

SUMMARY
Flux through the RAF-MEK-ERK protein kinase cascade is shaped by phosphatases acting on the core components of the pathway. Despite being an established drug target and a hub for crosstalk regulation, little is known about dephosphorylation of MEK, the central kinase within the cascade. Here, we identify PPP6C, a phosphatase frequently mutated or downregulated in melanoma, as a major MEK phosphatase in cells exhibiting oncogenic ERK pathway activation. Recruitment of MEK to PPP6C occurs through an interaction with its associated regulatory subunits. Loss of PPP6C causes hyperphosphorylation of MEK at activating and crosstalk phosphorylation sites, promoting signaling through the ERK pathway and decreasing sensitivity to MEK inhibitors. Recurrent melanoma-associated PPP6C mutations cause MEK hyperphosphorylation, suggesting that they promote disease at least in part by activating the core oncogenic pathway driving melanoma. Collectively, our studies identify a key negative regulator of ERK signaling that may influence susceptibility to targeted cancer therapies.
Graphical abstract
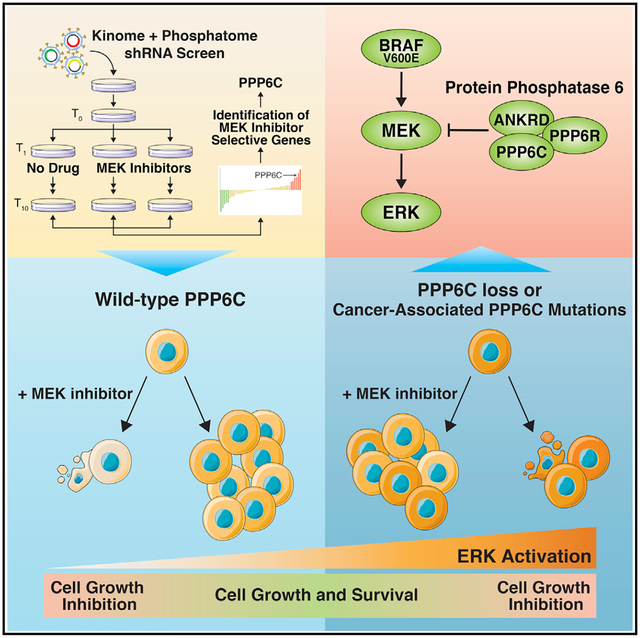
In brief
Through an shRNA screen, Cho et al. identify PPP6C as a phosphatase that inactivates the kinase MEK, sensitizing tumor cells to clinical MEK inhibitors. This study suggests that cancer-associated loss-of-function PPP6C mutations prevalent in melanoma serve to activate the core oncogenic RAF-MEK-ERK pathway that drives the disease.
INTRODUCTION
The RAF-MEK-ERK mitogen-activated protein kinase (MAPK) signaling cascade regulates essential cellular processes including cell proliferation, differentiation, and survival (Lavoie et al., 2020). Deregulation of ERK signaling, typically through mutations in core pathway components or upstream regulators, is among the most frequent driver events in human cancer (Burotto et al., 2014). Malignant melanoma, in particular, is dependent upon hyperactivated ERK signaling. About half of human melanomas harbor mutations in the BRAF gene, the most common being the V600E mutation that causes high level constitutive activity (Cancer Genome Atlas Network, 2015; Davies et al., 2002; Hayward et al., 2017; Hodis et al., 2012; Krauthammer et al., 2015). Most other melanoma tumors have either gain-of-function mutations in the NRAS GTPase, a direct activator of RAF kinases, or loss-of-function mutations in the RAS GTPase-activating protein NF1. The dependence of melanomas on the ERK pathway fueled the development and approval of selective BRAF inhibitors (BRAFi) and MEK inhibitors (MEKi) with clinical efficacy in treating tumors harboring BRAFV600E mutations (Samatar and Poulikakos, 2014). Unfortunately, responses to BRAFi and MEKi are almost invariably short lived due to the development of acquired resistance (Flaherty et al., 2012). While resistance to these agents can involve activation of alternative cell growth and survival pathways, it most commonly occurs through reactivation of the ERK pathway despite the continued presence of inhibitor (Lim et al., 2017). Mechanisms underlying ERK pathway reactivation include acquisition of RAS or MEK mutations, BRAF amplification or alternative splicing, disruption of negative feedback regulation, and induction of receptor tyrosine kinases (Johnson et al., 2015; Rizos et al., 2014; Shi et al., 2014; Van Allen et al., 2014). Understanding how tumor cells become resistant to BRAFi and MEKi can suggest additional therapeutic targets and thus contribute to the development of durable and more generally applicable cancer treatments. In addition, investigations into mechanisms of inhibitor resistance have provided insight into the basic wiring of the ERK pathway and how it participates in larger signaling networks.
Because proper control of the ERK pathway is important to normal physiology, the core cascade is positioned within a complex network involving extensive feedback and crosstalk regulation (Lavoie et al., 2020). By counteracting regulatory phosphorylation events, protein phosphatases play key roles in controlling the magnitude and duration of ERK signaling, and their dysregulation can contribute to disease and influence inhibitor sensitivity. For example, ERK signaling induces expression of dual-specificity MAPK phosphatases (DUSPs), which dephosphorylate and inactivate ERK (Bermudez et al., 2010). Disruption of this negative feedback loop through deletion or downregulation of ERK-selective DUSPs has been reported in some tumors and is associated with more advanced disease and poor patient prognosis (Cai et al., 2015; Okudela et al., 2009; Shin et al., 2013; Xu et al., 2005). Protein phosphatases also have important roles in positively regulating ERK signaling. For example, the tyrosine phosphatase SHP2 is important for relaying signals from receptor tyrosine kinases to RAS GTPases, and SHP2 inhibitors are currently in development as cancer therapeutics (Chen et al., 2016). In addition, the MRAS-SHOC2-PP1 complex dephosphorylates an inhibitory site on RAF and mediates ERK pathway reactivation induced by MEKi in KRAS mutant pancreatic and lung cancers (Jones et al., 2019; Sulahian et al., 2019). Protein phosphatase 2A (PP2A) can promote ERK signaling by dephosphorylating inhibitory feedback phosphorylation sites on RAF and the ERK pathway scaffold KSR1 (Ory et al., 2003). While these phosphatases regulating RAS, RAF, and ERK have established roles in normal and pathological signaling, less is known about phosphatases regulating MEK, the central component of the cascade. PP2A was initially identified as a MEK phosphatase in vitro and in non-transformed monkey kidney CV-1 cells (Gómez and Cohen, 1991; Sontag et al., 1993). However, other studies have suggested that PP2A restrains oncogenic MAPK signaling primarily through direct dephosphorylation of ERK.
Here, we have identified PPP6C as a MEK phosphatase that negatively regulates oncogenic ERK signaling and promotes sensitivity to MEKi. The potential significance of this observation is underscored by the presence of loss-of-function PPP6C mutations in 6%–9% of malignant melanomas, generally co-occurring with BRAF and NRAS mutations (Cancer Genome Atlas Network, 2015; Hodis et al., 2012; Krauthammer et al., 2015). PPP6C is a conserved essential serine-threonine metallophosphatase related to the catalytic subunit of PP2A (Shi, 2009). PPP6C functions within heterotrimeric PP6 holoenzymes consisting of a Sit4-associated protein (SAPS) domain regulatory subunit (PPP6R1, PPP6R2, or PPP6R3) that mediates substrate recruitment and an ankyrin repeat (ANKRD28, ANKRD44, or ANKRD52) subunit that may serve a scaffolding role (Stefansson and Brautigan, 2006; Stefansson et al., 2008). The presence of multiple regulatory and scaffolding subunits defines nine potential PP6 complexes that collectively participate in a variety of cellular processes, including cell-cycle progression (Douglas et al., 2014; Rusin et al., 2017; Zeng et al., 2010), the DNA damage response (Douglas et al., 2014; Hosing et al., 2012; Shen et al., 2011; Zhong et al., 2011), autophagy (Wengrod et al., 2015), miRNA processing (Golden et al., 2017), inflammatory response (Kajino et al., 2006; Ye et al., 2015), and antiviral immunity (Tan et al., 2017). Relevant to its role as a tumor suppressor, loss of PPP6C causes defects in mitotic spindle assembly owing to hyperphosphorylation of the kinase Aurora A, and consequent genomic instability has been proposed as an early event in tumorigenesis (Gold et al., 2014; Hammond et al., 2013). By contrast, connections between PPP6C and core oncogenic pathways have yet to be described.
We identified PPP6C as a MEK phosphatase through a short hairpin RNA (shRNA) screen to identify genes modulating the response of a BRAF mutant melanoma cell line to MEKi. Loss of PPP6C appears to decrease sensitivity to MEKi by inducing MEK hyperphosphorylation, thereby maintaining ERK activity in the presence of inhibitor. We found that negative regulation of MEK by PPP6C appears to be a general feature of cells characterized by hyperactive ERK signaling, including cell lines harboring NRAS and KRAS mutations. Furthermore, we find that melanoma-associated mutations impair the ability of PPP6C to dephosphorylate MEK, providing a likely explanation for the prevalence of PPP6C mutations in melanomas and potentially other tumors. These studies report a major negative regulator that dephosphorylates the central component in the core ERK signaling cascade.
RESULTS
Pooled shRNA library screen identifies PPP6C as a mediator of response to MEKi
To identify genes involved in modulating the response to MEK inhibitors, we performed a pooled shRNA screen in the BRAFV600E mutant melanoma cell line 501mel (Figure 1A). We used a custom-made lentiviral library of 7,649 shRNAs targeting 817 genes encoding annotated protein and lipid kinases and phosphatases (Table S1). After transduction with the shRNA library, we harvested a portion of the cells for a start time reference sample (T0) and divided the remaining cells into 5 populations, which were subsequently treated with either vehicle control or one of two concentrations of the MEKis trametinib (1 and 3.3 nM) or selumetinib (33 and 100 nM). MEKi concentrations were chosen to flank the approximate IC50 for inhibition of cell growth. Cells were propagated through 10 population doublings (T1–T10). The change in abundance of each shRNA under each growth condition was then determined by next-generation sequencing (Illumina HiSeq) following PCR amplification from genomic DNA preparations of cells collected at T0 and T10 (Table S1). As expected for a cell line carrying a BRAFV600E mutation, hairpins targeting BRAF were depleted in all populations over time (Figure 1B), providing an internal control for the screens.
Figure 1. Pooled shRNA library screen identifies PPP6C as a mediator of response to MEKi.
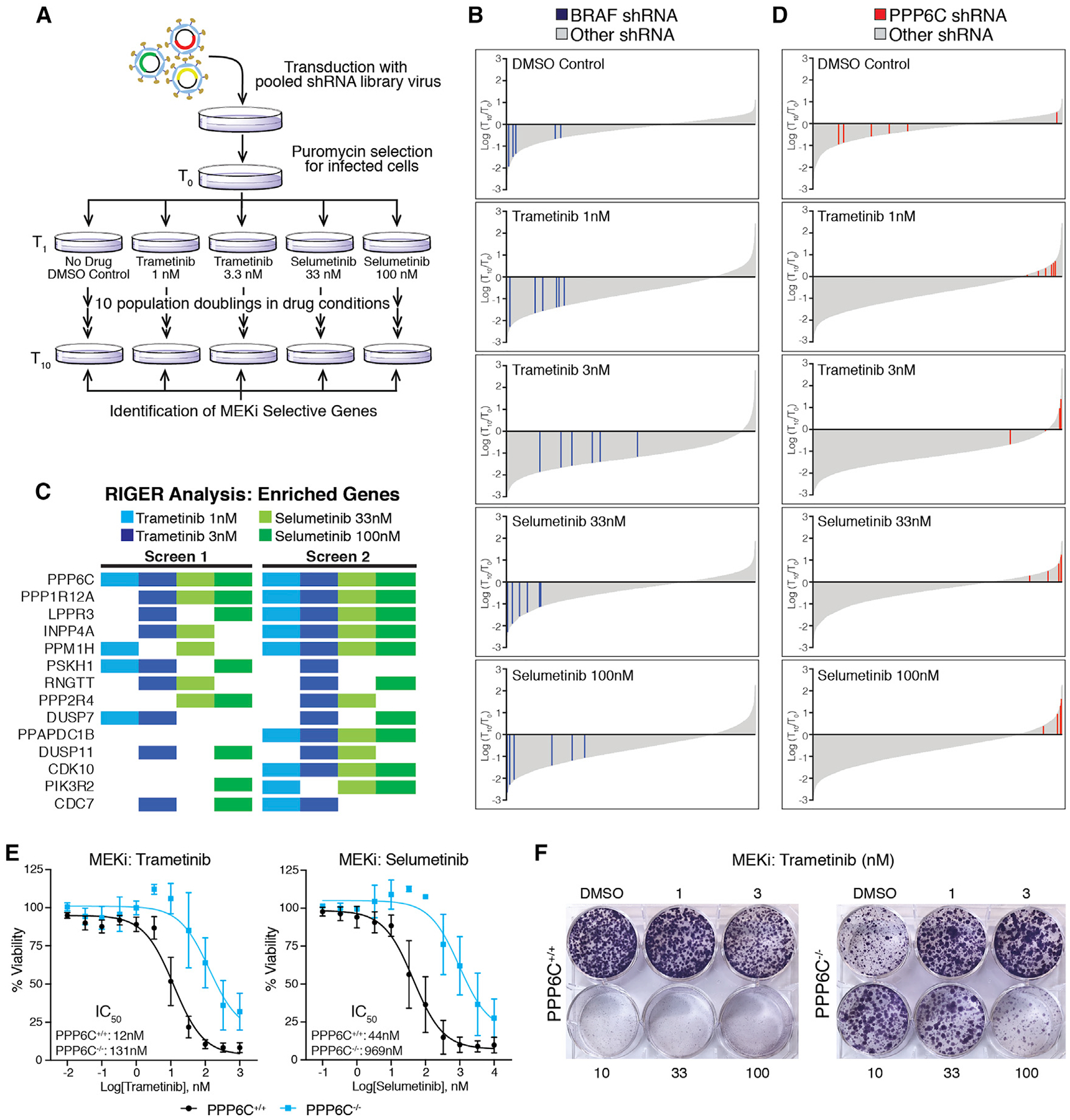
(A) Schematic of the pooled shRNA MEKi sensitivity screen.
(B) Changes in all shRNA hairpins shown as log2(T10/T0) from most depleted to most enriched for each drug condition. Bars representing shRNA hairpins targeting BRAF are shown in blue. All others are shown in gray.
(C) Top enriched genes for each drug condition from two replicates of the screen. Colored boxes indicate the genes ranked in the top 50 enriched genes by RIGER for that drug condition not found in the DMSO control condition.
(D) Changes in all shRNA hairpins shown as log2(T10/T0) arranged as in (B) but with red bars indicating shRNA hairpins targeting PPP6C. Ranking of each hairpin is shown in Figure S1A.
(E) PPP6C+/+ and PPP6C−/− 501mel cells were treated for 72 h with increasing concentrations of trametinib or selumetinib. Cell viability was determined and normalized to vehicle control for each cell line. Dose-response curves and IC50 values for PPP6C+/+ (black) and PPP6C−/− (blue) are shown. The 95% confidence intervals (n = 3) were 8.9–17 nM (PPP6C+/+, trametinib), 65–209 nM (PPP6C−/−, trametinib), 30–66 nM (PPP6C+/+, selumetinib), and 500–1,900 nM (PPP6C−/−, selumetinib). Error bars show SD.
(F) PPP6C+/+ and PPP6C−/− 501mel cells were cultured in media containing DMSO or the indicated concentration of trametinib for 2 weeks and stained with crystal violet. Quantification is shown in Figure S2A. n = 3.
See also Figures S1B, S2B, and S2C.
We used RNAi gene enrichment ranking (RIGER) analysis (Luo et al., 2008) to rank genes based on depletion or enrichment of their shRNAs in the screen. This analysis revealed that hairpins targeting PPP6C were the most consistently enriched in the presence of MEKi (Figures 1C and S1A). PPP6C was the top ranked gene in 5 of 8 MEKi conditions across the two screens and scored within the top 20 genes under all drug conditions (Table S2). While the six PPP6C-targeting hairpins were highly enriched in the presence of MEKi, they were generally depleted from the untreated culture (Figures 1D and S1A). These results suggest that PPP6C is required both for optimal cell growth and for a maximal cytostatic response to MEKi.
To independently verify PPP6C as a factor modulating sensitivity to MEKi, we generated clonal PPP6C knockout 501mel lines by CRISPR-Cas9-mediated gene disruption (Figure S1B). Notably, we were unable to expand PPP6C−/− clones unless we supplemented the growth media with a low concentration of the MEKi trametinib. PPP6C deletion (Figure 1E) or shRNA knockdown (Figure S2) caused rightward shifts to MEKi dose-response curves, increasing IC50 values for growth inhibition by trametinib and selumetinib (Figure 1E). In clonogenic assays, control PPP6C+/+ cells exhibited dose-dependent growth inhibition by both MEKi as anticipated (Figures 1F and S2A). PPP6C knockout alone substantially decreased cell growth, yet this growth defect was largely reversed in the presence of low concentrations of MEKi. In addition, cells in which PPP6C had been deleted grew better than control cells at higher MEKi concentrations. Growth suppression was also observed in cells transduced with individual shRNAs targeting PPP6C, though rescue by MEKi was less consistent than with complete deletion of the gene (Figure S2C). These observations verify that loss of PPP6C renders 501mel cells less sensitive to the cytostatic effects of MEKi, consistent with the behavior of shRNAs targeting PPP6C in the screens.
PPP6C negatively regulates ERK signaling
Resistance to ERK pathway inhibitors can occur through multiple mechanisms, for example, through activation of bypass pathways (Johnson et al., 2015; Lu et al., 2017; Shi et al., 2014). We suspected that loss of PPP6C led to ERK pathway hyperactivation, as this phenomenon can underlie inhibitor resistance and cause growth suppression (Hong et al., 2018; Moriceau et al., 2015). Indeed, we observed that silencing of PPP6C in 501mel cells elevated the levels of activating phosphorylation of ERK1/2 and MEK1/2 (Figure 2A). Cells expressing shPPP6C required higher concentrations of inhibitor to reduce MEK and ERK phosphorylation to levels seen in control cells (Figure 2B), likely explaining why PPP6C knockdown decreases sensitivity to MEKi. Likewise, low concentrations of MEKi attenuated hyperactivation of ERK signaling that is presumably toxic to these cells, explaining why decreasing PPP6C expression reduces growth. Furthermore, the ability of low concentrations of MEKi to rescue growth (Figure 1F) suggests that PPP6C is required for optimal growth of 501mel cells largely because it restrains ERK signaling. Analysis of PPP6C knockout 501mel cells revealed an even more pronounced increase in MEK and ERK phosphorylation than seen with partial loss of PPP6C via shRNA (Figure 2C). Re-expression of wild-type (WT) PPP6C, but not a phosphatase inactive mutant (D84N, PD), in these cells reversed ERK hyperactivation (Figure 2D) and rescued the growth defect (Figure S2D), indicating that negative regulation of ERK signaling requires PPP6C phosphatase activity. PPP6C knockout or downregulation by shRNA did not appear to induce apoptosis in 501mel cell lines as judged by levels of caspase-3 and PARP cleavage, suggesting that they cause slow growth rather than cell death (Figure S2E).
Figure 2. PPP6C negatively regulates ERK signaling.
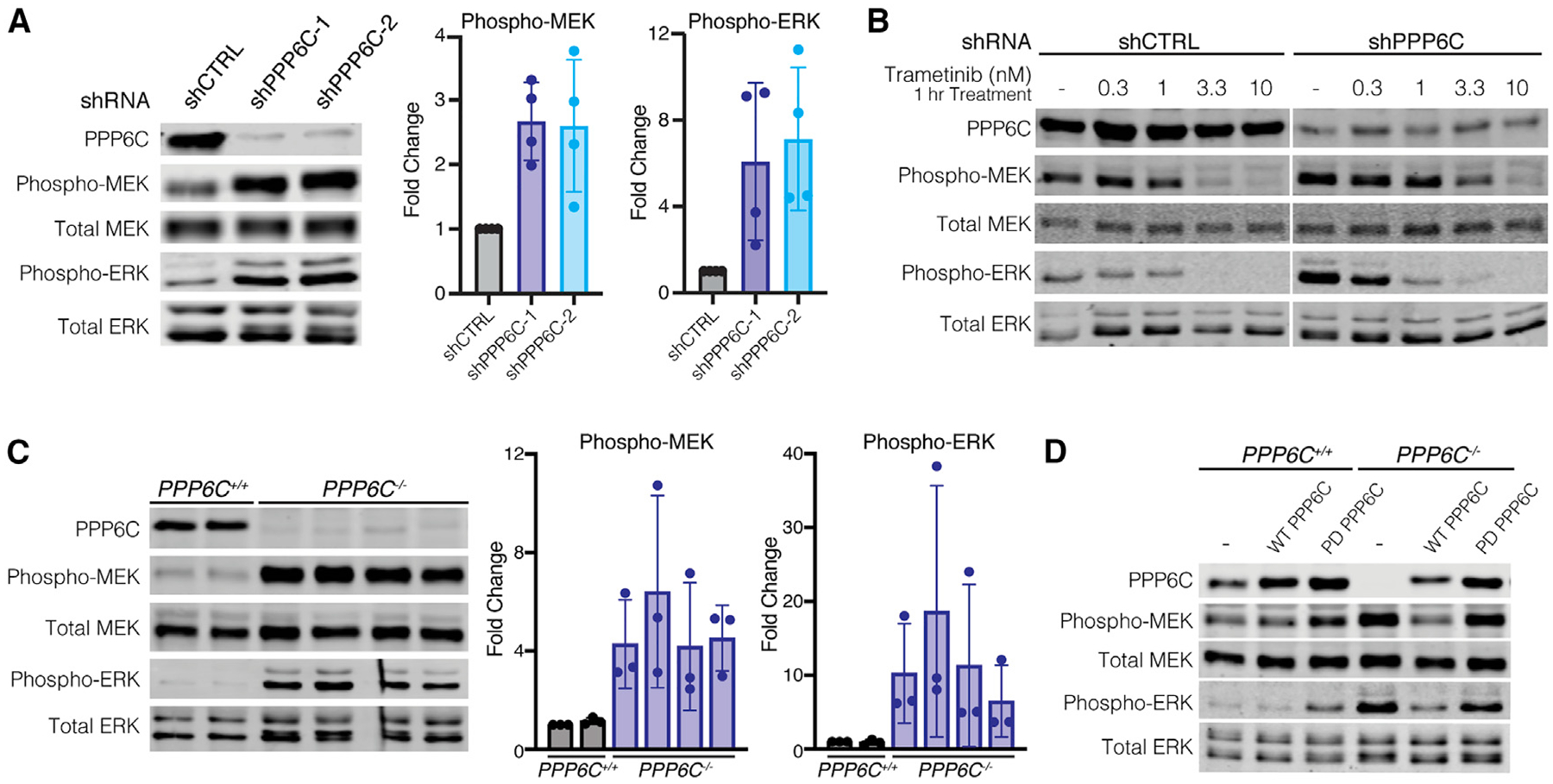
(A) 501mel cells stably expressing shCTRL, shPPP6C-1, and shPPP6C-2 were lysed, and levels of phosphorylated and total MEK and ERK were assessed by immunoblot. Quantification of Phospho/Total MEK and ERK was normalized to shCTRL. Data are represented as mean ± SD, n = 4.
(B) shCTRL and shPPP6C-expressing 501mel cells were treated with the indicated concentrations of trametinib for 1 h and lysed. Phosphorylated and total levels of MEK and ERK were detected by immunoblot.
(C) PPP6C+/+ and PPP6C−/− 501mel cell lines were lysed and assessed by immunoblot for levels of phosphorylated and total MEK and ERK. Quantification of Phospho/Total MEK and ERK was normalized to PPP6C+/+. Data are represented as mean ± SD, n = 3.
(D) PPP6C+/+ and PPP6C−/− 501mel cells stably expressing WT PPP6C, phosphatase inactive PPP6CD84N (PD) or GFP (–) as a control were lysed and assessed by immunoblot for phosphorylated and total MEK and ERK.
See also Figure S2E.
To determine whether PPP6C acts as a general regulator of ERK signaling, we examined the effect of PPP6C knockdown on a panel of cell lines of varying genotype and lineage (Figures 3A and S3). Silencing PPP6C expression led to MEK hyperphosphorylation in each of five additional BRAFV600 mutant melanoma cell lines tested, including YURIF cells heterozygous for a PPP6CS270L mutation. Among four NRASQ61 mutant melanoma cell lines, all but one (YUGASP) exhibited increased MEK phosphorylation upon PPP6C knockdown. We observed the same phenomenon with MEL-ST, a non-transformed immortalized melanocyte cell line. Additionally, in three colon carcinoma cell lines with BRAF or RAS mutations, we also observed increased MEK phosphorylation with PPP6C loss. We note that the impact of PPP6C knockdown on the level of ERK phosphorylation across this panel of cell lines was more variable, suggesting that feedback mechanisms acting directly on ERK may blunt regulation by PPP6C. The osteosarcoma cell line U2OS, which does not harbor BRAF or RAS mutations, and KRAS mutant A549 lung adenocarcinoma cells did not display consistent increases in phospho-MEK levels upon PPP6C knockdown. Overall, the majority of cell lines examined displayed PPP6C regulation of ERK signaling.
Figure 3. PPP6C regulation of ERK signaling is prominent in ERK pathway-driven cancer cells.
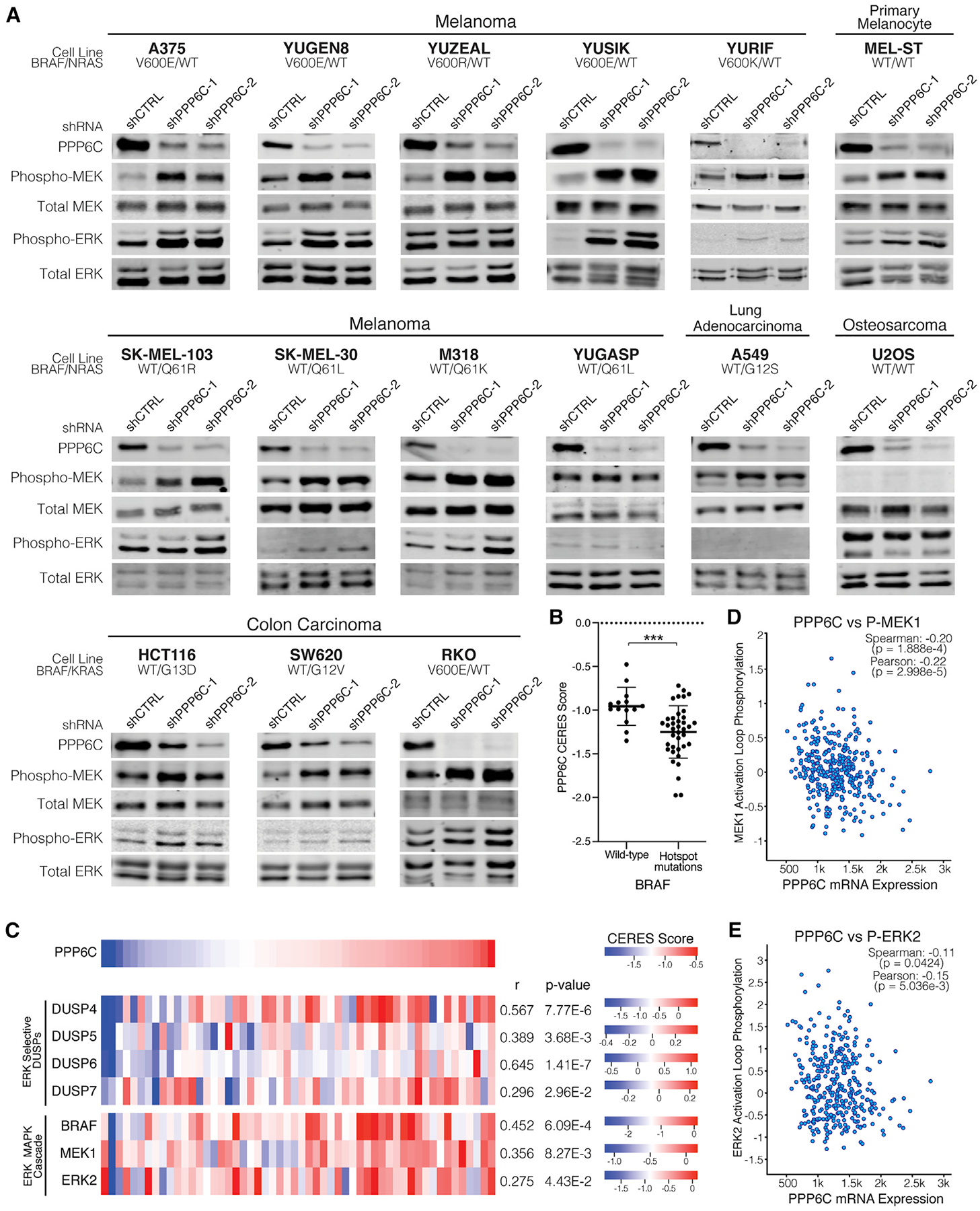
(A) The indicated cell lines were transduced to stably express control (shCTRL) or PPP6C-targeting (shPPP6C-1 or PPP6C-2) shRNAs. Cells were lysed and assessed by immunoblot for levels of phosphorylated and total MEK and ERK. Quantification is shown in Figure S3. n ≥ 2.
(B) PPP6C CERES scores for skin cancer cell lines with WT BRAF or hotspot BRAF mutations from the Cancer Dependency Map Project. Cell lines harboring BRAF variants of unknown significance were excluded. Data are represented as mean ± SD. ***p < 0.0005, Welch’s t test.
(C) Heatmaps depicting CERES scores of PPP6C, ERK-selective DUSPs, and ERK MAPK cascade components in skin cancer cell lines using data from the Cancer Dependency Map Project. Pearson’s correlation coefficients and p values from linear regression analysis of each gene with PPP6C in the DepMap portal are listed.
(D) PPP6C RNA-seq mRNA expression level plotted against MEK1 phosphorylation levels for TCGA tumor samples in cBioPortal. Correlation coefficients and associated p values from linear regression analyses are indicated.
(E) PPP6C mRNA levels plotted against ERK2 phosphorylation levels for TCGA tumor samples as in (D).
See also Figure S4.
We further examined a role for PPP6C as a regulator of ERK signaling by analyzing genome-wide CRISPR-Cas9 screening data across a large panel of cell lines from the Cancer Dependency Map Project (Dempster et al., 2019; DepMapBroad, 2020, 2020; Meyers et al., 2017). PPP6C is categorized as a common essential gene with an average CERES gene dependency score of −1.00 ± 0.28, where a more negative score indicates a larger effect on cell growth or survival (Meyers et al., 2017). Notably, in skin cancer cell lines, the mean CERES score for PPP6C is −1.16 ± 0.31, indicating these cell lines are in general more dependent on PPP6C. Among skin cancer cell lines, those with BRAF hotspot mutations were significantly more dependent on PPP6C than those that are WT for BRAF (Figure 3B). Taken together with our data in 501mel cells, these data suggest that cells characterized by hyperactive ERK signaling are more sensitive to loss of PPP6C.
We also compared the dependency of skin cancer cell lines on PPP6C and core components of the ERK signaling pathway by examining pairwise correlations between genes. In these data, co-dependency between two genes across cell lines can indicate that they participate in a common pathway (Boyle et al., 2018; Kim et al., 2019). Dependency on PPP6C significantly correlates with dependency on BRAF, MAP2K1 (encoding MEK1), and MAPK1 (encoding ERK2) (Figures 3C and S4A–S4D). Strikingly, strongest co-dependency with PPP6C was found among negative regulators of the pathway, the ERK-selective dual specificity protein phosphatases (DUSP4, DUSP5, DUSP6, and DUSP7). Consistent with a key role for PPP6C in dephosphorylating Aurora A (Hammond et al., 2013; Zeng et al., 2010), PPP6C dependency negatively correlated with that of AURKA among the full set of cell lines across all lineages (Pearson’s correlation coefficient =− 0.216, p = 1.40 × 10−9) (Figure S4F). However, this correlation was not significant in skin cancer cell lines (p = 0.44) (Figure S4E), suggesting that regulation of Aurora A is not the primary determinant of PPP6C dependency in these cells. Overall, these correlations suggest that in skin cancer cell lines, dependency on PPP6C is associated with its role as a regulator of ERK signaling.
To examine whether PPP6C might modulate ERK signaling in human melanoma, we analyzed reverse-phase protein array and RNA-seq data collected across a panel of tumors from The Cancer Genome Atlas. We observed a significant correlation between PPP6C mRNA expression and levels of both phospho-MEK and phospho-ERK among tumor specimens (Figures 3D and 3E). However, the extent of ERK pathway activation was not significantly higher in tumors harboring recurrent or truncating PPP6C mutations (Figures S4G and S4H). These data suggest that MEK phosphorylation in melanoma tumors may be more strongly influenced by PPP6C levels than by its mutation.
PPP6C regulates ERK signaling via MEK1/2
Hyperphosphorylation of MEK observed with PPP6C loss suggests PPP6C regulates ERK signaling either at the level of MEK or upstream of MEK. To determine which component of the ERK signaling cascade is regulated by PPP6C, we initially investigated the RAF kinases directly upstream of MEK. In BRAFV600E mutant melanoma, oncogenic signaling is driven primarily by mutant BRAF, (Karasarides et al., 2004), but ERK pathway reactivation in settings of BRAFi/MEKi resistance can depend instead on CRAF (Montagut et al., 2008; Shi et al., 2011; Villanueva et al., 2010). To determine whether increased MEK phosphorylation observed with PPP6C loss is due to compensation by ARAF or CRAF, we silenced each of the RAF isoforms by small interfering RNA (siRNA) in combination with shRNA knockdown of PPP6C in 501mel cells (Figures 4A and 4B). In both the shCTRL and shPPP6C cells, only BRAF knockdown decreased MEK phosphorylation, while silencing ARAF and CRAF alone or in combination (Figure S5A) did not. Thus, in the context of PPP6C loss, BRAF remains the principal activator of MEK in 501mel cells. We note that in cells harboring RAS mutations, other RAF isoforms such as CRAF likely have a predominant role in MEK phosphorylation.
Figure 4. PPP6C regulates ERK signaling via MEK1/2.
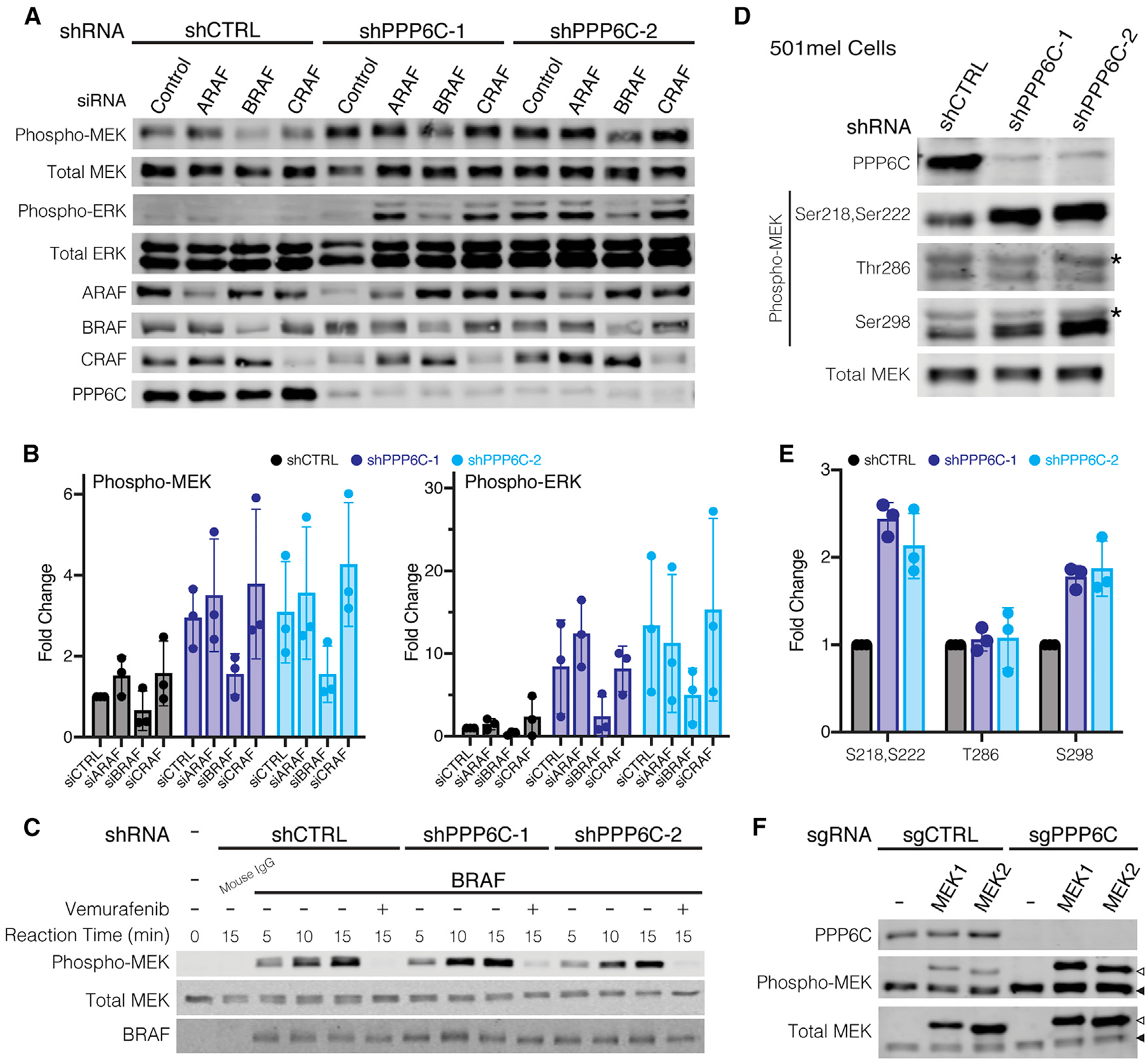
(A) shCTRL, shPPP6C-1, and shPPP6C-2-expressing 501mel cells were transfected with non-targeting control siRNA or siRNAs directed to ARAF, BRAF, or CRAF as indicated. Cells were lysed and assessed by immunoblot for phosphorylated and total MEK and ERK.
(B) Quantification of the relative levels of Phospho/Total MEK and ERK from (A) was normalized to shCTRL, siCONTROL. Data are represented as mean ± SD, n = 3.
(C) BRAF was immunoprecipitated from 501mel cells expressing shCTRL, shPPP6C-1, or shPPP6C and evaluated in vitro in kinase assays on MEK1 over the indicated time course. Vemurafenib (1 μM) was added to negative control reactions. Reactions were evaluated by immunoblot.
(D) 501mel cells expressing shCTRL, shPPP6C-1, and shPPP6C-2 were lysed and assessed by immunoblot for MEK phosphorylation at Ser218/Ser222, Thr286, and Ser298. Non-specific cross-reacting bands in the pThr286 and pSer298 blots are indicated with an asterisk.
(E) Quantification of the relative levels of Phospho/Total MEK from (D) was normalized to shCTRL. Data are represented as mean ± SD, n = 3.
(F) PPP6C+/+ and PPP6C−/− 501mel cell lines were transiently transfected to express His epitope-tagged MEK1 or MEK2. Cell lysates were analyzed by immunoblot for phosphorylated and total MEK. Open arrows indicate ectopically expressed His-tagged MEK1/2, and solid arrows indicate endogenous MEK1/2. See also Figure S5.
We next considered whether loss of PPP6C leads to increased BRAF activity. In immunoprecipitation kinase assays, BRAF isolated from both shCTRL and shPPP6C cells phosphorylated MEK1 at similar rates (Figure 4B), indicating PPP6C does not regulate BRAF activity. We noted that loss of PPP6C caused upward electrophoretic mobility shifts in BRAF and CRAF suggestive of a change in their phosphorylation states (Figures S5B and S5C). Treatment of cells with MEKi or BRAFi caused the multiple BRAF species to collapse into a lower, presumably less phosphorylated species (Figures S5B and S5C). Though we were unable to identify specific sites regulated by PPP6C (Figure S5C), this phenomenon likely reflects feedback phosphorylation from ERK hyperactivation (Lake et al., 2016; Ritt et al., 2010). As BRAFV600E is insensitive to such feedback phosphorylation (Ritt et al., 2010), it is unlikely to impact MEK phosphorylation in cells.
In addition to activation loop phosphorylation, we examined whether PPP6C affected sites of crosstalk phosphorylation on MEK. We found that MEK1 phosphorylation at Ser298, which is mediated by PAK1 to promote activation loop phosphorylation (Coles and Shaw, 2002; Lake et al., 2016), was elevated in cells lacking PPP6C (Figure 4C). In contrast, there was no effect on phosphorylation at Thr286, a negative regulatory site phosphorylated by CDK1 or CDK5 (Rossomando et al., 1994; Sharma et al., 2002). Because Ser298 phosphorylation is specific to MEK1 and not MEK2, we examined whether PPP6C selectively regulates MEK isoforms. Like endogenous MEK1/2, ectopically expressed MEK1 and MEK2 were both hyperphosphorylated when expressed in PPP6C−/− cells in comparison to WT cells (Figure 4D). PPP6C therefore does not preferentially regulate one isoform of MEK. This suggests that PPP6C regulates MEK activity by modulating activation loop phosphorylation independently of crosstalk pathways.
MEK1/2 is a direct substrate of PP6
As our findings above indicate PPP6C regulates MEK1/2 activation loop phosphorylation without affecting RAF activity, PPP6C likely promotes MEK1/2 dephosphorylation, possibly acting directly. To assess PPP6C dephosphorylation of MEK, we isolated PP6 complexes by affinity purification from HEK293T cells ectopically expressing FLAG epitope-tagged PPP6C with PPP6R3 and ANKRD28. Complexes containing WT PPP6C dephosphorylated the activation loop residues (pSer218 and pSer222) of MEK1 in a manner sensitive to the pan-PP2A family phosphatase inhibitor okadaic acid (Figure 5A), while phosphatase inactive PPP6CD84N complexes had no activity. We found that PP6 also dephosphorylated pSer298 on MEK1, albeit with slower kinetics than with the activation loop sites, while it did not detectably dephosphorylate pThr286. Thus, PP6 dephosphorylates MEK1 selectively at the same sites that are elevated in cells lacking PPP6C (Figure 4C). Furthermore, PP6 had no activity on phospho-ERK2, consistent with PPP6C acting as a regulator of MEK (Figure 5B). Overall, these studies demonstrate the direct dephosphorylation of MEK1 by PP6 with substrate and phosphorylation site specificity (Figure 5C).
Figure 5.
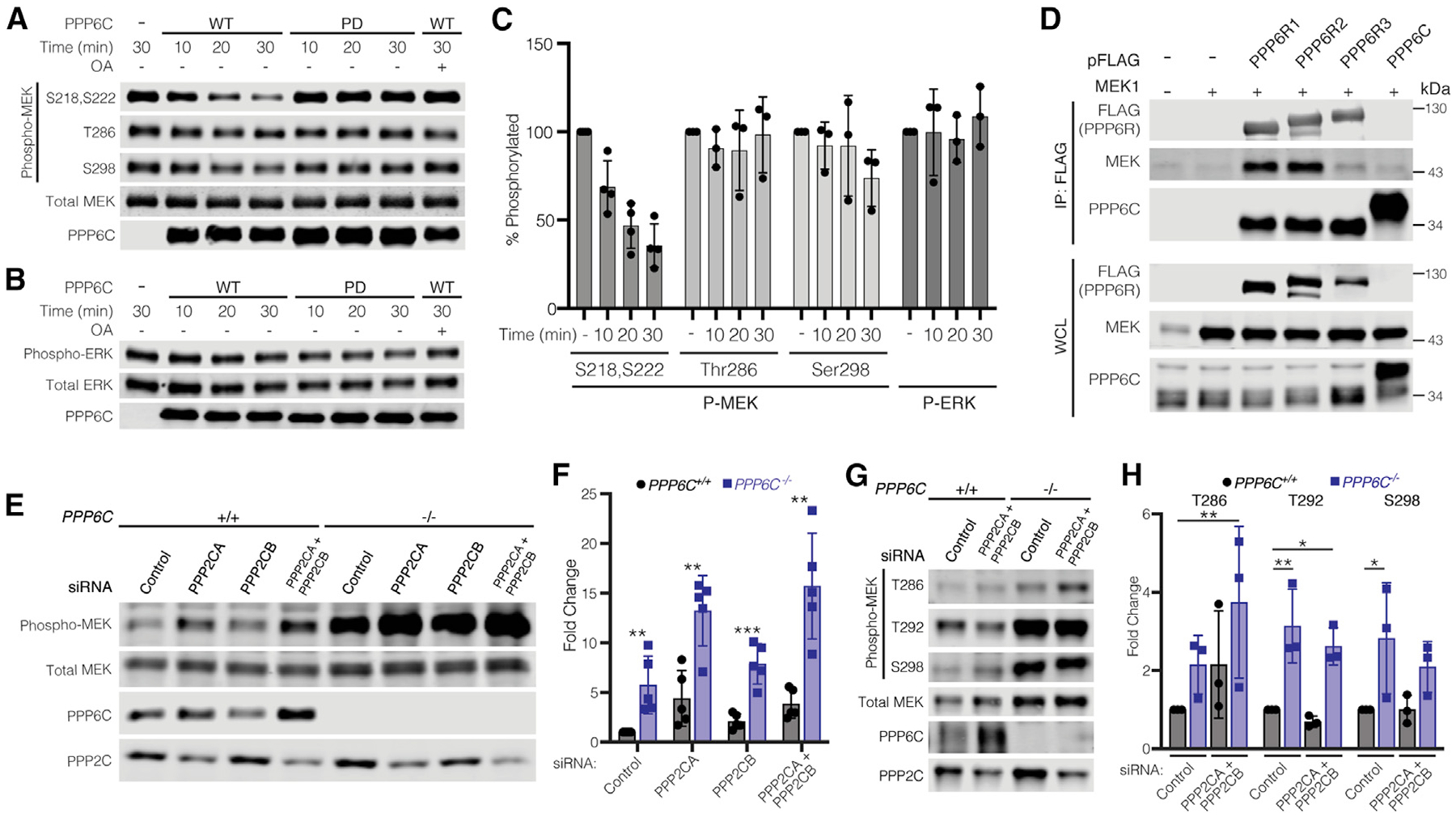
MEK1/2 is a direct substrate of PP6 (A) PP6 complexes with WT or phosphatase inactive PPP6C (PD) were partially purified from HEK293T cells and incubated with phosphorylated MEK1 in vitro for the indicated times. Okadaic acid (OA, 100 nM) was added where indicated. Reactions were evaluated by immunoblot.
(B) In vitro phosphatase assays with phospho-ERK2 were carried out as in (A).
(C) Quantification of in vitro phosphatase assays in (A) and (B). Remaining phosphorylation is shown relative to the 30 min control reaction. Data are represented as mean ± SD. For MEK1 pSer218/pSer222, n = 4; for all other data, n = 3.
(D) HEK293T cells were co-transfected to express the indicated FLAG epitope-tagged PP6 subunit and untagged MEK1. Anti-FLAG immunoprecipitates and whole-cell lysates (WCLs) were evaluated by immunoblot for MEK.
(E) PPP6C+/+ and PPP6C−/− 501mel cells were transfected with non-targeting control siRNA or siRNA SMARTpools targeting PPP2CA and/or PPP2CB. Cells were lysed and evaluated by immunoblot for phosphorylated and total MEK.
(F) Quantification of the relative level of Phospho/Total MEK for PPP2CA/PPP2CB knockdown in PPP6C+/+ and PPP6C−/− cells in (E). MEK phosphorylation was normalized to PPP6C+/+, siRNA control. Data are represented as mean ± SD, n = 5. **p < 0.01, ***p < 0.001, unpaired t test.
(G) PPP6C+/+ and PPP6C−/− 501mel cells were transfected with non-targeting control siRNA or siRNA SMARTpools targeting PPP2CA and PPP2CB. Cells were lysed and evaluated by immunoblot for phosphorylated and total MEK. See also Figure S6.
(H) Quantification of the relative levels of Phospho/Total MEK for PPP2CA/PPP2CB knockdown in PPP6C+/+ and PPP6C−/− cells in (G). MEK phosphorylation was normalized to PPP6C+/+, siRNA Control. Data are represented as mean ± SD, n = 5. *p < 0.05, **p < 0.01, unpaired t test.
We also performed co-immunoprecipitation experiments to determine whether PP6 can interact with MEK in cells. FLAG epitope-tagged PP6 subunits (PPP6C, PPP6R1, PPP6R2, or PPP6R3) were isolated from HEK293T cells co-transfected with untagged MEK1. We found that MEK1 co-immunoprecipitated with each of the PP6 regulatory subunits (Figure 5D), with significantly less associating with FLAG-tagged PPP6C. These results suggest that PP6 regulatory subunits serve to recruit MEK to the PP6 complex for dephosphorylation by PPP6C, consistent with a general role for non-catalytic subunits of PP6 and other PP2A family phosphatases in substrate binding (Brautigan and Shenolikar, 2018; Heo et al., 2020; Hosing et al., 2012; Stefansson and Brautigan, 2006; Zhong et al., 2011). In keeping with our observation that MEK binds to each of the regulatory subunits, we found that combined siRNA silencing of PPP6R1, PPP6R2, and PPP6R3, but not knockdown of individual subunits, significantly elevated MEK phosphorylation in 501mel cells (Figure S5D).
Our observations collectively suggest that PP6 has a general role as a MEK phosphatase across multiple cell types. Classic studies, however, had implicated PP2A as the major MEK phosphatase, suggesting that PP6 may act indirectly by regulating PP2A activity on MEK (Gómez and Cohen, 1991; Sontag et al., 1993). We therefore investigated the impact of PP6 and PP2A loss, alone and in combination, on MEK phosphorylation. We used siRNA SMARTpools to knockdown the two PP2A catalytic subunits (PPP2CA and PPP2CB) in PPP6C−/− and control 501mel cells (Figure 5E). In these cells, PPP2CA appeared to be the predominantly expressed isozyme, as PPP2CB siRNA alone did not detectably decrease total catalytic subunit levels. Knockdown of PPP2CA, but not PPP2CB, increased MEK activation loop phosphorylation levels in the control cell line. In PPP6C−/− cells, PP2A downregulation further increased MEK phosphorylation, with the two phosphatases having an apparently additive effect (Figure 5F). Notably, loss of either phosphatase increased expression levels of the other, suggestive of a compensatory mechanism (Figures 5E and 5G). Two other phosphorylation sites on MEK1, pThr292 and pSer298, were impacted by loss of PPP6C but not PP2A (Figures 5G and 5H). This experiment suggests that PP6 and PP2A act independently to dephosphorylate the activation loop of MEK, while PP6 also dephosphorylates additional sites.
Cancer-associated PPP6C mutations decrease phosphatase activity against MEK1/2
PPP6C mutations are found across multiple cancer types but are most common in melanoma and other skin cancers (Hodis et al., 2012; Krauthammer et al., 2015; Malicherova et al., 2019; Palmieri et al., 2018). Prior characterization of PPP6C mutations has focused primarily on non-recurrent mutations that cluster at the catalytic center, which reportedly reduce or eliminate phosphatase activity. Interestingly, there are several recurrently mutated PPP6C hotspot residues, R264C being the most common (Figure 6A). When modeled onto the X-ray crystal structure of a PPP5C-peptide complex (Oberoi et al., 2016), sites of recurrent mutations are generally located within or proximal to the catalytic cleft (Figure 6B). Of these, His55 appears critical for activity as it coordinates one of the bound metal ions.
Figure 6. Cancer-associated PPP6C mutations decrease phosphatase activity against MEK1/2.
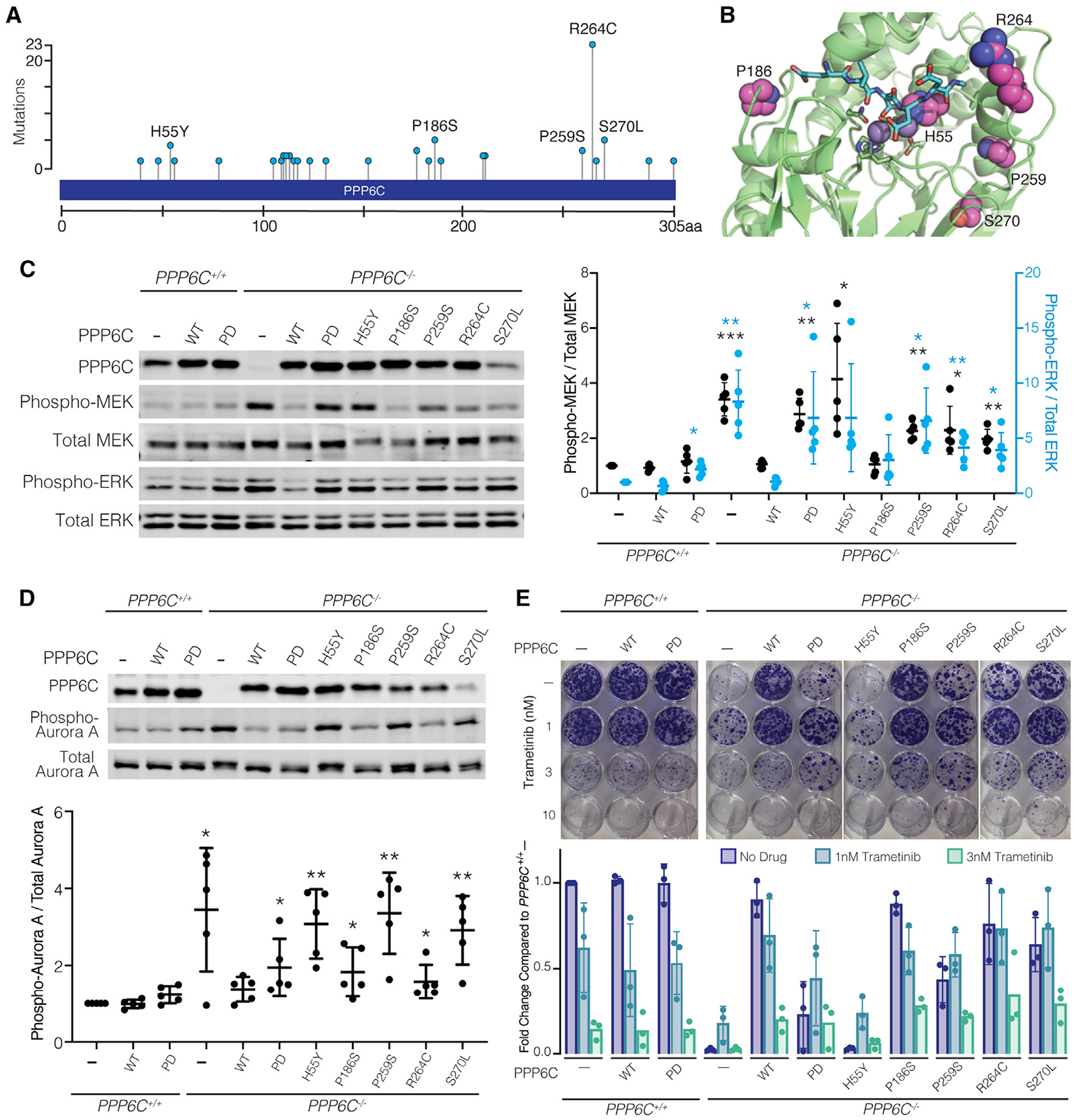
(A) Frequencies of PPP6C mutations reported in melanomas. Data are from nonoverlapping melanoma studies in cBioportal (Cerami et al., 2012; Gao et al., 2013).
(B) PPP6C residues mutated in cancer are shown in spacefill representation modeled on the X-ray crystal structure of PPP5C in complex with a peptide substrate (PDB: 5HPE). Bound peptide (cyan) and catalytic metal ions (gray spheres) are shown.
(C) PPP6C+/+ and PPP6C−/− 501mel cells were transduced to stably express GFP (–), WT PPP6C, or the indicated PPP6C mutants. Cells were lysed and assessed by immunoblot for phosphorylated and total MEK and ERK. Phospho/Total MEK (black) and ERK (blue) signal ratios were quantified and normalized to the GFP-expressing PPP6C+/+ samples. Mean values ± SD are shown, n = 5. Significance is shown in comparison to PPP6C+/+ cells expressing GFP. *p < 0.05, **p < 0.01, ***p < 0.001, paired t test.
(D) Cells from (C) were treated with 100 ng/mL nocodazole for 24 h. Mitotic cells were lysed and assessed by immunoblot for phosphorylated and total Aurora A. Phospho-Aurora A/Total Aurora A signal ratios were quantified (n = 5) and significance determined as in (C). *p < 0.05, **p < 0.01, paired t test.
(E) Cells from (C) were cultured in media containing DMSO vehicle alone or the indicated trametinib concentration for 2 weeks and stained with crystal violet. Clonogenic growth was analyzed by ColonyArea in ImageJ and normalized to GFP-expressing PPP6C+/+ samples, n = 3.
We examined the five most common mutations reported in melanomas (H55Y, P186S, P259S, R264C, and S270L) for their ability to regulate ERK signaling (Figure 6A). While re-expression of WT PPP6C in PPP6C−/− cells suppressed MEK and ERK phosphorylation to levels observed in parental cells, melanoma-associated PPP6C mutants varied in their impact on MEK and ERK phosphorylation (Figure 6C). Cells expressing the H55Y mutant exhibited the highest level of MEK phosphorylation, similar to that seen in empty vector control cells. Cells expressing the P259S, R264C, and S270L mutants had moderate but significant increases in MEK phosphorylation compared to cells expressing WT PPP6C, suggesting partial loss of activity. We note that the S270L mutant consistently expressed to lower levels than WT PPP6C or the other mutants, likely underlying its inability to promote MEK dephosphorylation. Unlike the other mutants, expression of PPP6CP186S reduced MEK phosphorylation to a similar extent as did the WT phosphatase. PPP6C mutants likewise impacted, to varying degrees, Aurora A phosphorylation in mitotically arrested cells (Figure 6D). We also examined the impact of PPP6C mutations on the levels of several transcriptional targets of ERK (DUSP6, ETV4, and SPRY2, Figure S6). Elevated expression of all three targets were significantly suppressed upon re-expression of WT PPP6C in PPP6C−/− cells. The effect of PPP6C mutants generally correlated with their impact on MEK and ERK phosphorylation, with only levels of ETV4 being significantly affected by all mutants.
We next performed clonogenic assays to examine the impact of PPP6C mutation on cell growth and MEKi sensitivity (Figure 6E). We found that growth of the cells in the absence of drug inversely correlated with the degree of ERK pathway activation. For example, cells expressing the PPP6CH55Y mutant, which had the highest levels of MEK and ERK phosphorylation, grew equivalently to PPP6C−/− cells, while other mutants that partially impacted MEK phosphorylation grew at an intermediate rate. In all cases, treatment with low concentrations of MEKi at least partially reversed the growth impairment observed in cells expressing PPP6C mutants. Collectively, these experiments indicate that cancer-associated PPP6C mutations generally cause partial loss of function, impacting both dephosphorylation of substrates and sensitivity to MEKi.
DISCUSSION
PPP6C is thought to be a tumor suppressor in melanoma (Cancer Genome Atlas Network, 2015; Hodis et al., 2012; Krauthammer et al., 2015; Malicherova et al., 2019; Palmieri et al., 2018), yet our understanding of how it modulates cancer relevant pathways is limited. Prior research on PPP6C in melanoma has focused on its roles in mitotic progression through dephosphorylation of the kinase DNA-dependent protein kinase (DNA-PK) (Douglas et al., 2010; Hosing et al., 2012), the condensin subunit NCAP-G (Rusin et al., 2015), and, in particular, Aurora A, an essential kinase regulating mitotic spindle assembly and chromosome segregation (Hammond et al., 2013; Zeng et al., 2010). Interestingly, Aurora A is reportedly a transcriptional target of oncogenic BRAF signaling in melanoma cells, suggesting that PP6 may coordinately regulate Aurora A through both direct dephosphorylation and through downregulation of ERK signaling (Puig-Butille et al., 2017). This phenomenon may offer a therapeutic vulnerability, as melanoma cells expressing mutant PPP6C are sensitized to Aurora A inhibitors (Gold et al., 2014).
Our identification of PPP6C as a MEK phosphatase suggests that it also acts as a negative regulator of the core pathway driving melanoma, likely underlying at least in part its role as a tumor suppressor. Impaired dephosphorylation of MEK may also be relevant to previous studies showing that loss of PPP6C promotes oncogenic RAS-driven tumors in mouse keratinocytes (Kurosawa et al., 2018) and in Drosophila (Ma et al., 2017). PPP6C mutations in melanoma almost exclusively co-occur with BRAF and NRAS mutations, suggesting that alone they do not provide oncogenic levels of ERK signaling. In this context, downregulation of PPP6C is likely to have a role in tuning flux through the ERK pathway to counteract negative feedback regulation. A similar phenomenon may drive selection for mutations in MEK1, MEK2, and ERK2 found at low frequency in melanomas that generally co-occur with other activating lesions (Cancer Genome Atlas Network, 2015; Gao et al., 2018; Hodis et al., 2012; Palmieri et al., 2018). We note that, like loss of PPP6C, these putatively oncogenic mutants suppress growth when delivered to cultured BRAF mutant melanoma cell lines, potentially reflecting different optimal levels of ERK signaling for cells in tumors in comparison with cells in culture.
The observation that loss or mutation of PPP6C is deleterious to cell growth may appear at odds with its role as a negative regulator of ERK signaling. Recent studies indicate, however, that in the context of activating BRAF and RAS mutations, further elevation of signaling through the ERK pathway is toxic (Hong et al., 2018; Kong et al., 2017; Leung et al., 2019; Moriceau et al., 2015). This phenomenon can give rise to inhibitor addiction, in which tumor cells treated with pathway inhibitors reactivate ERK signaling to re-establish signaling within an optimal range, or “fitness zone.” Subsequent inhibitor withdrawal results in a rebound signaling, leading to toxic hyperactive signaling outside of the fitness zone. In this context, even modest increases in ERK phosphorylation suppress growth, while further elevation induces cell death (Hong et al., 2018). Such toxicity has been associated with a phenotypic switch mediated in part by downregulation of the melanocyte lineage-specific transcription factor MITF and by transcriptional induction of genes encoding secreted cytostatic proteins (Kong et al., 2017; Leung et al., 2019). BRAFi and/or MEKi-addicted melanoma tumors grown in mice regress when treatment with inhibitors is ceased (Hong et al., 2018; Kong et al., 2017), suggesting periodic “drug holidays” could benefit patients who have progressed on BRAFi and MEKi. However, in clinical studies, cessation of BRAFi and/or MEKi therapy for several months re-sensitized to the inhibitors, though did not cause tumor regression (Rogiers et al., 2017; Seghers et al., 2012), revealing how the diversity of resistance mechanisms, tumor heterogeneity, and adaptability complicate response to drug withdrawal in patients. Our studies suggest downregulation or inactivation of PPP6C as an unappreciated mechanism influencing inhibitor sensitivity. This substantiates the identification of PPP6C in modulating sensitivity to BRAFi in an insertional mutagenesis screen in a mouse model of melanoma, and to MEKi in CRISPR-Cas9 screens conducted in NRAS and KRAS mutant cells in culture (Dompe et al., 2018; Hayes et al., 2019; Perna et al., 2015).
The toxicity associated with high-level ERK signaling indicates that tumor cells harboring hyperactivating BRAF or RAS mutations rely on negative feedback control to maintain signaling within the fitness zone. For example, silencing expression of the ERK phosphatase DUSP6 is toxic to BRAF mutant melanoma and KRAS mutant lung cancer cells (Unni et al., 2018; Wittig-Blaich et al., 2017). We hypothesize that, through dephosphorylation of MEK, PPP6C likewise contributes to negative feedback control of the ERK pathway. While we did not observe changes in levels of PPP6C upon inhibition of BRAF-MEK-ERK signaling, we cannot rule out transcriptional control of PP6 regulatory or scaffolding subunits as a mechanism of feedback regulation. Furthermore, because phosphorylation of PP6 regulatory subunits can mediate recruitment to substrates and other interaction partners (Heo et al., 2020; Kettenbach et al., 2018), targeting of MEK may be impacted by ERK-dependent phosphorylation or other modifications.
Atypically for a tumor suppressor, more than half of melanoma-associated PPP6C mutations occur recurrently in hotspots (Figure 6A). While frameshift/truncation mutations are less common, they do appear to be associated with reduced progression-free survival in melanoma (Gold et al., 2014). Consistent with prior reports (Gold et al., 2014; Hammond et al., 2013), we find that PPP6C mutations vary in their ability to suppress MEK phosphorylation. Given that PPP6C has been characterized as a “common essential” gene, it is possible that full loss of function is incompatible with cell proliferation. It has also been reported that PPP6C mutations weaken association with the PPP6R2 regulatory and ANKRD28 scaffolding subunits (Hammond et al., 2013). While the three-dimensional structure of a PP6 heterotrimer has not yet been determined, in the X-ray crystal structures of the PP2A-B56 holoenzyme, the residue analogous to R264 participates in interactions between catalytic and regulatory subunit (Xu et al., 2006). In contrast, the same residue is not at the catalytic-regulatory subunit interface in structures of other PP2A holoenzymes (Wlodarchak et al., 2013; Xu et al., 2008). This raises the possibility that PPP6C mutations might change the heterotrimer composition, favoring some regulatory subunits over others, which could favor selective dephosphorylation of substrates in a manner that preserves cell viability. By analogy, recurrent cancer-associated mutations in the PP2A scaffolding subunit PPP2R1A preferentially disrupt interactions with some regulatory B subunits rather than causing complete loss of function (O’Connor et al., 2020). The capacity for specific complexes to restrain cell proliferation is key to the activity of recently developed PP2A small-molecule activators, which stabilize specific holoenzymes (Leonard et al., 2020; Morita et al., 2020). As with PP2A, at least some PP6 substrates are recruited through interaction with individual regulatory subunits (Heo et al., 2020; Hosing et al., 2012; Stefansson and Brautigan, 2006; Zhong et al., 2011), suggesting the potential for developing PP6 activators with therapeutic benefit.
While our studies implicate PP6 asa MEK phosphatase, early reports suggested that MEK is dephosphorylated by PP2A (Gómez and Cohen, 1991; Sontag et al., 1993). Indeed, downregulation of PP2A activity by loss or mutation of its scaffolding subunit PPP2R1A causes resistance to MEK inhibitors in KRAS mutated lung and colorectal cancer cell lines (Kauko et al., 2018; O’Connor etal., 2020).Likewise, enhancing PP2A activity through downregulation of endogenous inhibitor proteins or through small-molecule activators sensitizes to MEK inhibition (Kauko et al., 2018). However, in these contexts PP2A promotes sensitivity to MEKi by restraining bypass PI3K/mTOR signaling and by direct dephosphorylation of MYC or ERK itself. Likewise, the tumor suppressor function of PP2A is suggested to involve other processes, such controlling the stability of MYC and β-catenin (Sablina et al., 2010; Yeh et al., 2004). Notably, in addition to restraining ERK signaling, PP2A can also promote MEK phosphorylation through dephosphorylation of inhibitory feedback phosphorylation sites on RAF and KSR (Ory et al., 2003). Because oncogenic mutant BRAF signals independently of KSR and is feedback resistant (Ritt et al., 2010), PP2A activity as a MEK phosphatase would not be counterbalanced by activation of upstream signaling. This phenomenon may explain our observation that like PP6, PP2A also contributes to MEK dephosphorylation in BRAF mutant 501mel cells. While we found that silencing PPP6C expression hyperactivates MEK in most cell lines we examined, this was not universally the case. The relative contributions of PP2A, PP6, and potentially other phosphatases to dephosphorylation of MEK is thus context dependent. Melanoma cells, in particular, are characterized by low PP2A activity, and the PP2A inhibitor protein CIP2A is an established transcriptional target of ERK signaling (Khanna et al., 2011; Mannava et al., 2012). Further work will be necessary to understand the lineage-specific, signaling, or genetic contexts that dictate PPP6C regulation of MEK1/2.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Benjamin E. Turk (ben.turk@yale.edu).
Materials availability
Deposition of plasmids to Addgene is in progress. Plasmids unavailable through Addgene and cell lines generated in this study are available upon request from the Lead Contact.
Data and code availability
The published article includes all datasets generated for this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
501mel, YUGEN8, YUZEAL, YUSIK, YURIF, and YUGASP cells were cultured in Opti-MEM medium (GIBCO) supplemented with 5% fetal bovine serum (FBS) (GIBCO) and 1% penicillin/streptomycin (P/S, GIBCO). A375 cells were cultured in Opti-MEM medium supplemented with 10% FBS and 1% P/S. MEL-ST, U2OS, and HEK293T cells were cultured in DMEM medium (GIBCO) supplemented with 10% FBS and 1% P/S. SK-MEL-103, SK-MEL-30, HCT116, and M318 cells were cultured in RPMI 1640 medium (GIBCO) supplemented with 10% FBS and 1% P/S. RKO were cultured in MEM medium (GIBCO) supplemented with 10% FBS and 1% P/S. All cell lines were cultured at 37°C under 5% CO2.
METHOD DETAILS
Plasmids, cloning, and mutagenesis
Plasmids harboring cDNAs of PPP6C, PPP6R1, PPP6R3 and ANKRD28 in pDONR223 were from the human ORFeome collection (v8.1), and the PPP6R2 cDNA was from Transomic. The lentiviral expression vector pLEX_305-PPP6C and the transient expression plasmid pVL1900-ANKRD28 (untagged) were generated by Gateway recombination cloning into their respective destination vectors. The untagged expression vector for PPP6R3 used for preparation of PP6 complexes was generated by Gateway recombination into pV1900 followed by QuikChange mutagenesis to insert a stop codon upstream of the FLAG tag. Transient expression vectors for N-terminally FLAG-tagged PPP6C, PPP6R1, PPP6R2 and PPP6R3 were made by PCR amplification of the coding sequence from the source plasmid and inserting into pcDNA3-FLAG by either Gibson assembly (PPP6R3) or restriction enzyme cloning (all others). The mammalian expression vector for N-terminally 6xHis-tagged MEK2 was generated by shuttling the entire coding sequence from pRSET-MEK2 (obtained from the laboratory of Natalie Ahn) into pcDNA3. All mutants were generated using QuikChange Site Directed Mutagenesis following standard protocols. Constructs were verified by Sanger sequencing through the entire open reading frame.
Recombinant lentivirus production and cell infection
shRNA lentiviruses were packaged in low passage HEK293T cells by polyethylenimine (PEI) co-transfection with packaging constructs dR8.91 and VsV-G (Addgene, 8454). PPP6C expression lentiviruses were packaged in low passage 293T cells by PEI co-transfection with packaging constructs psPAX2. (Addgene, 12260) and VsV-G (Addgene, 8454). For PEI co-transfection, lentiviral transfer plasmid:packaging plasmid:envelope plasmid ratio was at 10:10:1 with the PEI:DNA ratio at 3:1. Supernatant media containing virus was collected at 48 hours post transfection. Cells were infected with lentivirus at an MOI of 0.3–0.4 in the presence of 4 μg/mL polybrene for 24 hours and selected for > 48 hours in fresh media containing (1.5–2.5 μg/mL) puromycin.
shRNA screening
The shRNA library was custom generated by pooling human MISSION shRNA constructs (Sigma) targeting all annotated protein kinases and phosphatases (Table S2) and packaged into lentiviral particles as described above. To initiate the screen, 501mel cells were transduced for 24 hours with the lentiviral library in 0.4 μg/ml polybrene at an MOI of 0.3 to assure that most cells receive a single viral integration. A sufficient number (8 × 106) of cells were infected to ensure > 1,000-fold coverage for each unique shRNA in the library for a reference sequencing sample and for each drug condition. Infected cells were selected with 1.8 μg/mL puromycin for 48 hours, trypsinized, and 8 × 106 cells were reserved for the T0 reference sample. For the remainder, 8 × 106 cells were plated for each of the 5 conditions: 0.0001% DMSO vehicle control, 1 nM trametinib, 3.3 nM trametinib, 33 nM selumetinib, and 100 nM selumetinib. Every two doublings, cells were counted, and 8 × 106 cells were replated for propagation. The screen was carried out for 10 total population doublings (T10). Genomic DNA from the T0 and T10 samples for each of the drug conditions was extracted using QIAGEN DNeasy Blood and Tissue Kit (QIAGEN, Cat No. 69504), following the manufacturer’s protocol. For each drug condition/time point sample, the shRNA integrants were PCR-amplified from the genomic DNA with barcoded primers and sequenced on an Illumina HiSeq instrument. The RIGER algorithm in GENE-E (https://software.broadinstitute.org/GENE-E/) was used to rank each gene by their enrichment.
MEKi dose response assays
Cells (750 per well) were seeded in 96-well black/clear bottom plates, allowed to recover overnight, and treated with varying concentrations of trametinib or selumetinib (6 wells/concentration) in fresh media for 72 h. Media aspirated and replaced with fresh media containing 44 μM resazurin (alamarBlue Cell Viability Reagent, Fisher Scientific). Plates were incubated in the dark for 4 hours at 37°C, and fluorescence (excitation 560 nm; emission 590nm) was measured on a plate reader. When MEKi treatment was initiated, starting time reading was obtained on a separate plate containing untreated cells. Starting point readings were subtracted from the 72 h readings to measure overall growth inhibition. Dose response curves were generated with GraphPad Prism.
Clonogenic growth assays
Cells (1 × 103) were plated in each well of a six-well plate containing 3 mL of media with or without MEKi and cultured at 37°C under 5% CO2 undisturbed for 14 days. Media was removed, and cells were gently washed with PBS. The cells were stained with crystal violet staining solution (0.5% crystal violet, 6% formaldehyde, 1% methanol in PBS) for 15 min and washed 3 times with water. Plates were air-dried and imaged. For experiments characterizing PPP6C mutants, cells (2.5 × 103) were plated in 12 well plates containing 1 mL media with or without MEKi.
Cell lysis and immunoblot analysis
Cells were placed on ice, washed twice with cold PBS, and lysed in cold lysis buffer (20 mM Tris [pH 8.0], 137 mM NaCl, 10% glycerol, 1% Igepal CA-630, 1 mM PMSF, 1 mM Na3VO4, 10 μg/mL leupeptin, 2 μg/mL pepstatin A, 10 μg/mL aprotinin) for 15 min. Cell lysates were scraped into 1.5 mL tubes and clarified in a 4°C microcentrifuge at 13,000 rpm for 10 min. Cleared lysates were analyzed by BCA protein assay. 4X SDS-PAGE loading buffer was added to lysates to prepare immunoblot samples. Equal amounts of lysate (15 μg per lane) were fractionated by SDS-PAGE and transferred to polyvinyl difluoride (PVDF) (Sigma, IPFL85R) membrane. Membranes were blocked in Tris buffered saline (TBS) with 5% non-fat milk for 1 h and probed overnight at 4°C with primary antibodies diluted according to manufacturer’s recommendations. Membranes were incubated for 1 h in fluorescently-labeled secondary antibodies diluted 1:10,000 in TBS with 5% bovine serum albumin (BSA) and 0.1% Tween20. Western blots were imaged with an Odyssey CLx imaging system (LI-COR Biosciences), and densitometry-based quantification was carried out with Image Studio Lite.
Co-immunoprecipitation
HEK293T cells in 10 cm plates were transiently transfected with equal amounts of pREP-MEK1 (untagged) and FLAG tagged PP6 subunit plasmid precomplexed with polyethyleneimine (PEI) at a 3:1 ratio with DNA. After 48 h, cells were placed on ice and washed twice with cold PBS. On the second PBS wash, cells were scraped into 1.5 mL tubes and pelleted at 1000 rpm for 5 min. Cells were resuspended in 300 μL hypotonic lysis buffer (10 mM Tris-HCl [pH 8.0], 1 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, 0.05% Igepal CA-630, 1 mM PMSF, 1 mM Na3VO4, 10 μg/mL leupeptin, 2 μg/mL pepstatin A, 10 μg/mL aprotinin) and kept on ice for 5 min. Cell lysates were vortexed for 1 minute and run three times through a 25G needle with a syringe. Lysates were spun at 3500 rpm for 10 minutes in a 4°C microcentrifuge. A portion (30 μL) of the supernatant was reserved for analysis of the whole cell lysate sample. The remaining total supernatant was brought to a volume of 500 μL with additional hypotonic lysis buffer, and the [NaCl] was adjusted to 150 mM. Anti-FLAG M2 Affinity Gel beads (Sigma, A2220) were blocked in 5% BSA-TBS solution for 1 h, equilibrated to hypotonic lysis buffer, and 30 μL of the suspension was added to each supernatant. Samples were rotated at 4°C overnight. Beads were pelleted and washed with cold wash buffer 1 (20 mM Tris [pH 7.5], 150 mM NaCl, 1% Triton X-100, 2.5 mM Na4P2O7, 1 mM β-glycerophosphate, 3 mM β-mercaptoethanol, 1 mM PMSF, 1 mM Na3VO4, 10 μg/mL leupeptin, 2 μg/mL pepstatin A, 10 μg/mL aprotinin) for 10 minutes, followed by one quick and one 10 min wash with cold wash buffer 2 (50 mM HEPES [pH 7.4], 150 mM NaCl, 3 mM 2-mercaptoethanol, 0.1 mM Na3VO4, 0.01% Igepal CA-630, 10% glycerol). Beads were resuspended in 30 μL 2X SDS buffer (100mM Tris-Cl [pH 6.8], 4% SDS, 20% glycerol) and boiled for 5 min. Samples were centrifuged in Whatman UNIFILTER 0.45 μm plates (Sigma, WHA77002808) at 4000 rpm for 10 minutes to remove beads, and 4X SDS-PAGE loading buffer was added to filtrates. Samples were analyzed via immunoblot as described above.
Generation of CRISPR/Cas9 knockout cell lines
CRISPR/Cas9 constructs were generated by cloning sgRNA sequences into pSpCas9(BB)-2A-GFP (Addgene, 48138) according to the cloning protocol established by the Zhang lab (https://www.addgene.org/browse/article/7475/). Two sets of sgRNA oligos were used but only one sgRNA targeting exon 4 resulted in PPP6C knockout clones. pSpCas9(BB)-2A-GFP was used to generate negative control clones. 501mel cells were transfected with CRISPR/Cas9 constructs via PEI, and 48 hours post transfection, cells were trypsinized. After centrifugation and removal of media/trypsin, cells were resuspended in PBS and transferred to FACS tubes. Single GFP positive cells were sorted into 96 well plates via a BD FACSAria instrument. 96 well plates were treated with 0, 1, or 2 nM trametinib and incubated for several weeks until colonies were observed. PPP6C knockout 501mel cell colonies only grew out in the presence of trametinib and were maintained in 1–2 nM trametinib but withdrawn from trametinib ≥ 24 h before experiments. PPP6C knockout was confirmed by immunoblot and Sanger sequencing of PCR amplified target site.
Protein purification
MEK1 was expressed in HEK293T cells by PEI transfection with pcDNA3-His6-MEK1 alone or in a 4:1 ratio with pFLAG-BRAF-V600E to generate phosphorylated MEK1. After 40 h, plates were put on ice and washed once with ice-cold PBS. To lyse cells, 1 mL ice cold lysis buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 1% Triton X-100, 2.5 mM Na4P2O7, 1 mM β-glycerophosphate, 1mM Na3VO4, 3 mM β-mercaptoethanol, 1 mM PMSF, 10 μg/mL leupeptin, 2 μg/mL pepstatin A, 10 μg/mL aprotinin) was added to each plate. Lysates were scraped into 1.5 mL tubes, incubated on ice for 10 minutes, and clarified in a microcentrifuge at 13,000 rpm for 10 min at 4°C. Supernatants were transferred to fresh tubes, and 50 μL of Talon resin (Takara) was added. Samples were rotated for 2 hours at 4°C. Beads were pelleted for 2 min (4000 rpm) at 4°C in a microcentrifuge, washed twice with lysis buffer containing 10 mM imidazole, and transferred into a column. Beads were washed with 2 mL of wash buffer (50 mM HEPES [pH 7.4], 150 mM NaCl, 3 mM β-mercaptoethanol, 10 mM imidazole, 0.01% Igepal CA-630, 10% glycerol), and MEK1 was eluted in 150 μL fractions with wash buffer + 250 mM imidazole. The two most concentrated fractions as determined by Bradford assay (Bio-Rad, 5000006) were combined and dialyzed overnight at 4°C into 20 mM HEPES [pH 7.4], 150 mM NaCl, 1 mM DTT, 10% glycerol, 0.01% Igepal CA-630. Protein concentration was estimated from Coomassie-stained 10% polyacrylamide gels using a BSA standard curve
To prepare PP6 complexes, HEK293T cells were co-transfected in 15 cm plates with 4 μg pFLAG-PPP6C, 8.6 μg pV1900-PPP6R3 and 8.6 μg pV1900-ANKRD28 pre-complexed with 63.3 μg PEI. Cells were lysed 40 hours post-transfection after washing with cold PBS in 2.25 μL CHAPS lysis buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 0.3% CHAPS, 1 mM PMSF, 10 μg/mL leupeptin, 2 μg/mL pepstatin A, 10 μg/mL aprotinin) per plate. Lysates were cleared as above, and M2 anti-FLAG affinity gel (33 μL per plate) was added to the supernatant. Samples were rotated at 4°C for 1 hr, and beads were pelleted, washed three times with lysis buffer (0.3 mL per plate) and once with wash buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 10% glycerol). Protein was eluted in two rounds with 30 uL wash buffer + 0.5 mg/mL 3xFLAG peptide (Sigma F4799) per plate, snap frozen on dry ice/EtOH and stored at −80°C. Protein concentration was estimated from Coomassie-stained 10% polyacrylamide gels using a BSA standard curve.
ERK2 was purified in unphosphorylated from bacteria as described in Sheridan et al. (2008). ERK2 (21 μM) was phosphorylated in vitro by incubation with 0.2 μM bacterially-expressed active His6-MEK1ΔE102-I103 in kinase reaction buffer (50 mM Tris [pH 8.0], 50 mM NaCl, 0.5 mM ATP, 1mM DTT, 0.01% Igepal CA-630, 10% glycerol, 10 mM MgCl2) at 30°C for 30 min. MEK1 was removed by adding 20 μL Talon resin, rotating at 4°C for 1 h, and filtering through a chromatography column.
BRAF IP kinase assay
Protocol for BRAF IP kinase assays was adapted from Bondzi et al. (2000). Confluent 10 cm plates of shCTRL, shPPP6C-1, or shPPP6C-2 expressing 501mel cells were washed twice with cold PBS and lysed in RIPA buffer (20 mM Tris [pH 8.0], 137 mM NaCl, 10% glycerol, 1% NP-40, 1 mM PMSF, 1 mM Na3VO4, 10 μg/mL leupeptin, 2 μg/mL pepstatin A, 10 μg/mL aprotinin, 0.1% SDS, 0.5% sodium deoxycholate) on ice for 15 min. Cells were scraped into 1.5 mL tubes and lysates were passed through 22G needle with a syringe 3 times. Lysates were clarified in a 4°C microcentrifuge at 13,000 rpm for 10 min. Lysates were analyzed by BCA protein assay and equivalent amounts of protein were pre-cleared for 1 hour at 4°C with nProtein A Sepharose 4 Fast Flow beads (Sigma, GE17–5280–01) pre-equilibrated with lysis buffer. Beads were removed and lysates were divided into 500 μL aliquots containing 500 μg protein for each assay condition or time point. Antibody to BRAF (7.5 μL) was added to each sample, and tubes were rotated 2 h at 4°C. To precipitate immune complexes, 50 μL of a 1:1 suspension of nProtein A Sepharose in lysis buffer was added, and tubes again rotated for 2 h at 4°C. Beads were pelleted, washed three times with lysis buffer, and resuspended in kinase reaction buffer (20 mM Tris [pH 7.4], 20 mM NaCl, 1 mM DTT, 10 mM MgCl2, 1 mM MnCl2). Purified unphosphorylated MEK1 (0.5 μg) and/or vemurafenib (to 1 μM) were added as indicated. To initiate kinase reactions, ATP was added to a final concentration of 1 mM and volume of 40 μL, and tubes were transferred to a 30°C heat block for the indicated times. Reactions were quenched with 4X SDS-PAGE loading buffer and boiled for 5 minutes and then subjected to SDS-PAGE (10% acrylamide) and immunoblotting as described above.
siRNA transfection
Cells plated in 6-well plates were transfected with siRNA using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific, 13778100). Equal parts siRNA oligonucleotides (100 nM in 1X siRNA buffer, Horizon Discovery) and Lipofectamine RNAiMAX (diluted 1:100 in Opti-MEM Medium) were combined and incubated for 15 minutes at room temperature. Lipofectamine:siRNA complexes (400 μL) and complete media (600 μL) were added to each well. Cells were incubated for 72 h before being lysed for immunoblot analysis as described above. For cells transfected with two different siRNAs, half the amount of each siRNA was used.
In vitro phosphatase assays
For each reaction, PP6 complex containing 125 ng PPP6C and 500 ng phosphorylated MEK were mixed in 30 μL reaction buffer (50 mM Tris-HCl [pH8.0], 0.5 mM MnCl2, 2 mM DTT) with or without 100nM okadaic acid as indicated. Reactions were incubated at 30°C for the indicated time, quenched by the addition of 4X SDS-PAGE loading buffer, and boiled for 5 min. Samples were separated by SDS-PAGE (10% acrylamide) and analyzed by immunoblot.
QUANTIFICATION AND STATISTICAL ANALYSIS
Western blots were quantified using Image Studio™ Lite software (LI-COR Biosciences) and normalized to controls as indicated in figure legends. Clonogenic growth assays were quantified using the ColonyArea plugin on ImageJ. Statistical tests are indicated in the figure legends and were performed using GraphPad Prism 9 software. Data are shown as mean ± standard deviation (SD). P values < 0.05 are considered significant. Significance values are indicated as *p < 0.05, **p < 0.01, ***p < 0.0001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PPP6C, rabbit polyclonal | Bethyl Laboratories, Inc | Cat# A300-844A; RRID: AB_2168899 |
| Phospho-MEK1/2 (Ser217/221), rabbit polyclonal | Cell Signaling Technology | Cat# 9121; RRID: AB_331648 |
| Phospho-MEK1/2 (Ser217/221) (41G9), rabbit monoclonal | Cell Signaling Technology | Cat# 3958; RRID: AB_2138014 |
| Phospho-MEK1 (Ser298), rabbit polyclonal | Cell Signaling Technology | Cat# 9128; RRID:AB_330810 |
| Phospho-MEK1 (Thr286), rabbit polyclonal | Cell Signaling Technology | Cat# 9127; RRID: AB_331654 |
| MEK1/2, rabbit polyclonal | Cell Signaling Technology | Cat# 9122; RRID: AB_823567 |
| MEK1/2 (L38C12), mouse monoclonal | Cell Signaling Technology | Cat# 4694; RRID: AB_10695868 |
| p44/42 MAPK (ERK1/2), rabbit polyclonal | Cell Signaling Technology | Cat# 9102; RRID: AB_330744 |
| Phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (E10), mouse monoclonal | Cell Signaling Technology | Cat# 9106; RRID: AB_331768 |
| RAF-B (F-7), mouse monoclonal | Santa Cruz Biotechnology | Cat# sc-5284; RRID: AB_626760 |
| RAF-1 (C-12), rabbit polyclonal | Santa Cruz Biotechnology | Cat# sc-133; RRID: AB_632305 |
| A-RAF (D2P9P), rabbit monoclonal | Cell Signaling Technology | Cat# 75804; RRID: AB_2799875 |
| Phospho-Aurora A (Thr288) (CD39D8), rabbit monoclonal | Cell Signaling Technology | Cat# 3079; RRID: AB_2061481 |
| Aurora A (1F8), mouse monoclonal | Cell Signaling Technology | Cat# 12100; RRID: AB_2797820 |
| PP2A C Subunit (52F8), rabbit monoclonal | Cell Signaling Technology | Cat# 2259; RRID: AB_561239 |
| Caspase-3, rabbit polyclonal | Cell Signaling Technology | Cat# 9662; RRID: AB_331439 |
| PARP (46D11), rabbit monoclonal | Cell Signaling Technology | Cat# 9532; RRID: AB_659884 |
| ETV4, rabbit polyclonal | Proteintech | Cat# 10684-1-AP; RRID: AB_2100984 |
| DUSP6/MKP3, rabbit polyclonal | Cell Signaling Technology | Cat# 39441; RRID: AB_2799156 |
| SPRY2 (D3G1A), rabbit monoclonal | Cell Signaling Technology | Cat# 14954l RRID: AB_2798658 |
| Phospho-BRAF (Ser445), rabbit polyclonal | Cell Signaling Technology | Cat# 2696; RRID: AB_390721 |
| Phospho-BRAF (Thr401) (JJ08-72), rabbit monoclonal | Invitrogen | Cat# MA5-32430; RRID: AB_2809708 |
| Phospho-BRAF (Thr753), rabbit polyclonal | Invitrogen | Cat# PA5-37498; RRID: AB_2554107 |
| Phospho-MEK1 (Thr292) (D5L3K), rabbit monoclonal | Cell Signaling Technology | Cat# 26975; RRID: AB_2798935 |
| Normal mouse IgG | Santa Cruz Biotechnology | Cat# sc-2025; RRID: AB_737182 |
| FLAG M2, mouse monoclonal | Sigma | Cat# F3165; RRID: AB_259529 |
| Penta-HIS, mouse monoclonal | QIAGEN | Cat# 34660; RRID: AB_2619735 |
| Goat anti-Rabbit secondary antibody, Alexa Fluor 680 | Invitrogen | Cat# A32734; RRID: AB_2633283 |
| Goat anti-Mouse secondary antibody, Alexa Fluor 800 | Invitrogen | Cat# A32730; RRID: AB_2633279 |
| Bacterial and virus strains | ||
| MAX efficiency DH5α | ThermoFisher Scientific | Cat# 18258012 |
| One Shot Stbl3 E. coli | ThermoFisher Scientific | Cat# C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| Selumetinib (AZD6244) | SelleckChem | Cat# S1008 |
| Trametinib (GSK1120212) | SelleckChem | Cat# S2673 |
| Vemurafenib (PLX4032) | SelleckChem | Cat# S1267 |
| Puromycin | Thermo Fisher Scientific | Cat# A1113803 |
| Nocodazole | Sigma | Cat# M1404 |
| Okadaic Acid | Enzo Life Sciences | Cat# ALX-350-063 |
| 3xFLAG peptide | Sigma | Cat# F4799 |
| Anti-FLAG M2 affinity gel | Sigma | Cat# A2220 |
| Critical commercial assays | ||
| QuikChange II Kit | Agilent | Cat# 200521 |
| Gateway LR Clonase II Enzyme Kit | Thermo Fisher Scientific | Cat# 11791100 |
| Lipofectamine RNAiMAX Reagent | Thermo Fisher Scientific | Cat# 13778100 |
| DNeasy Blood and Tissue Kit | QIAGEN | Cat# 69504 |
| Pierce BCA Protein Assay | Thermo Fisher Scientific | Cat# 23250 |
| Experimental models: Cell lines | ||
| 501mel (human) | Yale SPORE in Skin Cancer Biospecimen Core | N/A |
| A375 (human) | Laboratory of Harriet Kluger | N/A |
| YUGEN8 (human) | Yale SPORE in Skin Cancer Biospecimen Core | N/A |
| YUZEAL (human) | Yale SPORE in Skin Cancer Biospecimen Core | N/A |
| YUSIK (human) | Yale SPORE in Skin Cancer Biospecimen Core | N/A |
| YURIF (human) | Yale SPORE in Skin Cancer Biospecimen Core | N/A |
| YUGASP (human) | Yale SPORE in Skin Cancer Biospecimen Core | N/A |
| SK-MEL-103 (human) | Laboratory of Narendra Wajapeyee | N/A |
| SK-MEL-30 (human) | Laboratory of Craig Crews | N/A |
| M318 (human) | Laboratory of Narendra Wajapeyee | N/A |
| MEL-ST (human) | Laboratory of Narendra Wajapeyee | N/A |
| A549 (human) | ATCC | Cat# CCL-185; RRID: CVCL_0023 |
| HCT116 (human) | ATCC | Cat# CCL-247; RRID: CVCL_0291 |
| SW620 (human) | ATCC | Cat# CCL-227; RRID: CVCL_0547 |
| RKO (human) | ATCC | Cat# CRL-2577; RRID: CVCL_0504 |
| U2OS (human) | ATCC | Cat# HTB-96; RRID: CVCL_0042 |
| HEK293T (human) | ATCC | Cat# CRL-11268; RRID: CVCL_1926 |
| Oligonucleotides | ||
| CRISPR PPP6C sgRNA 2a (CACCGTGAGAGTAGACAGATAACAC) | This paper | N/A |
| CRISPR PPP6C sgRNA 2b (AAACGTGTTATCTGTCTACTCTCAC) | This paper | N/A |
| CRISPR PPP6C Sequencing Primer F (CAGATTCTTGTAGATTTCCCTGGAATC) | This paper | N/A |
| CRISPR PPP6C Sequencing Primer R (CTTTGAGGCACAGATCTAGAAAGATG) | This paper | N/A |
| BRAF siRNA (UCUGUAAGGCUUUCACGUUAUA) | Horizon Discovery | N/A |
| ARAF siRNA (UUUCGUCCCUUGAUGAGUCGGU) | Horizon Discovery | N/A |
| CRAF siRNA (UCUCUGAAAACAUGUGUUCUGC) | Horizon Discovery | N/A |
| siGENOME Non-Targeting siRNA #2 | Horizon Discovery | Cat# D-001210-02-05 |
| ON-TARGETplus Human PPP2CA (5515) siRNA - SMARTpool | Horizon Discovery | Cat# L-003598-01-0005 |
| ON-TARGETplus Human PPP2CB (5516) siRNA - SMARTpool | Horizon Discovery | Cat# L-003599-00-0005 |
| ON-TARGETplus Human PPP6R1 siRNA (A) | Horizon Discovery | Cat# J-020420-09-0002 |
| ON-TARGETplus Human PPP6R1 siRNA (B) | Horizon Discovery | Cat# J-020420-10-0002 |
| ON-TARGETplus Human PPP6R2 siRNA (A) | Horizon Discovery | Cat# J-021331-09-0002 |
| ON-TARGETplus Human PPP6R2 siRNA (B) | Horizon Discovery | Cat# J-021331-11-0002 |
| ON-TARGETplus Human PPP6R3 siRNA (A) | Horizon Discovery | Cat# J-014646-09-0002 |
| ON-TARGETplus Human PPP6R3 siRNA (B) | Horizon Discovery | Cat# J-014646-10-0002 |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP (PX458) | Addgene | Cat# 48138 |
| pSpCas9(BB)-2A-GFP_hPPP6C-2 | This paper | N/A |
| pDONR223_PPP6C_WT | Addgene | Cat# 81811 |
| pLEX_305 | Addgene | Cat# 41390 |
| pLEX_305-PPP6C-WT | This paper | N/A |
| pLEX_305-PPP6C-D84N | This paper | N/A |
| pLEX_305-PPP6C-H55Y | This paper | N/A |
| pLEX_305-PPP6C-P186S | This paper | N/A |
| pLEX_305-PPP6C-P259S | This paper | N/A |
| pLEX_305-PPP6C-R264C | This paper | N/A |
| pLEX_305-PPP6C-S270L | This paper | N/A |
| pLEX_305-GFP | This paper | N/A |
| pLKO.1_shPPP6C-1 | Sigma | TRCN0000379835 |
| pLKO.1_shPPP6C-2 | Sigma | TRCN0000002767 |
| pREP4-MEK1 | Abbott and Holt, 1999 | N/A |
| pcDNA3-His6-MEK1 | Goldberg et al., 2017 | N/A |
| pcDNA3-His6-MEK2 | This paper | N/A |
| pGEX4T3_ERK2 | Sheridan et al., 2008 | N/A |
| pET22b-MEK1 | Laboratory of Titus Boggon | N/A |
| pET22b-MEK1-ΔE102_I103 | This paper | N/A |
| pFLAG-BRAF-V600E | Laboratory of Benjamin Turk | N/A |
| pFLAG-PPP6C | This paper | N/A |
| psPAX2 | Addgene | Cat# 12260 |
| pCMV-VsV-G | Addgene | Cat# 8454 |
| pCMV-dR8.91 | Laboratory of Benjamin Turk | N/A |
| pV1900 | Miller et al., 2019 | N/A |
| pV1900-PPP6R3 | This paper | N/A |
| pV1900-ANKRD28 | This paper | N/A |
| pFLAG-PPP6R1 | This paper | N/A |
| pFLAG-PPP6R2 | This paper | N/A |
| pFLAG-PPP6R3 | This paper | N/A |
| Software and algorithms | ||
| GENE-E | Broad Institute | https://software.broadinstitute.org/GENE-E/ |
| GraphPad Prism | GraphPad | https://www.graphpad.com |
| RIGER | Broad Institute | https://software.broadinstitute.org/GENE-E/extensions.html |
| PYMOL | Schrodinger | https://pymol.org/ |
| Image Studio Lite | LI-COR Biosciences | https://www.licor.com/bio/image-studio-lite/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| ImageJ plugin ColonyArea | EUDAT CDI | https://b2share.eudat.eu/records/39fa39965b314f658e4a198a78d7f6b5 |
Highlights.
The phosphatase PPP6C promotes sensitivity of melanoma cells to MEK inhibitors
PPP6C loss causes ERK pathway hyperactivation in cells harboring BRAF and RAS mutations
PP6 regulatory subunits recruit MEK for dephosphorylation by PPP6C
Recurrent melanoma-associated PPP6C mutants lead to elevated MEK and ERK activity
ACKNOWLEDGMENTS
We thank David Stern for helpful comments on the manuscript and Valia Mihaylova for assistance with the shRNA screen. We are grateful to Ruth Halaban and Antonietta Bacchiocchi for providing cell lines from the Yale SPORE in Skin Cancer Biospecimen Core and to Harriet Kluger, Craig Crews, and Narendra Wajapeyee for providing additional cell lines. We thank Titus Boggon, Derek Abbott, and Natalie Ahn for providing plasmids. This work was supported by NIH R01 GM135331 and a developmental research project from the Yale SPORE in Skin Cancer (NIH P50 CA121974) to B.E.T. Support for E.C. was provided by a Ruth L. Kirschstein Predoctoral National Research Service Award (NIH F31 CA220999) and Institutional National Research Service Award (T32 GM007324).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108928.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abbott DW, and Holt JT (1999). Mitogen-activated protein kinase kinase 2 activation is essential for progression through the G2/M checkpoint arrest in cells exposed to ionizing radiation. J. Biol. Chem 274, 2732–2742. [DOI] [PubMed] [Google Scholar]
- Bermudez O, Pagès G, and Gimond C (2010). The dual-specificity MAP kinase phosphatases: critical roles in development and cancer. Am. J. Physiol. Cell Physiol 299, C189–C202. [DOI] [PubMed] [Google Scholar]
- Bondzi C, Grant S, and Krystal GW (2000). A novel assay for the measurement of Raf-1 kinase activity. Oncogene 19, 5030–5033. [DOI] [PubMed] [Google Scholar]
- Boyle EA, Pritchard JK, and Greenleaf WJ (2018). High-resolution mapping of cancer cell networks using co-functional interactions. Mol. Syst. Biol 14, e8594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigan DL, and Shenolikar S (2018). Protein Serine/Threonine Phosphatases: Keys to Unlocking Regulators and Substrates. Annu. Rev. Biochem 87, 921–964. [DOI] [PubMed] [Google Scholar]
- Burotto M, Chiou VL, Lee J-M, and Kohn EC (2014). The MAPK pathway across different malignancies: a new perspective. Cancer 120, 3446–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C, Chen J-Y, Han Z-D, He H-C, Chen J-H, Chen Y-R, Yang S-B, Wu Y-D, Zeng Y-R, Zou J, et al. (2015). Down-regulation of dual-specificity phosphatase 5 predicts poor prognosis of patients with prostate cancer. Int. J. Clin. Exp. Med 8, 4186–4194. [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2015). Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-NP, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH-T, Chen Z, Cooke VG, et al. (2016). Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148–152. [DOI] [PubMed] [Google Scholar]
- Coles LC, and Shaw PE (2002). PAK1 primes MEK1 for phosphorylation by Raf-1 kinase during cross-cascade activation of the ERK pathway. Oncogene 21, 2236–2244. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. (2002). Mutations of the BRAF gene in human cancer. Nature 417, 949–954. [DOI] [PubMed] [Google Scholar]
- Dempster JM, Rossen J, Kazachkova M, Pan J, Kugener G, Root DE, and Tsherniak A (2019). Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv, 720243. [Google Scholar]
- DepMap, Broad (2020). DepMap 20Q2 Public
- Dompe N, Klijn C, Watson SA, Leng K, Port J, Cuellar T, Watanabe C, Haley B, Neve R, Evangelista M, and Stokoe D (2018). A CRISPR screen identifies MAPK7 as a target for combination with MEK inhibition in KRAS mutant NSCLC. PLoS ONE 13, e0199264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas P, Zhong J, Ye R, Moorhead GBG, Xu X, and Lees-Miller SP (2010). Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX. Mol. Cell. Biol 30, 1368–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas P, Ye R, Trinkle-Mulcahy L, Neal JA, De Wever V, Morrice NA, Meek K, and Lees-Miller SP (2014). Polo-like kinase 1 (PLK1) and protein phosphatase 6 (PP6) regulate DNA-dependent protein kinase catalytic subunit (DNA-PKcs) phosphorylation in mitosis. Biosci. Rep 34, 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al. (2012). Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med 367, 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Chang MT, McKay D, Na N, Zhou B, Yaeger R, Torres NM, Muniz K, Drosten M, Barbacid M, et al. (2018). Allele-Specific Mechanisms of Activation of MEK1 Mutants Determine Their Properties. Cancer Discov 8, 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold HL, Wengrod J, de Miera EV-S, Wang D, Fleming N, Sikkema L, Kirchhoff T, Hochman T, Goldberg JD, Osman I, and Gardner LB (2014). PP6C hotspot mutations in melanoma display sensitivity to Aurora kinase inhibition. Mol. Cancer Res 12, 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AB, Cho E, Miller CJ, Lou HJ, and Turk BE (2017). Identification of a Substrate-selective Exosite within the Metalloproteinase Anthrax Lethal Factor. J. Biol. Chem 292, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden RJ, Chen B, Li T, Braun J, Manjunath H, Chen X, Wu J, Schmid V, Chang T-C, Kopp F, et al. (2017). An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature 542, 197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez N, and Cohen P (1991). Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature 353, 170–173. [DOI] [PubMed] [Google Scholar]
- Hammond D, Zeng K, Espert A, Bastos RN, Baron RD, Gruneberg U, and Barr FA (2013). Melanoma-associated mutations in protein phosphatase 6 cause chromosome instability and DNA damage owing to dysregulated Aurora-A. J. Cell Sci 126, 3429–3440. [DOI] [PubMed] [Google Scholar]
- Hayes TK, Luo F, Cohen O, Goodale AB, Lee Y, Pantel S, Bagul M, Piccioni F, Root DE, Garraway LA, et al. (2019). A Functional Landscape of Resistance to MEK1/2 and CDK4/6 Inhibition in NRAS-Mutant Melanoma. Cancer Res 79, 2352–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, Patch A-M, Kakavand H, Alexandrov LB, Burke H, et al. (2017). Whole-genome landscapes of major melanoma subtypes. Nature 545, 175–180. [DOI] [PubMed] [Google Scholar]
- Heo J, Larner JM, and Brautigan DL (2020). Protein kinase CK2 phosphorylation of SAPS3 subunit increases PP6 phosphatase activity with Aurora A kinase. Biochem. J 477, 431–444. [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J-P, Nickerson E, Auclair D, Li L, Place C, et al. (2012). A landscape of driver mutations in melanoma. Cell 150, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong A, Moriceau G, Sun L, Lomeli S, Piva M, Damoiseaux R, Holmen SL, Sharpless NE, Hugo W, and Lo RS (2018). Exploiting Drug Addiction Mechanisms to Select against MAPKi-Resistant Melanoma. Cancer Discov 8, 74–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosing AS, Valerie NCK, Dziegielewski J, Brautigan DL, and Larner JM (2012). PP6 regulatory subunit R1 is bidentate anchor for targeting protein phosphatase-6 to DNA-dependent protein kinase. J. Biol. Chem 287, 9230–9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao S, Rizos H, Sucker A, Scolyer RA, Gutzmer R, et al. (2015). Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 51, 2792–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones GG, Del Río IB, Sari S, Sekerim A, Young LC, Hartig N, Areso Zubiaur I, El-Bahrawy MA, Hynds RE, Lei W, et al. (2019). SHOC2 phosphatase-dependent RAF dimerization mediates resistance to MEK inhibition in RAS-mutant cancers. Nat. Commun 10, 2532, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajino T, Ren H, Iemura S, Natsume T, Stefansson B, Brautigan DL, Matsumoto K, and Ninomiya-Tsuji J (2006). Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J. Biol. Chem 281, 39891–39896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasarides M, Chiloeches A, Hayward R, Niculescu-Duvaz D, Scanlon I, Friedlos F, Ogilvie L, Hedley D, Martin J, Marshall CJ, et al. (2004). B-RAF is a therapeutic target in melanoma. Oncogene 23, 6292–6298. [DOI] [PubMed] [Google Scholar]
- Kauko O, O’Connor CM, Kulesskiy E, Sangodkar J, Aakula A, Izadmehr S, Yetukuri L, Yadav B, Padzik A, Laajala TD, et al. (2018). PP2A inhibition is a druggable MEK inhibitor resistance mechanism in KRAS-mutant lung cancer cells. Sci. Transl. Med 10, eaaq1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenbach AN, Schlosser KA, Lyons SP, Nasa I, Gui J, Adamo ME, and Gerber SA (2018). Global assessment of its network dynamics reveals that the kinase Plk1 inhibits the phosphatase PP6 to promote Aurora A activity. Sci. Signal 11, eaaq1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna A, Okkeri J, Bilgen T, Tiirikka T, Vihinen M, Visakorpi T, and Westermarck J (2011). ETS1 mediates MEK1/2-dependent overexpression of cancerous inhibitor of protein phosphatase 2A (CIP2A) in human cancer cells. PLoS ONE 6, e17979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Dede M, Lenoir WF, Wang G, Srinivasan S, Colic M, and Hart T (2019). A network of human functional gene interactions from knockout fitness screens in cancer cells. Life Sci. Alliance 2, e201800278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Kuilman T, Shahrabi A, Boshuizen J, Kemper K, Song J-Y, Niessen HWM, Rozeman EA, Geukes Foppen MH, Blank CU, and Peeper DS (2017). Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature 550, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Bacchiocchi A, Evans P, Pornputtapong N, Wu C, McCusker JP, Ma S, Cheng E, Straub R, et al. (2015). Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet 47, 996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosawa K, Inoue Y, Kakugawa Y, Yamashita Y, Kanazawa K, Kishimoto K, Nomura M, Momoi Y, Sato I, Chiba N, et al. (2018). Loss of protein phosphatase 6 in mouse keratinocytes enhances K-rasG12D -driven tumor promotion. Cancer Sci 109, 2178–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake D, Corrêa SAL, and Müller J (2016). Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci 73, 4397–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H, Gagnon J, and Therrien M (2020). ERK signalling: a master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol 21, 607–632. [DOI] [PubMed] [Google Scholar]
- Leonard D, Huang W, Izadmehr S, O’Connor CM, Wiredja DD, Wang Z, Zaware N, Chen Y, Schlatzer DM, Kiselar J, et al. (2020). Selective PP2A Enhancement through Biased Heterotrimer Stabilization. Cell 181, 688–701.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung GP, Feng T, Sigoillot FD, Geyer FC, Shirley MD, Ruddy DA, Rakiec DP, Freeman AK, Engelman JA, Jaskelioff M, and Stuart DD (2019). Hyperactivation of MAPK Signaling Is Deleterious to RAS/RAF-mutant Melanoma. Mol. Cancer Res 17, 199–211. [DOI] [PubMed] [Google Scholar]
- Lim SY, Menzies AM, and Rizos H (2017). Mechanisms and strategies to overcome resistance to molecularly targeted therapy for melanoma. Cancer 123 (S11), 2118–2129. [DOI] [PubMed] [Google Scholar]
- Lu H, Liu S, Zhang G, Bin Wu, Zhu Y, Frederick DT, Hu Y, Zhong W, Randell S, Sadek N, et al. (2017). PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 550, 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, Hinkle G, Boehm JS, Beroukhim R, Weir BA, et al. (2008). Highly parallel identification of essential genes in cancer cells. Proc. Natl. Acad. Sci. USA 105, 20380–20385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Lu J-Y, Dong Y, Li D, Malagon JN, and Xu T (2017). PP6 Disruption Synergizes with Oncogenic Ras to Promote JNK-Dependent Tumor Growth and Invasion. Cell Rep 19, 2657–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malicherova B, Burjanivova T, Minarikova E, Kasubova I, Pecova T, Bobrovska M, Homola I, Lasabova Z, and Plank L (2019). Detection of driver mutations in FFPE samples from patients with verified malignant melanoma. Neoplasma 66, 33–38. [DOI] [PubMed] [Google Scholar]
- Mannava S, Omilian AR, Wawrzyniak JA, Fink EE, Zhuang D, Miecznikowski JC, Marshall JR, Soengas MS, Sears RC, Morrison CD, and Nikiforov MA (2012). PP2A-B56α controls oncogene-induced senescence in normal and tumor human melanocytic cells. Oncogene 31, 1484–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CJ, Lou HJ, Simpson C, van de Kooij B, Ha BH, Fisher OS, Pirman NL, Boggon TJ, Rinehart J, Yaffe MB, et al. (2019). Comprehensive profiling of the STE20 kinase family defines features essential for selective substrate targeting and signaling output. PLoS Biol 17, e2006540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, Dias-Santagata D, Stubbs H, Lee DY, Singh A, et al. (2008). Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res 68, 4853–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, Koya RC, Samatar AA, Khanlou N, Braun J, et al. (2015). Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 27, 240–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K, He S, Nowak RP, Wang J, Zimmerman MW, Fu C, Durbin AD, Martel MW, Prutsch N, Gray NS, et al. (2020). Allosteric Activators of Protein Phosphatase 2A Display Broad Antitumor Activity Mediated by Dephosphorylation of MYBL2. Cell 181, 702–715.e20. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- O’Connor CM, Leonard D, Wiredja D, Avelar RA, Wang Z, Schlatzer D, Bryson B, Tokala E, Taylor SE, Upadhyay A, et al. (2020). Inactivation of PP2A by a recurrent mutation drives resistance to MEK inhibitors. Oncogene 39, 703–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberoi J, Dunn DM, Woodford MR, Mariotti L, Schulman J, Bourboulia D, Mollapour M, and Vaughan CK (2016). Structural and functional basis of protein phosphatase 5 substrate specificity. Proc. Natl. Acad. Sci. USA 113, 9009–9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okudela K, Yazawa T, Woo T, Sakaeda M, Ishii J, Mitsui H, Shimoyamada H, Sato H, Tajiri M, Ogawa N, et al. (2009). Down-regulation of DUSP6 expression in lung cancer: its mechanism and potential role in carcinogenesis. Am. J. Pathol 175, 867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory S, Zhou M, Conrads TP, Veenstra TD, and Morrison DK (2003). Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14–3–3 binding sites. Curr. Biol 13, 1356–1364. [DOI] [PubMed] [Google Scholar]
- Palmieri G, Colombino M, Casula M, Manca A, Mandalà M, and Cossu A; Italian Melanoma Intergroup (IMI) (2018). Molecular Pathways in Melanomagenesis: What We Learned from Next-Generation Sequencing Approaches. Curr. Oncol. Rep 20, 86, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perna D, Karreth FA, Rust AG, Perez-Mancera PA, Rashid M, Iorio F, Alifrangis C, Arends MJ, Bosenberg MW, Bollag G, et al. (2015). BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc. Natl. Acad. Sci. USA 112, E536–E545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-Butille JA, Vinyals A, Ferreres JR, Aguilera P, Cabré E, Tell-Martí G, Marcoval J, Mateo F, Palomero L, Badenas C, et al. (2017). AURKA Overexpression Is Driven by FOXM1 and MAPK/ERK Activation in Melanoma Cells Harboring BRAF or NRAS Mutations: Impact on Melanoma Prognosis and Therapy. J. Invest. Dermatol 137, 1297–1310. [DOI] [PubMed] [Google Scholar]
- Ritt DA, Monson DM, Specht SI, and Morrison DK (2010). Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol. Cell. Biol 30, 806–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, et al. (2014). BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin. Cancer Res 20, 1965–1977. [DOI] [PubMed] [Google Scholar]
- Rogiers A, Wolter P, and Bechter O (2017). Dabrafenib plus trametinib rechallenge in four melanoma patients who previously progressed on this combination. Melanoma Res 27, 164–167. [DOI] [PubMed] [Google Scholar]
- Rossomando AJ, Dent P, Sturgill TW, and Marshak DR (1994). Mitogen-activated protein kinase kinase 1 (MKK1) is negatively regulated by threonine phosphorylation. Mol. Cell. Biol 14, 1594–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusin SF, Schlosser KA, Adamo ME, and Kettenbach AN (2015). Quantitative phosphoproteomics reveals new roles for the protein phosphatase PP6 in mitotic cells. Sci. Signal 8, rs12, rs12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusin SF, Adamo ME, and Kettenbach AN (2017). Identification of Candidate Casein Kinase 2 Substrates in Mitosis by Quantitative Phosphoproteomics. Front. Cell Dev. Biol 5, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablina AA, Hector M, Colpaert N, and Hahn WC (2010). Identification of PP2A complexes and pathways involved in cell transformation. Cancer Res 70, 10474–10484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samatar AA, and Poulikakos PI (2014). Targeting RAS-ERK signalling in cancer: promises and challenges. Nat. Rev. Drug Discov 13, 928–942. [DOI] [PubMed] [Google Scholar]
- Seghers AC, Wilgenhof S, Lebbé C, and Neyns B (2012). Successful rechallenge in two patients with BRAF-V600-mutant melanoma who experienced previous progression during treatment with a selective BRAF inhibitor. Melanoma Res 22, 466–472. [DOI] [PubMed] [Google Scholar]
- Sharma P, Veeranna, Sharma M, Amin ND, Sihag RK, Grant P, Ahn N, Kulkarni AB, and Pant HC (2002). Phosphorylation of MEK1 by cdk5/p35 down-regulates the mitogen-activated protein kinase pathway. J. Biol. Chem 277, 528–534. [DOI] [PubMed] [Google Scholar]
- Shen Y, Wang Y, Sheng K, Fei X, Guo Q, Larner J, Kong X, Qiu Y, and Mi J (2011). Serine/threonine protein phosphatase 6 modulates the radiation sensitivity of glioblastoma. Cell Death Dis 2, e241, e241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan DL, Kong Y, Parker SA, Dalby KN, and Turk BE (2008). Substrate discrimination among mitogen-activated protein kinases through distinct docking sequence motifs. J. Biol. Chem 283, 19511–19520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y (2009). Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484. [DOI] [PubMed] [Google Scholar]
- Shi H, Kong X, Ribas A, and Lo RS (2011). Combinatorial treatments that overcome PDGFRβ-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res 71, 5067–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. (2014). Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 4, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S-H, Park S-Y, and Kang GH (2013). Down-regulation of dual-specificity phosphatase 5 in gastric cancer by promoter CpG island hypermethylation and its potential role in carcinogenesis. Am. J. Pathol 182, 1275–1285. [DOI] [PubMed] [Google Scholar]
- Sontag E, Fedorov S, Kamibayashi C, Robbins D, Cobb M, and Mumby M (1993). The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the map kinase pathway and induces cell proliferation. Cell 75, 887–897. [DOI] [PubMed] [Google Scholar]
- Stefansson B, and Brautigan DL (2006). Protein phosphatase 6 subunit with conserved Sit4-associated protein domain targets IkappaBepsilon. J. Biol. Chem 281, 22624–22634. [DOI] [PubMed] [Google Scholar]
- Stefansson B, Ohama T, Daugherty AE, and Brautigan DL (2008). Protein phosphatase 6 regulatory subunits composed of ankyrin repeat domains. Biochemistry 47, 1442–1451. [DOI] [PubMed] [Google Scholar]
- Sulahian R, Kwon JJ, Walsh KH, Pailler E, Bosse TL, Thaker M, Almanza D, Dempster JM, Pan J, Piccioni F, et al. (2019). Synthetic Lethal Interaction of SHOC2 Depletion with MEK Inhibition in RAS-Driven Cancers. Cell Rep 29, 118–134.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan P, He L, Cui J, Qian C, Cao X, Lin M, Zhu Q, Li Y, Xing C, Yu X, et al. (2017). Assembly of the WHIP-TRIM14-PPP6C Mitochondrial Complex Promotes RIG-I-Mediated Antiviral Signaling. Mol. Cell 68, 293–307.e5. [DOI] [PubMed] [Google Scholar]
- Unni AM, Harbourne B, Oh MH, Wild S, Ferrarone JR, Lockwood WW, and Varmus H (2018). Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. eLife 7, 3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, et al. ; Dermatologic Cooperative Oncology Group of Germany (DeCOG) (2014). The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 4, 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al. (2010). Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 18, 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengrod J, Wang D, Weiss S, Zhong H, Osman I, and Gardner LB (2015). Phosphorylation of eIF2α triggered by mTORC1 inhibition and PP6C activation is required for autophagy and is aberrant in PP6C-mutated melanoma. Sci. Signal 8, ra27, ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig-Blaich S, Wittig R, Schmidt S, Lyer S, Bewerunge-Hudler M, Gronert-Sum S, Strobel-Freidekind O, Müller C, List M, Jaskot A, et al. (2017). Systematic screening of isogenic cancer cells identifies DUSP6 as context-specific synthetic lethal target in melanoma. Oncotarget 8, 23760–23774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarchak N, Guo F, Satyshur KA, Jiang L, Jeffrey PD, Sun T, Stanevich V, Mumby MC, and Xing Y (2013). Structure of the Ca2+-dependent PP2A heterotrimer and insights into Cdc6 dephosphorylation. Cell Res 23, 931–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Furukawa T, Kanai N, Sunamura M, and Horii A (2005). Abrogation of DUSP6 by hypermethylation in human pancreatic cancer. J. Hum. Genet 50, 159–167. [DOI] [PubMed] [Google Scholar]
- Xu Y, Xing Y, Chen Y, Chao Y, Lin Z, Fan E, Yu JW, Strack S, Jeffrey PD, and Shi Y (2006). Structure of the protein phosphatase 2A holoenzyme. Cell 127, 1239–1251. [DOI] [PubMed] [Google Scholar]
- Xu Y, Chen Y, Zhang P, Jeffrey PD, and Shi Y (2008). Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol. Cell 31, 873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Shi H, Shen Y, Peng C, Liu Y, Li C, Deng K, Geng J, Xu T, Zhuang Y, et al. (2015). PP6 controls T cell development and homeostasis by negatively regulating distal TCR signaling. J. Immunol 194, 1654–1664. [DOI] [PubMed] [Google Scholar]
- Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, et al. (2004). A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol 6, 308–318. [DOI] [PubMed] [Google Scholar]
- Zeng K, Bastos RN, Barr FA, and Gruneberg U (2010). Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. J. Cell Biol 191, 1315–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Liao J, Liu X, Wang P, Liu J, Hou W, Zhu B, Yao L, Wang J, Li J, et al. (2011). Protein phosphatase PP6 is required for homology-directed repair of DNA double-strand breaks. Cell Cycle 10, 1411–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated for this study.