

ABSTRACT
Although MIR516A has been reported to be downregulated and act as a tumor suppressor in multiple cancers, its expression and potential contribution to human bladder cancer (BC) remain unexplored. Unexpectedly, we showed here that MIR516A was markedly upregulated in human BC tissues and cell lines, while inhibition of MIR516A expression attenuated BC cell monolayer growth in vitro and xenograft tumor growth in vivo, accompanied with increased expression of PHLPP2. Further studies showed that MIR516A was able to directly bind to the 3′-untranslated region of PHLPP2 mRNA, which was essential for its attenuating PHLPP2 expression. The knockdown of PHLPP2 expression in MIR516A-inhibited cells could reverse BC cell growth, suggesting that PHLPP2 is a MIR516A downstream mediator responsible for MIR516A oncogenic effect. PHLPP2 was able to mediate BECN1/Beclin1 stabilization indirectly, therefore promoting BECN1-dependent macroautophagy/autophagy, and inhibiting BC tumor cell growth. In addition, our results indicated that the increased autophagy by attenuating MIR516A resulted in a dramatic inhibition of xenograft tumor formation in vivo. Collectively, our results reveal that MIR516A has a novel oncogenic function in BC growth by directing binding to PHLPP2 3′-UTR and inhibiting PHLPP2 expression, in turn at least partly promoting CUL4A-mediated BECN1 protein degradation, thereby attenuating autophagy and promoting BC growth, which is a distinct function of MIR516A identified in other cancers.
Abbreviation: ATG3: autophagy related 3; ATG5: autophagy related 5; ATG7: autophagy related 7; ATG12: autophagy related 12; BAF: bafilomycin A1; BC: bladder cancer; CHX: cycloheximide; Co-IP: co-immunoprecipitation; CUL3: cullin 3; CUL4A: cullin 4A; CUL4B: cullin 4B; IF: immunofluorescence: IHC-p: immunohistochemistry-paraffin; MIR516A: microRNA 516a (microRNA 516a1 and microRNA 516a2); MS: mass spectrometry; PHLPP2: PH domain and leucine rich repeat protein phosphatase.
KEYWORDS: Autophagy, BECN1, bladder cancer, MIR516A, PHLPP2
Introduction
MicroRNAs (miRNAs) are endogenous small RNA molecules that mediate post-transcriptional regulation of dozens to hundreds of target genes [1–3]. miRNAs have drawn increased attention because of their important roles in cancer development. Previous studies have reported that MIR516A is downregulated and is thought to act as a tumor suppressor in ovarian cancer and prostate cancer [4,5]. However, the results obtained from current studies revealed that MIR516A was upregulated in human BC and played an oncogenic role in the promotion of human BC cell growth both in vitro and in vivo. The MIR516A gene is located in chromosome l9 band 19q13.42, which contains two subtypes MIR516A1 and MIR516A2, with the production of the same matured miRNA nucleotides (https://www.ncbi. nlm.nih.gov/gene/). Aberrant expression of MIR516A is always observed in human diseases, and its downregulation has been reported in multiple cancers, including ovarian cancer [4], lung cancer [6], prostate cancer [5], and primary gastric cancer [7]. The tumor suppressor function of MIR516A has also been demonstrated in ovarian cancer and prostate cancer in terms of promoting cancer cell proliferation and metastasis [5]. A study also reports that the strong expression of MIR516A is associated with high-risk neuroblastoma [8]. However, we know nothing about the expression status and biological significance of MIR516A in human BC development.
PHLPP2 (PH domain and leucine rich repeats protein phosphatase 2) belongs to a family of novel protein phosphatases and has always been found to be downregulated in the malignancies, including lung [9], colon [10], breast [11], and gastric cancers [12]. PHLPP2 could dephosphorylate AKT, PRRT2, RPS6KB1, and JUN, by which it inhibits cancer cell proliferation, tumorigenesis, carcinogenesis, as well as cancer progression [13]. However, the mechanisms underlying the downregulation of PHLPP2 in human BC and its tumor suppressor properties are still largely unknown.
Autophagy is a stress response that was originally identified for its role in protecting cells from injury. Abnormalities in autophagy contribute to various disorders, including autoimmune, neurodegenerative, and viral diseases, and cancer [14,15]. However, the function of autophagy in human diseases, especially in various cancers’ development, remains controversial. Studies suggest that autophagy has a protective effect on cancer by improving sensitivity to chemotherapy [16], whereas other studies indicate that autophagy promotes cancer development by inhibiting apoptosis [17–19]. PHLPP2 is a tumor suppressor by catalyzing the de-phosphorylation of the AGC kinases. Our recent studies indicated that PHLPP2 protein could promote bladder cancer cell autophagy, which resulted in the degradation of MMP2 protein and inhibition of BC invasion [20].
The Cullin family is hydrophobic scaffold proteins that provide support for ubiquitin ligases (E3); consequently, they play important roles in multiple biological functions through the degradation of their target proteins [21,22]. The human genome contains eight CUL (cullin) genes, including CUL1, CUL2, CUL3 CUL4A, CUL4B, CUL5, CUL7, and CUL9. Cullin-RING ligases have been reported to be involved in the regulation of autophagy [23]. As a member of the cullin-RING ligase family, CUL4 has drawn much attention due to its contribution to tumorigenesis [24]. However, the functions and related mechanisms of CUL4A in the regulation of cancer cell autophagy and behavior remain largely unknown. Exploration of these issues will provide valuable information for a comprehensive understanding of autophagy in cancer cell behavior. In the present study, we identified PHLPP2 as a direct downstream target of MIR516A, therefore affecting CUL4A-participated and BECN1-mediated autophagic flux and BC cell growth.
Results
MIR516A is required for BC cell growth in vitro and in vivo
The downregulation of MIR516A was reported in human ovarian cancer [4]; however, the expression status of MIR516A in human BCs remains unexplored. To examine the expression pattern of MIR516A in human BCs, we analyzed MIR516A expression in fresh clinical BC tissues by quantitative PCR. As shown in Figure 1A, MIR516A expression was unexpectedly higher in BC tissues than in the paired adjacent non-tumor tissues in a significant manner (n = 30, *p < 0.05). The observation was consistent with the results obtained from the analyzes of 19 paired bladder tissues (cancer tissues vs. normal tissues; 10 male patients and 9 female patients) in TCGA database (Fig. S1A and S1B). Given the inconsistent alterations between human ovarian cancer and human BCs, we tested if there is any difference between the fresh male and female human BC tissues for MIR516A. As shown in Fig. S1 C and S1D, MIR516A level was elevated in BC tissues obtained from both males and females, which is consistent with the observation in TCGA database (Fig. S1A and S1B). Moreover, we also observed MIR516A overexpression in human BC cell lines (TCCSUP, UMUC3, J82, and RT112) in comparison to that in the human immortalized normal urinary epithelial cell line UROtsa (Figure 1B) (p < 0.05). To determine whether MIR516A upregulation plays a role in human BC growth, we stably transfected a MIR516A sponge inhibitor (MIR516Ai) plasmid into two BC cell lines, UMUC3 and J82. With puromycin antibiotic selection, we established the stable MIR516Ai-expressing BC transfectants, UMUC3-MIR516Ai, and J82-MIR516Ai, in which the MIR516A level was dramatically attenuated by the sponge inhibitor constructs (Figure 1C). The inhibition of MIR516A significantly decreased the monolayer growth of UMUC3 and J82 cells as compared with that in the vector control transfectants under the same experimental conditions (Figure 1D,E, p < 0.05), suggesting that MIR516A promotes BC cell growth. The results of soft agar assays showed that the anchorage-independent growth of UMUC3 and J82 cells was also impaired by inhibition of MIR516A (Figure 1F,G). These results suggested that MIR516A was not only overexpressed in human BCs but also played an oncogenic role in BC cell growth.
Figure 1.
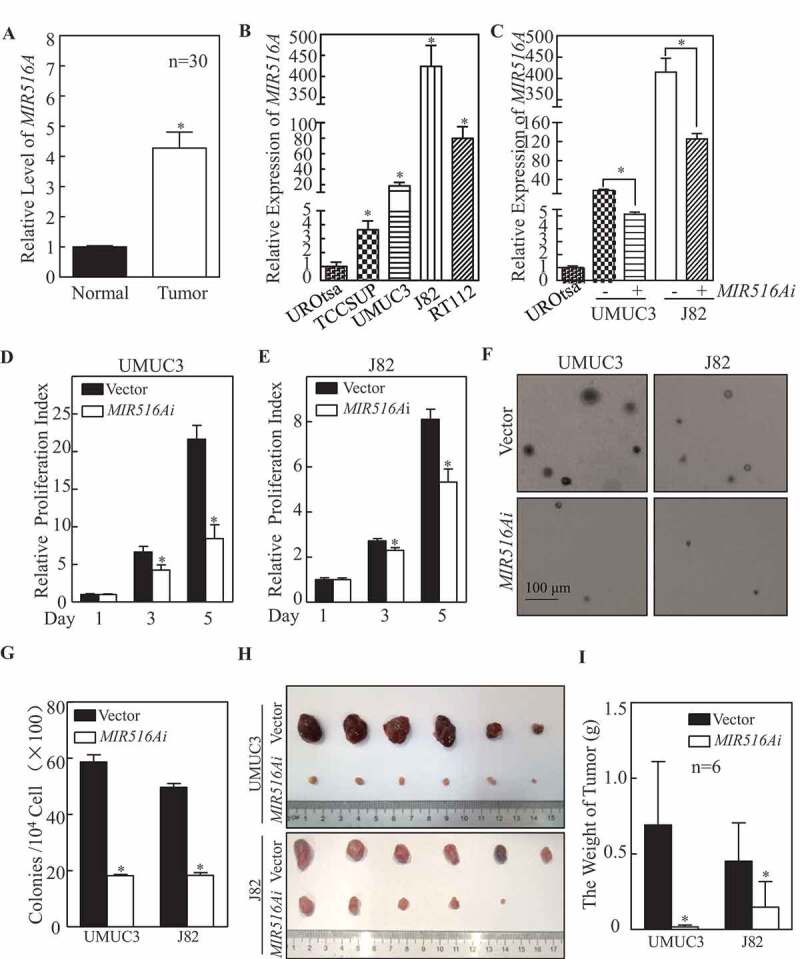
MIR516A suppression resulted in the inhibition of BC cell growth in vitro and in vivo. (A and B) The expression of MIR516A in fresh BC tissues (N, normal tissue; T, tumor tissue) and in indicated BC cells was evaluated by real-time PCR. (C) MIR516A inhibitor (MIR516Ai) was ectopically expressed in UMUC3 and J82 cells, and the expression of MIR516A was determined by qPCR. (D and E) UMUC3 and J82 cells were plated at a density of 200 cells per well in 96-well plates. After cell adhesion to the wells, the cells were cultured for 12 h with 0.1% FBS DMEM or 0.1% FBS MEM, followed by culture in 10% FBS medium for 1, 3, and 5 d. Subsequently, an ATP activity assay was performed as described in the section of “Materials and Methods.” An asterisk (*) indicates a significant decrease compared with the Vector control. (F and G) UMUC3-MIR516Ai, UMUC3-Vector, J82-MIR516Ai, and J82-Vector cells were subjected to soft agar assays. Colonies were counted under a microscope, and the results showed the colonies per 104 cells and are expressed as the mean ±SD. An asterisk (*) indicates a significant decrease in comparison to vector transfectants (p < 0.05). (H and I) Athymic nude mice were subcutaneously injected with UMUC3-Vector and UMUC3-MIR516Ai transfectants, or J82-Vector and J82-MIR516Ai transfectants (2.5 × 106 suspended in 100 μL PBS) into the right axillary region as described in the section of “Materials and Methods.” Four weeks after the injection, the mice were sacrificed (n = 6), and the tumors were surgically removed, photographed and weighed. Bars represent mean ± SD from each group of six mice. Student’s t-test was utilized to determine the p-value. Asterisks indicate a significant decrease (*p < 0.05)
In addition to in vitro experiments, we also evaluated the effect of MIR516A on the tumorigenicity of J82 and UMUC3 cells in a xenograft nude mouse model in vivo. We subcutaneously injected equal numbers of UMUC3-Vector and UMUC3-MIR516Ai cells into nude mice, as described in Materials and Methods. The results showed that inhibition of MIR516A expression also attenuated xenograft tumor growth of UMUC3 cells in nude mice (Figure 1H), and the weight of xenograft tumors was significantly lower in the UMUC3-MIR516Ai group than in the UMUC3-Vector control group (Figure 1H,I, p < 0.05). The regulatory effect of MIR516A on xenograft BC cell growth was also consistently observed in J82 cell transfectants (Figure 1H,I, p < 0.05), strongly indicating that MIR516A promotes bladder tumor growth in vivo.
MIR516A binds to the 3′-UTR of PHLPP2 mrna and downregulates its protein expression in human BC cells
miRNAs modulate target gene mRNA stability or suppress protein translation by binding to the 3′-UTR of target genes, thereby regulating various cellular biological functions [25,26]. To elucidate the mechanism underlying the effect of MIR516A on promoting BC cell growth, we used TargetScan 7.2 software (http://www.targetscan.org/vert_72/) to analyze the potential candidate genes that could be targeted by MIR516A. The comprehensive analysis results indicated that there might be binding sites for MIR516A in the 3′-UTR of mRNAs of ACTG2 (actin gamma 2, smooth muscle), ETS2 (ETS proto-oncogene 2, transcription factor), and PHLPP2 as shown in Figure 2A. To identify the genes involved in the regulatory function of MIR516A, we examined the expression of the corresponding proteins in both UMUC3 and J82 cell lines. As shown in Figure 2B, MIR516A inhibition by MIR516Ai upregulated PHLPP2, downregulated ACTG2, and had no effect on ETS2 expression in UMUC3 and J82 cells [27]. Given that PHLPP2 is a well-characterized tumor suppressor, we stably transfected HA-PHLPP2 into UMUC3 cells (Fig. S2A), and the ectopic expression of PHLPP2 remarkably inhibited anchorage-independent growth of UMUC3 cells (Figure 2C,D). To further explore the mechanisms underlying MIR516A regulation of PHLPP2 expression, we constructed both wild-type and mutant forms (MIR516A binding site mutation) of PHLPP2 mRNA 3′-UTR luciferase reporter as shown in Figure 2A. We transiently transfected the pGL3 control vector, WT and mutant of PHLPP2 3′-UTR luciferase reporter constructs together with pRL-TK into UMUC3-Vector, UMUC3-MIR516Ai, J82-Vector, and J82-MIR516Ai cells. Assessment of luciferase activity showed that inhibition of MIR516A by its inhibitor elevated PHLPP2 mRNA 3′-UTR luciferase activity in comparison to its vector control transfectants, while it did not show comparable inhibition on control reporter pGL3 transfectant (Figure 2E,F). Point mutation in MIR516A bindings sites completely abolished the effect of MIR516A inhibitor on the induction of PHLPP2 mRNA 3ʹ-UTR luciferase activity in both UMUC3 and J82 cells (Figure 2E,F, p < 0.05). These results indicated that MIR516A binding site in PHLPP2 mRNA 3′-UTR reporter was important for MIR516A regulating the activity of the PHLPP2 mRNA 3′-UTR. To further investigate the molecular mechanism underlying the regulation of PHLPP2 by MIR516A, we determined the mRNA level by real-time PCR (Figure 2G). The result led us to exclude the possibility that MIR516A affected PHLPP2 expression at the RNA degradation level because MIR516A did not affect PHLPP2 mRNA expression (Figure 2G). Thus, we anticipate that MIR516A downregulated PHLPP2 by inhibiting its protein translation.
Figure 2.
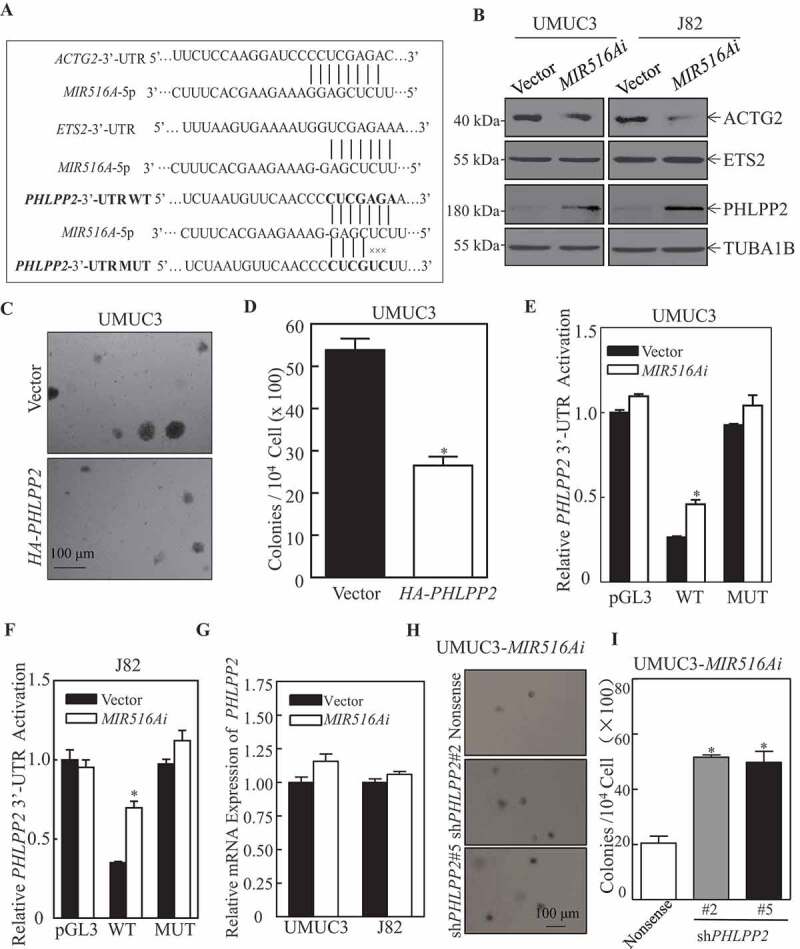
MIR516A bound to the 3′-UTR of PHLPP2 mRNA and downregulated its protein expression in human BC cells. (A) Potential MIR516A targeting sequences in the 3′-UTR of PHLPP2, ACTG2, and ETS2 mRNAs as identified by using TargetScan; and the mutation constructs of potential MIR516A binding sites of 3ʹ-UTR of PHLPP2. (B) The protein expression of ACTG2, ETS2, and PHLPP2 in UMUC3 and J82 cells were determined by western blotting in cells overexpressing the MIR516A inhibitor. (C and D) A soft agar assay was employed to determine the effect of PHLPP2 overexpression on UMUC3 anchorage-independent growth and the images were captured under microscopy (C); the number of colonies was counted and presented as colonies per 10,000 cells (D). (E and F) pGL3, wild-type and mutant of PHLPP2 3′-UTR mRNA luciferase reporters together with pRL-TK were transiently co-transfected into the indicated cells. The luciferase activity of each transfectant was evaluated, and the results were shown as relative pGL3 activity. The results were expressed as the mean ± SD obtained from the triplicates (*p < 0.05). (G) The indicated cells were extracted for total RNA with TRIzol reagent. qPCR was used to determine PHLPP2 mRNA expression. (H and I) The effects of PHLPP2 knockdown in MIR516Ai-expressed UMUC3 cells by a soft agar assay, representative images of colonies of the indicated cells were acquired under a microscope (H). (I) The results are expressed as the mean ± SD, and asterisks indicate a significant increase compared with nonsense control transfectants (*p < 0.05)
To determine whether PHLPP2 is a downstream mediator of MIR516A in the effect of MIR516Ai on the anchorage-independent growth of BC cells, PHLPP2 expression was knocked down in UMUC3-MIR516Ai cells by transfection of shRNAs targeting different PHLPP2 sequences. Western blot confirmed the efficiency of PHLPP2 knockdown (Fig. S2B) and knockdown of PHLPP2 increased anchorage-independent growth of UMUC3-MIR516Ai cells (Figure 2H,I), suggesting that PHLPP2 is a direct downstream target of MIR516A and MIR516A-induced PHLPP2 downregulation plays a role in MIR516A promotion of BC cell growth.
The inhibition of MIR516A results in autophagy induction and anchorage-independent growth inhibition in human BC cells
Autophagy plays a crucial role in the regulation of tumor growth [28]. To investigate the mechanisms underlying the regulation of MIR516A in human BC growth, we evaluated the status of autophagy in MIR516Ai cells in comparison to that in control vector cells. The results showed that the inhibition of MIR516A by MIR516Ai induced autophagy in both UMUC3 and J82 BC cells, as demonstrated by the convention of the autophagic marker from LC3-I to LC3-II (Figure 3A). This notion was supported by the formation of LC3 puncta (Figure 3B–D) and the autolysosome observed in MIR516Ai BC transfectants (Figure 3E,F). Consistently, knockdown of PHLPP2 in MIR516Ai cells substantially reduced autophagy level, as indicated by LC3-II conversion (Figure 3G), puncta formation (Figure 3H,I), and autolysosome formation (Figure 3J). To determine the role of the autophagic induction in the regulation of MIR516Ai–induced inhibition of BC cell growth, bafilomycin A1 (BAF), a lysosomal inhibitor of autophagic flux, was employed. The disruption of autophagy by BAF markedly accumulated LC3-II levels (Figure 3K) and reversed the inhibition of anchorage-independent growth in both UMUC3-MIR516Ai and J82-MIR516Ai cells (Figure 3L,M). These results indicated that MIR516A inhibited autophagy in human BC cells, which further mediates its promotion of BC cell growth.
Figure 3.
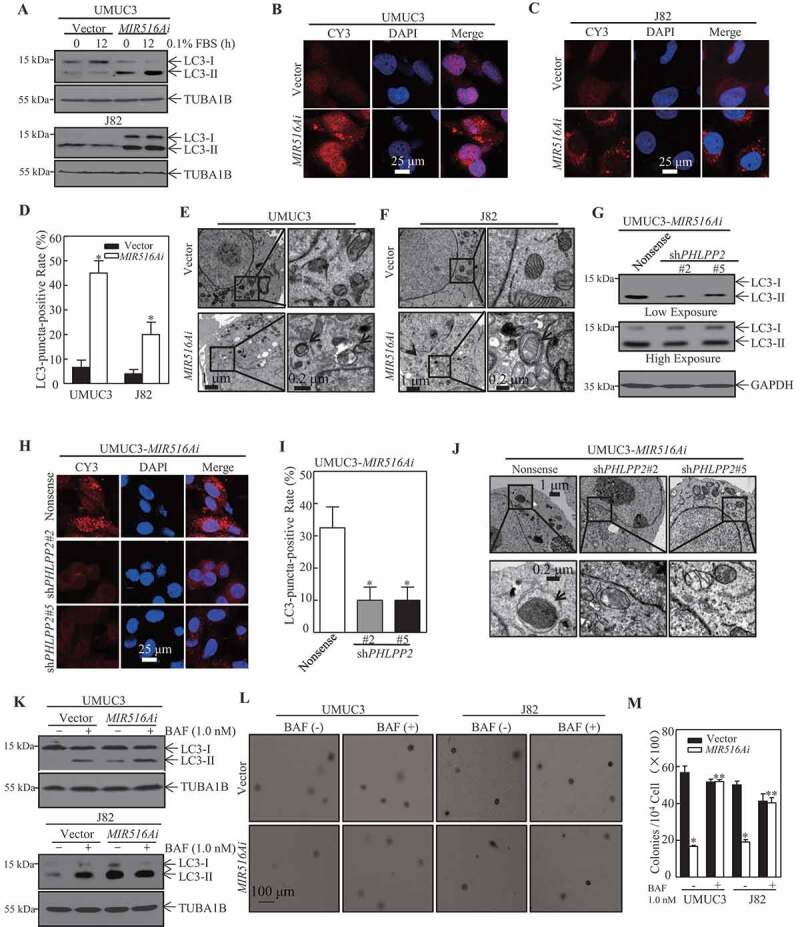
PHLPP2-mediated autophagy was involved in MIR516A regulation of the growth of BC cells. (A) UMUC3-MIR516Ai, UMUC3-Vector, J82-MIR516Ai, and J82-Vector cells were serum-starved for 12 h, and the protein levels of LC3-I and LC3-II were assessed by western blotting. (B, C, and H) The LC3 puncta formation of indicated cells was detected by immunofluorescence, and images were acquired using a microscope. (D and I) The LC3-puncta-positive cell rate was calculated as described in the section of “Materials and Methods.” An asterisk (*) indicates a significant change compared with the vector (or nonsense) control group (p < 0.05). (E, F and J) Electron microscopy analysis of autophagosome in the indicated BC cell transfectants. Black arrows, autophagosomes. (G) The protein levels of LC3-I and LC3-II of indicated cells were assessed by western blotting. (K) UMUC3-MIR516Ai, UMUC3-Vector, J82-MIR516Ai, and J82-Vector cells were seeded into each well of 6-well plates and pretreated with BAF (1.0 nM) for 12 h. The cell extracts were subjected to western blotting. (L and M) UMUC3-MIR516Ai, UMUC3-Vector, J82-MIR516Ai, and J82-Vector cells were subjected to soft agar assay with or without the presence of BAF (1.0 nM). Colonies were counted under a microscope and representative images of colonies were also captured (L). The results showed the colonies per 104 cells and are expressed as the mean ±SD (M). An asterisk (*) indicates a significant decrease in comparison to vector transfectants (p < 0.05). An asterisk (**) indicates a significant increase compared with the vehicle control (p < 0.05)
The inhibition of BECN1 mediates MIR516A attenuation of autophagy in human BC cells
ATGs-mediated autophagy plays an important role in the bulk degradation of cellular constituents [14,29]. To elucidate the mechanism underlying the MIR516A-mediated inhibition of autophagy, we examined the potential effect of MIR516Ai on ATG proteins. Overexpression of MIR516Ai specifically upregulated BECN1 with no comparable effect on other autophagic proteins, including ATG3, ATG5, ATG7, and ATG12 in BC cells (Figure 4A,B). Consistently, knockdown of PHLPP2 in UMUC3 cells and UMUC3-MIR516Ai transfectants attenuated BECN1 expression in comparison to nonsense transfectants (Figure 4C). These results revealed that BECN1 was inhibited by MIR516A, and BECN1 inhibition might play a role in autophagy inhibition due to MIR516A overexpression in human BC cells. Thus, GFP-BECN1 was introduced into UMUC3 -MIR516Ai-shPHLPP2#2 cells; and the stable transfectants were identified by western blotting (Figure 4D) and further employed for our further exploration. BECN1 overexpression markedly increased the conversion of LC3 from LC3-I to LC3-II (Figure 4D). Consistently, BECN1 overexpression also inhibited anchorage-independent growth in UMUC3-MIR516Ai-shPHLPP2#2 cells (Figure 4E,F). Next, shRNAs targeting human BECN1 were used to knock down the expression of endogenous BECN1 in J82-MIR516Ai cells (Figure 4G). The results showed that knockdown of BECN1 expression decreased the conversion of LC3-I to LC3-II, the formation of puncta and autolysosome formation (Figure 4H–J), suggesting that BECN1 does play a role in autophagy induced by ectopic expression of MIR516Ai in human BC cells. As shown in Figure 4K,L, BECN1 knockdown also rescued the anchorage-independent growth of J82-MIR516Ai cells. These results indicated that MIR516A inhibition resulted in BECN1 induction, which mediates autophagy and, in turn, inhibits the anchorage-independent growth of human BC cells.
Figure 4.
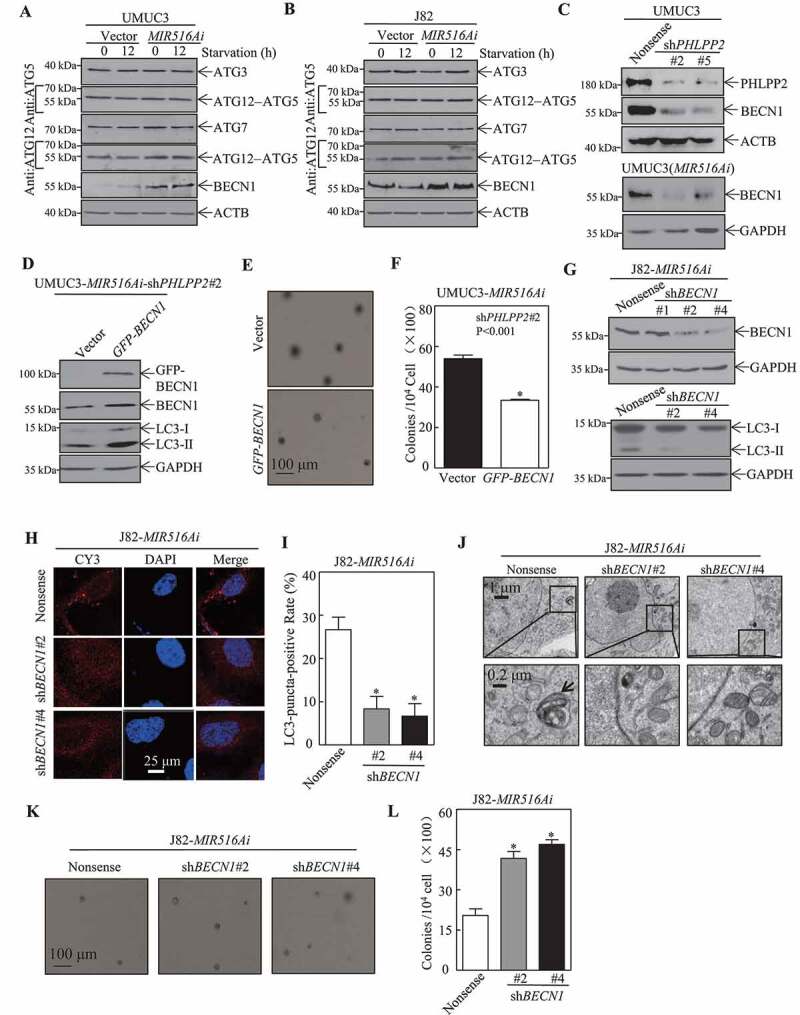
BECN1 induction by MIR516A contributed to autophagic response in human BC cells. (A and B) UMUC3-MIR516Ai, UMUC3-Vector, J82-MIR516Ai, and J82-Vector cells were seeded into 6-well plates, and the cells were then starved in 0.1% FBS DMEM for 12 h. Cell lysates were subjected to western blotting for determination of protein as indicated. (C) Western blot was employed to determine the effects of knockdown of PHLPP2 (transient) on BECN1 expression in UMUC3 cells or UMUC3-MIR516Ai stable cells. ACTB or GAPDH were used as an internal loading control. (D-F) Ectopic expression of GFP-BECN1 was detected by western blotting (D), and the cells were then subjected to the soft agar assay (E). The results are expressed as the mean ±SD. An asterisk (*) indicates a significant decrease (p < 0.05) (F). (G) shRNA BECN1#1, shRNA BECN1#2 and shRNA BECN1#4 were stably transfected into J82-MIR516Ai cells, stable transfectants were identified by determination of BECN1 expression and the conversion of LC3-II by western blotting. (H and I) The LC3 puncta formation of indicated cells was observed and images were captured under a microscope. An asterisk (*) indicates a significant decrease as compared with the nonsense control (p < 0.05). (J) Electron microscopy analysis of the autophagosome in the indicated BC cell transfectants. Black arrows, autophagosomes. (K and L) Representative images of colonies of J82-MIR516Ai-shBECN1#2, J82-MIR516Ai-shBECN1#4 and J82-MIR516Ai-Nonsense cells in soft agar assay were captured using a microscope. The results are expressed as the mean ±SD. An asterisk (*) indicates a significant increase compared with the nonsense control (p < 0.05)
The MIR516A-PHLPP2 cascade promotes cul4a-mediated BECN1 protein degradation
BECN1, also known as ATG6 in yeast, is a key protein for autophagy initiation and other biological processes [30,31]. As a prerequisite to evaluate the mechanisms underlying BECN1 downregulation in PHLPP2 knockdown cells, BECN1 mRNA levels were first determined by qRT-PCR. As shown in Figure 5A, PHLPP2 knockdown had no effect on BECN1 mRNA levels in MIR516Ai-expressing cells, suggesting that the MIR516A-PHLPP2 axis does not regulate BECN1 transcription or mRNA stability. We, therefore, examined the potential effect of MIR516Ai on BECN1 protein degradation. Knockdown of PHLPP2 increased the rate of BECN1 protein degradation compared with that in control UMUC3-MIR516Ai-Nonsense cells (Figure 5B–D). The introduction of MIR516Ai markedly reduced the rate of BECN1 protein degradation compared with that in vector-transfected J82 cells (Fig. S3), indicating that the MIR516A-PHLPP2 cascade inhibits BECN1 protein expression at least partially through increasing BECN1 protein degradation. To further investigate the molecular mechanisms underlying the PHLPP2-mediated inhibition of BECN1 protein degradation, we used UMUC3 cells, which stably expressed GFP-BECN1 or GFP vector, to perform co-immunoprecipitation (co-IP) and subsequent mass spectrometry (MS) analysis. Intriguingly, the results obtained from MS analysis showed that PHLPP2 might not directly interact with and affect BECN1. However, after careful investigation and preliminary validation of the immunoprecipitated protein candidates, we found several core components of E3 ubiquitin-protein ligase, including CUL3, CUL4A, and CUL4B, in our system and showed higher peptide-spectrum matching values (Figure 5E, S4 and Source file). It has been reported that CUL3 and CUL4 can regulate the stability and degradation of BECN1 protein through the ubiquitination-mediated proteasome pathway [32]. Therefore, to further validate whether the effects of MIR516A and its downstream PHLPP2 on BECN1 are actually dependent on CUL4B, CUL4A, or CUL3, we first detected the alteration in expression levels of these proteins. As shown in Figure 5F, MIR516A-PHLPP2 cascade could regulate CUL4A, but had no observable effect on CUL3 and CUL4B expression, suggesting CUL4A might be a key factor for PHLPP2 inhibition of BECN1 protein degradation. This notion was also supported by the results showing the CUL4A-BECN1 interaction observed in the Co-IP assay (Figure 5G).
Figure 5.
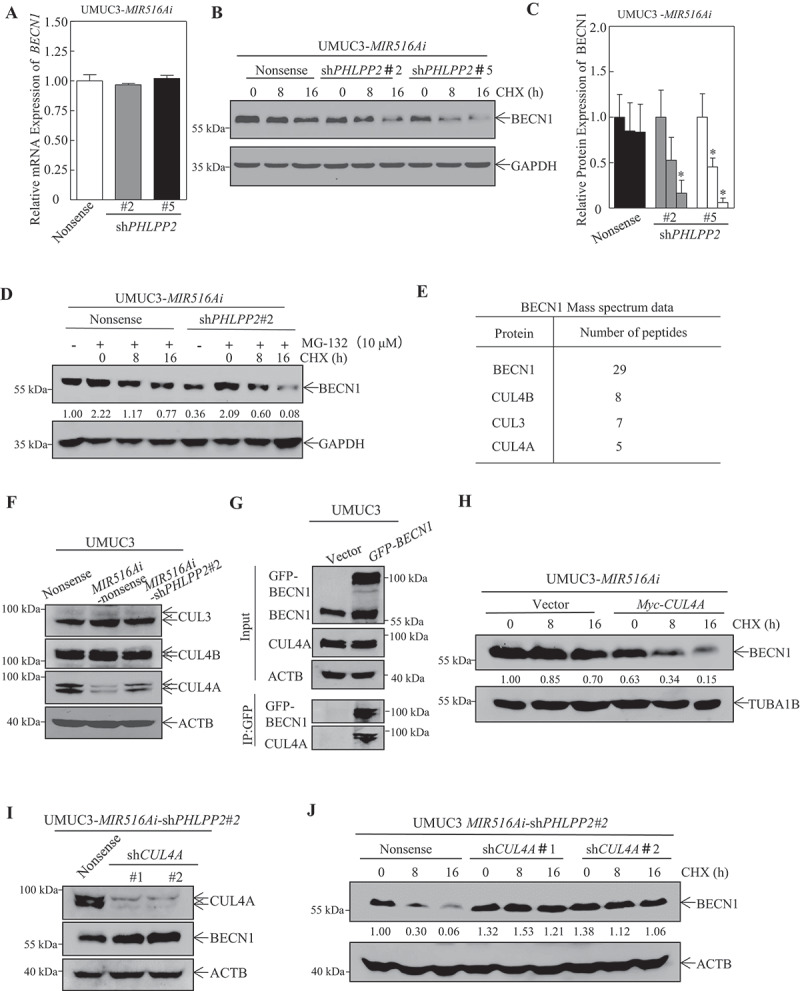
Knockdown of PHLPP2 promoted BECN1 protein degradation in UMUC3-MIR516Ai -Nonsense cells. (A) UMUC3-MIR516Ai-shPHLPP2#2, UMUC3-MIR516Ai -shPHLPP2#5, and UMUC3-MIR516Ai-Nonsense cells were cultured in 6-well plates until the cell density reached 70–80%. Following synchronization in 0.1% FBS DMEM for 12 h, the medium was replaced with 10% FBS DMEM for another 12 h. Then, total RNA was extracted using the TRIzol reagent, and the mRNA expression of BECN1 was detected by qPCR. (B and C) UMUC3-MIR516Ai-shPHLPP2#2, UMUC3-MIR516Ai-shPHLPP2#5, and UMUC3-MIR516Ai-Nonsense cells were subjected to determination of BECN1 protein degradation in the presence of the eukaryote protein synthesis inhibitor, CHX (50 μg/mL) by western blotting (B). The results are expressed as the mean ±SD from three independent experiments. An asterisk (*) indicates a significant decrease in comparison to UMUC3-MIR516Ai-Nonsense cells (p < 0.05) (C). (D) UMUC3-MIR516Ai-shPHLPP2#2 and UMUC3-MIR516Ai-Nonsense cells were pretreated with proteasome inhibitor MG-132 (10 μM) for 12 h, and then subjected to determination of BECN1 protein degradation in the presence of CHX (50 μg/mL) by western blotting. (E) UMUC3 cells expressing GFP-BECN1 or vector control GFP were co-immunoprecipitated with anti-GFP antibody and then subjected to MS analysis. Numbers of peptides identified by MS for BECN1 degradation-related proteins were listed as indicated. (F) Indicated UMUC3 cell transfectants were subjected to western blot for the determination of BECN1 degradation-related proteins. (G) UMUC3 cell transfectant lysates were subjected to Co-IP with an anti-GFP antibody. The binding proteins were determined by western blot. (H) UMUC3-MIR516Ai–Vector and UMUC3-MIR516Ai-Myc-CUL4A cells were subjected to the determination of BECN1 protein degradation in the presence of CHX (50 μg/ml) by western blot. (I) The knockdown of CUL4A in UMUC3-MIR516Ai-shPHLPP2#2 cells and BECN1 expression was assessed by western blot. (J) The BECN1 protein degradation was evaluated in UMUC3-MIR516Ai -shPHLPP2#2-Nonsense, UMUC3-MIR516Ai-shPHLPP2#2-shCUL4A#1 and UMUC3-MIR516Ai -shPHLPP2#2-shCUL4A#2 cells
To further confirm that CUL4A mediated BECN1 protein degradation by MIR516A, we transiently transfected Myc-CUL4A construct into UMUC3-MIR516Ai transfectants (Fig. S2 C) or shRNA targeting CUL4A construct (shCUL4A) into UMUC3-MIR516Ai-shPHLPP2#2 transfectants (Figure 5I). As shown in Figure 5H,J, ectopic CUL4A expression markedly promoted BECN1 protein degradation, while knockdown of CUL4A dramatically reduced BECN1 protein degradation as compared to their control transfectants. These results suggested that the MIR516A-PHLPP2 cascade promoted BECN1 protein degradation mainly through a CUL4A-dependent mechanism.
Inhibition of MIR516A increases PHLPP2 and BECN1 expression accompanied with MKI67 downregulation in vivo xenograft tumors
To determine whether MIR516A plays on the downregulation of PHLPP2 and BECN1 in vivo, we assessed the expression of PHLPP2 and BECN1 using IHC in J82-MIR516Ai xenograft tumors in comparison to J82-Vector xenograft tumors. Quantitative analysis of staining intensity showed remarkable PHLPP2 and BECN1 upregulation in J82-MIR516Ai xenograft tumors compared with the expression of these proteins in xenograft tumors generated by injection of J82-Vector cells (n = 6) (Figure 6A–C). PHLPP2 expression positively correlated with BECN1 expression in MIR516A inhibitor tissues obtained from xenograft nude mice (r = 0.8235, p = 0.0013) (Figure 6E). We also determined MKI67/Ki-67, which is a universally expressed nuclear non-histone protein among proliferating cells and absent in quiescent cells, as a biomarker for cancer cell proliferation [33]. As shown in Figure 6A,C, MKI67-positive BC cells consistently decreased in MIR516A inhibitor xenograft nude mice tissues. Collectively, these results demonstrated that MIR516A inhibition upregulated PHLPP2 by binding to the 3′-UTR of PHLPP2 mRNA, leading to a decrease in CUL4A-mediated BECN1 protein degradation and further increasing BECN1-mediated autophagy, resulting in the suppression of BC growth (Figure 6F).
Figure 6.
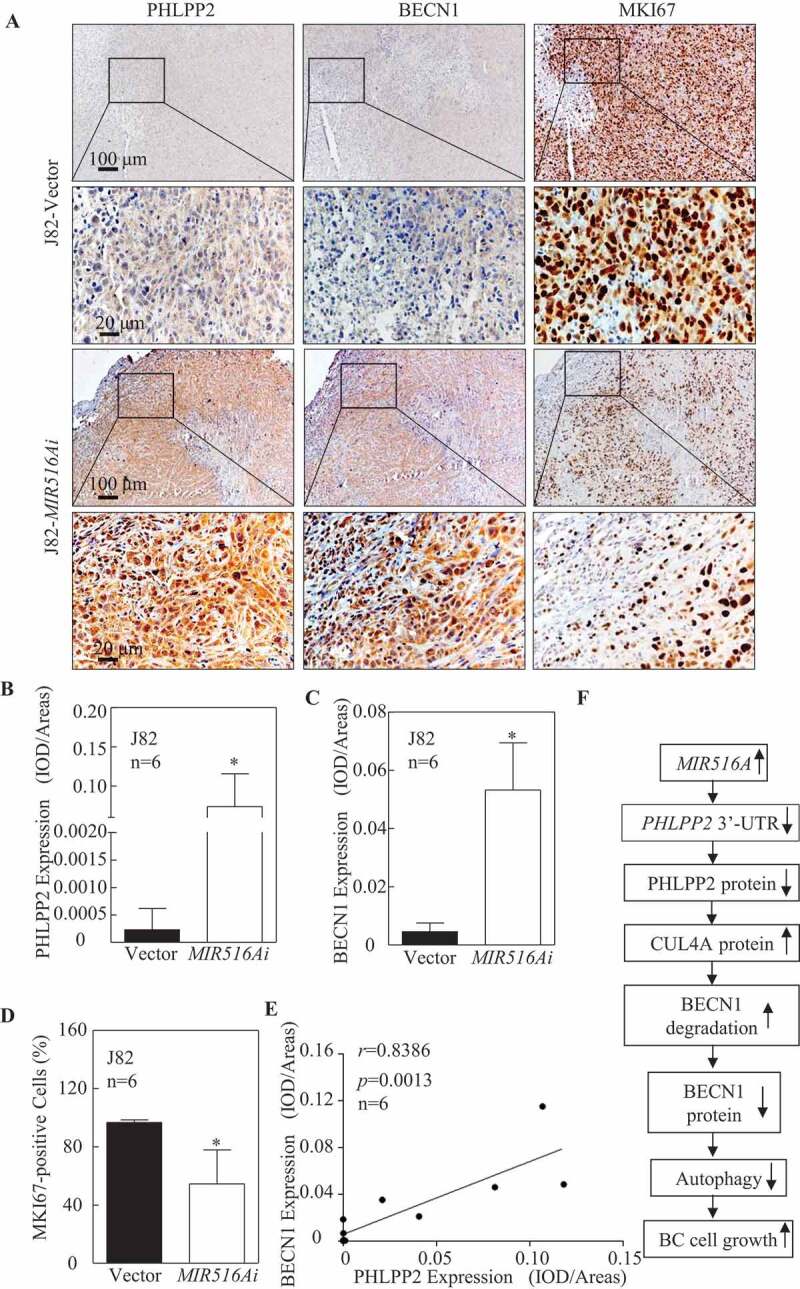
The expression of PHLPP2, BECN1, and MKI67 was determined by IHC staining in the xenograft tumor tissues. (A to D) Representative IHC images showing the expression of PHLPP2, BECN1, and MKI67 in xenograft tumor tissues. Protein expression levels of PHLPP2 (B) and BECN1 (C) were analyzed by calculating the integrated optical density per stained area (IOD/area) using Image-Pro Plus version 6.0, and the MKI67-positive cell rate was analyzed by as described in the section of “Materials and Methods” (D). Results are presented as the mean ± SD. Student′s t-test was used to determine the p-value (*p < 0.05). (E) Positive correlation between PHLPP2 and BECN1 expression in xenograft tumor tissues from nude mice. (F) Schematic mechanisms underlying the effect of MIR516A upregulation of promoting BC growth
Discussion
MIR516A has been reported to be downregulated in ovarian cancer, and overexpression of MIR516A suppresses ovarian cancer growth, revealing its tumor-suppressive function in cancers [4]. We showed here that MIR516A was upregulated in human BC tissues in comparison to their paired adjacent non-tumor tissues. Overexpression of MIR516A in human BCs was also greatly supported by the results obtained from analyzes of human BCs in TCGA database and human BC cell lines. The distinct expression status of MIR516A in human BCs reveals its potential differential biological function in comparison to its tumor-suppressive function that is identified in human ovarian cancers and prostate cancers. Our results showed that inhibition of MIR516A by its specific sponge constructs attenuated both BC cell monolayer growth and anchorage-independent growth in vitro and BC xenograft tumor growth in vivo, accompanied with a remarkable increase in expression of PHLPP2 and BECN1, as well as a decrease in expression of MKI67. MIR516A was further defined to bind to the 3′-UTR region of PHLPP2 mRNA, resulting in attenuating PHLPP2 expression. The suppression of PHLPP2 was further defined as a mediator for MIR516A promotion of CUL4A -mediated BECN1 protein degradation and in turn, leading to a reduction of BC cell autophagy and consequently promoting BC cell growth (Fig. S5). Together, our studies reveal that MIR516A overexpression in human BCs plays an important oncogenic role in promoting BC tumor growth by promoting CUL4A-mediated BECN1 protein degradation and consequently attenuating autophagy, thereby promoting BC growth both in vitro and in vivo as diagramed in Figure 6F. These findings provide significant insight into understanding the nature of MIR516A in human BC development, which is distinct from its suppressive function defined in ovarian cancers.
Although most cancers are associated with gene mutations and living environments, individual cancer always has its self-specificity. Bladder cancer develops when urothelial cells that line the inside of the human bladder begin to grow abnormally, and males are more likely to develop BCs than in females [34]. MIR516A has been reported to be downregulated in several category cancers and plays tumor-suppressive roles in ovarian cancer and prostate cancer by inhibiting cell proliferation and metastasis [4,5]. It has been reported most recent that higher MIR516A-3p expression contributes to breast cancer progression, although its expression status in breast cancers had not been known yet [35]. Unexpectedly, for the first time, we discovered that MIR516A was upregulated in human BC, while the upregulated MIR516A showed its oncogenic function in promoting BC cell growth both in vitro and in vivo.
miRNAs execute their functions by regulating the expression of their target genes [7,36]. In the present study, we first analyzed using bioinformatics the potential MIR516A-targeted genes and revealed that three genes, including ACTG2, ETS2, and PHLPP2, might be targeted by MIR516A. ACTG2 and ETS2 have been reported to play roles in the promotion of cell proliferation in small intestinal neuroendocrine tumors or lung cancers [27,37]. The results obtained from current studies by determining the effect of MIR516Ai overexpression on protein expression of ACTG2 and ETS2 excluded the potential involvement of ACTG2 and ETS2 in the mediation of MIR516A in promotion of BC growth, while PHLPP2 was markedly increased in MIR516Ai ectopic expressed cells. PHLPP2, as an important tumor suppressor involved in the regulation of cancer cell proliferation and inflammation, is downregulated in many cancer cell lines and tumor tissues [38–40]. Mechanisms underlying that of PHLPP2 expression most focus on protein degradation and post-transcription [41]. Knockdown of KCTD17 in primary hepatocytes increased the PHLPP2 protein level, but not the PHLPP2 mRNA level, revealing that KCTD17 mediates PHLPP2 protein degradation [41]. PHLPP2 expression is also negatively regulated by hypoxia in colon cancer cells via HIF1A-dependent cascade by promoting PHLPP2 protein degradation [42]. Several miRNAs including MIR3117, MIR760, MIR181, and MIR938 that post-transcriptionally regulate PHLPP2 have been identified in breast cancer, colon cancer, ovarian cancer, or liver cancer [43–46]. The results from the present study discovered that MIR516A targeted 3ʹ-UTR of PHLPP2 mRNA, which downregulates its protein translation in human BC; therefore, we defined the novel mechanism of MIR516A regulating PHLPP2 protein translation.
Due to the complexity of autophagy, it has been shown to function as a double-edged sword that either protect or suppress human bladder cancer, depending on the stimuli of autophagy, the stage of cancer, and the downstream mediators or effectors [47,48]. Our recent studies have shown that autophagic responses mediated by SESN2/Sestrin 2 mainly result in autophagic inhibition of human bladder cancer cells [48], whereas ATG7-mediated autophagic responses promote growth and invasion of human bladder cancer cells [47]. Therefore, it is essential and highly significant to identify the functional types of autophagy induced by different initiators as well as the underlying mechanisms prior to the application in clinical trials. In the current study, we found that autophagy mediation by BECN1 induction upon MIR516A overexpression acted as an inhibitory effect on tumor growth both in vitro and in vivo. As a phosphatase, PHLPP2 downstream substrates, including AKT, PRRT2, and RPS6KB1, have been discovered in many previous studies [49–51]. And the tumor suppressor role of PHLPP2 has been reported mainly through inhibiting AKT signaling pathway, which contributes to inhibiting cancer cell proliferation. In the present study, however, PHLPP2 inhibition of BC cell growth was achieved through increasing BECN1 protein expression, which in turn promoted autophagy flux and consequently leading to BC growth inhibition. Our results from mass spectrometry assay indicated that PHLPP2 was not a BECN1-interacting protein, suggesting that MIR516A-regulated PHLPP2 indirectly regulated BECN1 expression and its mediated autophagy. Given that the results from both mass spectrometry and Co-IP assays revealed the binding of CUL4A and BECN1 proteins, and the MIR516A-PHLPP2 axis showed a remarkable effect on CUL4A expression, which is responsible for BECN1 protein degradation. We concluded that MIR516A-inhibited PHLPP2 expression mediated CUL4A protein induction, which promotes BECN1 protein degradation, in turn resulting in autophagy reduction and consequently leading to BC growth.
In summary, the present results identified a new MIR516A-PHLPP2-CUL4A-BECN1 autophagy pathway involved in the MIR516A promoting BC growth in vitro and in vivo. We found that MIR516A downregulated PHLPP2 protein by directly binding to 3′-UTR of its mRNA, and the downregulated PHLPP2 further decreased BECN1 expression by promoting BECN1 protein degradation, which inhibited autophagy and thereby enhanced the growth of human BC cells. These findings suggested the crucial role of MIR516A in promoting BC cell growth and also reiterated the role of autophagy in the inhibition of tumor growth, which could lead to the potential development of MIR516A inhibitor and BECN1-mediated autophagy-based strategies for BC treatment.
Materials and methods
Chemical reagents, plasmids, and antibodies
BAF was purchased from Selleck.cn (S1413). Cycloheximide (CHX) was purchased from Calbiochem (508739). Small hairpin RNAs (shRNAs) specifically targeting PHLPP2 [9] and a set of shRNA plasmids specifically targeting BECN1 (human, RHS3979-201763046) were purchased from Open Biosystems (Thermo Fisher Scientific). MIR516A sponge inhibitor (MIR516Ai) and its control constructs were obtained from Shanghai GenePharma Co., Ltd. (C6133). The antibodies specific against LC3A/B (D3U4 C), ATG3 (3415), ATG5 (D5 F5 U), ATG7 (D12B11), ATG12 (D88H11) and BECN1 (D40C5) were purchased from Cell Signaling Technology. Antibody against ETS2 was bought from Santa Cruz Biotechnology (sc-351). The antibodies specific against MKI67 (ab16667), ACTG2 (ab133871), PHLPP2 (ab77665), BECN1 (ab62557), GAPDH (ab9484) and ACTB/β-Actin (ab8224) were bought from Abcam. Antibody against PHLPP2 was bought from Life Span BioSciences, Inc. (LS-B6340). GFP-BECN1 plasmid was constructed by using human BCEN1 cDNA cloned into pLenti-III-miR-GFP-Bank (abm, m001) with primers (F) 5ʹ- CCC AAG CTT CTA TGG AAG GGT CTA AGA CGT C-3ʹ and (R) 5ʹ- CGG GGT ACC TCA TTG TGA GGA CAC CCA AG −3ʹ.
Cell culture and transfection
The human BC cell lines UMUC3, J82, RT112 and TCCSUP and the immortalized human urinary epithelial cell line UROtsa were used in the study. The human bladder cancer cell lines, UMUC3 (CRL-1749) and J82 (HTB-1), were bought from ATCC. RT112 was bought from COBIOER (CBP60316). TCCSUP was bought from SCST.CN (SCSP-571). UROtsa cells were kindly provided by Dr. Scott H. Garrent (University of North Dakota, Grand Forks, ND, USA) [52]. Other cells were tested and identified before use in the study. All cells were maintained at 37°C in a 5% CO2 incubator. UMUC3 cells were cultured in Dulbecco′s Modified Eagle′s Medium (DMEM, Gibco, 11995–065) supplemented with 10% fetal bovine serum (FBS, Gibco, 10437–028). UROtsa cells were cultured in 1640 medium (Gibco, 11875–093) supplemented with 10% FBS, and TCCSUP, RT112, and J82 cells were cultured in minimum essential medium (MEM, Gibco, 11095–080) with 10% FBS. Stable transfections were performed with PolyJet™ DNA In Vitro Transfection Reagent (SignaGen Laboratories, SL100688) according to the manufacturer′s instructions. For stable transfectants, cells were selected with puromycin (0.2–0.3 μg/mL; Amresco Inc., J593) or hygromycin B (200–400 μg/mL; GOLDBIO.COM, H-270-1) for 3–4 weeks [53,54], and surviving cells after selection were pooled as stable mass transfectants. For the determination of PHLPP2 mRNA 3′-UTR activity, UMUC3-Vector, UMUC3-MIR516Ai, J82-Vector and J82-MIR516Ai cells were transiently transfected with pRL-TK (Promega, E2241) together with PHLPP2 mRNA 3′-UTR luciferase reporter or PHLPP2 mRNA 3′-UTR mutant (MIR516A binding sites mutation) luciferase reporter. At 24 h after transfection, luciferase activity was determined using a luciferase assay system kit (Promega, E1960). The internal TK signal was used for normalization. All experiments were performed in triplicate, and the results were expressed as the mean ± SD.
Western blot analysis
Whole-cell extracts were prepared using lysis buffer, and proteins were resolved by SDS-PAGE before transfer to a membrane. The membrane was probed with the indicated primary antibodies, followed by incubation with AP-conjugated secondary antibody. The enhanced ECF chemifluorescence system was used to detect the signals, and images were acquired by scanning with the phosphorimager (GE Healthcare, Typhoon FLA 7000) [55,56].
Lentivirus packaging and infection
Lentivirus packaging and infection were performed as described previously [57]. Briefly, stable GFP-BECN1 cells were established by lentivirus infection. First, 2.0 μg GFP-BECN1 plasmid and two packaging vectors [1.2 μg pMD2.G (addgene, 12259) and 1.2 μg psPAX2 (addgene, 12260)] diluted in 100 μL serum-free DMEM and 8 μL PolyJet™ reagent diluted in 100 μL serum-free DMEM were transfected into 293 T (ATCC, CRL-11268) cells in 6-well plates. Then, the viral supernatant fractions were used to transfect target cells. Stable cell lines were screened by hygromycin B.
Fluorescence microscopy
Immunofluorescence (IF) detection was performed as previously described [58]. Briefly, UMUC3 and J82 cell transfectants were cultured on cover slides in 10% FBS-supplemented DMEM or MEM in each well of 24-well plates. After synchronization, cells were cultured in complete medium for 12 h, followed by 0.1% FBS medium for another 12 h. The cells were fixed with 4% paraformaldehyde (Sigma Aldrich Corporation, 158127) in PBS (Gibco, 8117296) at room temperature (RT) for 15 min, followed by permeabilization in 0.25% Triton X-100 (Beyotime, ST795) at RT for 15 min and staining with LC3A/B (1:100; Cell Signaling Technology, D3U4 C) for 12 h at 4°C. The slides were washed three times with PBS and stained with 0.1 mg/mL DAPI (Sigma Aldrich Corporation, 9542) and CY3 (1:100; BOSTER, BA1032) for 60 min at RT. The slides were washed three times with PBS and once with ddH2O, and then fixed in glycerin. Cell images were captured using an inverted Nikon fluorescence microscope (A1 R). For the quantification of autophagic cells, puncta were determined in 20 random cells per slide.
Anchorage-independent growth assay
Cells (1 × 104 in 10% FBS-supplemented basal medium Eagle [BME] containing 0.33% soft agar) were seeded over the basal layer containing 0.5% agar in 10% FBS BME in each well of 6-well plates. The plates were incubated in a 5% CO2 incubator at 37°C for 2–3 weeks. Colonies were captured under a microscope (Leica, DMi1), and only colonies with over 32 cells were counted. The results are presented as the mean ± SD, as described in our previous study [59].
Qrt-PCR for regular PCR
Total RNA was extracted using the TRIzol reagent (Invitrogen, 15596018) as described in the manufacturer′s instructions, and cDNAs were synthesized with the SuperScript™ First-Strand Synthesis system (Invitrogen, 18091200). The target mRNA content in cells was measured by fluorescence quantitative PCR. The primers used in this study were as follows: human BECN1 (forward, 5′-TCA CCA TCC AGG AAC TCA C-3′; reverse, 5′-GGA TCA GCC TCT CCT CCT CT-3′), and human GAPDH (forward, 5′-GAC TCA TGA CCA CAG TCC ATG C-3′; reverse, 5′-CAG GTC AGG TCC ACC ACT GA-3′).
For miRNA determination, total miRNAs were extracted from cells using the miRNeasy Mini Kit (Qiagen, 218161), and miRNA expression was evaluated by using a Q6 real-time PCR system (Applied Biosystems) and the miScript PCR Kit (Qiagen). The primer for MIR516A (5′-TGC TTC CTT TCA GAG GGT-3′) was synthesized by Sunny Biotechnology, and RNU6 was used as an internal loading control with the primer provided in the miScript PCR Kit. Data were analyzed as described in a previous publication [60].
Transmission electron microscopy
The UMUC3 and J82 cell transfectants were harvested and centrifuged at 1000 g for 5 min. Cells were immersed in 2.5% glutaraldehyde for 12 h at 4°C and then fixed in 1% osmium tetroxide for 1 h. After dyeing in 2% uranyl acetate for 1 h, samples were dehydrated in acetone and embedded in epoxy resin (Sigma, Epon 812). The 50–60 nanometer slices were made using Microtome (RMC boeckeler, PowerTome-XL), and collected on copper grids. Sections were stained with 2% uranyl acetate for 5 min and lead citrate for 1 min. Grids were subjected to a transmission electron microscope (Hitachi, H-7500) at 80 kV for observation of autolysosome.
Immunoprecipitation
UMUC3 cells were transfected with empty vector or GFP-tagged BECN1 constructs. 36 h after transfection, cells were harvested, and total cell extracts were prepared in cell lysis buffer (Cell Signaling Technology, 9803) and complete protein cocktail inhibitors from Roche (04693116001) on ice. Lysates (1 mg) were then incubated with rotation at 4 °C for 30 min. After centrifugation, clarified cell lysates were incubated with 20 μL of anti-GFP mAb Agarose (MBL, D153-8) overnight at 4°C. The immunoprecipitates were washed four times with cell lysis buffer and then subjected to western blot or a mass spectrometry analysis by Guangzhou Fitgene Biotechnology.
Mass spectrometry (MS)
The protein solution was reduced with 0.05 M TCEP (Sigma, 646547) for 1 h at 60°C and then alkylated with 55 mM MMTS (Sigma, 208795) for 45 min at room temperature in darkness. The sample was added into a 10 kDa Millipore, and centrifuged at 12,000 g at 4°C for 20 min, discarded the filtrate. 100 μL UA [8 M urea (Amresco, 0568), 0.1 M Tris-HCl, pH 8.5] was added into Millipore and centrifuged at 12000 g at 4°C for 20 min twice, then 100 μL 0.25 M TEAB (Sigma, T418) was added into Millipore and centrifuged at 12000 g at 4°C for 20 min three times. Finally, replacing a new collection tube, 25 mM TEAB and trypsin were added at 1:50 trypsin-to-protein mass ratio for the first digestion overnight and 1:100 trypsin-to-protein mass ratio for a second 4 h-digestion. After centrifugation at 12,000 g at 4°C for 20 min, the filtrate was collected. Adding 50 μL/0.5 M TEAB into Millipore and centrifuging the filter units at 14,000 g for 10 min. Peptides were dissolved in 0.1% FA and 2% ACN, directly loaded onto a reversed-phase analytical column (75 μM i.d. x 150 mm, packed with Acclaim PepMap RSLC C18, 2 μM, 100 Å, nanoViper), The gradient was comprised of an increase from 5% to 50% solvent B (0.1% FA in 80% ACN) over 40 min, and climbing to 90% in 5 min, then holding at 90% for the 5 min. All at a constant flow rate of 300 nL/min. The MS analysis was performed on Q Exactive hybrid quadrupole-Orbitrap mass spectrometer (ThermoFisher Scientific). Protein identification was performed with MASCOT software (Matrix Science) by searching Uniprot_Aedis Aegypti.
Xenograft model in nude mice in vivo
Animal experiments were performed in the animal institute of Wenzhou Medical University according to the protocols approved by the Laboratory Animal Center of Wenzhou Medical University and Laboratory Animal Ethics Committee of Wenzhou Medical University. Female BALB/c athymic nude mice (3–4 weeks old) were obtained from Shanghai Silaike Experimental Animal Company, Ltd. (license no. SCXK, Shanghai 2010–0002). After 1 week, mice were randomly divided into four groups and subcutaneously injected with 0.1 mL J82-Vector and J82-MIR516Ai or UMUC3-Vector and UMUC3-MIR516Ai cells (2.5 × 106 suspended in 100 μL PBS) at the right side of the back region. After 4–5 weeks, the mice were sacrificed, and tumors were surgically removed, imaged, and weighed. Tumors were also stored at suitable conditions for further experiments.
Immunohistochemistry (IHC)
Tumor tissues removed from nude mice were fixed and embedded in paraffin. For IHC staining, antibodies specific against PHLPP2 (1:200; Abcam, ab77665), BECN1 (1:300; Abcam, ab62557) and MKI67 (1:300; Abcam, ab16667) were used. The staining was performed with Ready-to-use sab-pod (rabbit IgG) kit (BOSTER Biological Technology, SA1022) according to the manufacturer′s instructions. Immunostained images were captured using the Nikon Eclipse Ni microsystem (DS-Ri2). The PHLPP2 and BECN1 protein expression levels were analyzed using a previously described protocol [61], and the MKI67 expression was assessed by randomly analyzing 200 cells per image, and at least 5 images were counted.
Human BC tissue specimens
Clinical samples were collected as our previous study [57]. The study was approved by the Human Subjects Committee of Wenzhou Medical University. With the approval of the patients, human BC tissues, and adjacent normal tissues (n = 30) were collected as shown in Table S1. Adjacent normal bladder specimens were collected from a standard distance (3 cm) from the margin of resected neoplastic tissues of patients who endured radical cystectomy. All samples were immediately cryopreserved in liquid nitrogen and confirmed by histological and pathological diagnosis, and the specimens were classified by a certified clinical pathologist according to the 2004 World Health Organization Consensus Classification and Staging System for bladder neoplasms. Tissue samples were extracted from RNA to synthesize cDNA and stored at −80°C.
Statistical analysis
The Student′s t-test was used to determine significant differences. The differences were considered to be significant at p ≤ 0.05.
Supplementary Material
Acknowledgments
Bioinformatics results announced here based on data produced by the TCGA research network (http://cancergenome.nih.gov/). We also thank participants, specimen donors, and research groups who participated in the data set of the TCGA BC data set to contribute to the database construction. We thank Ms. Liangliang Pan (School of Laboratory Medicine and Life Sciences, Wenzhou Medical University) and Ms. Lijun Shen (School of Laboratory Medicine and Life Sciences, Wenzhou Medical University) for their technical assistance in Transmission electron microscopy assay. We thank Guangzhou FitGene Biotechnology Co., Ltd., China, for technical assistance in MS.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Funding Statement
This work was partially supported by the Natural Science Foundation of China NSFC81702530, NSFC81872587 and NSFC31970696]; Wenzhou Science and Technology Bureau [Y20170028 and Y20190065], Wenzhou Medical University [89216021]; Key Project of Science and Technology Innovation Team of Zhejiang Province [2013TD10], Key Discipline of Zhejiang Province in Medical Technology (First Class, Category A) and Xinmiao Talent Program of Zhejiang Province [2017R413064].
Disclosure Statement
No potential conflicts of interest were disclosed
Supplemental Material
Supplemental data for this article can be accessed here.
References
- [1].Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009. January 23;136(2):215–233. PMID: 19167326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cui R, Meng W, Sun HL, et al. MicroRNA-224 promotes tumor progression in nonsmall cell lung cancer. Proc Natl Acad Sci U S A. 2015. August 4;112(31):E4288–4297. PMID: 26187928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004. January 23;116(2):281–297. PMID: 14744438. [DOI] [PubMed] [Google Scholar]
- [4].White NM, Chow TF, Mejia-Guerrero S, et al. Three dysregulated miRNAs control kallikrein 10 expression and cell proliferation in ovarian cancer. Br J Cancer. 2010. April 13;102(8):1244–1253. PMID: 20354523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhang H, Lian Z, Sun G, et al. Loss of miR-516a-3p mediates upregulation of ABCC5 in prostate cancer and drives its progression. Onco Targets Ther. 2018;11:3853–3867. PMID: 30013366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Leidinger P, Keller A, Meese E. MicroRNAs - Important Molecules in Lung Cancer Research. Front Genet. 2011;2:104. PMID: 22303398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Takei Y, Takigahira M, Mihara K, et al. The metastasis-associated microRNA miR-516a-3p is a novel therapeutic target for inhibiting peritoneal dissemination of human scirrhous gastric cancer. Cancer Res. 2011. February 15;71(4):1442–1453. PMID: 21169410. [DOI] [PubMed] [Google Scholar]
- [8].Gattolliat CH, Le Teuff G, Combaret V, et al. Expression of two parental imprinted miRNAs improves the risk stratification of neuroblastoma patients. Cancer Med 2014. August;3(4):998–1009. PMID: 24931722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Huang H, Pan X, Jin H, et al. PHLPP2 downregulation contributes to lung carcinogenesis following B[a]P/B[a]PDE exposure. Clin Cancer Res. 2015. August 15;21(16):3783–3793. PMID: 25977341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liu J, Weiss HL, Rychahou P, et al. Loss of PHLPP expression in colon cancer: role in proliferation and tumorigenesis. Oncogene. 2009. February 19;28(7):994–1004. PMID: 19079341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xia H, Long J, Zhang R, et al. MiR-32 contributed to cell proliferation of human breast cancer cells by suppressing of PHLPP2 expression. Biomed Pharmacother. 2015;75:105–110. PMID: 26276160. [DOI] [PubMed] [Google Scholar]
- [12].Ding L, Zhang S, Xu M, et al. MicroRNA-27a contributes to the malignant behavior of gastric cancer cells by directly targeting PH domain and leucine-rich repeat protein phosphatase 2. J Exp Clin Cancer Res. 2017. March 21;36(1):45. PMID: 28327189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Grzechnik AT, Newton AC. PHLPPing through history: a decade in the life of PHLPP phosphatases. Biochem Soc Trans. 2016. December 15;44(6):1675–1682. PMID: 27913677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hale AN, Ledbetter DJ, Gawriluk TR, et al. Autophagy: regulation and role in development. Autophagy 2013. July;9(7):951–972. PMID: 3722331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Onorati AV, Dyczynski M, Ojha R, et al. Targeting autophagy in cancer. Cancer. 2018. August;124(16):3307–3318. PMID: 6108917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Comincini S, Allavena G, Palumbo S, et al. microRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells. Cancer Biol Ther. 2013. July;14(7):574–586. PMID: 23792642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004. December 23;432(7020):1032–1036. PMID: 15525940. [DOI] [PubMed] [Google Scholar]
- [18].Gallolu Kankanamalage S, Lee AY, Wichaidit C, et al. Multistep regulation of autophagy by WNK1. Proc Natl Acad Sci U S A. 2016. December 13;113(50):14342–14347. PMID: 5167150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kovaleva V, Mora R, Park YJ, et al. miRNA-130a targets ATG2B and DICER1 to inhibit autophagy and trigger killing of chronic lymphocytic leukemia cells. Cancer Res. 2012. April 1;72(7):1763–1772. PMID: 22350415. [DOI] [PubMed] [Google Scholar]
- [20].Peng M, Wang J, Zhang D, et al. PHLPP2 stabilization by p27 mediates its inhibition of bladder cancer invasion by promoting autophagic degradation of MMP2 protein. Oncogene. 2018. October;37(43):5735–5748. PMID: 29930380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sarikas A, Hartmann T, Pan ZQ. The cullin protein family. Genome Biol. 2011;12(4):220. PMID: 21554755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chen Z, Sui J, Zhang F, et al. Cullin family proteins and tumorigenesis: genetic association and molecular mechanisms. J Cancer. 2015;6(3):233–242. PMID: 25663940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cui D, Xiong X, Zhao Y. Cullin-RING ligases in regulation of autophagy. Cell Div. 2016;11:8. PMID: 27293474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cheng J, Guo J, North BJ, et al. The emerging role for Cullin 4 family of E3 ligases in tumorigenesis. Biochim Biophys Acta Rev Cancer. 2019. January;1871(1):138–159. PMID: 30602127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kim DH, Saetrom P, Snove O Jr., et al. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc Natl Acad Sci U S A. 2008. October 21;105(42):16230–16235. PMID: 18852463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight?. Nat Rev Genet. 2008. February;9(2):102–114. PMID: 18197166. [DOI] [PubMed] [Google Scholar]
- [27].Edfeldt K, Hellman P, Westin G, et al. A plausible role for actin gamma smooth muscle 2 (ACTG2) in small intestinal neuroendocrine tumorigenesis. BMC Endocr Disord. 2016. April 23;16:19. PMID: 27107594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].White E. The role for autophagy in cancer. J Clin Invest. 2015. January;125(1):42–46. PMID: 25654549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. Embo J. 2015. April 01;34(7):856–880. PMID: 25712477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhu H, Wu H, Liu X, et al. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy. 2014;5(6):816–823. PMID: 19535919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yue Z, Jin S, Yang C, et al. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003. December 09;100(25):15077–15082. PMID: 14657337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Boutouja F, Brinkmeier R, Mastalski T, et al. Regulation of the tumor-suppressor BECLIN 1 by distinct ubiquitination cascades. Int J Mol Sci. 2017. November 27;18(12):2541. PMID: 29186924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Weigel MT, Dowsett M. Current and emerging biomarkers in breast cancer: prognosis and prediction. Endocr Relat Cancer. 2010. December;17(4):R245–262. PMID: 20647302. [DOI] [PubMed] [Google Scholar]
- [34].Lopez-Beltran A, Henriques V, Montironi R, et al. Variants and new entities of bladder cancer. Histopathology. 2019. January;74(1):77–96. PMID: 30565299. [DOI] [PubMed] [Google Scholar]
- [35].Foekens JA, Sieuwerts AM, Smid M, et al. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci U S A. 2008. September 2;105(35):13021–13026. PMID: 18755890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Baek D, Villen J, Shin C, et al. The impact of microRNAs on protein output. Nature. 2008. September 4;455(7209):64–71. PMID: 18668037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kabbout M, Garcia MM, Fujimoto J, et al. ETS2 mediated tumor suppressive function and MET oncogene inhibition in human non-small cell lung cancer. Clin Cancer Res. 2013. July 01;19(13):3383–3395. PMID: 23659968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li X, Stevens PD, Yang H, et al. The deubiquitination enzyme USP46 functions as a tumor suppressor by controlling PHLPP-dependent attenuation of Akt signaling in colon cancer. Oncogene. 2013. January 24;32(4):471–478. PMID: 22391563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].O’Neill AK, Niederst MJ, Newton AC. Suppression of survival signalling pathways by the phosphatase PHLPP. Febs J. 2013. January;280(2):572–583. PMID: 22340730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cai J, Fang L, Huang Y, et al. miR-205 targets PTEN and PHLPP2 to augment AKT signaling and drive malignant phenotypes in non-small cell lung cancer. Cancer Res. 2013. September 1;73(17):5402–5415. PMID: 23856247. [DOI] [PubMed] [Google Scholar]
- [41].Kim K, Ryu D, Dongiovanni P, et al. Degradation of PHLPP2 by KCTD17, via a glucagon-dependent pathway, promotes hepatic steatosis. Gastroenterology. 2017. December;153(6):1568–1580 e1510. PMID: 28859855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wen YA, Stevens PD, Gasser ML, et al. Downregulation of PHLPP expression contributes to hypoxia-induced resistance to chemotherapy in colon cancer cells. Mol Cell Biol. 2013. November;33(22):4594–4605. PMID: 24061475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liao Y, Deng Y, Liu J, et al. MiR-760 overexpression promotes proliferation in ovarian cancer by downregulation of PHLPP2 expression. Gynecol Oncol. 2016. December;143(3):655–663. PMID: 27726922. [DOI] [PubMed] [Google Scholar]
- [44].Milbar N, Kates M, Chappidi MR, et al. Oncological Outcomes of Sequential Intravesical Gemcitabine and Docetaxel in Patients with Non-Muscle Invasive Bladder Cancer. Bladder Cancer. 2017. October 27;3(4):293–303. PMID: 29152553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Strotbek M, Schmid S, Sanchez-Gonzalez I, et al. miR-181 elevates Akt signaling by co-targeting PHLPP2 and INPP4B phosphatases in luminal breast cancer. Int J Cancer. 2017. May 15;140(10):2310–2320. PMID: 28224609. [DOI] [PubMed] [Google Scholar]
- [46].Cui X, Li Q, He Y. miR-3117 regulates hepatocellular carcinoma cell proliferation by targeting PHLPPL. Mol Cell Biochem. 2017. January;424(1–2):195–201. PMID: 27822662. [DOI] [PubMed] [Google Scholar]
- [47].Zhu J, Tian Z, Li Y, et al. ATG7 promotes bladder cancer invasion via autophagy-mediated increased ARHGDIB mRNA stability. Adv Sci (Weinh). 2019. April 17;6(8):1801927. PMID: 31016112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liang Y, Zhu J, Huang H, et al. SESN2/sestrin 2 induction-mediated autophagy and inhibitory effect of isorhapontigenin (ISO) on human bladder cancers. Autophagy. 2016. August 2;12(8):1229–1239. PMID: 27171279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Brognard J, Newton AC. PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol Metab. 2008;19(6):223–230. PMID: 18511290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Brognard J, Sierecki E, Gao T, et al. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007. March 23;25(6):917–931. PMID: 17386267. [DOI] [PubMed] [Google Scholar]
- [51].Liu J, Stevens PD, Li X, et al. PHLPP-mediated dephosphorylation of S6K1 inhibits protein translation and cell growth. Mol Cell Biol. 2011. December;31(24):4917–4927. PMID: 21986499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rossi MR, Masters JR, Park S, et al. The immortalized UROtsa cell line as a potential cell culture model of human urothelium. Environ Health Perspect. 2001. August;109(8):801–808. PMID: 11564615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Xu XL, Ye YL, Wu ZM, et al. Overexpression of PTK6 predicts poor prognosis in bladder cancer patients. J Cancer. 2017;8(17):3464–3473. PMID: 29151930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fang Y, Cao Z, Hou Q, et al. Cyclin d1 downregulation contributes to anticancer effect of isorhapontigenin on human bladder cancer cells. Mol Cancer Ther. 2013. August;12(8):1492–1503. PMID: 23723126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Yu Y, Zhang D, Huang H, et al. NF-kappaB1 p50 promotes p53 protein translation through miR-190 downregulation of PHLPP1. Oncogene. 2014. February 20;33(8):996–1005. PMID: 23396362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jin H, Yu Y, Hu Y, et al. Divergent behaviors and underlying mechanisms of cell migration and invasion in non-metastatic T24 and its metastatic derivative T24T bladder cancer cell lines. Oncotarget. 2015. January 1;6(1):522–536. PMID: 25402510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Jin H, Sun W, Zhang Y, et al. MicroRNA-411 downregulation enhances tumor growth by upregulating MLLT11 expression in human bladder cancer. Mol Ther Nucleic Acids. 2018. June 1;11:312–322. PMID: 29858066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Huang H, Zhu J, Li Y, et al. Upregulation of SQSTM1/p62 contributes to nickel-induced malignant transformation of human bronchial epithelial cells. Autophagy. 2016. October 02;12(10):1687–1703. PMID: 27467530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yan H, Ren S, Lin Q, et al. Inhibition of UBE2N-dependent CDK6 protein degradation by miR-934 promotes human bladder cancer cell growth. Faseb J. 2019 Nov 2;33(11):12112–12123. [DOI] [PubMed] [Google Scholar]
- [60].Huang C, Zeng X, Jiang G, et al. XIAP BIR domain suppresses miR-200a expression and subsequently promotes EGFR protein translation and anchorage-independent growth of bladder cancer cell. J Hematol Oncol. 2017. January 05;10(1):6. PMID: 28057023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jiang G, Wu AD, Huang C, et al. Isorhapontigenin (ISO) inhibits invasive bladder cancer formation In Vivo and human bladder cancer invasion In Vitro by TARGETING STAT1/FOXO1 axis. Cancer Prev Res (Phila) 2016. July;9(7):567–580. PMID: 27080594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.