

Abstract
Plectin, a highly versatile cytolinker protein, provides tissues with mechanical stability through the integration of intermediate filaments (IFs) with cell junctions. Here, we hypothesize that plectin-controlled cytoarchitecture is a critical determinant of the intestinal barrier function and homeostasis. Mice lacking plectin in an intestinal epithelial cell (IEC; PleΔIEC) spontaneously developed colitis characterized by extensive detachment of IECs from the basement membrane (BM), increased intestinal permeability, and inflammatory lesions. Moreover, plectin expression was reduced in the colons of ulcerative colitis (UC) patients and negatively correlated with the severity of colitis. Mechanistically, plectin deficiency in IECs led to aberrant keratin filament (KF) network organization and the formation of dysfunctional hemidesmosomes (HDs) and intercellular junctions. In addition, the hemidesmosomal α6β4 integrin (Itg) receptor showed attenuated association with KFs, and protein profiling revealed prominent downregulation of junctional constituents. Consistent with the effects of plectin loss in the intestinal epithelium, plectin-deficient IECs exhibited remarkably reduced mechanical stability and limited adhesion capacity in vitro. Feeding mice with a low-residue liquid diet that reduced mechanical stress and antibiotic treatment successfully mitigated epithelial damage in the PleΔIEC colon.
Introduction
The intestinal epithelium is composed of a single layer of tightly linked intestinal epithelial cells (IECs), forming a selective physical barrier that is critical for gut homeostasis. A breach in the intestinal barrier, referred to as “leaky gut”1, results in excessive exposure to luminal microbiota and in a concomitant innate immune response. Subsequent dysregulation of the finely-tuned interplay among gut microbiota, IECs, and immune cells accounts for uncontrolled inflammation and pathogenesis of intestinal disorders such as inflammatory bowel disease (IBD) and colorectal cancer (CRC)2,3.
The epithelial barrier function is secured by cell junctions that seal intercellular spaces and interlink IECs with the underlying basement membrane (BM) into a structural and functional continuum. Alterations in junctional proteins and BM components may lead to a breakdown of the barrier, and genetic studies identified multiple links between junction/BM-associated genes and the development of IBD4–6. While apical tight junctions (TJs) and subjacent adherens junctions (AJs) confer paracellular transport selectivity, desmosomes (Ds) together with BM-linked hemidesmosomes (HDs) provide the intestinal epithelium with resilience to mechanical stress generated by intestinal peristalsis7. It is noteworthy that recently reported mouse models demonstrate the protective role of Ds and HDs in the context of both intestinal inflammation8,9 and colitis-associated CRC8. Accumulating evidence suggests that fundamental features of Ds and HDs (such as stability, dynamics, and mechanotransduction capacity) heavily rely on their interconnection with keratin filament (KF) networks10,11. This places plakins12, a family of cytolinker proteins mediating physical linkage between KFs and cell junctions, at the very center of the processes controlling epithelial homeostasis.
Plectin, a highly versatile member of the plakin protein family, crosslinks intermediate filaments (IFs) of different types and anchors them at cellular junctions, including HDs and Ds of epithelial cells13. Multiple mutations in the plectin gene have been identified in epidermolysis bullosa (EB)14, a disorder characterized by excessive blister formation in skin15,16 with reported cases of concurrent IBD17,18. Previous studies have shown that plectin ablation disrupts highly organized epithelial KF networks and alters the structure and functionality of cell junctions19–22. For example, tissue-specific deletion of plectin in the mouse biliary epithelium has adverse effects on the formation of TJs, AJs, and Ds, with deleterious consequences for epithelial stability under cholestasis22. Likewise, analysis of knock-in mice recapitulating dominant EB simplex suggests that HD stability in basal keratinocytes depends on plectin-mediated recruitment of KFs20. Mechanistically, dysfunctional HDs account for epithelial fragility and lesional defects23 which resemble those seen in patients with IBD24. Although these observations suggest a linkage between plectin dysfunction and intestinal pathologies, plectin’s role in the intestinal epithelium remains unaddressed.
In this study, we found that plectin expression was reduced in patients with active ulcerative colitis (UC) and that plectin expression levels negatively correlated with the severity of colitis. To study the underlying molecular mechanisms, we generated two new mouse lines: one constitutive (PleΔIEC) and the other with tamoxifen (TMX)-inducible (PleΔIEC-ERT2) plectin ablation in IECs. The phenotypic characterization of these mice demonstrated that loss of plectin leads to spontaneous development of a colitic phenotype characterized by extensive detachment of IECs from the BM, increased intestinal permeability, and formation of inflammatory lesions. These results demonstrate the absolute indispensability of plectin for the maintenance of intestinal epithelium integrity, and moreover that both mouse lines provide a useful model system for investigating disease etiology and testing palliative therapies.
Results
Suppression of plectin in human patients with UC
To examine the role of plectin in the pathogenesis of UC, we screened for potential alterations of plectin expression in a cohort of ~100 UC patients. The analysis of immunolabeled biopsy samples taken from patients and healthy controls revealed discontinuous and rather patchy plectin staining patterns in UC biopsies. The gaps in the plectin staining pattern coincided with goblet cell openings heavily loaded with mucus. In healthy controls, plectin decorated both apical and basal membranes of IECs evenly (Figs. 1A and S1A), resembling plectin localization in mouse intestinal sections (Fig. S1B and published previously25). In addition, mRNA profiling showed significantly reduced expression levels of plectin in biopsies from patients with active UC (Fig. 1B). Histological analysis revealed that low mRNA levels of plectin were associated with higher inflammation (Fig. 1C) and higher C reactive protein levels in serum (not shown).
Fig. 1. Loss of plectin is associated with UC in human patients and leads to intestinal epithelial barrier dysfunction with concomitant inflammation in mouse.

A Paraffin-embedded colon sections from UC patients (UC) and healthy controls (healthy) were immunolabeled with antibodies to plectin (red), keratin 8 (K8; green), and mucin 2 (Muc2; magenta). Nuclei were stained with Hoechst (blue). Arrows, apical IEC membrane; arrowheads, basal IEC membrane. Scale bar, 40 μm. B Relative plectin mRNA levels in rectum biopsies collected from healthy controls and patients with active UC. Scattered boxplots show individual data points, median, 25th, and 75th percentile with whiskers reaching the last data point. The numbers of included participants per cohort are indicated in the graph. C Relative plectin mRNA expression in rectum biopsies collected from UC patients clustered based on inflammation scored in H&E-stained rectum sections. Scattered boxplots show individual data points, median, 25th, and 75th percentile with whiskers reaching the last data point. The numbers of included participants per cohort are indicated in the graph. D Bodyweight of Plefl/fl and PleΔIEC mice was monitored for 25 weeks, n = 7. E Representative images of the rectum of 30-week-old Plefl/fl and PleΔIEC mice. Kaplan–Meier graph shows age-related rectal prolapse incidence. F Intestinal transepithelial electrical resistance (TEER) measured ex vivo in both proximal and distal colons of 12-week-old Plefl/fl and PleΔIEC mice, n = 4. G In vivo permeability of mucosa of Plefl/fl and PleΔIEC mice (at the age indicated) measured by monitoring 40-kDa FITC-dextran levels in plasma 4 h after orogastric gavage, n = 3–7. H Representative image of PleΔIEC colon section from 30-week-old PleΔIEC mouse stained with H&E. Arrows, bacterial patches in the mucosa. Scale bar, 50 μm. I In vivo chemiluminescence images of 12-week-old PleΔIEC and Plefl/fl mice injected with myeloperoxidase (MPO) inflammation probe. J MPO activity (a marker of neutrophil infiltration) measured in colon lysates from 12-week-old PleΔIEC and Plefl/fl mice, n = 3. K, L Inflammation extent (percentage) (K) and the number of lymphatic follicles (L) assessed from H&E-stained sections of 12-week-old Plefl/fl and PleΔIEC colons, n = 4. Data are presented as mean ± SEM, n.s. not significant, *P < 0.05, **P < 0.01, †P < 0.001.
IEC-specific plectin-deficient mice develop a colitic phenotype due to intestinal barrier dysfunction
To explore the role of plectin in the intestinal epithelium in greater detail, we generated IEC-specific plectin knockout (PleΔIEC) mice. Successful ablation of plectin in IECs was confirmed by immunofluorescence microscopy (Fig. S1C). The newly generated PleΔIEC mice had a considerably lower bodyweight (Figs. 1D and S1D), suffered from persistent diarrhea with occasional rectal bleeding (Fig. S1E, and not shown), and frequently developed rectal prolapse (Fig. 1E). As the onset and progression of UC correlate with defects in the intestinal barrier function26,27, we assessed barrier integrity either by ex vivo measurements of intestinal transepithelial electrical resistance (TEER) or by in vivo orogastric gavage of FITC-dextran. We observed significantly lower TEER in the proximal and distal colon regions of 12-week-old PleΔIEC compared to Plefl/fl mice. Moreover, TEER values in Plefl/fl mice were three-times higher in their distal parts than in their proximal parts; by contrast, TEER values in PleΔIEC mice were equally low in distal and proximal colon segments (Fig. 1F). Compared to Plefl/fl mice, PleΔIEC mice consistently displayed a higher penetration rate of FITC-dextran into blood already at 4 weeks, and this difference became even more apparent in older animals (Fig. 1G).
In addition, a histological inspection of hematoxylin-eosin (H&E)-stained colon sections revealed extensive translocation of luminal bacteria into PleΔIEC mucosa in 30-week-old mice (Fig. 1H). Given the hampered barrier function in the PleΔIEC intestine, we screened Plefl/fl and PleΔIEC mice for signs of inflammation. Chemiluminescence-based whole body imaging28 showed positive abdominal areas in PleΔIEC mice (Fig. 1I), which correlated strongly with significantly higher myeloperoxidase activity (MPO; Fig. 1J). Moreover, mild inflammation of the PleΔIEC colon was confirmed by increased immune cell infiltration, extent (or intensity) of acute/chronic inflammation, and lymphatic follicle size (Figs. 1K, L and S2A), and a higher percentage of edema and ulceration indicated higher epithelial damage (Fig. S2B). Together, these results suggest that plectin is critical for the maintenance of the intestinal barrier and thus could be directly linked to the onset and progression of UC.
Loss of plectin leads to hyperproliferation and aberrant differentiation of IECs
Further histological inspection of H&E-stained colonic sections revealed thickening of the colonic mucosa and significant crypt damage with excessive sloughing of IECs detached from the subjacent BM in plectin-deficient specimens (Fig. 2A). In addition, the colon of PleΔIEC mice showed a higher rate of proliferation as determined from Ki-67-stained sections (Fig. 2B). Consistently, an increase in the number of proliferating transit-amplifying IECs in the crypts of PleΔIEC animals was evident from BrdU incorporation assessed 2, 24, and 48 h after a BrdU pulse (Fig. 2C). Interestingly, TUNEL staining indicated a minimal degree of spontaneous apoptosis in both Plefl/fl and PleΔIEC mice (Fig. S3A). In parallel with the prominent hyperplasia, the PleΔIEC colon contained a higher proportion of PAS-positive goblet cells (Fig. 2D), corresponding to a higher mucus discharge (Fig. 2E). Immunolabeled PleΔIEC colonic sections also showed a lower percentage of chromogranin A (ChgA)-positive enteroendocrine cells (Fig. S3B) and an extended keratin 20 (K20)-positive zone (Fig. S3C). Similar, albeit less pronounced, trends were observed in the PleΔIEC small intestine (Fig. S4). Plectin deficiency thus results in hyperproliferation and aberrant differentiation of IECs, affecting the spatiotemporal organization of the intestinal epithelium.
Fig. 2. Plectin-deficient IECs exhibit aberrant proliferation and differentiation, resulting in altered crypt organization.

A, B Representative images of H&E staining (A) and Ki-67 immunohistochemistry (proliferating cells) (B) of Plefl/fl and PleΔIEC paraffin-embedded colon sections. Scale bar, 100 μm. Graphs show quantification of colonic crypt damage given as a percentage of crypts with >5% of IECs detached from BM (A) and percentage of the Ki-67-positive (Ki-67+) IECs per crypt (B), n = 3–4. C Histograms showing the percentage of BrdU-positive (BrdU+) cells in given positions of Plefl/fl and PleΔIEC colonic crypts at 2, 24, and 48 h after BrdU pulse. Cells were numbered sequentially from crypt base to lumen, with cell position 0 assigned to the first cell at the base of each crypt. At least nine crypts per mouse were analyzed from three mice per time point and genotype. D, E Representative images of PAS staining (goblet cells) (D) and mucin-2 (Muc2) immunofluorescence in mucus layer (E) of Plefl/fl and PleΔIEC distal colon sections (D) and colon whole mounts (E). Scale bars, 100 μm (D), and 200 μm (E). Graphs show quantification of percentage of PAS-positive (PAS+) IECs per crypt (D) and percentage of mucin-2-positive (Muc2+) area per whole mount area examined (E), n = 3–4. Data are presented as mean ± SEM, *P < 0.05, **P < 0.01, †P < 0.001.
Plectin-deficient IECs form aberrant cell junctions and disordered KF networks
As the structural and functional integrity of epithelia is secured by cell junctions2,29, we compared the morphology of cell-ECM (HDs) and cell–cell (TJs, AJs, and Ds) adhesions formed by Plefl/fl and PleΔIEC IECs, using transmission electron microscopy (TEM). A quantitative analysis of the HD size revealed an extended cross-sectional length of seemingly less electrodense HD plaques in the PleΔIEC colon; furthermore, the space between HDs and the BM was significantly dilated (Fig. 3A). Similar to HDs, we also found significantly dilated intercellular spaces of TJs, AJs, and Ds between adjacent PleΔIEC IECs (Fig. 3A). These morphological alterations coincided with generally lower expression levels of the hemidesmosomal constituents Itgα6 and Itgβ4 and the following cell–cell junctional markers: zonula occludens 1 (ZO-1; TJs), E-cadherin (E-cad; AJs), desmoglein 2 (Dsg2; Ds), and desmoplakin 1/2 (Dsp1/2; also Ds) at both mRNA and protein levels (Fig. 3B–E). These results clearly show that plectin deficiency leads to the formation of aberrant intestinal junctional complexes, which likely accounts for breached epithelial barrier integrity.
Fig. 3. Formation of aberrant cell junctions in PleΔIEC IECs.

A Representative TEM micrographs of Plefl/fl and PleΔIEC IEC junctional complexes. Braces (white) indicate hemidesmosomes (HD), tight junctions (TJ), adherens junctions (AJ), and desmosomes (Ds). Scale bar, 500 nm. Graphs show quantitative analyses of junctional complex widths (measured as the distance from IEC to BM (HD) or distance from IEC to IEC membrane (TJ, AJ, and Ds)). Five to fifteen junctions were measured (two mice per genotype). B Relative mRNA levels of integrin (Itg) α6 and β4 in scraped mucosa from Plefl/fl and PleΔIEC distal colons, n = 4–5. C Quantification of Itgβ4 and Itgα6 in scraped distal colon mucosa from Plefl/fl and PleΔIEC mice by immunoblotting. GAPDH, loading control. The graph shows relative band intensities normalized to average Plefl/fl values, n = 3. D Relative mRNA levels of ZO-1, E-cadherin (E-cad), desmoglein 2 (Dsg2), and desmoplakin 1/2 (Dsp1/2) in Plefl/fl and PleΔIEC distal colons, n = 5. E Quantification of ZO-1, E-cad, and Dsg2 in Plefl/fl and PleΔIEC colon mucosa by immunoblotting. GAPDH, loading control. The graph shows relative band intensities normalized to average Plefl/fl values, n = 3. Data are presented as mean ± SEM, n.s. not significant, *P < 0.05, **P < 0.01, †P < 0.001.
In previous studies, we showed that plectin controls cell junctions through anchorage of IF networks20,22,30. Therefore, we next compared the appearance of KFs in Plefl/fl and PleΔIEC colon sections using immunofluorescence microscopy. Although the general appearance of K8 and K19 networks did not significantly differ in the two cell types (Fig. S5), super-resolution microscopy of pan-K-labeled sections revealed less pronounced apical staining of PleΔIEC IECs (Fig. 4A). Moreover, in PleΔIEC IECs, pan-K positive filaments formed less-ordered and rather coarse meshworks, while Plefl/fl IECs displayed typical staining patterns with filaments regularly aligned along the apicobasal axis (Fig. 4A). The changes in KF network organization were not caused by altered keratin (K8, K18, and K19) expression, as no differences were found at either the mRNA or the protein level (Fig. 4B, C). No apparent abnormalities were seen in actin filament and microtubule organization (Figs. S5 and S6A).
Fig. 4. Plectin organizes KFs in IECs.
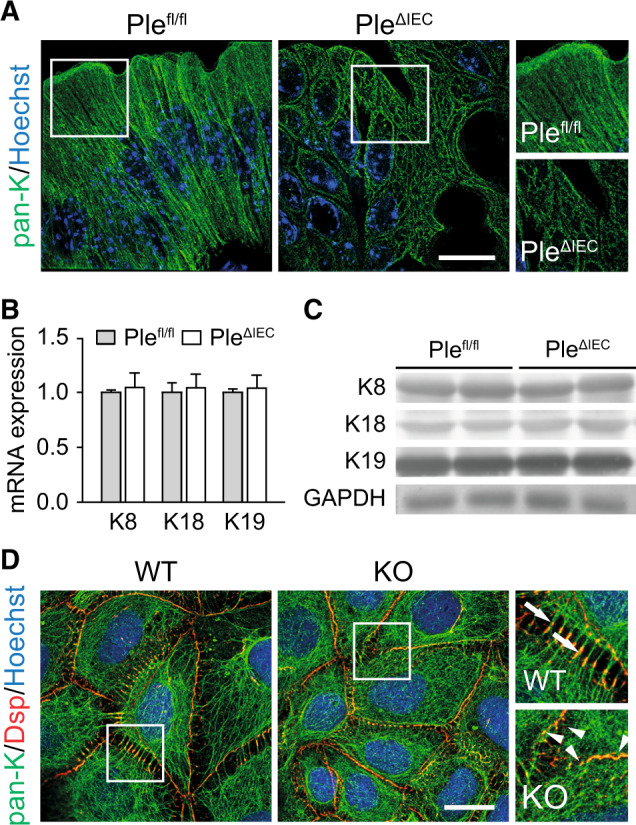
A Representative super-resolution STED images of Plefl/fl and PleΔIEC distal colon sections immunolabeled for pan-keratin (pan-K; green) with nuclei stained with Hoechst (blue). Scale bar, 10 μm. Boxed areas show ×1.3 images. B, C Relative mRNA (B) and protein (C) levels of K8, 18, and 19 in Plefl/fl and PleΔIEC distal colon, n = 3–5. Data are presented as mean ± SEM, P > 0.05 by unpaired Student t test. D Representative immunofluorescence images of WT and KO Caco-2 cell monolayer cultures immunolabeled for pan-K (green) and desmoplakin (Dsp; red). Nuclei were stained with Hoechst (blue). Arrows, straight K8 filaments anchored to Dsp-positive desmosomes; arrowheads, tangled K8 filaments. Scale bar, 20 μm. Boxed areas show ×2.5 images.
Aberrant KF cytoarchitecture was also clearly discernible in pan-K immunolabeled monolayers of plectin-deficient (KO) human IECs (Caco-2). To mimick the in vivo situation, mature differentiated Caco-2 cells 16 days after the confluency were used. In wild-type (WT) cells, the KF network was densely packed around the cell center, from which individual KFs extended towards the cell periphery delineated by clearly defined desmoplakin-positive Ds (Fig. 4D). In contrast, KO cells showed tangled KFs, which were evenly distributed throughout the cytoplasm and seemingly overlapped with rather continuous desmoplakin-positive structures at the cell–cell borders (Fig. 4D). Similar to PleΔIEC IECs, actin organization in KO cells appeared inconspicuous (Fig. S6B). Collectively, these findings indicate that plectin ablation in IECs results in altered keratin network organization and aberrant KF anchorage to desmosomal junctions.
Plectin preserves intestinal epithelial integrity through HD stabilization
Plectin-mediated attachment of the keratin network to Itgα6β4-containing HDs plays a crucial role in stabilizing the adhesion of keratinocytes to the matrix and hence imparts mechanical stability to the skin20,31. To examine whether the PleΔIEC intestine phenotypically follows the same paradigm, we scrutinized colon and small intestine sections immunolabeled for K8 and Itgα6 (Fig. 5A) or collagen (Col) IV (Fig. S7). In line with the observations from H&E- and Sirius red-stained colon sections (Figs. 2A and S7), PleΔIEC IECs partially lost their polarized orientation (Fig. S8A–C); they were misaligned and largely detached from the BM at the luminal surface of the crypts (Fig. 5A, upper panels). Despite a partial loss of apicobasal polarity of PleΔIEC IECs (Fig. S8A–C), the epithelium retained a characteristic polarized distribution of the apical markers villin and ezrin (Fig. S8D, E). The extensive detachment of PleΔIEC IECs was even more apparent in the small intestine, where we often found the whole epithelial sheet physically separated from underlying structures (Fig. 5A, lower panels). Remarkably, in both the PleΔIEC colon and the small intestine, Itgα6-positive patches remained confined to the BM, while detached IECs were entirely devoid of Itgα6 signals. Thus we conclude that plectin ablation abrogates the functional link between KFs and HDs.
Fig. 5. Plectin stabilizes IEC hemidesmosomes through KF recruitment.

A Representative immunofluorescence images of Plefl/fl and PleΔIEC distal colon (upper panels) and small intestine (lower panels) sections immunolabeled for K8 (green) and Itgα6 (red); Hoechst-stained nuclei (blue). Arrowheads, Itgα6-positive clusters. Scale bar, 25 μm. Boxed areas show ×1.5 images. Drawn schematics depict aligned, BM-attached Plefl/fl IECs (upper panel) and mislocalized, detaching PleΔIEC IECs (lower panel). B Cell lysates, cytosol fractions, and keratin-enriched high salt extracts (HSE) were prepared from WT and KO Caco-2 cells and subjected to immunoblotting with antibodies to Itgβ4 and K8. GAPDH, loading control. Graphs show relative band intensities normalized to average Plefl/fl values, n = 4–6. C Viability of WT and KO Caco-2 cells exposed to uniaxial cyclic stretch presented as a percentage of dead (PI-positive; PI+) cells, n = 9–11. D Quantification of WT and KO Caco-2 cell viability (left) and adhesion (right) under radial shear flow shown as a percentage of dead and detached cells, respectively, n = 6. Boxplot data represent median, 25th, and 75th percentile with whiskers reaching the last data point. E Adhesion strength between ECM-coated superparamagnetic beads and WT and KO Caco-2 cells was quantified using magnetic tweezers that generated forces ramps at a speed of 1 nN/s up to a maximum force of 15 nN. Image and schematic depict magnetic tweezer setup. Arrowhead, paramagnetic bead; asterisk, magnetic tweezer tip; dotted circular line, cell border. Scale bar, 20 μm. The graph shows the percentage of beads (n = 103 WT, 109 KO cells) that remained adherent at a given pulling force. The boxplot shows the distribution of the median detachment force (calculated from bootstrapping by sampling with replacement, n = 1000 runs) and its distribution (25th, and 75th percentile with whiskers reaching the minimum and maximum sampled values). Bar graph data in all other subplots represent mean ± SEM, n.s. not significant, *P < 0.05, †P < 0.001.
To address the effects of plectin ablation biochemically, we prepared keratin-enriched cell fractions19 from human WT and KO IEC Caco-2 lines and compared their integrin (Itg) content by immunoblotting using antibodies to Itgβ4. As expected, such cell fractions were highly enriched in keratins 8 (Fig. 5B), 18, and 19 (not shown). Although Itgβ4 levels were comparable in cell lysates, the Itgβ4 content of insoluble keratin fractions was significantly reduced in KO cells compared to WT cells (Fig. 5B). These observations correlated well with the histological data (Fig. 5A).
To assess whether plectin deficiency affects the biomechanical properties of IECs, we performed a series of quantitative assays with human IEC lines Caco-2 and hCC. Monitoring cell viability under mechanical stress on a stretched flexible membrane (uniaxial cyclic stretch) revealed the higher mechanical vulnerability of both KO cell lines, as the proportion of PI-positive (dead) cells significantly increased with stretch amplitudes ranging from 10% to 50% (Figs. 5C and S9A). Reduced mechanical resistance of KO cells was confirmed by fluid shear stress assay using a spinning disc device. When exposed to constant radial flow, KO cells displayed death rates about twice as high as that of their WT counterparts (Figs. 5D and S9B). Moreover, the fraction of detached Caco-2 (but not hCC) cells was higher for KO than WT cells. This suggests that plectin ablation weakens their adhesion to the underlying substratum.
To confirm this hypothesis, we quantified adhesion strength between ECM-coated superparamagnetic beads and cell adhesions using magnetic tweezers. We applied increasing forces of up to 15 nN to magnetic beads (force increase at 1 nN/s) and recorded the force at which each bead detached from the cell. From a total of >100 detachment events for each cell type, we calculated the cumulative detachment probability as a function of pulling force and report the force at which 50% of the beads detached from the cells (Figs. 5E and S9C). We measured lower detachment forces in both Caco-2 and hCC KO compared to WT cells, which confirms our hypothesis of weaker Itg-mediated adhesions in plectin-deficient cells. Hence, like for skin type I HDs20, plectin loss is deleterious for the stability of type II HDs present in the intestine, leading to compromised mechanical resilience of IECs and intestinal epithelia.
IEC-specific plectin deficiency exacerbates experimental colitis
The spontaneous colitic phenotype in PleΔIEC mice (Fig. 1) suggests that plectin deletion can contribute substantially to the pathogenesis of UC. To assess whether loss of plectin increases the susceptibility to colitis, we induced experimental colitis in Plefl/fl and PleΔIEC mice. Even a short exposure (3–4 days) to low DSS doses (1.5–2%) resulted in a dramatic bodyweight loss of PleΔIEC mice, in sharp contrast to similarly treated Plefl/fl which experienced only insignificant weight losses (Fig. 6A). The weight loss of mutant mice was associated with a higher disease activity index (DAI; Fig. 6A), a decreased survival rate (not shown), and a significant reduction in colon length (Fig. 6B). The severity of induced colitis in PleΔIEC mice coincided with larger inflamed areas at days 4 and 6 after the initiation of DSS-treatment (Fig. 6C), corresponding to a higher influx of MPO-positive neutrophils and intestinal epithelial injury. Further, histological evaluation of “Swiss rolls” of the entire colon confirmed these results and revealed clear signs of inflammation and epithelial damage in all DSS-treated animals. However, large regions with heavy ulceration, crypt damage, and inflammatory response in PleΔIEC mice were in striking contrast to fewer lesions in Plefl/fl mice (Figs. 6D and S10A).
Fig. 6. PleΔIEC mice are more susceptible to DSS-induced colitis.

A Relative bodyweight and disease activity index (DAI) of untreated and DSS-treated Plefl/fl and PleΔIEC mice during experimental colitis. Four to seven mice per genotype and time point were analyzed. B Representative images of colon and caecum of DSS-treated Plefl/fl and PleΔIEC mice. The graph shows colon length, n = 4–6. C In vivo chemiluminescence images and signal quantification (graph) of DSS-treated Plefl/fl and PleΔIEC mice injected with the myeloperoxidase substrate luminol on days 4 and 6 of DSS treatment, n = 3–4. D Representative hematoxylin-stained sections of Swiss roll mounts from untreated (control) and DSS-treated (DSS) mice. Scale bars, 2 mm, magnified boxed areas, 100 μm. Insets, outlines of lesions (in red) distributed along mucosa (black lines) in corresponding panels. Graphs show quantification of colonic tissue damage given as the percentage of ulceration and crypt damage, n = 3, E, F Fecal microbiota beta diversity in 4-, 12-, and 20-week-old untreated Plefl/fl and PleΔIEC mice as determined by 16S rDNA sequencing. Principal coordinate analysis plot (E), constructed with unweighted UniFrac distance metric, shows clustering of microbial beta diversity. PC1, PC2, and PC3 represent the top three principal coordinates that captured most of the diversity (given as a percentage). Global composition (F) of bacterial microbiota at phyla level shown as relative operational taxonomic unit (OTUs) abundance per time point and genotype, n = 4–6. Data are presented as mean ± SEM, *P < 0.05, **P < 0.01, †P < 0.001.
Since gut microbial dysbiosis is a typical finding in UC patients32–34, we compared the composition of fecal microbiota in unchallenged Plefl/fl and PleΔIEC mice at the ages of 4, 12, and 20 weeks. Surprisingly, despite the impaired intestinal barrier and concomitant inflammation phenotype of PleΔIEC mice (Fig. 1F–L), we observed no significant differences in alpha (Fig. S10B) and beta (Fig. 6E) diversities between both genotypes. In all animals, bacterial microbiota was dominated by bacteria belonging to families S24-7 (bacteroidetes), lactobacillaceae (firmicutes), and lachnospiraceae (firmicutes) (Fig. 6F). Together, these data show higher susceptibility of PleΔIEC mice to DSS-induced colitis, accompanied by severe epithelial damage and inflammation in the absence of microbial dysbiosis.
Reduced mechanical stability of epithelia accounts for intestinal injury in PleΔIEC mice
To identify the onset and time course of intestinal injury in PleΔIEC mice, we assessed intestinal epithelial damage scores in newborn, 21-day-old, and 12-week-old mice (Figs. S11A and 7A–C). While newborn mice were histologically inconspicuous, the colon and the small intestine displayed first signs of damage in 21-day-old PleΔIEC mice (see also Fig. S12B), which coincided with weaning and transition to solid chow. The onset of the epithelial breach was accompanied by the subsequent development of inflammatory response as shown by increased immune cell infiltration, extent (or intensity) of inflammation, and lymphatic follicle number/size (Fig. S12A).
Fig. 7. Intestinal epithelial damage in PleΔIEC mice results from mechanical stress.

A–C Plefl/fl and PleΔIEC mice were sacrificed on postnatal day 0 (P0), postnatal day 21 (P21), and at 12 weeks (12w) of age, and epithelial damage scores were assessed from colon and small intestine sections. Schematic illustrates the experimental setup (A). Solid, transition to solid chow at P21. Graphs show quantification of epithelial damage in the colon (B) and small intestine (C) at the age indicated. D–F Nine-week-old Plefl/fl and PleΔIEC- ERT2 mice were either kept on solid chow or provided with a liquid diet for 14 days. Plectin inactivation was induced by three consecutive applications of tamoxifen (TMX) on days 6, 8, and 10; mice were sacrificed on day 14. The schematic illustrates the experimental setup (D). Solid, transition to solid chow at P21; arrows, TMX application; red bar, period on a liquid diet. Graphs show quantification of epithelial damage in the colon (E) and small intestine (F) on solid chow and liquid diet. G–I Nine-week-old Plefl/fl and PleΔIEC-ERT2 mice were kept either untreated or treated with broad-spectrum antibiotics. Plectin inactivation and sample collection were identical to (B). Schematics illustrate experimental setup (G). Chow, the transition to solid chow at P21; arrows, TMX application; red bar, period of antibiotics (ATB) treatment. Graphs show quantification of epithelial damage in the colon (H) and small intestine (I) on solid chow and liquid diet. Data are presented as mean ± SEM, n.s. not significant, *P < 0.05, **P < 0.01, †P < 0.001.
To gain better control over plectin inactivation timing, we generated TMX-inducible IEC-specific plectin knockout (PleΔIEC-ERT2) mice. Three consecutive applications of TMX in 9-week-old PleΔIEC-ERT2 mice resulted in recombination efficiency comparable to that of constitutive PleΔIEC mice, and a distinctive intestinal phenotype developed as early as 5 days post-TMX administration (not shown). To determine the effect of a diet change on the intestinal injury, PleΔIEC-ERT2 and Plefl/fl control mice were either kept on solid chow or provided with a low-residue liquid diet 6 days before TMX administration. The liquid diet significantly attenuated epithelial damage in the colon of PleΔIEC-ERT2 mice; however, the histological score indicates more severe injury of the small intestine (Figs. 7D–F and S11B). The beneficial effects of the liquid diet also manifested as less prominent colon swelling (not shown).
As previous studies linked the severity of colitis and intestinal injury with commensal microbiota8,35, next we treated Plefl/fl and PleΔIEC-ERT2 mice with well-established broad-spectrum antibiotics8. In our experimental setup, the apparent milder colitic phenotype consistently coincided with lower epithelial damage in the colon of PleΔIEC-ERT2 mice. This treatment however did not affect epithelial injury of the PleΔIEC-ERT2 small intestine (Figs. 7G–I and S11C). Collectively, these data support the notion that the increased susceptibility of the plectin-deficient intestinal epithelium to mechanical strain impinged by luminal content is caused by a lack of KF attachment to Itg clusters and destabilization of intestinal HDs. Further, the fact that antibiotics also partially alleviate epithelial damage suggests that luminal bacteria significantly contribute to intestinal injury in PleΔIEC-ERT2 mice.
Discussion
The intestinal epithelium faces substantial mechanical stress36, which is inextricably linked to gut physiology. Although several studies suggest the importance of intestinal KF networks35,37,38 and KF-associated cell junctions (Ds and HDs)8,9 for protection against intestinal inflammation and CRC, the contribution of altered epithelial mechanics to observed phenotypes remain unexplored. Here, we focus on the role of KF-cell junction linker plectin in the maintenance of intestinal homeostasis, and we provide a comprehensive analysis of molecular mechanisms governing the mechanical stability of intestinal epithelia.
The most notable phenotype of both plectin-deficient mouse models (PleΔIEC and PleΔIEC-ERT2) is the detachment of IECs from the underlying BM, resulting in extensive epithelial injury and eventually in the spontaneous development of a colitic phenotype. Strikingly, this is accompanied by loss of the hemidesmosomal ECM receptor Itgα6 from detached IECs, while Itgα6 patches remain on a collagen-stained BM, likely indicating their inefficient linkage to cytoskeletal structures. Indeed, our TEM analysis revealed that less electrodense HDs formed by PleΔIEC IECs were somewhat elongated, and gaps between HD plaques and the BM were significantly wider compared to Plefl/fl IECs. The formation of morphologically abnormal HDs was paralleled with reduced expression levels of both HD-forming integrins (α6 and β4) in the PleΔIEC mucosa. Moreover, the content of Itgβ4 was also significantly diminished in keratin-enriched fractions prepared from plectin-deficient IECs, suggesting the reduced association of Itgα6β4 complexes with intestinal keratins (K8, 18, and 19).
Our observations are concordant with a recently published model for skin type I HD10, which proposed that plectin (along with BPAG, another plakin family member) fortifies HD plaques both horizontally (by a lateral association of Itgβ4) and vertically (by interlinking Itgβ4 with KFs). Accordingly, ablation of plectin, the only plakin present in intestinal-type II HD39, would fully abrogate a functional link between KFs and HDs and would result in their overall destabilization. In line with this hypothesis, we in vitro show a trend towards higher detachment (paralleled with a higher death rate) of plectin-deficient IECs exposed to a uniaxial cyclic stretch and a constant radial flow compared to their WT counterparts. As both Plefl/fl and PleΔIEC IECs display a minimal degree of spontaneous apoptosis in vivo, the observed excessive cell death in our experiments in vitro can likely be attributed to incomparable force magnitude and cell context under these two conditions. Consistent with our results from cell stretching and radial shear assays, we also determine significantly lower adhesion strength between ECM-coated superparamagnetic beads and plectin-deficient IECs using magnetic tweezers. Hence, by combining in vivo and in vitro approaches, we provide evidence that plectin is essential for the stability of intestinal HD type II, a structure preventing colitis8 and presumably also the risk of colitis-associated CRC8,40,41.
Previous studies demonstrated that the deletion of plectin has adverse effects on the formation of intercellular junctions, with consequences for the epithelial barrier function21,22. It has been shown that plectin-deficient cholangiocytes form dysfunctional Ds and fail to upregulate some desmosomal proteins, such as desmoplakin, a putative binding partner of plectin42, in response to bile stasis22. This failure results in mechanical weakening of the biliary epithelium and contributes to plectin-related familial intrahepatic cholestasis43. In terms of mechanistic parallels between plectin-deficient biliary and intestinal epithelia, we found that apart from destabilizing HDs, plectin deficiency also leads to the prominent broadening of Ds, AJs, and TJs. Moreover, PleΔIEC IECs exhibit downregulation of corresponding junctional constituents (ZO-1, E-cad, Dsg2, and Dsp1/2). Although the resulting dilatation of intercellular spaces would per se suffice to explain the observed increase in intestinal permeability and bacterial penetration, the “leaky gut” in PleΔIEC mice seems ultimately rooted in the less firm IEC/BM connection, given the extent of IEC detachment. On the other hand, proper anchorage of KFs (determining cell mechanics) to Ds (ensuring intercellular cohesion) is known to provide load-bearing tissues with mechanical stability11. Showing altered KF cytoarchitecture and aberrant D formation in both in vitro plectin-deficient IEC systems and PleΔIEC mice, our results suggest that D-keratin complex abnormality substantially contributes to the compromised mechanics of the PleΔIEC intestinal epithelium.
We propose that the lack of functional plectin at HDs (in combination with its effects on KFs and Ds) and the resulting mechanical epithelial fragility favor an impaired intestinal barrier function and are ultimately responsible for colitis. Importantly, comparable mucosal deterioration was observed upon plectin ablation during development and in the adult intestine with its fully mature immune system. Furthermore, we also demonstrate that loss of plectin can exacerbate experimental colitis in mice. Consistent with these results, lower expression levels of plectin correlate with UC development in human patients, suggesting that defects in cytoskeleton coordination mediated through plectin contribute to IBD pathogenesis in humans by affecting IEC/BM adhesion, IEC cohesion, and mechanical properties. However, it is well recognized that properly organized KF networks38,44, HDs8, and intercellular junctions2,3,9 exert numerous non-mechanical functions, providing the intestinal epithelium with protection against microbial infection and uncontrolled inflammation. Our data do not rule out similar functions in the PleΔIEC intestine. Further studies will be required to investigate how plectin deficiency affects cell-autonomous (barrier function-independent) mechanisms involved in the interplay between IECs, gut microbiota, and immune cells.
We observed a prominent hyperproliferation of plectin-deficient IECs paralleled by a dramatically higher proportion of PAS-positive goblet cells. This phenotype closely resembles the situation in mice lacking hemidesmosomal α6 integrin8 or K845,46. Together, these findings suggest that the keratin/plectin/integrin axis is essential for balanced proliferation and differentiation of IECs. Interestingly, intestinal K8 and K18 were shown to promote Notch1 signaling, a major pathway of colonic cell fate specification46. Since Notch-mediated signal transduction depends on cytoskeletal tension47,48, it is tempting to speculate that intact plectin-controlled KF cytoarchitecture facilitates mechano-regulation of intestinal cell fate. Moreover, plectin anchors the cytoskeleton to the nuclear envelope via interaction with nesprin-349 and mediates transmission of mechanical stimuli directly to the nucleus. Multiple studies provide evidence that loss of plectin results in nuclear phenotypes, including altered nuclear positioning22, nuclear deformations50,51, chromatin modification, and gene expression51. To elucidate whether and how plectin regulates specific transcriptional programs in IECs and to find their contribution to the proper spatiotemporal proliferation/differentiation pattern within colonic crypts is a goal of our ongoing studies.
The rapid deterioration of the PleΔIEC intestinal mucosa following weaning (i.e., a switch to a solid diet and the amplification of muscle contractions) indicates that the origin of colitis in the absence of plectin is primarily associated with a reduced capacity of IECs to resist mechanical stress. In addition, the most severe epithelial injury was found in the distal colon, which is the region most intensely subjected to such stress. Comparably devastating epithelial instability has been well documented for epidermal layers in EB patients15,16. As there is no causal therapy for EB available15, the current treatment focuses mainly on the prevention of tissue destruction. Following the same rationale, we demonstrate that a low-residue liquid diet significantly attenuates colonic epithelial damage, thus protecting its barrier function. Surprisingly, this approach aggravates IEC detachment in the PleΔIEC small intestine, which might suggest augmented susceptibility of the small intestine to plectin loss. Therefore, future studies should investigate whether differential expression of plectin along the gastrointestinal tract might have an impact on regional differences in disease manifestations in patients. In line with the previous observations8,35, antibiotic treatments markedly decrease not only mucosal inflammation but, intriguingly, also epithelial damage, which implies that host-microbiota interactions contribute to excessive IEC sloughing in the PleΔIEC intestine. Although dietary factors can likely ameliorate only less extensive trauma, our results suggest that a low-residue liquid diet combined with antibiotic treatment might be a useful palliative modality. To translate our findings into clinical medicine: it remains to be determined whether such a strategy (i) is suitable for long-term treatment and (ii) is effective with respect to systemic disease manifestation.
Methods
Patients
Colon biopsy samples were collected from patients diagnosed with UC (n = 97) and from healthy controls (n = 20) admitted to the Hepatogastroenterology Department at the Institute for Clinical and Experimental Medicine (Prague, Czech Republic) for a colonoscopy from July 2016 to May 2019. Subjects were assigned to the healthy control group only after all clinical examinations excluded any signs of autoimmune disease, inflammatory disease, and colon cancer. All UC patients with concurrent primary sclerosing cholangitis (PSC) were excluded from the study. Endoscopic UC activity at the time of a standard optical colonoscopy was categorized according to the Mayo endoscopic subscore and confirmed by histology examinations of the grade of inflammation. Clinical characteristics of patients are shown in Supplementary Table S1. Standard endoscopic biopsies were extracted from the inflamed non-dysplastic mucosa of the left colon (rectum) and immediately placed in an RNAlater solution. Total RNA was extracted according to the manufacturer’s instructions.
Mice
Plectinflox/flox (Plefl/fl) mice23 were crossed with villin-Cre transgenic mice (MGI 2448639) to generate Plefl/fl/villin-Cre mice (PleΔIEC) and with villin-creERT2 transgenic mice (MGI 3053826; both Cre strains were kindly provided by S. Robine52) to generate Plefl/fl/villin-creERT2 mice (PleΔIEC-ERT2). Age-matched littermate male mice were used in all experiments. Unless stated otherwise, mice were 12–14 weeks old. Animals were housed under specific pathogen-free conditions with regular access to chow and drinking water and a 12 h light/12 h dark regime.
Cells and CRISPR-mediated targeting of plectin
Caco-2 cells were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 20% fetal bovine serum (FBS) in a 5% CO2/air humidified atmosphere at 37 °C. Human colonic cells (hCC; T0570, Applied biological materials, Inc.) were cultured in DMEM supplemented with 10% FBS in 5% CO2/air humidified atmosphere at 37 °C. Plectin knockout (KO) cell lines were generated by targeting genomic sequences of intron 25–26 and exon 31 of Plectin using CRISPR/Cas9 plasmid pX330 Cas9-Venus (a kind gift of B. Schuster, IMG CAS, Prague, Czech Republic) as described previously22. The potential off-target sites were predicted using CRISPOR (http://crispor.tefor.net/). The four top-ranking potential off-target sites for each guide RNA were selected for validation. The genomic DNA sequences surrounding the potential off-target sites were amplified by PCR using gene-specific primers (Supplementary Table 3). PCR products were analyzed by direct sequencing (Figs. S12 and S13).
Statistics
All results are presented as mean ± SEM. All normally distributed parametric data were analyzed by two-tailed unpaired Student t test. Comparisons of multiple groups to controls were performed using two-tailed one-way ANOVA. Comparisons of frequency distributions of BrdU-positive cells were analyzed with Mann–Whitney test. Survival curves were analyzed by Mantel–Cox test. Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA). Comparisons of detachment forces were done with bootstrapping (sampling with replacement) with 1000 replicates. Statistical significance was determined at the levels of *P < 0.05, **P < 0.01, †P < 0.001; n values are specified in the figure legends.
Study approval
This study was approved by the Ethics Committee of the Institute for Clinical and Experimental Medicine and Thomayer Hospital with Multi-Center Competence (G16-06-25). Written informed consent was obtained from all subjects before the study. All animal studies were performed in accordance with European Directive 2010/63/EU and were approved by the Czech Central Commission for Animal Welfare (48/2014 and 23/2020).
Supplementary information
Acknowledgements
We would like to thank L. Macurek (IMG CAS, Prague) for generously providing the hCC cell line, S. Robine (Institut Curie, Paris) for Cre mouse lines, and B.Schuster (IMG CAS, Prague) for pX330 Cas9-Venus plasmid; H. Havelkova (IMG CAS, Prague) and I. Muricova (Institute of Physiology CAS, Prague) for their outstanding technical assistance; S. Reipert (University of Vienna, Vienna), M. Efenbergova, J. Tureckova (both IMG CAS, Prague), and Z. Jiraskova (Institute of Microbiology CAS, Prague) for their expertize; and T. Epp, M. Prechova (both IMG CAS, Prague) and A.I. Ivanov (Lerner Research Institute of Cleveland Clinic Foundation, Cleveland) for critical reading of the manuscript. We acknowledge the Light and Electron Microscopy Core Facilities, IMG CAS, Prague, Czech Republic, for their support with advanced microscopy imaging.
Author contributions
Study concept and design: M.G.; acquisition of data: A.K., P.B., L.S., G.O.-E., G.K., J. Prochazka, M.S., C.D., Z.S., M.V., N.Z.; analysis and interpretation of data: A.K., M.G., J.S., E.S., L.B., J. Pacha, J. Prochazka, D.J., M.K., B.F.; drafting of the manuscript: A.K, M.G.; critical revision of the manuscript for important intellectual content: all authors. Funding: M.G., M.K., G.W., B.F. technical and material support: P.W., L.B., V.K., R.S., G.W.
Funding
This work was supported by the Grant Agency of the Ministry of Health of the Czech Republic (17-31538A); the Czech Academy of Sciences (L200521601; postdoctoral fellowship to AK); the Grant Agency of the Czech Republic (17-09869S and 20-16520Y); the Institutional Research Project of the Czech Academy of Sciences (RVO 68378050); COST Action CA15214-EuroCellNet, Strategy AV21—QUALITAS; the Grant Agency of Charles University (192119); TACR GAMA 2 (TP01010031); MH CZ—DRO (Institute for Clinical and Experimental Medicine—IKEM, IN 00023001); MEYS CR projects (LQ1604 NPU II, LTC17063, LM2015062, LO1419, and LM2015040); MEYS CR/ERDF projects (OP RDI CZ.1.05/2.1.00/19.0395 and CZ.1.05/1.1.00/02.0109); MEYS CR/ESIF project OP RDE CZ.02.1.01/0.0/0.0/16_013/0001775; the Operational Program Prague–Competitiveness project (CZ.2.16/3.1.00/21547); the German Science Foundation (DFG FA 336/12-1); and the Austrian Science Research Fund (FWF grant I413-B09).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Alzbeta Krausova, Petra Buresova.
Change history
10/22/2021
A Correction to this paper has been published: 10.1038/s41385-021-00463-x
Change history
3/17/2022
A Correction to this paper has been published: 10.1038/s41385-022-00501-2
Supplementary information
The online version contains supplementary material available at 10.1038/s41385-021-00380-z.
References
- 1.Camilleri M. Leaky gut: mechanisms, measurement and clinical implications in humans. Gut. 2019;68:1516–1526. doi: 10.1136/gutjnl-2019-318427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front. Immunol. 2013;4:1–22. doi: 10.3389/fimmu.2013.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014;14:141–153. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- 4.Consortium UIG, et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat. Genet. 2009;41:1330–1334. doi: 10.1038/ng.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGovern DP, et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet. 2010;42:332–337. doi: 10.1038/ng.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson CA, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011;43:246–252. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia MA, Nelson WJ, Chavez N. Cell–cell junctions organize structural and signaling networks. Cold Spring Harb. Perspect. Biol. 2018;10:1–27. doi: 10.1101/cshperspect.a029181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Arcangelis A, et al. Hemidesmosome integrity protects the colon against colitis and colorectal cancer. Gut. 2017;66:1748–1760. doi: 10.1136/gutjnl-2015-310847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gross A, et al. Desmoglein 2, but not desmocollin 2, protects intestinal epithelia from injury. Mucosal Immunol. 2018;11:1630–1639. doi: 10.1038/s41385-018-0062-z. [DOI] [PubMed] [Google Scholar]
- 10.Walko G, Castanon MJ, Wiche G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2015;360:529–544. doi: 10.1007/s00441-015-2216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hatzfeld M, Keil R, Magin TM, Magin TM. Desmosomes and intermediate filaments: their consequences for tissue mechanics. Cold Spring Harb. Perspect. Biol. 2017;9:1–20. doi: 10.1101/cshperspect.a029157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruhrberg C, Watt FM. The plakin family: versatile organizers of cytoskeletal architecture. Curr. Opin. Genet. Dev. 1997;7:392–397. doi: 10.1016/s0959-437x(97)80154-2. [DOI] [PubMed] [Google Scholar]
- 13.Wiche G, Osmanagic-Myers S, Castanon MJ. Networking and anchoring through plectin: a key to IF functionality and mechanotransduction. Curr. Opin. Cell Biol. 2015;32:21–29. doi: 10.1016/j.ceb.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Rezniczek GA, Walko G, Wiche G. Plectin gene defects lead to various forms of epidermolysis bullosa simplex. Dermatol. Clin. 2010;28:33–41. doi: 10.1016/j.det.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 15.Uitto J, Bruckner-Tuderman L, McGrath JA, Riedl R, Robinson C. EB2017-progress in epidermolysis bullosa research toward treatment and cure. J. Investig. Dermatol. 2018;138:1010–1016. doi: 10.1016/j.jid.2017.12.016. [DOI] [PubMed] [Google Scholar]
- 16.Has C, Fischer J. Inherited epidermolysis bullosa: new diagnostics and new clinical phenotypes. Exp. Dermatol. 2018;28:1146–1152. doi: 10.1111/exd.13668. [DOI] [PubMed] [Google Scholar]
- 17.Smith PK, et al. Epidermolysis bullosa and severe ulcerative colitis in an infant. J. Pediatr. 1993;122:600–603. doi: 10.1016/s0022-3476(05)83545-0. [DOI] [PubMed] [Google Scholar]
- 18.Freeman EB, et al. Gastrointestinal complications of epidermolysis bullosa in children. Br. J. Dermatol. 2008;158:1308–1314. doi: 10.1111/j.1365-2133.2008.08507.x. [DOI] [PubMed] [Google Scholar]
- 19.Osmanagic-Myers S, et al. Plectin-controlled keratin cytoarchitecture affects MAP kinases involved in cellular stress response and migration. J. Cell Biol. 2006;174:557–568. doi: 10.1083/jcb.200605172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walko G, et al. Targeted proteolysis of plectin isoform 1a accounts for hemidesmosome dysfunction in mice mimicking the dominant skin blistering disease EBS-Ogna. PLoS Genet. 2011;7:e1002396. doi: 10.1371/journal.pgen.1002396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Osmanagic-Myers S, et al. Plectin reinforces vascular integrity by mediating crosstalk between the vimentin and the actin networks. J. Cell Sci. 2015;128:4138–4150. doi: 10.1242/jcs.172056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jirouskova M, et al. Plectin controls biliary tree architecture and stability in cholestasis. J. Hepatol. 2018;68:1006–1017. doi: 10.1016/j.jhep.2017.12.011. [DOI] [PubMed] [Google Scholar]
- 23.Ackerl R, et al. Conditional targeting of plectin in prenatal and adult mouse stratified epithelia causes keratinocyte fragility and lesional epidermal barrier defects. J. Cell Sci. 2007;120:2435–2443. doi: 10.1242/jcs.004481. [DOI] [PubMed] [Google Scholar]
- 24.Huang BL, Chandra S, Shih DQ. Skin manifestations of inflammatory bowel disease. Front. Physiol. 2012;3:13. doi: 10.3389/fphys.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiche G, Krepler R, Artlieb U, Pytela R, Denk H. Occurrence and immunolocalization of plectin in tissues. J. Cell Biol. 1983;97:887–901. doi: 10.1083/jcb.97.3.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kiesslich R, et al. Local barrier dysfunction identified by confocal laser endomicroscopy predicts relapse in inflammatory bowel disease. Gut. 2012;61:1146–1153. doi: 10.1136/gutjnl-2011-300695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang J, et al. Impaired intestinal permeability contributes to ongoing bowel symptoms in patients with inflammatory bowel disease and mucosal healing. Gastroenterology. 2017;153:723–731 e721. doi: 10.1053/j.gastro.2017.05.056. [DOI] [PubMed] [Google Scholar]
- 28.Brauer R, et al. MMP-19 deficiency causes aggravation of colitis due to defects in innate immune cell function. Mucosal Immunol. 2015;9:974–985. doi: 10.1038/mi.2015.117. [DOI] [PubMed] [Google Scholar]
- 29.Luissint AC, Parkos CA, Nusrat A. Inflammation and the intestinal barrier: leukocyte-epithelial cell interactions, cell junction remodeling, and mucosal repair. Gastroenterology. 2016;151:616–632. doi: 10.1053/j.gastro.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gregor M, et al. Mechanosensing through focal adhesion-anchored intermediate filaments. FASEB J. 2014;28:715–729. doi: 10.1096/fj.13-231829. [DOI] [PubMed] [Google Scholar]
- 31.Kostan J, Gregor M, Walko G, Wiche G. Plectin isoform-dependent regulation of keratin-integrin {alpha}6{beta}4 anchorage via Ca2+/calmodulin. J. Biol. Chem. 2009;284:18525–18536. doi: 10.1074/jbc.M109.008474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pr. Gastroenterol. Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 33.Pascal V, et al. A microbial signature for Crohn’s disease. Gut. 2017;66:813–822. doi: 10.1136/gutjnl-2016-313235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bajer L, et al. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017;23:4548–4558. doi: 10.3748/wjg.v23.i25.4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Habtezion A, et al. Absence of keratin 8 confers a paradoxical microflora-dependent resistance to apoptosis in the colon. Proc. Natl. Acad. Sci. USA. 2011;108:1445–1450. doi: 10.1073/pnas.1010833108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gayer CP, Basson MD. The effects of mechanical forces on intestinal physiology and pathology. Cell Signal. 2009;21:1237–1244. doi: 10.1016/j.cellsig.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baribault H, Penner J, Iozzo RV, Wilson-Heiner M. Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev. 1994;8:2964–2973. doi: 10.1101/gad.8.24.2964. [DOI] [PubMed] [Google Scholar]
- 38.Habtezion A, Toivola DM, Butcher EC, Omary MB. Keratin-8-deficient mice develop chronic spontaneous Th2 colitis amenable to antibiotic treatment. J. Cell Sci. 2005;118:1971–1980. doi: 10.1242/jcs.02316. [DOI] [PubMed] [Google Scholar]
- 39.Litjens SH, de Pereda JM, Sonnenberg A. Current insights into the formation and breakdown of hemidesmosomes. Trends Cell Biol. 2006;16:376–383. doi: 10.1016/j.tcb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 40.Beaulieu JF. Integrin I+/-6 variants and colorectal cancer. Gut. 2018;67:1747–1748. doi: 10.1136/gutjnl-2017-315415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Arcangelis A, Chamaillard M, Simon-Assmann P, Labouesse M. Integrin a6 loss promotes colitis-associated colorectal cancer. Response to: “Integrin a6 variants and colorectal cancer” by Beaulieu JF. Gut. 2018;67:2227–2228. doi: 10.1136/gutjnl-2017-315727. [DOI] [PubMed] [Google Scholar]
- 42.Eger A, Stockinger A, Wiche G, Foisner R. Polarisation-dependent association of plectin with desmoplakin and the lateral submembrane skeleton in MDCK cells. J. Cell Sci. 1997;110:1307–1316. doi: 10.1242/jcs.110.11.1307. [DOI] [PubMed] [Google Scholar]
- 43.Wu SH. Plectin mutations in progressive familial intrahepatic cholestasis. Hepatology. 2019;70:2221–2224. doi: 10.1002/hep.30841. [DOI] [PubMed] [Google Scholar]
- 44.Geisler F, Leube RE. Epithelial intermediate filaments: guardians against microbial infection? Cells. 2016;5:1–18. doi: 10.3390/cells5030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toivola DM, Krishnan S, Binder HJ, Singh SK, Omary MB. Keratins modulate colonocyte electrolyte transport via protein mistargeting. J. Cell Biol. 2004;164:911–921. doi: 10.1083/jcb.200308103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lahdeniemi IAK, et al. Keratins regulate colonic epithelial cell differentiation through the Notch1 signalling pathway. Cell Death Differ. 2017;24:984–996. doi: 10.1038/cdd.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luca VC, et al. Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science. 2017;355:1320–1324. doi: 10.1126/science.aaf9739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hunter GL, et al. A role for actomyosin contractility in Notch signaling. BMC Biol. 2019;17:12. doi: 10.1186/s12915-019-0625-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ketema M, Kreft M, Secades P, Janssen H, Sonnenberg A. Nesprin-3 connects plectin and vimentin to the nuclear envelope of Sertoli cells but is not required for Sertoli cell function in spermatogenesis. Mol. Biol. Cell. 2013;24:2454–2466. doi: 10.1091/mbc.E13-02-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Almeida FV, et al. The cytolinker plectin regulates nuclear mechanotransduction in keratinocytes. J. Cell Sci. 2015;128:4475–4486. doi: 10.1242/jcs.173435. [DOI] [PubMed] [Google Scholar]
- 51.Staszewska I, Fischer I, Wiche G. Plectin isoform 1-dependent nuclear docking of desmin networks affects myonuclear architecture and expression of mechanotransducers. Hum. Mol. Genet. 2015;24:7373–7389. doi: 10.1093/hmg/ddv438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.el Marjou F, et al. Tissue-specific and inducible cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.