

ABSTRACT
Introduction
SARS-CoV-2, the causative agent of COVID-19, attacks the immune system causing an exaggerated and uncontrolled release of pro-inflammatory mediators (cytokine storm). Recent studies propose an active role of coagulation disorders in disease progression. This hypercoagulability has been displayed by marked increase in D-dimer in hospitalized patients.
Areas Covered
This review summarizes the pathogenesis of SARS-CoV-2 infection, generation of cytokine storm, the interdependence between inflammation and coagulation, its consequences and the possible management options for coagulation complications like venous thromboembolism (VTE), microthrombosis, disseminated intravascular coagulation (DIC), and systemic and local coagulopathy. We searched PubMed, Scopus, and Google Scholar for relevant reports using COVID-19, cytokine storm, and coagulation as keywords.
Expert Opinion
A prophylactic dose of 5000–7500 units of low molecular weight heparin (LMWH) has been recommended for hospitalized COVID-19 patients in order to prevent VTE. Treatment dose of LMWH, based on disease severity, is being contemplated for patients showing a marked rise in levels of D-dimer due to possible pulmonary thrombi. Additionally, targeting PAR-1, thrombin, coagulation factor Xa and the complement system may be potentially useful in reducing SARS-CoV-2 infection induced lung injury, microvascular thrombosis, VTE and related outcomes like DIC and multi-organ failure.
KEYWORDS: Coagulation, coagulation complications management, covid-19, cytokine storm
1. Introduction
An outbreak of pneumonia of unknown cause was detected in Wuhan, China in late December 2019 and began spreading across the world at an alarming rate [1]. Chinese researchers and scientific workers discovered a new, previously unknown strain of betacoronavirus, also known as the 2019-new coronavirus (2019-nCoV), using the method of unbiased sequencing of samples obtained from the pneumonia patients [2]. This nCoV, known as the SARS-CoV-2, was found to cause a highly contagious and infectious disease called the Coronavirus Disease 2019 (COVID-19), which could easily and rapidly be transmitted from person to person*** [3]***. Owing to the alarming rate of transmission and severity of the disease*** [4]***, on January 30, 2020, COVID-19 was declared as an international public health emergency and on March 11, 2020, it was placed as a global pandemic by the World Health Organization (WHO) (Statement on the second meeting of the International Health Regulations (2005) Emergency Committee regarding the outbreak of novel coronavirus (2019-nCoV)). According to the COVID-19 epidemiological update given by WHO, as of March 9, 2021 the number of corona positive cases globally is 116.2 million with a global death toll of 2.6 million deaths [5].
Coronaviruses are RNA viruses which are enveloped and are widely distributed across various species of birds and mammals, including humans. They cause enteric, neurologic, respiratory and hepatic diseases [6]. Seven strains of coronavirus, which include OC43, 229E, HKU1, NL63, SARS-CoV, MERS-CoV, and SARS-CoV-2, are known to cause diseases in humans and new strains are emerging [7]. Out of these, four coronavirus strains, OC43, 229E, HKU1 and NL63, have been reported to affect the upper respiratory tract, causing mild-moderate and seasonal symptoms similar to common cold in individuals with an incompetent immune system [8]. The other two strains, severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), have zoonotic origin, affect the lower respiratory tract and have been causative of some fatal illnesses like severe pneumonia, acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) [9–11]. ARDS is also known as respiratory distress syndrome or wet lung. It is a condition in which the lung air sacs get filled with fluid, thus depriving organs of oxygen [12]. The seventh strain of coronaviruses affecting humans is SARS-CoV-2, which is the causative agent of COVID-19 [7]. SARS-CoV was identified as the causative agent for the outbreaks of severe acute respiratory syndrome in the years 2002 and 2003 in the Guangdong province of China [13–15]. MERS-CoV caused the outbreak of severe respiratory distress disorders in the Middle East in the year 2012 [16]. The wide distribution and prevalence of coronaviruses, their genetic diversity along with their genomes’ ability of frequent recombination and the increase in activities involving human-animal interfaces make the periodic emergence of novel strains of human coronaviruses highly possible as a result of cross-species infections [9,17].
SARS-CoV-2 enters the human lungs via angiotensin-converting enzyme 2 (ACE2) and the CD147 receptors present on the alveolar epithelial cells [18–20]. Upon entry into the respiratory tract, the virus may cause mild – moderate respiratory infections or severe acute respiratory distress syndrome (ARDS) consequently accompanied with inflammatory responses [21–23]. Recent clinical observations are indicative that the severity of the SARS-CoV-2 infection can range from a non-symptomatic, unapparent infection, to a respiratory disorder with fever spikes and dry cough, which is accompanied by a rapid and high transmission rate. Atypical pneumonia of the upper respiratory tract, which is one of the most severe complications of COVID-19, poses a great challenge in the management of the disease. Critically ill COVID-19 pneumonia patients have displayed an abnormally high production of inflammatory cytokines [24]. The production of GM-CSF leads to further activation of CD14+ or CD16+ inflammatory monocytes, producing an excessive amount of IL-6 and other pro-inflammatory factors via a positive feedback mechanism [25]. Additionally, high levels of extracellular neutrophil traps can also be a contributing factor to the release of cytokines. Ultimately, an uncontrolled and exaggerated inflammatory response leads to a condition, commonly known as the ‘cytokine storm’, resulting in damage to heart, kidney and liver tissues along with respiratory or multi-organ failure. This has proved to be fatal in patients suffering from severe SARS-CoV-2 infection [26]. This uncontrolled ‘cytokine storm’ was found to have central involvement in symptom exacerbation and disease progression, thus becoming a major contributing factor in COVID-19 – related mortality [26].
Furthermore, the correlation between viral infections and non-communicable co-morbid diseases has previously been observed in other viral infections, such as the influenza [27]. Similarly, it has been well established that individuals above 60 years of age with co-morbid conditions like cardiovascular diseases, diabetes, chronic respiratory and inflammatory diseases are at a higher risk of either developing COVID-19 or of progressing toward a severe disease state due to the presence of a low-grade inflammatory condition [7]. There are implications of unresolved chronic inflammation in development, onset and progression of these diseases, which may cause exacerbation of SARS-CoV-2 infection [28]. Diabetic patients may get infected due to impairment in their phagocyte cell capabilities. The Mendelian randomization analysis revealed a causal relationship between high levels of ACE-2 receptors and diabetes, which may prejudice diabetic patients to a higher chance of developing SARS-CoV-2 infection [29]. Furin, which is a membrane-bound protease and is highly expressed in diabetics, facilitates the pre-activation of the S protein of SARS-CoV-2, and promotes the entry of the virus by helping it escapes the host immune system [30,31]. Uncontrolled blood pressure has found association with COVID-19 and has led to a high case fatality rate (CFR). In China, 23% hypertensive COVID-19 patients reported 6% CFR [32]. In hypertensive patients, angiotensin receptor blockers (ARBs) and ACE-2 inhibitors are frequent therapeutic medications. When these medications are used in high concentrations, they cause upregulation of ACE-2 receptor expression, which leads to an increased susceptibility to SARS-CoV-2 infection [33]. Moreover, SARS and MERS showed a strong association with cardiovascular disorders [34,35]. A similar prevalence was observed in COVID-19 patients, mainly among those with severe disease. The exact mechanism of this association is not elusive; however, it may be due to the presence of ACE-2 receptors on cardiac muscle cells, which is suggestive of its involvement in the SARS-CoV-2 infection. Patients having cardiovascular disorders commonly have a compromised immune system, and may have high risk of developing acute coronary syndrome, procoagulant activation, atherosclerosis, and hemodynamic instability, causing thrombosis and ischemia [28,36].
The hyper-responsiveness of the immune system, exhibited as the cytokine storm, has been considered as the predominant cause of clinical deterioration of symptoms and mortality of COVID-19 patients [37]. However, there have been several recent reports of an increase in the number and severity of thrombotic events in severely ill COVID-19 patients [38–41]. Giuseppe Magro suggests that the key to lower mortality rates of these severely-ill COVID-19 patients may be related to the coagulation disorder [42]. This hypercoagulability has been displayed by the marked increase in D-dimer in nearly all hospitalized patients [43]. D-dimer is a product of fibrin degradation, measurable in plasma or whole blood, which is formed due to thrombi breakdown by the system of fibrinolysis [44]. It is a marker of the activation of fibrinolysis and coagulation, and it is used for quick assessment of thrombotic activities. Its role has been well established in diagnosing venous thromboembolism (VTE), thus causing a reduction in the need for imaging studies for many patients having deep vein thrombosis (DVT) and pulmonary embolism (PE) [44]. Venous thromboembolism is a condition characterized by the formation of a blood clot in the deep veins of the arms, legs and groin, which then circulates through blood, and may lodge itself in the lungs. It is further classified into DVT (arms, groin and legs) and PE (lungs) based on the location of the clot [45]. Hence, the most prevalent and vital question in the recent events of progression of COVD-19 lies in determining the link between the immune response and coagulation, the extremes of both of which have resulted in the cytokine storm, and clinical presentation of venous and arterial thrombo-embolism, respectively.
This review summarize the pathogenesis of SARS-CoV-2 infection and the generation of the cytokine storm, the interdependence and interplay between inflammation and coagulation, its consequences and the possible management options for the coagulation complications like venous thromboembolism, microthrombosis, disseminated intravascular coagulation (DIC), and systemic and local coagulopathy.
2. Pathogenesis of ‘cytokine storm’ in COVID-19
It has been widely believed and an accepted fact that a well-coordinated and rapid response of the innate immune system is the first-line defense mechanism of the human body against a viral infection [46]. Cytokine release plays a vital role in the immune-pathology against a virus [47–49]. However, over-expressions and responses of immune cells and mediators may cause severe damage to the human organ systems. Patients suffering from COVID-19 have been diagnosed with overexpression of monocyte chemo-attractant protein (MCP-1), IFN-γ, IP-10, and IL-1β [50]. These pro-inflammatory cytokines may cause activation of responses from the T-helper type 1 cells (Th1) [50]. This marks a key event in activating specific immune responses [51]. However, unlike patients with SARS, those with COVID-19 have also shown high levels of cytokines secreted by Th2 cells like IL-10 and IL-4 [52]. These two cytokines are involved in inhibition of the inflammatory response. Furthermore, the levels of IL-6 and IL-2 R, obtained from serum, depict a positive correlation with the disease severity, which means they are highest in patients who are critically ill and lowest in ordinary patients [52]. Another study on patients in Wuhan, China, compared the COVID-19 patients from the general wards and the intensive care units (ICUs). The latter were reported to exhibit increased levels of serum IP-10, macrophage inflammatory protein-1A, MCP-1, granulocyte colony-stimulating factor and TNF-α [50]. The above studies act as evidence of the positive correlation between the ‘cytokine storm’ and COVID-19 progression.
A retrospective, single-centered, observational study on critically ill COVID-19 patients by Yang et al reported that 71.2% patients required mechanical ventilation and 67.3% patients suffered from ARDS. This study highlighted a significant increase in mortality of geriatric ARDS patients [53]. A major pathological factor in ARDS is the damage to the interstitial and pulmonary tissue, which is caused due to infiltration of nonspecific inflammatory cells [54]. The induction of this damage and its clinical manifestation is caused by elevated localized release of pro-inflammatory cytokines [55]. In COVID-19, close relation exists between the generation of cytokine storm, development of ARDS and the progression of the disease. Elevated serum cytokine levels are observed in the case of ARDS and this increase is directly related to the rate of mortality [56]. The clinical progression of extrapulmonary multi-organ failure is also closely dependent on the cytokine storm [57].
Several clinical symptoms in patients with COVID-19 have been identified which may have developed as a result of the ‘Cytokine Release Syndrome’ (CRS). CRS is the consequence of the uncontrolled cytokine storm caused by the SARS-CoV2 infection. CRS is an acute condition of systemic inflammation and it characterized by fever and multi-organ dysfunction [58]. IFN-γ may cause fever with chills, headaches, fatigue and dizziness. TNF-α may also exhibit flu-like symptoms along with cardiomyopathy, vascular leakage, synthesis of acute phase proteins and lung injury [58]. IL-6 is thought to be an important target against adoptive cell therapy-induced CRS. IL-6 can cause vascular leakage, complement activation and activation of the coagulation cascade. This leads to diffuse/disseminated intravascular coagulation (DIC), which is a characteristic symptom of severe CRS [59,60]. IL-6 is also highly likely to lead to cardiomyopathy via promotion of myocardial dysfunction, which is a common symptom in patients having CRS [61]. Another major clinical hallmark of severe CRS is endothelial activation, which can result in leakage of capillaries, coagulopathy and hypotension [62]. Figure 1 summarizes the generation and clinical consequences of cytokine storm induced by the SARS-CoV-2 infection.
Figure 1.
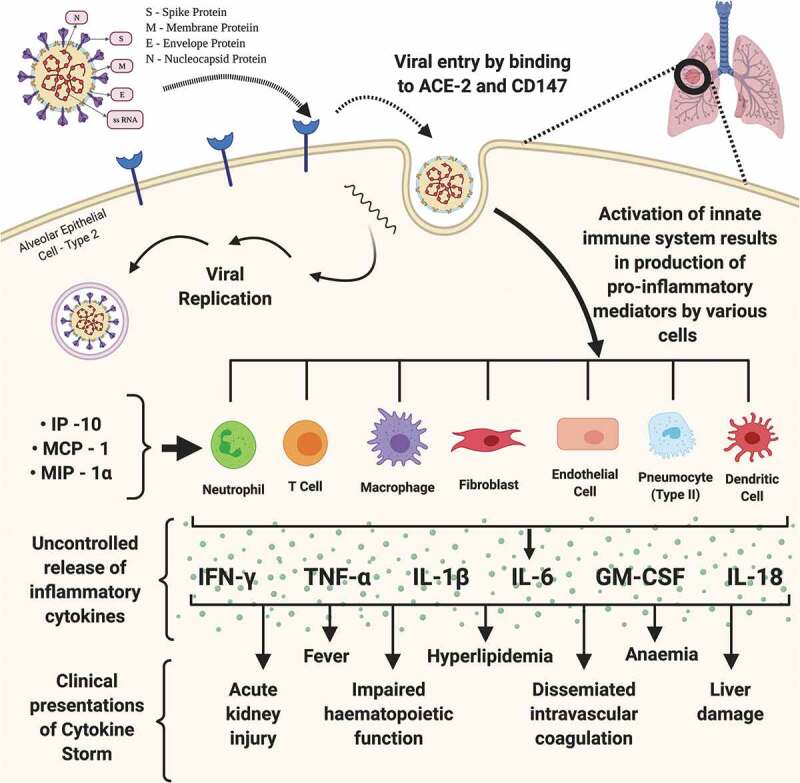
Pathogenesis of Cytokine Storm. The figure depicts the structure of SARS-CoV-2, its viral genome comprising of single stranded RNA and various viral proteins responsible for binding, entry and replication of the virus. Process of replication of SARS-CoV-2 begins with binding of viral S protein with the Angiotensin-Converting-Enzyme and CD147 receptors of the host leading to release of viral genome followed by its genomic and sub-genomic replication, and subsequent exocytosis of mature viruses. SARS-CoV-2 infection in the host lungs, upon its entry through the type 2 alveolar epithelial cells and interaction with the blood vessels at the pneumocyte – capillary interface, causes the activation of host innate immunity leading to the release of pro-inflammatory cytokines, which eventually results in an uncontrolled and exaggerated response, the cytokine storm. IL – interleukin; IP – interferon gamma-induced protein; MCP – monocyte chemoattractant protein; MIP – macrophage inflammatory protein; IFNγ – interferon gamma; TNFα – tumor necrosis factor alpha
3. Interplay between cytokine storm and coagulation
There have been reports of the existence of a common immune and hemostatic system, known as the hemolymph, in embryological organisms like the horse-shoe crab [63]. In these cases, the hemostatic system formed a blood clot resulting in the prevention of loss of nutrients along with formation of a barrier to the pathogenic invasion. This function of the immune system has been studied and explored by a number of researchers, calling it an immune-thrombosis concept [64]. Here, various hemostatic system components like thrombocytes, thrombin and coagulation factors all act as immune workers. Their function is to act as chemotactic for the immune system cells, resulting in the stimulation of the components of immune system. They also themselves get activated by the immune system components [65–67]. In simpler terms, the activation of pathways of coagulation during the inflammatory immune response to the infection leads to an over-production of various pro-inflammatory cytokines and ultimately results in multi-organ injury. IL-1α, which is expressed widely by activated platelets, endothelial cells, and circulating monocytes during pro-inflammatory conditions, functions as a link between the coagulation cascade and inflammatory response [68,69]. Mechanistically, during hemorrhage or wounding, thrombin causes activation of pro – IL-1α, which shows expression on the endothelial cell surfaces. Human conditions involving activation of the thrombo-inflammatory system, like sepsis, show the presence of thrombin-cleaved IL-1α. IL-1α plays a role in thrombosis by causing an increase in clot lysis time, an increase in platelet activity, and endothelial cell activation [70–72]. The pathogenesis of the SARS-CoV-2 infection recapitulates many key steps of inflammation mediated by IL-1α. IL-1α release by epithelial cells as an alarmin is followed by the sensing of inflammatory myeloid cells and inflammasome activation, ultimately resulting in amplification of inflammatory cascade. Simultaneously, IL-1α expression by endothelial cells causes recruitment of granulocytes and inflammation-mediated thrombosis [73]. The primary function of thrombin is promotion of clot formation by activation of thrombocytes and by conversion of fibrinogen to fibrin. However, there are multiple cellular effects of thrombin, which can cause further augmentation of inflammatory responses by activating the proteinase-activated receptors (PARs), especially PAR-1 [74]. PARs are transmembrane GPCRs and have a unique activation code. There are collectively four PARs, named PAR1-4. The proteinases of the cascade of coagulation are able to target all four PARs. Thrombin is one of the major activators of PAR1-3. Other activators of PAR1 are matrix metalloproteinase-1 (MMP-1) and activated protein C (APC). In cases of lung injury and inflammation, receptors PAR1 and PAR2 are mostly expressed on fibroblasts and macrophages in the alveoli, and on the pulmonary endothelium and epithelium. PARs1-4 are also expressed by various cells, like neutrophils, platelets and monocytes, upon their recruitment to the lungs. Multiple effects are exhibited upon activation of PARs and these effects depend on the type of cells involved, concentration and nature of the proteinases involved inside the microenvironment of the tissue [75]. Elevation of tissue factor (TF), thrombin, and D-dimer are observed in the bronchoalveolar fluid of ARDS patients. This is indicative of an on-going and activated coagulation pathway [76]. The protective effects of inflammation and coagulation processes have been considered in cases of infection. However, excessive activity of these processes may result in an augmentation of tissue injury, causing disruption to the alveolar-endothelial capillary barrier along with protein-rich fluid accumulation in alveolar spaces. Current evidence is suggestive of an important role of PAR1 in experimental models of ARDS [77]. PAR1 signaling exerts influence on many important features of ARDS like recruitment of neutrophils, alveolar leak and fibrosis in cases of lung injury. The alveolar leak is likely to be mediated via direct effects of PAR1 but it may also occur as a result of release of chemokines from neutrophils, ultimately leading to further recruitment of neutrophils and tissue injury [77,78]. The pathogenesis of pulmonary fibrosis, including both systemic sclerosis and idiopathic pulmonary fibrosis (IPF), has also been strongly correlated with PAR1 mediated coagulation signaling [79]. It is evident that PAR1 plays a vital role in the interplay between coagulation and inflammation. Generation of thrombin is controlled tightly by negative feedback mechanisms and anticoagulants like tissue factor pathway inhibitor, protein C system, and antithrombin III. The prevalence of inflammation can impair all of these feedback control mechanisms and there is a reduction in the concentrations of these anti-coagulants due to their increased consumption and reduced production. The defect in this procoagulant – anticoagulant balance leads to the development of DIC, micro-thrombosis and multi-organ failure, as observed in COVID-19 patients with severe pneumonia. The increased levels of D-dimer act as a poor prediction factor for DIC, which is common in non-survivors of this infection [43,80]. The activation of coagulation by the inflammatory responses occurs via many pro-coagulant pathways. Microorganism-derived polyphosphates cause activation of mast cells, coagulation factor XII (also known as Hageman factor or FXII), and platelets present in the contact pathway of the process of coagulation. These polyphosphates also play downstream roles in amplification of responses of pro-coagulants in the intrinsic pathway of coagulation [81]. There is contribution from complement pathways too in activating various factors of coagulation [82]. The neutrophil extracellular traps (NETs) show presence inside thrombi [83,84]. However, activation of the contact and other prothrombotic pathways is caused by the individual NET components of cell-free histones and DNA, which ultimately results in the generation of thrombin [83,84]. In case of sepsis, the pathogen-associated molecular mechanisms (PAMPs) play vital roles in the interactions between coagulation and the inflammatory responses [84,85]. Cytokines exert certain inflammatory effects which lead to activation of vascular endothelial cells and injury to the endothelium, ultimately resulting in pro-thrombotic properties [84,86]. The existence of this bidirectional relationship between coagulation and the immune system, as depicted in Figure 2, is being widely studied and correlated with some common observations in cases of COVID-19.
Figure 2.
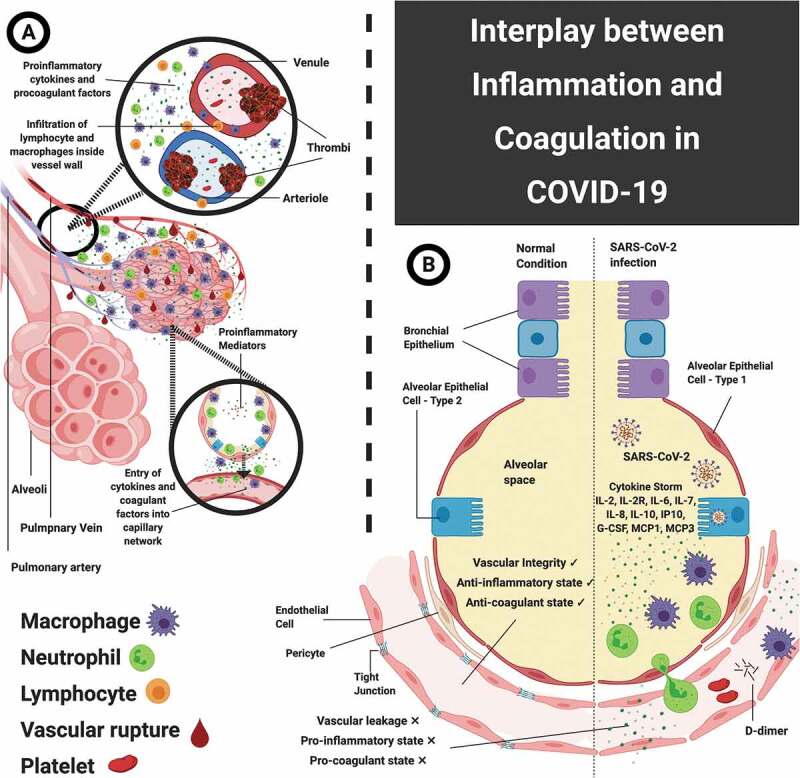
Interplay between inflammation and coagulation. A: Pulmonary intravascular coagulopathy – figure shows the interface between infected type 2 alveolar cells, the activation of immune and inflammatory cells, and the pulmonary micro-vascular network. This infected inflammatory state triggers the coagulation cascade, which in-turn activates production and release of pro-inflammatory mediators via positive feedback mechanism. B: Comparative pneumocyte-capillary interface with and without SARS-CoV-2 infection – the left side depicts normal anatomy and physiology of this interface, whereas the right side depicts vascular leakage, pro-inflammaotry and pro-coagulant state, which are effects of infection – mediated activation of inflammatory cells and exaggerated release of cytokines
Treatment of COVID-19 patients warrants the use of anti-viral and supportive therapeutic agents. Cytokine storm has been commonly observed in severe cases, and often leads to disease exacerbation, using anti-inflammatory therapeutic agents has proved to be helpful in preventing further injury. There is a wide range of anti-inflammatory drugs available for use, which includes glucocorticoids, immunosuppressants, antagonists of inflammatory cytokines (like, TNF inhibitors, monoclonal antibodies of IL-6 R, IL-1 antagonists, and janus kinase (JAK) inhibitors). Siddiqi and Mehra suggest the use of immunomodulatory agents for tailored therapy in stage III of the disease, resulting in reduced systemic inflammation and preventing multi-organ dysfunction. In this phase, glucocorticoids may be used along with cytokine inhibitors like anakinra (IL-1 R antagonist) and tocilizumab (IL-6 inhibitor). The prognosis and recovery at these stages has been poor and prompt recognition is critical, which warrants the use of this therapy [87]. However, there is a significant dilemma regarding the use of anti-inflammatory therapy due to the criticality of their benefit/risk ratio. A major concern regarding anti-inflammatory drugs like corticosteroids is that they may delay viral elimination and increase secondary infection risk in individuals with an impaired immune system. Additionally, biological antagonists or inhibitors of certain pro-inflammatory cytokines can act only against a specific inflammatory factor, which may prove to be ineffective in curbing the overall cytokine storm in COVID-19. It has been observed that IL-1α inhibitors, like anakinra, show more beneficial effects than IL-6 inhibiting monoclonal antibodies [73,88]. Furthermore, JAK inhibitors are known to cause an inhibition of interferons, which may be significant in fighting the viral infection. Therefore, it is critical to explore the advantages and limitations of anti-inflammatory therapies as adjunct alternatives for the treatment of severe COVID-19. Table 1 summarizes the beneficial effects and limitations of some commonly investigated anti-inflammatory drugs in the treatment of COVID-19.
Table 1.
Anti-inflammatory agents explored for management of COVID-19
| Drug Class | Drug/Monoclonal antibody | Mechanism of Action | Conventional Use | Dose in COVID-19 | Inclusion criteria for administration | Effects observed – % of patients | Limitations | Reference |
|---|---|---|---|---|---|---|---|---|
| IL-6 inhibitors | Tocilizumab | Binds to both trans-membrane receptor (mIL-6 R) and soluble bound receptor (sIL-6 R) and blocks both classic and trans-signaling pathways of IL-6 | Rheumatoid arthritis Juvenile idiopathic arthritis Crohn’s disease |
400/800 mg once or twice (Intravenous infusion) |
High level of IL-6 (> 40 pg/mL) Increased D-dimer and ferritin Progressively increased fibrinogen |
Temperature returned to normal Improved oxygenation – 75% Normalized % of peripheral lymphocytes – 52.6% Improved respiratory function Absorption of primary lesions, decreased lung opacity – 90.5% Decreased IL-6 levels |
Contraindicated in patients with tuberculosis Hepatotoxicity, hypertriglyceridemia, diverticulitis Inhibition of both protective and inflammatory pathways of IL-6 |
[89–95] |
| Siltuximab | Binds to both mIL-6 R and sIL-6 R Inhibits IL-6 dimerization with gp130, causing inactivation of downstream signaling Forms high-affinity complexes with soluble human IL-6 |
Castleman’s disease | 700–1200 mg Single dose (intravenous infusion) |
Patients with diagnosed ARDS Patients requiring ventilation High levels of IL-6 |
Reduction of serum CRP to normal range – 76.2% Reduction in need for ventilation – 33% Stabilized condition – 43% |
Worsened condition (related to mortality or cerebrovascular events) and intubation required Side effects – hypertension, fatigue, nausea, neutropenia, infection Contraindicated in patients having diminished cytochrome P450 activity |
[96–98] | |
| JAK/STAT inhibitors | Baricitinib | Selective inhibition of JAK1 and JAK2 kinase activity Inhibits adaptor associated protein kinase 1 (AAK1) Binds to cyclin G-associated kinase (GAK) |
Rheumatoid arthritis Psoriatic arthritis |
2–4 mg Once daily for 10 days – 2 weeks (oral) |
Moderate to severe COVID-19 Hospitalized patient SpO2 ≤ 94% on room air Required mechanical ventilation Temperature ≥ 36.6°C armpit |
Improved respiratory function Decreased cytokine storm Reduction of SARS-CoV-2 particle entry in host lung cells Decreased IL-6 and GM-CSF levels |
Inhibition of IFN-γ Ongoing clinical trials |
[99–101] |
| IL-1 inhibitors | Anakinra | Prevents binding of IL-1α and IL-1β, thus reducing availability of ligand for endogenous IL-1 receptor | Rheumatoid arthritis Macrophage activation syndrome Hemophagocytic lymphohistiocytosis Still’s disease Cryopyrin associated periodic syndrome |
5 mg/kg twice a day (intravenous – high dose) 100–200 mg twice a day (subcutaneous – low dose) |
Moderate to severe ARDS and COVID-19 Hyperinflammation (CRP ≥ 100 mg/L, Ferritin ≥ 900 ng/mL) Noninvasive ventilation required Presence of severe sepsis |
Decreased CRP and improved respiratory function (72%) Reduced mortality Reduced need for mechanical ventilation |
Associated with injection site reactions Majorly well tolerated No beneficial effects observed in patients having thromboembolism |
[102–108] |
4. Coagulation complications in COVID-19
So far in this pandemic, the occurrence of severe symptoms has been rare in cases of children with proven infection. However, there have been certain reports of hyper-inflammatory responses in a very small number of child patients [109]. This could be due to the fact that in the absence of a central venous access device or an underlying cancer, pediatric thrombotic complications are a rare occurrence [110]. Similarly, milder symptoms have been observed in infected pregnant women despite being at risk for coagulation abnormalities. This may be due to the suppression of their immune system during the period of gestation in order to prevent fetal rejection. Hence, the concept of immune-thrombosis is not evident in the cases of pregnant females [111]. On the contrary, geriatric patients have been found to display severe symptoms if they had been prone to developing thrombotic complications before the SARS-CoV-2 infection [112]. There have also been differences in mortality rates between cohorts from South East Asia and the Western population. This may be due to a lack of tendency of pro-thrombotic complications in the case of south East Asian population [113]. However, the reason for some Western patients displaying a severe respiratory disorder requiring critical care while others are displaying relatively milder symptoms still remains unknown.
A critical issue in case of COVID-19 patients is the emergence of various thrombotic complications. Preliminary reports on the outcomes of patients of this pandemic have displayed the development of thrombocytopenia in 36.2% of the 46.4% infected patients showing elevated levels of D-dimer. These rates have been higher in severely ill COVID-19 patients (57.7% patients developed thrombocytopenia out of 59.6% patients showing elevated D-dimer levels) [114]. Recent data supports the risk of development of DIC in patients suffering from SARS-CoV-2 infection [43,114,115]. DIC is disseminated intravascular coagulopathy. DIC is a condition in which blood clots are formed throughout the small vessels in the body [116]. However, poor prediction of thrombotic events in these patients have been associated with increase in D-dimer levels, increased levels of fibrin degradation products and a prolonged pro-thrombin time [43]. A report by Tang and coworkers states that out of 21 non-survivors of COVID-19, 15 patients (which was 8% of the total cohort) had shown development of an overt DIC (≥ 5 points), based on the criteria laid down by the International Society on Thrombosis and Hemostasis [43]. Lippi et al. conducted a meta-analysis, identifying significantly low platelet counts in cases of patients suffering from severe infection (mean difference: −31 × 109/L, 95% CI: −35 to −29 × 109/L). They also report that thrombocytopenia has been associated with a fivefold higher risk in cases of severe infection (OR: 5.13; 95% CI: 1.81–14.58) [117].
As discussed above, the SARS-CoV-2 enters mainly through lungs where there is presence of a localized coagulation system, also known as broncho-alveolar hemostasis. This coagulation system counters the infection alongside the immune system [118]. There were two proofs for this clot formation specifically in the lungs. First, the inclusion of fibrinogen and platelets in the acute phase response along with other markers like ferritin and C-reactive protein (CRP). These patients exhibited an increase in the thrombocyte count in the initial phase, which is not commonly reported in cases of infectious diseases. This count is not found to be severely low even in critically-ill COVID-19 patients [119]. Similarly, a marked rise in levels of fibrinogen has been a historic marker for hypercoagulability [120]. Second, examinations carried out postmortem have reported the presence of micro-thrombi in COVID-19 patients [121]. These micro-thrombi are thought to be vital factors for the clinical deterioration of these patients [122]. In cases of non-severe infection, these micro-thrombi undergo degradation by the fibrinolytic function of the lungs, which are highly active to allow exchange of gases. This is exhibited by elevated levels of D-dimers. On the contrary, in cases of severe infection, the coagulation system of the lungs undergoes activation, which is clinically manifested by increase in oxygen demand and possibly, an increase in the risk of renal impairment. In critical cases of infection, venous and arterial thrombosis along with ischemia of the limbs, intestines and cerebrum, and a multi-organ failure has been reported [116,123,124]. Understanding this spectrum of the localized to the more widespread systemic pulmonary coagulopathy is essential for adequate management of critically-ill COVID-19 patients.
The presence of an associated coagulopathy and/or the presence of certain anti-phospholipid antibodies have been recently observed in some cases of COVID-19 patients with multiple infarcts [125]. These patients have displayed evidences of ischemia in hands and lower limbs, and bilateral cerebral infarcts in many vascular areas [125]. On hospitalization, laboratory findings of these patients included thrombocytopenia, increased prothrombin time and partial thromboplastin time, leucocytosis, increased level of D-dimer and fibrinogen, and the presence of anti-beta2 glycoprotein IgA and IgG antibodies and anti-cardiolipin IgA antibodies. An important finding was the negative detection of Lupus anticoagulant [125]. These observations point toward a systemic coagulopathy. However, there is a possibility of the lungs being the point of origin of this coagulopathy, which eventually spreads into other vital organs. Hence, it suggests that the etiology of mortality may not be restricted to an uncontrolled inflammatory response but may also include a localized coagulation disorder, which eventually takes systemic effect. Figure 3 highlights the generation and types of coagulation complications, which develop as a result of SARS-CoV-2 mediated cytokine storm.
Figure 3.
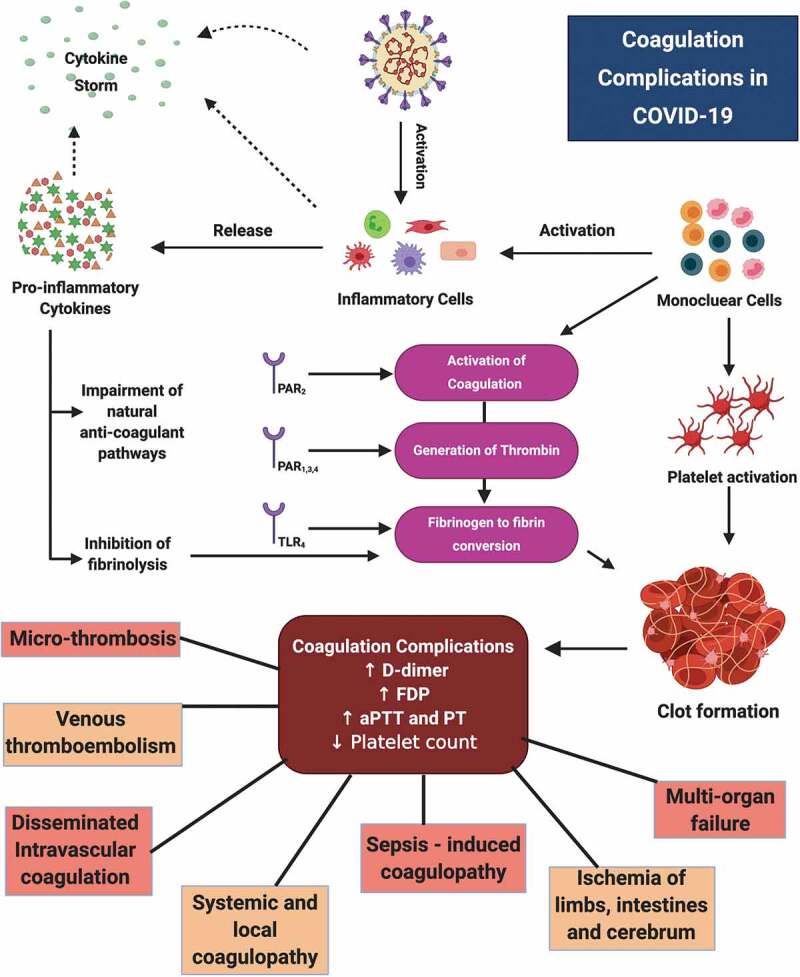
Pathogenesis of coagulation complications in COVID-19 The figure depicts the pathogenesis of inflammation-associated coagulation complications, highlighting the roles of PARs, TLR, mononuclear cells, inflammatory cells and pro-inflammatory cytokines in activation of coagulation cascade resulting in manifestations of severe complications
5. Management of coagulation complications
On the basis of multiple reports on hypercoagulability, it has now been recognized that all hospitalized COVID-19 patients require prophylactic anti-coagulant therapy, when absolute contraindications are absent [126,127]. However, in cases where observations of localized coagulopathy progressing or becoming systemic are prevalent, the prophylactic dose of anti-coagulant has been deemed inadequate. Several clinical trials have looked at increasing the dose of anti-coagulant as well as using therapeutic doses in such patients. However, it may not be necessary to administer a full-dose of anti-coagulant to treat systemic coagulopathy in all cases. Furthermore, more aggressive therapies against thrombosis (such as thrombolysis or anti-platelet drugs) and therapies against immune-thrombosis (such as anti-complement and immune-modulatory drugs) may be warranted in cases of extra-pulmonary coagulopathy [128–131]. However, in these cases, it is ideal to be dependent on pathological markers for prediction of the transition from activated pulmonary coagulation, which may be beneficial, to the more harmful systemic spread of coagulopathy.
5.1. Surveillance of laboratory tests
Newly confirmed cases as well as presumptive cases of hospitalized patients with COVID-19 infection are warranted to have coagulation tests performed on their admission. These tests include D-dimer, activated partial thromboplastin time (aPTT), prothrombin time (PT), platelet count, and fibrinogen, and can be productive of prognostic information useful for management and treatment of these patients. Typically, D-dimer levels have been found to increase parallel with age. This leads to a large number of geriatric patients exhibiting levels of D-dimer which are higher than the conventional values, which have been set at 500 µg/L FEU. FEU is a type of unit used to measure D-dimer, and represents a comparison of D-dimer mass to that of fibrinogen. Using cutoffs adjusted according to age, calculated as µg/L FEU = [(age in years) x 10], is now recommended universally for reporting test results for D-dimer [132]. Risk of coagulation complications has reportedly been higher among elderly patients as compared to young patients in COVID-19. Hence, for a 60 year old patient, threshold value of D-dimer would be considered as 600 µg/L FEU instead of 500 µg/L. Therefore, in such cases, age-adjusted threshold values find more clinical applicability to prevent obtaining false positive results related to risk of VTE [132]. Within 7–11 days following the onset of symptoms or 4–10 days following hospitalization of COVID-19 patients, a rapid decrease in fibrinogen, which may be associated with DIC, and a rise in D-dimer, which is associated with non-survivors of the infection, has been observed [43,80,133]. It is noteworthy that the timing for laboratory findings of increases in aPTT, D-dimer, PT, and decreases in platelet count and fibrinogen clearly coincides with the hospitalization duration, commencing between 7–10 days on admission. However, increase in D-dimer can begin by the 4th day. Such patients are found to be critically ill having a septic physiology. This progressive coagulation may be indicative of developing DIC, which can be independent of the effects of COVID-19 due to the prolongation of hospitalization, superinfection, mechanical ventilation, and other types of ICU etiologies [43,80,133]. Additionally, elevated serum levels of C-reactive protein (CRP), procalcitonin (PCT), and ferritin may be used as pathological biomarkers of risk of poor outcome or mortality in COVID-19. Cutoff points for PCT (≥ 0.5 ng/mL), CRP (≥ 10 mg/L), and D-dimer (> 0.5 mg/L) have been suggested and higher cutoff values may be indicative of a poorer outcome in severely ill COVID-19 patients [134]. Furthermore, serum CRP has been proposed as a marker to monitor the improvement of disease in COVID-19. Elevated PCT has been proposed as useful marker for guiding therapeutic strategies to combat bacterial co-infections with COVID-19 [134]. Table 2 summarizes threshold values for important laboratory biomarkers, which are useful in determining disease severity and potential of developing coagulation complications in COVID-19 patients.
Table 2.
Important laboratory parameters and their significance in COVID-19
| Parameter | Normal/threshold value | Pathological observations in COVID-19 | Effect on disease progression | References |
|---|---|---|---|---|
| D-dimer | 500 µg/L FEU (age-adjusted cutoffs recommended) |
Marked increase | Increased risk of VTE Presence of low-grade DIC May be associated with severe COVID-19 |
[132] |
| aPTT | 21–35 seconds | Mild prolongation | [135] | |
| PT | 11–12.5 seconds | Mild prolongation | [43] | |
| Platelet count | 150 x 109–450 × 109 cells/L | Mild decrease | [50,126] | |
| Fibrinogen | 200–400 mg/dL | At upper limits of normal | [43] | |
| Lactate Dehydrogenase (LDH) | 140–280 units/L | Increase | Thrombotic microangiopathy | [80,135] |
| Ferritin | Men: 24–336 µg/L Women: 11–307 µg/L |
Striking increase | ||
| CRP | 10–1000 mg/L | Increased | Marker of inflammation in severe COVID-19 | [136,137] |
| PCT | 0.15 ng/mL | Normal to slight increase | Presence of bacterial co-infection in severe COVID-19 | [134] |
5.2. Venous thromboembolism
Clarity has been gained on the fact that the DIC associated with COVID-19 is predominantly of prothrombotic origin having high rates of venous thromboembolism (VTE), elevated levels of fibrinogen and D-dimer, and low levels of anti-thrombin. A report by Tang et al. investigated the predictive factors associated with 28-day death of severely ill COVID-19 patients and stated that the usage of heparin as anti-coagulant for at least seven days has been associated with a decreased mortality. Higher death rate found association with elevation in D-dimer, prolongation of prothrombin time and increased age, whereas an increase in the platelet count was found to be associated with a relatively lower death rate. Heparin, more commonly available as unfractionated heparin (UFH), is a natural polysaccharide that inhibits coagulation. Low molecular weight heparin (LMWH) is derived from UFH by chemical or enzymatic digestion of longer heparin chains to shorter chains. Decreasing the length of heparin chains increases its half-life and makes its pharmacodynamics more predictable. Some examples of commercially available LMWHs, employed for VTE prophylaxis and the treatment of DVT and PE, are dalteparin and enoxaparin [138]. The positive effect of administration of low molecular weight heparin (LMWH) was observed especially in cases of patients meeting the criteria of sepsis-induced coagulopathy (SIC ≥ 4; LMWH: 40.0% v/s No-LMWH: 64.2%, p = 0.029) and in cases of patients showing a marked six-fold elevation in D-dimer (LMWH: 32.8% v/s No-LMWH: 52.4%, P = 0.017) [40]. This information is suggestive of considering the usage of low molecular weight heparin (LMWH) at its prophylactic doses for patients having high D-dimer and meeting the SIC criteria. The finding of an increase in levels of D-dimer in COVID-19 patients has prompted curiosity regarding the exacerbation of the ventilation – perfusion match due to prevalence of venous thrombo-embolism, and certain evidences of the existence of pulmonary embolism (PE) have also been observed [139]. However, there is an increased risk of bleeding, which has been found in previously conducted negative sepsis-related trials of endogenous anti-coagulants.
All suspected or confirmed cases of hospitalized COVID-19 patients should be administered pharmacological treatment for prophylaxis of VTE due to the prevalence of elevated inflammation, unless there is a specific contraindication. However, in coronavirus infections apart from COVID-19, VTE incidences have been reportedly low in Asian population, thus the routine prophylaxis treatment of VTE is not used frequently in COVID-19 [140,141]. Reports of severely ill ICU patients with the SARS-CoV-2 infection are suggestive of a higher incidence of VTE even with the usage of the standard procedures for VTE prophylaxis. A report from the Netherlands found a cumulative VTE incidence of 27% in ICU patients. A second report found symptomatic VTE incidences at 11% and 23% on the seventh and fourteenth days, respectively, having an SHR of 3.8 on comparing findings of ICU patients and ward patients [142]. An increased prophylactic VTE dosage was given to the ICU patients based on these findings in the Netherlands. An increase in the VTE risk in ICU patients has also been reported in France. One center reported an increase in VTE on comparing cohorts of patients suffering from COVID-19 ARDS and those without COVID-19, with incidences of 11.7% and 2.1% respectively [39,143]. Another center reported similar increase in PE prevalence having incidence of 20.4% at fifteen days. Of the 22 cases of PE occurring in 107 ICU patients, 20 cases occurred in patients while they were on standard dosage for VTE prophylaxis [144]. This increase in the VTE incidences has led to the discussion regarding an increment in the intensity of anti-coagulant therapy for VTE prophylaxis. Multiple centers have reportedly increased the anti-coagulation doses for prophylaxis to doses of ‘intermediate intensity’ like 0.5 mg/kg b.i.d of enoxaparin by using strategies adapted for risk stratification based on D-dimer and fibrinogen levels, the location of ICU and other risk-associated factors. A document using the Delphi method of consensus for centers involved in treatment of hospitalized patients having moderate to severe COVID-19 and showing a lack of DIC reported that 31.6% of the participants were in support of dosing at intermediate intensity and 5.2% of participants preferred a therapeutic dosing, while the others supported usage of standard prophylactic dosage for VTE [127].
In the case of obese patients, it has been suggested that a daily dose of 40 mg of enoxaparin is insufficient under post-operative settings as it does not achieve adequate plasma levels. Higher dosage based on individual weight was found to be well tolerated with 7500 units of unfractionated heparin (UFH) t.i.d or 40 mg of enoxaparin b.i.d [145,146]. Therefore, an imperative requirement for prophylaxis of VTE is the individualized patient assessment approach incorporating risk factors for VTE and bleeding with clinical judgment.
However, beside the management of venous thromboembolism, it has become evidently clear that activation of coagulation cascades have effects which are beyond clotting disorders and the interplay between inflammation and coagulation can affect progression of the disease significantly leading to a poor outcome. Along with its known primary use, heparin has been found to have anti-inflammatory properties [147]. These could find therapeutic application in patients suffering severe inflammation of lungs and impairment in the pulmonary exchange.
Another common complication of severe SARS-CoV-2 infection is ARDS, which is associated with the activation of the coagulation system. Comparative analysis of laboratory findings of ARDS patients with non-ARDS patients showed significantly higher levels of median plasma concentrations of plasminogen activator inhibitor – 1 (PAI-1) and tissue factor (TF) at seventh day. Localized TF-mediated generation of thrombin and a plasminogen activator-mediated fibrinolysis depression, which is linked with increase in PAI-1, gives rise to coagulopathy [148]. This suggests the usefulness of LMWH against this coagulopathy.
Another therapeutic property of interest is heparin’s antiviral activity [149]. A study conducted by Vicenzi et al. reports the inhibitory activity of heparin (100 mg/kg) in vitro in experimental vero cells, infected with sputum from a SARS-CoV pneumonia patient [150]. Collectively, all this evidence is suggestive of the potential application of low dose of heparin in early stages in the SARS-CoV-2 infection in order to prevent the progression of coagulation toward systemic spread.
5.3. Microvascular thrombosis
The theoretical basis for the management strategy for SIC is the quick and timely treatment of the causative and underlying infection. However, there is still an unavailability of a specific anti-viral therapy for combating the SARS-CoV-2 infection. Overall management should consider inclusion of assessment of prevalence of concomitant infections, like ARDS and acute lung injury in case of patients who are critically ill. Despite their importance in the management of VTE, heparin and its derivatives have found to have limited efficacy in cases of SIC. Previous randomized clinical trials of physiologic anti-coagulants like APC, anti-thrombin and thrombomodulin have demonstrated their limited efficacy. However, these trials were not specifically conducted for patients having SIC and DIC. Post-hoc database analyses which examined patients having sepsis and laboratory proven DIC have reported a decrease in mortality in cases of thrombomodulin and anti-thrombin supplementation tending toward an improvisation in septic patient survival [151–153]. However, the levels of anti-thrombin have not been observed to markedly decreased in COVID-19 patients [43].
Microvascular thrombosis has proven to cause multi-organ failure in patients having a prolonged SARS-CoV-2 infection. Patients having sepsis should undergo standard treatment with supportive care. Despite the possible mitigation of microvascular thrombosis and end-organ dysfunction by the use of anti-coagulants, there have been no observable survival advantages for patients with sepsis. Therefore, such patients should receive prophylactic treatment with anti-coagulants as previously discussed. However, for prevention of microvascular thrombosis the usage of full dosage of anti-coagulants has been considered in cases of severe infection using theoretical correlation based on evidence from cases of DIC-induced skin necrosis and previous SARS-CoV infections [150,154–157]. These mechanisms look theoretically interesting but there is no concrete evidence for the interaction of heparin and SARS-CoV-2, and there is no established role for the clinical application of heparin and derivatives for the purpose of lowering the infection level in COVID-19 patients.
6. Expert opinion
6.1. Current approaches for treatment of COVID-19
The first-line treatment options for COVID-19 are the classic antiviral drugs, which are important in reducing mortality in mild to severe cases along with simultaneous organ function support [158]. The antiviral drugs used in SARS-CoV-2 infection include Remdesivir, Ribavarin, Galidesivir, Tenofovir, Lopinavir, Ritonavir and Sofosbuvir [158]. There have been previous reports of the anti-inflammatory properties of these antiviral drugs, when used individually or in combination as highly active antiviral therapies [159]. The anti-malarial drugs like chloroquine and hydroxychloroquine have also shown beneficial effects against SARS-CoV-2 infection. Hydroxychloroquine was found to possess immunomodulatory effects, which have been useful in targeting the cytokine storm in moderate to severe cases [37,160]. Additionally, these antiviral medications have been effectively combined with several biological agents targeting specific cytokines, like IL-6 R monoclonal antibodies, IL-1 inhibitors, TNF inhibitors, JAK inhibitors, etc. Tocilizumab has also effectively reduced microthrombosis. Adopting drugs or compounds that work against a specific cytokine is associated with the risk of targeting only a specific part of the cytokine storm. Furthermore, immunosuppressive medication should be used rationally as it may result in a compromise of host innate immunity [159].
There are many points in the pathophysiology of SARS-CoV-2 infection, from initial infection to development of cytokine storm, ARDS, and multi-organ failure, which can be controlled with effective therapeutic intervention. Dexamethasone plays a role against ARDS in moderate to severe COVID-19. It reduces mortality and has therefore been incorporated in the standard treatment regimen alongside immunomodulatory and antiviral therapies. Supplementation with a combination of zinc, oral vitamin D and intravenous vitamin C has been implicated as these levels may be diminished due to systemic inflammation. Aspirin, Azithromycin, and N-acetylcysteine have shown roles in inhibition of NF-κB with effective reduction in activation of coagulation cascade in moderate to severe COVID-19 [161].
6.2. Clinical indications for anti-coagulation
In view of the thrombotic risk in the progression of COVID-19, anti-coagulation treatment has gained importance for both, prophylactic as well as therapeutic purposes. However, the choice of anticoagulant and intensity of anticoagulation must be varied depending on the patient condition. Patients having other indications which require anti-coagulation, like recently diagnosed VTE, mechanical cardiac valves, or atrial fibrillation, must be treated with a full – or therapeutic dose of anticoagulant, or with dosage pertaining to their on-going therapy. In case of critically ill in-patients, LMWH or UFH usage has been preferred over oral anticoagulants due to the former having shorter half-lives and can be administered via parenteral route. Certain COVID-19 patients have displayed an increase in fibrinogen levels, which is one of the causes of heparin resistance as well as hypercoagulability. Hence, in case of concerns with respect to aPTT measurement, levels of anti-factor Xa heparin should be evaluated for monitoring such patients [162]. In cases of PE presumptions, problems regarding mobility of mechanically ventilated patients, and limiting staff exposure have been encountered for CT scanning procedures. Here, D-dimers have proven to provide little help due to being significantly elevated in these patients. In such cases, evidences of right ventricular strain in ECG or deep vein thrombosis (DVT), and clinical manifestations of sudden respiratory decompensation, have been used for increasing the dosage of therapeutic anticoagulants. Hence, the pragmatic requirement of administration of therapeutic anticoagulants in such cases cannot be denied [163]. In the absence of clinical trials proving the anticoagulant therapy for COVID-19 patients, these decisions will be empirically made and will be mainly based on opinions. Figure 4 depicts the guidelines for anticoagulation in COVID-19 patients at low, intermediate and high risk of developing coagulation abnormalities. The risk is determined by a number of factors like the presence or absence of VTE and bleeding, the D-dimer levels, the SIC score and other factors. Along with the BMI and weight of individual patients, another important player in determining the type and dose of anticoagulant medication is creatinine clearance (CrCl). CrCl is an important marker of kidney impairment. Owing to the risk of anticoagulant accumulation, LMWH is contraindicated in patients having CrCl of less than 30 mL/min [164]. Thus, Figure 4 gives an empirical idea about how an individualized anticoagulant therapy can look for COVID-19 patients based on their disease severity and risk of developing coagulation complications.
Figure 4.
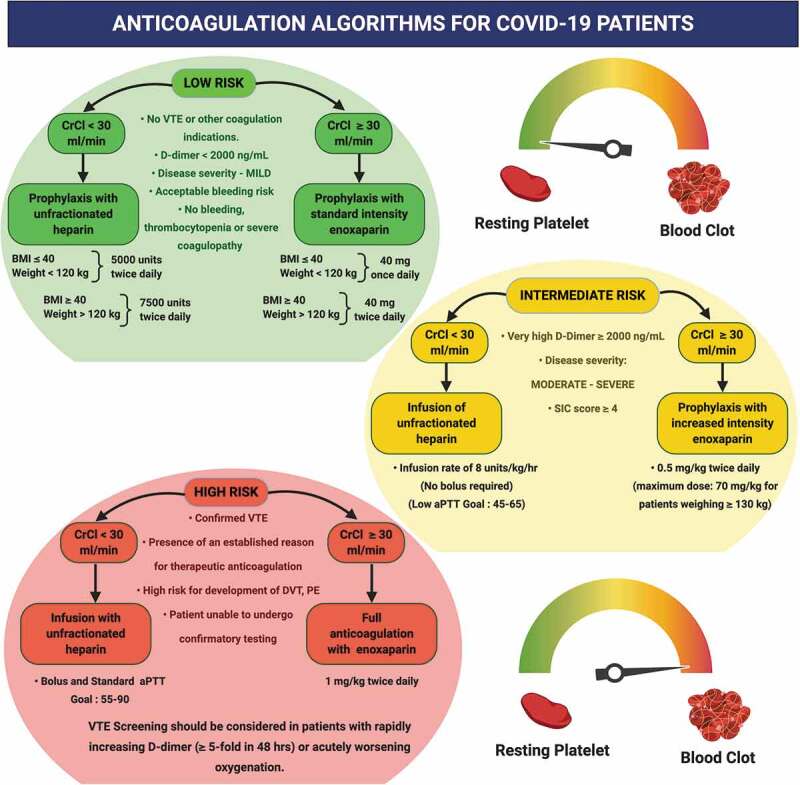
Algorithm for Anticoagulation in COVID-19 patients. The figure represents possible protocols and algorithms for management of coagulation complications using suitable anti-coagulant therapy. Varied approaches have been laid down for patients with low, moderate and high risk of development of these complications. CrCL – creatinine clearance
6.3. Future perspective
A prophylactic dose of LMWH or UFH has been recommended for hospitalized COVID-19 patients in order to prevent VTE. Treatment dose of LMWH is being contemplated for patients showing a marked rise in levels of D-dimer due to possible pulmonary thrombi. Furthermore, LMWH also has some anti-inflammatory properties like inhibition of chemokine synthesis and function, inhibition of NF-κB, TNF-α and other cytokines, which could prove to be useful against COVID-19. Heparin’s anti-inflammatory effects are associated with vasculature and the airway tracts. Heparin has found to bind and interact with many pro-inflammatory mediators like IL-8, stromal-derived factor 1a, L – and P-selectins, neutrophil elastase, platelet growth factor 4 (PGF4), etc [165]. Moreover, it is important to understand the role and activity of PAR antagonists and inhibitors of coagulation proteases. PAR-1 has been established as the primary thrombin receptor and is involved in mediating platelet aggregation induced by thrombin and the interplay between fibrotic, inflammatory and coagulation responses, which are all vital aspects in the COVID-19 pathophysiology [74]. PAR-1 antagonists have been developed for treating CVDs as anti-platelet agents. Despite being less likely to ameliorate VTE, these drugs may cause attenuation of the deleterious effects linked with formation of thrombin and coagulation cascade activation [166]. A PAR-1 antagonist drug, approved for clinical application, was found to cause a reduction in murine levels of neutrophilic lung inflammation, pro-inflammatory cytokines and alveolar leakage in cases of bacterial pneumonia and LPS-induced lung injury [167,168]. Anti-thrombin and antifactor Xa directed oral anticoagulants have been established for prophylaxis and management of VTE. Thrombin is considered as the primary PAR-1 activator, and production of pro-inflammatory cytokines by activating PAR-2, PAR-1 and PAR-5 is induced by the coagulation factor Xa [74]. Therefore, these drugs display a promising potential in amelioration of the severity and attenuation of the progression of COVID-19 infection. Moreover, targeting the complement system is another frontier being investigated for effective management of severe COVID-19 hyperinflammation associated hypercoagulability [169]. The complement system is responsible for mediating innate immune responses promoting inflammation, and its activation may be critically related to thrombosis and cytokine storm in SARS-CoV-2 infection. Here, a possible pharmacological solution could be the use of biological agents like Eculizumab or Ravulizumab, which are monoclonal antibodies responsible for blocking the complement protein C5, which ultimately inhibits activation of downstream terminal complement cascade involved in pro-inflammatory and prothrombotic activities [170,171]. Despite the risk of bleeding posed by the usage of these drugs, their benefits in this pro-coagulant state outweigh these risks. Furthermore, the existence of reversal drugs against the anti-coagulant effects of these agents helps in consideration of these approaches for clinical application. Hence, targeting PAR-1, thrombin, coagulation factor Xa and the complement system may be a potentially useful approach in reducing SARS-CoV-2 infection induced lung injury, microvascular thrombosis, VTE and related outcomes like DIC and multi-organ failure.
Funding Statement
This paper was not funded.
Article highlights
Coronavirus disease 2019 (COVID-19) was first detected in China in December 2019 and declared as a pandemic by WHO on 11 March 2020.
This review summarises the pathogenesis of SARS-CoV-2 infection – mediated generation of cytokine storm, the interdependence between inflammation and coagulation, its consequences and the possible management options for coagulation complications.
The most vital question in the progression of COVD-19 lies in determining the link between immune response and coagulation, the extremes of both of which have resulted in cytokine storm, and the clinical presentation of venous and arterial thromboembolism, respectively.
In view of the thrombotic risk in progression of COVID-19, anti-coagulation treatment has gained importance for both, prophylactic as well as therapeutic purposes. However, the choice of anticoagulant and intensity of anticoagulation must be varied depending on the patient condition.
Targeting PAR-1, thrombin, coagulation factor Xa and the complement system may be a potentially useful approach in reducing coagulation complications associated with COVID-19.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants, or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
- 1.Lu H, Stratton CW, Tang YW.. Outbreak of pneumonia of unknown etiology in Wuhan, China: the mystery and the miracle. J Med Virol. 2020;92(4):401–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Statement on the second meeting of the International Health Regulations (2005) Emergency Committee regarding the outbreak of novel coronavirus (2019-nCoV) https://www.who.int/news-room/detail/30-01-2020-statement-on-the-second-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-outbreak-of-novel-coronavirus-(2019-ncov). (Accessed 2020 May27
- 4.WHO-audio-emergencies-coronavirus-press-conference-full-and-final . https://www.who.int/docs/default-source/coronaviruse/transcripts/who-audio-emergencies-coronavirus-press-conference-full-and-final-11mar2020.pdf?sfvrsn=cb432bb3_2 (accessed 2020 May27
- 5.Weekly epidemiological update - 9 March 2021. https://www.who.int/publications/m/item/weekly%2depidemiological%2dupdate—10%2dmarch%2d2021 (accessed 2021 March11
- 6.Weiss SR, Leibowitz JL. Coronavirus pathogenesis. Adv Virus Res. 2011;81:85–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baviskar T, Raut D, Bhatt LK. Deciphering Vaccines for COVID-19: where do we stand today? Immunopharmacol. Immunotoxicol. 2021;43(1):8–21. [DOI] [PubMed] [Google Scholar]
- 8.Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24(6):490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17(3):181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peiris JSM, Lai ST, Poon LLM, et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361(9366):1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuiken T, Fouchier RAM, Schutten M, et al. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet. 2003;362(9380):263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med. 2017;377(6):562–572. [DOI] [PubMed] [Google Scholar]
- 13.Drosten C, Günther S, Preiser W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1967–1976. [DOI] [PubMed] [Google Scholar]
- 14.Ksiazek TG, Erdman D, Goldsmith CS, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1953–1966. [DOI] [PubMed] [Google Scholar]
- 15.Zhong NS, Zheng BJ, Li YM, et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in guangdong, people’s republic of China, in february, 2003. Lancet. 2003;362(9393):1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zaki AM, Van Boheemen S, Bestebroer TM, et al. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–1820. [DOI] [PubMed] [Google Scholar]
- 17.Wong G, Liu W, Liu Y, et al. MERS, SARS, and ebola: the role of super-spreaders in infectious disease. Cell Host Microbe. 2015;18(4):398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gheblawi M, Wang K, Viveiros A, et al. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res. 2020;126(10):1456–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wan Y, Shang J, Graham R, et al. Receptor recognition by the novel coronavirus from wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 2020;94(7):e00127–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou P, Lou YX, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W, Zhao Y, Zhang F, et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): the experience of clinical immunologists from China. Clin Immunol. 2020;214:108393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conti P, Ronconi G, Caraffa A, et al. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents. 2020;34(2):327–331. [DOI] [PubMed] [Google Scholar]
- 23.Li G, Fan Y, Lai Y, et al. Coronavirus infections and immune responses. J Med Virol. 2020;92(4):424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu JT, Leung K, Leung GM. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study. Lancet. 2020;395(10225):689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thevarajan I, Nguyen THO, Koutsakos M, et al. Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat Med. 2020;26(4):453–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao X. COVID-19: immunopathology and its implications for therapy. Nat Rev Immunol. 2020;20(5):269–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Kerkhove MD, Vandemaele KAH, Shinde V, et al. Risk factors for severe outcomes following 2009 influenza a (H1N1) infection: a global pooled analysis. PLoS Med. 2011;8(7):e1001053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J, Zheng Y, Gou X, et al. Prevalence of comorbidities and its effects in coronavirus disease 2019 patients: a systematic review and meta-analysis. Int J Infect Dis. 2020;94:91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rao S, Lau A, Hc S, Traits . Exploring diseases/traits and blood proteins causally related to expression of ACE2, the putative receptor of SARS-CoV-2: a mendelian randomization analysis highlights tentative relevance of diabetes-related. Diabetes Care. 2020;43(7):1416–1426. [DOI] [PubMed] [Google Scholar]
- 30.Fernandez C, Rysä J, Almgren P, et al. Plasma levels of the proprotein convertase furin and incidence of diabetes and mortality. J Intern Med. 2018;284(4):377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shang J, Wan Y, Luo C, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci U S A. 2020;117(21):11727–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma LY, Chen WW, Gao RL, et al. China cardiovascular diseases report 2018: an updated summary.. J Geriatr Cardiol. 2020;17(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection?. Lancet Respir Med. 2020;8(4):e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Badawi A, Ryoo SG. Prevalence of comorbidities in the Middle East respiratory syndrome coronavirus (MERS-CoV): a systematic review and meta-analysis. Int J Infect Dis. 2016;49:129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan JWM, Ng CK, Chan YH, et al. Short term outcome and risk factors for adverse clinical outcomes in adults with severe acute respiratory syndrome (SARS). Thorax. 2003;58(8):686–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonow RO, Fonarow GC, O’Gara PT, et al. association of coronavirus disease 2019 (COVID-19) with myocardial injury and mortality. JAMA Cardiol. 2020;5(7):751–753. [DOI] [PubMed] [Google Scholar]
- 37.Yao X, Ye F, Zhang M, et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin Infect Dis. 2020;71(15):732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui S, Chen S, Li X, et al. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost. 2020;18(6):1421–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klok FA, Kruip MJHA, Van Der Meer NJM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang N, Bai H, Chen X, et al. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020;18(5):1094–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Llitjos J, Leclerc M, Chochois C, et al. High incidence of venous thromboembolic events in anticoagulated severe COVID‐19 patients. J Thromb Haemost. 2020;18(7):1743–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cytokine Storm: MG. Is it the only major death factor in COVID-19 patients? Coagulation role. Med Hypotheses. 2020;142:109829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang N, Li D, Wang X, et al. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18(4):844–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weitz JI, Fredenburgh JC, Eikelboom JW. A test in context: d-dimer [internet]. J Am Coll Cardiol. 2017;70(19):2411–2420. [DOI] [PubMed] [Google Scholar]
- 45.Phillippe HM. Overview of venous thromboembolism [Internet]. Am J Manag Care. 2017;23(20 Suppl):S376–82. [PubMed] [Google Scholar]
- 46.Bavishi C, Bonow RO, Trivedi V, et al. Acute myocardial injury in patients hospitalized with COVID-19 infection: a review. Prog Cardiovasc Dis. 2020;63(5):682–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davidson S, Maini MK, Wack A. Disease-promoting effects of type i interferons in viral, bacterial, and coinfections. J Interf Cytokine Res. 2015;35(4):252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat Rev Immunol. 2013;13(12):875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Channappanavar R, Fehr AR, Vijay R, et al. Dysregulated type i interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19(2):181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marchingo JM, Sinclair LV, Howden AJ, et al. Quantitative analysis of how myc controls t cell proteomes and metabolic pathways during t cell activation. Elife. 2020;9:e53725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen L, Liu HG, Liu W, et al. [Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia]. Zhonghua Jie He He Hu Xi Za Zhi. 2020;43:E005. [DOI] [PubMed] [Google Scholar]
- 53.Yang X, Yu Y, Xu J, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8(5):475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ranieri VM, Rubenfeld GD, Thompson BT, et al. Acute respiratory distress syndrome: the Berlin definition. J Am Med Assoc. 2012;307:2526–2533. [DOI] [PubMed] [Google Scholar]
- 55.Douda DN, Jackson R, Grasemann H, et al. Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. J Immunol. 2011;187(4):1856–1865. [DOI] [PubMed] [Google Scholar]
- 56.Parsons PE, Eisner MD, Thompson BT, et al. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit Care Med. 2005;33(1):1–6. [DOI] [PubMed] [Google Scholar]
- 57.Wang H, Ma S. The cytokine storm and factors determining the sequence and severity of organ dysfunction in multiple organ dysfunction syndrome. Am J Emerg Med. 2008;26(6):711–715. [DOI] [PubMed] [Google Scholar]
- 58.Shimabukuro-Vornhagen A, Gödel P, Subklewe M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16(5):448–457. [DOI] [PubMed] [Google Scholar]
- 60.Tanaka T, Narazaki M, Kishimoto T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy. 2016;8(8):959–970. [DOI] [PubMed] [Google Scholar]
- 61.Pathan N, Hemingway CA, Alizadeh AA, et al. Role of interleukin 6 in myocardial dysfunction of meningococcal septic shock. Lancet. 2004;363(9404):203–209. [DOI] [PubMed] [Google Scholar]
- 62.Hay KA, Hanafi LA, Li D, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T-cell therapy. Blood. 2017;130(21):2295–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iwanaga S, Kawabata SI, Muta T. New types of clotting factors and defense molecules found in horseshoe crab hemolymph: their structures and functions. J Biochem. 1998;123(1):1–15. [DOI] [PubMed] [Google Scholar]
- 64.Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13(1):34–45. [DOI] [PubMed] [Google Scholar]
- 65.Herter JM, Rossaint J, Zarbock A. Platelets in inflammation and immunity. J Thromb Haemost. 2014;12(11):1764–1775. [DOI] [PubMed] [Google Scholar]
- 66.Maas C, Renne T. Coagulation factor XII in thrombosis and inflammation. Blood. 2018;131(17):1903–1909. [DOI] [PubMed] [Google Scholar]
- 67.Conway EM. Reincarnation of ancient links between coagulation and complement. J Thromb Haemost. 2015;13:S121–32. [DOI] [PubMed] [Google Scholar]
- 68.Burzynski LC, Humphry M, Pyrillou K, et al. The coagulation and immune systems are directly linked through the activation of interleukin-1α by thrombin. Immunity. 2019;50(4):1033–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thornton P, McColl BW, Greenhalgh A, et al. Platelet interleukin-1α drives cerebrovascular inflammation. Blood. 2010;115(17):3632–3639. [DOI] [PubMed] [Google Scholar]
- 70.Bester J, Matshailwe C, Pretorius E. Simultaneous presence of hypercoagulation and increased clot lysis time due to IL-1β IL-6 and IL-8. Cytokine. 2018;110:237–242. [DOI] [PubMed] [Google Scholar]
- 71.Brunn GJ, Saadi S, Platt JL. Constitutive repression of interleukin-1α in endothelial cells. Circ Res. 2008;102(7):823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tunjungputri RN, Li Y, De Groot PG, et al. The inter-relationship of platelets with interleukin-1β-mediated inflammation in humans. Thromb Haemost. 2018;118(12):2112–2125. [DOI] [PubMed] [Google Scholar]
- 73.Cavalli G, Colafrancesco S, Emmi G, et al. Interleukin 1α: a comprehensive review on the role of IL-1α in the pathogenesis and treatment of autoimmune and inflammatory diseases. Autoimmun Rev. 2021;20:102763. [DOI] [PubMed] [Google Scholar]; •• This review illustrates the clinical relevance of IL-1α to the pathogenesis of inflammatory diseases, as well as the rationale for the targeted inhibition of this cytokine for treatment of these conditions. Three biologics are available to reduce the activities of IL-1α; the monoclonal antibody bermekimab, the IL-1 soluble receptor rilonacept, and the IL-1 receptor antagonist anakinra. These advances in mechanistic understanding and therapeutic management make it incumbent on physicians to be aware of IL-1α and of the opportunity for therapeutic inhibition of this cytokine in a broad spectrum of diseases.
- 74.José RJ, Williams AE, Chambers RC. Proteinase-activated receptors in fibroproliferative lung disease. Thorax. 2014;69(2):190–192. [DOI] [PubMed] [Google Scholar]
- 75.Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105(8):3178–3184. [DOI] [PubMed] [Google Scholar]
- 76.Günther A, Mosavi P, Heinemann S, et al. Alveolar fibrin formation caused by enhanced procoagulant and depressed fibrinolytic capacities in severe pneumonia: comparison with the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2000;161(2):454–462. [DOI] [PubMed] [Google Scholar]
- 77.Howell DCJ, Johns RH, Lasky JA, et al. Absence of proteinase-activated receptor-1 signaling affords protection from bleomycin-induced lung inflammation and fibrosis. Am J Pathol. 2005;166(5):1353–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mercer PF, Williams AE, Scotton CJ, et al. Proteinase-activated receptor-1, CCL2, and CCL7 regulate acute neutrophilic lung inflammation. Am J Respir Cell Mol Biol. 2014;50(1):144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chambers RC, Scotton CJ. Coagulation cascade proteinases in lung injury and fibrosis. Proc Am Thorac Soc. 2012;9(3):96–101. [DOI] [PubMed] [Google Scholar]
- 80.Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smith SA, Mutch NJ, Baskar D, et al. Polyphosphate modulates blood coagulation and fibrinolysis. Proc Natl Acad Sci U S A. 2006;103(4):903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Subramaniam S, Jurk K, Hobohm L, et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood. 2017;129(16):2291–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Noubouossie DF, Reeves BN, Strahl BD, et al. Neutrophils: back in the thrombosis spotlight. Blood. 2016;133(20):2186–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Iba T, Levy JH. Inflammation and thrombosis: roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J Thromb Haemost. 2018;16(2):231–241. [DOI] [PubMed] [Google Scholar]
- 85.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8(10):776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Iba T, Levy JH. Derangement of the endothelial glycocalyx in sepsis. J Thromb Haemost. 2019;17(2):283–294. [DOI] [PubMed] [Google Scholar]
- 87.Siddiqi HK, Mehra MR. COVID-19 illness in native and immunosuppressed states: a clinical–therapeutic staging proposal. J Heart Lung Transplant. 2020;39(5):405–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cavalli G, Larcher A, Tomelleri A, et al. Interleukin-1 and interleukin-6 inhibition compared with standard management in patients with COVID-19 and hyperinflammation: a cohort study. Lancet Rheumatol. 2021. (online ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]; ٠ IL-1 inhibition, but not IL-6 inhibition, was associated with a significant reduction of mortality in patients admitted to hospital with COVID-19, respiratory insufficiency, and hyperinflammation. IL-6 inhibition was effective in a subgroup of patients with markedly high C-reactive protein concentrations, whereas both IL-1 and IL-6 inhibition were effective in patients with low lactate dehydrogenase concentrations.
- 89.Fu B, Xu X, Wei H. Why tocilizumab could be an effective treatment for severe COVID-19? J Transl Med. 2020;18(1):164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang C, Wu Z, Li JW, et al. Cytokine release syndrome in severe COVID-19: interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int J Antimicrob Agents. 2020;55(5):105954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xu X, Han M, Li T, et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci U S A. 2020;117(20):10970–10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Luo P, Liu Y, Qiu L, et al. Tocilizumab treatment in COVID-19: a single center experience. J Med Virol. 2020;92(7):814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fontana F, Alfano G, Mori G, et al. COVID-19 pneumonia in a kidney transplant recipient successfully treated with tocilizumab and hydroxychloroquine. Am J Transplant. 2020;20(7):1902–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mihai C, Dobrota R, Schröder M, et al. COVID-19 in a patient with systemic sclerosis treated with tocilizumab for SSc-ILD. Ann Rheum Dis. 2020;79(5):668–669. [DOI] [PubMed] [Google Scholar]
- 95.Zhong J, Tang J, Ye C, et al. The immunology of COVID-19: is immune modulation an option for treatment? Lancet Rheumatol. 2020;2(7):e428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Deisseroth A, Ko CW, Nie L, et al. FDA approval: siltuximab for the treatment of patients with multicentric castleman disease. Clin Cancer Res. 2015;21(5):950–954. [DOI] [PubMed] [Google Scholar]
- 97.Palanques-Pastor T, López-Briz E, Poveda Andrés JL. Involvement of interleukin 6 in SARS-CoV-2 infection: siltuximab as a therapeutic option against COVID-19. Eur J Hosp Pharm. 2020;27(5):297–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Van Rhee F, Casper C, Voorhees PM, et al. Long-term safety of siltuximab in patients with idiopathic multicentric Castleman disease: a prespecified, open-label, extension analysis of two trials. Lancet Haematol. 2020;7(3):e209–17. [DOI] [PubMed] [Google Scholar]
- 99.Richardson P, Griffin I, Tucker C, et al. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet. 2020;395(10223):e30–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Russell B, Moss C, George G, et al. Associations between immune-suppressive and stimulating drugs and novel COVID-19 - A systematic review of current evidence. Ecancermedicalscience. 2020;14:1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schett G, Manger B, Simon D, et al. COVID-19 revisiting inflammatory pathways of arthritis. Nat Rev Rheumatol. 2020;16(8):465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dimopoulos G, De Mast Q, Markou N, et al. Favorable anakinra responses in severe covid-19 patients with secondary hemophagocytic lymphohistiocytosis. Cell Host Microbe. 2020;28:117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huet T, Beaussier H, Voisin O, et al. Anakinra for severe forms of COVID-19: a cohort study. Lancet Rheumatol. 2020;2(7):e393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Navarro-Millán I, Sattui SE, Lakhanpal A, et al. Use of anakinra to prevent mechanical ventilation in severe COVID-19: a case series. Arthritis Rheumatol. 2020;72(12):1990–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cavalli G, De Luca G, Campochiaro C, et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. 2020;2(6):e325–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Monteagudo LA, Boothby A, Gertner E. Continuous intravenous anakinra infusion to calm the cytokine storm in macrophage activation syndrome. ACR Open Rheumatol. 2020;2(5):276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.King A, Vail A, O’Leary C, et al. Anakinra in COVID-19: important considerations for clinical trials. Lancet Rheumatol. 2020;2(7):e379–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aouba A, Baldolli A, Geffray L, et al. Targeting the inflammatory cascade with anakinra in moderate to severe COVID-19 pneumonia: case series. Ann Rheum Dis. 2020;79(10):1381–1382. [DOI] [PubMed] [Google Scholar]
- 109.Riphagen S, Gomez X, Gonzalez-Martinez C, et al. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet. 2020;395(10237):1607–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ak C PM. Updates in thrombosis in pediatrics: where are we after 20 years? Hematol Am Soc Hematol Educ Program. 2012;439–43:2012. [DOI] [PubMed] [Google Scholar]
- 111.Della Gatta AN, Rizzo R, Pilu G, et al. Coronavirus disease 2019 during pregnancy: a systematic review of reported cases. Am J Obstet Gynecol. 2020;223(1):36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nikolich-Zugich J, Knox KS, Rios CT, et al. SARS-CoV-2 and COVID-19 in older adults: what we may expect regarding pathogenesis, immune responses, and outcomes. GeroScience. 2020;42(2):505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fogarty H, Townsend L, Ni Cheallaigh C, et al. COVID19 coagulopathy in Caucasian patients. Br J Haematol. 2020; 189(6): 1044–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guan W, Ni Z, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang YD, Zhang SP, Wei QZ, et al. COVID-19 complicated with DIC: 2 cases report and literatures review. Zhonghua Xue Ye Xue Za Zhi. 41, 245–247 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.AlSubaie AM. Coagulopathies in novel coronavirus (SARS-CoV-2) pandemic: emerging evidence for hematologists. Saudi J Biol Sci. 2020;28(1):956–961. [DOI] [PMC free article] [PubMed] [Google Scholar]; • The COVID-19 patients are showing varied coagulopathies and are at high risk for venous thromboembolisms (VTE) which demands an early intervention patients. This paper reviews the evolving data for the proposed pathogenesis models and management of coagulopathies in COVID-19 patients.
- 117.Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin Chim Acta. 2020;506:145–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Glas GJ, Van Der Sluijs KF, Schultz MJ, et al. Bronchoalveolar hemostasis in lung injury and acute respiratory distress syndrome. J Thromb Haemost. 2013;11(1):17–25. [DOI] [PubMed] [Google Scholar]
- 119.Yang X, Yang Q, Wang Y, et al. Thrombocytopenia and its association with mortality in patients with COVID‐19. J Thromb Haemost. 2020;18(6):1469–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Machlus KR, Cardenas JC, Church FC, et al. Causal relationship between hyperfibrinogenemia, thrombosis, and resistance to thrombolysis in mice. Blood. 2011;117(18):4953–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dolhnikoff M, Duarte-Neto AN, De Almeida Monteiro RA, et al. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J Thromb Haemost. 2020;18(6):1517–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gattinoni L, Chiumello D, Caironi P, et al. COVID-19 pneumonia: different respiratory treatment for different phenotypes? Intensive Care Med. 2020;46(6):1099–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Khandait H, Gandotra G, Sachdeva S, et al. COVID-19 and hematology—what do we know so far? SN Compr Clin Med. 2020;2(12):2631–2636. online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Asakura H, Ogawa H. COVID-19-associated coagulopathy and disseminated intravascular coagulation. Int J Hematol. 2021;113(1):45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• The pathology of coronavirus disease 2019 (COVID-19) is exacerbated by the progression of thrombosis, and disseminated intravascular coagulation (DIC), and cytokine storms. The most frequently reported coagulation/fibrinolytic abnormality in COVID-19 is the increase in D-dimer, and its relationship with prognosis has been discussed.
- 125.Zhang Y, Xiao M, Zhang S, et al. Coagulopathy and antiphospholipid antibodies in patients with covid-19. N Engl J Med. 2020;382(17):e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Thachil J, Tang N, Gando S, et al. ISTH interim guidance on recognition and management of coagulopathy in COVID‐19. J Thromb Haemost. 2020;18(5):1023–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bikdeli B, Madhavan MV, Jimenez D, et al. COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow-up. J Am Coll Cardiol. 2020;75(23):2950–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]; ٠ The authors review the current understanding of the pathogenesis, epidemiology, management, and outcomes of patients with COVID-19 who develop venous or arterial thrombosis, of those with pre-existing thrombotic disease who develop COVID-19, or those who need prevention or care for their thrombotic disease during the COVID-19 pandemic.
- 128.Karasu E, Nilsson B, Köhl J, et al. Targeting complement pathways in polytrauma- And sepsis-induced multiple-organ dysfunction. Front Immunol. 2019;10:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Licciardi F, Giani T, Baldini L, et al. COVID-19 and what pediatric rheumatologists should know: a review from a highly affected country. Pediatr Rheumatol. 2020;18(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Whyte CS, Morrow GB, Mitchell JL, et al. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID‐19. J Thromb Haemost. 2020;18(7):1548–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ranucci M, Ballotta A, Di Dedda U, et al. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J Thromb Haemost. 2020;18(7):1747–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Favresse J, Lippi G, Roy PM, et al. D-dimer: preanalytical, analytical, postanalytical variables, and clinical applications. Crit Rev Clin Lab Sci. 2018;55(8):548–577. [DOI] [PubMed] [Google Scholar]
- 133.Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. J Am Med Assoc. 2020;323(11):1061–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Huang I, Pranata R, Lim MA, et al. C-reactive protein, procalcitonin, D-dimer, and ferritin in severe coronavirus disease-2019: a meta-analysis. Ther Adv Respir Dis. 2020;14:1753466620937175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Levi M, Thachil J, Iba T, et al. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020;7(6):e438–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liu F, Li L, Da XM, et al. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J Clin Virol. 2020;127:104370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Koozi H, Lengquist M, Frigyesi A. C-reactive protein as a prognostic factor in intensive care admissions for sepsis: a Swedish multicenter study. J Crit Care. 2020;56:73–79. [DOI] [PubMed] [Google Scholar]
- 138.Solari F, Varacallo M. Low molecular weight heparin (LMWH). StatPearls. 2020. [PubMed] [Google Scholar]
- 139.Rotzinger DC, Beigelman-Aubry C, Von Garnier C, et al. Pulmonary embolism in patients with COVID-19: time to change the paradigm of computed tomography. Thromb Res. 2020;190:58–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zakai NA, Mcclure LA. Racial differences in venous thromboembolism. J Thromb Haemost. 2011;9(10):1877–1882. [DOI] [PubMed] [Google Scholar]
- 141.Stein PD, Kayali F, Olson RE, et al. Pulmonary thromboembolism in asians/pacific islanders in the United States: analysis of data from the national hospital discharge survey and the United States bureau of the census. Am J Med. 2004;116(7):435–442. [DOI] [PubMed] [Google Scholar]
- 142.Middeldorp S, Coppens M, Van Haaps TF, et al. Incidence of venous thromboembolism in hospitalized patients with COVID‐19. J Thromb Haemost. 2020;18(8):1995–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Helms J, Tacquard C, Severac F, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020;46(6):1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Poissy J, Goutay J, Caplan M, et al. Pulmonary Embolism in COVID-19 Patients: awareness of an Increased Prevalence. Circulation. 2020;142(2):184–186. [DOI] [PubMed] [Google Scholar]
- 145.Wang TF, Milligan PE, Wong CA, et al. Efficacy and safety of high-dose thromboprophylaxis in morbidly obese inpatients. Thromb Haemost. 2013;111(1):88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pannucci CJ, Fleming KI, Holoyda K, et al. Enoxaparin 40 mg per day is inadequate for venous thromboembolism prophylaxis after thoracic surgical procedure. Ann Thorac Surg. 2018;106(2):404–411. [DOI] [PubMed] [Google Scholar]
- 147.Young E. The anti-inflammatory effects of heparin and related compounds. Thromb Res. 2008;122(6):743–752. [DOI] [PubMed] [Google Scholar]
- 148.Ozolina A, Sarkele M, Sabelnikovs O, et al. Activation of coagulation and fibrinolysis in acute respiratory distress syndrome: a prospective pilot study. Front Med. 2016;3:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shukla D, Spear PG. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J Clin Invest. 2001;108(4):503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Vicenzi E, Canducci F, Pinna D, et al. Coronaviridae and SARS-associated coronavirus strain HSR1. Emerg Infect Dis. 2004;10(3):413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yamakawa K, Levy JH, Iba T. Recombinant human soluble thrombomodulin in patients with sepsis-associated coagulopathy (SCARLET): an updated meta-analysis. Crit Care. 2019;321(20):1993–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kienast J, Juers M, Wiedermann CJ, et al. Treatment effects of high-dose antithrombin without concomitant heparin in patients with severe sepsis with or without disseminated intravascular coagulation. J Thromb Haemost. 2006;4(1):90–97. [DOI] [PubMed] [Google Scholar]
- 153.Iba T, Arakawa M, Ohchi Y, et al. Prediction of early death in patients with sepsis-associated coagulation disorder treated with antithrombin supplementation. Clin Appl Thromb. 2018;24(9_suppl):145S–9S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lin L, Lu L, Cao W, et al. Hypothesis for potential pathogenesis of SARS-CoV-2 infection–a review of immune changes in patients with viral pneumonia. Emerg Microbes Infect. 2020;9(1):727–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Iba T, Levy JH, Raj A, et al. Advance in the management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J Clin Med. 2019;8(5):728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Du L, Kao RY, Zhou Y, et al. Cleavage of spike protein of SARS coronavirus by protease factor Xa is associated with viral infectivity. Biochem Biophys Res Commun. 2007;359(1):174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kam Y-W, Okumura Y, Kido H, et al. Cleavage of the SARS coronavirus spike glycoprotein by airway proteases enhances virus entry into human bronchial epithelial cells in vitro. PLoS One. 2009;4(11):e7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Elfiky AA. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci. 2020;253:117592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zabetakis I, Lordan R, Norton C, et al. COVID-19: the inflammation link and the role of nutrition in potential mitigation. Nutrients. 2020;12(5):1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhang C, Huang S, Zheng F, et al. Controversial treatments: an updated understanding of the coronavirus disease 2019. J Med Virol. 2020;92(9):1441–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Karim S, Islam A, Rafiq S, et al. The COVID-19 pandemic: disproportionate thrombotic tendency and management recommendations. Trop Med Infect Dis. 2021;6(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Smyth SS, Mcever RP, Weyrich AS, et al. Platelet functions beyond hemostasis. J Thromb Haemost. 2009;7(11):1759–1766. [DOI] [PubMed] [Google Scholar]
- 163.Connors J, States U, Levy J, et al. COVID-19 and its implications for thrombosis and anticoagulation COVID-19 and its implications for thrombosis and anticoagulation. J Blood. 2020;2:1–21. [Google Scholar]
- 164.Nagge J, Crowther M, Hirsh J. Is impaired renal function a contraindication to the use of low-molecular-weight heparin? Arch Intern Med. 2002;162(22):2605–2609. [DOI] [PubMed] [Google Scholar]
- 165.Hippensteel JA, LaRiviere WB, Colbert JF, et al. Heparin as a therapy for COVID-19: current evidence and future possibilities. Am J Physiol - Lung Cell Mol Physiol. 2020;319(2):L211–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Morrow DA, Braunwald E, Bonaca MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366(15):1404–1413. [DOI] [PubMed] [Google Scholar]
- 167.José R, Williams A, Sulikowski M, et al. Regulation of neutrophilic inflammation in lung injury induced by community-acquired pneumonia. Lancet. 2015;385:S52. [DOI] [PubMed] [Google Scholar]
- 168.José RJ, Williams AE, Mercer PF, et al. Regulation of neutrophilic inflammation by proteinase-activated receptor 1 during bacterial pulmonary infection. J Immunol. 2015;194(12):6024–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jiang Y, Zhao G, Song N, et al. Blockade of the C5a–C5aR axis alleviates lung damage in hDPP4 -transgenic mice infected with MERS-CoV. Emerg Microbes Infect. 2018;7(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Reis ES, Berger N, Wang X, et al. Safety profile after prolonged C3 inhibition. Clin Immunol. 2018;197:96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Vitiello A, La Porta R, D’Aiuto V, et al. Pharmacological approach for the reduction of inflammatory and prothrombotic hyperactive state in COVID-19 positive patients by acting on complement cascade. Hum Immunol. 2021;82(4):264–269. online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]