

Abstract
Purpose:
Neratinib is an irreversible, pan-HER tyrosine kinase inhibitor that is FDA-approved for HER2-overexpressing/amplified (HER2+) breast cancer. In this preclinical study, we explored the efficacy of neratinib in combination with inhibitors of downstream signaling in HER2+ cancers in vitro and in vivo.
Experimental Design:
Cell viability, colony formation assays, and western blotting were used to determine effect of neratinib in vitro. In vivo efficacy was assessed with patient-derived xenografts (PDXs): two breast, two colorectal and one esophageal cancer; two with HER2 mutations. Four PDXs were derived from patients who received previous HER2-targeted therapy. Proteomics were assessed through Reverse Phase Protein Arrays (RPPA) and network level adaptive responses were assessed through Target Score algorithm.
Results:
In HER2+ breast cancer cells, neratinib was synergistic with multiple agents, including mTOR inhibitors everolimus and sapanisertib, MEK inhibitor trametinib, CDK4/6 inhibitor palbociclib, and PI3Kα inhibitor alpelisib. We tested efficacy of neratinib with everolimus, trametinib, or palbociclib in five HER2+ PDXs. Neratinib combined with everolimus or trametinib led to a 100% increase in median event-free survival (EFS; tumor doubling time) in 25% (1 of 4) and 60% (3 of 5) models respectively while neratinib with palbociclib increased EFS in all five models. Network analysis of adaptive responses demonstrated upregulation of EGFR and HER2 signaling in response to CDK4/6, mTOR and MEK inhibition, possibly providing an explanation for the observed synergies with neratinib.
Conclusion:
Taken together, our results provide strong preclinical evidence for combining neratinib with CDK4/6, mTOR and MEK inhibitors for the treatment of HER2+ cancer.
STATEMENT OF TRANSLATIONAL RELEVANCE
Neratinib is an irreversible, pan-HER tyrosine kinase inhibitor that is FDA-approved for HER2-overexpressing/amplified (HER2+) breast cancer in the adjuvant and metastatic setting. This preclinical study explored the combinatorial therapies of neratinib. Results from our in vitro HER2 amplified cell lines showed that neratinib was synergistic with the mTOR inhibitors everolimus and sapanisertib, CDK4/6 inhibitor palbociclib MEK inhibitor trametinib, and PI3Kα inhibitor alpelisib. In vivo tumor growth study of HER2 amplified tumor PDXs further confirmed the enhanced therapeutic efficacy when neratinib was combined with everolimus, trametinib, or palbociclib. RPPA assay and network-level adaptive response analysis revealed potential molecular mechanisms for the observed synergies with neratinib. Taken together, the promising outcomes of this preclinical study provide strong rationale for combining neratinib with a number of pathway inhibitors.
INTRODUCTION
Human epidermal growth factor receptor 2 (HER2/neu), also known as ERBB2, is a member of EGFR/ERBB family of four structurally related transmembrane HER receptors – HER1 (EGFR), HER2, HER3 and HER4 [1]. By autophosphorylation, receptor dimerization causes activation of their intrinsic intracellular receptor-tyrosine kinase activity, leading to ultimate activation of downstream signaling cascades, principally through the mitogen-activated protein kinases (MAPK) and the PI3K/Akt pathways [2, 3], resulting in the activation of gene expression, proliferation, cell migration, differentiation, and regulation of apoptosis. Amplification or overexpression of HER2 is found in about 15-25% of breast cancers, which is associated with aggressive biology and worse overall survival in the absence of HER2-targeted therapy [4–6].
Neratinib (HKI-272, Puma Biotechnology) is an oral, small-molecule, irreversible pan-inhibitor of the EGFR, HER2, and HER4 members of the ERBB tyrosine kinase family [7–9]. Neratinib covalently binds to cysteine residues (Cys773 and Cys805) which are conserved in these receptors, preventing receptors from phosphorylation, thus blocking activation of tyrosine kinase activity and subsequent downstream signaling cascades [8, 10]. In addition to the MAPK and PI3K/Akt pathways, neratinib also induces cell cycle arrest through cyclin D and Rb [11]. Neratinib is FDA-approved for extended adjuvant treatment of high risk HER2-overexpressing or amplified (HER2+) breast cancer in monotherapy, and in combination therapy with capecitabine for advanced or metastatic breast cancer who have received two or more anti-HER2 agents. Combination therapies of neratinib with trastuzumab or other chemotherapeutic agents such as paclitaxel have demonstrated clinical benefit [12–15]. Further, neratinib has been shown to have antitumor efficacy alone and in combination with endocrine therapy for selected tumors with activating HER2 mutations [16, 17].
Although many patients benefit from neratinib, as with other targeted therapies patients often ultimately progress. Findings from a recent study indicated that an acquired ERBB2T798I mutation may contribute to the acquired resistance to neratinib [18]. Therefore, pursuing new therapeutic strategies that can enhance initial efficacy of neratinib therapy is necessary.
Several cell signaling inhibitors targeting HER2 downstream signaling have already been FDA approved for indications other than HER2 positive cancers. MEK inhibitor trametinib in combination with dabrafenib is approved for melanoma as well as lung cancer with BRAF_V600E mutations [19]. PI3K inhibitor alpelisib in combination with fulvestrant is approved for hormone receptor positive breast cancers bearing PIK3CA mutations [20]. Everolimus is approved for multiple types of solid tumors including hormone receptor positive breast cancer (in combination with exemestane) [21], neuroendocrine tumors, renal cell carcinoma, and subependymal giant cell astrocytoma associated with tuberous sclerosis. Of three FDA-approved CDK4/6 inhibitors, palbociclib is approved in combination with aromatase inhibitors as initial endocrine-based therapy for metastatic hormone receptor positive cancer, and in combination with fulvestrant in patients with disease progression on endocrine therapy [22].
In this study we sought to explore effective combination therapies with neratinib for HER2+ cancer. We examined potential synergistic combinations of neratinib with targeted inhibitors of multiple downstream adaptive cell survival pathways, including the MAPK and PI3K/Akt pathways by testing in vitro cell line models and in vivo patient-derived xenografts (PDXs) of multiple histologies.
MATERIALS AND METHODS
Cell lines, drugs and other reagents
Breast cancer cell lines, including BT-474, SK-BR-3, HCC-1954, MDA-MB-361, MDA-MDA-MB-453, and CAMA-1 were obtained from the American Type Culture Collection (ATCC). All the cell lines were tested mycoplasma negative. Cells were cultured in Dulbecco’s modified Eagle’s medium/F-12 (DMEM) supplemented with 10% fetal bovine serum at 37° and humidified 5% CO2. Neratinib was obtained from Puma Biotechnology Inc. Everolimus (NSC733504) and trametinib (NSC758246) were obtained from NCI-Developmental Therapeutics Program. Palbociclib was obtained as a gift from Pfizer as well as from the NCI-Developmental Therapeutic Program (https://dtp.cancer.gov). Alpelisib was obtained through the AACR PI3K SU2C Dream Team. Sapanisertib was purchased from Selleck Chemicals (Houston TX, USA). The following antibodies for Western blotting were purchased from Cell Signaling Technology (CST), including anti-HER2 (#2242), anti-phospho-Akt/T308 (#4056), anti-Akt (#9272), anti-phospho-S6K1/T389 (#9234), anti-S6K1 (#9202), anti-phospho-S6/S235/236 (#4858), anti-S6 (#2217), anti-phospho-4E-BP1/S65 (#9456), anti-4E-BP1 (#9452), anti-phospho-ERK1/2/T202/Y204 (#4370), anti-ERK1/2 (#9102), anti-phospho-MEK1/2/S217/221 (#9154), anti-MEK1/2 (#9126), anti-phospho-Rb/S780 (#9307). Anti-β-actin antibody (#A5441) was purchased from Sigma. Second antibodies Goat-anti-Rabbit-Alexa Fluor-680 (#A21076) and Goat-anti-Mouse- Dylight-800 (#610145-121) were purchased from Life Tech and Rockland Immunochemicals respectively.
Cell viability assay
Cells were seeded in 96-well plates at densities of 0.5-1.0 x 104 cells/100 μl per well in triplicates for each treatment dose. After adhering overnight, for single drug treatment, 100 μl of drug at serially diluted concentrations were added to the wells and incubated at 37°C for 72 hours. Cells were then fixed with 50% trichloroacetic (TCA) followed by staining with 0.4% sulforhodamine B (SRB) solution. OD values were read at 490 nm by plate reader Synergy 4 (BioTek). The half maximal inhibitory concentration (IC50) was determined based on the sigmoid drug-inhibition curve using GraphPad Prism v6.05 software. For combinatorial drug treatment, the cells were treated with neratinib together with individual drugs at individual combinatorial dose ratios based on single drug sigmoid dose-response curves. Each combination treatment group has 6 doses with a fixed combination ratio and a fixed serial dilution. For example, in combination of neratinib + alpelisib, we chose 10,000 nM and 90,000 nM as the highest doses for neratinib and alpellisib respectively (combination ratio: 0.11). Then we had 5 serial dilution at 10-fold from the highest combo doses. In combination of neratinib with everolimus, 10,000 nM and 20 nM were chosen as the highest doses for neratinib and everolimus respectively (combination ratio: 500). (Suppl Table 3). To evaluate combination efficacy, combination index (CI) was determined using CalcuSyn program based on Chou-Talalay IC50 model. CI<1.0 (curve left-shift): synergistic; CI=1.0, additive; CI>1.0 (curve right-shift): antagonistic) [23].
Colony formation assay
Cells were seeded in 6-well plates at a density of 1000 cells per well in triplicates for each treatment group. Next day, cells were treated with the individual drugs, or in combination at the different concentrations. Culture medium was changed with fresh drugs twice a week. Cells were cultured for 3 weeks. Cell colonies were then fixed in 10% formalin and stained with 0.05% crystal violet in 25% methanol. The stained colonies were scanned and total colony area was quantitated using NIH ImageJ v.1.48 software. Effect-based Combination Index (CI) was calculated by inhibition percentage of single drug and combination treatments using Bliss combination model. CI = ((EA + EB) − (EA*EB))/EAB, where EA, EB, and EAB are effects of drug A, B and combination AB inhibition percentage. Here the effect is inhibition percentage of colony formation compared to vehicle controls. (CI <1.0: synergistic; CI = 1.0: additive; CI > 1.0: antagonistic) [24].
Western blot analysis
In immunoblotting assay to investigate the effect of drug combination on cell signaling, we first tested single agents at different doses and selected a proper dose for each agent for combination. After treated with single drug or combination for 24 hours, cells were washed with cold PBS and lysed in 2x Laemmli buffer. The protein concentrations in the cell lysates were measured using Pierce BCA protein assay Kit (ThermoFisher). The same amount protein for each group was loaded to the gel (20-50 μg/lane). After SDS-PAGE, the protein was transferred to a 0.2μm nitrocellulose membrane (Bio-Rad Laboratories). Membranes were blocked with 0.1% casein blocking buffer at room temperature for 1 hour, followed by immunoblotting with the primary antibodies at room temperature overnight. After washing, the immunoblotting membrane was probed with the secondary antibodies with fluorescence conjugation. The immunoblots were visualized and the immunoblotting signal intensity quantitated using the Odyssey IR imaging system (Li-Cor Biosciences) [25, 26]. Several drug treatment experiments were performed and Figure 2A is one of these immunoblotting.
Figure 2. Effects of neratinib combination on downstream signaling in HER2+ cells.
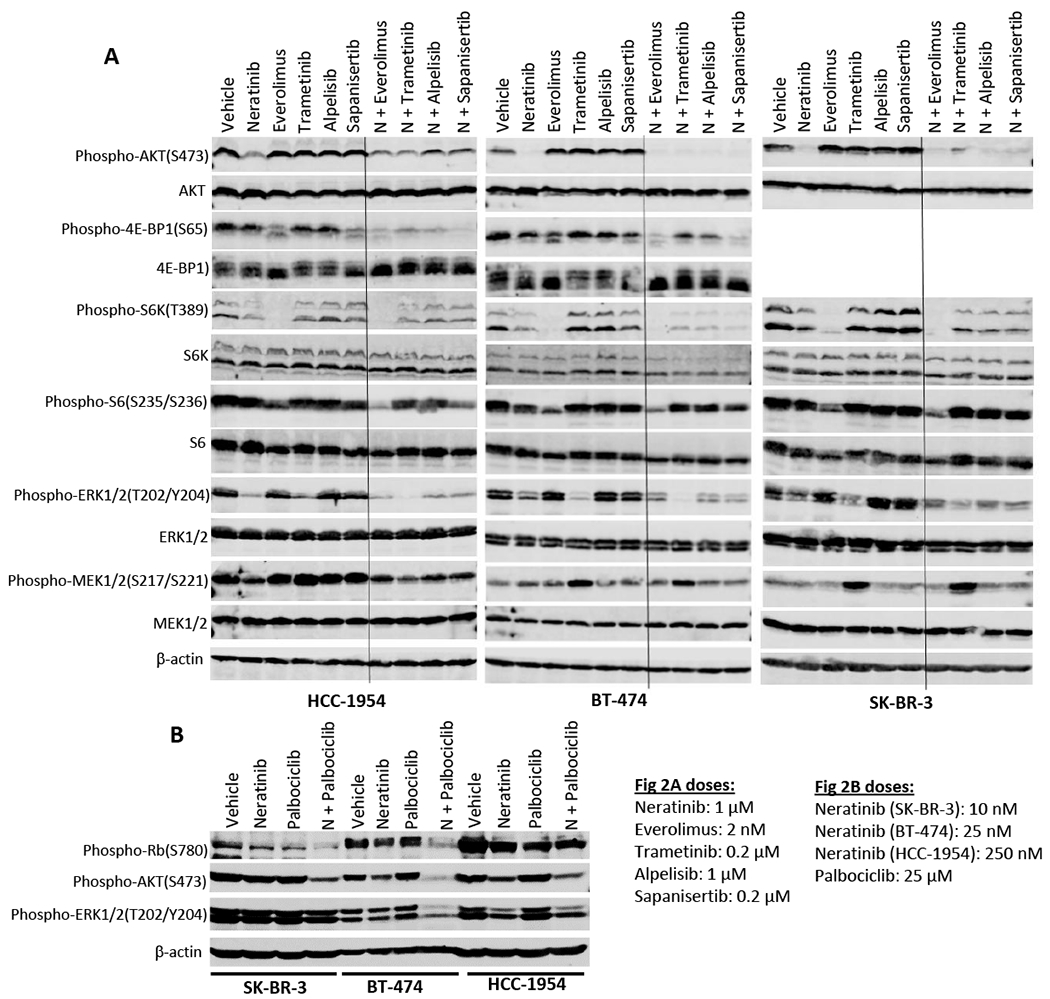
(A) Combination of neratinib with everolimus, trametinib, alpelisib and sapanisertib. HCC-1954, BT-474 and Sk-BR-3 cells were incubated with single drug or combination drugs for 24 hours. Treatment groups include (1) vehicle; (2) neratinib at 1 μM; (3) everolimus at 2 nM; (4) trametinib at 0.2 μM; (5) alpelisb at 1 μM; and (6) sapanisertib at 0.2 μM; or (7) neratinib + everolimus; (8) neratinib + trametinib; (9) neratinib + alpelisb; and (10) neratinib + sapanisertib at the same concentrations as single treatment. (B) Combination of neratinib with palbociclib. Neratinib was used at 10, 25, and 250 nM on SK-BR-3, BT-474, and HCC-1954 cell lines respectively. Palbociclib was used at 25 μM on all three cell lines. Cell lysates were analyzed by western blot. Phosphorylated protein levels of the key signaling molecules in the MAPK and PI3K/Akt pathways, including phospho-Akt, phospho-4E-BP1, phospho-S6K, phospho-S6, phospho-ERK1/2, phospho-MEK1/2 and phospho-Rb, were detected by their specific antibodies. Their total protein levels and β-actin levels were also detected for controls.
In vivo studies
PDX TMR-248 was developed by implantation of post-neoadjuvant therapy surgical samples through a collaboration with Champions Oncology. The other four PDXs (MDA-PDX.003.025, B8086, MDA-PDX.003.087, MDA-PDX.003.164) were generated from core biopsy samples obtained from patients with metastatic cancer as previously described [27, 28]. Alterations shown in Table 1 are selected actionable genes. Genomic alterations for TMR-248, B8086, and PDX.003.164 were identified in lab based whole exome sequencing of developed PDXs. Genomic alterations for PDX.003.025 and PDX.003.087 were identified in patient samples prior to PDX development. After implantation in NSG mice, with subsequent passaging, models underwent short tandem repeat (STR) testing to confirm genomic match to the parental tumor, with subsequent passaging in nu/nu mice. Fragments (3x3x3mm) of the PDXs were surgically implanted on to the flank of nude mice as previously describe [27, 28]. Once tumors grew to approximately 200 mm3, they were randomized for treatment. Neratinib, palbociclib, trametinib, and everolimus were suspended in water containing 0.5% methyl cellulose and were administered via oral gavage (100 ul) daily for the length of experiment. Tumor sizes were assessed twice a week and body weight was measured once a week. Tumor volumes were calculated using the formula (length x width2)/2. All experiments were approved by Institutional Animal Care and Use Committee (IACUC).
Table 1.
Clinical and molecular annotation of patient-derived xenografts.
| Model | Tumor Type | ER/PR | Selected Genomic Co-alterations | Prior to Collection |
|---|---|---|---|---|
| MDA-PDX.003.025 | Breast, Invasive Lobular Carcinoma | ER(−), PR(−) | PIK3CA H1047R, RPTOR amplification | *Docetaxel/Carboplatin/Trastuzumab/Pertuzumab *T-DM1 *AC *Carboplatin *Capecitabine |
| TMR-248 | Breast, Invasive Ductal Carcinoma | ER(+), PR(−) | ERBB2 V777L, PIK3CA H1047Q,AKT1 Deletion, BRAF E26D, | *Pertuzumab *Docetaxel/Pertuzumab/Trast uzumab |
| MDA-PDX.003.087 | Colorectal cancer | N/A | ERBB2 R678Q, IDH1 R132C,PIK3CA H1047R TSC2 T1462I, R73Q, MET Amplification | *Folfox/Bevacizumab *Capecitabine/Bevacizumab *5-FU/Bevacizumab *Folfiri/Panitumumab *Folfox *Lapatinib/Trastuzumab |
| B8086 | Colorectal Adenocarcinoma | N/A | KRAS G12V, PTEN 79+1G>A, MAP2K4 Q163* | *5FU/Oxaliplatin/Irinotecan |
| MDA-PDX.003.164 | Gastroesophageal junction (GEJ) adenocarcinoma | N/A | CDKN2A A97V, TP53Y236C | *Carboplatin/Taxol *Capecitabine/Oxaliplatin/Trastuzumab *Nivolumab *Taxotere *Investigational HER2 Ab, *Investigational HER2 small molecule inhibitor |
RPPA Analysis
Tumor tissues of PDXs (TMR-248 [n=4-5], PDX-003.025 [n=5], B8086 [n=3-4], PDX-003.087 [n-5-7]) were harvested from mice treated with neratinib and its combinations. The extracted tumor proteins were detected by reverse-phase protein array (RPPA), which was conducted by the MD Anderson Functional Proteomics core facility as previously described [29]. The protein levels were normalized for protein loading and then log-transformed. The two group comparisons were performed by fitting linear mixed effect model (LMEM) using “Group” as the fixed effect and “Model” as the random effect. The differentially expressed proteins were identified with specified FDR.
The network-level adaptive responses were computed using the Target Score algorithm as described in Yan et al [30]. The method is developed on the rationale that collective molecular responses of functionally associated molecule have a higher likelihood of therapeutic relevance than changes in abundances of individual molecules. Based on the collective upregulation of network modules in response to targeted therapies, the algorithm nominates combination therapies, involving the agent that induces the adaptive response and a second agent that targets vulnerabilities induced by the adaptive response. An adaptive response score, termed target score is quantified for each molecular entity as a sum of the self-change and cumulative change in its network neighborhood. The highest target scores correspond to potential adaptive responses (drug-induced activation of oncogenic processes or deactivation of tumor suppressors). In summary, the method uses molecular profiles of response to a perturbation as input. In parallel, a reference network which captures potential relations between all measured molecular entities of interest is constructed. Here, the interactions between the measured entities are extracted from the SignedPC module of the Pathway Commons database [31]. The collective adaptive responses to a perturbation are quantified as a sample/context-specific adaptation score (target score) that links protein interactions to drug response on the reference network using proteomic drug response data. Next, we identified the network modules that have a significantly high target score (i.e., collectively participate in adaptive responses) in each sample. The actionable targets that participate in sample specific adaptive responses and drug combinations are selected.
Statistical analysis
For in vitro studies, student’s t-test was performed to compare groups. Pearson test was used for correlation between groups. For in vivo studies, one-way ANOVA tests followed by Tukey was used by Dunnett’s multiple comparison. Data was presented as means ± SEM. Log-rank test was used for comparison of Kaplan-Meier survival curves.
RESULTS
Neratinib combination magnified inhibition of cell growth
First, we tested the efficacy of neratinib in a panel of breast cancer cell lines that are known to express HER2 protein at various levels, along with varying genomic alterations in the PI3K/Akt and MAPK pathways (Suppl Table 1). Immunoblotting analysis showed that BT-474, SK-BR-3, and HCC-1954 cells that are known to be HER2 amplified cell lines [32, 33], strongly expressed HER2 protein, while MDA-MB-361 and MDA-MB-453 cells displayed lower HER2 expression levels (Suppl Figure 1A, 1B). Cell viability assay showed that IC50s of neratinib in these cell lines ranged from 4.5 nM to 2,024 nM (Suppl Figure 1C, Suppl Table 2). Among these, HER2+ BT-474, SK-BR-3, and HCC-1954 cells were the most sensitive cell lines to neratinib with low nanomolar IC50s. Pearson correlation analysis showed that that cell sensitivity to neratinib is significantly and positively correlated with HER2 levels (p = 0.042) (Suppl Figure 1D). We have also examined in parallel the sensitivity of these breast cancer cells to multiple kinase inhibitors targeting the MAPK and PI3K/Akt pathways. The results showed varying sensitivities in these cell lines to different inhibitors (Suppl Table 2).
The activity of neratinib in combination therapy was then evaluated in the three HER2+ breast cancer cell lines. Cell viability assay showed that in HCC-1954 and BT-474 cell lines, combinatorial treatments of neratinib with multiple targeting agents, including the MEK inhibitor trametinib, the PI3Kα inhibitor alpelisib, the mTOR inhibitors everolimus (allosteric mTOR inhibitor) and sapanisertib (catalytic mTOR inhibitor), and the CDK4/6 inhibitor palbociclib, produced anti-proliferative activity. Combination index (CI) analysis using a Chou-Talalay model revealed that adding these drugs markedly synergized with neratinib-induced growth inhibition with low CI values ranging from 0.003 to 0.228 (Figure 1A, 1B). Dose-response curves in HCC-1954 cells showed that neratinib combinations caused a left-shift with substantially reduced IC50s, compared to single drug treatment (Suppl Figure 2A–E). On the other hand, the combinatorial efficacy in SK-BR-3 cells was relatively less potent in general than that observed in other two cell lines. We did not observe a synergy between neratinib and trametinib in this cell line (Figure 1C).
Figure 1. Effects of neratinib combination on cell growth of HER2+ cells.
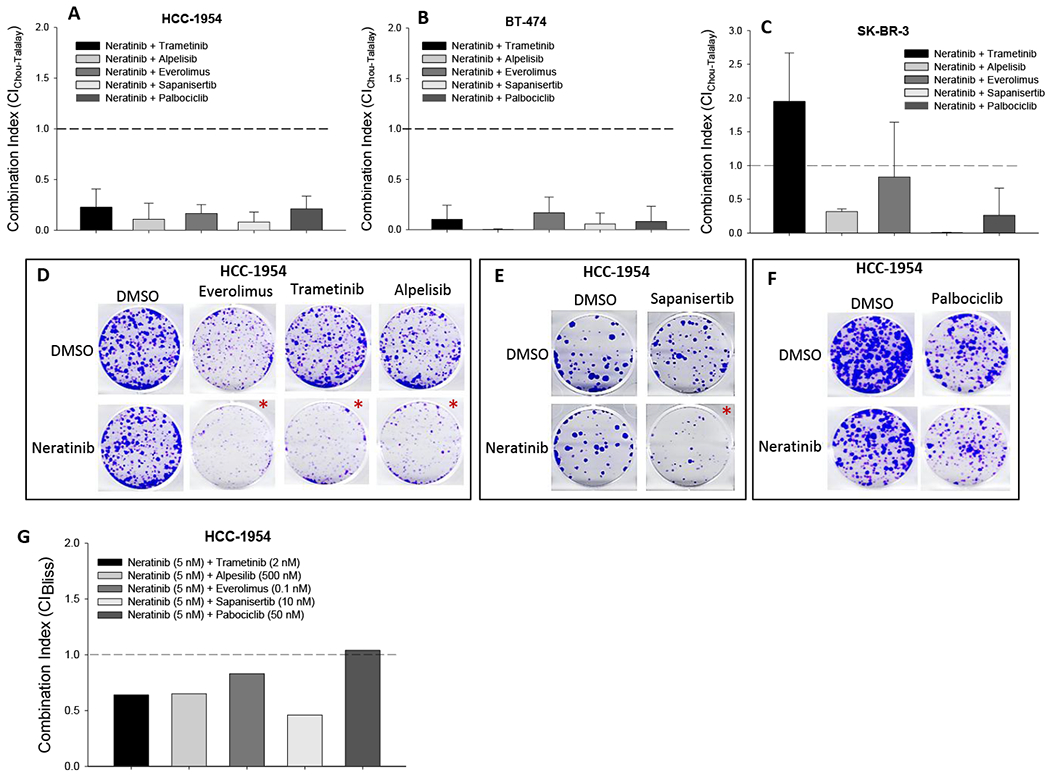
(A-C) Cell viability assay. HER2+ HCC-1954 (A), BT-474 (B), and SK-BR-3 (C) cells, seeded in 96-well plates, were treated with neratinib together with individual drugs (trametinib, alpelisib, everolimus, sapanisertib, and palbociclib) at combinatorial dose ratios (0.11 ~ 500). 72 hours after treatment, cell viability was determined by SRB staining. IC50s of each drug in both single and combination treatment were calculated from each sigmoid curve using the GraphPad Prism program. Triplicate wells were used for each concentration. Combination index (CI) was calculated using the Chou-Talalay drug combination model based on IC50 of single and combination treatments. Average CI was obtained from 3-5 cell viability assays as described above. (CI < 1.0: synergistic; CI = 1.0: additive; CI > 1.0: antagonistic). The depicted values represent average ± standard deviation from biological triplicate. (D-F): Colony formation assay. HCC-1954 cells, seeded in 6-well plates, were cultured for 3 weeks with single drug or combinatorial treatment, including neratinib (5 nM), everolimus (0.1 nM), trametinib (2 nM) and alpelisib (500 nM), sapanisertib (10 nM) and palbociclib (50 nM) and combinations. (*: synergistic combination) Data were from triplicate wells. (G) CI of colony formation. Total colony area was quantitated using ImageJ program and normalized with vehicle controls. Effect-based CI was calculated using Bliss combination analysis model based on inhibition percentage of single drug and combination treatment. (CIBliss < 1.0: synergistic; CIBliss = 1.0: additive; CIBliss > 1.0: antagonistic).
Next, we examined the combination effects on cell capacity of colony formation using HCC-1954 cells. First, we determined dose response of single drug treatment for all the six agents. After 3-week exposure to individual drugs at different doses, colony quantitation showed that neratinib and other targeting agents dose-dependently inhibited colony formation at appropriate dose ranges (Suppl Figure 3A–F). In the subsequent combination assay, combination dose for each drug was selected based on the response of single agent treatment. Cell colony staining showed that the cell ability to form colony was further reduced by combination of neratinib (5 nM) with trametinib (2 nM), alpelisib (500 nM), everolimus (0.1 nM), and sapanisertib (10 nM), compared to the single agent treatment (Figure 1D, 1E). These results were confirmed by colony area quantitation (Suppl Figure 4A–D). Combination efficacy was analyzed using a Bliss combination model. Combination index values (CIBliss) proved that neratinib was capable of synergizing with trametinib, alpelisib, everolimus and sapanisertib at their relatively lower doses with CIBliss ranging from 0.46 to 0.83 (Figure 1G, Suppl Figure 4A–D). For combination of neratinib plus palbociclib, although the colony staining revealed a further reduction in colony formation with this combination compared to single agent treatment (Figure 1F), Bliss analysis showed a CIBliss value of 1.04, indicating an additive combinatorial effect between these two agents (Figure 1G, Suppl Figure 4E).
Neratinib combination intensified inhibition of downstream signaling
We next evaluated the impact of neratinib and its combination on intracellular signaling. We treated three HER2+ cell lines, HCC-1954, BT-474 and SK-BR-3, with single agent or combinations for 24 hours, followed by western blot analysis. Neratinib itself reduced phosphorylation levels of ERK1/2, Akt, and S6K in all three cell lines, compared to vehicle controls (Figure 2A, Suppl Figure 5A, C, E), indicating that the HER2-targeting drug neratinib inhibited HER2 signaling through both MAPK and PI3K/Akt pathways. Neratinib also mildly inhibited MEK1/2 phosphorylation in HCC-1954 cells, while increased it in BT-474 cells (Figure 2A, Suppl Figure 5F). All three cell lines displayed a substantial reduction of phospho-ERK1/2, a downstream substrate of MEK kinase, when they were subjected to MEK inhibitor trametinib, in comparison with vehicle controls (Figure 2A, Suppl Figure 5F). However, trametinib was shown to increase phosphorylated MEK1/2 levels in all the three cell lines. In parallel, single drug treatment with mTOR inhibitors everolimus or sapanisertib attenuated phospho-4E-BP1 expression in HCC-1954 and BT-474 cells (Figure 2A, Suppl Figure 5B). Everolimus alone also almost completely abolished phosphorylation of S6K protein in all three cell lines, with consequently reduced phospho-S6 protein levels (Figure 2A, Suppl Figure 5B, D). However, the PI3K inhibitor alpelisib did not show signaling inhibition activity at the dose tested in these cells (Figure 2A, Suppl Figure 5).
In combination treatment, neratinib plus trametinib showed a further reduction in phosphorylation of ERK1/2 in all three cell lines, compared to single drug treatment (Figure 2A, Suppl Figure 5E). The data also revealed that when neratinib was combined with mTOR inhibitors everolimus or sapanisertib, they produced an enhanced effect in inhibiting phosphorylation of 4E-BP1 and S6 over the single drug treatment in BT-474 and HCC-1954 cells (Figure 2A, Suppl Figure 5B, D). Combinations of neratinib with mTOR inhibitors also further reduced phospho-ERK1/2. Finally, neratinib combination with PI3K inhibitor alpelisib exhibited an enhanced inhibition of phosphorylation of 4E-BP1 in HCC-1954 and BT-474 and ERK1/2 in all three cell lines (Figure 2A, Suppl Figure 5B, E). In separate immunoblotting assays we examined combination efficacy of neratinib with palbociclib. We found that when neratinib was combined with palbociclib, it substantially enhanced palbociclib-induced inhibition of Rb phosphorylation in SK-BR-3 and BT-474 cells (Figure 2B, Suppl Figure 5G). Neratinib/palbociclib combination also revealed greater inhibition of AKT phosphorylation in all three cell lines (Figure 2B, Suppl Figure 5H). However, this combination showed enhanced reduction of phospho-EKR1/2 levels only in BT-474 cells (Figure 2B, Suppl Figure 5I).
Neratinib combination enhanced antitumor efficacy on tumor growth of HER2+ PDXs
In vivo studies were designed to explore optimal therapeutic regimens of neratinib combination on tumor growth. The efficacy of the combinations was tested in five HER2+ PDXs that have been recently established (Table 1) from patients with HER2 positive as defined by IHC score of 3+ and/or gene amplification by FISH. This PDX panel consisted of two breast cancer PDXs (TMR-248 and MDA-PDX.003.025), two colorectal cancer (CRC) PDXs (B8086 and MDA-PDX.003.087), and one gastroesophageal junction cancer (GEJ) PDX (MDA-PDX.003.164). Both breast cancer models and one colorectal cancer model (MDA-PDX.003.087) had PIK3CA mutations, reported to confer relative resistance to trastuzumab-based therapy in several preclinical as well as clinical studies [34, 35]. In addition, two of the models had HER2 missense mutations (ERBB2_V777L in TMR-248 and ERBB2_R678Q in MDA-PDX.003.087) which are both known to be activating mutations [36, 37]. It is also notable that four PDXs was generated from tumors of HER2+ patients who previously were treated with standard of care HER2-targeted therapy. TMR-248 was generated from a patient who did not achieve pathological complete response with neoadjuvant pertuzumab/trastuzumab and paclitaxel therapy. PDX.003.025 was derived from a locally recurrent breast cancer in a patient previously treated with trastuzumab/pertuzumab, docetaxel and carboplatin as well as T-DM1. PDX-003-087 had already been treated with standard of care CRC treatments as well as lapatinib/trastuzumab, a HER2 directed therapy that has been shown to have efficacy in HER2+, KRAS wild-type CRC [38] and that has recently been incorporated into National Cancer Center Network guidelines [39]. PDX-003-164 was derived from an esophageal cancer from a patient previously treated with not only standard therapies including trastuzumab, but also two investigational HER2-targeted therapies. While B8086 had not been previously treated with prior HER2-targeted therapy, concomitant KRAS mutations have been shown to limit the efficacy of HER2-targeted therapy [40].
Nude mice bearing these PDXs were treated with orally administered single agent neratinib (10 mg/kg) or combinations with trametinib (0.3 mg/kg), everolimus (5 mg/kg) or palbociclib (50 mg/kg) daily. The combinations were well tolerated as evidenced by monitoring of body weight and lack of treatment related deaths. The therapeutic benefit of these combinations was evidenced by lower tumor volumes in these PDXs, compared to the single agent alone. The combination of neratinib with palbociclib significantly reduced tumor volume with T/C ratios from 0.03 to 0.16 across all five PDXs (Figure 3A–E, Suppl Figure 6A–E). The neratinib combination with trametinib showed enhanced efficacy to reduce tumor volume in PDX.003.164 (Figure 3E, Suppl Figure 6E), while neratinib plus everolimus further reduced tumor volume in both PDX.003.025 and PDX.003.164 models (Figure 3B, E, Suppl Figure 6B, E).
Figure 3. Effects of neratinib combination on tumor growth of HER2+ PDXs.
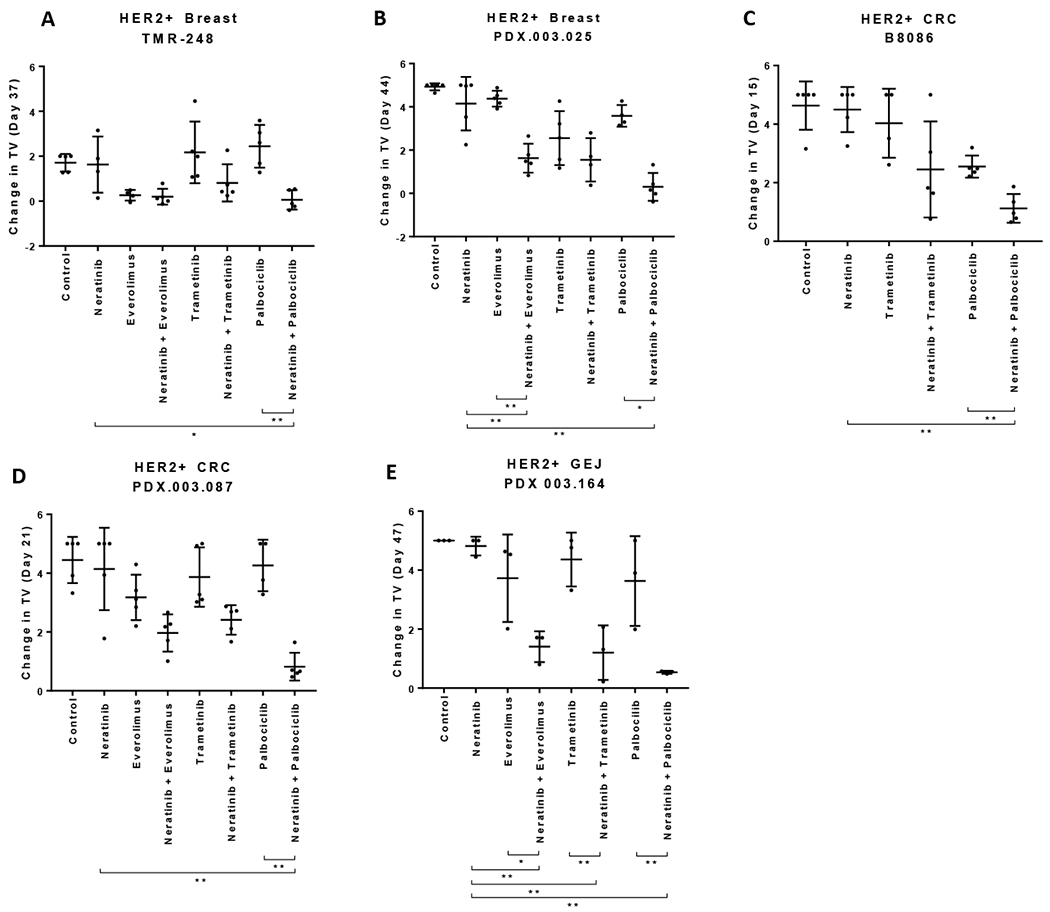
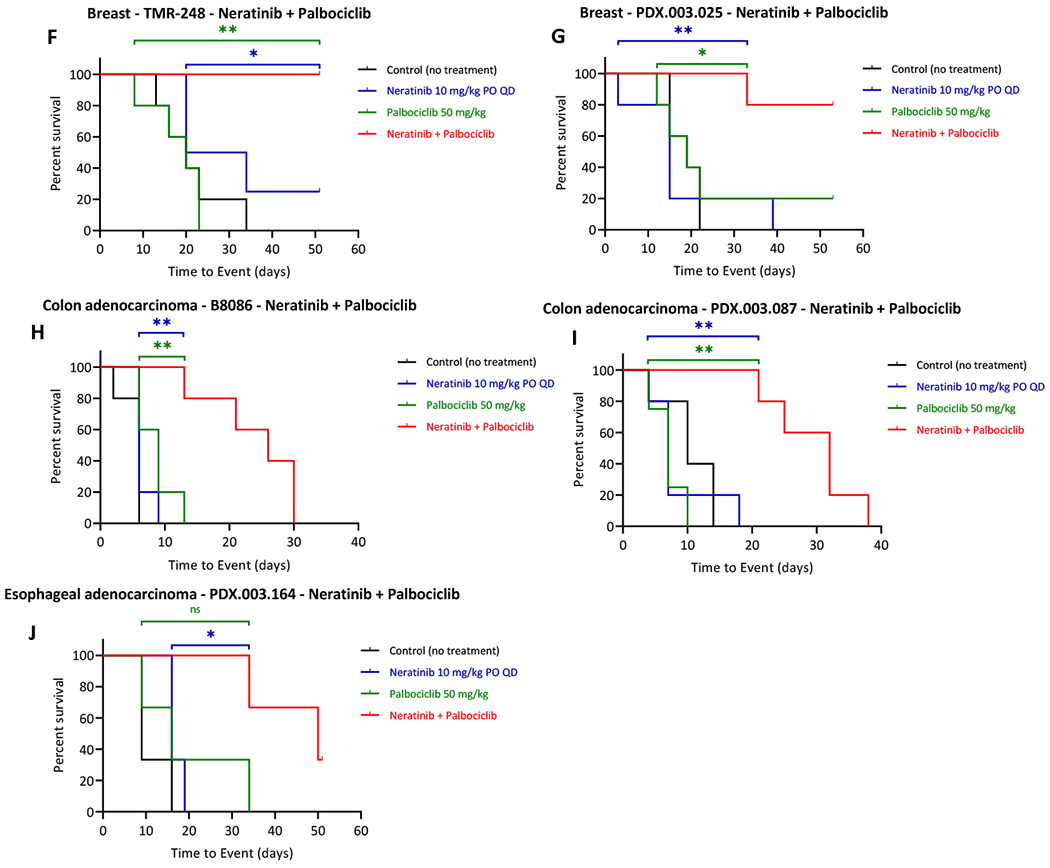
Nude mice bearing the individual PDX tumors,TMR-248 (A, F), PDX.003.025 (B, G), B8086 (C, H), PDX.003.087 (D, I), and PDX.003.164 (E, J) were orally administered with neratinib, everolimus, trametinib, and palbociclib at 10, 5, 0.3 and 50 mg/kg respectively or their combinations, daily for the length of experiment. (A-E) Change in tumor volume (TV). Tumor volume were measured at the end time points. Y axis value are relative change in TV calculated as (TVDayX – TVDay0) /TVDay0. (F-J) Kaplan-Meier event-free survival curves responding to neratinib combination with palbociclib. Tumor volume was measured longitudinally. Kaplan-Meier curves were generated with time to event (tumor doubling in size or meeting criteria for euthanasia per protocol) vs percent survival rate. Statistics are shown for the comparison showing significance combination versus single agent (ns: p>0.05; *: p≤0.05; **: p≤0.01). For in vivo studies, one-way ANOVA tests followed by Tukey was used by Dunnett’s multiple comparison. The depicted values represent mean ± SEM.
We also compared event free survival (EFS) defined as the tumor doubling using Kaplan-Meier survival curves between treatment groups [41]. The survival curves showed that combination treatment of neratinib with palbociclib significantly extended EFS in all five PDX models, compared to the single agent treatment and control groups (Figure 3F–J). The combinatorial regimen of neratinib with everolimus significantly increased the EFS in PDX.003.025 when compared to the everolimus single agent group but not the neratinib group. In contrast, the combination of neratininib with everolimus showed a significant increase in EFS in PDX.003.164 when compared to the neratinib single agent group but not the everolimus group (Suppl Figure 7B, 7D). The neratinib plus trametinib combination significantly increased the PFS in PDX.003.164 (Suppl Figure 7I). For TMR-248 the combination of neratinib and tramentinib also increased the EFS in this group when compared to the trametinib treatment group but not to the neratinib group. (Suppl Figure 7E). In summary, (1) combination of neratinib with palbociclib demonstrated substantially enhanced antitumor efficacy in all five HER2+ PDX models; (2) Combinations of neratinib with everolimus or with trametinib provided varying therapeutic benefit; (3) the GEJ cancer PDX (PDX.003.164) had the best responses to all the three combinatorial treatments.
Effects of neratinib combination on functional proteomics of HER2+ PDXs
We also performed a proteomic study to explore the potential mechanisms underlying the enhanced combinatorial efficacy of neratinib therapeutics. RPPA was applied on 136 tumor tissue samples harvested from the treated PDX models as described above. When normalized protein expression was analyzed by unsupervised hierarchical clustering, tumors were clustered by PDX model, rather than treatment, demonstrating that the treatment related differences were less robust than the intertumoral proteomic heterogeneity (Suppl Figure 8). With FDR 0.05, we assessed the differentially expressed proteins (DEPs) with single agent treatment compared with controls as well as single drug to combination treatment (Figure 4). We found that neither neratinib nor trametinib induced significant DEPs. Everolimus treatment was associated with 4 differentially expressed proteins compared to untreated control PDXs (Figure 4A). When everolimus was added to neratinib, 13 proteins were differentially expressed compared to neratinib alone (Figure 4B). Everolimus treatment alone and in combination with neratinib led to upregulation of IR-b and phospho-NDRG1_T346, but downregulation of phospho-S6, a well-established pharmacodynamic marker of mTOR signaling.
Figure 4. Effects of neratinib combination on proteomics of HER2+ PDXs.
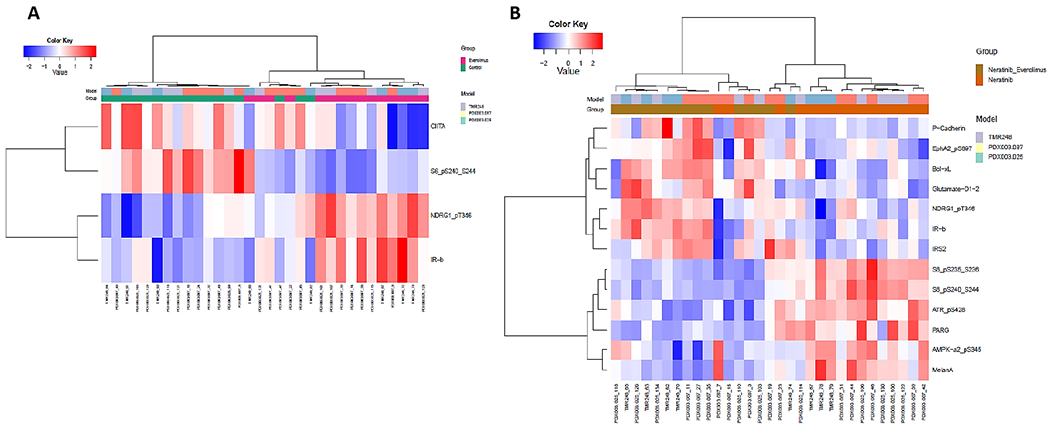
Proteins were extracted from PDX tumor tissues after single drug or combination treatment as described in Figure 3. Normalized protein expression levels detected by RPPA assay were analyzed. Heat maps (A-D) of DEPs were generated by unsupervised hierarchical cluster analysis with FDR 0.05. (A) Everolimus vs control; (B) Neratinib + everolimus vs neratinib; (C) Palbociclib vs control; (D) Neratinib + palbociclib vs neratinib. Color key from −2 to +2 represents the relative protein expression levels of DEPs.
Palbociclib treatment was associated with 26 DEPs compared to untreated controls (Figure 4C), There were 44 DEPs identified in the group of neratinib/palbociclib combination, compared to neratinib alone (Figure 4D). Both palbociclib single and combination treatments downregulated expression of Wee1 and phospho-Wee1_S642. Interestingly, some DEPs were presented in both everolimus and palbociclib combination groups, such as increased Bcl-XL, Glutamate-D1-2, IRS2, and IR-b, but decreased phospho-ATR_S428 and MelanA, suggesting an involvement of these DEPs in mechanisms or action of neratinib combination therapy.
We further analyzed adaptive responses with the Target Score algorithm, which quantifies network modules of functionally related molecular entities involved in adaptive responses to targeted perturbations [30]. Using the Target Score algorithm we analyzed the network-level adaptive responses to neratinib, palbociclib, everolimus and trametinib (Figure 5, Suppl Figure 9). The adaptation scores were significantly higher for EGFR and HER2 signaling in response to CDK4, MEK and mTOR inhibitors possibly providing an explanation for the observed synergies with neratinib. Both palbociclib and everolimus led to statistically significant (Q<0.05) increases in adaptive responses in EGFR, AKT, SHP2, SRC, RAF, STAT1/3, JNK, PRAS, FAK and others. Trametinib treatment resulted in a unique adaptive response compared to palbociclib and everolimus with low scores for MAPK and AKT pathways and yet high scores associated with EphA2,PRAS40 and PEA15 phosphorylation as well as total levels of the kinases, EGFR, FAK and DAPK2. In neratinib treatment, we did not observe a significant change in any actionable target involved in the adaptive responses. Albeit statistically insignificant, heregulin was increased in response to neratinib possibly suggesting a negative feedback loop activating other HER or other RTK family members. Overall, the adaptive responses to trametinib, palbociclib and everolimus provided the potential predictive markers of synergistic interactions with neratinib particularly evidenced by increases in receptor tyrosine kinase levels and phosphorylation while the downstream events differed remarkably in trametinib compared to the other agents.
Figure 5. Network analysis of adaptive responses.
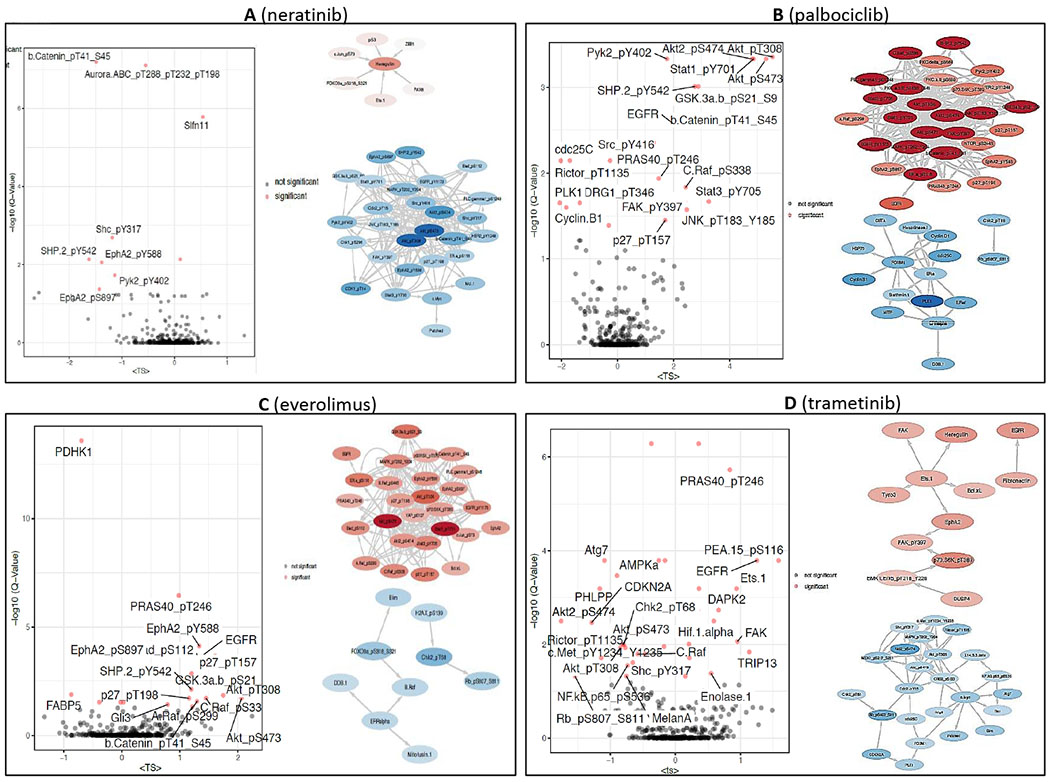
(A) Neratinib, (B) Palbociclib, (C) Everolimus, (D) Trametinib. The volcano-plots (left) show the significantly high adaptive responses as quantified by the Target Score method and statistical assessment. Q-values are the FDR-adjusted P-values based on a null model of scores generated from randomized data labels and fixed network topology. The proteins with target score > 0.5 and Q-val <0.05 are labeled on the plots. The network diagrams (right) demonstrate the collective adaptive response modules, where the red nodes correspond to proteins upregulated and blue nodes correspond to downregulated protein entities in response to drug treatment.
DISCUSSION
Overexpression/amplification of the HER2 oncogene in approximately 25% of human breast cancers predicts response to therapies targeting HER2. HER2 signaling is primarily mediated through downstream PI3K/Akt and MAPK axis which govern cell proliferation and apoptosis. The importance of the PI3K/Akt and MAPK pathways in oncogenic signaling is becoming increasingly apparent, especially in the case of HER2-amplified breast cancer [42, 43]. Meanwhile the contribution of aberrations in these pathways to resistance of HER2-targeted therapies has been evidenced. For example, PIK3CA hotspot mutations are found in approximately 25% of breast cancers and can overlap with HER2 amplification [44, 45]. The presence of these mutations confers relative resistance to trastuzumab or lapatinib [46–48]. Hence, the PI3K/Akt and MAPK signaling pathways are potential targets for HER2+ breast cancer with drug resistance due to activating mutations in these pathways. A number of clinical trials have suggested that HER2-directed therapies in combination with agents targeting PI3K or MAPK signaling, or cell cycle, have clinical efficacy in patients, particularly those with hyperactivity in these pathways. For examples, combinations of trastuzumab or lapatinib with everolimus have been proven encouraging antitumor activity in HER2-overexpressing tumors [49–52]. A recent report demonstrated that trastuzumab in combination with palbociclib exhibits promising survival outcomes in trastuzumab pretreated ER-positive/HER2-positive advanced breast cancer [53]. As a novel HER2 inhibitor, neratinib has demonstrated powerful therapeutic efficacy. In view of this and the mechanisms potentially involved in its resistance as described above, this study is designed to examine the synergism between neratinib and inhibitors targeting the PI3K/Akt and MAPK pathways, establishing potential optimal combination regiments for HER2+ breast cancer.
In cell viability assay, we found that neratinib displayed powerful synergistic efficacy when combined with all these targeted inhibitors in HCC-1954 and BT-474 cell lines, compared to SK-BR-3 cell line which showed less synergistic response to these combinations (Figure 1). The discrepancy of combination efficiency could be presumably resulted from the varying genomic mutations in the PI3K/Akt and MAPK pathways. As shown in Supplemental Table 1, HCC-1954 and BT-474 cells have multiple mutations in MAPK signaling molecules. This could reasonably account for the less synergistic actions in SK-BR-3 cells than the other two cell lines. These phenotypic differences of cell line viabilities in response to the treatment are correlated with cell signaling activity. For example, in HCC-1954 and BT-474 cells when neratinib was combined with these MEK and mTOR inhibitors, they demonstrated enhanced signaling inhibition in both MAPK and PI3K/Akt pathways in all these combination groups. On the other hand, SK-BR-3 cells had less response to most of these combinations in both pathways, except for the neratinib/trametinib combination which only affected phospho-ERK. Compared to other two cell lines, SK-BR-3 cells have much fewer mutations in both pathways which may lead to less responses in cell survival and signaling. We also realized that variations of combination index values in cell survival assay are bigger in SK-BR-3 cells than those in the other two cell lines. Whether this is a biological or technical variation needs further study.
We noticed that, in contrast to trametinib, everolimus, or sapanisertib all of which diminished phosphorylation of their own downstream kinase target proteins, the PI3Kα inhibitor alpelisib lacked activity on signaling inhibition across the cell lines. This could be possibly attributed to the occurrence of activating PIK3CA mutations in these HER2+ cell lines, except SK-BR-3 cells. Intriguingly, in BT-474 cells, combinations of neratinib with mTOR inhibitors synergized not only on the mTOR targets 4E-BP1 and S6, but also on the MEK target ERK1/2, demonstrating signaling cross-talk between the PI3K/Akt and MAPK pathways. Signaling cross-talk has been previously described to contribute to resistance in HER2-targeted therapies [54, 55]. We also noticed that palbociclib was able to reduce Rb phosphorylation in HCC-1954 and SK-BR-3 cell lines, but not in BT-474 cells. Both HER2 and ER are found to ultimately impinge activity of CDK4/6. While all three cell lines are HER2+, only BT-474 cells are ER+. Whether different ER status plays a role in Rb phosphorylation by CDK4/6 needs to be further clarified. DNA damage repair pathway is also known to be involved in regulation of Rb activity. Unlike the other two cell lines, BT-474 has a deleterious BRCA2 mutation. Further study is needed to determine if this differential BRCA2 mutational status may play a role in the differential response of cell lines to the CDK4/6 inhibitor on Rb phosphorylation.
From multiple drug combinations that were demonstrated synergistic in vitro on cell lines, we tested three combination regimens in vivo on PDX models. The tumor growth results showed enhanced antitumor efficacy by these combinatorial therapies which are generally consistent with the in vitro results. However, we observed diverse responses to combination therapies across the 5 PDX models. As shown in Table 1, in addition to HER2 amplification status, these PDXs have a variety of genomic alterations in multiple pathways, many of which are known to be cancer drivers. Some of these alterations overlapped between the PDXs, such as both breast models and one colorectal cancer PDX have PIK3CA mutations. Some PDXs have MAPK pathway mutations including BRAF and KRAS while some have activating ERBB2 mutations. These diverse genomic backgrounds may contribute to the observed inconsistency in therapeutic outcomes in these in vivo models. Therefore, the association between genomics and therapeutic efficacy needs to be further established.
To elucidate the potential mechanisms responsible for enhanced combinatorial efficacy, in addition to the cell signaling study on cell lines as described above, we have also performed RPPA assay on in vivo tumor samples. Previous studies have demonstrated that neratinib and other targeting agents such as palbociclib and everolimus are capable of inducing cell cycle arrest and apoptosis [56–58]. The evidence from our RPPA data suggests that the enhanced antitumor efficacy of combinatorial neratinib therapeutics could be attributed at least to boosted cell apoptosis. For example, two combination treatments (neratinib + everolimus and neratinib + palbociclib) share the same apoptosis-related DEPs, including Bcl-XL, Glutamate-D1-2, IRS2, and IR-b, phospho-ATR, and MelanA. Bcl-xL and Glutamate-D1-2 are known to be required for apoptosis [59–62]. Increased IR-b expression was also found to induce apoptosis [63]. It is interesting that while palbociclib alone was able to decrease Wee1 and Wee1_pS642 levels, combination of palbociclib and neratinib downregulated expression of multiple molecules in the DNA repair pathway, including Wee1, Wee1_pS642, ATR_pS428, CHK2_pT68, Cdc25C, and CDK1_pT114. The DNA repair pathway is well known to play an important role in regulating cell apoptosis [64, 65]. Thus, the enhanced cell apoptosis induced by these shared DEPs could be a potential mechanism responsible for the synergistic efficacy of combinatorial neratinib therapeutics. We noticed that single neratinib or trametinib did not induced DEPs, suggesting that two drugs do not have powerful impact on DEPs as when they act alone at the doses and at the time points we tested at the FDR cut-off of 0.05.
We also performed network-level adaptive response analysis using RPPA data. The results demonstrated feedback loops between drug inhibitions and HER2/EGFR signaling as well as its downstream pathways, providing the rationale for drug combinations. The results also verified the involvement of an apoptosis mechanism in the combinatorial therapy which we identified in RPPA assay. Data showed that combination treatments, such as neratinib with palbociclib or neratinib with everolimus, triggered survival stress response, as evidenced by enhanced Bcl-2 and Bcl-xL. Moreover, findings of DNA damage checkpoint molecule SLFN11 with a high Target Score, indicates that modulation of DNA repair pathways may contribute to functional mechanisms for the combination therapy. Overall, the results indicate that multiple signaling pathways are associated with the enhanced antitumor efficacy of neratinib combination therapeutics. The functional involvement of these pathways and their sophisticated network in neratinib therapy warrant further investigation. Future study is also needed to determine whether individual adaptive responses can be used to help personalize optimal combination therapy.
In summary, while the in vitro cell line and in vivo PDX approaches consistently demonstrated therapeutic benefits from these neratinib combinations in this study, the in silico assays identified potential molecular mechanisms and predictive biomarkers. In particular, from these in vivo results, we have several take-home messages: (1) combination of neratinib with palbociclib demonstrated substantially enhanced antitumor efficacy in all these HER2-amplified PDX models; (2) Combinations of neratinib with everolimus or with trametinib provided varying therapeutic benefits; (3) esophageal adenocarcinoma (PDX.003.164) had the best responses to all three combinatorial treatments. Although the combination of HER2 targeted therapy with ER-targeted therapy or a triplet could make sense for ER+/HER2+ breast cancer, we believe that simply based on HER2+ status the combination of HER2 inhibitor and palbociclib may be worth exploring across tumor types. The promising outcomes of this preclinical study provide guidance of clinical application of combinatorial therapies of neratinib for HER2-amplified cancer. A phase I trial evaluating the safety and clinical activity of neratinib in combination with everolimus, trametinib, or palbociclib in patients with solid tumors is ongoing (NCT03065387).
Supplementary Material
ACKNOWLEDGEMENT
This work was funded by PUMA Biotechnology Inc. (Los Angeles, CA), U54-CA224065, and Cancer Prevention and Research Institute Precision Oncology Decision Support Core (RP150535), Nellie B. Connally Breast Cancer Endowment, CTSA (1UL1TR003167), and the National Institutes of Health through MD Anderson’s Cancer Center Support Grant (CA016672). We appreciate the financial support from these funders. We also thank Susanna Brisendine for coordination in submission of this manuscript.
S. Kopetz reports receiving commercial research grants from Sanofi, Biocartis, Guardant Health, Array BioPharma, Genentech/Roche, EMD Serono, MedImmune, Novartis, Amgen, and Lilly, He also served as a consultant or advisor for Roche, Genentech, EMD Serono, Merck, Karyopharm Therapeutics, Amal Therapeutics, Navire Pharma, Symphogen, Holy Stone, Biocartis, Amgen, Novartis, Lilly, Boehringer Ingelheim, Boston Biomedical, AstraZeneca/MedImmune, Bayer Health, Pierre Fabre, EMD Serono, Redx Pharma, Ipsen, Daiichi Sankyo, Natera. He has stock and other ownership Interests in MolecularMatch and Navire.
I. Diala and A.S. Lalani are employees and shareholders for Puma Biotechnology, Inc.
S. Piha-Paul reports receiving commercial research grants from AbbVie, Inc.; ABM Therapeutics, Inc.; Acepodia, Inc; Alkermes; Aminex Therapeutics; Amphivena Therapeutics, Inc.; BioMarin Pharmaceutical, Inc; Boehringer Ingelheim; Bristol Myers Squib; Cerulean Pharma Inc.; Chugai Pharmaceutical Co., Ltd; Curis, Inc.; Daichi Sanko; Eli Lilly; ENB Therapeutics; Five Prime Therapeutics; Gene Quantum; Genmab A/S; GlaxoSmithKline; Helix BioPharma Corp.; Incyte Corp.; Jacobio Pharmaceuticals Co., Ltd.; Medimmune, LLC.; Medivation, Inc.; Merck Sharp and Dohme Corp.; Novartis Pharmaceuticals; Pieris Pharmaceuticals, Inc.; Pfizer; Principia Biopharma, Inc.; Puma Biotechnology, Inc.; Rapt Therapeutics, Inc.; Seattle Genetics; Silverback Therapeutics; Taiho Oncology; Tesaro, Inc.; TransThera Bio; NCI/NIH; P30CA016672 – Core Grant (CCSG Shared Resources).
F. Meric-Bernstam reports receiving commercial research grants from Aileron Therapeutics, Inc. AstraZeneca, Bayer Healthcare Pharmaceutical, Calithera Biosciences Inc., Curis Inc., CytomX Therapeutics Inc., Daiichi Sankyo Co. Ltd., Debiopharm International, eFFECTOR Therapeutics, Genentech Inc., Guardant Health Inc., Millennium Pharmaceuticals Inc., Novartis, Puma Biotechnology Inc., and Taiho Pharmaceutical Co. She also served as a consultant for Aduro BioTech Inc., Alkermes, DebioPharm, eFFECTOR Therapeutics, F. Hoffman-La Roche Ltd., Genentech Inc., IBM Watson, Jackson Laboratory, Kolon Life Science, OrigiMed, PACT Pharma, Parexel International, Pfizer Inc., Samsung Bioepis, Seattle Genetics Inc., Tyra Biosciences, Xencor, and Zymeworks. Additionally, she has served on advisory committees for Immunomedics, Inflection Biosciences, Mersana Therapeutics, Puma Biotechnology Inc., Seattle Genetics, Silverback Therapeutics, Spectrum Pharmaceuticals, and Zentalis. She has also received honoraria for speaking engagements from Chugai Biopharmaceuticals, Mayo Clinic, Rutgers Cancer Institute of New Jersey and travel related support from Beth Israel Deaconess Medical Center.
Footnotes
Conflict of Interest Statement:
M. Zhao, S. Scott, K. Evans, E. Yuca, T. Saridogan, X. Zheng, H. Wang, A. Korkut, C. Cruz-Pico, M. Demirhan, B. Kirby declare no potential conflict of interest.
REFERENCES
- 1.Linggi B and Carpenter G, ErbB receptors: new insights on mechanisms and biology. Trends Cell Biol, 2006. 16(12): p. 649–56. [DOI] [PubMed] [Google Scholar]
- 2.Citri A and Yarden Y, EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol, 2006. 7(7): p. 505–16. [DOI] [PubMed] [Google Scholar]
- 3.Moasser MM, The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene, 2007. 26(45): p. 6469–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slamon DJ, et al. , Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science, 1987. 235(4785): p. 177–82. [DOI] [PubMed] [Google Scholar]
- 5.Varga Z and Noske A, Impact of Modified 2013 ASCO/CAP Guidelines on HER2 Testing in Breast Cancer. One Year Experience. PLoS One, 2015. 10(10): p. e0140652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross JS and Fletcher JA, The HER-2/neu oncogene in breast cancer: prognostic factor, predictive factor, and target for therapy. Stem Cells, 1998. 16(6): p. 413–28. [DOI] [PubMed] [Google Scholar]
- 7.Tsou HR, et al. , Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J Med Chem, 2005. 48(4): p. 1107–31. [DOI] [PubMed] [Google Scholar]
- 8.Wissner A and Mansour TS, The development of HKI-272 and related compounds for the treatment of cancer. Arch Pharm (Weinheim), 2008. 341(8): p. 465–77. [DOI] [PubMed] [Google Scholar]
- 9.Feldinger K and Kong A, Profile of neratinib and its potential in the treatment of breast cancer. Breast Cancer (Dove Med Press), 2015. 7: p. 147–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kourie HR, et al. , Pharmacodynamics, pharmacokinetics and clinical efficacy of neratinib in HER2-positive breast cancer and breast cancer with HER2 mutations. Expert Opin Drug Metab Toxicol, 2016. 12(8): p. 947–57. [DOI] [PubMed] [Google Scholar]
- 11.Rabindran SK, et al. , Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res, 2004. 64(11): p. 3958–65. [DOI] [PubMed] [Google Scholar]
- 12.Awada A, et al. , Safety and efficacy of neratinib (HKI-272) plus vinorelbine in the treatment of patients with ErbB2-positive metastatic breast cancer pretreated with anti-HER2 therapy. Ann Oncol, 2013. 24(1): p. 109–16. [DOI] [PubMed] [Google Scholar]
- 13.Gandhi L, et al. , Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J Clin Oncol, 2014. 32(2): p. 68–75. [DOI] [PubMed] [Google Scholar]
- 14.Saura C, et al. , Safety and efficacy of neratinib in combination with capecitabine in patients with metastatic human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol, 2014. 32(32): p. 3626–33. [DOI] [PubMed] [Google Scholar]
- 15.Park JW, et al. , Adaptive Randomization of Neratinib in Early Breast Cancer. N Engl J Med, 2016. 375(1): p. 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smyth LM, et al. , Efficacy and Determinants of Response to HER Kinase Inhibition in HER2-Mutant Metastatic Breast Cancer. Cancer Discov, 2020. 10(2): p. 198–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hyman DM, et al. , HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature, 2018. 554(7691): p. 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanker AB, et al. , An Acquired HER2T798I Gatekeeper Mutation Induces Resistance to Neratinib in a Patient with HER2 Mutant-Driven Breast Cancer. Cancer Discov, 2017. 7(6): p. 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.FDA Approves dabrafenib plus trametinib for adjuvant treatment of melanoma with BRAF V600E or V600K mutations. 2018. p. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-dabrafenib-plus-trametinib-adjuvant-treatment-melanoma-braf-v600e-or-v600k-mutations.
- 20.FDA approves alpelisib for metastatic breast cancer. 2019. p. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-alpelisib-metastatic-breast-cancer.
- 21.FDA Approves Everolimus for Advanced Breast Cancer. 2012. p. https://www.onclive.com/view/fda-approves-everolimus-for-advanced-breast-cancer.
- 22.Walker AJ, et al. , FDA Approval of Palbociclib in Combination with Fulvestrant for the Treatment of Hormone Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Clin Cancer Res, 2016. 22(20): p. 4968–4972. [DOI] [PubMed] [Google Scholar]
- 23.Chou TC, Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res, 2010. 70(2): p. 440–6. [DOI] [PubMed] [Google Scholar]
- 24.Delaney W.E.t., et al. , Combinations of adefovir with nucleoside analogs produce additive antiviral effects against hepatitis B virus in vitro. Antimicrob Agents Chemother, 2004. 48(10): p. 3702–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arango NP, et al. , Selinexor (KPT-330) demonstrates anti-tumor efficacy in preclinical models of triple-negative breast cancer. Breast Cancer Res, 2017. 19(1): p. 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meric-Bernstam F, et al. , PIK3CA/PTEN mutations and Akt activation as markers of sensitivity to allosteric mTOR inhibitors. Clin Cancer Res, 2012. 18(6): p. 1777–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McAuliffe PF, et al. , Ability to Generate Patient-Derived Breast Cancer Xenografts Is Enhanced in Chemoresistant Disease and Predicts Poor Patient Outcomes. PLoS One, 2015. 10(9): p. e0136851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evans KW, et al. , A Population of Heterogeneous Breast Cancer Patient-Derived Xenografts Demonstrate Broad Activity of PARP Inhibitor in BRCA1/2 Wild-Type Tumors. Clin Cancer Res, 2017. 23(21): p. 6468–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xing Y, et al. , Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res, 2019. 21(1): p. 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonghong Yan HW, Augustin Luna, Behnaz Bozorgui, Xubin Li, Maga Sanchez, Zeynep Dereli, Nermin Kahraman, Goknur Kara, Xiaohua Chen, Yiling Lu, Ozgun Babur, Murat Cokol, Bulent Ozpolat, Chris Sander, Gordon B. Mills, Anil Korkut, Adaptive response to BET inhibition induces therapeutic vulnerability to MCL1 inhibitors in breast cancer. BioRxiv, 2020. [Google Scholar]
- 31.Ozgun Babur AL, Anil Korkut, Funda Durupinar, Metin Can Siper, and Ugur Dogrusoz JEA, Chris Sander, and Emek Demir, Causal interactions from proteomic profiles: molecular data meets pathway knowledge. BioRxiv, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bartel CA and Jackson MW, HER2-positive breast cancer cells expressing elevated FAM83A are sensitive to FAM83A loss. PLoS One, 2017. 12(5): p. e0176778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sabbaghi M, et al. , Defective Cyclin B1 Induction in Trastuzumab-emtansine (T-DM1) Acquired Resistance in HER2-positive Breast Cancer. Clin Cancer Res, 2017. 23(22): p. 7006–7019. [DOI] [PubMed] [Google Scholar]
- 34.Untch M, et al. , Survival Analysis After Neoadjuvant Chemotherapy With Trastuzumab or Lapatinib in Patients With Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer in the GeparQuinto (G5) Study (GBG 44). J Clin Oncol, 2018. 36(13): p. 1308–1316. [DOI] [PubMed] [Google Scholar]
- 35.Krop IE, et al. , Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): final overall survival results from a randomised open-label phase 3 trial. Lancet Oncol, 2017. 18(6): p. 743–754. [DOI] [PubMed] [Google Scholar]
- 36.Kavuri SM, et al. , HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov, 2015. 5(8): p. 832–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pahuja KB, et al. , Actionable Activating Oncogenic ERBB2/HER2 Transmembrane and Juxtamembrane Domain Mutations. Cancer Cell, 2018. 34(5): p. 792–806 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinelli E, et al. , Sequential HER2 blockade as effective therapy in chemorefractory, HER2 gene-amplified, RAS wild-type, metastatic colorectal cancer: learning from a clinical case. ESMO Open, 2018. 3(1): p. e000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benson AB 3rd, et al. , Colon Cancer, Version 1.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw, 2017. 15(3): p. 370–398. [DOI] [PubMed] [Google Scholar]
- 40.Meric-Bernstam F, et al. , Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): an updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol, 2019. 20(4): p. 518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Einarsdottir BO, et al. , A patient-derived xenograft pre-clinical trial reveals treatment responses and a resistance mechanism to karonudib in metastatic melanoma. Cell Death Dis, 2018. 9(8): p. 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rexer BN and Arteaga CL, Optimal targeting of HER2-PI3K signaling in breast cancer: mechanistic insights and clinical implications. Cancer Res, 2013. 73(13): p. 3817–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tao JJ, et al. , Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci Signal, 2014. 7(318): p. ra29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saal LH, et al. , PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res, 2005. 65(7): p. 2554–9. [DOI] [PubMed] [Google Scholar]
- 45.Stemke-Hale K, et al. , An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res, 2008. 68(15): p. 6084–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Junttila TT, et al. , Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell, 2009. 15(5): p. 429–40. [DOI] [PubMed] [Google Scholar]
- 47.Berns K, et al. , A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell, 2007. 12(4): p. 395–402. [DOI] [PubMed] [Google Scholar]
- 48.Eichhorn PJ, et al. , Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res, 2008. 68(22): p. 9221–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hurvitz SA, et al. , Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2-positive advanced breast cancer (BOLERO-1): a phase 3, randomised, double-blind, multicentre trial. Lancet Oncol, 2015. 16(7): p. 816–29. [DOI] [PubMed] [Google Scholar]
- 50.Morrow PK, et al. , Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J Clin Oncol, 2011. 29(23): p. 3126–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jerusalem G, et al. , Phase I trial of oral mTOR inhibitor everolimus in combination with trastuzumab and vinorelbine in pre-treated patients with HER2-overexpressing metastatic breast cancer. Breast Cancer Res Treat, 2011. 125(2): p. 447–55. [DOI] [PubMed] [Google Scholar]
- 52.Gadgeel SM, et al. , Phase I study evaluating the combination of lapatinib (a Her2/Neu and EGFR inhibitor) and everolimus (an mTOR inhibitor) in patients with advanced cancers: South West Oncology Group (SWOG) Study S0528. Cancer Chemother Pharmacol, 2013. 72(5): p. 1089–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ciruelos E, et al. , Palbociclib and Trastuzumab in HER2-Positive Advanced Breast Cancer: Results from the Phase II SOLTI-1303 PATRICIA Trial. Clin Cancer Res, 2020. 26(22): p. 5820–5829. [DOI] [PubMed] [Google Scholar]
- 54.Yamaguchi H, et al. , Signaling cross-talk in the resistance to HER family receptor targeted therapy. Oncogene, 2014. 33(9): p. 1073–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nahta R, Pharmacological strategies to overcome HER2 cross-talk and Trastuzumab resistance. Curr Med Chem, 2012. 19(7): p. 1065–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang G, et al. , Palbociclib triggers apoptosis in bladder cancer cells by Cdk2-induced Rad9-mediated reorganization of the Bak.Bcl-xl complex. Biochem Pharmacol, 2019. 163: p. 133–141. [DOI] [PubMed] [Google Scholar]
- 57.Cai Y, et al. , mTOR inhibitor RAD001 (everolimus) induces apoptotic, not autophagic cell death, in human nasopharyngeal carcinoma cells. Int J Mol Med, 2013. 31(4): p. 904–12. [DOI] [PubMed] [Google Scholar]
- 58.Ciolczyk-Wierzbicka D, et al. , mTOR inhibitor Everolimus-induced apoptosis in melanoma cells. J Cell Commun Signal, 2019. 13(3): p. 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X, et al. , Targeting anti-apoptotic BCL-2 family proteins for cancer treatment. Future Med Chem, 2020. [DOI] [PubMed] [Google Scholar]
- 60.Lee EF, et al. , BCL-XL and MCL-1 are the key BCL-2 family proteins in melanoma cell survival. Cell Death Dis, 2019. 10(5): p. 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Caruso C, et al. , Glutamate induces apoptosis in anterior pituitary cells through group II metabotropic glutamate receptor activation. Endocrinology, 2004. 145(10): p. 4677–84. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y and Bhavnani BR, Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci, 2006. 7: p. 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nevado C, Benito M, and Valverde AM, Role of insulin receptor and balance in insulin receptor isoforms A and B in regulation of apoptosis in simian virus 40-immortalized neonatal hepatocytes. Mol Biol Cell, 2008. 19(3): p. 1185–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Zio D, Cianfanelli V, and Cecconi F, New insights into the link between DNA damage and apoptosis. Antioxid Redox Signal, 2013. 19(6): p. 559–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Norbury CJ and Zhivotovsky B, DNA damage-induced apoptosis. Oncogene, 2004. 23(16): p. 2797–808. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.