasmid #12457) with 0.1 μg of pRL-TK (Promega) to normalize transfection efficiency and assayed using the Dual-Glo Luciferase Assay System (Promega).
RNA isolation and quantitative reverse transcription–PCR
Cells in 6-well plates at 50% confluency were treated with BVES siRNA or transiently transduced with BVES shRNA lentivirus overnight. Thirty-six to forty-eight hours after knockdown, cells were stimulated with 50% L-cell-conditioned media or 50% Wnt3a-conditioned media overnight. Cells were harvested in TRIzol Reagent (Invitrogen) and RNA purified using the Direct-zol RNA Miniprep kit (Zymo Research). RNA (2 μg) was reverse transcribed to cDNA using qSCRIPT XLT cDNA SuperMix (QuantaBio) according to manufacturer’s protocol. The 20 μl cDNA reaction was diluted in 380 μl H2O and 2 μl was used as a template in each subsequent PCR. Taqman reactions were performed using TaqMan Universal PCR Master Mix (Applied Biosystems, #4304437) with probes for BVES (Hs00362584_m1) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Hs02786624_g1). SYBR Green reactions were performed using PerfeCTa SYBR Green FastMix (QuantaBio) and the following primers: AXIN2: (F: CAACACCAGGCGGAACGAA and R: GCCCAATAAGGAGTGTAAGGACT), β-catenin/CTNNB1: (F: AAAGCGGCTGTTAGTCACTGG and R: CGAGTCATTGCATACTGTCCAT) and GAPDH (F: GGCCTCCAAGGAGTAAGACC and R: AGGGGTCTACATGGCAACTG). All qPCRs were normalized to GAPDH and fold changes calculated using the delta–delta Ct method.
Murine adenoma tumoroid isolation, maintenance and treatments
Tumoroids were cultured as described previously (22). Briefly, colons and small intestines of Lrig1-CreER;Apcfl/+;BvesWTor Bves–/– mice were flushed with ice-cold PBS and multiple macroscopic tumors from each mouse were resected and pooled. Tissue was digested with constant agitation at 37°C in 10 ml pre-warmed digestion buffer (9.7 ml DMEM + 250 μl fetal bovine serum + 100 μl Pen Strep + 1 mg Collagenase XI + 1.25 mg Dispase II) for 30 min. Digested tumor fragments were then shaken 2–3× to loosen cell clusters. Larger fragments were allowed to settle to the bottom of the tube and the supernatant was collected, spun at 200 g for 5 min and then washed with 5 ml cold PBS to remove remaining digestion buffer. Samples were pelleted at 200 g for 5 min, the supernatant discarded, and the tumor fragments resuspended in 300–500 µl of growth-factor-reduced Matrigel™ (Corning) depending on the size of the pellet.
50 µl Matrigel™ plugs were spotted in the middle of a 12-well plate and allowed to polymerize at 37°C for 30 min before overlaying with 500 µl of epidermal growth factor/Noggin/R-spondin media that consists of basal media [(Advanced DMEM/F12 (Gibco), 1× Glutamax (Gibco), 100 U/ml Pen-Strep (Invitrogen #15140-148), 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) (Cellgro MT25060CI), 1× N2 Supplement (Gibco), 1× B27 Supplement (Gibco)] supplemented with 20% R-Spondin conditioned media (produced from R-spondin-expressing cells kindly provided by Dr Jeff Whitsett, Cincinnati Children’s Hospital Medical Centre and The University of Cincinnati College of Medicine, Cincinnati, OH), 10% Noggin conditioned media (made from Noggin-producing cells kindly provided by G.R. van den Brink, Amsterdam, The Netherlands [described in (23)], and 50 ng/ml mouse recombinant epidermal growth factor (R&D 2028EG200)]. Media was replenished every 3–4 days. For splitting, Matrigel™ plugs were scraped into 1 ml of ice-cold PBS and sheared by pulling through a 25G needle 1–2×. Sheared tumoroids were pelleted at 200 g for 5 min and remaining PBS and Matrigel™ aspirated off before resuspending in fresh Matrigel™ and spotting new 50 µl plugs in 12-well plates.
For Wnt3a and R-Spondin treatment of WT adenoma tumoroids, tumoroids were split and maintained in basal or basal media supplemented with 100 ng/ml mouse recombinant Wnt3a (Vanderbilt Antibody and Protein Resource, VAPR Core) and 250 ng/ml mouse recombinant R-Spondin (VAPR Core) for 4 days before imaging and size quantification using ImageJ.
Human colonoid and tumoroid isolation and maintenance
Deidentified human normal colon tissue was acquired from the Vanderbilt Cooperative Human Tissue Network in accordance with the Vanderbilt Institutional Review Board. Human tumor organoids were established according to previously published protocols (22). Briefly, minced colon tissue was incubated in PBS with 2 mM ethylenediaminetetraacetic acid for 30 min at 4°C. Tissue fragments were then washed and resuspended in 5 ml PBS containing with 43.4 mM sucrose and 54.9 mM d-sorbitol. Tissue was then gently shaken for 1–2 min to release intestinal crypts that were then collected, resuspended in Matrigel™ and spotted in 10 µl plugs in the bottom of a 12-well plate. After polymerization, Matrigel™ plugs were overlaid with human culture media containing Advanced DMEM F12 (Gibco), 50% LWRN conditioned media (produced from L-cells expressing Wnt, R-spondin, and Noggin, ATCC), 1× B27 (Gibco), 1× N2 (Gibco), 1× Glutamax (Gibco), 1 mM HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 ng/ml epidermal growth factor (R&D), 10 nM [Leu15]-Gastrin 1 (Sigma–Aldrich), 1 mM n-acetylcysteine (Sigma–Aldrich), 10 μM SB202190 (Sigma–Aldrich), 10 mM nicotinamide (Sigma–Aldrich), 500 nM A83-01 (Tocris) and 10 nM prostaglandin E2.
Adenoma tumoroid embedding and staining
Tumoroids were collected in ice-cold PBS and fixed in 10% neutral buffered formalin on ice for 20 min. Fixed enteroids were then pelleted at 200 g for 3 min and washed 3× with ice-cold PBS. After the final centrifugation, the supernatant was aspirated and the enteroid pellet was resuspended in 2% agarose dissolved in PBS. The agarose was allowed to solidify for 10 min then placed in 70% ethanol and paraffin embedded by the Vanderbilt TPSR Core.
Immunoblots and immunoprecipitation
For cytoplasmic fractionation, cells were incubated on ice for 15 min in cytoplasmic fractionation lysis buffer [10 mM HEPES (pH 7.8), 10 mM KCl, 2 mM MgCl2, 0.1 mM ethylenediaminetetraacetic acid with phosphatase inhibitor cocktail 2 (Sigma–Aldrich], phosphatase inhibitor cocktail 3 (Sigma–Aldrich), and protease inhibitor (Sigma–Aldrich), then scraped and transferred to microfuge tubes. NP-40 was added to 0.5% and lysates were vortexed then sheared through a 23-gauge needle 6 times, and spun at 16 000g for 2 min. This protocol enriches for cytoplasmic and membrane proteins but significantly reduces nuclear protein contamination. Supernatants (cytoplasmic fractions) were recovered and protein concentrations determined using the Pierce BCA Protein Assay Kit (Thermo Scientific). Samples were combined with 5× Laemmli buffer and then boiled for 5 min. Protein (20–30 μg) was loaded per lane followed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Lysates were transferred to a 0.2 µm Nitrocellulose membrane (PerkinElmer) and then blocked using Odyssey Tris-buffered saline blocking buffer (LI-COR) for 30 min. Membranes were probed overnight at 4°C with the following antibodies diluted in Odyssey blocking buffer with 0.1% Tween-20: β-catenin (1:2000, BD Biosciences #610153), LRP6 (1:1000, Cell Signaling Technologies #3395), phospho-LRP6 Ser1490 (1:750, Cell Signaling Technologies #2568), LRP5 (1:1000, Cell Signaling Technologies #5731), DVL-2 (1:1000, Cell Signaling Technologies #3224), DVL-3 (1:1000, Cell Signaling Technologies #5731), Flag (1:1000, Sigma–Aldrich #F1804), GFP (1:1000, Cell Signaling Technologies #2956), β-Actin (1:5000, Sigma A5441) and GAPDH (1:4000, Cell Signaling Technologies #5174). IRDye Goat-anti rabbit 800 (LI-COR #827-08365) or IRDye Goat anti-Mouse 680LT (LI-COR #925-68020) secondary antibodies were diluted 1:10 000 in Tris-buffered saline + 0.1% Tween-20 and incubated for 30 min at room temperature. Blots were imaged on a LI-COR Clx Near-infrared system and quantification of western blot band intensity was conducted using the LI-COR Image Studio with samples normalized to GAPDH. All western blots are representative of at least three independent experiments unless otherwise noted.
For co-immunoprecipitations, cell lysates were prepared using CellLytic M (Sigma–Aldrich) with protease and phosphatase inhibitors. One milligram of lysate was incubated with anti-GFP binding protein magnetic beads (VAPR Core) or Anti-flag M2 affinity gel (Sigma–Aldrich) for 2 h at 4°C with constant agitation. Beads/resin were washed 3× with lysis buffer and proteins were eluted from beads with 2× Laemmli buffer. The Flag-BVES IPs probed for endogenous LRP5 and LRP6 was developed using Supersignal West Femto (Sigma–Aldrich) chemiluminescence and HyBlot CL film (Denville Scientific).
Tumoroids were harvested based on standard passage protocols described earlier with the addition of a 30-min incubation step in Cell Recovery Solution (Corning #354253) to digest away the Matrigel™. Tumoroids were then washed 1× in PBS, pelleted and resuspended in radioimmunoprecipitation assay buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.1% sodium dodecyl sulfate).
Immunohistochemistry
Protocols for fluorescent staining of paraffin-embedded sections were described previously (15). Primary antibodies include anti-phospho Histone H3 (1:400, Millipore #06-570), cleaved caspase 3 (1:400, Cell Signaling Technologies #9661), β-catenin (1:400, BD Biosciences #610153) and E-cadherin (1:500, BD Biosciences #610181).
Statistical analysis
Statistical analysis comparing was performed in Graphpad Prism Software as indicated in the figure legends. For all studies, error is represented by standard deviation and P value of less than 0.05 is considered significant.
Results
BVES knockdown increases Wnt pathway activation
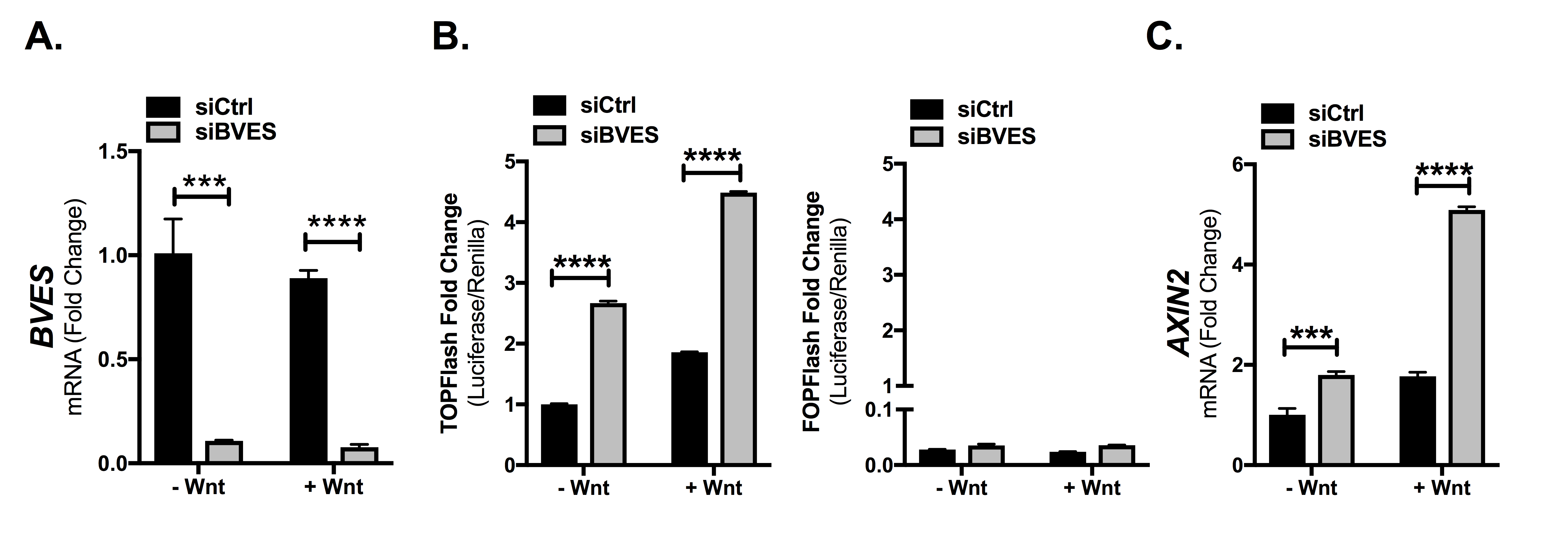
Previous reports have associated BVES loss with Wnt pathway activation, but the mechanisms by which this occurred were unclear (12–14). To elucidate these mechanisms, we first turned to in vitro systems using HEK293 cells stably expressing the SuperTopFlash Wnt reporter (293STF) (24). 293STF cells respond to canonical Wnt signaling and are a sensitive tool to determine the impact of BVES on Wnt pathway activation. BVES knockdown in these cells using a lentivirally delivered shRNA yielded a 90% reduction in BVES transcript and increased Wnt reporter activity at both baseline and following stimulation with Wnt3a conditioned media (Figure 1A, 15.6 ± 0.1 versus 108.1 ± 1.1-fold activation over unstimulated control, P < 0.0001 and Supplementary Figure 1A, available at Carcinogenesis Online). A concomitant increase in the expression of the Wnt target gene AXIN2 was also observed (Figure 1B). As β-catenin is the key effector of Wnt pathway activity, we next tested whether BVES knockdown increased β-catenin protein levels. Indeed, cytoplasmic fractionation followed by immunoblotting demonstrated increased β-catenin protein levels (Figure 1C). Pooled siRNAs targeting BVES also increased Wnt reporter activity and Wnt target gene expression both at baseline and following Wnt stimulation (Figure 1D and E). Increases in β-catenin protein levels following BVES knockdown and cytoplasmic fractionation were also observed (Figure 1F). Similar results were obtained using individual siRNAs (Supplementary Figure 1B and C, available at Carcinogenesis Online) and using the HCT116 CRC cell line (Supplementary Figure 2A–C, available at Carcinogenesis Online). Together, these results support a role for BVES in modulating Wnt pathway activation through regulating cytoplasmic β-catenin protein levels.
Figure 1.

BVES loss increases Wnt signaling and β-catenin protein levels. (A) TopFlash activity in 293STF cells following BVES shRNA knockdown. (B) AXIN2 fold change by qPCR in 293STF cells with BVES shRNA knockdown. Cells were treated with 50% L-cell-conditioned media or 50% Wnt3a-conditioned media for 16 h before isolation. (C) Cytoplasmic fractions of 293STF cells following shRNA knockdown of BVES. Cells were treated with 50% L-cell-conditioned media or 50% Wnt3a-conditioned media for 2 h before harvest. (D) TopFlash activity in 293STF cells following siRNA knockdown. (E) AXIN2 fold change by qPCR in 293STF cells with siRNA knockdown. Cells were treated with 50% L-cell-conditioned media or 50% Wnt3a-conditioned media for 16 h before isolation. (F) Cytoplasmic fractions of 293STF cells following siRNA knockdown of BVES. Cells were treated with 50% Wnt3a-conditioned media for 2 h before harvest. (G) β-catenin (CTNNB1) transcript levels in 293STF cells 56–72 h following BVES siRNA knockdown. (H) 293STF cells were treated with 20 μM MG132 for 4 h followed by cytoplasmic fractionation. Black arrow indicates β-catenin, and the relative intensity of only this lower band is quantified below. Bracket indicates ubiquitinated species. Western blot is representative of two independent experiments. (I) 293STF cells were treated with 100 μg/ml cycloheximide (CHX) dissolved in dimethyl sulfoxide (DMSO) or DMSO vehicle control for the indicated time points followed by cytoplasmic fractionation. Western blot is representative of two independent experiments. For (A), (B), (D) and (E), data are representative of at least four independent experiments. **P < 0.01, ****P < 0.0001 by Student’s t-test in the minus Wnt and plus Wnt conditions. For (C) and (F), data are pooled from n = 5 independent experiments, each normalized to the control minus Wnt3a condition. **P < 0.01 by Mann–Whitney test in the minus Wnt and plus Wnt conditions. For (G), data are pooled from n = 4 independent experiments. ns = non-significant by Mann–Whitney test in the minus Wnt and plus Wnt conditions.
BVES loss leads to stabilization of β-catenin protein
β-catenin levels can be regulated transcriptionally or post-translationally, where it is sequentially phosphorylated by two kinases, GSK3β and CK1α (5,25,26). This targets the protein for ubiquitination by β-TrCP and leads to its subsequent proteasomal degradation (6). We tested whether the observed increases in β-catenin protein levels following BVES loss were due to alterations in β-catenin transcript levels or an effect on β-catenin ubiquitination and proteasomal degradation. BVES knockdown did not alter β-catenin transcript levels in 293STF or HCT116 cells (Figure 1G and Supplementary Figure 3, available at Carcinogenesis Online). Treatment of 293STF cells with the proteasome inhibitor MG132 led to an accumulation of higher molecular weight, ubiquitinated β-catenin species, as has been described previously (Figure 1H, Lane 3) (27). In the setting of BVES knockdown, there was an increase in non-ubiquitinated β-catenin (Figure 1H, arrow) and a reduction in ubiquitinated species (Figure 1H, bracketed bands). To further these findings, we performed cycloheximide chase experiments to assess β-catenin stability. BVES knockdown cells maintained increased β-catenin protein levels after cycloheximide treatment (Figure 1I). Even when accounting for the overall increase in β-catenin protein levels in the setting of BVES knockdown, higher relative proportions of β-catenin were observed at each time point. Overall, this is consistent with BVES loss enhancing Wnt pathway activation through the accumulation of β-catenin due to reduced ubiquitination and proteasomal degradation leading to enhanced protein stability.
BVES inhibits Wnt receptor activation
Reductions in β-catenin ubiquitination lead to its stabilization and this can occur through multiple mechanisms, such as Wnt ligand binding and receptor activation, or through alterations in β-catenin destruction complex components (i.e. APC truncations) (28). Because BVES rapidly traffics to the membrane on cells reaching confluency, we hypothesized that BVES stabilization of β-catenin may be orchestrated via modulating Wnt receptor levels (20,29). LRP6, and its homologue LRP5, are Wnt ligand co-receptors that, in the presence of Wnt ligand, bind the seven-transmembrane-domain receptor Frizzled and stabilize cytoplasmic β-catenin (30). siRNA knockdown of BVES led to a reproducible 1.5-fold increase in LPR6 receptor levels in 293STF cells with concomitant increases in cytoplasmic β-catenin levels (Figure 2A). This increase in LRP6 protein levels was not due to an increase in LRP6 mRNA transcript (Figure 2B), suggesting that BVES is affecting LRP6 protein stability.
Figure 2.

LRP6 levels and phosphorylation are increased with BVES loss. (A) Cytoplasmic fractionation of 293STF cells following siRNA knockdown of BVES. Cells were treated with 50% Wnt3a conditioned media for 2 h before harvest. pLRP6 quantification is pooled from n = 5 independent experiments, and total LRP6 quantification is pooled from n = 7 independent experiments, each normalized to the control minus Wnt stimulation. (B) LRP6 fold change by qPCR in 2923STF cells with BVES siRNA knockdown. Cells were treated with 50% L-cell-conditioned media or 50% Wnt3a-conditioned media for 8 h before isolation. (C) Cytoplasmic fractionations of 293STF cells with Wnt stimulation time course following BVES knockdown. Cells were treated with 50% Wnt3a-conditioned media for indicated time points. (D) Cytoplasmic fractionations of 293STF or 293 Dishevelled Triple-Knockout (293DVL TKO) cells following BVES knockdown. Cells were stimulated with 50% Wnt3a-conditioned media for 2 h and immunoblotted. β-catenin quantification in the 293DVL TKO is pooled from n = 3 independent experiments. Black line between lanes 6 and 7 is debris on the membrane. (E) Cytoplasmic fractionations of 293STF cells stimulated with 50% control or 50% Wnt3a-conditioned media for 2 h. At the time of Wnt3a stimulation, cells were concurrently treated with dimethyl sulfoxide (DMSO), okadaic acid (OA) at 100 nM or ceramide at 50 μM and probed for pLRP6 (S1490), pGSK3β (S9) and pCREB (S133). Data are representative of two independent experiments. For (A), (B) and (D), *P < 0.05, **P < 0.01, ***P < 0.001 by Mann–Whitney test in the minus Wnt and plus Wnt conditions. ns = non-significant by Mann–Whitney test.
We next determined if increases in total LRP6 levels also correlated with increased receptor activation. Cells were treated with or without Wnt3a conditioned media and immunoblotted for phospho-LRP6 (pLRP6, Ser1490), which is indicative of LRP6 receptor activation. BVES knockdown increased LRP6 phosphorylation both at baseline (without Wnt ligand) and with Wnt stimulation (Figure 2A and C). Rapid phosphorylation of LRP6, within 1 h, was observed with cytoplasmic levels of β-catenin peaking around 2 h post-stimulation (Figure 2C). Consistently, pLRP6 levels were higher at baseline following BVES knockdown and remained elevated throughout the 3 h of Wnt stimulation. Minimal change in LRP5 receptor levels was observed. Comparable results were observed in HCT116 cells (Supplementary Figure 4A, available at Carcinogenesis Online) and mouse embryonic fibroblasts isolated from littermate-controlled WT and Bves–/– embryos (Supplementary Figure 4B and C, available at Carcinogenesis Online).
Interestingly, a mobility shift in DVL2 and DVL3 was also observed (Figure 2C). DVL proteins are key signal transducers that have roles in both canonical and non-canonical Wnt pathways (31). DVL is phosphorylated in response to Wnt stimulation, which leads to a mobility shift (32), and it is these higher molecular weight, phosphorylated DVL species that are increased with BVES knockdown. To further analyze the level at which BVES alters Wnt signaling, we used the HEK 293 DVL knockout cell line (293DVL TKO), which is null for all three DVL genes (19). In agreement with a prior study that used a combination of genetic knockout and DVL shRNA knockdown (33), the 293DVL TKO cells failed to phosphorylate LRP6 in response to Wnt3a (Figure 2D). Furthermore, BVES knockdown in this cell line led to small but reproducible increases in levels of the LRP6 receptor but did not result in changes in β-catenin levels, suggesting that the effects of BVES on Wnt signaling are upstream of DVL. These results indicate that DVL is not required for BVES-dependent changes in total LRP6, but is required for the downstream effects on β-catenin.
As we have shown previously that BVES directs PP2A phosphatase activity to dephosphorylate, and subsequently destabilize c-Myc (13), we next asked the question of whether BVES directed PP2A phosphatase activity reduces LRP6 receptor activation. 293STF cells with BVES knockdown were treated with Wnt3a conditioned media and an inhibitor of PP2A (okadaic acid, OA) or an activator of PP2A (Ceramide). Phospho-GSK3β (S9) and phospho-CREB (S133) are known PP2A targets and were included as reagent controls (34,35). Reproducibly, BVES knockdown increased LRP6 phosphorylation both at baseline and with Wnt3a conditioned media (Figure 2E and Supplementary Figure 4D, available at Carcinogenesis Online). This effect was further augmented in the context of PP2A inhibition with OA, possibly owing to incomplete BVES knockdown or an indirect effect of BVES/PP2A on the activity of an intermediary protein such as GSK3β. In the setting of Wnt3a stimulation, activation of PP2A phosphatase activity with ceramide reduced LRP6 phosphorylation in the presence of BVES. The ceramide mediated reduction in pLRP6 levels was abrogated following BVES knockdown, suggesting that BVES directs PP2A mediated dephosphorylation and subsequent inactivation of LRP6.
Overall, these data support a model in which the presence of BVES destabilizes the LRP6 co-receptor and reduces receptor activation, conceivably through directing the phosphatase activity of PP2A.
BVES interacts with LRP6
As BVES is recognized to orchestrate changes in cellular state via protein interactions in the carboxy-terminus, we next hypothesized that BVES regulates LRP6 levels through protein–protein interaction and performed co-immunoprecipitation experiments. Reciprocal co-immunoprecipitations of overexpressed Flag-tagged chick BVES (Flag-BVES) and GFP-tagged LRP6 (GFP-LRP6) demonstrated a BVES-LRP6 interaction (Figure 3A). Overexpression of BVES also reduced exogenous levels of LRP6 in the input samples, complementing our previous observations using BVES knockdown. Overexpression of BVES followed by immunoprecipitation with anti-Flag resin also demonstrated an interaction with endogenous LRP6 as well as LRP5 (Figure 3B) and again confirmed that BVES levels are inversely correlated with LRP6 levels. To narrow the putative BVES-LRP6 interaction domain, we used an LRP6 construct spanning amino acids 1364 to 1539, which comprises the transmembrane and proximal intracellular domain along with an N-terminal Myc tag (Myc-LRP6ICD). These mapping studies demonstrate that the extracellular domain is dispensable for the BVES-LRP6 interaction and narrow the interaction domain to around 175 amino acids (Figure 3C). To test whether Wnt3a augments the BVES-LRP6 interaction, cells were stimulated for 2 h with Wnt3a conditioned media before immunoprecipitation (Figure 3D). A subtle increase in the BVES-LRP6 interaction on ligand stimulation was observed but overall, BVES and LRP6 appear to associate quite readily even in the absence of ligand. Together, these data demonstrate that BVES interacts with LRP6 and that BVES overexpression leads to reductions in LRP6 protein levels.
Figure 3.

BVES interacts with LRP6 and LRP5. (A) Reciprocal co-immunoprecipitation of Flag-tagged BVES (Flag-BVES) and GFP-tagged LRP6 (GFP-LRP6). HEK293T cells were transiently transfected with 4 μg of each plasmid using polyethylenimine. Filler plasmid was used to maintain equal DNA quantities. GFP-LRP6 was immunoprecipitated with GFP-binding protein magnetic beads following by immunoblotting with anti-Flag antibodies. Flag agarose resin was used to immunoprecipitate BVES followed by immunoblotting with anti-GFP antibodies. (B) Immunoprecipitation of Flag-BVES followed by immunoblotting for endogenous LRP5 and LRP6. 4 μg of vector or Flag-BVES was transfected into HEK293T cells using polyethylenimine. The LRP5 and LRP6 blots were developed using enhanced chemiluminescence and film, whereas the rest of the blots were immunoblotted using Odyssey infrared reagents as discussed in Materials and methods. (C) Schematic of the N-terminal myc-tag LRP6 (Myc-LRP6ICD) construct that spans amino acids 1364–1539 and contains the LRP6 transmembrane domain (TM) and proximal intracellular domain (ICD). Myc-LRP6ICD was transfected into HEK293T cells along with Flag-BVES as in (A) and immunoprecipitation experiments performed 48 h later. (D) Cells were transfected with 4 μg of Flag-BVES and then 48 h later treated with 50% L-cell-conditioned media or 50% Wnt3a-conditioned media for 2 h before flag immunoprecipitation.
BVES regulates LRP6 and Wnt activation in human colonoids
To expand our studies into the intestinal epithelium, we performed BVES knockdown using lentivirally delivered shRNAs in a primary human colonoid line. Human colonoids more faithfully recapitulate intestinal physiology and are an excellent tool for studying junctional biology (36). Following BVES knockdown, levels of both pLRP6 and total LRP6 increased (Figure 4A). This occurred despite only about a 50% reduction in BVES message (Figure 4B). Concomitant increases in AXIN2 transcript levels were also observed with BVES knockdown (Figure 4C). Using robust human intestinal models, these data further demonstrate that BVES loss enhances Wnt signaling and increases LRP6 receptor levels and phosphorylation.
Figure 4.

BVES knockdown increases LRP6 levels and Wnt activity in human colonoids. (A) Whole-cell lysates from human colonoids maintained in human culture media (see Materials and methods). Analysis was performed 96 h following lentiviral shRNA knockdown. (B) BVES fold change by qPCR in human colonoids from (A). (C) AXIN2 fold change by qPCR in human colonoids from (A). Data are representative of n = 2 independent experiments. ****P < 0.0001 by Student’s t-test.
BVES loss increases Wnt receptor activation in Apc-deficient adenoma tumor organoids
As our data support a role for BVES in modulating Wnt receptor activation in non-transformed cells, we next sought to determine whether BVES loss can alter Wnt receptor activation in pathologic conditions such as in the setting of APC loss. We used the Lrig1-CreER driver to induce loss of one allele of Apc in intestinal stem cells. Lrig1 is a pan-ErbB family inhibitor that marks stem/progenitor cells in the intestinal crypt base (37). Heterozygous loss of Apc driven by Lrig1-CreER leads to the development of intestinal adenomas on stochastic loss of the second Apc allele (37). Intestinal adenomas were isolated from tumor-bearing Lrig1-CreER;Apcfl/+;BvesWT mice and grown in 3D-Matrigel™ cultures. Importantly, BvesWT adenoma tumoroids grown in basal media supplemented with purified Wnt3a and R-spondin grew larger than adenoma tumoroids grown without growth supplementation (Figure 5A) indicating that adenoma tumoroids, despite homozygous loss of Apc, were still responsive to Wnt stimulation. These data support prior observations by Saito-Diaz et al. (17) and demonstrate that even in the absence of functional APC, modulating upstream Wnt receptors further activates the Wnt pathway and increases organoid growth.
Figure 5.

BVES loss increases LRP6 receptor activation and proliferation in Lrig1-CreER;Apcfl/+ adenoma tumoroids. BvesWT and Bves–/– mice were crossed with Lrig1-CreER;Apcfl/+ mice and injected with 2 mg Tamoxifen for 3 consecutive days. Tumors were harvested at 100 days. (A) Established WT adenoma tumoroid lines were passaged and plated in basal media (Untx) or basal media supplemented with purified Wnt3a and R-Spondin for 4 days. Representative 4× brightfield images are shown, and organoid area is quantified to the right. Data are from n = 2 WT mouse tumoroid lines and representative of n = 3 independent experiments. Quantification was performed on five images per condition. (B) Adenoma tumoroids from three BvesWT and Bves–/– mice were maintained in ENR media and harvested for western blot. (C) Adenoma tumoroids were fixed, embedded and stained for phospho-Histone H3 (pH3) to measure proliferation and cleaved caspase 3 (CC3) to measure apoptosis. Data are from n = 2 WT mouse tumoroid lines and n = 2 Bves–/– tumoroid lines. Quantification was performed on five images per line. *P < 0.05, ***P < 0.001 by Student’s t-test.
We then isolated adenoma tumoroids from additional Lrig1-CreER;Apcfl/+;BvesWT and Lrig1-CreER;Apcfl/+;Bves–/– mice. As expected, the Bves–/– tumoroids exhibited increases in both total LRP6 and activated pLRP6 levels (Figure 5B). Immunofluorescence analysis of formalin-fixed paraffin embedded adenoma tumoroids demonstrated an increased percentage of phospho-Histone H3 (pH3) positive proliferative cells with no change in cleaved caspase 3 positive apoptotic cells (Figure 5C). These data demonstrate that BVES loss increases LRP6 receptor levels and activation in the setting of APC loss with concurrent increases in adenoma tumoroid proliferation.
BVES loss augments intestinal tumorigenesis
Given that mutations in Wnt signaling components are among the most common initiating events in CRC, we next determined whether BVES could modify Wnt-dependent tumorigenesis (3). Using a purely Wnt-driven genetic model, we analyzed tumor burden in Lrig1-CreER;Apcfl/+;BvesWT and Lrig1-CreER;Apcfl/+;Bves–/– mice. Apc loss was induced with three consecutive daily doses of Tamoxifen administered intraperitoneally. Mice were killed after 100 days and analyzed for tumor burden. There was no difference in survival between cohorts, and all mice developed tumors independent of Bves genotype. There was no difference in tumor size; however, Bves–/– mice displayed an almost 2-fold increase in tumor multiplicity over littermate-matched controls (Figure 6A; 35.1 ± 5.1 versus 68 ± 13.1; P < 0.05) and an increased incidence of high-grade dysplasia on histological review by a pathologist (M.K.W.) who was blinded to the genotypes of the samples (Figure 6B and C).
Figure 6.

BVES loss increases tumorigenesis. (A) Tumor multiplicity in small and large intestine of Lrig1-CreER;Apcfl/+ mice crossed with Bves–/– mice. (B) Representative histology from Lrig1-CreER;Apcfl/+ and Lrig1-CreER;Apcfl/+;Bves–/– tumor bearing mice. (C) Dysplasia scoring in Lrig1-CreER;Apcfl/+ and Lrig1-CreER;Apcfl/+;Bves–/– mice. (D) Representative images from AOM-treated BvesWT and Bves–/– colons at killing. Tumors indicated by white arrows. (E) Tumor incidence, (F) tumor multiplicity and (G) dysplasia scoring in AOM-treated mice. (H) Schematic and proposed model for the role of BVES in the regulation of Wnt signaling. For (A–C), n = 11 WT and n = 10 Bves–/– mice. For (D–G), n = 11 WT and n = 10 Bves–/– mice. *P < 0.05 by Student’s t-test.
We also used a chemically induced model of tumorigenesis to confirm these findings. Cohorts of 8-week-old WT and Bves–/– mice were treated with 5 weekly injections of AOM, which can induce β-catenin mutations but can also promote Apc loss (16,38). Mice were harvested at 30 weeks of age and assessed for tumor burden. Increased tumor incidence (Figure 6D and E) and tumor multiplicity (Figure 6F) were observed in Bves–/– mice. The tumors in AOM-treated Bves–/– mice also demonstrated higher degrees of dysplasia on histological review. Together, these tumor models are in agreement in supporting a role for BVES as a tumor suppressor and highlight that BVES loss can increase both tumor multiplicity as well as histological progression.
Discussion
Multiple mechanisms regulate β-catenin levels and activity within the cell. Classically, the transmembrane glycoprotein E-cadherin sequesters β-catenin at the cell membrane, thereby preventing its nuclear translocation and repressing Wnt signaling (39). However, there is little evidence that tight junctions can regulate Wnt signaling and β-catenin levels. Here, we demonstrate a novel role for BVES, a tight junction-associated molecule, in modulating Wnt signaling by controlling LRP6 receptor levels and activation state, thereby determining cellular β-catenin levels.
As a tight junction protein, BVES is implicated in regulating a variety of diverse cellular phenotypes such as cell migration and epithelial–mesenchymal transition. BVES is downregulated in multiple epithelial malignancies and, notably, is reduced in the earliest stages of CRC (12). Furthermore, BVES interacts with a PP2A regulatory subunit to reduce c-Myc protein levels, and loss of BVES increases tumor formation in a mouse model of colitis-associated cancer (13). BVES also demonstrates high affinity for cyclic adenosine 3′,5′-monophosphate, and aberrant cyclic nucleotide signaling has been implicated in tumorigenesis (40,41). This suggests that BVES suppression may cooperate with multiple oncogenic factors in promoting tumorigenesis. Our previous studies have also associated BVES loss with Wnt pathway activation, a foundational step in the initiation of CRC (3). We now demonstrate that BVES directly regulates Wnt signaling by altering cellular β-catenin levels via an effect on LRP6 activity and that such regulation increases APC-dependent tumorigenesis.
We show that BVES loss increases TopFlash reporter activity, Wnt target gene transcription and β-catenin protein levels at baseline, in the absence of Wnt ligand. In addition, BVES loss coordinates with Wnt ligand to robustly increase pathway activation. This suggests cells lacking BVES are “primed” and more responsive to Wnt ligand binding. Thus, it follows that BVES may influence receptor level dynamics, and indeed, we observed that BVES loss increased LRP6 receptor levels in the absence and in the presence of Wnt ligand. No changes in Frizzled 8 or the E3 ubiquitin-ligase RNF43 were observed (data not shown), suggesting that this is a direct effect on LRP6. Across multiple experiments and models, BVES loss increased Ser1490 phosphorylation of the LRP6 receptor even in the absence of Wnt ligand, suggesting that BVES inhibits ligand-independent receptor activation and Wnt signalosome formation. Phosphorylated LRP6 can subsequently recruit AXIN2 to the plasma membrane that ultimately leads to stabilization of β-catenin. This effect appears to be upstream of the scaffolding DVL proteins because no changes in β-catenin levels were observed in the DVL TKO cells following BVES loss. Because DVL is required for LRP6 receptor phosphorylation and signalosome formation (42), one working model is that BVES prevents LRP6 receptor activation by disrupting the DVL-LRP6 interaction. Interestingly, BVES interacts with ZO-1, a tight junction protein that, similar to DVL, contains a PDZ domain (43,44). Although this protein motif could link BVES and DVL, an interaction between BVES and DVL2 was not observed (data not shown). Instead, immunoprecipitation studies demonstrate that BVES interacts with both LRP5 and LRP6. Chick BVES was used for these experiments because it is much more readily expressed than full-length human BVES, is nearly 80% homologous with even higher homology in key functional domains (45) and has been extensively used for many key studies describing BVES function (12,43,46). LRP5 and LRP6 share a high degree of sequence homology (47), therefore it is not surprising that BVES could interact with both proteins. However, why BVES knockdown differentially increases LRP6 levels with minimal changes to LRP5 levels will require further exploration. Mapping studies place the BVES-LRP6 interaction within the transmembrane/intracellular domain of LRP6 and confirm that the extracellular domain of LRP6 is not required for the interaction. As the effects of BVES loss on LRP6 occur even in the absence of Wnt ligand, it is unlikely that BVES is altering ligand-receptor dynamics, which is consistent with a BVES–LRP6 interaction that is independent of the LRP6 extracellular domain. Although a direct interaction between BVES and LRP5/6 cannot be ruled out, we believe that this interaction exists within a larger complex and is indirect as BVES–LRP6 direct interactions have not been observed in prior yeast two hybrid screens (12,48). Interestingly, we also observed increases in BVES transcript following stimulation with Wnt3a conditioned media, suggesting that BVES itself is a Wnt target gene. Overall, this supports a potential model whereby BVES is upregulated in response to Wnt stimulation in a negative feedback loop that subsequently attenuates Wnt signaling, a well-described phenomena in Wnt signaling exemplified by studies of AXIN2 (10).
Our studies using OA and ceramide highlight a novel and unexplored mechanism of Wnt receptor regulation. A model by which BVES-directed, PP2A-mediated, dephosphorylation of the LRP6 co-receptor occurs at the membrane would not be without precedent as PP2A dephosphorylates a number of Wnt signaling components and appears to have dual (both activating and inhibiting) roles in this regulation similar to other pathway components such as GSK3β (49). The role of BVES in directing PP2A activity appears to be more critical in the setting of Wnt3a stimulation when phosphorylated LRP6 levels are highest. This is consistent with the dramatically enhanced TopFlash activity in the setting of BVES loss and Wnt3a stimulation and suggests that while the BVES–LRP6 interaction itself appears to be ligand independent, some components of this regulatory mechanism may be ligand dependent.
In the setting of tumorigenesis, the question remains as to whether or not modulating Wnt receptor activity upstream of mutations in APC is functionally relevant. Inactivating mutations in APC are potent drivers of CRC development and lead to stabilization of β-catenin. Teleological thinking would suggest that downstream APC mutations subsequently render cells insensitive to upstream ligand stimulation; however, a recent study by Saito-Diaz et al. (17) highlights a novel role for APC in preventing ligand-independent LRP6 activation and proposes a model by which the APC homolog APC2 functions within the destruction complex to degrade β-catenin. We also know that despite APC mutations, cellular β-catenin levels do not rise indefinitely, which may be in part due to APC2’s function in the destruction complex (17,50). Therefore, in the setting of APC loss, ligand stimulation may enhance Wnt signaling by preventing APC2-dependent destruction complex formation. In support of this, we show that WT adenoma tumoroids derived from Lrig1-CreER;Apcfl/+;BvesWT tumors are still responsive to Wnt ligand stimulation, growing significantly larger in the setting of Wnt3a and R-Spondin.
Tumoroids derived from ApcMin adenomas are sensitive to LRP6 knockdown (17), highlighting a critical dependency of Wnt receptor signaling in the context of APC loss. Our studies reveal that adenoma tumoroids derived from Lrig1-CreER;Apcfl/+;Bves–/– adenomas display increased LRP6 and pLRP6 levels. These changes in receptor activation were associated with an increase in the number of pH3-positive proliferating cells. Expanding these observations, we show that BVES knockdown increases LRP6 phosphorylation in non-transformed human colonoids with associated increases in AXIN2 mRNA. Using the HCT116 CRC cell line, we also observed increases in Wnt pathway activation and LRP6 and β-catenin protein levels following BVES knockdown despite a stabilizing mutation in one allele of β-catenin (51). The more nuanced effect on β-catenin protein levels in this cell line is probably due to the reduced pool of modifiable β-catenin. Together, these data demonstrate that BVES loss increases LRP6 receptor levels and activation in the setting of an intact destruction complex, in adenoma tumoroid models with homozygous APC truncations and in CRC cell lines.
Given the preponderance of APC mutations in CRC, it was important to show that BVES loss can alter tumorigenic phenotypes in the context of these strong pro-tumorigenic mutations. Using the Lrig1-CreER;Apcfl/+ tumor model, we confirm that BVES loss leads to increased tumor multiplicity and higher degrees of dysplasia. Importantly, this model mimics human CRC progression where BVES loss and APC truncations occur early in the tumorigenic cascade. Surprisingly, we did not observe an overall change in tumor size but the increased tumor multiplicity suggests that BVES loss increases tumor initiation, a process associated with higher levels of Wnt tone (12,52,53). We suspect that the strong pro-proliferative signal generated by loss of both alleles of Apc in these tumors masks the increase Wnt signaling observed in the Bves–/– setting. AOM is a known chemical mutagen and tumor initiator and we again observed increased tumor multiplicity and dysplasia in the setting of BVES loss. Together, these tumor models support a role for BVES as a tumor suppressor and highlight that BVES loss can increase both tumor multiplicity and tumor progression. Although the effects of BVES loss on tumorigenesis are multifactorial, it is likely that the disinhibition of Wnt signaling plays a prominent role.
This report has focused on BVES in regulating canonical Wnt signaling; however, a role for BVES in regulating non-canonical Wnt signaling cannot be excluded. In fact, previous reports have demonstrated that BVES reduces RhoA activity by sequestering GEF-H1 and Wnt ligands can activate Rho signaling through the non-canonical Planar Cell Polarity pathway (28). Future studies will be required to determine whether BVES coordinately inhibits RhoA through multiple mechanisms.
In summary, our current working model is that BVES tunes Wnt signaling at the receptor level by interacting with LRP6 and subsequently inhibiting signalosome formation (Figure 6H). This prevents ligand-independent LRP6 receptor and Wnt pathway activation. In the absence of BVES, the LRP6 receptor is phosphorylated, the β-catenin destruction complex is inhibited and β-catenin protein levels increase. Given this primed receptor state, Wnt stimulation then further increases pathway activation. Because of the increased Wnt tone and sensitivity to ligand stimulation, the loss of BVES early in tumorigenesis facilitates growth of transformed cells, thereby augmenting tumorigenesis and progression.
Funding
National Institute of Health grants (R01DK099204 to C.S.W.; R35CA197570 and P50CA95103 to R.J.C.; R35GM122516 to E.L.; P50CA095103 to M.K.W.; T32GM007347 to J.J.T., B.P., R.E.B.; F30DK111107 to J.J.T.; F30DK096718 to B.P.; F32DK108492 to S.P.S.; F30DK120149 to R.E.B.), P30DK058404 (Vanderbilt Digestive Disease Research Center), UL1TR000445 (Vanderbilt’s Clinical and Translational Science Award). Merit Review Grant from the Office of Medical Research, Department of Veterans Affairs (1I01BX001426 to C.S.W.).
Conflict of Interest Statement
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We appreciate the many helpful discussions with members of the Williams, Lee, and Coffey laboratories. J.J.S. is supported by the Joel J. Roslyn Faculty Research Award from the Association for Academic Surgery, a Limited Project Grant and a Career Development Grant from the American Society of Colon and Rectal Surgeons, a Research Grant from the Society of Memorial Sloan Kettering, and the Franklin Martin, MD, FACS Faculty Research Fellowship from the American College of Surgeons, the Department of Surgery Faculty Research Award and the Wasserman Colon and Rectal Cancer Fund. The funders had no role in the decision to publish, or in preparation of the manuscript.
Glossary
Abbreviations
- AOM
azoxymethane
- APC
adenomatous polyposis coli
- BVES
blood vessel epicardial substance
- cDNA
complementary DNA
- CRC
colorectal cancer
- CK1α
casein kinase 1 alpha
- DMEM
Dulbecco’s modified Eagle’s medium
- DVL
Dishevelled
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GSK3
glycogen synthase kinase 3
- HEPES
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- OA
okadaic acid
- PBS
phosphate-buffered saline
- PP2A
protein phosphatase 2A
- siRNA
small interfering RNA
- shRNA
short hairpin RNA
- WT
wild-type
References
- 1. Siegel, R.L. et al. (2017) Colorectal cancer statistics, 2017. CA. Cancer J. Clin., 67, 177–193. [DOI] [PubMed] [Google Scholar]
- 2. Siegel, R.L. et al. (2018) Cancer statistics, 2018. CA. Cancer J. Clin., 68, 7–30. [DOI] [PubMed] [Google Scholar]
- 3. The Cancer Genome Atlas, N. (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature, 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clevers, H. et al. (2012) Wnt/β-catenin signaling and disease. Cell, 149, 1192–1205. [DOI] [PubMed] [Google Scholar]
- 5. Liu, C. et al. (2002) Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell, 108, 837–847. [DOI] [PubMed] [Google Scholar]
- 6. Marikawa, Y. et al. (1998) beta-TrCP is a negative regulator of Wnt/beta-catenin signaling pathway and dorsal axis formation in Xenopus embryos. Mech. Dev., 77, 75–80. [DOI] [PubMed] [Google Scholar]
- 7. Wehrli, M. et al. (2000) Arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature, 407, 527–530. [DOI] [PubMed] [Google Scholar]
- 8. Pinson, K.I. et al. (2000) An LDL-receptor-related protein mediates Wnt signalling in mice. Nature, 407, 535–538. [DOI] [PubMed] [Google Scholar]
- 9. Niehrs, C. et al. (2010) Regulation of Lrp6 phosphorylation. Cell. Mol. Life Sci., 67, 2551–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jho, E.H. et al. (2002) Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell. Biol., 22, 1172–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reese, D.E. et al. (1999) bves: a novel gene expressed during coronary blood vessel development. Dev. Biol., 209, 159–171. [DOI] [PubMed] [Google Scholar]
- 12. Williams, C.S. et al. (2011) BVES regulates EMT in human corneal and colon cancer cells and is silenced via promoter methylation in human colorectal carcinoma. J. Clin. Invest., 121, 4056–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parang, B. et al. (2017) BVES regulates c-Myc stability via PP2A and suppresses colitis-induced tumourigenesis. Gut, 66, 852–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reddy, V.K. et al. (2016) BVES regulates intestinal stem cell programs and intestinal crypt viability after radiation. Stem Cells, 34, 1626–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choksi, Y.A. et al. (2018) BVES is required for maintenance of colonic epithelial integrity in experimental colitis by modifying intestinal permeability. Mucosal Immunol., 11, 1363–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takahashi, M. et al. (2000) Frequent mutations of the beta-catenin gene in mouse colon tumors induced by azoxymethane. Carcinogenesis, 21, 1117–1120. [PubMed] [Google Scholar]
- 17. Saito-Diaz, K. et al. (2018) APC inhibits ligand-independent Wnt signaling by the clathrin endocytic pathway. Dev. Cell, 44, 566–581.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu, Q. et al. (2004) Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell, 116, 883–895. [DOI] [PubMed] [Google Scholar]
- 19. Gammons, M.V. et al. (2016) Essential role of the dishevelled DEP domain in a Wnt-dependent human-cell-based complementation assay. J. Cell Sci., 129, 3892–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Osler, M.E. et al. (2005) Bves modulates epithelial integrity through an interaction at the tight junction. J. Cell Sci., 118(Pt 20), 4667–4678. [DOI] [PubMed] [Google Scholar]
- 21. Kategaya, L.S. et al. (2009) Bili inhibits Wnt/beta-catenin signaling by regulating the recruitment of axin to LRP6. PLoS One, 4, e6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sato, T. et al. (2011) Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology, 141, 1762–1772. [DOI] [PubMed] [Google Scholar]
- 23. Heijmans, J. et al. (2013) ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Rep., 3, 1128–1139. [DOI] [PubMed] [Google Scholar]
- 24. Thorne, C.A. et al. (2010) Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol., 6, 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bandapalli, O.R. et al. (2009) Transcriptional activation of the beta-catenin gene at the invasion front of colorectal liver metastases. J. Pathol., 218, 370–379. [DOI] [PubMed] [Google Scholar]
- 26. Freeman, T.J. et al. (2012) Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of β-catenin. Gastroenterology, 142, 562–571.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Su, Y. et al. (2003) Eradication of pathogenic beta-catenin by Skp1/Cullin/F box ubiquitination machinery. Proc. Natl. Acad. Sci. USA, 100, 12729–12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhan, T. et al. (2017) Wnt signaling in cancer. Oncogene, 36, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wada, A.M. et al. (2001) Bves: prototype of a new class of cell adhesion molecules expressed during coronary artery development. Development, 128, 2085–2093. [DOI] [PubMed] [Google Scholar]
- 30. Saito-Diaz, K. et al. (2013) The way Wnt works: components and mechanism. Growth Factors, 31, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gao, C. et al. (2010) Dishevelled: the hub of Wnt signaling. Cell. Signal., 22, 717–727. [DOI] [PubMed] [Google Scholar]
- 32. González-Sancho, J.M. et al. (2013) Functional consequences of Wnt-induced dishevelled 2 phosphorylation in canonical and noncanonical Wnt signaling. J. Biol. Chem., 288, 9428–9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zeng, X. et al. (2008) Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development, 135, 367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hernández, F. et al. (2010) Regulation of GSK3 isoforms by phosphatases PP1 and PP2A. Mol. Cell. Biochem., 344, 211–215. [DOI] [PubMed] [Google Scholar]
- 35. Wadzinski, B.E. et al. (1993) Nuclear protein phosphatase 2A dephosphorylates protein kinase A-phosphorylated CREB and regulates CREB transcriptional stimulation. Mol. Cell. Biol., 13, 2822–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zachos, N.C. et al. (2016) Human enteroids/colonoids and intestinal organoids functionally recapitulate normal intestinal physiology and pathophysiology. J. Biol. Chem., 291, 3759–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Powell, A.E. et al. (2012) The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell, 149, 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maltzman, T. et al. (1997) AOM-induced mouse colon tumors do not express full-length APC protein. Carcinogenesis, 18, 2435–2439. [DOI] [PubMed] [Google Scholar]
- 39. Orsulic, S. et al. (1999) E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J. Cell Sci., 112 (Pt 8), 1237–1245. [DOI] [PubMed] [Google Scholar]
- 40. Froese, A. et al. (2012) Popeye domain containing proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J. Clin. Invest., 122, 1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fajardo, A.M. et al. (2014) The role of cyclic nucleotide signaling pathways in cancer: targets for prevention and treatment. Cancers (Basel)., 6, 436–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bilic, J. et al. (2007) Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science, 316, 1619–1622. [DOI] [PubMed] [Google Scholar]
- 43. Russ, P.K. et al. (2011) Bves modulates tight junction associated signaling. PLoS One, 6, e14563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee, H.J. et al. (2010) PDZ domains and their binding partners: structure, specificity, and modification. Cell Commun. Signal., 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Osler, M.E. et al. (2006) Bves, a member of the Popeye domain-containing gene family. Dev. Dyn., 235, 586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Knight, R.F. et al. (2003) Membrane topology of Bves/Pop1A, a cell adhesion molecule that displays dynamic changes in cellular distribution during development. J. Biol. Chem., 278, 32872–32879. [DOI] [PubMed] [Google Scholar]
- 47. Joiner, D.M. et al. (2013) LRP5 and LRP6 in development and disease. Trends Endocrinol. Metab., 24, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hager, H.A. et al. (2010) Identification of a novel Bves function: regulation of vesicular transport. EMBO J., 29, 532–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thompson, J.J., et al. (2018) Protein phosphatase 2A in the regulation of Wnt signaling, stem cells, and cancer. Genes (Basel), 9:121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goentoro, L. et al. (2009) Evidence that fold-change, and not absolute level, of beta-catenin dictates Wnt signaling. Mol. Cell, 36, 872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ilyas, M. et al. (1997) Beta-catenin mutations in cell lines established from human colorectal cancers. Proc. Natl. Acad. Sci. USA, 94, 10330–10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Giles, R.H. et al. (2003) Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta, 1653, 1–24. [DOI] [PubMed] [Google Scholar]
- 53. Fearon, E.R. et al. (1990) A genetic model for colorectal tumorigenesis. Cell, 61, 759–767. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.