

Summary
Multiple sclerosis (MS) is an immune-mediated disease whose precise etiology is unknown. Several studies found alterations in the microbiome of individuals with MS, but the mechanism by which it may affect MS is poorly understood. Here we analyze the microbiome of 129 individuals with MS and find that they harbor distinct microbial patterns compared with controls. To study the functional consequences of these differences, we measure levels of 1,251 serum metabolites in a subgroup of subjects and unravel a distinct metabolite signature that separates affected individuals from controls nearly perfectly (AUC = 0.97). Individuals with MS are found to be depleted in butyrate-producing bacteria and in bacteria that produce indolelactate, an intermediate in generation of the potent neuroprotective antioxidant indolepropionate, which we found to be lower in their serum. We identify microbial and metabolite candidates that may contribute to MS and should be explored further for their causal role and therapeutic potential.
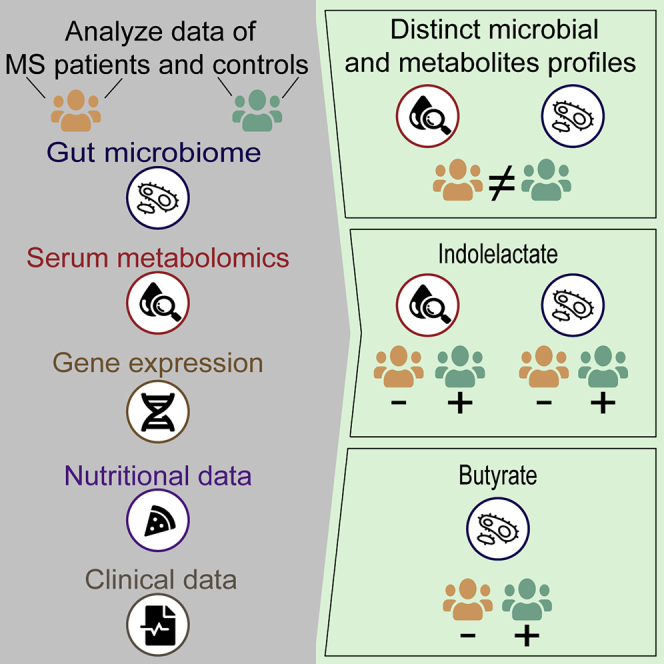
Graphical abstract
Highlights
Gut microbiota and metabolites profiles of individuals with MS differ from healthy controls
Individuals with MS have reduced serum levels of indolepropionate
Individuals with MS have reduced serum levels of indolelactate and its producing bacteria
Individuals with MS have lower levels of species and genes involved in butyrate production
Levi et al. perform a coupled analysis of gut microbiota, untargeted metabolomics, gene expression, nutritional records, and clinical data of individuals with MS. The results suggest that increasing the levels of butyrate-producing species and the levels of the tryptophan metabolites indolepropionate and indolelactate in individuals with MS may have potential benefits.
Introduction
Multiple sclerosis (MS) is a CNS disease affecting young adults with a world-wide prevalence of 60–120 per 100,000. MS etiology involves genetic and environmental factors as well as immune-mediated mechanisms.1, 2, 3 The gut microbiota, defined as the set of microorganisms that reside in the mucous membranes of the human intestine, has been suggested to play a role in the pathogenesis of neurological diseases, including MS. It has been shown that the gut microbiota can interact with the brain and cause neurochemical changes, regulates different behaviors, and modulates brain development, suggesting a role of the gut microbiota in MS pathogenesis.4, 5, 6, 7, 8 The gut microbiota can interact with the brain by immune activation; by endocrine and neural pathways, including vagal afferents; and by microbial metabolites acting directly or indirectly on the brain, in some cases passing the blood-brain-barrier.9, 10, 11 The effects of the microbiota on the brain can also be mediated by diet.12,13 Alterations in the gut microbiota and damaged intestinal barrier, also known as leaky gut syndrome, can cause dysfunction in the cerebellum and hippocampus.14,15 Conversely, changes in the gut can also positively affect the clinical course and symptoms of CNS disorders,16 and it has been shown in studies using experimental autoimmune encephalomyelitis (EAE), the most widely used animal model of MS, that treatment with probiotics suppressed development of EAE and reduced the severity of clinical symptoms.17, 18, 19
Several recent case-control studies found alterations in the gut microbiota of individuals with MS and suggested immunological links between dysbiosis and MS pathogenesis. Their main findings are shown in Table S1.20, 21, 22, 23, 24, 25, 26, 27, 28, 29 These studies vary in the number of individuals with MS who were followed from 722 to 60.27 Other studies used targeted or untargeted serum metabolomics to identify metabolites that are altered in individuals with MS compared with healthy controls (their main findings are shown in Table S130, 31, 32, 33, 34, 35, 36, 37, 38) or to distinguish MS from other neurological diseases.39, 40, 41
Here we performed a coupled analysis of gut microbiotas, untargeted metabolomics, peripheral blood gene expression, and clinical neurological data in a large cohort of individuals with MS with various disease types and different clinical stages of the disease. We used a machine-learning algorithm that accurately classified individuals with MS from controls using only microbiome data and found that butyrate-producing bacteria had the highest influence on the predictive models. Consistent with this result, we found that individuals with MS have significant depletion of butyrate-producing species and bacterial genes, even when aggregating all butyrate-producing bacteria.42 Similarly, a logistic regression model we developed using only serum metabolite data classified affected individuals from controls with nearly perfect accuracy and uncovered specific metabolites that were associated with the microbiome, some of which have been described previously as having a role in inflammatory processes. Differences in these metabolites, such as indolelactate, were also reflected in bacterial species that produce these metabolites, suggesting a role of microbiota-derived metabolites in MS. We further searched for associations between changes in bacterial species abundance and host gene expression levels and found associations that may be related to inflammation pathways.
Our coupled analysis allowed us to find altered bacteria and associations between the microbiota and metabolite products that may affect MS pathogenesis and provide a comprehensive source for potential therapeutic candidates.
Results
Cohort description and study design
To study the role of the gut microbiome and serum metabolites in MS, we recruited 187 individuals aged 18–75, comprising 129 individuals with MS followed at the Multiple Sclerosis Center, Sheba Medical Center, with different types of the disease and with different clinical manifestations, and 58 healthy controls (Figure 1A; Table S2). We performed metagenomic sequencing of rectal stool samples from the 187 participants and randomly sampled 5 million reads from each metagenomic sample to allow fair comparison across subjects (Table S3). The age and gender distributions differed between individuals with MS and controls (mean ± SD of 38.3 ± 11.8 versus 45.8 ± 12.5 for age of individuals with MS versus controls, respectively [Kolmogorov-Smirnov test, p < 0.004] and 27.9% versus 50% for percentages of males with MS versus controls, respectively [chi-square test, p < 0.004]). We accounted for these differences throughout our analyses.
Figure 1.

The gut microbiota of individuals with MS differs from that of healthy controls
(A) Illustration of our experimental design.
(B) Phylogenetic tree of analyzed bacteria, from phylum level (inner layer) to family, genus, and species (outer layer). Orange and green dots represent significant enrichment in individuals with MS and control participants, respectively. Background colors represent different phyla.
(C) Volcano plot of bacterial species that appear in at least 5% of the participants. The data are plotted as log (base 2) fold change versus the negative log (base 10) of the adjusted p value (computed with Mann-Whitney U test). Each dot represents one species. Orange dots represent species enriched in individuals with MS, and green dots represent species enriched in control participants. The dashed horizontal blue line indicates the threshold for statistical significance (p ≤ 0.05 after 5% FDR correction).
(D) SHAP summary plot showing the distribution of the effect of each feature on the model output for a prediction of each group (MS versus control). The y axis indicates the feature name in order of importance (defined by the sum of SHAP value magnitudes over all samples) from top to bottom. Shown on the x axis is the SHAP value, indicating whether the effect of that feature is associated with MS (higher values) or a control prediction (lower values). Each point represents one subject. The color represents the feature value (red, high; blue low). Low levels of Coprococcus catus (s4669) and another unknown species (which is close to Roseburia inulinivorans, s4939), increase the probability of MS group prediction by the model.
(E and F) Receiver operating characteristic curve (E) and precision recall curve (F) for distinguishing individuals with MS from control participants using gradient boosting decision trees; average of 100 models. Light lines represent results of individual models, and dark lines represent the mean of 100 models. The gray curve represents baseline predictions using only age, gender, and BMI as features. Area under curve = 0.58 (95% CI = [0.573, 0.582]), average precision = 0.74 (95% CI = [0.741, 0.746]). The Blue line represents prediction results using microbial features. Area under curve = 0.70 (95% CI = [0.695, 0.705]), average precision = 0.82 (95% CI = [0.81, 0.82]). The pink line represents prediction results using bacterial abundance and age, gender, and BMI features. Area under curve = 0.83 (95% CI = [0.825, 0.835]), average precision = 0.92 (95% CI = [0.919, 0.928]). C. catus, Coprococcus catus (4669); R. inulinivorans∗, unknown species closest to Roseburia inulinivorans (4939); C. sp.CAG, Clostridium species CAG 264 (5121); R. sp, Ruminococcus species (4552); G. pamelaeae, Gordonibacter pamelaeae (14824).
For a partial subgroup of 90 individuals with MS (Table S2), we used untargeted mass spectrometry to profile 1,251 metabolites from their serum samples (Tables S4 and S5), covering a wide range of biochemicals, including lipids, amino acids, xenobiotics, carbohydrates, peptides, nucleotides, and ∼30% unknown compounds. We compared it with the metabolite profile of 90 age-, gender-, and BMI-matched healthy controls from another study43 by using a quantile normalization process for which we used 35 healthy controls from our study (STAR Methods).
For a subgroup of 49 participants (36 individuals with MS and 13 controls), we also used mRNA sequencing (mRNA-seq) to measure the expression of 22,929 human genes (Table S6). Furthermore, 44 of the individuals with MS recorded their entire nutritional intake for at least 3 days and in real time using a specialized app provided to them (Table S7; STAR Methods). Together, they logged 4,806 different food items. We also collected a comprehensive profile from all participants, including food frequency, lifestyle, and medical background questionnaires (STAR Methods).
The gut microbiota of individuals with MS differs from that of healthy controls
To test for differences between the microbiome of individuals with MS and healthy controls, we computed the relative abundances of species (Table S3) using a much-expanded set of genomes published recently44 and performed a Mann-Whitney U test. Of 686 species tested, we found 23 species that differed significantly between the two groups (p < 0.05 after 5% false discovery rate [FDR] correction; Figures 1B and 1C; Table S3). These differences are unlikely to occur as a result of the different age, gender, and BMI distributions between the two groups because 19 of these 23 species do not correlate significantly with these parameters across a 1,361-healthy person cohort,43 and the remaining 4 species have a low correlation (Pearson R2 < 0.02; Table S3).
Next we wanted to find out whether a machine-learning algorithm based only on microbiome data can distinguish individuals with MS from healthy controls. We used gradient boosting decision trees45 because it is a powerful model that can capture nonlinear interactions between bacteria. As microbiome features, we used species, genus, and family level relative abundances computed from the metagenomics data. Performance was evaluated using a standard 10-fold cross-validation scheme where the labels (MS/control) of subjects in each fold were predicted using a model trained on the microbial data of the participants from all other folds. We obtained significant predictive power (ROC [receiver operating characteristics] AUC [area under the curve] = 0.7, AUPR [area under the precision-recall curve] = 0.74, using species- and family-level relative abundances as features; permutation test, p < 0.002 for ROC AUC and AUPR for 1,000 label permutations), suggesting differences between the microbiomes of individuals with MS and controls. To verify that our model’s predictive power was not reliant solely on age, gender, and BMI differences between the cohorts, we compared a model based only on age, gender, and BMI with a model that combines both of these features and microbiome-based features and indeed found the latter to perform better (ROC AUC of 0.58 versus 0.83, respectively; Mann-Whitney U test, p < 10−34; Figures 1E and 1F).
Individuals with MS have lower levels of species and genes involved in butyrate production
To interpret our machine-learning models and find the features that were important for driving its predictions, we used Shapley additive explanations (SHAP), a feature attribution analysis tool that assigns each feature an importance value (SHAP value) for a particular prediction (STAR Methods). Notably, bacteria that produce butyrate, a short-chain fatty acid (SCFA) suggested to have a role in MS pathogenesis,46 had the highest influence on the model predictions. The two bacteria with the highest SHAP values were Coprococcus catus (s4669) and an unknown species whose closest other species is Roseburia inulinivorans (s4939). Both bacteria have been shown previously to produce butyrate.42 Low levels of both species result in higher predictions of the model for the MS group (Figure 1D). More broadly, checking for a difference in the sum of the abundances of all butyrate-producing bacteria, we found lower levels of butyrate-producing bacteria in individuals with MS (mean relative abundance of 0.056 versus 0.083 for individuals with MS versus controls, respectively; Mann-Whitney U test, p < 0.004; Figure 2A). Six butyrate-producing species were depleted significantly in comparison with the control group (Mann-Whitney U test, p < 0.05 after 5% FDR correction). Here, too, we validated that these results are not due to age, gender, and BMI differences between affected individuals and controls (STAR Methods). Among individuals with MS, no correlation was found between levels of butyrate-producing bacteria and being treated with disease-modifying drugs (DMDs) or treatment duration (STAR Methods).
Figure 2.

Individuals with MS have lower levels of butyrate-producing bacteria and genes involved in butyrate production
(A) Boxplots (center, median; box, interquartile range [IQR]; whiskers, 1.5 × IQR) showing bacterial relative abundance for 129 individuals with MS and 58 controls for butyrate-producing species. The sum of the abundance of all butyrate-producing species is shown on the left. Other species are sorted from left to right by their significance level, which is shown at the top of the boxplot (Mann-Whitney U test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n.s., not significant).
(B) Boxplots (center, median; box, IQR; whiskers, 1.5 × IQR) showing relative abundance for 129 individuals with MS and 58 controls for butyrate-producing genes, sorted from left to right by their significant level, which is shown at the top of the boxplot (same significant markings as in A).
(C) The metabolic pathways that convert proteins and carbohydrates into butyrate. The bacterial genes (with their KO [KEGG orthology] identifiers) in the green boxes were enriched in controls (Mann-Whitney U test, p < 0.05). The bacterial genes (with their KO identifiers) in the orange boxes were enriched in individuals with MS (Mann-Whitney U test, p < 0.05). The bacterial genes in the striped boxes were not enriched in any of the groups.
K00656, formate C-acetyltransferase; K00244, fumarate reductase flavoprotein subunit; K00626, acetyl-CoA C-acetyltransferase; K18119, succinate-semialdehyde dehydrogenase; K00074, 3-hydroxybutyryl-CoA dehydrogenase; K00043, 4-hydroxybutyrate dehydrogenase; K01715, enoyl-CoA hydratase; K18122, 4-hydroxybutyrate CoA-transferase; K00209, enoyl-[acyl-carrier protein] reductase/trans-2-enoyl-CoA reductase; K14534, 4-hydroxybutyryl-CoA dehydratase/vinylacetyl-CoA-Delta-isomerase; K01034, butyryl-CoA:acetate CoA transferase; K00929, butyrate kinase; B. crossotus, Butyrivibrio crossotus; F. prausnitzii, Faecalibacterium prausnitzii; R. sp CAG 18, Roseburia sp. CAG 18; R. inulinivorans CAG 15, Roseburia inulinivorans CAG 15; E. rectale CAG 36, Eubacterium rectale CAG 36; E. hallili CAG 12, Eubacterium hallili CAG 12; E. ventriosum, Eubacterium ventriosum; A. hadrus, Anaerostipes hadrus; R. intestinalis CAG 13, Roseburia intestinalis CAG 13; L. bacterium CAG 25, Lachnospiraceae bacterium CAG 25; E. sp CAG 146, Eubacterium sp. CAG 146; C. sp CAG 146, Coprococcus sp. CAG 131; F. sp, Faecalibacterium sp.; O. sp CAG 241, Oscillibacter sp. CAG 241.
To gain further mechanistic insight into the butyrate association, we next searched for differences at the level of butyrate-producing bacterial genes. We used the DIAMOND algorithm47 applied to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database48 to infer the functions of the microbial communities of the MS and control groups (STAR Methods; Table S3). We first compared the abundances of two known bacterial gene biomarkers for butyrate-producing communities, butyryl-CoA:acetate CoA transferase (K01034) and butyrate kinase (K00929).49 Consistent with our results, we found lower levels of the sum of these two gene groups in individuals with MS compared with controls (Mann-Whitney U test, p < 0.021; Figures 2B and 2C).
Notably, lower relative abundance in individuals with MS was not reflected in serum levels of butyrate, which were similar between individuals with MS and controls (Mann-Whitney U test, p > 0.1). We further sought to test whether diet is correlated with the sum of the abundances of all butyrate-producing bacteria. Of the 129 individuals with MS, 44 recorded their nutritional intake (Table S7; STAR Methods). Although high-fiber diets may increase butyrate levels,50 no correlation was found between fiber consumption per day or per calorie and the sum of the abundances of all butyrate-producing bacteria (Spearman R = 0.003, p > 0.97; Spearman R = −0.1, p > 0.53). No significant difference was found in the sum of the abundances of all butyrate-producing bacteria between individuals with a high-fiber diet (at least 25 g of fiber per day51 compared with other affected individuals; Mann-Whitney U test, p > 0.1). We also found no correlation between caloric consumption from specific food categories that are high in fiber (vegetables and their products, nuts, and seeds and their products) and the sum of the abundances of all butyrate-producing bacteria (Spearman P > 0.05). In a much larger cohort of 23,191 individuals from Israel and the United States who sent their samples to a consumer microbiome company and signed an appropriate consent form,52 we also did not find any correlation between the sum of the abundances of all butyrate-producing bacteria and fiber consumption per day (Spearman R = −0.02, p > 0.015). Finally, no correlation was found between the sum of the abundances of all butyrate-producing bacteria and any other major diet components (Spearman p > 0.05 for both cohorts).
Serum metabolites of individuals with MS differ from those of healthy controls
We next examined the metabolomics data we collected (Tables S4 and S5). Following quantile normalization and its conservative cutoffs (STAR Methods), we were left with 517 metabolites of 1,251 metabolites and found that levels of 42 metabolites differed significantly between individuals with MS and controls (Mann-Whitney U test, p < 0.05 after 5% FDR correction; Figure 3A). These metabolites mostly comprise lipids and amino acids but also include xenobiotics, carbohydrates, peptides, nucleotides, and unknown compounds (Figure 3B). Notably, the most significantly different metabolites were not reported in previous studies. For example, the most significantly differing metabolite, β-hydroxyasparagine (Figure S1; Mann-Whitney U test, p < 0.004 after 5% FDR correction), a modified asparagine amino acid, has so far not been associated directly with MS, to the best of our knowledge. Sphingosine 1-phosphate (S1P), known to be involved in immunological and neurological processes through interaction with the S1P receptor (S1PR),53 was significantly lower in individuals with MS (Mann-Whitney U test, p < 0.007 after 5% FDR correction); fingolimod, which modifies this signaling pathway by S1PR internalization, is an approved treatment for MS. The difference in SP1 could not be associated with treatment in our cohort because only one individual with MS received the medication. Some of the differing metabolites agree with previous studies. For example, carnitine, which has anti-inflammatory and antioxidative properties, showed significantly lower levels in individuals with MS (Figure S1; Mann-Whitney U test, p < 0.007 after 5% FDR correction), as shown previously by Kasakin et al.36
Figure 3.

Global differences between serum metabolites of individuals with MS and healthy controls
(A) Volcano plot of serum metabolites. The data are plotted as log (base 2) fold change versus the −log (base 10) of the adjusted p value (computed with Mann-Whitney U test). The threshold for significant adjusted p values is shown as a dashed blue line (p ≤ 0.05 after 5% FDR correction). Orange and green dots represent different metabolites that are significantly higher in individuals with MS or controls, respectively.
(B) Pie chart of pathways of the significantly different metabolites. The outer layer represents biochemical groups, and the inner layer represents sub-pathways (Table S6). Significantly different metabolites belong to 6 biochemical groups.
(C) Heatmap showing the explained variance of metabolite levels (R2) by different features. The 12 shown metabolites were significantly different between individuals with MS and healthy controls and highly correlated with one of the feature groups (e.g., age, gender, long-term dietary habits [diet], and microbiome). Feature groups are ordered from left to right, from the feature with the highest number of associated metabolites (gender) to the feature with the lowest number of associated metabolites (cardiometabolic).
(D and E) Receiver operating characteristic curve (D) and precision recall curve (E) for prediction of each group (MS versus control) using a logistic regression model; average of 100 models. Light lines represent results of individual models, and dark lines represent the mean of 100 models. The gray curve represents baseline prediction with covariates (age, gender, and BMI) as features. Area under the curve = 0.488 (95% CI = [0.486, 0.489]), average precision = 0.513 (95% CI = [0.511, 0.515]). The blue curve represents prediction results using serum metabolites as features. Area under curve = 0.965 (95% CI = [0.963, 0.966]), average precision = 0.97 (95% CI = [0.969, 0.971]). The pink curve represents prediction results using age, gender, and BMI together with microbial features. Area under the curve = 0.965 (95% CI = [0.964, 0.966]), average precision = 0.97 (95% CI = [0.969, 0.971]). M, metabolites.
See also Figure S1.
We next searched for feature groups (e.g., long-term dietary habits [“diet”], microbiome composition, lifestyle, age, gender, and time of day) associated with the differential metabolites to understand which of the feature groups could explain a large fraction of the variance of the altered metabolites and highlight the potential of specific feature groups as long-term dietary habits and microbiome as their key determinants. The associations were calculated based on a large cohort of 521 serum samples from 491 healthy individuals, for whom data regarding host genetics, the gut microbiome, cardiometabolic parameters, long-term dietary habits, lifestyle, anthropometrics measurements, and medications were collected54 (STAR Methods). Of the 42 significantly altered metabolites (Figure 3A), 12 were associated with one of the feature groups (based on the ability of a gradient boosting model to significantly predict the metabolite levels using only data from the relevant feature group, with R2 > 0.1;54 Table S5). Notably, indolepropionate is associated with the microbiome, which explains 15% of its variance (R2 = 0.15; Figure 3C), and four metabolites are associated with long-term dietary habits (stachydrine, R2 = 0.22; 1-stearoyl-2-docosahexaenoyl-GPC, R2 = 0.12; 3-hydroxyhippurate, R2 = 0.11; arabonate/xylonate, R2 = 0.1).
Finally, we tested the ability of a cross-validation logistic regression model based only on metabolomics data to distinguish individuals with MS from healthy controls and found nearly perfect separation (AUC ROC = 0.965, 95% confidence interval [CI] = [0.963, 0.966]; Figures 3D and 3E). Overall, these results suggest that metabolite profiles are highly different between individuals with MS and controls.
Influence of the gut microbiome via serum metabolites
To further understand how metabolites produced by the gut microbiota may exert an effect, we focused on metabolites that we found previously to be associated with the microbiome based on the ability of a gradient boosting model to significantly predict the metabolite using only microbiome data.54 A total of 26 metabolites of our 517 are associated with the microbiome by this analysis (R2 > 0.1, p < 10−25). To test for differences between each of the 26 metabolite levels of individuals with MS and healthy controls, we performed a Mann-Whitney U test and applied FDR correction for multiple-hypothesis correction. We found that indolepropionate, a potent neuroprotective antioxidant,55 which is significantly predicted by the microbiome (R2 = 0.15, p < 10−49), was significantly lower in the serum of individuals with MS (Figures 4A and 4B; Mann-Whitney U test, p < 0.03 after 5% FDR correction). Indolelactate, an intermediate in the process of degrading tryptophan to indolepropionate,56 was also significantly lower in individuals with MS (Figures 4A and 4B; Mann-Whitney U test, p < 0.03 after 5% FDR correction). Summation of the relative abundance of the nine bacterial species that produce indolepropionate56 did not show any difference between individuals with MS and controls (Mann-Whitney U test, p > 0.45), but the total relative abundance of the 24 species that produce indolelactate56 was significantly lower in individuals with MS (mean relative abundance of 0.073 versus 0.094 for individuals with MS versus controls, respectively; Mann-Whitney U test, p < 0.01; Figure 4C). This result suggests that the lower abundance of indolelactate-producing species in individuals with MS may result in lower serum levels of indolelactate and indolepropionate because lower serum levels of these metabolites may contribute to the ongoing inflammatory processes in individuals with MS. We further evaluated whether a high-protein diet correlates with the total abundance of all bacteria that produce indolelactate or indolepropionate but did not find significant correlations (Spearman p > 0.2 for both).
Figure 4.

Individuals with MS have lower levels of bacterial species that produce indolelactate
(A) Tryptophan degradation pathway.56 Gray, similar levels in individuals with MS and controls; green, significantly higher levels in controls; white, not measured. Indolelactate and indolepropionate serum levels were higher in controls compared with individuals with MS. Bacterial species that produce indolelactate are lower in abundance in individuals with MS.
(B) Boxplots (center, median; box, IQR; whiskers, 1.5 × IQR) showing indolepropionate serum levels for 90 individuals with MS and 90 controls.
(C) Boxplots (center, median; box, IQR; whiskers, 1.5 × IQR) showing bacterial relative abundance for 129 individuals with MS patients and 58 controls for bacterial species that produce indolelactate.56 The sum of the relative abundance of all the species is shown on the left. Other species are sorted from left to right by their significance level, which is shown at the top of the boxplot (Mann-Whitney U test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). ILA, indolelactate.
We also found that p-cresol sulfate, the major component of urinary myelin basic protein (MBP)-like material (MBPLM), which is the immunochemically homolog to MBP peptide57 and is also associated with EAE,58 was characterized by high microbiome association (R2 = 0.38, p < 10−208) and had higher levels in individuals with MS (Mann-Whitney U test, p < 0.12 after 5% FDR correction). We did not find significant differences between individuals with MS and controls in the relative abundances of bacterial species associated with p-cresol (STAR Methods). There was also no significant correlation between the relative abundances of bacterial species associated with p-cresol and consumption of a vegetarian diet (Spearman p > 0.2).
To further test for associations between microbiome composition and the metabolite profile, we searched for abundances of bacterial species that correlate with metabolite levels in individuals with MS. We did not find significant differences (Pearson p > 0.1 for all correlations).
Associations between gut microbiota and expression of human genes
Next we examined peripheral blood gene expression data (Table S6) in search for genes that correlate with bacterial species we found to significantly differ between individuals with MS and controls (p < 0.05 after 5% FDR correction; Figures 1B and 1C). Following the normalization process and conservative cutoffs, we remained with 16,837 genes (STAR Methods). After correction for multiple hypothesis, we found that BTF3L4, a homolog of the basic transcription factor 3 (BTF3), significantly correlated with Lawsonella (s3665) which we found to have higher levels in individuals with MS (p < 0.03 after 5% FDR correction); Spearman R2 > 0.47, p < 0.02 after 5% FDR correction; Figure 5).
Figure 5.

Significant correlation between Lawsonella (s3665) relative abundance and BTF3L4 gene expression levels (Spearman R = 0.68, p < 0.02 after 5% FDR correction)
Shown on the x axis is the expression level of the BTF3L4 gene. Shown on the y axis is the relative abundance of Lawsonella (s3665). Each point represents one subject, 49 in total.
No gut microbiota differences between MS disease subtypes and stages
Because individuals with MS are heterogeneous in terms of clinical outcomes and disease progression, we searched for differences in the gut microbiota of individuals with a specific disease type or stage. We first searched for differences along disease progression by comparing (1) early-stage individuals defined as having radiologically isolated syndrome (RIS), who are people with brain and/or spinal cord MRI findings suggestive of MS but without clinical symptoms and with normal neurological findings;59 (2) individuals defined as having clinically isolated syndrome (CIS), who present with the first clinical neurological episode suggestive of MS;60,61 (3) individuals with relapsing remitting MS (RRMS), characterized by relapses and remissions;62 and (4) individuals with a progressive disease course, characterized by steadily increasing neurological disability independent of relapses. We examined whether pairs of individuals from the same group are more similar, in terms of their microbiome composition, than pairs of individuals where each person is from a different group but found no significant differences (Mann-Whitney U test, p > 0.05 for all possible comparisons). Second, each individual received a predictive score from the MS-control microbiome-based-predictor we described earlier, and we examined whether there was a difference in peoples’ scores between the various disease types groups. Here, too, we did not find any significant differences between the different groups, suggesting that no MS group was significantly more distinct than the control group compared with any other MS group (Mann-Whitney U test, p > 0.1 after 5% FDR correction for all possible comparisons). We also compared the microbiome of a group of individuals during an acute relapse with the other groups and repeated the analysis but found no significant differences.
Next we wanted to find out whether deterioration across time in clinical status is related to gut microbiome measurements taken at baseline. We defined deterioration as a relapse during the 1-year follow-up (defined as a monophasic clinical episode with symptoms reported by the affected individual and objective neurological findings typical of MS;62 n = 22), an increase in neurological disability 1 year after the microbiome sample was taken, as measured by expanded disability status scale (EDSS);63 n = 26), or both (n = 10). We defined individuals as being without deterioration when they had no relapses and were neurologically stable 1 year after their microbiome sample was taken (n = 55). We did not find any difference between the two groups (ROC AUC < 0.52).
Finally, because immunomodulatory treatment can influence the microbiome profile and the metabolite levels,22,64,65 we tested whether the observed differences in microbiome composition and the metabolite profiles between individuals with MS and healthy controls were unrelated to MS treatment. Our results suggest that the effect of immunomodulatory treatment on the altered microbiota and on the significantly different metabolites we found in individuals with MS is negligible (STAR Methods; Figures S2 and S3).
Discussion
In the current study, we profiled the microbiome, the serum metabolome, and peripheral blood gene expression together with clinical and demographic variables of individuals with MS with diverse disease stages and types and found significant differences across all of these profiles compared with healthy participants. We used machine-learning algorithms that accurately distinguish MS patients from controls using microbiome data alone or using only metabolomics data. We uncovered significantly different bacterial species and metabolites, and one correlation between human gene expression levels and bacteria was altered in individuals with MS. Our findings suggest potential immunopathogenic mechanisms by which the gut microbiota may affect MS.
Key findings: Indolelactate and butyrate differences
One of the key differences we found was that indolepropionate and indolelactate, an intermediate in the process of degrading tryptophan to indolepropionate, were lower in the serum of individuals with MS. Indolepropionate protects neurons against oxidative damage and death55 and has a negative association with inflammation markers.66 It is a product of tryptophan degradation by the gut microbiome.56 The tryptophan metabolism pathway influences inflammatory and neurodegenerative processes and has been suggested to play a role in MS, although the focus has been on the kynurenine pathway of tryptophan degradation.67 Tryptophan catabolites have been reported to suppress CNS inflammation in EAE via aryl hydrocarbon receptor (AHR), which is expressed widely by immune cells and causes naive T cells to differentiate to regulatory T (Treg) cells or TH17 cells.68 Activation of AHR by tryptophan metabolites induces expression of the interleukin-10 (IL-10) receptor on intestinal epithelial cells, suggesting an anti-inflammatory phenotype.69 Reduced serum levels of indolelactate in pediatric individuals with MS compared with controls have been reported,30 but here we found that indolelactate was also significantly lower in adults with MS, as were bacteria that produce indolelactate. This provides independent support for the validity of this finding from the serum metabolite and gut microbiota aspects.
Another key difference we found was depletion of the levels of butyrate producing bacteria. Butyrate enhances intestinal barrier function70,71 and has anti-inflammatory properties that are realized through promotion of peripheral Treg cell generation,72, 73, 74, 75 and inhibition of activation of nuclear factor κB (NF-κB), the transcription factor that regulates production of proinflammatory enzymes and cytokines,76,77 induces secretion of the anti-inflammatory cytokine IL-10 in dendritic cells and macrophages. Butyrate-induced Treg cell differentiation was driven by augmented histone H3 acetylation at the FoxP3 promoter,74,75 a transcription factor that induces differentiation of CD4+ T cells toward Treg cells. In a mouse model of MS, treatment with butyrate suppressed demyelination and enhanced remyelination,78 implying that butyrate-producing bacteria may have beneficial effects in treatment or prevention of MS. These results agree with several studies that found depletion in several butyrate producing bacteria in individuals with MS22,24,27,29,79 as well as in other autoimmune and inflammatory diseases.80,81, 82, 83 We further empowered these findings by showing that this alteration holds when summing the abundance of all butyrate-producing bacteria as well as when considering only butyrate-producing genes and by showing that two butyrate-producing bacteria also had the highest SHAP value and were thus the main drivers in the machine-learning algorithm we used for distinguishing individuals with MS from controls. The fact that there were no significant differences in serum levels of butyrate between individuals with MS and controls might be related to butyrate utilization by endothelial cells in the colon.84 One possible way to further explore the differences in butyrate producers is to perform targeted liquid chromatography-mass spectrometry (LC-MS) on fecal samples that show enriched or depleted butyrate producers to see whether there is an associated change in colonic butyrate levels.
Notably, secretion of these metabolites may be impaired by excess consumption of dietary components. Indolepropionate is a product of gut microbiota degradation of the essential amino acid tryptophan. Although most of our dietary protein intake is absorbed in the small intestine, some of it reaches the colon, where it can be degraded by the microbiome.85 If the diet contains low levels of tryptophan, then not enough tryptophan will reach the colon, low levels of tryptophan catabolites will be produced, and the anti-inflammatory advantages of these compounds will be minimal. As for butyrate, a high-fiber diet has been suggested to be related to higher levels of butyrate-producing-species50 and to increase fecal butyrate levels.86 Despite the plausible role of diet in determining the levels of these metabolites, we did not find correlations between their levels and diet, which may also reflect the lower number of participants who recorded their nutritional intake for at least 3 days (n = 44) or inherent difficulties and noise with the collection of accurate dietary information.
Because tryptophan metabolites may induce anti-inflammatory processes and suppress inflammatory responses, our results suggest that increasing the levels of butyrate-producing species or tryptophan metabolites in individuals with MS may have potential benefits. Alternatively, prebiotics such as fiber could be used to enrich strains that are deficient in the host.
Other differentially abundant metabolites as possible attractive therapeutic options
Some of the metabolites we found to be significantly different between individuals with MS and healthy controls have so far not been associated with MS. For example, β-hydroxyasparagine, which was lower in serum of individuals with MS, is a modified asparagine amino acid that appears in posttranslational modification of calcium-binding EGF-like domains. The modified amino acid residue is found in fibrillin-1,87 which is the major constituent of microfibrils affecting an autoimmune response.88
We found choline phosphate to be significantly lower in individuals with MS. Ottenlinger et al.89 have shown that conjugation of a choline phosphate group to ceramide leads to formation of sphingomyelin, which may play an important role in MS.
p-Cresol sulfate, the major component of urinary MBPLM, which is immunochemically homologous to MBP peptide,57 is higher in serum of individuals with MS, and we have reported previously that its levels may be partly determined by the gut microbiota.54 Elevated levels of MBPLM have been observed in individuals with primary progressive MS and secondary progressive MS.90, 91, 92 p-Cresol sulfate can also cause release of endothelial microparticles (EMPs), which, in turn, activate several inflammatory and oxidative stress pathways and can lead to cellular dysfunction.93 Although p-cresol forms from the non-essential amino acids phenylalanine and tyrosine, it can represent the importance of diet because it is generated by gut bacteria acting on food components that are not absorbed in the small intestine. A study that examined p-cresol sulfate levels in omnivores compared with vegetarians found that this metabolite is significantly lower in vegetarians. In an intervention study of individuals with chronic kidney disease, fiber supplementation was found to decrease p-cresol sulfate levels in plasma.94 Because the number of vegetarians in our cohort was very small (n = 2), dietary effects on p-cresol levels should be explored further.
Some of the metabolites we detected here to be significantly different in individuals with MS replicated previous results, supporting the validity of our results. These include carnitine, which we found to be lower in individuals with MS36 and which is an essential component in mitochondrial energy production and involved in transporting lipids into mitochondria for β-oxidation. People with a mutation in carnitine palmitoyl transferase 1A (CPT1A) are at lower risk of developing MS, possibly because of downregulation of lipid metabolism.95 Accordingly, a high-fat diet is associated with an increased risk of MS, and inhibition of lipid metabolism has been found to ameliorate MS in an EAE model.96 With respect to treatment, several drugs for MS interfere with the carnitine network.97,98 Moreover, this metabolite was investigated as a potential therapeutic agent for fatigue in MS, although there was no clear evidence of its efficacy.99 S1P, which we found to be lower in individuals with MS, is a lipid mediator that regulates many cellular processes. It is formed from sphingomyelin or glycosphingolipids, which are part of the cell membrane. Fingolimod, an approved treatment for MS, modifies this signaling pathway by leading to S1PR internalization. This internalization eliminates the ability of immune cells in the lymph nodes to respond to S1P and leads to their sequestration, and the final outcome is a reduction in infiltration of inflammatory cells into the CNS.100
Some of the significantly different metabolites we found or the bacterial species that produce them may be partly determined by diet. Long-term dietary habits explain 10% or more of the variance in the levels of 4 metabolites (stachydrine, 1-stearoyl-2-docosahexaenoyl-GPC, 1-stearoyl-2-docosahexaenoyl-GPC 3-hydroxyhippurate, and arabonate/xylonate) and there is potential to affect their levels by changing dietary habits; a further investigation is required here.
BTF3L4 gene-Lawsonella correlation
Interestingly, the BTF3L4 gene, which we found to be correlated with Lawsonella (s3665), is a homolog of BTF3, and overexpression of BTF3 has been associated with a variety of malignancies, including different types of cancer.101, 102, 103, 104 Lawsonella (s3665), whose closest known species is Lawsonella clevelandensis, has been found to be significantly higher in CNS tissue of individuals with amyotrophic lateral sclerosis (ALS).105 Further investigation is required to understand the potentials of the correlation we found.
Limitations of study
Our study has a few potential shortcomings. First, MS and control groups differed in age, gender, and BMI. To overcome such confounders, we used several measures to ensure that our results are not due to age, gender, and BMI differences between affected individuals and controls but due to associations with disease status (STAR Methods). Second, although this is the largest integrated metabolomics-metagenomic study of MS, the sample size remains rather limited especially because of the only partially overlapping groups that underwent metabolomics and metagenomics (36 of the 90 participants with MS from the metabolomics group [40%] were also participants in the microbiome group; Table S2). Third, a consequence of the choice to include participants with different subtypes and stages is the small size of the studied subgroups, which can explain the lack of significant differences between them. Although the differences in butyrate abundance in MS using metagenomics analyses are solid, the results are not supported by the serum metabolomics analysis.
Another possible drawback is that our microbiome analyses were performed from rectal swab samples and not from stool samples, as typically done in microbiome studies. However, previous studies have shown that rectal swabs are a reliable proxy for fecal samples.106, 107, 108 Finally, because of our study is observational, we cannot attribute causality to the alterations we describe in the gut microbiome and metabolomics. The altered microbes and metabolites may play a role in disease-related immunological changes or vice versa.
Our results unravel a comprehensive list of potential microbial and metabolite therapeutic candidates. Although some of the candidates we detected here replicate previous results, supporting the validity of our results, many others require further investigation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Software and algorithms | ||
| GEM mapper build 1.376 | Marco-Sola et al.109 | https://sourceforge.net/projects/gemlibrary/ |
| Segata DB | Pasolli et al.44 | https://github.com/SegataLab |
| MetaPhlAn2 | Truong et al.110 | https://bitbucket.org/biobakery/metaphlan2; RRID: SCR_004915 |
| Bowtie2 | Langmead et al.111 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Diamond | Buchfink et al.47 | https://github.com/bbuchfink/diamond |
| STAR | Dobin et al.112 | N/A |
| htseq-count | Anders et al.113 | N/A |
| Other | ||
| Study participants data | Table S2 | |
| Untargeted mass spectrometry | Metabolon, Inc., Durham, North Carolina, USA | https://www.metabolon.com/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Eran Segal (eran.segal@weizmann.ac.il).
Materials availability
This study did not generate new unique reagents
Data and code availability
All data about participants, microbiome, metabolomics and human gene expression measurements, and nutritional values in the paper are available in Tables S2, S3, S4, S6, and S7, accordingly.
Experimental model and subject details
All information about study participants can be found in Table S2. All MS patients followed at the Multiple Sclerosis Center, Sheba Medical Center. Exclusion criteria included diseases such as HIV, jaundice or taking antibiotics within 3 months before the recruitment. The study was approved by the Sheba Institutional Review Board. All participants signed written informed consent forms.
Notably, we performed metagenomic sequencing of rectal stool samples from 187 MS participants followed at the Multiple Sclerosis Center, Sheba Medical Center and 58 healthy controls. After collecting the data on participants until October 2017, we processed the metabolomic samples on 125 of them (35 controls and 90 MS patients with different types of the disease and with different clinical manifestations) and then collected the rest of the participants. So, for a partially subgroup of 90 MS patients (Table S2), we used untargeted mass spectrometry to profile 1,251 metabolites from their serum samples, covering a wide range of biochemicals including lipids, amino acids, xenobiotics, carbohydrates, peptides, nucleotides and ∼30% unknown compounds (Tables S4 and S5). We compared it with the metabolites profile of 90 age-, gender- and BMI-matched healthy controls from another study43, by using a quantile normalization process, for which we used 35 healthy controls from our study (see more details about the quantile normalization process under the Metabolomics profiling and preprocessing in the Method details).
For a subgroup of 49 participants (36 MS patients and 13 controls), we also used mRNA-Seq to measure the expression of 22,929 human genes (Table S6).
As some of the patients were only partially compliant to record all their nutritional intake for at least 3 days only a subgroup of 44 MS patients with complete data was selected for the diet analysis; These 44 MS patients recorded all of their nutritional intake for at least 3 days and in real-time using a specialized app provided to them, Together, they logged 4,806 different food items (Table S7; STAR Methods; Nutritional data).
We also collected a comprehensive profile from all participants, including food frequency, lifestyle and medical background questionnaires.
Method details
Microbiome preprocessing
Sample collection, DNA extraction, and sequencing of the samples was performed as described previously in Rothschild et al.114 and Zeevi et al.43 with one difference to use rectal swabs for samples collection. We filtered metagenomic reads containing Illumina adapters, low-quality reads and trimmed low-quality read edges. We detected host DNA by mapping with GEM109 to the human genome (hg19) and removed human reads. We subsampled all samples to have 5 million reads. Bacterial relative abundance estimation was performed by mapping bacterial reads to species-level genome bins (SGB) representative genomes.44 We selected all SGB representatives with at least 5 genomes in the group, and for these representatives genomes kept only unique regions as a reference dataset. Mapping was performed using bowtie2 and abundance was estimated by calculating the mean coverage of unique genomic regions across the 50 percent most densely covered areas. Feature names including the lowest taxonomy level were identified. We selected only species that observed in more than 5% of the individuals.
Metabolomics profiling and preprocessing
Metabolite concentrations were measured in serum samples by an untargeted LC/MS platform (Metabolon, Inc., Durham, North Carolina, USA) as previously described.115, 116, 117 Metabolon Inc. performed all preprocessing and profiling. In this study, we handled the processed metabolite intensities, and all annotations were provided by Metabolon. Samples were sent to profiling in two different runs. A total of 540 serum samples belonging to 491 healthy participants from a different study43 were profiled in the first run. In the second run, we profiled 90 MS patients together with 35 healthy controls from our study. Quantile normalization was performed for each metabolite based on 35 samples from the first run and 35 controls from the second run; all 70 samples belonged to healthy subjects and the two groups were matched by gender, age and BMI. The matching was performed with MatchIt R package for age and BMI in males and females separately, using nearest neighbor method and Mahalanobis distance. 35 quantiles were calculated for each metabolite, and the mean of the two groups was taken as a reference distribution. Interpolation was performed based on the reference distribution in order to get the corrected values of the metabolites for 90 MS samples from the second run and 90 matched healthy samples from the first run. The matching procedure here, as before, was performed with MatchIt R package for age and BMI in males and females separately, using nearest neighbor method and Mahalanobis distance. Using imputations models while using quantile normalization can cause unreliable results on the imputed metabolites, so only metabolites that were present in 100% of the samples were included.
Gene expression preprocessing
Sequencing Libraries were prepared using INCPM mRNA Seq. SR60 reads were sequenced on 8 lane(s) of an Illumina HiSeq High Output V4. The output was ∼21 million reads per sample. Poly-A/T stretches and Illumina adapters were trimmed from the reads using cutadapt;118 resulting reads shorter than 30bp were discarded. Reads were mapped to the H. sapiens reference genome GRCh38 using STAR,112 supplied with gene annotations downloaded from Ensembl (with EndToEnd option and outFilterMismatchNoverLmax was set to 0.04). Expression levels for each gene were quantified using htseq-count,113 using the gtf above. For each gene, we truncated outliers, which were defined as values not within three standard deviations of the data between the 5 percentile and the 95 percentile. For the computation of the correlation with the different species, we used only genes, which had a value higher than five in at least 50% of the 49 participants. The data was normalized for each sample by the sum of all the genes expression values.
Nutritional data
We obtained a very large nutritional data from 44 patients, which recorded their entire nutritional intake for at least 3 days and in real-time using a specialized app provided to them (Table S7). They logged 4,806 different food items. Each food item within every meal was logged along with its weight by selecting it from a database of 6,401 foods with full nutritional values based on the Israeli Ministry of Health database that was further improved and expanded with additional items from certified sources.
Feature groups definitions
The feature groups for association with metabolites analysis (Figure 3C) were previously described by Bar et al.54; The “diet” feature group includes answers for a detailed food frequency questionnaire (FFQ) aimed at capturing long term dietary habits, and the daily mean consumption of different food types, computed over a week based on real-time logging. In both cases only items that were reported to be consumed at least once by at least 1% of our participants were kept, resulting in 670 different food types from logging, and 141 different items from the FFQ. The “anthropometrics” feature group includes weight, BMI, waist and hips circumference, and waist to hips ratio (WHR). The “cardiometabolic” feature group includes systolic and diastolic blood pressure, heart rate in beats per minute and a glycemic status as previously described in Rothschild et al.114 The “drugs” feature group includes 30 binary features representing the intake of 20 common medications as reported in questionnaires, in addition to 10 medication groups as previously described. The “lifestyle” feature group includes smoking status (current, past), stress levels obtained from questionnaires, and the daily mean sleeping time, exercise time and midday sleep time based on real time logging. The “time of day” feature is a binary feature indicating whether the sample was taken during the first half of the day. The “seasonal effects” feature is the month in which the sample was taken. In some analyses months were grouped by season (winter: December - February; spring: March - May; summer: June - August; fall: September - November). The “microbiome” feature group includes bacterial relative abundance calculated both by considering coverage and by MetaPhlAn2, as well as the first 10 principal components computed over the log transformed relative abundance of a bacterial gene catalog119 as previously described.114,120 Bar et al.54 built gradient boosting trees models for predicting the level of each metabolite based on the different feature groups. The R2 of each model was considered as the importance of the feature that the model was based on in explaining the variability of that metabolite (Figure 3C).
MS treatment effects
Modifying-disease treatments for MS have mainly immunomodulatory effects. Their goal is to reach a target of no evident disease activity (NEDA), meaning that the patient will have no relapses, no increase in neurological disability (as measured by EDSS63) and no new or active brain or spinal cord lesions (on MRI scans). The strong connection between the gut microbiome and the immune system can suggest that immunomodulatory treatment will influence microbiome composition.22,64,65
In order to assess the effect of the different immunomodulatory treatments (Figures S1A, S1B, S2A, and S2B) on the altered microbiota and on the altered metabolites profile we found in MS patients, we first asked whether a machine learning algorithm based on microbiome data can classify treated patients from untreated patients. No separation was observed between treated and untreated MS patients based on microbiome data (Figures S2C and S2D). A machine-learning algorithm based on metabolomics data yielded a moderate separation between the two groups (Figures S3C and S3D). Using SHAP, we identified the contribution of each metabolite to this prediction. Only one metabolite had a high impact on the model (6-hydroxyindole sulfate, SHAP value > 0.05) and none of the significantly different metabolites between MS patients and controls had a high impact on the model (SHAP value < 0.05), meaning that the effect of immunomodulatory treatment on these metabolites is negligible.
To further ensure that the observed effects between MS and healthy controls is not solely based on MS treatment, we repeated the main analyses for a group of untreated MS patients (N = 65), which did not receive any kind of treatment. To test for differences between the microbiome of untreated patients and healthy controls, we performed Mann-Whitney U test on the relative abundances of species. We found 11 species that significantly differed between the two groups (p < 0.05 after 5% FDR correction). Out of these 11 species, 10 were significantly different also between all the MS patients to the controls.
Next, we examined for a difference in the sum of the abundances for all butyrate-producing bacteria between untreated MS patients and controls, and we found a lower level in the untreated MS patients (mean relative abundance of 0.06 versus 0.083 for untreated MS versus controls respectively, Mann-Whitney U p < 0.03). No significant difference was found for levels of butyrate-producing bacteria, between patients under treatment (N = 64) to patients which were not under treatment (N = 65) (Mann-Whitney U p > 0.26). We also searched for differences of butyrate-producing bacteria, between patients that received a DMD treatment for at least 90 or 180 days to patients that were not under treatment or received treatment for less than 90 or 180 days respectively. No significant difference was found also in these cases (N = 78 or N = 64 respectively versus N = 51 or N = 85 respectively; Mann-Whitney U p > 0.43 or p > 0.24 respectively). No correlation was found between levels of butyrate-producing bacteria to treatment duration (Pearson R = −0.12 p > 0.15).
We then examined for difference in the sum of the abundances of all indolelactate-producing bacteria between untreated MS patients and controls (mean relative abundance of 0.073 versus 0.094 for untreated MS versus control respectively, Mann-Whitney U p < 0.04). To test for differences between the metabolomics of untreated MS patients and healthy controls, we performed Mann-Whitney U test on the relative abundances of metabolites and we found 31 metabolites that significantly differed between the two groups (p < 0.05 after 5% FDR correction; Table S5). We found lower serum levels of indolelactate and indolepropionate in MS patients (Mann-Whitney U p < 0.04 after 5% FDR correction).
Overall, we have found for the untreated MS patients group the same trends throughout the analyses. Consistent with the previous results (Figures S2C, S2D, S3C, and S3D), our findings suggest that the effect of immunomodulatory treatment on the altered microbiota and on the altered metabolites profile we found in MS patients is negligible.
Experimental design
We did not replicate or blind the data at any stage of the study. Randomization for cross validation was used with Python Numpy random.seed function. All the statistical methods we used for the analyses are described through all the main text and the STAR Methods.
Quantification and statistical analysis
All of the statistical details of this work can be found in the Results section. Further details can be found in this section. Significance was defined in this work as p val. < 0.05, after 5% FDR correction.
Functional analysis
To infer the functions of the microbial communities of the MS and control groups we applied DIAMOND algorithm47 for each metagenomic sample after subsampling it to 8 million reads. We set our subsampling threshold to 8M reads for the microbiome gene analysis to balance between the number of samples and the number of reads in each of the samples, so it can conserve a large amount of the study participants together with a large number of reads for each sample. The reference database was comprised from both the KEGG genes database48 and the IGC database,119 resulting in 11,245 different KOs (KEGG Orthology) (Table S3). For each metagenomic read in a sample, we assigned the KO with the highest-scoring annotated hit, only if the e value of the score was smaller than 0.0001, otherwise no KO was assigned to the read. Lastly, a relative abundance was assigned to each KO by accumulating its hits and divided by the total number of reads, which was 8 million for our samples. The abundances of the enzymes were inspected, and not a binary presence/absence. The read coverage per KO is large enough for the known bacterial gene biomarkers for butyrate-producing communities, so we can reliably rely on the results we achieved (Table S3).
Predictive models
Our microbiome predictor is based on code adapted from the 0.72.1 xgboost library.45 Our metabolomics predictor is based on a logistic regression model; code is adapted from the sklearn library. 100 predictors were built, each with ten-fold cross-validation, and prediction accuracy was measured as the mean of area-under-curves.
Feature attribution analysis
For interpreting our predictions (Figure 1D), we used SHAP (SHapley Additive exPlanations), a recently introduced framework, which assigns each feature an importance value for a particular prediction. Briefly, for a specific prediction, a feature’s SHAP value is defined as the change in the expected value of the model’s output when this feature is observed versus when it is missing. It is computed using a sum that represents the impact of each feature being added to the model averaged over all possible orderings of features being introduced.
Individual SHAP values were computed for held-out subjects in 10-fold CV using the module XGBoost (version 0.72.1),45 based on models trained only on features from the respective feature group. For every feature, we computed the mean absolute SHAP value across all instances in a specific model, reflecting the mean impact of each feature on the predictions and serving as a feature importance measure.
Possible confounders effects on the results
First, as there are significant differences in the males/females proportions, ages distribution and BMI levels between MS and controls, we wanted to test if the differences we obtained for the butyrate-producing bacteria and indolelactate-producing bacteria levels are not because one or a combination of those possible confounders. We relied on previous knowledge for estimating the effect of the covariates on the outcome (bacteria levels). We examined if there are any correlations between the sum of butyrate-producing bacteria levels to the covariates in a large cohort of healthy participants (N = 1361). We did not found a significant difference in sum of butyrate-producing bacteria levels between genders (542 males and 819 females, Mann-Whitney U p > 0.38), and a weak negative correlation with age (Pearson R = −0.05, p > 0.047) and with BMI (Pearson R = −0.002, p > 0.93). We also tried to predict butyrate-producing levels using gradient boosting decision trees using age, gender and BMI as features, and we achieved low accuracy (Pearson R = 0.02, p > 0.3). In addition, we examined correlation for each of the butyrate-producing bacteria separately, and then removed the species which were significantly correlated to one of the covariates (Mann-Whitney U p < 0.05 after 5% FDR correction). After we removed the correlated species, we still achieved a significant difference in the sum of the remaining butyrate-producing species levels, between MS and controls (mean relative abundance of 0.021 versus 0.034 for MS versus control respectively, Mann-Whitney U p < 0.001). We repeated this process for indolelactate-producing bacteria; Also here, after removing the species with significant correlation to age, gender or BMI (Mann-Whitney U p < 0.05 after 5% FDR correction), we still achieved a significant difference between MS and controls (mean relative abundance of 0.023 versus 0.037 for MS versus control respectively, Mann-Whitney U p < 0.001).
Second, we wanted to test for the effect of disease type on butyrate-producing bacteria levels. No differences were found between the different disease groups (Mann-Whitney U p > 0.3 after 5% FDR correction for all pairwise-comparisons for the following 5 groups: RIS, CIS, RRMS, progressive, patients during a relapse).
No differences in bacteria associated with P-cresol
We examined any differences in levels of bacteria which are associated with p-cresol between MS and controls. The associated bacteria were defined as ones that have the ability to produce p-cresol from tyrosine (C. difficile121,122) or contribute to p-cresol formation either by converting tyrosine to p-hydroxyphenylacetate (pHPA) (C. difficile, C. scatologenes,123 P. vulgaris,124 or converting pHPA to p-cresol (Lactobacillaceae species.125,126 For all the above species, only L. ruminis (s7061) appeared in more than 5% of the individuals, and no significant difference was found for this species (0.0009 ± 0.003 versus 0.0005 ± 0.001 for MS versus control respectively, Mann-Whitney U p > 0.34).
Acknowledgments
We thank that the individuals who willingly participated in the study and contributed to a better understanding of the role of the microbiome in multiple sclerosis, the Segal group members for fruitful discussions, Yaara Unger for technical assistance, and Martin Mikl and Ron Sender for insights. E.S. is supported by the Crown Human Genome Center; the Larson Charitable Foundation New Scientist Fund; the Else Kroener Fresenius Foundation; the White Rose International Foundation; the Ben B. and Joyce E. Eisenberg Foundation; the Nissenbaum family; Marcos Pinheiro de Andrade and Vanessa Buchheim; Lady Michelle Michels; Aliza Moussaieff; and grants from the Minerva Foundation with funding from the Federal German Ministry for Education and Research, the European Research Council, and the Israel Science Foundation. A.A. received grants from the Erez MS Fund, IL (40427) and Peleg MS Fund, IL (40428).
Author contributions
Conceptualization, I.L., M.G., G.P., E.S., and A.A.; formal analysis, I.L., M.G., G.P., N.B., A.G., L.Z., E.Y., D.R., S.L., A.A., and E.S.; software, I.L., M.G., G.P., and A.G..; resources and data curation, I.L., G.P., M.G., N.K., G.M., B.C.W., A.T., M. Didkin, S.D., H.E., Y.T., S.M., P.S., A.W., M.L.-P., and A.A.; project administration, I.L., A.W., S.M., M.G., E.S., and A.A.; writing, I.L., G.P., A.A., and E.S.; visualization, I.L. and G.P.; supervision of nutritional decisions, D.C.; supervision of clinical aspects, D.M., S.M., M. Dolev, Y.S., and A.A.; funding acquisition, A.A. and E.S.
Declaration of interests
The authors declare no competing interests.
Published: April 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100246.
Contributor Information
Anat Achiron, Email: anat.achiron@sheba.health.gov.il.
Eran Segal, Email: eran.segal@weizmann.ac.il.
Supplemental information
References
- 1.Olsson T., Barcellos L.F., Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat. Rev. Neurol. 2017;13:25–36. doi: 10.1038/nrneurol.2016.187. [DOI] [PubMed] [Google Scholar]
- 2.Ascherio A. Environmental factors in multiple sclerosis. Expert Rev. Neurother. 2013;13(12, Suppl):3–9. doi: 10.1586/14737175.2013.865866. [DOI] [PubMed] [Google Scholar]
- 3.Oksenberg J.R. Decoding multiple sclerosis: an update on genomics and future directions. Expert Rev. Neurother. 2013;13(12, Suppl):11–19. doi: 10.1586/14737175.2013.865867. [DOI] [PubMed] [Google Scholar]
- 4.Riordan S.M., Williams R. Treatment of hepatic encephalopathy. N. Engl. J. Med. 1997;337:473–479. doi: 10.1056/NEJM199708143370707. [DOI] [PubMed] [Google Scholar]
- 5.Bravo J.A., Forsythe P., Chew M.V., Escaravage E., Savignac H.M., Dinan T.G., Bienenstock J., Cryan J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA. 2011;108:16050–16055. doi: 10.1073/pnas.1102999108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sudo N., Chida Y., Aiba Y., Sonoda J., Oyama N., Yu X.-N., Kubo C., Koga Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004;558:263–275. doi: 10.1113/jphysiol.2004.063388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diaz Heijtz R., Wang S., Anuar F., Qian Y., Björkholm B., Samuelsson A., Hibberd M.L., Forssberg H., Pettersson S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA. 2011;108:3047–3052. doi: 10.1073/pnas.1010529108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neufeld K.M., Kang N., Bienenstock J., Foster J.A. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol. Motil. 2011;23:255–264e119. doi: 10.1111/j.1365-2982.2010.01620.x. [DOI] [PubMed] [Google Scholar]
- 9.Collins S.M., Surette M., Bercik P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 2012;10:735–742. doi: 10.1038/nrmicro2876. [DOI] [PubMed] [Google Scholar]
- 10.Rhee S.H., Pothoulakis C., Mayer E.A. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat. Rev. Gastroenterol. Hepatol. 2009;6:306–314. doi: 10.1038/nrgastro.2009.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones M.P., Dilley J.B., Drossman D., Crowell M.D. Brain-gut connections in functional GI disorders: anatomic and physiologic relationships. Neurogastroenterol. Motil. 2006;18:91–103. doi: 10.1111/j.1365-2982.2005.00730.x. [DOI] [PubMed] [Google Scholar]
- 12.Li W., Dowd S.E., Scurlock B., Acosta-Martinez V., Lyte M. Memory and learning behavior in mice is temporally associated with diet-induced alterations in gut bacteria. Physiol. Behav. 2009;96:557–567. doi: 10.1016/j.physbeh.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Ohland C.L., Kish L., Bell H., Thiesen A., Hotte N., Pankiv E., Madsen K.L. Effects of Lactobacillus helveticus on murine behavior are dependent on diet and genotype and correlate with alterations in the gut microbiome. Psychoneuroendocrinology. 2013;38:1738–1747. doi: 10.1016/j.psyneuen.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 14.Grochowska M., Laskus T., Radkowski M. Gut microbiota in neurological disorders. Arch. Immunol. Ther. Exp. (Warsz) 2019;67:375–383. doi: 10.1007/s00005-019-00561-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farhadi A., Banan A., Fields J., Keshavarzian A. Intestinal barrier: an interface between health and disease. J. Gastroenterol. Hepatol. 2003;18:479–497. doi: 10.1046/j.1440-1746.2003.03032.x. [DOI] [PubMed] [Google Scholar]
- 16.Julio-Pieper M., Bravo J.A., Aliaga E., Gotteland M. Review article: intestinal barrier dysfunction and central nervous system disorders--a controversial association. Aliment. Pharmacol. Ther. 2014;40:1187–1201. doi: 10.1111/apt.12950. [DOI] [PubMed] [Google Scholar]
- 17.Lavasani S., Dzhambazov B., Nouri M., Fåk F., Buske S., Molin G., Thorlacius H., Alenfall J., Jeppsson B., Weström B. A novel probiotic mixture exerts a therapeutic effect on experimental autoimmune encephalomyelitis mediated by IL-10 producing regulatory T cells. PLoS ONE. 2010;5:e9009. doi: 10.1371/journal.pone.0009009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forbes J.D., Van Domselaar G., Bernstein C.N. The Gut Microbiota in Immune-Mediated Inflammatory Diseases. Front. Microbiol. 2016;7:1081. doi: 10.3389/fmicb.2016.01081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ochoa-Repáraz J., Mielcarz D.W., Ditrio L.E., Burroughs A.R., Foureau D.M., Haque-Begum S., Kasper L.H. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J. Immunol. 2009;183:6041–6050. doi: 10.4049/jimmunol.0900747. [DOI] [PubMed] [Google Scholar]
- 20.Ventura R.E., Iizumi T., Battaglia T., Liu M., Perez-Perez G.I., Herbert J., Blaser M.J. Gut microbiome of treatment-naïve MS patients of different ethnicities early in disease course. Sci. Rep. 2019;9:16396. doi: 10.1038/s41598-019-52894-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kozhieva M., Naumova N., Alikina T., Boyko A., Vlassov V., Kabilov M.R. Primary progressive multiple sclerosis in a Russian cohort: relationship with gut bacterial diversity. BMC Microbiol. 2019;19:309. doi: 10.1186/s12866-019-1685-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cantarel B.L., Waubant E., Chehoud C., Kuczynski J., DeSantis T.Z., Warrington J., Venkatesan A., Fraser C.M., Mowry E.M. Gut microbiota in multiple sclerosis: possible influence of immunomodulators. J. Investig. Med. 2015;63:729–734. doi: 10.1097/JIM.0000000000000192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cosorich I., Dalla-Costa G., Sorini C., Ferrarese R., Messina M.J., Dolpady J., Radice E., Mariani A., Testoni P.A., Canducci F. High frequency of intestinal TH17 cells correlates with microbiota alterations and disease activity in multiple sclerosis. Sci. Adv. 2017;3:e1700492. doi: 10.1126/sciadv.1700492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tremlett H., Fadrosh D.W., Faruqi A.A., Hart J., Roalstad S., Graves J., Lynch S., Waubant E., US Network of Pediatric MS Centers Gut microbiota composition and relapse risk in pediatric MS: A pilot study. J. Neurol. Sci. 2016;363:153–157. doi: 10.1016/j.jns.2016.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tremlett H., Fadrosh D.W., Faruqi A.A., Hart J., Roalstad S., Graves J., Spencer C.M., Lynch S.V., Zamvil S.S., Waubant E. Associations between gut microbiota and immune markers in pediatric multiple sclerosis and controls. BMC Neurol. 2016;16:182. doi: 10.1186/s12883-016-0703-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tremlett H., Fadrosh D.W., Faruqi A.A., Zhu F., Hart J., Roalstad S., Graves J., Lynch S., Waubant E., US Network of Pediatric MS Centers Gut microbiota in early pediatric multiple sclerosis: a case-control study. Eur. J. Neurol. 2016;23:1308–1321. doi: 10.1111/ene.13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jangi S., Gandhi R., Cox L.M., Li N., von Glehn F., Yan R., Patel B., Mazzola M.A., Liu S., Glanz B.L. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016;7:12015. doi: 10.1038/ncomms12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J., Chia N., Kalari K.R., Yao J.Z., Novotna M., Paz Soldan M.M., Luckey D.H., Marietta E.V., Jeraldo P.R., Chen X. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 2016;6:28484. doi: 10.1038/srep28484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyake S., Kim S., Suda W., Oshima K., Nakamura M., Matsuoka T., Chihara N., Tomita A., Sato W., Kim S.W. Dysbiosis in the Gut Microbiota of Patients with Multiple Sclerosis, with a Striking Depletion of Species Belonging to Clostridia XIVa and IV Clusters. PLoS ONE. 2015;10:e0137429. doi: 10.1371/journal.pone.0137429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nourbakhsh B., Bhargava P., Tremlett H., Hart J., Graves J., Waubant E. Altered tryptophan metabolism is associated with pediatric multiple sclerosis risk and course. Ann. Clin. Transl. Neurol. 2018;5:1211–1221. doi: 10.1002/acn3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Del Boccio P., Pieragostino D., Di Ioia M., Petrucci F., Lugaresi A., De Luca G., Gambi D., Onofrj M., Di Ilio C., Sacchetta P., Urbani A. Lipidomic investigations for the characterization of circulating serum lipids in multiple sclerosis. J. Proteomics. 2011;74:2826–2836. doi: 10.1016/j.jprot.2011.06.023. [DOI] [PubMed] [Google Scholar]
- 32.Lim C.K., Bilgin A., Lovejoy D.B., Tan V., Bustamante S., Taylor B.V., Bessede A., Brew B.J., Guillemin G.J. Kynurenine pathway metabolomics predicts and provides mechanistic insight into multiple sclerosis progression. Sci. Rep. 2017;7:41473. doi: 10.1038/srep41473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andersen S.L., Briggs F.B.S., Winnike J.H., Natanzon Y., Maichle S., Knagge K.J., Newby L.K., Gregory S.G. Metabolome-based signature of disease pathology in MS. Mult. Scler. Relat. Disord. 2019;31:12–21. doi: 10.1016/j.msard.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sylvestre D.A., Slupsky C.M., Aviv R.I., Swardfager W., Taha A.Y. Untargeted metabolomic analysis of plasma from relapsing-remitting multiple sclerosis patients reveals changes in metabolites associated with structural changes in brain. Brain Res. 2020;1732:146589. doi: 10.1016/j.brainres.2019.146589. [DOI] [PubMed] [Google Scholar]
- 35.Villoslada P., Alonso C., Agirrezabal I., Kotelnikova E., Zubizarreta I., Pulido-Valdeolivas I., Saiz A., Comabella M., Montalban X., Villar L. Metabolomic signatures associated with disease severity in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2017;4:e321. doi: 10.1212/NXI.0000000000000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kasakin M.F., Rogachev A.D., Predtechenskaya E.V., Zaigraev V.J., Koval V.V., Pokrovsky A.G. Targeted metabolomics approach for identification of relapsing-remitting multiple sclerosis markers and evaluation of diagnostic models. MedChemComm. 2019;10:1803–1809. doi: 10.1039/c9md00253g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poddighe S., Murgia F., Lorefice L., Liggi S., Cocco E., Marrosu M.G., Atzori L. Metabolomic analysis identifies altered metabolic pathways in Multiple Sclerosis. Int. J. Biochem. Cell Biol. 2017;93:148–155. doi: 10.1016/j.biocel.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 38.Bhargava P., Fitzgerald K.C., Calabresi P.A., Mowry E.M. Metabolic alterations in multiple sclerosis and the impact of vitamin D supplementation. JCI Insight. 2017;2:95302. doi: 10.1172/jci.insight.95302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sinclair A.J., Viant M.R., Ball A.K., Burdon M.A., Walker E.A., Stewart P.M., Rauz S., Young S.P. NMR-based metabolomic analysis of cerebrospinal fluid and serum in neurological diseases--a diagnostic tool? NMR Biomed. 2010;23:123–132. doi: 10.1002/nbm.1428. [DOI] [PubMed] [Google Scholar]
- 40.Moussallieh F.M., Elbayed K., Chanson J.B., Rudolf G., Piotto M., De Seze J., Namer I.J. Serum analysis by 1H nuclear magnetic resonance spectroscopy: a new tool for distinguishing neuromyelitis optica from multiple sclerosis. Mult. Scler. 2014;20:558–565. doi: 10.1177/1352458513504638. [DOI] [PubMed] [Google Scholar]
- 41.Dickens A.M., Larkin J.R., Griffin J.L., Cavey A., Matthews L., Turner M.R., Wilcock G.K., Davis B.G., Claridge T.D., Palace J. A type 2 biomarker separates relapsing-remitting from secondary progressive multiple sclerosis. Neurology. 2014;83:1492–1499. doi: 10.1212/WNL.0000000000000905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vital M., Karch A., Pieper D.H. Colonic Butyrate-Producing Communities in Humans: an Overview Using Omics Data. mSystems. 2017;2:e00130-17. doi: 10.1128/mSystems.00130-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeevi D., Korem T., Zmora N., Israeli D., Rothschild D., Weinberger A., Ben-Yacov O., Lador D., Avnit-Sagi T., Lotan-Pompan M. Personalized nutrition by prediction of glycemic responses. Cell. 2015;163:1079–1094. doi: 10.1016/j.cell.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 44.Pasolli E., Asnicar F., Manara S., Zolfo M., Karcher N., Armanini F., Beghini F., Manghi P., Tett A., Ghensi P. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell. 2019;176:649–662.e20. doi: 10.1016/j.cell.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen T., Guestrin C. XGBoost: A Scalable Tree Boosting System. arXiv. 2016 https://arxiv.org/abs/1603.02754 arXiv:1603.02754. [Google Scholar]
- 46.Quintana F.J., Pérez-Sánchez S., Farez M.F. [Immunopathology of multiple sclerosis] Medicina (B. Aires) 2014;74:404–410. [PubMed] [Google Scholar]
- 47.Buchfink B., Xie C., Huson D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 48.Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vital M., Penton C.R., Wang Q., Young V.B., Antonopoulos D.A., Sogin M.L., Morrison H.G., Raffals L., Chang E.B., Huffnagle G.B. A gene-targeted approach to investigate the intestinal butyrate-producing bacterial community. Microbiome. 2013;1:8. doi: 10.1186/2049-2618-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bourassa M.W., Alim I., Bultman S.J., Ratan R.R. Butyrate, neuroepigenetics and the gut microbiome: Can a high fiber diet improve brain health? Neurosci. Lett. 2016;625:56–63. doi: 10.1016/j.neulet.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson J.W., Randles K.M., Kendall C.W.C., Jenkins D.J.A. Carbohydrate and fiber recommendations for individuals with diabetes: a quantitative assessment and meta-analysis of the evidence. J. Am. Coll. Nutr. 2004;23:5–17. doi: 10.1080/07315724.2004.10719338. [DOI] [PubMed] [Google Scholar]
- 52.Rothschild D., Leviatan S., Hanemann A., Cohen Y., Weissbrod O., Segal E. An atlas of robust microbiome associations with phenotypic traits based on large-scale cohorts from two continents. bioRxiv. 2020 doi: 10.1101/2020.05.28.122325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Proia R.L., Hla T. Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J. Clin. Invest. 2015;125:1379–1387. doi: 10.1172/JCI76369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bar N., Korem T., Weissbrod O., Zeevi D., Rothschild D., Leviatan S., Kosower N., Lotan-Pompan M., Weinberger A., Le Roy C.I. A reference map of potential determinants for the human serum metabolome. 2020;588:135–140. doi: 10.1038/s41586-020-2896-2. [DOI] [PubMed] [Google Scholar]
- 55.Chyan Y.J., Poeggeler B., Omar R.A., Chain D.G., Frangione B., Ghiso J., Pappolla M.A. Potent neuroprotective properties against the Alzheimer beta-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J. Biol. Chem. 1999;274:21937–21942. doi: 10.1074/jbc.274.31.21937. [DOI] [PubMed] [Google Scholar]
- 56.Roager H.M., Licht T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018;9:3294. doi: 10.1038/s41467-018-05470-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cao L., Kirk M.C., Coward L.U., Jackson P., Whitaker J.N. p-Cresol sulfate is the dominant component of urinary myelin basic protein like material. Arch. Biochem. Biophys. 2000;377:9–21. doi: 10.1006/abbi.2000.1764. [DOI] [PubMed] [Google Scholar]
- 58.Poisson L.M., Suhail H., Singh J., Datta I., Denic A., Labuzek K., Hoda M.N., Shankar A., Kumar A., Cerghet M. Untargeted Plasma Metabolomics Identifies Endogenous Metabolite with Drug-like Properties in Chronic Animal Model of Multiple Sclerosis. J. Biol. Chem. 2015;290:30697–30712. doi: 10.1074/jbc.M115.679068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moore F., Okuda D.T. Incidental MRI anomalies suggestive of multiple sclerosis: the radiologically isolated syndrome. Neurology. 2009;73:1714. doi: 10.1212/WNL.0b013e3181bd69a9. [DOI] [PubMed] [Google Scholar]
- 60.Miller D.H., Weinshenker B.G., Filippi M., Banwell B.L., Cohen J.A., Freedman M.S., Galetta S.L., Hutchinson M., Johnson R.T., Kappos L. Differential diagnosis of suspected multiple sclerosis: a consensus approach. Mult. Scler. 2008;14:1157–1174. doi: 10.1177/1352458508096878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller D.H., Chard D.T., Ciccarelli O. Clinically isolated syndromes. Lancet Neurol. 2012;11:157–169. doi: 10.1016/S1474-4422(11)70274-5. [DOI] [PubMed] [Google Scholar]
- 62.Lublin F.D., Reingold S.C., Cohen J.A., Cutter G.R., Sørensen P.S., Thompson A.J., Wolinsky J.S., Balcer L.J., Banwell B., Barkhof F. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83:278–286. doi: 10.1212/WNL.0000000000000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kurtzke J.F. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS) Neurology. 1983;33:1444–1452. doi: 10.1212/wnl.33.11.1444. [DOI] [PubMed] [Google Scholar]
- 64.Katz Sand I., Zhu Y., Ntranos A., Clemente J.C., Cekanaviciute E., Brandstadter R., Crabtree-Hartman E., Singh S., Bencosme Y., Debelius J. Disease-modifying therapies alter gut microbial composition in MS. Neurol. Neuroimmunol. Neuroinflamm. 2018;6:e517. doi: 10.1212/NXI.0000000000000517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Castillo-Álvarez F., Pérez-Matute P., Oteo J.A., Marzo-Sola M.E. The influence of interferon β-1b on gut microbiota composition in patients with multiple sclerosis. Neurologia. 2018 doi: 10.1016/j.nrleng.2020.05.006. S0213-4853(18)30158-0. [DOI] [PubMed] [Google Scholar]
- 66.de Mello V.D., Paananen J., Lindström J., Lankinen M.A., Shi L., Kuusisto J., Pihlajamäki J., Auriola S., Lehtonen M., Rolandsson O. Indolepropionic acid and novel lipid metabolites are associated with a lower risk of type 2 diabetes in the Finnish Diabetes Prevention Study. Sci. Rep. 2017;7:46337. doi: 10.1038/srep46337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lovelace M.D., Varney B., Sundaram G., Franco N.F., Ng M.L., Pai S., Lim C.K., Guillemin G.J., Brew B.J. Current evidence for a role of the kynurenine pathway of tryptophan metabolism in multiple sclerosis. Front. Immunol. 2016;7:246. doi: 10.3389/fimmu.2016.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rothhammer V., Mascanfroni I.D., Bunse L., Takenaka M.C., Kenison J.E., Mayo L., Chao C.C., Patel B., Yan R., Blain M. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016;22:586–597. doi: 10.1038/nm.4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lanis J.M., Alexeev E.E., Curtis V.F., Kitzenberg D.A., Kao D.J., Battista K.D., Gerich M.E., Glover L.E., Kominsky D.J., Colgan S.P. Tryptophan metabolite activation of the aryl hydrocarbon receptor regulates IL-10 receptor expression on intestinal epithelia. Mucosal Immunol. 2017;10:1133–1144. doi: 10.1038/mi.2016.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kelly C.J., Zheng L., Campbell E.L., Saeedi B., Scholz C.C., Bayless A.J., Wilson K.E., Glover L.E., Kominsky D.J., Magnuson A. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe. 2015;17:662–671. doi: 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roediger W.E. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut. 1980;21:793–798. doi: 10.1136/gut.21.9.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thangaraju M., Cresci G.A., Liu K., Ananth S., Gnanaprakasam J.P., Browning D.D., Mellinger J.D., Smith S.B., Digby G.J., Lambert N.A. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009;69:2826–2832. doi: 10.1158/0008-5472.CAN-08-4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kimura I., Inoue D., Maeda T., Hara T., Ichimura A., Miyauchi S., Kobayashi M., Hirasawa A., Tsujimoto G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41) Proc. Natl. Acad. Sci. USA. 2011;108:8030–8035. doi: 10.1073/pnas.1016088108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arpaia N., Campbell C., Fan X., Dikiy S., van der Veeken J., deRoos P., Liu H., Cross J.R., Pfeffer K., Coffer P.J., Rudensky A.Y. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Furusawa Y., Obata Y., Fukuda S., Endo T.A., Nakato G., Takahashi D., Nakanishi Y., Uetake C., Kato K., Kato T. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 76.Fu S.-P., Wang J.-F., Xue W.-J., Liu H.-M., Liu B.R., Zeng Y.-L., Li S.N., Huang B.X., Lv Q.K., Wang W., Liu J.X. Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J. Neuroinflammation. 2015;12:9. doi: 10.1186/s12974-014-0230-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Aguilar E.C., Leonel A.J., Teixeira L.G., Silva A.R., Silva J.F., Pelaez J.M.N., Capettini L.S., Lemos V.S., Santos R.A., Alvarez-Leite J.I. Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFκB activation. Nutr. Metab. Cardiovasc. Dis. 2014;24:606–613. doi: 10.1016/j.numecd.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 78.Chen T., Noto D., Hoshino Y., Mizuno M., Miyake S. Butyrate suppresses demyelination and enhances remyelination. J. Neuroinflammation. 2019;16:165. doi: 10.1186/s12974-019-1552-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zeng Q., Junli Gong, Liu X., Chen C., Sun X., Li H., Zhou Y., Cui C., Wang Y., Yang Y. Gut dysbiosis and lack of short chain fatty acids in a Chinese cohort of patients with multiple sclerosis. Neurochem. Int. 2019;129:104468. doi: 10.1016/j.neuint.2019.104468. [DOI] [PubMed] [Google Scholar]
- 80.de Goffau M.C., Fuentes S., van den Bogert B., Honkanen H., de Vos W.M., Welling G.W., Hyöty H., Harmsen H.J. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia. 2014;57:1569–1577. doi: 10.1007/s00125-014-3274-0. [DOI] [PubMed] [Google Scholar]
- 81.Wang W., Chen L., Zhou R., Wang X., Song L., Huang S., Wang G., Xia B. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J. Clin. Microbiol. 2014;52:398–406. doi: 10.1128/JCM.01500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Scher J.U., Sczesnak A., Longman R.S., Segata N., Ubeda C., Bielski C., Rostron T., Cerundolo V., Pamer E.G., Abramson S.B. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202. doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takahashi K., Nishida A., Fujimoto T., Fujii M., Shioya M., Imaeda H., Inatomi O., Bamba S., Sugimoto M., Andoh A. Reduced Abundance of Butyrate-Producing Bacteria Species in the Fecal Microbial Community in Crohn’s Disease. Digestion. 2016;93:59–65. doi: 10.1159/000441768. [DOI] [PubMed] [Google Scholar]
- 84.Stilling R.M., van de Wouw M., Clarke G., Stanton C., Dinan T.G., Cryan J.F. The neuropharmacology of butyrate: The bread and butter of the microbiota-gut-brain axis? Neurochem. Int. 2016;99:110–132. doi: 10.1016/j.neuint.2016.06.011. [DOI] [PubMed] [Google Scholar]
- 85.Oliphant K., Allen-Vercoe E. Macronutrient metabolism by the human gut microbiome: major fermentation by-products and their impact on host health. Microbiome. 2019;7:91. doi: 10.1186/s40168-019-0704-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McOrist A.L., Miller R.B., Bird A.R., Keogh J.B., Noakes M., Topping D.L., Conlon M.A. Fecal butyrate levels vary widely among individuals but are usually increased by a diet high in resistant starch. J. Nutr. 2011;141:883–889. doi: 10.3945/jn.110.128504. [DOI] [PubMed] [Google Scholar]
- 87.Glanville R.W., Qian R.Q., McClure D.W., Maslen C.L. Calcium binding, hydroxylation, and glycosylation of the precursor epidermal growth factor-like domains of fibrillin-1, the Marfan gene protein. J. Biol. Chem. 1994;269:26630–26634. [PubMed] [Google Scholar]
- 88.Zhou X., Tan F.K., Milewicz D.M., Guo X., Bona C.A., Arnett F.C. Autoantibodies to fibrillin-1 activate normal human fibroblasts in culture through the TGF-beta pathway to recapitulate the “scleroderma phenotype”. J. Immunol. 2005;175:4555–4560. doi: 10.4049/jimmunol.175.7.4555. [DOI] [PubMed] [Google Scholar]
- 89.Ottenlinger F.M., Mayer C.A., Ferreirós N., Schreiber Y., Schwiebs A., Schmidt K.G., Ackermann H., Pfeilschifter J.M., Radeke H.H. Interferon-Beta Increases Plasma Ceramides of Specific Chain Length in Multiple Sclerosis Patients, Unlike Fingolimod or Natalizumab. Front. Pharmacol. 2016;7:412. doi: 10.3389/fphar.2016.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Whitaker J.N., Williams P.H., Layton B.A., McFarland H.F., Stone L.A., Smith M.E., Kachelhofer R.D., Bradley E.L., Burgard S., Zhao G. Correlation of clinical features and findings on cranial magnetic resonance imaging with urinary myelin basic protein-like material in patients with multiple sclerosis. Ann. Neurol. 1994;35:577–585. doi: 10.1002/ana.410350511. [DOI] [PubMed] [Google Scholar]
- 91.Whitaker J.N. The presence of immunoreactive myelin basic protein peptide in urine of persons with multiple sclerosis. Ann. Neurol. 1987;22:648–655. doi: 10.1002/ana.410220516. [DOI] [PubMed] [Google Scholar]
- 92.Bashir K., Whitaker J.N. Clinical and laboratory features of primary progressive and secondary progressive MS. Neurology. 1999;53:765–771. doi: 10.1212/wnl.53.4.765. [DOI] [PubMed] [Google Scholar]
- 93.Favretto G., Cunha R.S.D., Dalboni M.A., Oliveira R.B., Barreto F.C., Massy Z.A., Stinghen A.E.M. Endothelial microparticles in uremia: biomarkers and potential therapeutic targets. Toxins (Basel) 2019;11:E267. doi: 10.3390/toxins11050267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Salmean Y.A., Segal M.S., Palii S.P., Dahl W.J. Fiber supplementation lowers plasma p-cresol in chronic kidney disease patients. J. Ren. Nutr. 2015;25:316–320. doi: 10.1053/j.jrn.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mørkholt A.S., Trabjerg M.S., Oklinski M.K.E., Bolther L., Kroese L.J., Pritchard C.E.J., Huijbers I.J., Nieland J.D.V. CPT1A plays a key role in the development and treatment of multiple sclerosis and experimental autoimmune encephalomyelitis. Sci. Rep. 2019;9:13299. doi: 10.1038/s41598-019-49868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shriver L.P., Manchester M. Inhibition of fatty acid metabolism ameliorates disease activity in an animal model of multiple sclerosis. Sci. Rep. 2011;1:79. doi: 10.1038/srep00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wishart D.S., Feunang Y.D., Marcu A., Guo A.C., Liang K., Vázquez-Fresno R., Sajed T., Johnson D., Li C., Karu N. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018;46(D1):D608–D617. doi: 10.1093/nar/gkx1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Laffon-Pioger M., Rocher F., Caruba C., Cohen M., Thomas P., Lebrun C. Carnitine serum levels and levocarnitine administration in multiple sclerosis patients treated with natalizumab. Eur. J. Neurol. 2011;18:e63–e64. doi: 10.1111/j.1468-1331.2010.03332.x. [DOI] [PubMed] [Google Scholar]
- 99.Tejani A.M., Wasdell M., Spiwak R., Rowell G., Nathwani S. Carnitine for fatigue in multiple sclerosis. Cochrane Database Syst. Rev. 2012;2012:CD007280. doi: 10.1002/14651858.CD007280.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chaudhry B.Z., Cohen J.A., Conway D.S. Sphingosine 1-Phosphate Receptor Modulators for the Treatment of Multiple Sclerosis. Neurotherapeutics. 2017;14:859–873. doi: 10.1007/s13311-017-0565-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hu J., Sun F., Chen W., Zhang J., Zhang T., Qi M., Feng T., Liu H., Li X., Xing Y. BTF3 sustains cancer stem-like phenotype of prostate cancer via stabilization of BMI1. J. Exp. Clin. Cancer Res. 2019;38:227. doi: 10.1186/s13046-019-1222-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Symes A.J., Eilertsen M., Millar M., Nariculam J., Freeman A., Notara M., Feneley M.R., Patel H.R., Masters J.R., Ahmed A. Quantitative analysis of BTF3, HINT1, NDRG1 and ODC1 protein over-expression in human prostate cancer tissue. PLoS ONE. 2013;8:e84295. doi: 10.1371/journal.pone.0084295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang C.-J., Frånbergh-Karlson H., Wang D.-W., Arbman G., Zhang H., Sun X.-F. Clinicopathological significance of BTF3 expression in colorectal cancer. Tumour Biol. 2013;34:2141–2146. doi: 10.1007/s13277-013-0745-8. [DOI] [PubMed] [Google Scholar]
- 104.Kusumawidjaja G., Kayed H., Giese N., Bauer A., Erkan M., Giese T., Hoheise J.D., Friess H., Kleeff J. Basic transcription factor 3 (BTF3) regulates transcription of tumor-associated genes in pancreatic cancer cells. Cancer Biol. Ther. 2007;6:367–376. doi: 10.4161/cbt.6.3.3704. [DOI] [PubMed] [Google Scholar]
- 105.Alonso R., Pisa D., Carrasco L. Searching for bacteria in neural tissue from amyotrophic lateral sclerosis. Front. Neurosci. 2019;13:171. doi: 10.3389/fnins.2019.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bassis C.M., Moore N.M., Lolans K., Seekatz A.M., Weinstein R.A., Young V.B., Hayden M.K., CDC Prevention Epicenters Program Comparison of stool versus rectal swab samples and storage conditions on bacterial community profiles. BMC Microbiol. 2017;17:78. doi: 10.1186/s12866-017-0983-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Biehl L.M., Garzetti D., Farowski F., Ring D., Koeppel M.B., Rohde H., Schafhausen P., Stecher B., Vehreschild M.J.G.T. Usability of rectal swabs for microbiome sampling in a cohort study of hematological and oncological patients. PLoS ONE. 2019;14:e0215428. doi: 10.1371/journal.pone.0215428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Reyman M., van Houten M.A., Arp K., Sanders E.A.M., Bogaert D. Rectal swabs are a reliable proxy for faecal samples in infant gut microbiota research based on 16S-rRNA sequencing. Sci. Rep. 2019;9:16072. doi: 10.1038/s41598-019-52549-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Marco-Sola S., Sammeth M., Guigó R., Ribeca P. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods. 2012;9:1185–1188. doi: 10.1038/nmeth.2221. [DOI] [PubMed] [Google Scholar]
- 110.Truong D.T., Franzosa E.A., Tickle T.L., Scholz M., Weingart G., Pasolli E., Tett A., Huttenhower C., Segata N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods. 2015;12:902–903. doi: 10.1038/nmeth.3589. [DOI] [PubMed] [Google Scholar]
- 111.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rothschild D., Weissbrod O., Barkan E., Kurilshikov A., Korem T., Zeevi D., Costea P.I., Godneva A., Kalka I.N., Bar N. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555:210–215. doi: 10.1038/nature25973. [DOI] [PubMed] [Google Scholar]
- 115.Shin S.-Y., Fauman E.B., Petersen A.-K., Krumsiek J., Santos R., Huang J., Arnold M., Erte I., Forgetta V., Yang T.P., Multiple Tissue Human Expression Resource (MuTHER) Consortium An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014;46:543–550. doi: 10.1038/ng.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mitchell M., Evans A., Bridgewater B., Liu Q., Stewart S., Dai H., Dehaven C.D., Miller L. High Resolution Mass Spectrometry Improves Data Quantity and Quality as Compared to Unit Mass Resolution Mass Spectrometry in High-Throughput Profiling Metabolomics. Metabolomics. 2014;4 [Google Scholar]
- 117.Jain A., Li X.H., Chen W.N. An untargeted fecal and urine metabolomics analysis of the interplay between the gut microbiome, diet and human metabolism in Indian and Chinese adults. Sci. Rep. 2019;9:9191. doi: 10.1038/s41598-019-45640-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10. [Google Scholar]
- 119.Li J., Jia H., Cai X., Zhong H., Feng Q., Sunagawa S., Arumugam M., Kultima J.R., Prifti E., Nielsen T., MetaHIT Consortium. MetaHIT Consortium An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014;32:834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 120.Zeevi D., Korem T., Godneva A., Bar N., Kurilshikov A., Lotan-Pompan M., Weinberger A., Fu J., Wijmenga C., Zhernakova A., Segal E. Structural variation in the gut microbiome associates with host health. Nature. 2019;568:43–48. doi: 10.1038/s41586-019-1065-y. [DOI] [PubMed] [Google Scholar]
- 121.Berg J.D., Mills R.G., Coleman D.J. Improved gas-liquid chromatography method for the identification of Clostridium difficile. J. Clin. Pathol. 1985;38:108–110. doi: 10.1136/jcp.38.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Levett P.N., Phillips K.D. Gas chromatographic identification of Clostridium difficile and detection of cytotoxin from a modified selective medium. J. Clin. Pathol. 1985;38:82–85. doi: 10.1136/jcp.38.1.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Elsden S.R., Hilton M.G., Waller J.M. The end products of the metabolism of aromatic amino acids by Clostridia. Arch. Microbiol. 1976;107:283–288. doi: 10.1007/BF00425340. [DOI] [PubMed] [Google Scholar]
- 124.Ward L.A., Johnson K.A., Robinson I.M., Yokoyama M.T. Isolation from swine feces of a bacterium which decarboxylates p-hydroxyphenylacetic acid to 4-methylphenol (p-cresol) Appl. Environ. Microbiol. 1987;53:189–192. doi: 10.1128/aem.53.1.189-192.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yokoyama M.T., Carlson J.R., Holdeman L.V. Isolation and characteristics of a skatole-producing Lactobacillus sp. from the bovine rumen. Appl. Environ. Microbiol. 1977;34:837–842. doi: 10.1128/aem.34.6.837-842.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yokoyama M.T., Carlson J.R. Production of Skatole and para-Cresol by a Rumen Lactobacillus sp. Appl. Environ. Microbiol. 1981;41:71–76. doi: 10.1128/aem.41.1.71-76.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data about participants, microbiome, metabolomics and human gene expression measurements, and nutritional values in the paper are available in Tables S2, S3, S4, S6, and S7, accordingly.