

SUMMARY
Vertebrates have evolved three paralogs, termed LUC7L, LUC7L2, and LUC7L3, of the essential yeast U1 small nuclear RNA (snRNA)-associated splicing factor Luc7p. We investigated the mechanistic and regulatory functions of these putative splicing factors, of which one (LUC7L2) is mutated or deleted in myeloid neoplasms. Protein interaction data show that all three proteins bind similar core but distinct regulatory splicing factors, probably mediated through their divergent arginine-serine-rich domains, which are not present in Luc7p. Knockdown of each factor reveals mostly unique sets of significantly dysregulated alternative splicing events dependent on their binding locations, which are largely non-overlapping. Notably, knockdown of LUC7L2 alone significantly upregulates the expression of multiple spliceosomal factors and downregulates glycolysis genes, possibly contributing to disease pathogenesis. RNA binding studies reveal that LUC7L2 and LUC7L3 crosslink to weak 5′ splice sites and to the 5′ end of U1 snRNA, establishing an evolutionarily conserved role in 5′ splice site selection.
Graphical abstract
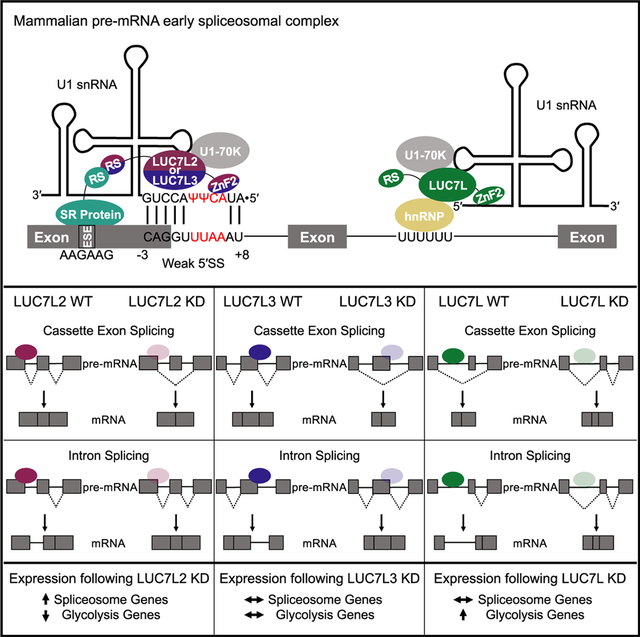
In brief
Mammals have three paralogs of the yeast pre-mRNA splicing factor Luc7p, termed LUC7L, LUC7L2, and LUC7L3, of which one (LUC7L2) is mutated in myeloid neoplasms. Daniels et al. provide functional characterizations of these spliceosomal proteins and elucidation of their distinct roles in alternative splicing and in potential disease pathogenesis.
INTRODUCTION
RNA binding proteins (RBPs) are critical to the processing of RNA at every step of gene expression and function. The number and diversity of RBPs are related to organismal complexity, reflecting their roles in processes such as alternative pre-mRNA splicing. Understanding the roles of individual RBPs requires a multi-omic approach, as applied in recent reports from the ENCODE project (Van Nostrand et al., 2020). Here, we focused our investigations on the functions of a small group of poorly characterized human RBPs, related to a single yeast paralog, that play distinct roles in the regulation of splicing.
Early in the process of pre-mRNA splicing, the 5′ splice site (5′SS) sequence of an intron is bound by the U1 small nuclear RNA (snRNA) and its associated proteins that form the U1 snRNP (Wilkinson et al., 2020). In yeast, this interaction is driven mainly by strong base pairing between the 5′ end of U1 snRNA and the consensus 5′SS. In vertebrates, the choice of a 5′SS is driven by a complex interplay of cis-acting RNA elements within nearby exonic and intronic regions and RBPs that bind to these elements both directly and through protein-protein contacts. Together, these interactions result in the recruitment of U1 snRNP to the appropriate 5′SS, with the mechanistic complexity being the substrate for the regulation of alternative splice site choice and subsequent mRNA diversity.
Recent cryoelectron microscopy (cryo-EM) analyses of the yeast early spliceosomal (E) complex, in which U1 snRNP is bound to the 5′SS, have revealed the function of Luc7p, a poorly studied member of the extended set of U1 snRNP-associated proteins. Yeast Luc7p binds U1 snRNP with its N-terminal alpha helix through Sm protein interactions, and the second zinc finger (ZnF2) stabilizes the binding of weak 5′SSs by directly contacting U1 snRNA at the 5′SS-U1 snRNA duplex (Plaschka et al., 2018; Puig et al., 2007).
Vertebrates have three Luc7p paralogs, namely, LUC7L, LUC7L2, and LUC7L3 (Figure 1A). The N-terminal alpha helix domain and both ZnF domains are conserved (α-helix 20%, Znf1 36%, and Znf2 50%) among the yeast Luc7p and mammalian LUC7-like proteins (Figures 1B and S1A). In Luc7p, these domains have distinct functions in pre-mRNA splicing. Altering the N-terminal domain or ZnF1 by mutation or deletion resulted in impaired in vivo splicing, whereas mutations in ZnF2 resulted in a lethal phenotype (Agarwal et al., 2016). Although the functions of the mammalian LUC7-like family of proteins have not yet been characterized, LUC7L2 has been shown to co-localize with components of the U1 snRNP in the nucleus, suggesting an evolutionarily conserved function in the process of splicing (Howell et al., 2007).
Figure 1. LUC7-like proteins interact with components of the spliceosome.
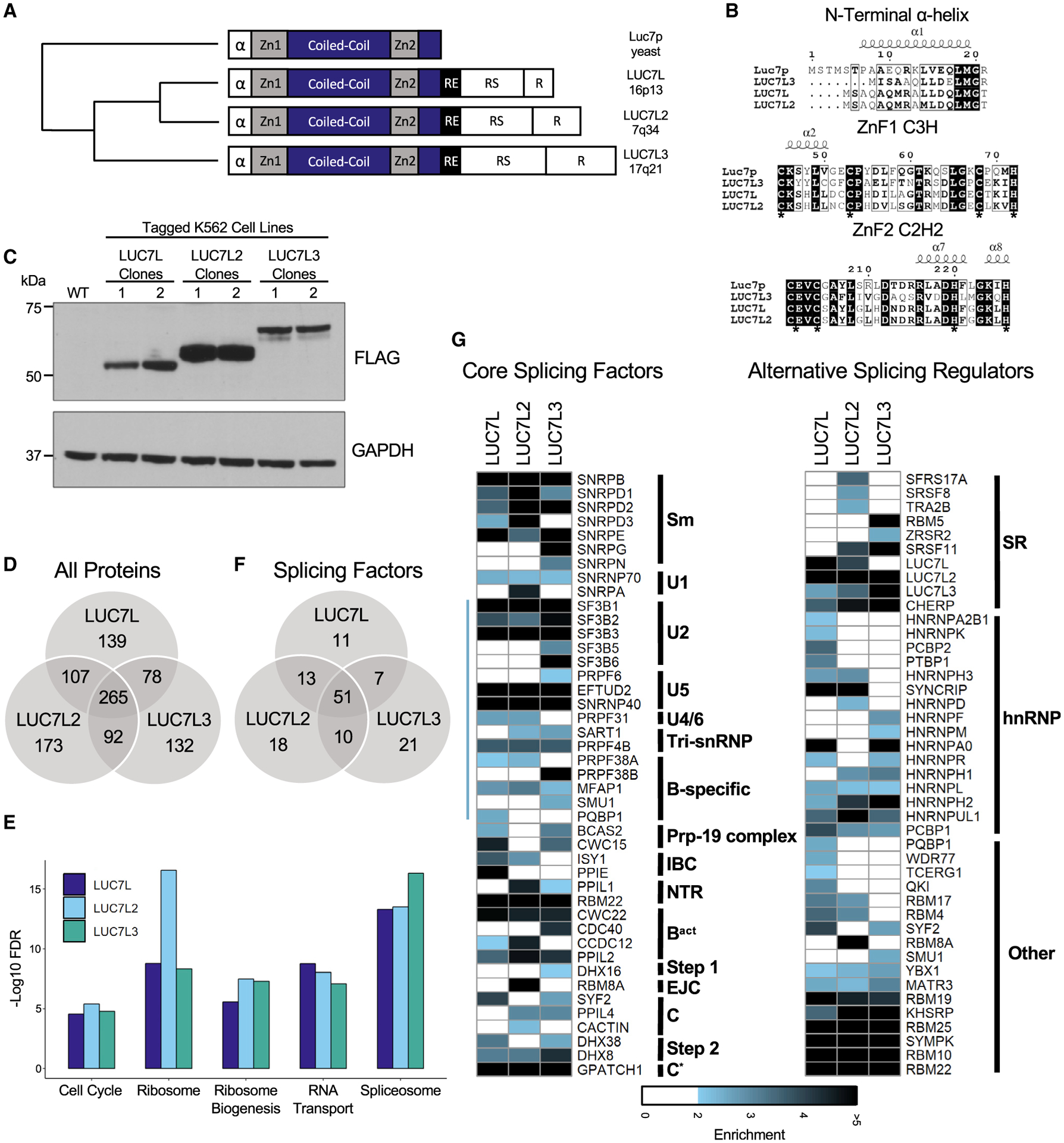
(A) Protein domain structure of yeast Luc7p and the mammalian LUC7-like family (zinc finger 1 [ZnF1], coiled-coil domain, zinc finger 2 [ZnF2], arginine-glutamic acid rich domain [RE], arginine-serine-rich [RS] domain, and arginine-rich [R] domain) with chromosomal locations and phylogenetic tree showing amino acid conservation (adapted from Howell et al., 2007).
(B) Conserved N-terminal α-helix and zinc finger domains of the LUC7-like family generated using CLUSTAL OMEGA 1.2.4 and ESPript 3 (Robert and Gouet, 2014). White letters with black background represent 100% conservation among the four proteins. Black letters with black frame represent conservation among three proteins. Asterisks depict essential cysteines and histidines of zinc fingers. Conserved α-helices are depicted above amino acid sequences and were determined by using the crystal structure of Luc7p from Plaschka et al. (2018).
(C) LUC7-like genes homozygously CRISPR tagged with V5, FLAG, and HA in individual K562 clones shown by western blot (WB) using an anti-FLAG antibody.
(D) Number of common and distinct co-immunoprecipitated (coIP’d) proteins that were ≥ 1.9-fold enriched in both replicates compared to WT K562 FLAG IP.
(E) Top five significantly enriched KEGG pathways.
(F) Number of common and distinct co-IP’d SFs that were ≥ 1.9-fold enriched in both replicates compared to WT K562 FLAG IP.
(G) Fold enrichment of SFs ordered by their appearance in sub-spliceosomal complexes (left) as well as factors involved in alternative splicing (right). Blue line on left-hand side of core spliceosomal proteins depicts proteins found in the human B complex (Bertram et al., 2017; Zhan et al., 2018).
In addition to the ZnF domains, the metazoan LUC7-like proteins contain additional protein domains. They include arginine-serine-rich (RS) domains, which are common features of the SR family of regulatory splicing factors (SFs) (Manley and Krainer, 2010). SR proteins act in the early steps of spliceosome formation and are involved in both constitutive and alternative splicing (AS), but their roles in splicing are context dependent. They often bind exonic splicing enhancers (ESEs) and recruit U1 snRNP and/or the U2AF proteins, assisting in the formation of the E complex (Cho et al., 2011; Zhu and Krainer, 2000). SR proteins are also involved in later steps of splicing, promoting the interaction of U2 snRNP with the branch site sequence and recruiting the U4/U6.U5-tri-snRNP. However, it is important to note that the functional consequences of the binding of SR proteins are highly dependent on the position of cis-regulatory ESEs and exonic splicing silencing (ESS) sequences (Graveley et al., 2001). Therefore, it may be the position of the ESE and ESS sequences that determines the differing functions of the SR proteins for each exon. Although the LUC7-like proteins may interact with RNA in a similar manner to Luc7p, their distinct RS domains (Figure S1B) suggest that they may have evolutionarily diverged to interact with different or additional proteins to regulate unique AS events.
LUC7L2, along with several other SFs, such as early-acting spliceosomal proteins (SF3B1, U2AF1, SRSF2, and ZRSR2) and later-acting core spliceosomal proteins (PRPF8 and DDX41), are frequently mutated in myeloid neoplasms (Makishima et al., 2012; Papaemmanuil et al., 2011; Yoshida et al., 2011). With the exception of ZRSR2, the most common types of identified mutations are single-amino acid change-of-function mutations. For example, mutations in SF3B1 most often occur at the K700E position in heat repeat 7, resulting in altered 3′ splice site (3′SS) selection as well as dysregulated intron splicing due to the increased recognition of cryptic 3′SSs that occur between the branch point and the canonical 3′SS (Darman et al., 2015). For LUC7L2, however, a loss of function due to frameshift and nonsense mutations as well as deletions that encompass LUC7L2 in the q arm of chromosome 7 are common (Chen et al., 2014; Jerez et al., 2012). Low expression of LUC7L2 in myeloid neoplastic patient bone marrow was shown to cause 5′SS dysregulation and an increase in splicing of normally retained introns (RIs) (Hershberger et al., 2020b). Interestingly, despite the homology between the LUC7-like family of putative SFs, LUC7L and LUC7L3 are not frequently targeted by mutations and deletions in patients with myeloid neoplasms, suggesting that LUC7L2 uniquely regulates a subset of genes that are important for the initiation or progression of disease.
Here, we characterized the functional interactions and roles in splicing of the human LUC7-like family of proteins by using crosslinking and immunoprecipitation followed by high-throughput sequencing (CLIP-seq), RNA sequencing (RNA-seq), and co-immunoprecipitation (coIP) mass spectrometry and compared the results to data from the ENCODE project on SFs with well-established functions. Notably, the LUC7-like proteins appear to share common interactions with core spliceosomal proteins, including the U1 snRNP-specific U1-70K protein, but largely differ in their interactions with spliceosomal regulatory proteins. All three LUC7-like family members bind snRNAs with enrichment at the 5′ ends of U1 and U11 snRNAs. LUC7L2 and LUC7L3 interact with pre-mRNAs predominantly in exons with enriched binding at weak 5′SSs. In contrast, LUC7L binds predominantly to intronic regions. Comparing AS events among the three knockdowns (KDs) revealed significant changes in AS, with the majority being specific to a single LUC7-like protein, consistent with their non-overlapping binding sites and tissue-specific expression patterns. Our data suggest that the mammalian LUC7-like proteins are components of the U1 snRNP that interact with distinct AS factors to regulate unique AS profiles.
RESULTS
The LUC7-like proteins interact with components of the spliceosome
The function of the mammalian LUC7-like family of proteins is largely unexplored. However, being paralogs of a U1 snRNP component in yeast (Luc7p) suggests that they may also function as SFs in metazoans (Fortes et al., 1999). Additionally, the LUC7-like genes are ubiquitously expressed but display tissue-specific relative expression patterns, with notably high expression of LUC7L3 in brain tissues and LUC7L2 in the bone marrow and thymus (Figure S1C). This raises the possibility that these proteins have some overlapping and distinct functions. To further understand these functions, we compared the protein interaction partners of LUC7L, LUC7L2, and LUC7L3. We elected to perform these experiments in the human K562 cell line because it is derived from leukemic cells and therefore would more closely resemble the gene expression and splicing patterns found in myeloid neoplasms.
Due to the homology among the LUC7-like proteins, most available antibodies are cross-reactive and unsuitable for IP. To study the binding patterns of the endogenous proteins, we used CRISPR-Cas9 to insert V5, hemagglutinin (HA), and FLAG tags at the C terminus of each LUC7-like protein (Van Nostrand et al., 2017a) (Figures 1C and S2A). Lysates from two homozygously tagged clones for each protein were used in anti-FLAG IP followed by nucleic acid degradation and extensive washing. Liquid chromatography with tandem mass spectrometry (LC-MS/MS) was performed to identify coIP’d proteins (Figure S2).
We identified 589, 637, and 567 proteins that were greater than 1.9-fold enriched over the wild-type (WT) control in both replicates for LUC7L, LUC7L2, and LUC7L3, respectively (Figure 1D; Table S1). Out of these enriched proteins, we identified 82, 92, and 89 to be components of the spliceosome for LUC7L, LUC7L2, and LUC7L3, respectively, making the spliceosome one of the top five enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways for each of the LUC7-like proteins (Figures 1E and 1F). We separated these spliceosomal proteins into two groups containing either core spliceosomal components or factors involved with AS. We found that all three LUC7-like proteins share common interactions with core spliceosomal proteins, such as the U1 snRNP-specific protein U1-70K (SNRNP70) (Figure 1G). Additionally, U1 snRNP-specific protein A (SNRPA) was enriched in the LUC7L2 coIP. Furthermore, all three LUC7-like proteins interacted with the E and A complex proteins PRPF40A and RBM25, similar to known interactions of Luc7p with their yeast paralogs PRP40 and SNU71 in the yeast E complex (Ester and Uetz, 2008; Plaschka et al., 2018). In addition to binding U1 snRNP-specific proteins, we identified a large number of B complex proteins that were enriched in the LUC7-like coIPs (Figure 1G, blue line at left; Bertram et al., 2017; Zhan et al., 2018). This finding suggests that the LUC7-like proteins are components of the earliest forming spliceosomal complexes and are present during the transition from the A to B spliceosomal complex. The LUC7-like proteins are not modeled in the B complex structures nor are they detected by crosslinking studies (Bertram et al., 2017; Zhan et al., 2018).
Although the LUC7-like proteins shared common interactions with core spliceosomal components, they differed in their interactions with factors involved in AS (Figure 1G). LUC7L2 and LUC7L3 IPs were enriched with SR proteins, whereas LUC7L predominantly associated with hnRNP proteins. Interestingly, LUC7L and LUC7L2 each brought down the other two LUC7-like proteins, while LUC7L3 only brought down LUC7L2, suggesting potential functional cooperativity or redundancy among the LUC7-like proteins (Figure 1G). We confirmed this interaction by identifying unique peptide sequences for each of the LUC7-like proteins in our proteomics data. In summary, all three LUC7-like proteins interact with E complex core proteins similar to the yeast paralog Luc7p but differ in binding AS regulators.
CLIP-seq assays reveal common and distinct RNA binding sites
To identify RNA binding profiles of the LUC7-like proteins, we performed single-end enhanced crosslinking and immunoprecipitation followed by high-throughput sequencing (seCLIP-seq) on the K562 epitope-tagged cell lines (Figure S3A). This protocol uses the sequences of the input library in conjunction with peak height to calculate enrichment scores and identify binding sites with high confidence (Van Nostrand et al., 2017a and Van Nostrand et al., 2017b). Additionally, we obtained data for 37 SF eCLIP experiments from the ENCODE project that were performed in the K562 cell line (Sloan et al., 2016).
All CLIP-seq experiments were analyzed for crosslinking site enrichment by using the CLIPper pipeline (Van Nostrand et al., 2017a). For LUC7L, LUC7L2, and LUC7L3, we identified 385, 260, and 4,473 highly reproducible and significant crosslinking sites shared between biological replicates and enriched over the input controls (log2 fold change ≥ 3; −log10 p ≥ 3; irreproducible discovery rate [IDR], ≤ 0.01) (Table S2). A less stringent threshold for significantly shared CLIP peaks between biological replicates (log2 fold change ≥1; −log10 p ≥ 3) revealed a more extensive coverage of the transcriptome for LUC7L (850), LUC7L2 (959), and LUC7L3 (8913). The number of highly significant and reproducible peaks varied heavily between the proteins that were analyzed, which was likely a result of differences in crosslinking efficiency, IP efficiency, variation in expression, and/or RNA binding specificity in K562 cells (Figure S3B).
For each CLIP experiment, we categorized the significant peaks by transcriptomic location. Peaks were classified as binding to constitutive or alternative exons, binding in non-coding RNAs (microRNA [miRNA] and small nucleolar RNA [snoRNA]), and binding to introns or splice junctions, and we ordered the CLIP experiments based on similarity by using unsupervised hierarchical clustering (Figure 2A). This analysis showed that LUC7L2 and LUC7L3 bound predominantly to exonic sequences with very few intronic peaks (Figure 2A). This pattern was also observed for known exon-binding proteins, such as the RS-domain-containing SFs SRSF1 and SRSF7. LUC7L, however, crosslinked to mostly intronic sequences, a pattern that resembled the binding profiles of the hnRNP proteins (Figure 2A).
Figure 2. CLIP-seq assays reveal common and distinct RNA binding sites.
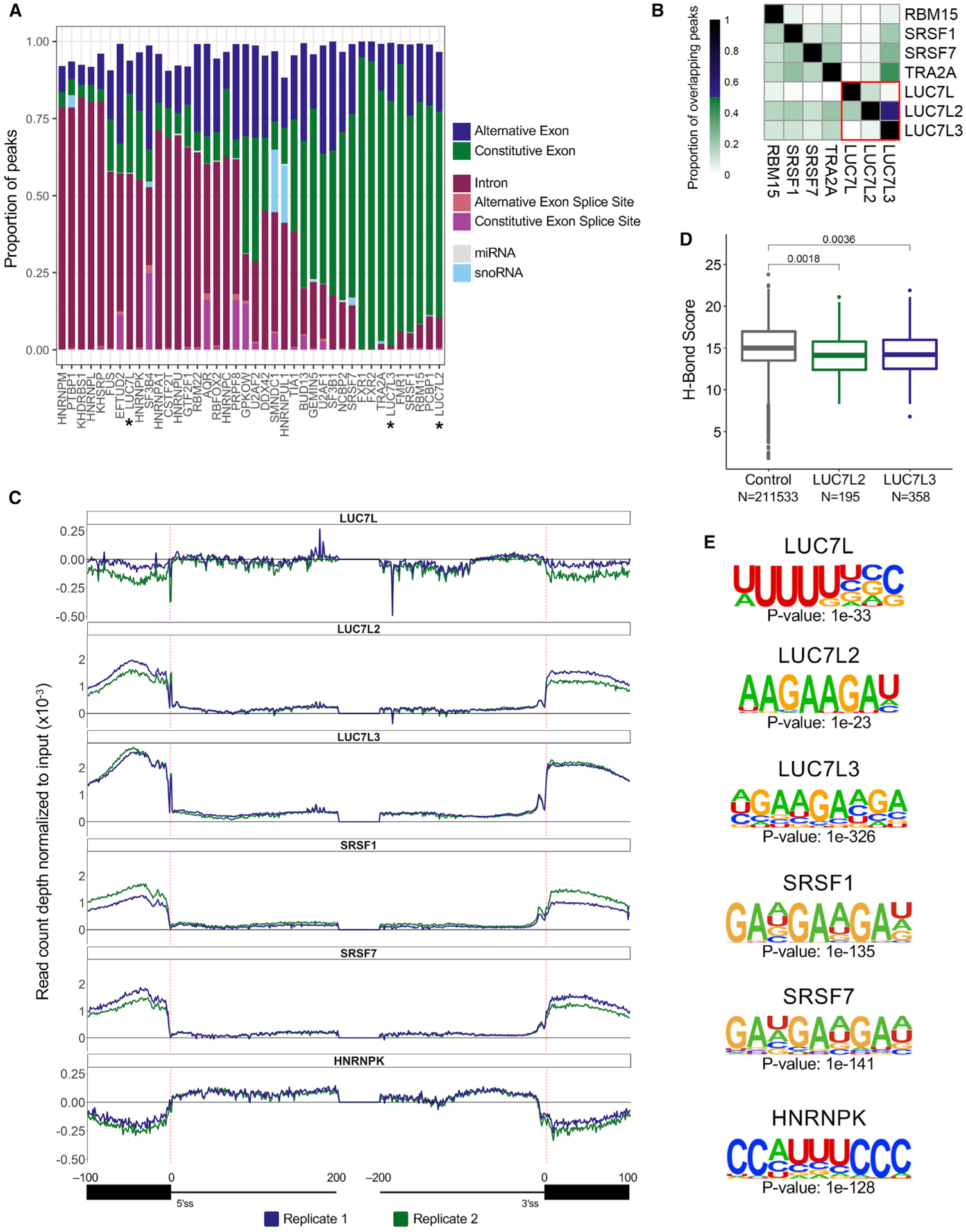
(A) Proportion of significant CLIP peaks (log2 fold change [FC] ≥ 3; −log10 p ≥ 3; IDR ≤ 0.01) that overlap a transcriptomic feature.
(B) ENCODE CLIP-seq experiments that have ≥ 10% CLIP peak overlap with at least one of the LUC7-like proteins.
(C) The distribution of LUC7-like crosslink sites, normalized to input crosslink sites at each nucleotide of all annotated human splice junctions. Depicted are binding data for 100 nucleotides into the exon and 200 nucleotides into the intron downstream and upstream of the 5′SS and 3′SS, respectively. Values above the 0-y-axis threshold depicted by a bold black line have enriched binding over the input control.
(D) U1 snRNA/5′SS hydrogen bonding score using the H-Bond tool (Freund et al., 2003). Control group contains all 5′SSs used in CLIP-seq mapping. LUC7L2 and LUC7L3 groups contain 5′SSs where there is an enriched crosslinking site at either position −1 or +1. A Wilcoxon rank-sum test was performed to compute p values.
(E) Binding motifs enriched in the significant CLIP peaks identified in the LUC7-like seCLIP-seq and ENCODE CLIP-seq experiments.
The LUC7-like proteins bound mostly to distinct locations on pre-mRNA, with only eight CLIP peaks that overlapped among all three proteins. LUC7L2 shared 151 of its binding sites with LUC7L3, primarily in exons, but there was little overlap with LUC7L (Figure 2B, bottom right quadrant, red box). To determine if the binding sites of the LUC7-like proteins coincided with the binding sites of any other SFs, we intersected the coordinates of CLIP peaks for all 40 CLIP-seq experiments (Figure S3C). We identified six SFs (RBM15, SRSF1, SRSF7, TRA2A, LUC7L, and LUC7L3), of which most are SR proteins that shared a high proportion (≥10%) of overlapping CLIP peaks with LUC7L2 (Figure 2B). SRSF1 and TRA2A shared a significant proportion (≥10%) of CLIP peaks with LUC7L3, whereas LUC7L only shared >10% of binding locations with LUC7L2 (47/385 CLIP peaks).
The CLIP-seq experiment preferentially preserves the crosslinking sites of proteins in direct contact with RNA. This allows for the identification at a single-nucleotide resolution of RNA crosslinking sites that we used to compare the binding of the LUC7-like family and the 37 other SFs across all splice junctions (Figures 2C and S4). For SFs with well-established functions, we observed the expected patterns. For example, U2AF1 and U2AF2 both show a dramatic peak at or near the 3′SS (Figure S4). The hnRNP family of proteins show enrichment in intronic sequences near splice sites. Two U2 snRNP-associated proteins, namely, SF3B1 and SF3B4, show peaks at the branch site sequence. These results allow us to interpret the LUC7-like family binding patterns with confidence.
We found that LUC7L binding is slightly enriched in intronic sequences and depleted in exonic sequences (Figure 2C). Conversely, LUC7L2 and LUC7L3 show high enrichment over input in exonic regions that peaks near or at the 5′SS (Figure 2C). The overall distribution is similar to the binding pattern of SRSF1 and SRSF7 (Figure 2C) but differs in an important way. Both the LUC7L2 and LUC7L3 patterns show a dramatic spike in binding at the 5′SS, whereas many of the other SR and hnRNP binding patterns are more symmetrical (Figure 2C). This finding implicates LUC7L2 and LUC7L3 in the regulation of 5′SS selection, similar to the Luc7p yeast paralog.
Prior studies of early yeast spliceosomal cryo-EM structures suggest that Luc7p stabilizes the 5′SS-U1 snRNA duplex of weak 5′SSs (Plaschka et al., 2018). This was shown to be mediated through direct binding of the second ZnF of Luc7p (Plaschka et al., 2018). In our data, we observed a dramatic enrichment of crosslinking to the −1 and +1 positions of 5′SSs for LUC7L2 and LUC7L3 (Figures 2C and S3D), suggesting that this may be an evolutionarily conserved function of these proteins. To test if the human paralogs preferentially bind to weak 5′SSs, we analyzed U1 snRNA hydrogen bond strength to the 5′SS nucleotides that had enriched crosslinking over input control in both replicates at the −1 and +1 positions of 5′SSs. The U1 snRNA hydrogen binding scores for the 5′SSs of LUC7L2 (median = 14.1) and LUC7L3 (median = 14.2) were significantly lower than that of our control set (median = 15.0), which included all 5′SSs used in our CLIP-seq mapping pipeline (Figure 2D). A less stringent binding threshold revealed a larger and more significant difference (Figure S3E). This result supports the conservation of this role for LUC7L2 and LUC7L3 in selection of weak 5′SSs.
The zinc fingers are highly conserved between the yeast and human paralogs, but the human paralogs diverge from yeast Luc7p by the addition of C-terminal RS domains. These domains allow for protein-protein interactions with other SR proteins, and these types of interactions are known to influence splice site selection and AS. We hypothesized that LUC7L2 and LUC7L3 are helping to bridge U1 snRNP with SR proteins binding to ESEs in exons upstream of weak 5′SSs. We tested for a correlation between binding at the 5′SS and binding within the upstream exon (within 99 nucleotides of the −1 position) and found that there was a significant correlation for both LUC7L2 and LUC7L3. For LUC7L2, we found that 47/194 (24%) of enriched 5′SSs had a co-occurring crosslinking enrichment in the upstream exon out of 1,399 exons, with at least 1 enriched crosslinking site (p ≤ 0.0001, chi-square test). For LUC7L3, we found that 145/358 (40.5%) of enriched 5′SSs had a co-occurring crosslinking enrichment in the upstream exon out of 2,947 exons (p ≤ 0.0001, chi-square test). This establishes a possible function of LUC7L2 and LUC7L3 in which weaker 5′SS/U1 snRNA base pairing is further stabilized by bridging SR protein binding in the upstream exon through their RS domains.
Binding motifs of the LUC7-like proteins
In the CLIP-seq experiments, the LUC7-like proteins were enriched for pre-mRNA binding (Figure 2A), allowing us to test for over-represented motifs in the LUC7-like peaks. This showed that the LUC7L peaks were enriched for uridine-rich sequences that are commonly found in introns and are often bound by proteins such as hnRNPK (Figure 2E). This motif is also similar to the binding site of TIA1, a SF that binds AU-rich sequences downstream of 5′SSs (Del Gatto-Konczak et al., 2000). TIA1 binds in the presence of U1 snRNA, activating the adjacent 5′SS from an intronic binding position (Del Gatto-Konczak et al., 2000; Figures S3F and S4, black box). LUC7L2 and LUC7L3, in contrast, were enriched for AAGAAG sequences, a motif of ESEs that are commonly bound by SR proteins, such as SRSF1 and SRSF7 (Bradley et al., 2015; Figures 2E and S3F). In addition to the predominant exonic binding, this indicates that, in vivo, LUC7L2 and LUC7L3 either bind ESE sequences or are binding the RNA in proximity to the SR proteins with which they interact. Network connectivity analysis of SFs has shown that many SR proteins are highly interconnected with other SFs by protein-protein interactions that may be RNA dependent or RNA independent, whereas many hnRNP proteins more often bind RNA directly (Akerman et al., 2015).
Interactions with snRNAs
Our seCLIP-seq analysis captured non-coding RNAs as well as coding RNAs. A total of 3% to 10% of all reads mapped to non-coding RNAs. To map binding to multi-copy snRNAs, we generated custom reference genomes for alignment. After normalizing the number of aligned CLIP reads to the input library, we determined that LUC7L2 had the highest enrichment of U1 snRNA binding of the three LUC7-like proteins (Figure 3A). LUC7L3 interacted with U1, U2, and U6 snRNAs, whereas LUC7L had enrichment of U2 and U6 snRNAs. Adding in the data from the ENCODE eCLIPs, we found that apart from LUC7L2 and LUC7L3, only FUS, GEMIN5, FMR1, and U2AF1 were similarly enriched for binding to U1 snRNA. U2AF1 is a known component of the E complex, present prior to the binding of catalytic snRNAs. This result suggests that LUC7L2 and LUC7L3 are also present in the earliest complexes of the spliceosome. Furthermore, all three LUC7-like proteins interacted with U6 snRNA (Figure 3A), suggesting that the proteins are present and in close proximity during the transfer of the 5′SS from U1 snRNA to U6 snRNA in the transition to the B complex of the spliceosome (Kastner et al., 2019; Plaschka et al., 2018).
Figure 3. LUC7-like family members bind snRNAs, including the 5′ ends of U1 and U11 snRNAs.
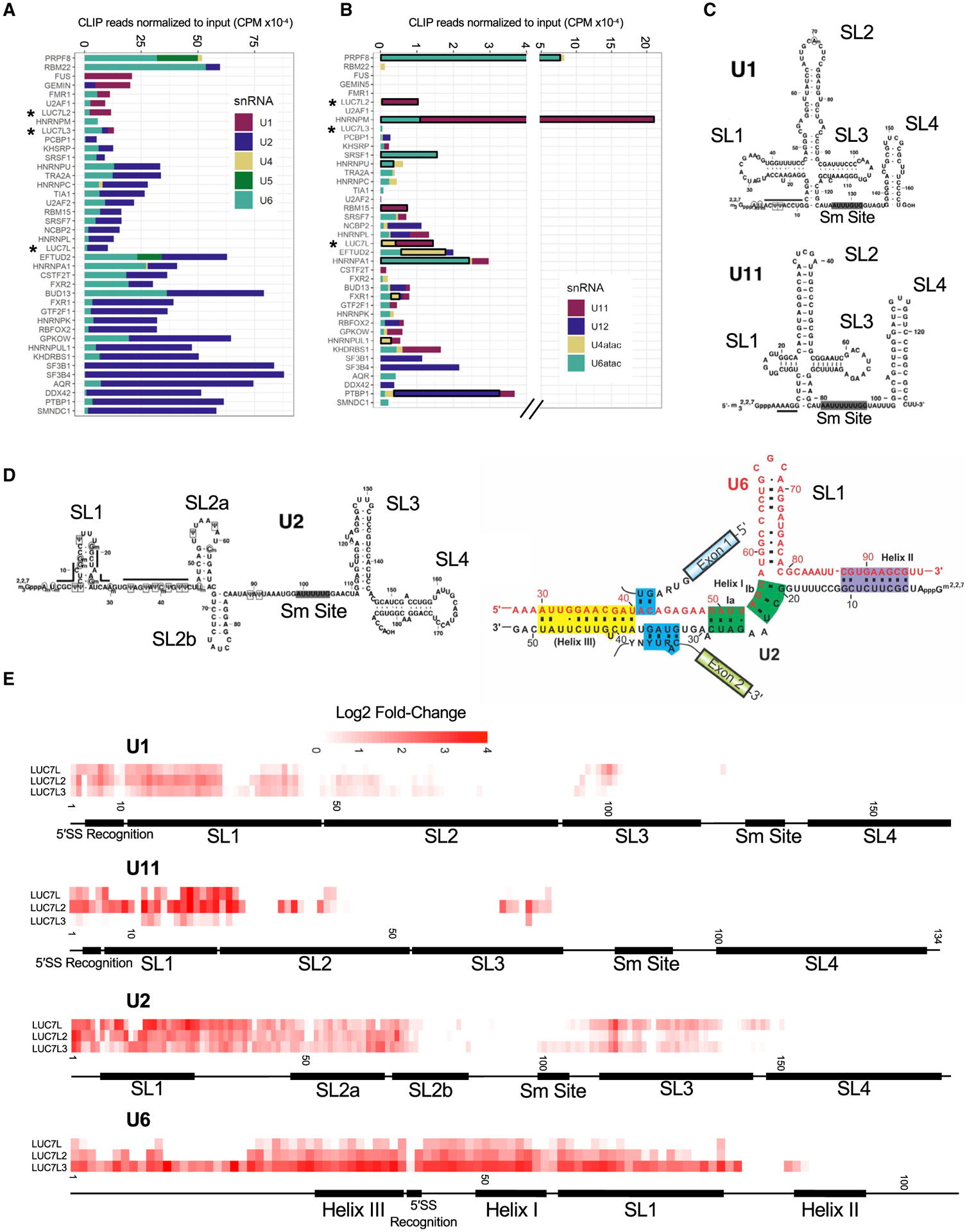
(A) The number of CLIP reads (normalized for library size) enriched over the input control in counts per million (CPM) that map to each major spliceosomal snRNA.
(B) The number of CLIP reads (normalized for library size) enriched over the input control in CPM that map to each minor spliceosomal snRNA. Significantly enriched minor snRNAs are boxed in black. snRNA enrichment was considered significant if the normalized minor snRNA was above the 90th percentile of the distribution in both experimental replicates for each RBP (see STAR Methods).
(C) Secondary structures of U1 and U11 snRNAs with labeled domains (modified from Zhao et al., 2018).
(D) Secondary structures of U2 snRNA (left) and U2/U6 snRNA interactions in the B complex of the spliceosome (right) with labeled domains (modified from Turunen et al., 2013; Zhao et al., 2018).
(E) Single-nucleotide resolution crosslinking maps for the LUC7-like proteins on U1, U11, U2, and U6 snRNAs shown as the averaged replicate log2 fold change enrichment over the input control.
Our data also show that LUC7L and LUC7L2 interact with U11 snRNA as a whole, which is an early-acting minor class snRNA that is a component of the minor spliceosome and is analogous to U1 snRNA in the major spliceosome. Therefore, these proteins may aid 5′SS selection in both U2- and U12-dependent splicing (Figure 3B).
To determine where the LUC7-like proteins crosslink on each snRNA, we generated single-nucleotide resolution protein-snRNA binding maps (Figures 3E and S5). Although only LUC7L2 and LUC7L3 were enriched for binding to U1 snRNA sequences as a whole, we find that all three LUC7 proteins are enriched for binding near the 5′ end of U1 and U11 snRNAs (Figures 3C and 3E). This binding site corresponds to the location where these snRNAs base pair with the 5′SS to form the 5′SS-U1 snRNA duplex as well as the known interaction of Luc7p at the 5′ end of yeast U1 snRNA, suggesting that this is a conserved function between Luc7p and the mammalian LUC7-like family (Plaschka et al., 2018). Following 5′SS base pairing with U1 snRNA and recognition of the branch point by U2 snRNA, extensive RNA-RNA rearrangements occur within the spliceosome (Kastner et al., 2019). U6 snRNA displaces the 5′SS/U1 snRNA duplex to base pair with the 5′SS while forming extensive RNA-RNA interactions with U2 snRNA (Figure 3D). Our data indicate substantial interactions with U2 and U6 snRNAs at and near these regions (Figure 2E). Additionally, we identified a considerable number of B-complex-specific proteins enriched in our proteomics data that are known to interact near the 5′ end of U6 snRNA where it base pairs with the 5′SS (Figure 1G, blue line; Bertram et al., 2017; Zhan et al., 2018). Furthermore, the yeast paralog Luc7p has been modeled to be in close proximity during this exchange in the Pre-B complex of the spliceosome (Plaschka et al., 2018). These data provide further evidence that the LUC7-like proteins are present and in close proximity during the exchange of the 5′SS from U1 snRNA to U6 snRNA.
KD of LUC7-like proteins results in dysregulated AS
To identify genome-wide constitutive and AS events regulated by the LUC7-like family, we knocked down LUC7L, LUC7L2, and LUC7L3 expression in K562 cells (Figure 4A) and performed rRNA-depleted RNA-seq. StringTie was used to identify all observed splice junctions (novel and canonical), followed by rMATS to categorize the types of AS events (alternative 3′SS [A3SS] = 9,275, alternative 5′SS [A5SS] = 7,197, mutually exclusive exons [MXEs] = 3,962, RIs = 20,069, skipped exons [SEs] = 51,224) and quantify the inclusion levels using percent spliced in (PSI) for each AS event; for each sample, p values were adjusted for multiple hypothesis testing with Bonferroni correction. Differential splice-site usage analyses among LUC7L, LUC7L2, and LUC7L3 depletions and non-targeting shGFP control identified 922, 1,061, and 1,535 dysregulated AS events, respectively (|ΔPSI| ≥ 10%; q value ≤ 0.05) (Figures 4B–4D; Table S3).
Figure 4. Decreased expression of LUC7-like proteins results in dysregulated AS.
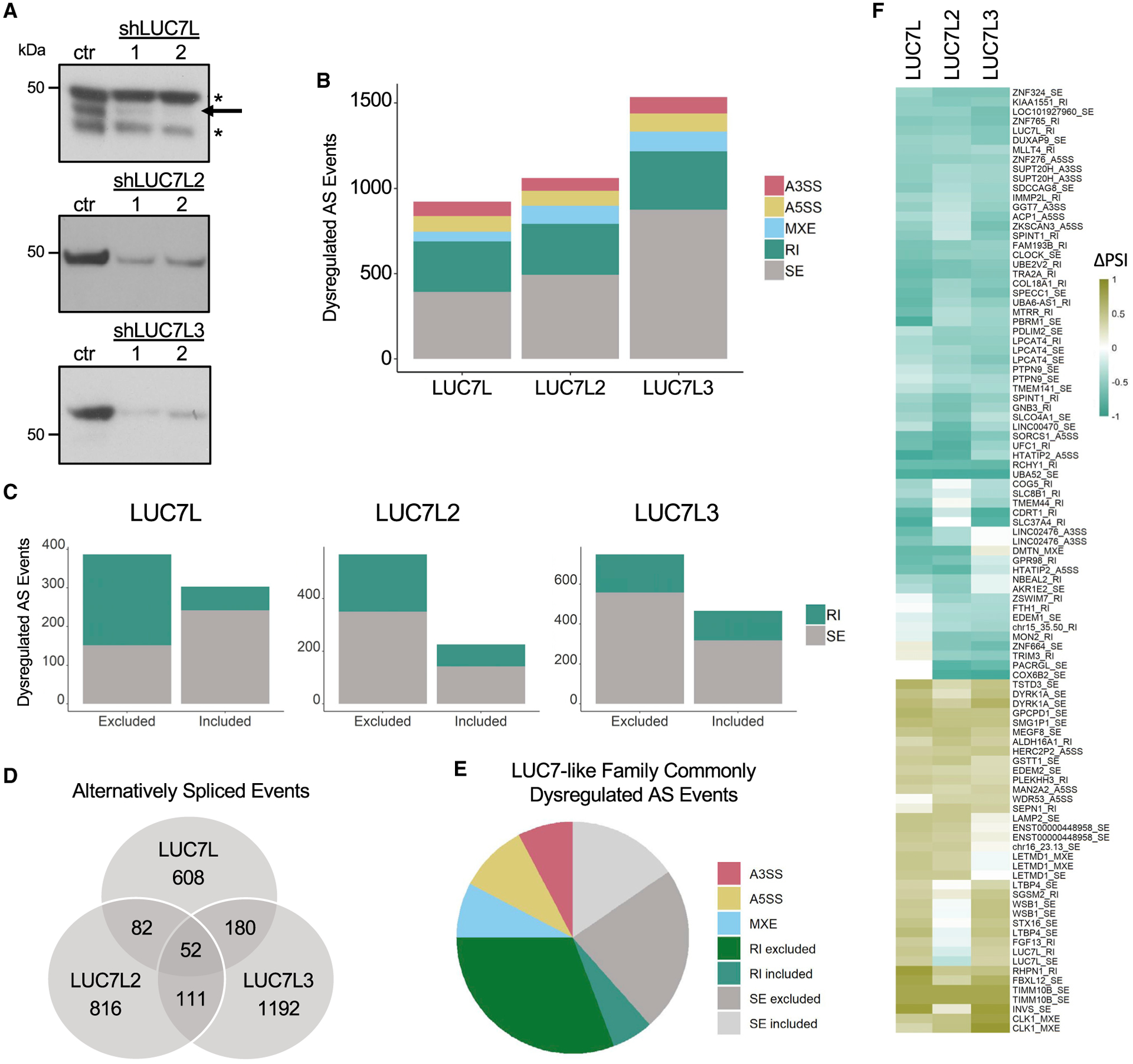
(A) Expression levels of LUC7L, LUC7L2, and LUC7L3 proteins in KD K562 cell lines and shGFP control shown as technical replicates.
(B) Significantly dysregulated AS changes (|ΔPSI| ≥ 10%; q value ≤ 0.05) in KD cell lines compared to that of shGFP control.
(C) Significantly dysregulated skipped exons [SEs] and retained introns [RIs] stratified by inclusion or exclusion (|ΔPSI| ≥ 10%; q value ≤ 0.05) in KD cell lines compared to those of the shGFP control.
(D) Number of common and distinctly dysregulated AS events in the LUC7-like KD cell lines.
(E) Distribution of AS type in the 52 commonly dysregulated AS events.
(F) Strong mis-splicing events (|ΔPSI| ≥ 40%; q value ≤ 0.05) that were dysregulated in at least two LUC7-like KD cell lines. AS events are labeled by the gene that they occur in followed by type of AS event (alternative 3′SS [A3SS], alternative 5′SS [A5SS], mutually exclusive exons [MXEs], RIs, and SEs).
We tested for over-representation of any specific type of mis-splicing in our LUC7-like depletions by using all of the splice junctions measured by rMATs in our K562 datasets as our comparison group. All three cell lines showed an unexpected amount of mutually exclusive exon dysregulation (Fisher’s exact test; LUC7L, p = 0.007; LUC7L2, p ≤ 0.001; LUC7L3, p ≤ 0.001). The LUC7L and LUC7L2 depletion showed an over-representation of alternatively removed or included introns (LUC7L, p ≤ 0.001; LUC7L2, p ≤ 0.001), and the LUC7L depletion alone resulted in more A5SS dysregulation than expected (p = 0.015) (Figure 4B).
We grouped the RI and SE events by increased inclusion or exclusion (Figure 4C). Interestingly, LUC7L2 and LUC7L3 depletion resulted in roughly twice as many SEs as included exons, whereas LUC7L depletion showed the opposite trend. However, KD of LUC7L and LUC7L2 both showed increased levels of intron exclusion as opposed to intron retention (Figure 4C). This phenomenon of increased intronic splicing efficiency has also been identified in myeloid neoplastic patient bone marrow samples with LUC7L2 deficiency (Hershberger et al., 2020b). Although yeast Luc7p is known to stabilize the interaction of U1 snRNA and the 5′SS, the mammalian LUC7-like proteins seem to have additional functions in AS, including the repression of a subset of alternatively spliced introns and regulation of SEs. Furthermore, the mostly non-overlapping dysregulated AS events (Figure 4D) suggest that the LUC7-like family has diverged evolutionarily to regulate unique AS profiles.
When we compared the dysregulated AS events among the 3 KD cell lines, the majority were unique to a single LUC7-like depletion and only 52 were dysregulated in all 3 lines (Figure 4D). This finding agrees with our CLIP-seq data that reveal mostly non-overlapping binding sites for these proteins (Figure 2B). However, further assessment of the 52 commonly mis-spliced AS events reveals a large number of excluded introns suggesting that a common function of all 3 LUC7-like proteins is to repress splicing of the included introns (Figure 4E; Table S4).
We identified 99 strong mis-splicing events (|ΔPSI| ≥ 40%) that occurred in at least 2 of the LUC7-like KDs, further revealing potential functional redundancy or cooperativity between pairs (Figure 4F). It is known that other SR proteins have a large overlap in targets (Bradley et al., 2015; Pandit et al., 2013), suggesting either redundant or cooperative functions. AS analysis of double SR protein KD RNA-seq data supports a model of cooperative splicing regulation (Bradley et al., 2015). Like many other SR proteins, loss of any of the LUC7-like proteins results in a moderate to severe growth defect in K562 cells (Wang et al., 2015; Jourdain et al., 2021), indicating that the other LUC7-like family members are incapable of fully compensating for the loss of either of these proteins.
LUC7-like protein functions are dependent on their binding sites
The CLIP-seq experiments identified pre-mRNA binding sites for the LUC7-like proteins, and the KD RNA-seq experiments categorized AS events that were dysregulated (directly or indirectly) by altering the expression levels of LUC7-like proteins. Combining the CLIP-seq and RNA-seq data, we looked for LUC7-like protein binding near dysregulated AS events (Figure 5A).
Figure 5. Comparison to the effects of KD of other splicing factors.
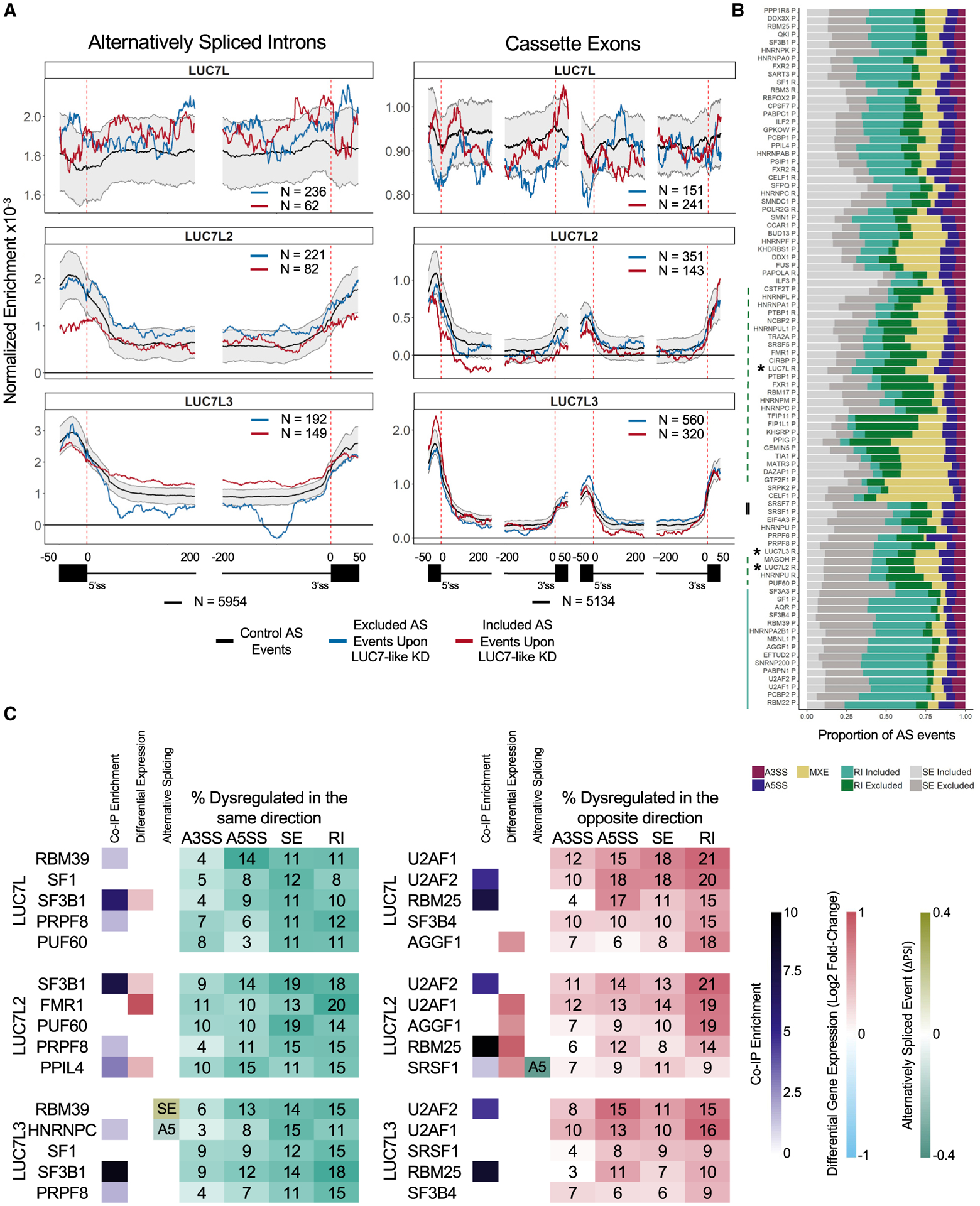
(A) Binding profiles of the LUC7-like proteins on alternatively spliced RIs and cassette exons (included AS event of ≥ 5% ΔPSI; excluded AS event of ≤ −5% ΔPSI; q value ≤ 0.05). Black line indicates binding on alternatively spliced events (PSI of ≥ 0.05 and ≤ 0.95 in the shGFP control K562 cells) in control K562 cells with 90th and 10th percentiles in gray generated from 2,000 random samplings, blue line indicates binding on excluded AS events, and red line indicates binding on included AS events identified from their respective LUC7-like KD experiments. The numbers of AS events are depicted in each map.
(B) Proportion of AS events dysregulated (|ΔPSI| ≥10%; q value ≤ 0.05) in each SF KD experiment and clustered by similarity using unsupervised hierarchical clustering. R indicates total RNA and P indicates polyA(+)-selected RNA used for library preparation. The LUC7-like experiments are depicted with an asterisk. Green dashed line depicts SF KD experiments with a large proportion of intron exclusion. Solid blue line indicates SF KD experiments with a large proportion of intron retention. Double-solid black line depicts SRSF1 and SRSF7.
(C) The five SF KD experiments with the highest overlap of dysregulated AS events as the LUC7-like proteins stratified by direction. Dysregulated in the same direction (teal table) or the opposite direction (red table). Each row includes whether that gene/protein was an enriched immunoprecipitated protein, significantly differentially expressed (FDR ≤ 0.05), and/or contains a significant AS event specified (|ΔPSI| ≥ 10%; q value ≤ 0.05; SE = skipped exon, A5 = alternative 5′SS) in the LUC7-like epitope-tagged line or LUC7-like KD line depicted.
We found that LUC7L2 and LUC7L3 have significantly enriched binding across exons near dysregulated 5′SSs, suggesting that they influence 5′SS selection (Figure 5A). In addition, LUC7L2 and LUC7L3 proteins show distinct binding profiles near mis-spliced introns and cassette exons. LUC7L2 binding enrichment just downstream of the 5′SS correlated with more efficient intron removal upon LUC7L2 KD, whereas binding on the downstream constitutive exon adjacent to an alternative exon was associated with inclusion of the alternative exon. These data suggest that LUC7L2 has repressive splicing properties when binding near or in exons. LUC7L3 binding showed position-dependent splicing effects on cassette exons, where binding at the upstream exon correlated with alternative exon inclusion, whereas binding to the cassette exon was associated with skipping after KD. These data suggest that binding location determines whether LUC7L3 acts as a splicing enhancer (cassette exon binding) or repressor (upstream exon binding) for the cassette exons that it regulates.
Comparing SF KDs reveals cooperative and antagonistic interactions with the LUC7-like family
We obtained RNA-seq data from a collection of 276 shRNA-targeted KDs performed in K562 cell lines from the ENCODE project (Sloan et al., 2016). We downloaded aligned reads for 2 biological replicates of 85 SF KD experiments and 50 control samples. To identify AS events that are co-regulated by multiple SFs, including the LUC7-like KDs, we quantified the number of AS events that were significantly dysregulated in more than one of the SF KD experiments. Of the 36,621 AS events that were dysregulated (|ΔPSI| ≥ 10%; q value ≤ 0.05), 26,248 were dysregulated in at least 2 experiments and 216 AS events were significantly dysregulated in 30 of the 88 experiments, which also included the LUC7-like KDs.
We categorized the dysregulated AS events identified in the 88 experiments by the type of mis-splicing and ordered them based on similarity by using unsupervised hierarchical clustering. We observed various patterns among the SF KDs (Figure 5B). A large number of SF KDs showed significant over-representation of mis-spliced introns. For example, a large number of hnRNP depletions resulted in elevated levels of intron exclusion (Figure 5B, dashed line), whereas a majority of the depletions of core spliceosomal components showed large amounts of intron retention (Figure 5B, solid line). Furthermore, we observed an even distribution of included and excluded introns and more excluded cassette exons in depletions of the SR proteins SRSF1 and SRSF7 (Figure 5B, double line). From this comparison, we see that the LUC7-like proteins (asterisks) are more functionally similar to the hnRNP family of splicing repressors in regard to intron removal than the SR family of splicing activators. In addition, the overall AS patterns were more similar between LUC7L2 and LUC7L3 than either of them with LUC7L in the clustering analysis (Figure 5B). This finding agrees with the binding site results of our CLIP-seq analyses (Figure 2).
We also identified SFs that regulate AS events cooperatively or antagonistically with the LUC7-like family (Figure 5C). We identified significantly dysregulated AS events (|ΔPSI| ≥ 10%; q value ≤ 0.05) in the LUC7-like KDs that were also dysregulated in other SF KDs. Mis-splicing is measured as the increased or decreased inclusion of a sequence of RNA. Accounting for this directionality, we separated the shared events into those mis-spliced in the same direction (potential cooperative interaction) and those mis-spliced in the opposite direction (potential antagonistic interaction).
We ranked the SFs by the percentage of commonly and oppositely dysregulated AS events. For example, we found that 20% of LUC7L2-regulated introns were commonly dysregulated in the same direction, as seen in the FMR1 KD (Figure 5C). Other SFs that shared high overlap with the LUC7-like family included SF3B1, SF1, and PRPF8 (Figure 5C). We found that 21% of LUC7L2-regulated introns were also mis-spliced in the U2AF2 KD but in the opposite direction. U2AF1, U2AF2, RBM25, SF3B4, and SRSF1 had the highest percentage of opposite dysregulation compared to the LUC7-like proteins (Figure 5C). These observations suggest some possible cooperative and antagonistic SF pairings that are further supported by protein-protein interactions identified from our coIP data (Figure 5C). For a subset of introns, U2AF1 and SRSF1 promote splicing, whereas the LUC7-like proteins repress it. Alternatively, the LUC7-like proteins and catalytic core proteins like PRPF8 share commonly dysregulated splicing events. However, we note that many of these SFs with high overlap are core components of the spliceosome, and therefore, they likely regulate a much larger collection of AS events than regulatory factors like the LUC7-like family.
Differential gene expression patterns in the LUC7-like KDs
To investigate the cellular role(s) of the factors, we performed differential gene expression analyses on the LUC7-like KDs. The three KDs showed a similar number of differentially expressed genes (LUC7L = 1,033, LUC7L2 = 1,336, LUC7L3 = 1,059); however, only 128 genes were dysregulated by all 3 LUC7-like KDs (Figure 6A; Table S5). Furthermore, a majority of shared differentially expressed genes were dysregulated in the same direction except for the LUC7L and LUC7L2 comparison (Figure 6B). We performed gene set enrichment analysis (GSEA) to determine if the LUC7-like deficiencies impacted similar pathways. Far more pathways were dysregulated in the LUC7L2 KD than in the LUC7L and LUC7L3 KDs, and there was little overlap of the dysregulated pathways (LUC7L2 = 1,341, LUC7L = 701, LUC7L3 = 92, false discovery rate [FDR] ≤ 0.05) (Figure 6C; Table S6). The glycolysis reactome pathway was one of the most significantly downregulated pathways identified by GSEA in the LUC7L2 KD, suggesting a potential role for LUC7L2 in cellular metabolism (Figure 6E) (Jourdain et al., 2021). Several pathways related to pre-mRNA splicing and the spliceosome, including the top dysregulated KEGG pathway, were found to be upregulated in the LUC7L2 KD (Figures 6D and 6E). This elevated expression of SFs (Figure 6F) includes a majority of the core and AS regulators that LUC7L2 interacts with, suggesting a complex interplay of these factors in pre-mRNA splicing (Figure S6), and may partially explain the increase in intron splicing. Of note, LUC7L is alternatively spliced in all three LUC7-like KDs (Figure S6). Interestingly, the upregulated SFs also included LUC7L and LUC7L3 in the LUC7L2 KD (Figure 6G). Conversely, LUC7L and LUC7L3 KDs did not impact the expression of the other LUC7-like transcripts in K562 cells, suggesting that LUC7L and LUC7L3 may partially compensate for the lowered expression of LUC7L2.
Figure 6. Differential gene expression patterns in the LUC7-like KDs.
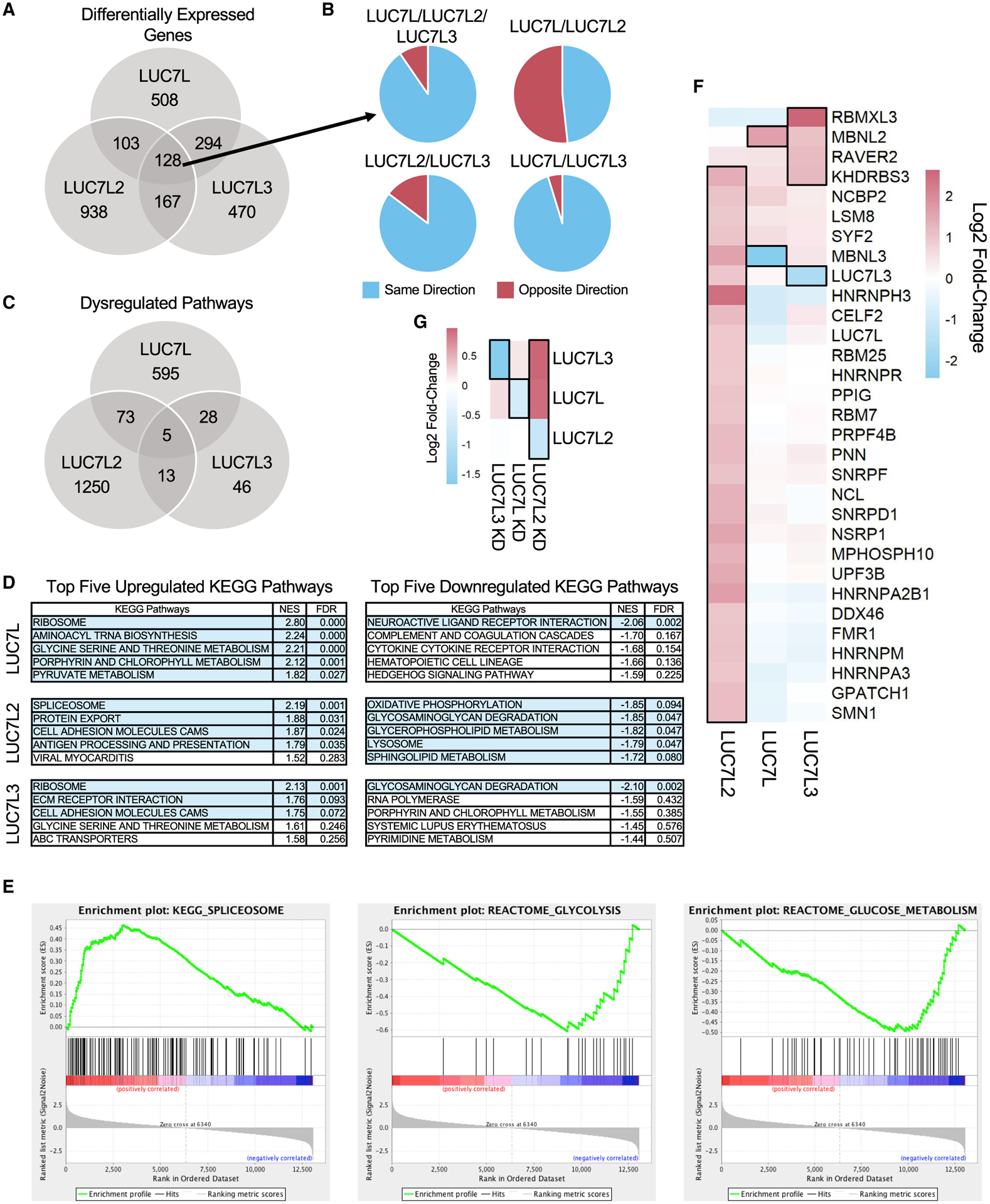
(A) Number of common and distinct significant differentially expressed genes in the LUC7-like KD cell lines (log2FC ≥ 1 or ≤ −1; FDR ≤ 0.05) compared to those of the shGFP control.
(B) Direction and proportion of the commonly differentially expressed genes shared between two or more LUC7-like KD cell lines depicted in (A).
(C) Overlap of significant GSEA pathways (FDR ≤ 0.05).
(D) Top five upregulated and downregulated KEGG pathways by normalized enrichment score (NES) identified by GSEA. Rows highlighted in light blue depict significant pathways with an FDR ≤ 0.1.
(E) GSEA enrichment plots of spliceosomal and glycolytic gene sets in the LUC7L2 KD that are significantly upregulated and downregulated, respectively. Genes are ranked by most upregulated in the LUC7L2 KD dataset at the far-left red bar to most downregulated at the far-right blue bar. The vertical black lines indicate where members of the gene set being tested fall on the ranked list. The green line is the running enrichment score that increases if a gene is identified in the ranked list that is in the gene set or decreases if it is not.
(F) Significantly differentially expressed SFs are boxed with a black outline (log2FC ≥ 0.9 or ≤ −0.9; FDR ≤ 0.05) in the LUC7-like KD experiments and clustered by similarity using unsupervised hierarchical clustering.
(G) Expression of the LUC7-like genes. Genes boxed with a black outline depict significantly differentially expressed genes in each specific LUC7-like KD (log2FC ≥ 0.6 or ≤ −0.6; FDR ≤ 0.05).
DISCUSSION
The complexity of metazoans is paralleled by the diversification of AS pathways that allow individual genes to generate multiple protein isoforms. This increase in splicing complexity is mirrored by the elaboration of splicing regulatory proteins, such as SR and hnRNP proteins. An example of this increase in regulatory factor complexity is seen in the case of the essential yeast SF Luc7p that has three paralogs in metazoans, namely, LUC7L, LUC7L2, and LUC7L3. Notably, these three paralogs share with the yeast protein a conserved N-terminal alpha helix and two zinc finger domains and have divergent C-terminal arginine and serine-rich domains.
Recent structural analysis of the early yeast splicing complex showed that the N-terminal alpha helix of Luc7p binds to the Sm protein complex of U1 snRNP, and the ZnF2 domain binds to the helix formed by the 5′ end of U1 snRNA and the 5′SS of the pre-mRNA (Plaschka et al., 2018). This study also suggests that the yeast protein is particularly important for the splicing of weak 5′SSs. Little has been previously published on the roles of the mammalian proteins, although a loss of LUC7L2 activity has been linked to a role in the development of myeloid neoplasms, including myelodysplastic syndrome and acute myeloid leukemia (Kotini et al., 2015; Haferlach et al., 2013; Hosono et al., 2014; Singh et al., 2013). In these diseases, the LUC7L2 gene, along with several other spliceosomal factors, acquires somatic mutations that introduce stop codons or frameshifts, which are predicted to reduce LUC7L2 protein levels, as well as frequent deletions of one copy of the 7q chromosomal arm that contains LUC7L2, leading to haploinsufficiency (Chen et al., 2014; Makishima et al., 2012). These features suggest that a reduction in LUC7L2 is permissive for a leukemic state in the bone marrow.
Here, we have investigated the three human LUC7-like paralogs LUC7L, LUC7L2, and LUC7L3, with respect to their protein binding partners, RNA crosslinking sites, and effects on gene expression and splicing caused by KD of each factor. In general, we see a combination of both common and distinct binding activities and functions of the three factors.
For protein and RNA binding studies, each of the three genes was endogenously epitope tagged in human erythroleukemic K562 cells by using CRISPR-Cas9. Using IP followed by mass spectrometry, we found that all three proteins interact with multiple spliceosomal factors, notably including the U1-70K protein (SNRNP70) and the Sm core proteins, as well as with each other. Previous RNA pull-downs of in-vitro-assembled splicing complexes suggested that the LUC7-like proteins are mainly found in early forming complexes that include U1 snRNP but not in the later catalytic complexes (Makarov et al., 2012; Sharma et al., 2008; Zhou et al., 2002). The proteins differ in their interactions with splicing regulatory factors such that LUC7L binds more to hnRNP proteins, whereas LUC7L2 and LUC7L3 bind more to SR family proteins.
This pattern was maintained in the seCLIP analysis of RNA binding of each protein. LUC7L bound predominantly to intronic sequences, whereas LUC7L2 and LUC7L3 bound mainly to exons near 5′ and 3′SSs. Again, the LUC7L binding pattern resembled that of hnRNP proteins, whereas the LUC7L2 and LUC7L3 patterns resembled those of SR proteins. Finally, analyses of crosslinking sites showed a pyrimidine-rich hnRNP-like binding motif for LUC7L and purine-rich SR-like motifs for LUC7L2 and LUC7L3.
Like the yeast Luc7p paralog, all of the human proteins showed crosslinking to U1 snRNA, particularly to the 5′ end. This pattern extended to U11 snRNA, the metazoan minor spliceosomal analog of U1 snRNA. If this crosslinking reflects a similar binding to the U1 snRNA/5′SS duplex, as seen in the yeast structure, one might expect to see crosslinking to the 5′SS of pre-mRNAs. Indeed, when crosslinking sites were mapped to 5′SSs, both LUC7L2 and LUC7L3 show distinct positive spikes, whereas LUC7L shows a contrasting but weaker negative spike (Figure 2C). In the case of LUC7L2 and LUC7L3, most crosslinking is seen in exonic regions near the 5′ and 3′SSs. This result could be due to interactions between the RS domains of these factors with other splicing regulatory factors, thus helping to bridge or integrate the exonic factors with the U1 snRNP binding at the 5′SS. Indeed, there is a significant correlation of exon binding and binding to the adjacent 5′SS. The significance of this binding may be in the activation of less consensus 5′SSs, as they are enriched in sites with LUC7L2 and LUC7L3 crosslinking. This finding would agree with evidence that the yeast Luc7p protein is required for splicing of weak 5′SSs (Plaschka et al., 2018; Puig et al., 2007).
KDs of each factor individually caused the disruption of many AS events, although there was little overlap in the introns affected by each factor. This result suggests that the three paralogs have evolved to regulate different groups or classes of splicing events. The types and directions of splicing alterations again were more similar for LUC7L2 and LUC7L3 than for LUC7L. An analysis of related altered gene sets revealed that LUC7L2 appears to regulate the expression of many other spliceosomal factor genes. Reduced LUC7L2 expression in K562 cells leads to increased expression of several other spliceosomal factors, which include LUC7L and LUC7L3. Interestingly, LUC7L and LUC7L3 do not share this function. In addition, reduced LUC7L2 expression also uniquely inhibits the expression of genes involved in glycolysis.
The unique role of LUC7L2 mutations or deletions in myelodysplastic syndrome and related neoplasms distinguishes it from the other paralogs. In a large set of myeloid disease patient bone marrows, expression of LUC7L2 was reduced with or without concomitant mutation or deletion compared with normal bone marrow (Hershberger et al., 2020b). In addition, low LUC7L2 expression or mutation is associated with reduced patient survival in these diseases (Hosono et al., 2014). In in vitro work, induced pluripotent stem cells (iPSCs) derived from the hematopoietic cells of patients harboring a deletion of chromosome 7q recapitulate the hematopoietic differentiation defects observed in myelodysplastic syndrome, with LUC7L2 being one of four genes able to partially rescue this phenotype (Kotini et al., 2015). These results suggest that the contribution of mutated LUC7L2 to the pathology of myelodysplastic syndrome may be through impairment of hematopoietic stem cell differentiation.
Several other spliceosomal factors are also characteristically mutated in these diseases, including U2AF1, SF3B1, and SRSF2 that are well-known splicing regulatory factors (reviewed in Hershberger et al., 2020a and Visconte et al., 2019). Although the prevailing hypothesis is that mutations in these factors affect the splicing of one or more key genes, giving rise to a diseased cell state, no clear common pathway has yet to clearly emerge. Here, we describe a role for LUC7L2 as a component of the human U1 snRNP that likely contributes to the activation of less consensus 5′SSs. The observation that the expression of spliceosome factor genes shows the highest dysregulation upon LUC7L2 KD, of which many are direct protein interactors of LUC7L2, suggests a complex interplay of these factors in pre-mRNA splicing and that its disease-related function might be in part due to an indirect effect on other SFs. On the other hand, LUC7L2 appears to regulate the expression of glycolysis genes such that reduction of LUC7L2 function inhibits these genes, possibly altering cellular metabolism in a pro-neoplastic direction. Future studies seeking to further understand the mechanistic role of LUC7L2 in pre-mRNA splicing as well as dysregulated pathways that contribute to disease pathogenesis are warranted.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Richard A. Padgett (padgetr@ccf.org).
Materials availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Richard A. Padgett (padgetr@ccf.org).
Data and code availability
The RNA-seq and seCLIP-seq data reported in this paper have been deposited to ArrayExpress with the dataset identifier ArrayExpress: E-MTAB-9709. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PRIDE: PXD022152.
EXPERIMENTAL MODEL DETAILS
Generation of knockdown cell lines
Generation of shRNA lentiviral stocks.
HEK293T cells were cultured in 10 cm plates, in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 4.5 g/L glucose, L-glutamine, 1 mM pyruvate, 10% FBS, and 100 U/mL penicillin-streptomycin. Cells were cultured at 37°C in incubators maintaining 5% CO2. Cells were sub-cultured using trypsin when they reached 80% confluency.
Viral stocks were generated using the Lipofectamine PLUS protocol. Optimem (750 μl), Lipofectamine (Thermo Fisher, A12621) (30 μl), pCMV8.2 (1.5 μg), VSV-G (1 μg), and shRNA vector (3 μg) were combined according to the manufacturer’s protocol. Virus components were added to cells with 5 mL Optimem and 7 mL DMEM supplemented with 10% FBS. Viral titers were collected on days 3, 4, and 5 post-infection. The shRNA targeting sequences were obtained from SigmaAldrich: LUC7L 5′-CCGGCCAGACAGAGGGTCAAGTTTACTCGAGTAAACTTGACCCTCTGTCTGGTTTTTTG-3′ (TRCN0000195589), LUC7L2 5′- CCGGGTAATGGATGAAGTAGAGAAACTCGAGTTTCTCTACTTCATCCATTACTTTTTG –3′ (TRCN0000320721), LUC7L3 5′- CCGGCCGGGATCGAAAGTCATATAACTCGAGTTATATGACTTTCGATCCCGGTTTTTG –3′ (TRCN000075115), and non-targeting shGFP control (SHC004).
K562: LUC7L, LUC7L2 and LUC7L3 knockdowns.
K562 cells were seeded between 5–8×104 cells/mL in 10 cm plates. The cells were resuspended in RPMI media (Cytiva, SH30027LS) with 10% FBS and 100 U/mL penicillin-streptomycin, viral titer, and poly-brene (8 mg/ml). Media was replaced after 24 hours. The cells were placed in selection media (RPMI, 2 μg/ml puromycin) after an additional 24 hours. The selection media was replaced every 48 hours for 8 days, at which point the control cells were dead. The knockdowns were confirmed by testing the expression levels of the LUC7-like family by western blot using anti-LUC7L (Protein Tech, 17085-1-AP), anti-LUC7L2 (Bethyl, A304-504A-M), and anti-LUC7L3 (Protein Tech, 14504-1-AP).
K562 V5-HA-FLAG-tagged endogenous LUC7L, LUC7L2, and LUC7L3
Homology arm vector cloning
Blue Heron Biotech pUC MinusMCS vectors were synthesized to include homology arms for LUC7L, LUC7L2 and LUC7L3, separated by a short sequence that contained two restriction digest sites (EcoRI and BamHI). The gRNA targeted-PAM sequence was mutated in the homology arms to prevent cutting of the edited gene (if necessary). The HR130 vector, courtesy of the Yeo lab and published in the CRISPR-tagging protocol (Van Nostrand et al., 2017a), contained the V5,HA,FLAG tag with self-cleavable GFP and puromycin resistance gene sequence, which was flanked by restriction digest sites (EcoRI and BamHI). The vectors were linearized and ligated together, inserting the tag and selectable marker sequences between the two homology arms. This process was performed to generate LUC7L, LUC7L2 and LUC7L3 homology arm vectors.
gRNA vector
The pX459 vector courtesy of the Yeo lab (Van Nostrand et al., 2017a) was linearized with BbsI. The gRNA sequences were synthesized as 100 μM forward and reverse oligos by IDT. The oligos were annealed and diluted (1:200) before being ligated into the pX459 vector using NEB T4 ligase.
Nucleofection
The CRISPR plasmids were introduced into the cells using the Amaxa® Cell Line Nucleofector® Kit V following the manufacturer’s protocol. One million K562 cells with 2.5 μg of the appropriate vectors were transfected using nucleofector program T-016 on a Nucleofector I device (Lonza).
The cells were sorted by GFP expression (Sony MA900 Single Cell Sorter) and those with the highest expression were plated as single cells in 96 well plates. The clones were screened by western blot (as previously described) using the anti-LUC7L, anti-LUC7L2, anti-LUC7L3, anti-V5 (anti-V5, Bethyl, A190-120A), and FLAG M2 antibody (Sigma, F3165).
METHOD DETAILS
Western blot
Cells (6–7 million) were collected and suspended in cold RIPA buffer with HALT Protease Inhibitor (1:100)(Thermo Fisher, 78429). Tubes were incubated on ice for 30 minutes and mixed by vortex every 10 minutes. The lysates were spun in a pre-chilled microcen-trifuge (21,000 × g, 20 minutes, 4°C). Sample concentration was determined by BCA assay. NuPage Reducing agent and NuPage LDS were added and the samples were incubated at 70°C for 10 minutes before being loaded onto a 10% Bis-Tris gel. The gel was run in MOPS-SDS running buffer (200V, 1 hour, room temperature) and the proteins were transferred from the gel to PVDF membrane using a Novex wet transfer apparatus (30V, 1 hour, 4°C). The membrane was blocked in 5% milk (TBST 20 mM Tris, 150 mM NaCl, 0.1% Tween 20) for 30 minutes and then rinsed in TBST. The membrane was incubated on a rocker with primary antibodies in 5% milk overnight at 4°C. This was followed by washing the membrane three times with TBST, incubated with secondary antibody, and washed five times with TBST. SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher, 34077) was added to the membrane and exposed to film.
Immunoprecipitation mass spectrometry
Protein Extraction.
100 million cells from each cell line were pelleted and washed in cold PBSW (1x PBS supplemented with Protease Inhibitor Cocktail at 1:100 (Sigma, P8340)) and re-pelleted. To separate the nuclei from the cytoplasmic fraction, 1:50 NP-40 was added to cells and the mixture was shaken and centrifuged (8800 rpm, 10 minutes). The supernatant containing the cytoplasmic fraction was removed and the cells were treated with benzonase to degrade RNA and DNA.
The nuclei were incubated on ice for 90 minutes and vortexed periodically. 500mM NaCl, 2% NP-40, PBSW were added to the nuclear pellet and the mixture was homogenized and spun down. The supernatant was collected and the process was repeated. An additional 250mM NaCl, 1% NP-40, PBSW was added to the pellet, homogenized, incubated on ice, and spun down followed by collecting the supernatant. A final wash of the nuclear pellet was performed with PBSW. Nuclear extracts were collected and stored at −80°C.
Immunoprecipitation.
Protein A/G beads (SCBT, sc2003) were washed and incubated with FLAG M2 antibody (Sigma, F3165) for 1 hour at room temperature. Then, the antibody-bound protein A/G beads were incubated with 1% BSA in 1x PBS to block non-specific binding sites. Upon the third wash, 25mg of Dimethyl pimelimidate dihydrochloride powder (DMP) (Sigma, D8388-1G) in 1 mL of 200mM N-ethylmaleimide (NEM, Thermo Fisher Scientific) was added to the bound beads and incubated at room temperature for 30 minutes. The reaction was repeated 2 more times. 50mM Glycine/HCl was added after the third wash and the beads were washed extensively with PBSW + 2% NP-40 before immunoprecipitation.
The protein extracts were incubated with Protein A/G beads for 30 minutes at room temperature and then spun to clear the supernatant. The supernatants were incubated with the antibody-bound Protein A/G beads (4°C, overnight) and the bead-antibody-protein complexes were washed three times with IP buffer. 10% SDS was added to the beads and incubated (15 minutes, 37°C) followed by collecting the supernatant. The process was repeated twice with 1% SDS and the washes were combined.
Sample Preparation for Mass Spectrometry Analysis.
The immunoprecipitated samples were run on an SDS-polyacrylamide gel and stained with Coomassie Blue (Gel Code Blue, Pierce Chemical). Each lane was cut into eight sections for processing. Proteins in each section were reduced with 10mM dithiothreitol (Sigma-Aldrich, D0632) and alkylated with 55mM iodoacetamide (Sigma-Aldrich, I1149) then digested with trypsin. Peptides were extracted from gel slices three times with 60% acetonitrile and 5% formic acid/water. The peptide mixtures were dissolved in 1% formic acid and submitted for liquid chromatography-tandem mass spectrometry (LC-MS/MS) on an Orbitrap mass spectrometer.
RNA-Seq
RNA was isolated from LUC7-like knockdown and control cell lines as follows. Cells (6–7 million) were collected, pelleted, and kept on ice. High Pure RNA Isolation Kit (Roche, 11828665001) protocol was used following manufacturer’s instructions to purify the RNA.
Ribosomal RNA was depleted using the Illumina RiboZero Plus kit (Illumina, 20040525) and libraries were prepared for high throughput sequencing using the Illumina TruSeq kit according to manufacturer’s protocols. 100bp paired-end sequencing was performed on three biological replicates for LUC7-like knockdown and control cell lines on the Illumina HiSeq2500 at a depth of 60 million reads.
seCLIP-Seq
Experiments were performed using the single-end crosslinking immunoprecipitation (seCLIP) protocol (Van Nostrand et al., 2017b). K562 cells with CRISPR-tagged LUC7L, LUC7L2 and LUC7L3 were transferred to 10cm plates for crosslinking using a Stratalinker 2400 with 254 nm light at 400 mJ/cm2. Samples were sonicated using the Biorupter (Diagenode) on the “low” setting for 30 s intervals followed by 30 s pauses for 5 minutes at 4°C. DNase I (2 μl, Invitrogen AM2239) and RNase I (10 μl, 1:25 RNase I:PBS solution, Ambion AM2295) were added to the sample and mixed in the Thermomixer (1200 rpm, 37°C, 5 minutes). LUC7-like protein-RNA complexes were immunoprecipitated with V5 antibody (anti-V5, Bethyl, A190-120A) followed by stringent washes using high salt wash buffer (50 mM Tris-HCl pH 7.4, 1 M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, in RNase/DNase free H2O) and wash buffer (20 mM Tris-HCl pH 7.4, 10 mM MgCl2, 0.2% Tween-20, in RNase/DNase free H2O). Following washing, RNA was dephosphorylated using FastAP and T4 PNK and a 3′ RNA adaptor was ligated onto the RNA. Samples were denatured and resolved using SDS-PAGE on a 4%–12% Bis-Tris gel. The gel was transferred to PVDF membrane in NuPAGE transfer buffer and the region on the membrane corresponding to the respective LUC7-like proteins and 75 kDa above were isolated for RNA extraction. The isolated RNA was reverse transcribed (AffinityScript Kit 600107) followed by the addition of a 5′ linker to the cDNA. The final libraries were amplified using Q5 PCR mix (NEB) and the 175–350bp region corresponding to the library was excised from a 3% low-melting temp agarose gel. The libraries were quantified on the BioAnalyzer D1000 then sequenced on the Illumina HiSeq2500 at a depth of 100–150 million single-end reads for all pooled samples. This provides a depth of 16.7–25 million reads per sample.
QUANTIFICATION AND STATISTICAL ANALYSIS
Immunoprecipitation-mass spectrometry
The sequenced peptides from each gel slice were aligned to the human proteome (Uniprot database, September 2017 release) using MASCOT Daemon software (version 2.3.2) (Perkins et al., 1999). MASCOT peaks for each gel slice were filtered and downloaded: file = ../data/20191108/F008409.dat do_export = 1 prot_hit_num = 1 prot_acc = 1 pep_query = 1 pep_rank = 1 pep_isbold = 1 pe-p_isunique = 1 pep_exp_mz = 1 export_format = CSV _sigthreshold = 0.05 report = AUTO _server_mudpit_switch = 0.000000001 _ requireboldred = 1 search_master = 1 show_header = 1 show_mods = 1 show_params = 1 show_format = 1 protein_master = 1 prot_score = 1 prot_desc = 1 prot_mass = 1 prot_matches = 1 prot_cover = 1 prot_empai = 1 peptide_master = 1 pep_exp_mr = 1 pep_exp_z = 1 pep_calc_mr = 1 pep_delta = 1 pep_miss = 1 pep_score = 1 pep_expect = 1 pep_seq = 1 pep_var_mod = 1 pep_scan_title = 1 show_unassigned = 1 query_master = 1
The MASCOT peaks were further filtered by a minimum MASCOT ion score of 25 and peptide rank of 1. Data from all gel slices were combined for each biological sample. The proteins that displayed a 1.9-fold or greater difference in spectral counts between experimental conditions and input controls for both replicates were considered to be enriched. Enriched KEGG pathways were identified by inputting the enriched protein lists for each LUC7-like protein in STRING v11.0 (Szklarczyk et al., 2019).
seCLIP-Seq
Quality Control, Alignment, and CLIP-Peak Identification.
seCLIP fastq files were assessed for quality using FastQC and adapters were trimmed using Cutadapt 2.8 on python 3.6.2 (Martin, 2011). Repetitive elements were removed from the fastq files by alignment to the RepBase human repetitive genome using STAR 2.5.2b (Dobin and Gingeras, 2015). Fastq files were aligned to the hg19 genome (GRCh37), downloaded from ENCODE Reference Sequences, using STAR 2.5.2b. PCR duplicates were removed from aligned bam files using UMI_tools dedup 1.0.0 on python 3.3.5 (Smith et al., 2017). Aligned and de-duplicated eCLIP bam files downloaded from ENCODE and seCLIP LUC7-like bam files were run through the Yeo lab CLIPper pipeline for peak identification and normalization to the SMInput samples. P values generated from the normalization pipeline were calculated by Yates’ Chi-Square Test or Fisher Exact Test if the read number was below 5. This is described in more detail in (Van Nostrand et al., 2017a). Reproducible-shared peaks between CLIP replicates were identified using the IDR pipeline (IDR 2.0.4.2) as documented on GitHub Kundajelab/idr (Li et al., 2011) by ranking CLIP-peak fold-enrichment over input controls and described in more detail in (Van Nostrand et al., 2017a). The full CLIP pipeline including scripts and commands can be found at Github Yeolab/eclip (Van Nostrand et al., 2017b).
CLIP-Peak Annotation.
CLIP-Peak bed files were intersected with transcriptomic annotations using bedtools 2.29.0 (bedtools intersect -wao -a $peak.bed -b $annotation.bed) (Quinlan, 2014). Exons, miRNAs, lnRNAs, and snoRNAs were downloaded from the USCS table browser for hg19. The introns were downloaded from the IAOD database (Moyer et al., 2020). If a peak overlapped an exon-intron junction then it was categorized as binding to a constitutive or alternative exon splice site. Otherwise, if a peak overlapped with more than one annotation then the annotation with the highest proportion of overlapping CLIP-Peaks was used.
To determine shared binding sites between RNA-binding proteins, each RNA-binding protein specific CLIP-Peak bed file was intersected with all others using bedtools intersect. CLIP-Peaks that were less than 20 nucleotides in length were considered in cases of at least 25% overlap whereas peaks with more than 20 nucleotides needed at least five overlapping nucleotides to be considered shared binding sites between two CLIP-Peaks. We performed unsupervised hierarchical clustering using the complete-linkage measures of similarity and Euclidean measurement of distance.
Motif analysis
Enriched motifs were identified with homer 4.9.1 (Heinz et al., 2010) using the highly reproducible and shared CLIP-peaks between replicates that passed through the IDR pipeline (CLIP-peaks: log2fc ≥ 3, −log10 p value ≥ 3, and IDR value ≤ 0.01). (homer find-MotifsGenome.pl hg19 -rna -S 10 -len 5,6,7,8,9 -preparsedDir).
Meta-splice site analysis
′SS and 3′SS coordinates including 100 nucleotides in the exon and 200 nucleotides in the intron were isolated using the intron annotation file from IAOD (Moyer et al., 2020) and then split by strand to make splice site windows using bedtools 2.29.0 (makewindows -b $splice_site_pos_strand.bed -w 1 -s 1 -i winnum for positive stranded beds and makewindows -b $splice_site_neg_ strand.bed -w 1 -s 1 -i winnum – reverse for negative stranded bed files). Crosslink sites were obtained by trimming mapped reads to the first nucleotide using deeptools 3.1.2 (bamCoverage -b $CLIP.bam -Offset 1 -binSize 1–effective 2864785220–exactScaling -of bedgraph) (Ramírez et al., 2014). Crosslink bed files were intersected with splice site windows using bedtools intersect and each annotated splice site was then condensed into one meta 5′ and 3′ splice site by summing crosslink sites at each position. Each metagene was normalized to library size and then background binding of the SMInput was subtracted from the experimental CLIP to generate final metagenes.
RBP splicing maps were influenced by Yee et al. (2019). To generate RBP splicing maps, 5′SS and 3′SS of all splicing events measured by rMATS were split by strand and type of AS event and used to generate splice site windows using bedtools 2.29.0 (makewindows -b $splice_site_pos_strand.bed -w 1 -s 1 -i winnum for positive stranded beds and makewindows -b $splice_site_neg_ strand.bed -w 1 -s 1 -i winnum – reverse for negative stranded bed files. Reads mapping to significant AS events including 50 nucleotides in the exon and 200 nucleotides in the intron as well as flanking regions measured using rMATs were isolated for each type of AS event (Shen et al., 2014). These events were split based on inclusion and exclusion of a particular splicing event compared to control samples. Significant AS events were defined as having an |ΔPSI| ≥ 0.05 and a q-value of ≤ 0.05. Following isolation of significant AS events, experiments were normalized to mapped library size in counts per million.
Final metagenes were produced by subtracting the SMInput from the experimental CLIP and then normalized the binding profiles on the splicing map by dividing the value at each nucleotide position by the sum of the values at all of the positions in the splicing map. The top and bottom 2.5% binding value outliers at each nucleotide were removed to then calculate the mean at each nucleotide position to generate the final values for the splicing map. Control events were picked by isolating alternatively spliced events in the control K562 scrambled shRNA experiments measured by rMATs. These alternatively spliced events needed to have a Percent Spliced In (PSI) of ≥ 0.05 and ≤ 0.95 and had to occur in at least 39/52 control experiments. To generate percentiles for significance, we performed 1000 random samplings of the control events using the number of significantly excluded and included AS events upon LUC7-like KD respectively. These combined 2000 permutations were used to generate the 90th and 10th percentiles. For cassette exons, splicing maps consisted of the 5′SS of the upstream exon, 3′SS and 5′SS of the cassette exon being measured, and the 3′SS of the downstream exon. For retained introns, the 5′SS of the upstream exon and the 3′SS of the downstream exon were used.
CLIP-tag mapping to snRNAs
Following initial QC and adaptor processing, CLIP fastq files were aligned to a customized snRNA reference genome using STAR 2.5.2b. PCR duplicates were removed from LUC7-like seCLIP using UMI_tools dedup 1.0.0, while ENCODE eCLIP PCR duplicates were removed using a custom python script from the Yeo lab (Github Yeolab/eCLIP). Summed reads to each individual snRNA was normalized to mapped library size and the SMInput was then subtracted from the experimental CLIP to generate enriched snRNA binding values. Due to the limited number of reads mapped to the minor snRNAs, we performed a permutation test to test for significance of enrichment. Following mapping to the snRNAs, minor snRNAs were isolated and normalized to mapped library size. All of the samples were randomly assigned labels and randomly sampled 100,000 times to generate a distribution of binding to each individual minor snRNA. snRNA enrichment was considered significant if the normalized minor snRNA was above the 90th percentile of the distribution in both experimental replicates for each RBP.
Crosslink fold change enrichment heatmaps were made by generating crosslink site bed files for each individual snRNA using deeptools 3.1.2 (bamCoverage -b $snRNA_CLIP.bam -Offset 1 -binSize 1-effective 1259-exactScaling -of bedgraph). Each nucleotide position was normalized to mapped library size and then a log2 fold-change was generated by comparing experimental CLIPs to SMInput to determine nucleotide specific enrichment on snRNAs.
Enriched 5′ splice site strength and exon correlation
Nucleotide positions analyzed were isolated from crosslinking sites generated in the Meta-Splice Site Analysis. For the main figure, the −1 and +1 positions for each 5′SS were normalized to mapped library size in counts per million. Due to the relatively smaller library sizes for the input controls, a pseudo count of 1 was added to each nucleotide position that did not have any mapped reads. This was done to avoid a large number of false positives. 5′SSs were isolated that were enriched over the input control at the same nucleotide position, either −1 or +1 separately, in both replicates. Nucleotide sequences for each individual 5′SS were isolated for position −3 – +8 using bedtools fasta command. This was performed for the enriched LUC7L2, enriched LUC7L3, and Control 5′ splice sites which were all of the 5′SSs that were used for CLIP mapping. To generate U1 snRNA hydrogen binding scores, the H-Bond tool was used as described in Freund et al. (2003). Significance in difference of scores was determined using a Wilcoxon rank-sum test. For the supplementary figure, we used less stringent parameters. In this case no pseudo count was added to the input control before determining 5′SS enrichment.
To determine associations between enriched 5′ splice site crosslinking described above and upstream exons, the following was performed. The 99 nucleotides in the exon upstream of the −1 splice site position were isolated from crosslinking sites generated in the Meta-Splice Site Analysis. Enriched exons were determined using the method in the above paragraph for the main and supplementary figure respectively to generate an enriched exon list. This was compressed to a list of individual exons with at least enrichment at one nucleotide position. The coordinates for the 5′ splice site and upstream exon list were intersected to determine co-occurring crosslinking enrichment for the 5′ splice site and upstream exon. A Chi-square test of independence was performed to test for significant association between the two groups.
RNA-Seq
Quality Control and Alignment.
Fastq files were assessed for quality using FastQC then trimmed for adaptor content and clipped to a uniform length of 100bp using with Bbduk 36.92 (Bushnell, 2014). Bam files were produced by aligning the fastq sequences to the hg19 genome using Hisat2 2.0.4 and Samtools 1.9 (Kim et al., 2015; Li et al., 2009). Pre-processed Bam files generated from RNA-Seq of splicing factor knockdowns from the ENCODE database were downloaded for comparison analyses (Sloan et al., 2016).
Alternative Splicing Analysis.
To identify novel splice junctions that are unique to our LUC7-like knockdowns and the ENCODE splicing factor-knockdown transcriptomes, we created a custom splice-junction annotation (GTF file format) using StringTie (Kovaka et al., 2019). A custom annotation was created for each individual file using the Ensemble GRCh37 gene annotation as a guide. We then merged the annotation files to create a single annotation that includes known and novel splice junctions from all samples and the reference genome.
To generate a single file containing PSI values for each AS event for each sample, rMATS 4.0.1 was run on all samples (rmat.py–b1 halfbamfiles.txt–b2 restofbamfiles.txt–gtf all_stringtie_merged.gtf–od $outputdirectory–readLength 100–statoff -t paired–nthread 20) (Shen et al., 2014). rMATS AS events were filtered for transcript coverage (10 reads across both the skipped and included junctions in at least two samples), junction coverage (three reads over each junction in at least two samples) and alternative splicing potential (PSI between 10 and 90 in two samples). The creation of a custom gene annotation introduced some AS events that were not biologically relevant and/or were duplicated in multiple AS event categories. We filtered out SE events that were duplicate labeled as A3SS or A5SS. We also removed AS events labeled SE where we saw the incorrect pairing of 5′ and 3′ splice sites (the 5′ splice site from an upstream exon and the 3′ splice site from a different exon). Finally, non-canonical MXE events were removed. 91,728 of 294,227 AS events remained.
rMATS STAT was run to identify significantly dysregulated AS events for each splicing factor knockdown. ENCODE shRNA-Seq experiments were performed in batches, therefore although each experiment was paired with two control samples, many experiments shared control samples. rMATS STAT was run twice: rMATS STAT 1 paired each set of shRNA-Seq knockdowns with their ENCODE defined controls (LUC7-like samples were paired with shGFP controls) and was used to generate Figures 4 and 5A, rMATS STAT 2 compared shRNA-Seq experiments to grouped controls (polyA(+) selected controls, or total RNA controls) and was used to generate Figures 5B and 5C. Q-values were obtained by adjusting P values for multiple hypothesis testing using Bonferroni correction. Unsupervised hierarchical clustering (pheatmap) of ΔPSI values was used to identify batch effects from the shRNA-Seq experiments.
Differential gene expression analysis
Counts per gene were calculated from bam alignment files with featureCounts (subread v1.5.3) (featureCounts -s 2 -T 20 -p -t exon -g gene_id -a genes.gtf -o featureCounts.txt input.bam) (Liao et al., 2014). Differential expression analysis was performed using the R Bioconductor package edgeR 3.10 (Robinson et al., 2010). 13,396 genes expressed at ≥ 1 counts per million were kept for analysis and normalized using weighted trimmed mean of M-values (TMM). Benjamini-Hochberg procedure was applied to the list of p values generated by edgeR to generate False Discovery Rate (FDR) values to correct for multiple hypothesis testing. Significant differentially expressed genes were detected with a cutoff of a log2FC of ≥ 1 or ≤ −1 and FDR ≤ 0.05 or otherwise specified in figure legends.
Gene set enrichment analysis (GSEA)
GSEA was performed on input files that were generated using library normalized counts per million (CPM) expression data (natural scale) (Subramanian et al., 2005). (“java -cp gsea-3.0.jar -Xmx2G xtools.gsea.Gsea -res,” expressiondataset, “-cls,” phenotype_ experiment, “-gmx gseaftp.broadinstitute.org://pub/gsea/gene_sets_final/h.all.v6.1.symbols.gmt -collapse false -mode Max_ probe -norm meandiv -nperm 1000 -permute gene_set -rnd_type no_balance -scoring_scheme weighted -rpt_label $output -metric Signal2Noise -sort real -order descending -create_gcts false -create_svgs false -include_only_symbols true -make_sets true - median false -num 100 -plot_top_x 20 -rnd_seed timestamp -save_rnd_lists false -set_max 500 -set_min 15 -zip_report false - out,” outputdirectory, “-gui false,”sep = ” ”).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-LUC7L | Protein Tech | Cat#17085-1AP; RRID:AB_2878346 |
| Anti-LUC7L2 | Bethyl | Cat#A304-540A; RRID:AB_2620735 |
| Anti-LUC7L3 | Protein Tech | Cat#14504-1-AP; RRID:AB_2229967 |
| Anti-V5 | Bethyl | Cat#A190-120A; RRID:AB_67586 |
| Anti-FLAG M2 | Sigma | Cat#F3165; RRID:AB_259529 |
| Anti-GapDH | Santa Cruz | Cat#47724; RRID:AB_627678 |
| Anti-Histone H3 | Cell Signaling | Cat#9715; RRID:AB_331563 |
| Chemicals, peptides, and recombinant proteins | ||
| Dimethyl pimelimidate dihydrochloride | Sigma | Cat#D8388 |
| N-ethylmaleimide | Thermo Fisher Scientific | Cat#04500 |
| Critical commercial assays | ||
| Amaxa® Cell Line Nucleofector® Kit V | Lonza | Cat#VCA-1003 |
| Protein A/G beads | Santa Cruz | Cat#sc2003 |
| High Pure RNA Isolation Kit | Roche | Cat#11828665001 |
| Illumina RiboZero Plus kit | Illumina | Cat# 20040525 |
| AffinityScript | Agilent | Cat#600107 |
| Q5 PCR mix | NEB | Cat#M0492 |
| Reverse Transcription System | Promega | Cat#A3500 |
| Deposited data | ||
| LUC7-like seCLIP-seq | This Paper | ArrayExpress: E-MTAB-9709 |
| LUC7-like Knockdown RNA-seq | This Paper | ArrayExpress: E-MTAB-9709 |
| LUC7-like Co-IP Mass Spectrometry | This Paper | PRIDE: PXD022152 |
| ENCODE Knockdown RNA-seq | Sloan et al., 2016 | https://encodeproject.org |
| ENCODE CLIP-seq | Sloan et al., 2016 | https://encodeproject.org |
| Experimental models: Cell lines | ||
| K562 | ATCC | Cat#CCL-243 |
| K562 LUC7-like Knockdown Lines | This Paper | N/A |
| K562 LUC7-like (V5,FLAG,HA) Tagged Lines | This Paper | N/A |
| Oligonucleotides | ||
| seCLIP-seq adapters, See Table S7 | This Paper | N/A |
| Recombinant DNA | ||
| LUC7L shRNA Targeting Vector | SigmaAldrich | Cat# TRCN0000195589 |
| LUC7L2 shRNA Targeting Vector | SigmaAldrich | Cat# TRCN0000320721 |
| LUC7L3 shRNA Targeting Vector | SigmaAldrich | Cat# TRCN000075115 |
| Non-Targeting shRNA Vector | SigmaAldrich | Cat#SHC004 |
| pX459 vector | Van Nostrand et al., 2017a | N/A |
| HR130 vector | Van Nostrand et al., 2017a | N/A |
| Software and algorithms | ||
| MASCOT Daemon Software | Perkins et al., 1999 | https://www.matrixscience.com/daemon.html |
| STRING v11.0 | Szklarczyk et al., 2019 | https://string-db.org/ |
| Cutadapt 2.8 | Martin, 2011 | https://github.com/marcelm/cutadapt |
| STAR 2.5.2b | Dobin and Gingeras, 2015 | https://github.com/alexdobin/STAR |
| UMI Tools 1.0.0 | Smith et al., 2017 | https://github.com/CGATOxford/UMI-tools |
| IDR 2.0.4.1 | Li et al., 2011 | https://github.com/kundajelab/idr |
| Bedtools 2.29.0 | Quinlan, 2014 | https://github.com/arq5x/bedtools2 |
| Homer 4.9.1 | Heinz et al., 2010 | https://github.com/IGBIllinois/HOMER |
| Deeptools 3.1.2 | Ramírez et al., 2014 | https://github.com/deeptools/deepTools |
| rMATs 4.0.1 | Shen et al., 2014 | https://rmats.sourceforge.io/ |
| H-Bond | Freund et al., 2003 | https://www2.hhu.de/rna/index.php |
| Bbduk 36.92 | Bushnell., 2014 | https://github.com/BioInfoTools/BBMap |
| Hisat2 2.0.4 | Kim et al., 2015 | https://github.com/DaehwanKimLab/hisat2 |
| Samtools 1.9 | Li et al., 2009 | https://samtools.sourceforge.net/ |
| StringTie | Kovaka et al., 2019 | https://github.com/gpertea/stringtie |
| featureCounts v1.5.3 | Liao et al., 2014 | https://subread.sourceforge.net/ |
| edgeR 3.10 | Robinson et al., 2010 | https://www.bioconductor.org/packages//2.7/bioc/html/edgeR.html |
| GSEA | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
Highlights.
LUC7-like proteins are U1 snRNP components and alternative splicing regulators
LUC7L2 and LUC7L3 share an evolutionarily conserved role in 5′ splice site selection
LUC7-like proteins bind distinct factors regulating unique alternative splicing events
Loss of LUC7L2 alters spliceosome and glycolytic genes that may contribute to disease
ACKNOWLEDGMENTS
This work was funded by US NIH grants R01HL132071 (to R.A.P. and J.P.M.); R35HL135795 (to J.P.M.); F31HL131140 (to C.E.H.); P01HL146372, R01CA204373, and P30CA043703 (to Y.S); as well as a grant from the Vera and Joseph Dresner Foundation (to R.A.P.). We thank R.C. Dietrich for constructive criticism of the manuscript and technical advice, the Cleveland Clinic Lerner Research Institute Genomics and Proteomics cores for the sequencing of samples, and Dr. Gene Yeo for providing us with the HR130 and px459 vectors for the seCLIP-seq.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108989.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following reference appears in the Supplemental Information: Uhlén et al., 2015.
REFERENCES
- Agarwal R, Schwer B, and Shuman S (2016). Structure-function analysis and genetic interactions of the Luc7 subunit of the Saccharomyces cerevisiae U1 snRNP. RNA 22, 1302–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerman M, Fregoso OI, Das S, Ruse C, Jensen MA, Pappin DJ, Zhang MQ, and Krainer AR (2015). Differential connectivity of splicing activators and repressors to the human spliceosome. Genome Biol. 16, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram K, Agafonov DE, Dybkov O, Haselbach D, Leelaram MN, Will CL, Urlaub H, Kastner B, Lührmann R, and Stark H (2017). Cryo-EM Structure of a Pre-catalytic Human Spliceosome Primed for Activation. Cell 170, 701–713. e11. [DOI] [PubMed] [Google Scholar]
- Bradley T, Cook ME, and Blanchette M (2015). SR proteins control a complex network of RNA-processing events. RNA 21, 75–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B (2014). BBMap: a fast, accurate, splice-aware aligner (Ernest Orlando Lawrence Berkeley National Laboratory).
- Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, Shroff AS, Dickins RA, Vakoc CR, Bradner JE, et al. (2014). MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 25, 652–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Hoang A, Sinha R, Zhong XY, Fu XD, Krainer AR, and Ghosh G (2011). Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1-70K snRNP protein determines early spliceosome assembly. Proc. Natl. Acad. Sci. USA 108, 8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. (2015). Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 13, 1033–1045. [DOI] [PubMed] [Google Scholar]
- Del Gatto-Konczak F, Bourgeois CF, Le Guiner C, Kister L, Gesnel MC, Stévenin J, and Breathnach R (2000). The RNA-binding protein TIA-1 is a novel mammalian splicing regulator acting through intron sequences adjacent to a 5′ splice site. Mol. Cell. Biol 20, 6287–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, and Gingeras TR (2015). Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinformatics 51, 11.14.1–11.14.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester C, and Uetz P (2008). The FF domains of yeast U1 snRNP protein Prp40 mediate interactions with Luc7 and Snu71. BMC Biochem. 9, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortes P, Bilbao-Cortés D, Fornerod M, Rigaut G, Raymond W, Séraphin B, and Mattaj IW (1999). Luc7p, a novel yeast U1 snRNP protein with a role in 5′ splice site recognition. Genes Dev. 13, 2425–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund M, Asang C, Kammler S, Konermann C, Krummheuer J, Hipp M, Meyer I, Gierling W, Theiss S, Preuss T, et al. (2003). A novel approach to describe a U1 snRNA binding site. Nucleic Acids Res. 31, 6963–6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Hertel KJ, and Maniatis T (2001). The role of U2AF35 and U2AF65 in enhancer-dependent splicing. RNA 7, 806–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, et al. (2013). Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28, 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger CE, Daniels NJ, and Padgett RA (2020a). Spliceosomal factor mutations and mis-splicing in MDS. Best Pract. Res. Clin. Haematol 33, 101199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger CE, Moyer DC, Adema V, Kerr CM, Walter W, Hutter S, Meggendorfer M, Baer C, Kern W, Nadarajah N, et al. (2020b). Complex landscape of alternative splicing in myeloid neoplasms. Leukemia, Published online August 4, 2020. 10.1038/s41375-020-1002-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosono N, Makishima H, Jerez A, Yoshida K, Przychodzen B, McMahon S, Shiraishi Y, Chiba K, Tanaka H, Miyano S, et al. (2014). Recurrent genetic defects on chromosome 7q in myeloid neoplasms. Leukemia 28, 1348–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell VM, Jones JM, Bergren SK, Li L, Billi AC, Avenarius MR, and Meisler MH (2007). Evidence for a direct role of the disease modifier SCNM1 in splicing. Hum. Mol. Genet 16, 2506–2516. [DOI] [PubMed] [Google Scholar]
- Jerez A, Gondek LP, Jankowska AM, Makishima H, Przychodzen B, Tiu RV, O’Keefe CL, Mohamedali AM, Batista D, Sekeres MA, et al. (2012). Topography, clinical, and genomic correlates of 5q myeloid malignancies revisited. J. Clin. Oncol 30, 1343–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdain AA, Begg BB, Mick E, Shah H, Calvo SE, Skinner OS, Sharma R, Blue SM, Yeo GW, Burge CB, and Mootha VK (2021). Loss of LUC7L2 and U1 snRNP subunits shifts energy metabolism from glycolysis to OXPHOS. Molecular Cell 81. 10.1016/j.molcel.2021.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner B, Will CL, Stark H, and Lührmann R (2019). Structural Insights into Nuclear pre-mRNA Splicing in Higher Eukaryotes. Cold Spring Harb. Perspect. Biol 11, a032417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360, Published online March 9, 2015. 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotini AG, Chang CJ, Boussaad I, Delrow JJ, Dolezal EK, Nagulapally AB, Perna F, Fishbein GA, Klimek VM, Hawkins RD, et al. (2015). Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat. Biotechnol 33, 646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaka S, Zimin AV, Pertea GM, Razaghi R, Salzberg SL, and Pertea M (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Sub-group (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H, and Bickel PJ (2011). Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat 5, 1752–1779. [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Makarov EM, Owen N, Bottrill A, and Makarova OV (2012). Functional mammalian spliceosomal complex E contains SMN complex proteins in addition to U1 and U2 snRNPs. Nucleic Acids Res. 40, 2639–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, et al. (2012). Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 119, 3203–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley JL, and Krainer AR (2010). A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins). Genes Dev. 24, 1073–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J 17, 10–12. [Google Scholar]
- Moyer DC, Larue GE, Hershberger CE, Roy SW, and Padgett RA (2020). Comprehensive database and evolutionary dynamics of U12-type introns. Nucleic Acids Res. 48, 7066–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit S, Zhou Y, Shiue L, Coutinho-Mansfield G, Li H, Qiu J, Huang J, Yeo GW, Ares M Jr., and Fu XD (2013). Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol. Cell 50, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, et al. ; Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium (2011). Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med 365, 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, and Cottrell JS (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567. [DOI] [PubMed] [Google Scholar]
- Plaschka C, Lin PC, Charenton C, and Nagai K (2018). Prespliceosome structure provides insights into spliceosome assembly and regulation. Nature 559, 419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O, Bragado-Nilsson E, Koski T, and Séraphin B (2007). The U1 snRNP-associated factor Luc7p affects 5′ splice site selection in yeast and human. Nucleic Acids Res. 35, 5874–5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR (2014). BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinformatics 47, 11.12.1–11.12.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T (2014). deep-Tools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X, and Gouet P (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kohlstaedt LA, Damianov A, Rio DC, and Black DL (2008). Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat. Struct. Mol. Biol 15, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 111, E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H, Lane AA, Correll M, Przychodzen B, Sykes DB, Stone RM, Ballen KK, Amrein PC, Maciejewski J, and Attar EC (2013). Putative RNA-splicing gene LUC7L2 on 7q34 represents a candidate gene in pathogenesis of myeloid malignancies. Blood Cancer J. 3, e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC, Gabdank I, Narayanan AK, Ho M, Lee BT, et al. (2016). ENCODE data at the ENCODE portal. Nucleic Acids Res. 44, D726–D732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen JJ, Niemelä EH, Verma B, and Frilander MJ (2013). The significant other: splicing by the minor spliceosome. Wiley Interdiscip. Rev. RNA 4, 61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Van Nostrand EL, Gelboin-Burkhart C, Wang R, Pratt GA, Blue SM, and Yeo GW (2017a). CRISPR/Cas9-mediated integration enables TAG-eCLIP of endogenously tagged RNA binding proteins. Methods 118–119, 50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Nguyen TB, Gelboin-Burkhart C, Wang R, Blue SM, Pratt GA, Louie AL, and Yeo GW (2017b). Robust, Cost-Effective Profiling of RNA Binding Protein Targets with Single-end Enhanced Crosslinking and Immunoprecipitation (seCLIP). Methods Mol. Biol 1648, 177–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, Blue SM, Chen JY, Cody NAL, Dominguez D, et al. (2020). A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visconte V, O Nakashima M, and J Rogers H (2019). Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features. Cancers (Basel) 11, E1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson ME, Charenton C, and Nagai K (2020). RNA Splicing by the Spliceosome. Annu. Rev. Biochem 89, 359–388. [DOI] [PubMed] [Google Scholar]
- Yee BA, Pratt GA, Graveley BR, Van Nostrand EL, and Yeo GW (2019). RBP-Maps enables robust generation of splicing regulatory maps. RNA 25, 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. (2011). Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69. [DOI] [PubMed] [Google Scholar]
- Zhan X, Yan C, Zhang X, Lei J, and Shi Y (2018). Structures of the human pre-catalytic spliceosome and its precursor spliceosome. Cell Res. 28, 1129–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Dunker W, Yu YT, and Karijolich J (2018). The Role of Noncoding RNA Pseudouridylation in Nuclear Gene Expression Events. Front. Bioeng. Biotechnol 6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Licklider LJ, Gygi SP, and Reed R (2002). Comprehensive proteomic analysis of the human spliceosome. Nature 419, 182–185. [DOI] [PubMed] [Google Scholar]
- Zhu J, and Krainer AR (2000). Pre-mRNA splicing in the absence of an SR protein RS domain. Genes Dev. 14, 3166–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq and seCLIP-seq data reported in this paper have been deposited to ArrayExpress with the dataset identifier ArrayExpress: E-MTAB-9709. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PRIDE: PXD022152.