

Abstract
Obesity, a growing pandemic, is a risk factor for many cancers and causes increased bone marrow adipose tissue (BMAT). In vitro studies and obese animal models suggest that BMAT contributes to cancer progression, but there is a lack of preclinical models to directly test BMAT’s role in cancer. Over activation of PPARγ (peroxisome-proliferator-activated receptor-γ) can skew bone formation and resorption rates, resulting in increased BMAT and trabecular bone loss. Thiazolidinediones (e.g., rosiglitazone) are anti-diabetic therapies that promote adipogenesis through PPARγ activation. We investigated if rosiglitazone increases BMAT in an immunocompromised model, commonly used in cancer research, and if these effects could be reversed by co-administering a bone anabolic agent (sclerostin-neutralizing antibody, SOST Ab), which has been shown to inhibit adipogenesis, using DEXA, μCT, OsO4 μCT, and dynamic histomorphometry. Four weeks of rosiglitazone in female SCID Beige mice (cohort 1) significantly decreased trabecular bone volume (BV/TV) by about half, through increased osteoclast and suppressed osteoblast activity, and significantly increased BMAT. In cohort 2, mice were administered rosiglitazone ± SOST Ab for 4 weeks, and then rosiglitazone was discontinued and SOST Ab or vehicle was continued for 6 weeks. SOST Ab significantly increased bone parameters (eg. BV/TV, N.Ob/B.Pm, and MS/BS) in both groups. SOST Ab also overcame many negative effects of rosiglitazone (eg. effects on trabecular bone parameters, increased MLT, and decreased BFR). Interestingly, SOST Ab significantly decreased rosiglitazone induced BMAT in the femur, mostly due to a reduction in adipocyte size, but had a much weaker effect on tibial BMAT. These data suggest targeting sclerostin can prevent rosiglitazone induced bone loss and reduce BM adiposity, in some, but not all BMAT locations. Collectively, our data demonstrate that rosiglitazone increases BMAT in SCID Beige mice, but concomitant changes in bone may confound its use to specifically determine BMAT’s role in tumor models.
Keywords: Preclinical Studies, anabolics, sclerostin, rosiglitazone, Bone QCT/μCT, Bone histomorphometry, DXA
2. Introduction
Obesity is a major risk factor for developing, or having a worse prognosis, for cancers that grow in or metastasize to the bone marrow, including multiple myeloma, prostate cancer, and breast cancer (1–5). Bone marrow adipose tissue (BMAT) has been shown to play a role in supporting tumor cell proliferation and drug resistance through in vitro studies and obese in vivo models(1,6–10), but the inability to specifically modulate BMAT, rather than whole body adipose tissue, has limited the field’s ability to study BMAT’s role in cancer preclinically. Thus, we aimed to determine if rosiglitazone, a PPARγ agonist, could be used to induce BMAT in an immunocompromised mouse model typically used for cancer research, and if a bone anabolic agent, sclerostin (SOST)-neutralizing antibody, SOST Ab, could reduce BMAT.
The relationship between bone and adiposity is intricate and bidirectional, and when unbalanced contributes to diseases including osteoporosis, obesity and diabetic bone disease, cancer-induced osteolysis, and potentially cancer directly(11–13). The osteocyte-derived protein SOST, which functions as an antagonist of canonical Wnt signaling, has traditionally been characterized as a key regulator of bone formation. In humans(14,15) and rodents(16,17), inactivating mutations in the Sost/SOST gene results in increased bone mass due to elevated bone formation rates(16). As such, targeting sclerostin via SOST Ab increases trabecular and cortical bone formation by stimulating osteoblast differentiation and decreases bone resorption by reducing osteocyte production of RANKL(18,19). We and others have shown that SOST Ab also can reverse bone damage induced by a variety of models (eg. ovariectomy(20), cancer-induced bone disease(12), osteogenesis imperfecta(20), and osteopenia due to deletion of TGFβ inducible early gene-1 (TIEG)(20)). Data from our group and others have also implicated sclerostin as a contributor for whole-body metabolism by regulating adipose depots, such as BMAT(21,22) and white adipose tissue (WAT), and influencing fat mass and glucose tolerance (23). Recently, SOST Ab been approved for osteoporosis treatment by the U.S. FDA and the European commission, and endorsed by the Endocrine Society for treatment of postmenopausal women at very high risk for osteoporotic fracture(24–26). However, to date, it has not been reported if SOST Ab can recover bone loss, or reduce BMAT, induced by excessive PPARγ signaling.
In both humans and rodent models, TZDs have been shown to function as PPARγ agonists in adipose, liver, and skeletal tissues that re-sensitize adipocytes to uptake circulating free fatty acids, alleviating insulin resistance(27–29). Rosiglitazone-induced bone loss is the result of unbalanced bone formation and resorption via activation of PPARγ signaling that negatively regulates osteoblastogenesis via decreased expression of osteoblast transcription factors, Runx2 and Osterix(28,30–33). We hypothesized that SOST Ab may be useful to combat TZD-induced bone loss, and potentially attenuate TZD-induced BMAT, since TZDs increase SOST expression due to osteocyte apoptosis(34).
Typically, TZDs decrease bone volume and increase BMAT in rodent models, but there are different responses in different models(33,35–37). Five-month old male and female Swiss Webster mice fed a rosiglitazone-supplemented diet for four weeks had increased BMAT and decreased bone volume(33). Similarly, C57BL/6J male mice aged 1, 6 and 24 months old that were fed a rosiglitazone-supplemented diet for 7 weeks showed significant bone loss and increased BMAT, compared to age-matched control diet mice, in the 6 and 24-month-old mice cohorts, but not the 1-month old cohort(36). Ackert-Bicknell et al. observed differences in response to 8 weeks of rosiglitazone exposure in C3H/HeJ, A/J, DBA/2J, and C57BL/6J mice and concluded that strain/genetic background differences governed their responses. C3H/HeJ mice had decreased BMD and no impact on BM adiposity while C57BL/6J had increased BM adiposity but no impact on trabecular bone volume. There was no apparent skeletal effect of rosiglitazone in A/J mice(37). Similarly, 2-month-old ApoE−/− mice that were placed on a diet containing trigolitazone for 10 months had increased overall adipose tissue but no impact on bones(38). This suggests that the bones of ApoE−/− mice may be more resistant to rosiglitazone-induced bone loss than other mice, and may not accurately represent what occurs in humans. Collectively, these studies suggest that the relationship between BM adipocytes and other cells within the BM microenvironment is still unknown and may be influenced by genetic background. Thus, we aimed to determine the effect of a TZD in a SCID Beige mouse model because this model is commonly used for researching cancer in xenograft models. By creating an immunocompromised, elevated BMAT model, we can accelerate research into how BMAT contributes to cancer, which is a novel question at the forefront of the field(20,39,40). This work sheds light on the connection between PPARγ and Wnt signaling in the bone and develops a novel model of BMAT and bone manipulation in a mouse used for cancer xenografts.
3. Research Design and Methods
Animals and experimental design:
All experimental studies and procedures involving mice were performed in accordance with protocols approved by the governing Institutional Animal Care and Use Committee (IACUC) and all state and federal laws. In experiment one (cohort 1), female Fox Chase SCID Beige (eight-weeks-old, Charles River Laboratory, Wilmington, MA) were fed 20 mg/kg/day rosiglitazone-supplemented diet (D16041209 containing 100 mg rosiglitazone/kg diet, Research Diets, New Brunswick, NJ, USA) or control diet (D10012M, Research Diets) ad libitum for four weeks (randomly assorted into cages and group housed), weighed, and then humanely euthanized and analyzed. Then, in a second study (cohort 2), we tested if SOST Ab could reverse the effects of rosiglitazone. Female Fox Chase SCID Beige mice (five-weeks-old, Charles River Laboratory) were fed rosiglitazone or control diets for four weeks, then put on the control diet (recovery period) for another six weeks. Once a week, mice were administered vehicle or SOST Ab (100 mg/kg, Mereo BioPharma, Austin, TX) via intraperitoneal (IP) injections for the full ten-week duration based on prior literature(41,42). Calcein (20 mg/kg; Sigma-Aldrich, St. Louis, MO) and xylenol orange (90 mg/kg, Sigma-Aldrich) were injected IP at 8 days and 2 days prior to euthanasia, respectively. Upon euthanasia, visceral and inguinal adipose depots were collected and weighed. Researchers working with the mice were not blinded to the groups or treatments.
Dual-Energy X-ray Absorptiometry (DEXA):
Mice were measured prior to the start of the diet and at sacrifice for lean mass, fat mass, and bone mineral density using a PIXImus dual-energy X-ray densitometer (GE Lunar, Boston, MA). The PIXImus was calibrated daily with a mouse phantom provided by the manufacturer. Mice were anesthetized using 2% isoflurane via a nose cone. They were placed ventral side down with each limb and tail positioned away from the body. Full-body scans were obtained and DEXA data were gathered and processed (Lunar PIXImus 2, version 2.1) as previously described(43). Briefly, BMD and BMC were calculated by extrapolating from a rectangular region of interest (ROI) drawn around one femur of each mouse, using the same ROI for every mouse, and lean and fat mass were also calculated for the entire mouse, exclusive of the head, using Lunar PIXImus 2.1 software default settings.
Static and Dynamic Bone Histomorphometry:
Bone histomorphometric analysis was performed on distal femurs (n=4 or 5). Dissected, cleaned, formalin-fixed (10%, 24 h) femurs were washed in PBS and transferred to 70% EtOH, dehydrated in graded ethanol and subsequently infiltrated and embedded in methylmethacrylate. Longitudinal sections (5 μM) were cut using a microtome (RM2255, Leica) and stained with Goldner’s Trichrome for measurements of cellular parameters and trabecular bone microarchitecture. Dynamic bone parameters were evaluated on unstained sections under UV light by measuring the extent and the distance between double labels; no single labels were considered. All parameters, including adipocyte number and area, were evaluated 0.2 mm below the epiphyseal growth plate under 200X magnification. The areas considered for histomorphometry were 1800 mm2 for the trabecular bone, and 1500 mm2 for adipocyte quantification. BM adipose volume was normalized to total volume (AV/TV, %) within a field of view. All histomorphometric analyses were performed using the OsteoMeasure analyzing system (Osteometrics Inc., Atlanta, GA)(44), in a blinded manner by the Harvard Center for Skeletal Research and reported according to the criteria established by the American Society for Bone and Mineral Research(45).
Histology and Histological Image Acquisition:
Inguinal and visceral fat pad tissues were fixed in 10% formalin for 3 days, paraffin embedded, and processed for staining using hematoxylin and eosin (H&E). Histology slides were photographed using a Keyence Fluorescence Microscope BZ-X700 (4x plan fluor PhL, Brightfield/Phase contrast) using an automated image capture software, BZ-X Analyzer, to create high resolution images. Adipocyte size and number were analyzed based on our imaging method, described(22).
Quantitative NMR:
Mice at the endpoint of the study were placed into a thin-walled plastic cylinder with freedom to turn around. A Bruker minispec L750 mz 7.5NMR analyzer (Billerica, MA) was used to measure fat, lean and total mass, according to the manufacturer’s instructions(46). Data was gathered and analyzed using the minispec Plus NF software (Billerica, MA).
Microcomputed tomography:
Trabecular microarchitecture and cortical morphology of the right tibia (n=10) were assessed in the proximal metaphysis and mid-diaphysis, respectively, as previously described (47). Briefly, a high-resolution desktop micro-tomographic imaging system (μCT40, Scanco Medical AG, Brüttisellen, Switzerland) acquired whole tibial scans using a 10 μm3 isotropic voxel size, 70 kVp peak x-ray tube potential, 114 μA x-ray intensity, 200 ms integration time, and were subjected to Gaussian filtration and segmentation. Trabecular architecture was analyzed in the endocortical region of a volume of interest beginning 100 μm inferior to the proximal growth plate and extending distally 1000 μm (100 transverse slices). Cortical bone morphology was analyzed in a region of the diaphysis beginning 2 mm superior to the distal tibiofibular junction and extending distally 500 μm (50 transverse slices). The trabecular and cortical regions were then analyzed using the standard cortical and trabecular morphometry scripts in the Scanco Evaluation Program using thresholds of 650 mgHA/cm3 and 700 mgHA/cm3 to segment trabecular and cortical bone, respectively (based on adaptive interative thresholding of the Control diet mouse bones). Image acquisition and analysis protocols adhered to guidelines for μCT assessment of rodent bone microstructure(48).
Osmium Tetroxide (OsO4) μCT:
Quantification and visualization of BMAT was performed as previously described (49). Briefly, following μCT scanning for trabecular and cortical structure, the tibiae were demineralized in 4.1% EDTA, pH 7.4, for 14 days, washed with cold water and then incubated at room temperature for 48 hours in an aqueous solution containing 2.5% potassium dichromate and 1% OsO4 (Electron Microscopy Sciences, Hatfield, PA). The tibiae were washed in cool tap water for 2 h to remove unbound OsO4 and then were transferred to fresh tubes containing phosphate buffered saline. μCT scans of the OsO4 stained tibiae were acquired and analyzed as previously described(47). Briefly, the proximal diaphysis region started 100 μm inferior to the proximal growth plate and extended to 2mm superior to the distal tibiofibular junction. The proximal metaphysis is the 1 mm long region beginning 100 μm inferior to the proximal growth plate and extending distally. The distal diaphysis region extended from the bottom of the proximal diaphysis region to the distal tibiofibular junction. Adipose volume was normalized by the total volume (AV/TV, %) or normalized to the marrow volume [measurement independent of bone] (AV/MV, %).
Glucose Tolerance Test (GTT):
GTT was performed after 4 weeks of treatments in both cohorts of mice. Mice were deprived of food for 3 hours and then injected IP with 1 g glucose/kg body weight (10 μL of a 100 mg/mL glucose stock solution per gram of mouse weight). Blood was taken from lancet-punctured mouse tail veins after 0, 20, 40, 60, and 120 minutes and tested using OneTouch UltraMini test strips (LifeScan, Inc. Milpitas, CA, USA) with the OneTouch UltraMini Blood Glucose Monitoring System (LifeScan).
mRNA Isolation and qRT-PCR:
mRNA was isolated from whole BM of left tibia using the miRNeasy mini-kit with On-Column DNase I digestion (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Total mRNA samples were quantified and tested for quality and contamination using a spectrophotometer machine (Nanodrop 2000, ThermoScientific) prior to cDNA synthesis using the High Capacity cDNA Kit from Applied Biosciences (Thermo-Fisher). qRT-PCR was performed using SYBR Green Master Mix (Bio-Rad) and thermocycling reactions were completed using a CFX-96 (Bio-Rad). Data was analyzed using Bio-Rad CFX Manager 3.1 and Excel (Microsoft). Relative gene expression of Adipoq (Fwd 5’-TGTTCCTCTTAATCCTGCCCA-3’; Rev 5’-CCAACCTGCACAAGTTCCCTT-3’), Fabp4 (Fwd 5’-GCTGCAGCCTTTCTCACC-3’; Rev 5’-CACTTTCCTTGTGGCAAAGC-3’), Axin2 (Fwd 5’-AAAACGGATTCAGGTCCTTCAA-3’; Rev 5’-GTCAGTGCGTCGCTGGATAAC-3’) and Smad6 (Fwd 5’ -GCGCGAGCTCCCTCATGTT-3’; Rev 5’-ACCTGAACATACGATACCCTT-3’) were normalized to the housekeeping gene Actb (Fwd 5’-CTCTGGCTCCTAGCACCATGAAGA-3’; Rev5’GTAAAACGCAGCTCAGTAACAGTCCG-3’) as previously described(21,50).
Statistical Analysis:
Data are expressed as data points with the error bar representing the mean ± standard error on the mean (SEM), unless otherwise noted. Student’s T-test, or ordinary one-way or two-way ANOVA tests were used to determine significance, using p<0.05 as the cut-off with Tukey’s multiple comparison post-hoc testing. Significance was denoted as: Student’s T-test #p<0.05; ANOVA ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05 unless otherwise stated. All statistical analyses were performed in GraphPad Prism 7.0 software unless otherwise noted.
4. Results
Four-week exposure to rosiglitazone negatively affects overall bone density and bone mineral content, and trabecular and cortical bone parameters in the femur and tibia
We first characterized the body composition and skeletal effects of four weeks of rosiglitazone treatment in SCID Beige mice (Supp Fig. 1A). Rosiglitazone induced no difference in overall fat mass (Supp Fig. 1B), but induced a small yet significant increase in lean mass (Supp Fig. 1C). Additionally, rosiglitazone caused a significant reduction in areal bone mineral density (aBMD) (Fig. 1A) and bone mineral content (Fig. 1B), as revealed by DEXA. No significant effects on body weight or glucose tolerance (Supp Fig. 1D, E) were observed. Tibial bone was then analyzed with μCT analysis. Bone volume fraction (BV/TV) was significantly reduced in rosiglitazone-treated mice, to about half the value of control mice (Fig. 1C). Trabecular number (Fig. 1D) and thickness (Fig. 1E) were also significantly reduced with rosiglitazone treatment, resulting in a significant increase in trabecular spacing (Fig. 1F). Rosiglitazone also significantly reduced cortical thickness (Fig. 1G) and increased cortical porosity (Fig. 1H), culminating in extensive bone loss after four weeks, as illustrated in the representative μCT images (Supp Fig. 2A).
Figure 1: Four weeks of rosiglitazone mice negatively affects bone in female SCID Beige mice.
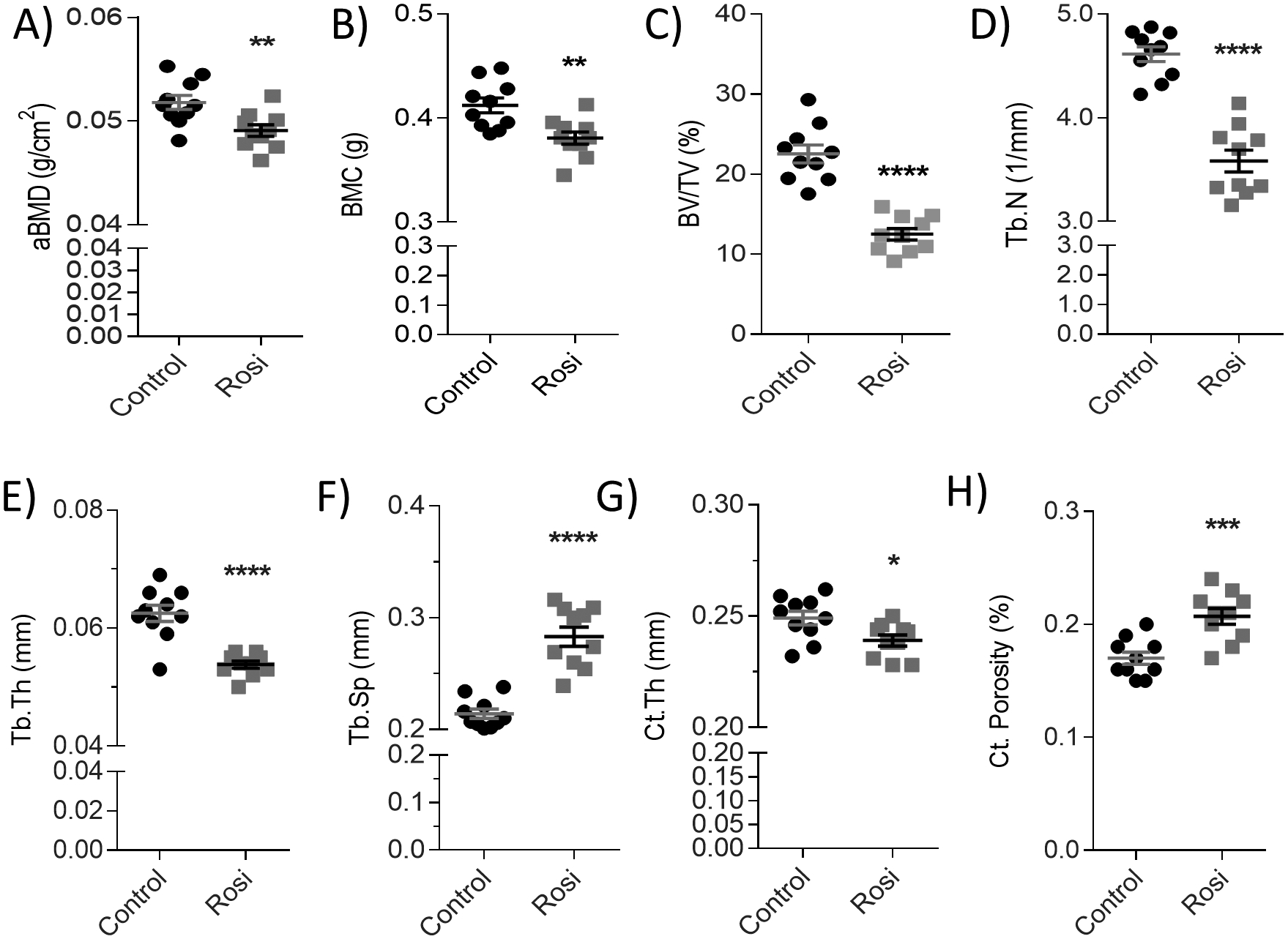
(A) Areal bone mineral density (g/cm2) and (B) bone mineral content (g). μCT data of tibial (C) bone volume per total volume (BV/TV, %), (D) trabecular number (Tb.N, 1/mm), (E) trabecular thickness (Tb.Th, mm), (F) trabecular spacing (Tb.Sp, mm) (n=10). (G) Cortical thickness (mm) and (H) cortical porosity (%) (n=10). Statistical significance: ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05. Data shown as data points with the error bar representing the mean ± S.E.M. Analyses were performed as unpaired Student’s t test within Prism.
We next performed high resolution characterization of the femora using static and dynamic histomorphometry to confirm the bone loss phenotype. Femoral BV/TV was significantly reduced by rosiglitazone to about 50% of the controls (Fig. 2A). The large changes in bone volume were attributed to significantly decreased trabecular number (Fig. 2B) and thickness (Fig. 2C), and significantly increased trabecular spacing (Fig. 2D). On a cellular level, there was a slight decrease in osteoblast number per bone perimeter (N.Ob/B.Pm) (Fig. 2E) and a significant increase in osteoclast number per bone perimeter (N.Oc/B.Pm) (Fig. 2F) in the rosiglitazone-treated mice. Mineral apposition rate (MAR) (Supp Fig. 2B) and mineralizing surface per bone surface (MS/BS) (equivalent to dlS/BS%) (Supp Fig. 2C) were not significantly changed. The eroded surface per bone surface (ES/BS) was slightly increased (Fig. 2G) and the bone formation rate per bone surface (BFR/BS) was significantly decreased after rosiglitazone treatment (Fig. 2H), demonstrating that the bone formation/resorption balance was adversely skewed, as visualized in the representative femoral dynamic histomorphometry images (Supp Fig. 2D). Overall, these data demonstrate that rosiglitazone treatment negatively affects bone.
Figure 2: Acute rosiglitazone treatment reduces bone volume and trabecular architecture in the femora.
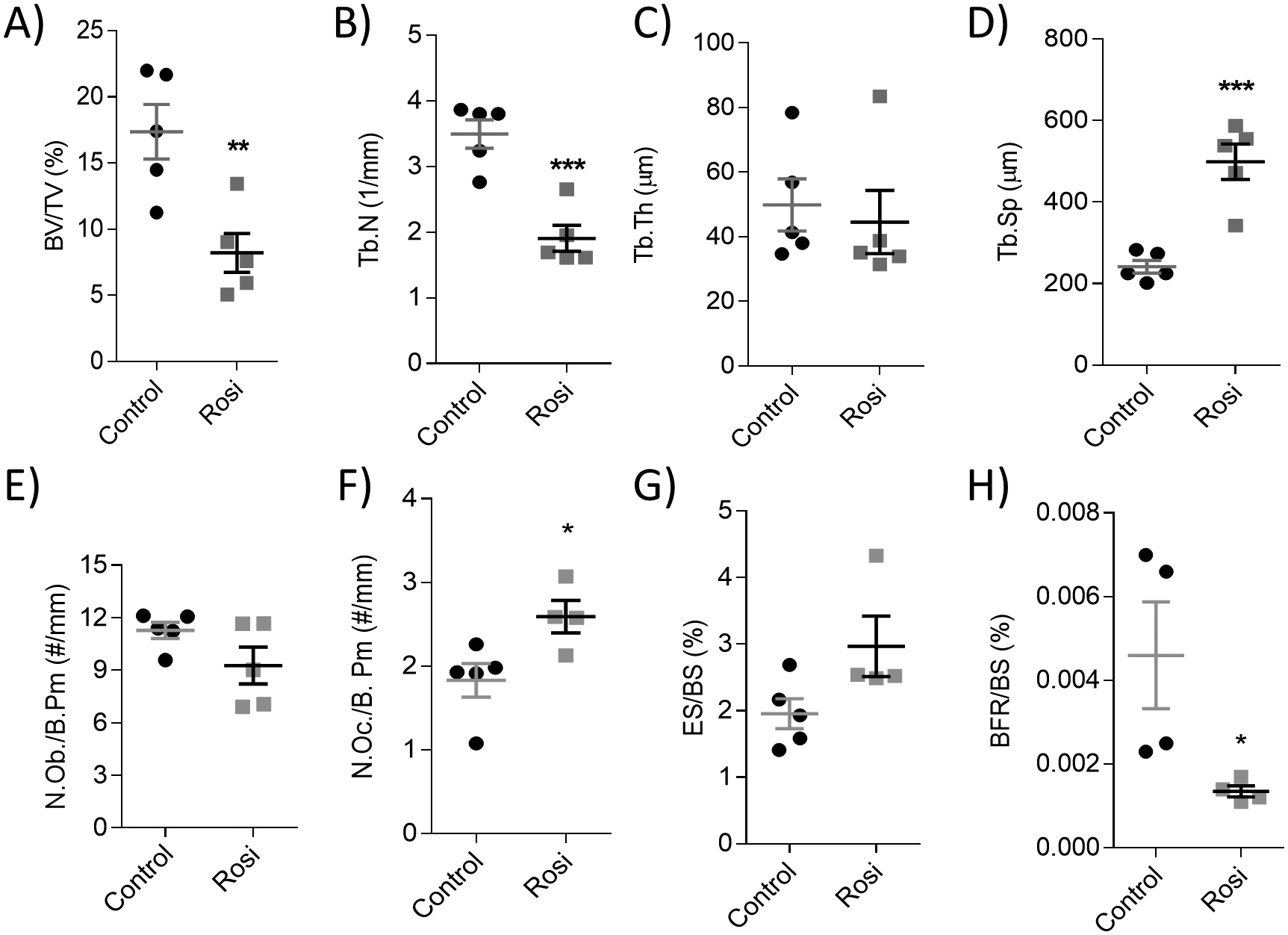
Static and dynamic histomorphometric analysis of the femoral (A) bone volume per total volume (BV/TV, %), (B) trabecular number (Tb.N, 1/mm), (C) trabecular thickness (Tb.Th, μm), (D) trabecular spacing (Tb.Sp, μm), (E) osteoblast number per bone perimeter (N.Ob/B.Pm, #/mm), (F) osteoclast number per bone perimeter (N.Oc/B.Pm, #/mm), (G) eroded surface per bone surface (ES/BS, %) and (H) bone formation rate per bone surface (BFR/BS, %), (n=4–5). Scale bar = 300 μm. Statistical significance: ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05. Data shown as data points with the error bar representing the mean ± S.E.M. Analyses were performed as unpaired Student’s t test within Prism.
Rosiglitazone increases bone marrow adiposity in the femur and tibia
Concurrently with extensive bone loss, rosiglitazone significantly increased femoral adipose volume fraction (AV/TV) (Fig. 3A) with a significant increase in the overall number of adipocytes (N.Ad) (Fig. 3B) within the BM, easily visualized in representative femoral histology sections (Fig. 3C). These phenotypic changes were accompanied by increases in adipocyte-specific gene expression in whole BM, with significant increases in both adiponectin (Adipoq, Supp Fig. 3E) and fatty acid binding protein 4 (Fabp4, Supp Fig. 3F). Similarly, tibial OsO4 μCT revealed significantly increased AV/TV and adipose volume per marrow volume (AV/MV) within the distal diaphysis (Fig. 3D and E) regions, proximal metaphysis (Supp Fig. 3A and B), and the proximal diaphysis (Supp Fig. 3C and D). Representative OsO4 μCT images demonstrate the increased BMAT in rosiglitazone-treated mice (Fig. 3F). Collectively, these data demonstrate that rosiglitazone increases adiposity at the expense of both bone and red marrow (based on AV/MV calculation); this aligns well with prior findings in other mouse models showing that rosiglitazone increases BMAT, decreases myelopoiesis and impairs the maintenance and differentiation of hematopoietic stem/progenitor cells(51).
Figure 3: Rosiglitazone significantly increases bone marrow adiposity after four weeks.
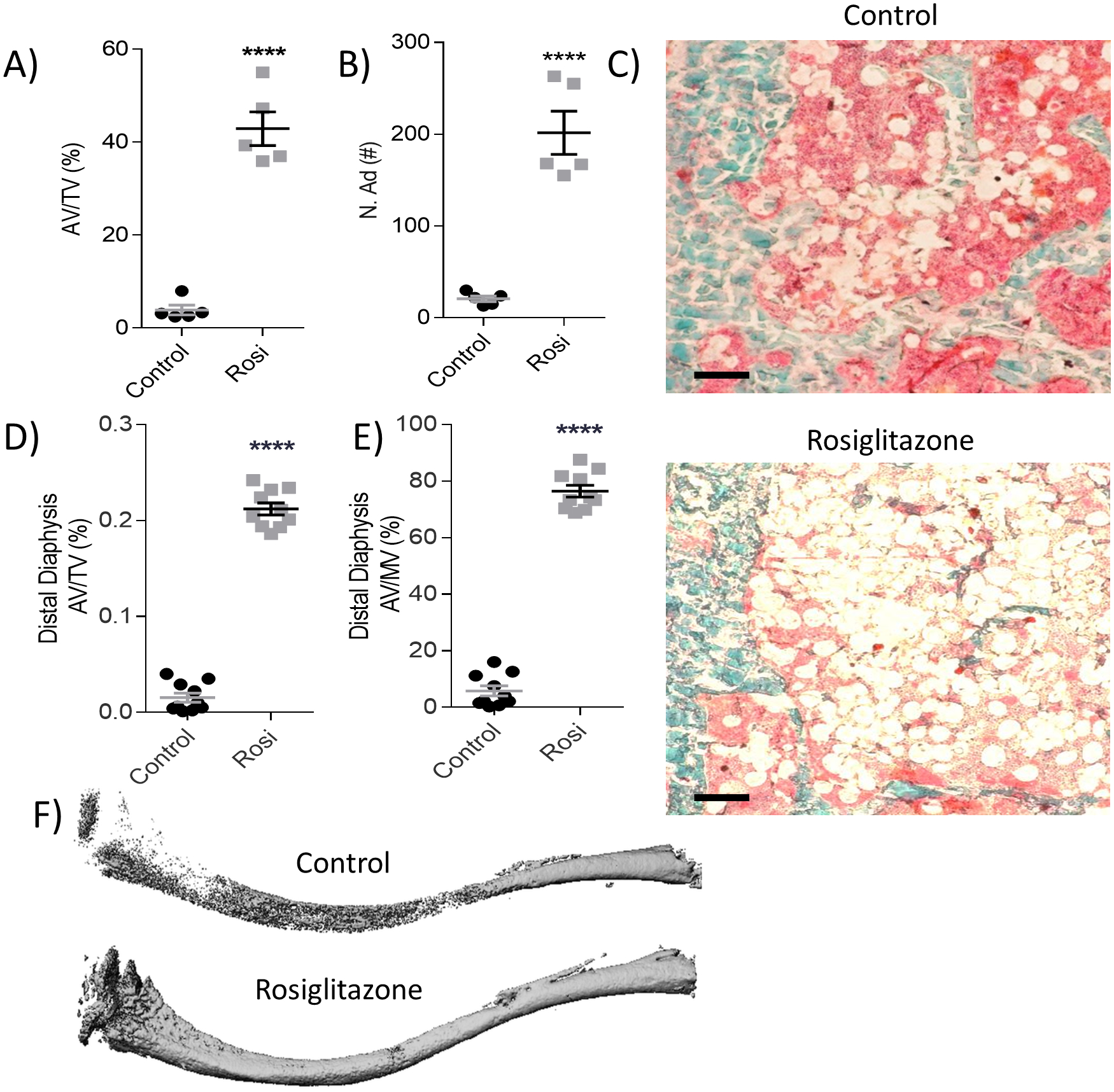
OsteoMeasure analysis of the femur (A) adipose tissue volume per total volume (AV/TV, %) and (B) adipose number (N.Ad, #) quantification, (n=5). (C) Representative histology images of femoral Goldner’s Trichrome in control (left) and rosiglitazone-treated mice (right). Scale bar = 100 μm. (D, E) Tibial osmium tetroxide (OsO4) μCT quantification of AV/TV (%) and adipose volume per marrow volume (AV/MV, %) in the distal diaphysis (n=10). (F) Representative reconstructions of OsO4 stained tibia of the control and rosiglitazone- treated mice. Statistical significance: ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05. Data shown as data points with the error bar representing the mean ± S.E.M. Analyses were performed as unpaired Student’s t test within Prism.
Effects of rosiglitazone and sclerostin-neutralizing antibody differ based on WAT depot
We then tested the effects of SOST Ab on rosiglitazone-treated and control mice, with the hypothesis that rosiglitazone-induced bone loss could be alleviated by ten weeks of SOST Ab treatments(12). In this study (cohort 2, Supp Fig. 4A), to characterize the metabolic effects of SOST Ab and rosiglitazone in WAT, visceral and inguinal WAT depots were analyzed. SOST Ab significantly reduced visceral fat mass, independent of rosiglitazone treatment (Supp Fig. 4B), while SOST Ab had no effect on inguinal fat mass (Supp Fig 4C). H&E histology (Supp Fig. 4D) illustrated no gross phenotypic changes in adipocytes, and adipocyte size was not significant between any of the groups for visceral (n=6) or inguinal (n=3) adipose depots (data not shown), suggesting that the difference in fat masses were due to number rather than size of adipocytes. No significant changes in body weight or blood glucose were observed throughout the ten weeks (Supp Fig. 5A, B). After four weeks of SOST Ab treatment, there was no significant change in fat mass or lean mass in the rosiglitazone-treated or control mice (Supp Fig. 5C and D). However, after ten weeks, fat mass and lean mass were significantly reduced by the addition of SOST Ab in the rosiglitazone-treated and control mice, as assessed by qNMR (Supp Fig. 5E and F).
Sclerostin-neutralizing antibodies reduce adverse effects of rosiglitazone on femoral and tibial bone properties
We then determined the lasting effects of rosiglitazone on trabecular bone and assessed if SOST Ab could rescue these effects. In cohort 2, characterization with bone histomorphometry and μCT analyses was completed on femur and tibia, respectively. Despite 6 weeks of recovery after stopping rosiglitazone treatment, the decreased trabecular parameters induced by rosiglitazone persisted in the femur (Fig. 4). In the femur, we observed a trend towards decreased BV/TV (Fig. 4A), significantly decreased trabecular number (Fig. 4B) and no change in trabecular thickness (Fig. 4C), leading to a significant increase in trabecular spacing (Fig. 4D), demonstrating the long-lasting detrimental effects of rosiglitazone on bone. Importantly, SOST Ab treatment significantly increased BV/TV (an average fold increase of 3.18 in control and 3.71 in rosiglitazone-treated mice), trabecular number, and trabecular thickness, and decreased trabecular spacing in rosiglitazone-treated and control mice (Fig. 4A–D). The mechanism of action of SOST Ab appears to be both anabolic and anti-resorptive, as seen by the significantly increased N.Ob/B.Pm, and a trend towards a decreased N.Oc/B.Pm in both groups (Fig. 4E and Supp Fig. 6A). The mineralization lag time (MLT) was increased by >2 fold in the rosiglitazone-treated mice, and SOST Ab significantly rescued this (Fig. 4F). In conjunction with the effects on MLT, SOST Ab significantly reduced osteoid volume per bone volume (OV/BV) in the rosiglitazone-treated mice and showed modest reductions in the ES/BS (Supp Fig. 6B and C). However, SOST Ab had no effect on CTX blood serum levels (data not shown). The prolonged negative effects of rosiglitazone on mineralizing surface per bone surface (MS/BS) (Fig. 4G) and BFR/BS (Fig. 4H) were reversed with SOST Ab by increasing mineralization and bone formation by an average fold increase of 2.46 and 5.34, respectively. The overall effects on trabecular bone can be seen in representative femoral dynamic histomorphometry images (Supp Fig. 6D). Overall, femoral histomorphometry data provided strong evidence of the negative impact rosiglitazone had on bone formation, in large part due to decreased osteoblast function, and the compensatory capabilities of SOST Ab to increase osteoblast number and efficacy to prevent bone loss.
Figure 4: Sclerostin-neutralizing antibodies prevent adverse effects of rosiglitazone in the femur of female SCID Beige mice.
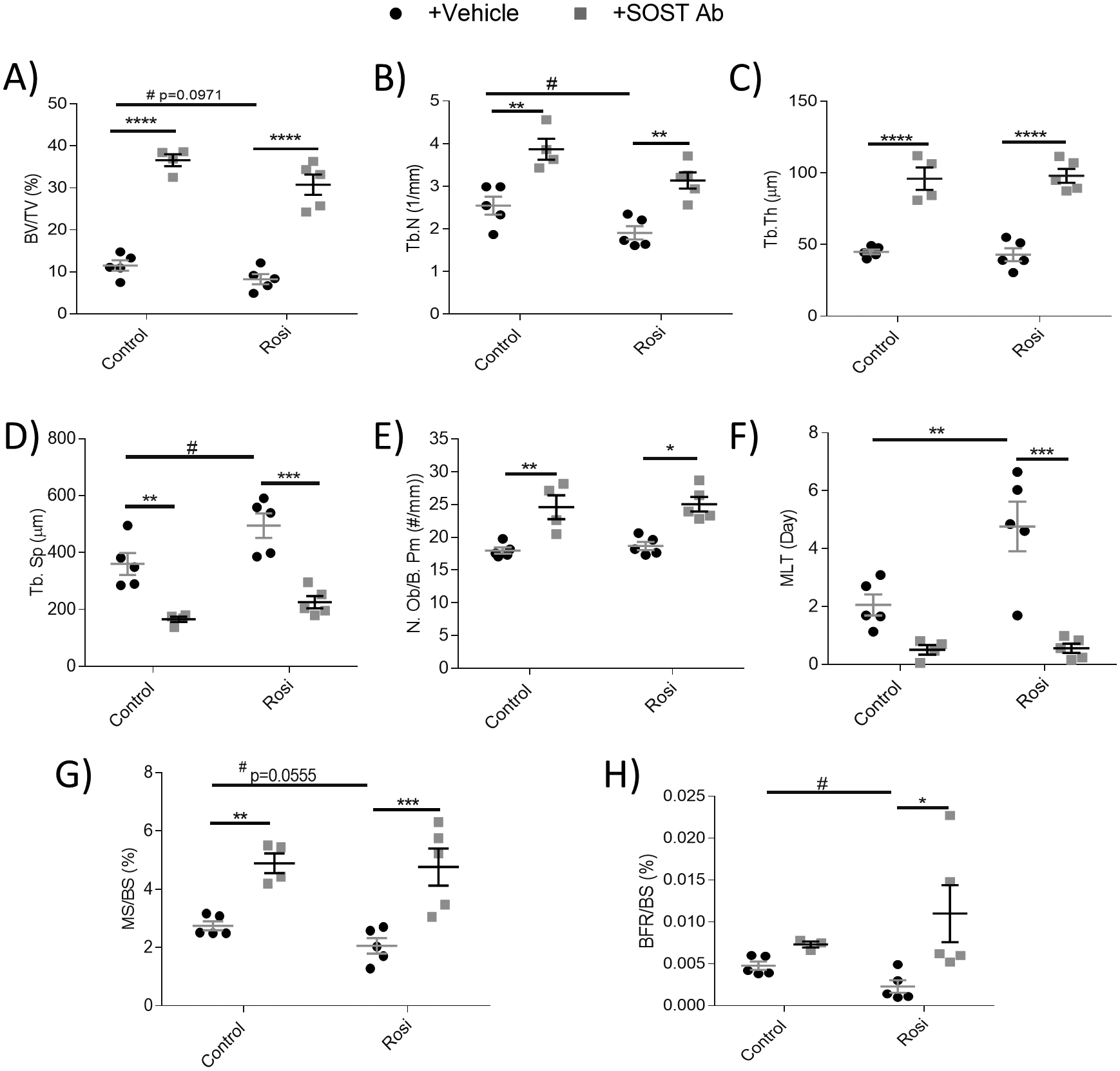
Static and dynamic histomorphometric analysis of the femoral (A) bone volume per total volume (BV/TV, %), (B) trabecular number (Tb.N, 1/mm), (C) trabecular thickness (Tb.Th, μm), (D) trabecular spacing (Tb.Sp, μm), (E) osteoblast number per bone perimeter (N.Ob/B.Pm, #/mm), (F) mineralization lag time (MLT/day), (G) mineralizing surface/bone surface (MS/BS, %), and (H) bone formation rate per bone surface (BFR,BS, %) (n=4–5). Data shown as data points with the error bar representing the mean ± S.E.M. Analyses were performed as unpaired Student’s t test or as 2-way ANOVA + Tukey’s multiple comparison within Prism. Statistical significance denoted as: t test #p<0.05; 2-way ANOVA ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05.
Tibial μCT data supported femoral data, as seen in representative images (Supp Fig. 7A). Here, rosiglitazone showed significantly decreased BV/TV and trabecular number (although no effect on trabecular thickness) which led to significantly increased trabecular spacing (Supp Fig. 7B–E). Rosiglitazone also caused a decrease in overall BMD, despite the recovery period (Supp Fig. 7F). SOST Ab significantly reversed this damage and improved all these parameters in rosiglitazone-treated mice. Further, the same trend of improved bone parameters was observed with SOST Ab treatment in controls. SOST Ab also significantly increased cortical area per total area in both rosiglitazone-treated and control mice (Supp Fig. 7G). Thus, in both tibia and femur analyses, using multiple analysis methods, we observed that SOST Ab can prevent bone loss caused by rosiglitazone, which was most notable in the trabecular region where rosiglitazone caused the greatest bone loss.
Sclerostin-neutralizing antibodies normalize rosiglitazone-induced BMAT expansion in some, but not all, bone marrow locations
Bone volume and BMAT are often considered to be inversely correlated, and while this is often true, the relationship between these tissues is in fact much more complicated and context-specific(22). Thus, we first interrogated if osteoanabolic SOST Ab affected BMAT via histology. In the femur, rosiglitazone induced an approximate 40–50% increase in AV/TV (Fig. 5A), as seen in representative histological sections (Fig. 5B). SOST Ab significantly decreased AV/TV in rosiglitazone-treated mice; this was due to significantly reducing adipocyte size (Supp Fig. 8A) and partly by adipocyte number (Fig. 5C).
Figure 5: Sclerostin-neutralizing antibodies modulate total adiposity, but not marrow adiposity, in mouse femur and tibia.
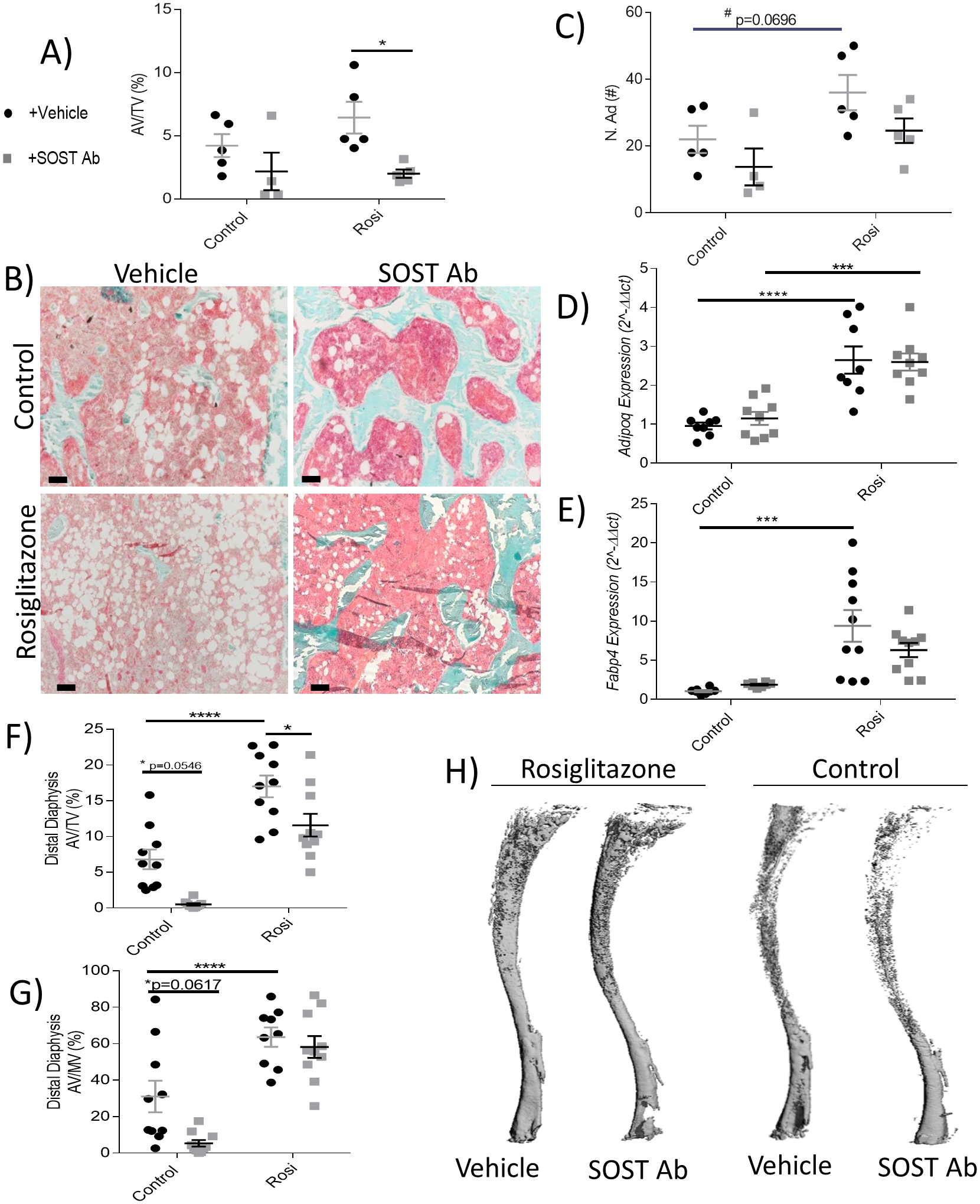
(A) Femoral adipose volume per total volume (AV/TV, %) quantification with OsteoMeasure after ten weeks (n=4–5). (B) Representative images of Goldner’s Trichrome-stained femora illustrate the effects of SOST Ab on bone (blue), marrow space (pink), and adipocytes (white) in the control and rosiglitazone- treated groups. Scale bar = 100 μm. (C) Number of adipocytes (N.Ad, #). Tibial BM gene expression of (D) adiponectin (Adipoq) and (E) fatty acid binding protein 4 (Fapb4) (n=10). Tibial OsO4 μCT of adipose volume in the (F) distal diaphysis AV/TV (%) and (G) AV/MV (%) (n=8–10). (H) Representative images of the control and rosiglitazone tibial OsO4 μCT, representing location of BMAT. Data shown as data points with the error bar representing the mean ± S.E.M. Analyses were performed as unpaired Student’s t test or as 2-way ANOVA + Tukey’s multiple comparison within Prism. Gene expression data analyses performed as 2-way ANOVA + Holm-Sidak multiple comparison within Prism. Statistical significance denoted as: t test #p<0.05; 2-way ANOVA ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05.
To gain a deeper understanding of BMAT responses to SOST Ab, we examined the tibia. BM from the tibia of rosiglitazone-treated mice had significant increases in adiponectin (Adipoq, Fig. 5D) and fatty acid binding protein 4 (Fabp4, Fig. 5E). However, SOST Ab was unable to reduce these levels similar to controls. We then examined the tibia with OsO4 μCT; there were no significant effects of rosiglitazone or SOST Ab in the proximal metaphysis (Supp Fig. 8B, C), but rosiglitazone significantly increased AV/TV and AV/MV within the proximal (Supp Fig. 8D, E) and distal diaphysis (Fig. 5 F, G). SOST Ab significantly reduced AV/TV, but not AV/MV, within the distal diaphysis of the rosiglitazone-treated group and reduced BM adiposity in the controls (AV/TV, p=0.0546 and AV/MV, p=0.0617) (Fig. 5 F, G). Representative tibial OsO4 μCT images, which show BMAT specifically, are shown in Fig. 5H. To understand the mechanism of action of SOST Ab, we explored changes in Wnt signaling by examining BM gene expression of Axin2 and Smad6, two indicators of Wnt signaling. Rosiglitazone induced no significant changes in Axin2 (Supp. Fig. 8F) or Smad6 (Supp. Fig. 8G). However, SOST Ab significantly increased Smad6, indicating an increase in Wnt signaling as expected, which could contribute to its effects on BM adiposity. In sum, SOST Ab reduced or had no effect on BMAT in the femur and regions of tibia, suggesting that different BMAT depots may respond differently to SOST Ab.
5. Discussion
In summary, we demonstrated the negative effects of rosiglitazone on mouse skeletal health (worse bone parameters and increased BMAT) after 4 weeks of treatment and found that some of these detrimental effects remained even after a 6-week recovery period. We showed many of the negative effects of rosiglitazone on bone could be reversed with SOST Ab intervention, and demonstrated that some of the BMAT depots increased by rosiglitazone could be decreased with SOST Ab. This is the first work to show rosiglitazone’s effects in an immunocompromised mouse, which allows researchers to better understand if the immune system plays any role in the adipogenic signaling of TZDs. This research also builds upon prior pre-clinical work demonstrating the efficacy of SOST Ab in various bone and metabolic models(12,17,21), by being the first to characterize the combination or SOST Ab and rosiglitazone in any mouse. The SCID Beige mouse model is commonly used in xenograft studies as a platform to model osteotropic cancers, such as multiple myeloma (MM), or bone-metastatic breast, prostate or lung cancer. Performing our research in this mouse is thus also useful, because within the last decade, the role of BM adipocytes in cancer and the role of SOST Ab as a potential therapy for bone cancer patients have become two emerging areas of research(52–54). Looking forward, our study provides a basis for future research using rosiglitazone and SOST Ab in SCID Beige to modulate both bone and bone marrow adiposity to investigate the effects of these drugs, the pathways they affect, and these tissue types on cancer progression (initiation, bone marrow homing, progression, and drug resistance).
Recently, BMAT has emerged as a dynamic, responsive member of the bone marrow microenvironment that contributes to many diseases, including osteoporosis, diabetic bone disease, and cancer growth within the marrow(11). BM adipocytes release many factors that create a supportive environment that many other cell types, benign or malignant, take advantage of. Elucidating the role that BM adipocytes play in progression of different diseases will allow for generation or repurposing of pharmaceutical treatments to improve bone health (eg. reduce diabetic bone disease, estrogen-deficiency driven osteoporosis, or tumor cell proliferation). Interestingly, our work illuminated the fact that adipocytes within WAT and BMAT respond differently both to rosiglitazone and SOST Ab, providing more evidence that these are distinct adipose depots and suggesting that even within the femur and tibia, BM adipocytes respond differently to stimuli. This interesting finding raises the question: are adipocytes/adipocyte progenitors in different adipose depots inherently different, or do they respond differently to stimuli due to differences in location in the body or along the bone? Future work is necessary to answer this and gain a deeper understanding of adipogenesis and mature adipocyte regulation in the marrow and in white adipose depots.
TZDs are acute, specific inhibitors of the mitochondrial pyruvate carrier, that effectively cause insulin sensitivity and enhance glucose uptake into cells(55). Mieczkowska and colleagues found TZDs act on osteocytes in two distinct mechanisms: TZDs can cause an increase in sclerostin production via PPARγ and they can trigger osteocyte apoptosis via GPR40, Erk1/2, and p38 pathways, causing a release of RANKL and sclerostin(34,56). This over-production of PPARγ and RANKL enhances adipocyte differentiation and decreases osteoblast differentiation(30,50). Our data showed short-term rosiglitazone treatment can significantly alter the functionality of osteoblasts and that these effects were still prevalent despite a six-week recovery period. However, since sclerostin can also regulate MSC lineage commitment, SOST Ab treatment can create a shift towards osteoblastogenesis through increased Wnt signaling(21). This change in progenitor cell fate would increase bone formation while decreasing adipogenesis. Supporting this, Smad6 gene expression was significantly increased in SOST Ab treated mice, even with rosiglitazone, suggesting increased Wnt signaling. ZBP1 has been shown to be instrumental in the fate of MSCs and plays a role in trans-differentiation between adipocytes and osteoblasts(57). However, ZBP1 gene expression in tibia BM was not significantly altered by rosiglitazone or SOST Ab at the end of cohort two (data not shown). It is possible that ZBP1 was modulated at earlier timepoints than investigated here.
Rosiglitazone-treated mice in combination with SOST Ab had dramatically improved femoral trabecular parameters through increased bone mineralization and formation rates. The increased osteoblast numbers and efficacy as a result of SOST Ab may have mitigated the damaging PPARγ-agonist properties of rosiglitazone, ultimately preventing bone loss. Overall, SOST Ab proved to be an effective course of treatment to prevent rosiglitazone-induced bone loss. To further support our experiment, it would be useful to test the addition of SOST Ab only after the 4 weeks of rosiglitazone in vivo, or only during the rosiglitazone treatments. This experiment would illustrate the effectiveness of SOST Ab treatment specifically at inhibiting bone loss or at improving bone recovery.
Another important finding was that SOST Ab decreased BM adiposity in the femur and specific regions of the tibia, based on the area measured and normalization used (total volume vs marrow volume). The decrease in BMAT may have contributed to the trend of decreased osteoclast numbers we observed in SOST Ab treatment, as BM adipocytes have been shown to express RANKL(50). However, we did not observe a large impact on osteoclast numbers with SOST Ab, which aligns with our prior findings that the major influence of SOST Ab is on osteoblasts, rather than osteoclasts(12). Within the femur, we did show SOST Ab reduced adipocyte size significantly, which contributed to the overall decrease in AV/TV. We speculate the reduction in adipocyte size may be the result of delipidation or fatty acid transfer, which could in turn increase osteoblast differentiation or function, which we observed in SOST Ab-treated mice. The spatial variation in tibial response may be due to different types of BMAT depots in these regions(58). The distal tibia is typically considered a more constitutive adipose depot, yet in the distal diaphysis, rosiglitazone and SOST Ab affected BM adiposity, demonstrating BMAT responsiveness in an adipose depot previously thought to be non-regulated. In rats, we have shown SOST Ab and human parathyroid hormone (1–34) (hPTH) can decrease total adiposity (AV/TV), while hPTH can also decrease marrow adiposity (AV/MV)(22,50). The mode of function differs from SOST Ab in that hPTH stimulates osteoblast and osteoclast activity simultaneously, although the former is more pronounced. Overall, these data highlight a need in the field to delve into the direct effects osteoanabolic agents have on BM adipocytes/pre-adipocytes (unrelated to bone accrual).
Clinically, novel mechanisms for counteracting the detrimental bone effects of PPARγ activation would benefit patients suffering from bone loss from excessive PPARγ signaling (either from genetic mutation or for those prescribed TZDs). Interestingly, the effects of rosiglitazone on bone mass and fragility were found to be attenuated with an anti-resorptive agent (alendronate)(35), suggesting that an SOST Ab and an anti-resorptive agent combination might be the ideal treatment for rosiglitazone-induced bone loss. Still, this remains speculative, especially since the alendronate + rosiglitazone study described was performed in an ovariectomized rat model, complicating the interpretation of the bone loss mechanism (and SOST Ab recovery effect) in this model.
In conclusion, herein we contributed to the understanding of how different tissues respond to osteogenic (Wnt) and adipogenic (PPARγ) signals in vivo. By characterizing the effects of rosiglitazone and SOST Ab on bone and BMAT in an immunocompromised mouse model, we had laid the framework for using these agents to modulate these tissues in cancer xenograft models. Cells and tissues are bombarded continuously with contradictory signals; how they simultaneously process these signals and determine how to respond is an enigma. In our work, we exposed which tissues were able to respond to osteogenic and adipogenic signals and determined that enhanced osteogenic signaling can overcome the inhibitory effects of PPARγ agonism in bone, and counteract the adipogenic signal in certain adipose locations, as well. Our results have implications for patients, researchers developing cancer mouse models, and researchers interested in how cells respond when given both a pro-adipogenic and pro-osteogenic signal.
Supplementary Material
6. Acknowledgments
Funding was supplied by the NIH/NIGMS (P20GM121301) and the NIH/NCI 1R37CA245330-01A1. The authors’ work is also supported by start-up funds from the Maine Medical Center Research Institute and the American Cancer Society (Research Grant #IRG-16-191-33 and #133077-RSG-19-037-01-LIB). This research utilized services of the U54GM115516 Core, which is supported by NIH/NIGMS P20GM121301, U54GM115516, and P30GM106391. This work was also partially supported by a pilot grant from the Massachusetts General Hospital (NIH/NIAMS P30AR066261). We thank Dr. Mary Bouxsein and Michael Bruce at the Massachusetts General Hospital Center for Skeletal Research (NIH P30AR066261) for μCT and OsO4 μCT analysis and Drs. Marie Demay and Janaina Da Silva Martins for assisting with the OsteoMeasure analysis. We thank Peter Cardonna of the Histology and Imaging Core at the University of New England for imaging assistance. We thank Mereo for providing the sclerostin-neutralizing antibodies. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Portions of this work were presented in abstract form as a poster at the American Society of Bone and Mineral Research (ASBMR) Annual Meeting in Orlando, Florida, Sept 2019.
Footnotes
Disclosure Statement: Authors have nothing to disclose.
8. References
- 1.Lwin ST, Olechnowicz SWZ, Fowler JA, Edwards CM. Diet-induced obesity promotes a myeloma-like condition in vivo. Leukemia. 2015. February;29(2):507–10. [DOI] [PubMed] [Google Scholar]
- 2.Marinac CR, Birmann BM, Lee I-M, Rosner BA, Townsend MK, Giovannucci E, Rebbeck TR, Buring JE, Colditz GA. Body mass index throughout adulthood, physical activity, and risk of multiple myeloma: a prospective analysis in three large cohorts. Br. J. Cancer. Springer US; 2018;(March):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beason TS, Chang S-H, Sanfilippo KM, Luo S, Colditz GA, Vij R, Tomasson MH, Dipersio JF, Stockerl-Goldstein K, Ganti A, Wildes T, Carson KR. Influence of Body Mass Index on Survival in Veterans With Multiple Myeloma. Oncologist. 2013. October 1;18(10):1074–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Picon-Ruiz M, Morata-Tarifa C, Valle-Goffin JJ, Friedman ER, Slingerland JM. Obesity and adverse breast cancer risk and outcome: Mechanistic insights and strategies for intervention. CA. Cancer J. Clin. Wiley; 2017. September;67(5):378–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Cobelli O, Terracciano D, Tagliabue E, Raimondi S, Galasso G, Cioffi A, Cordima G, Musi G, Damiano R, Cantiello F, Detti S, Victor Matei D, Bottero D, Renne G, Ferro M. Body mass index was associated with upstaging and upgrading in patients with low-risk prostate cancer who met the inclusion criteria for active surveillance. Urol. Oncol. Semin. Orig. Investig. Elsevier Inc; 2015. May 1;33(5):201.e1–201.e8. [DOI] [PubMed] [Google Scholar]
- 6.Bullwinkle EM, Parker MD, Bonan NF, Falkenberg LG, Davison SP, DeCicco-Skinner KL. Adipocytes contribute to the growth and progression of multiple myeloma: Unraveling obesity related differences in adipocyte signaling. Cancer Lett. 2016. September 28;380(1):114–21. [DOI] [PubMed] [Google Scholar]
- 7.Liu Z, Xu J, He J, Liu H, Lin P, Wan X, Navone NM, Tong Q, Kwak LW, Orlowski RZ, Yang J, Nora M, Tong Q, Kwak LW, Orlowski RZ, Yang J. Mature adipocytes in bone marrow protect myeloma cells against chemotherapy through autophagy activation. Oncotarget. 2015. October 27;6(33):34329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caers J, Deleu S, Belaid Z, De Raeve H, Van Valckenborgh E, De Bruyne E, Defresne M-P, Van Riet I, Van Camp B, Vanderkerken K. Neighboring adipocytes participate in the bone marrow microenvironment of multiple myeloma cells. Leukemia. 2007. July;21(7):1580–4. [DOI] [PubMed] [Google Scholar]
- 9.Campbell HF, Marinac CR, Masarwi M, Birmann B, Reagan M. Investigation of the relationship between obesity, weight cycling, and tumor progression in a myeloma xenograft model. Clin. Lymphoma Myeloma Leuk. Elsevier; 2019. October 1;19(10):e88. [Google Scholar]
- 10.Farrell M, Falank C, Campbell HF, Costa S, Bowers D, Reagan M. Targeting bone marrow adipose tissue and the FABP family increases efficacy of dexamethasone in MM. Clin. Lymphoma Myeloma Leuk. Elsevier; 2019. October 1;19(10):e89–90. [Google Scholar]
- 11.Veldhuis-Vlug AG, Rosen CJ. Clinical implications of bone marrow adiposity. J. Intern. Med 2018. February;283(2):121–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDonald MM, Reagan MR, Youlten SE, Mohanty ST, Seckinger A, Terry RL, Pettitt JA, Simic MK, Cheng TL, Morse A, Le LMT, Abi-Hanna D, Kramer I, Falank C, Fairfield H, Ghobrial IM, Baldock PA, Little DG, Kneissel M, Vanderkerken K, Bassett JHD, Williams GR, Oyajobi BO, Hose D, Phan TG, Croucher PI. Inhibiting the osteocyte-specific protein sclerostin increases bone mass and fracture resistance in multiple myeloma. Blood. 2017. June 29;129(26):3452–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reagan MR, Liaw L, Rosen CJ, Ghobrial IM. Dynamic Interplay between Bone and Multiple Myeloma: Emerging Roles of the Osteoblast. Bone. 2015. February 25;75:161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Q-Y, Li GHY, Kung AWC. The −9247 T/C polymorphism in the SOST upstream regulatory region that potentially affects C/EBPalpha and FOXA1 binding is associated with osteoporosis. Bone. 2009. August;45(2):289–94. [DOI] [PubMed] [Google Scholar]
- 15.Yerges LM, Klei L, Cauley JA, Roeder K, Kammerer CM, Moffett SP, Ensrud KE, Nestlerode CS, Marshall LM, Hoffman AR, Lewis C, Lang TF, Barrett-Connor E, Ferrell RE, Orwoll ES, Zmuda JM. High-density association study of 383 candidate genes for volumetric BMD at the femoral neck and lumbar spine among older men. J. Bone Miner. Res 2009. December;24(12):2039–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Ominsky MS, Niu Q-T, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res 2008. June;23(6):860–9. [DOI] [PubMed] [Google Scholar]
- 17.Ominsky MS, Brown DL, Van G, Cordover D, Pacheco E, Frazier E, Cherepow L, Higgins-Garn M, Aguirre JI, Wronski TJ, Stolina M, Zhou L, Pyrah I, Boyce RW. Differential temporal effects of sclerostin antibody and parathyroid hormone on cancellous and cortical bone and quantitative differences in effects on the osteoblast lineage in young intact rats. Bone. 2015. December;81:380–91. [DOI] [PubMed] [Google Scholar]
- 18.Ren Y, Han X, Jing Y, Yuan B, Ke H, Liu M, Feng JQ. Sclerostin antibody (Scl-Ab) improves osteomalacia phenotype in dentin matrix protein 1(Dmp1) knockout mice with little impact on serum levels of phosphorus and FGF23. Matrix Biol. 2016. May;52–54:151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyce RW, Niu Q-T, Ominsky MS. Kinetic reconstruction reveals time-dependent effects of romosozumab on bone formation and osteoblast function in vertebral cancellous and cortical bone in cynomolgus monkeys. Bone. 2017. August;101:77–87. [DOI] [PubMed] [Google Scholar]
- 20.Cardinal M, Dessain A, Roels T, Lafont S, Ominsky MS, Devogelaer JP, Chappard D, Mabilleau G, Ammann P, Nyssen-Behets C, Manicourt DH. Sclerostin-Antibody Treatment Decreases Fracture Rates in Axial Skeleton and Improves the Skeletal Phenotype in Growing oim/oim Mice. Calcif. Tissue Int. Springer; 2020. February 6; [DOI] [PubMed] [Google Scholar]
- 21.Fairfield H, Falank C, Harris E, Demambro V, McDonald M, Pettitt JA, Mohanty ST, Croucher P, Kramer I, Kneissel M, Rosen CJ, Reagan MR. The skeletal cell-derived molecule sclerostin drives bone marrow adipogenesis. J. Cell. Physiol 2018. February;233(2):1156–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costa S, Fairfield H, Reagan MR. Inverse correlation between trabecular bone volume and bone marrow adipose tissue in rats treated with osteoanabolic agents. Bone. 2019. June 4;123:211–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim SP, Frey JL, Li Z, Kushwaha P, Zoch ML, Tomlinson RE, Da H, Aja S, Noh HL, Kim JK, Hussain MA, Thorek DLJ, Wolfgang MJ, Riddle RC. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc. Natl. Acad. Sci. U. S. A 2017. December 26;114(52):E11238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.FDA Approves EVENITY™ (romosozumab-aqqg) For The Treatment Of Osteoporosis In Postmenopausal Women At High Risk For Fracture - National Osteoporosis Foundation [Internet]. [cited 2019 Apr 29]. Available from: https://www.nof.org/news/fda-approves-evenity-romosozumab-aqqg-for-the-treatment-of-osteoporosis-in-postmenopausal-women-at-high-risk-for-fracture/
- 25. European Commission Approves EVENITY romosozumab For The Treatment Of Severe Osteoporosis In Postmenopausal Women At High Risk Of Fracture.
- 26.Shoback D, Rosen CJ, Black DM, Cheung AM, Murad MH, Eastell R. Pharmacological Management of Osteoporosis in Postmenopausal Women: An Endocrine Society Guideline Update. J. Clin. Endocrinol. Metab 2020. March 1;105(3). [DOI] [PubMed] [Google Scholar]
- 27.Lupi R, Del Guerra S, Marselli L, Bugliani M, Boggi U, Mosca F, Marchetti P, Del Prato S. Rosiglitazone prevents the impairment of human islet function induced by fatty acids: Evidence for a role of PPARγ2 in the modulation of insulin secretion. Am. J. Physiol. - Endocrinol. Metab 2004. April;286(4 49–4). [DOI] [PubMed] [Google Scholar]
- 28.Liu C, Feng T, Zhu N, Liu P, Han X, Chen M, Wang X, Li N, Li Y, Xu Y, Si S. Identification of a novel selective agonist of PPARγ with no promotion of adipogenesis and less inhibition of osteoblastogenesis. Sci. Rep. Nature Publishing Group; 2015. August;5(1):9530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kahn CR, Chen L, Cohen SE. Unraveling the mechanism of action of thiazolidinediones. J. Clin. Invest. The American Society for Clinical Investigation; 2000. p. 1305–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stechschulte LA, Czernik PJ, Rotter ZC, Tausif FN, Corzo CA, Marciano DP, Asteian A, Zheng J, Bruning JB, Kamenecka TM, Rosen CJ, Griffin PR, Lecka-Czernik B. PPARG Post-translational Modifications Regulate Bone Formation and Bone Resorption. EBioMedicine. Elsevier B.V; 2016. August;10:174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beck GR, Khazai NB, Bouloux GF, Camalier CE, Lin Y, Garneys LM, Siqueira J, Peng L, Pasquel F, Umpierrez D, Smiley D, Umpierrez GE. The effects of thiazolidinediones on human bone marrow stromal cell differentiation in vitro and in thiazolidinedione-treated patients with type 2 diabetes. Transl. Res 2013. March;161(3):145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sulston RJ, Learman BS, Zhang B, Scheller EL, Parlee SD, Simon BR, Mori H, Bree AJ, Wallace RJ, Krishnan V, MacDougald OA, Cawthorn WP. Increased Circulating Adiponectin in Response to Thiazolidinediones: Investigating the Role of Bone Marrow Adipose Tissue. Front. Endocrinol. (Lausanne) 2016. September 21;7:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ali AA, Weinstein RS, Stewart SA, Parfitt AM, Manolagas SC, Jilka RL. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology. 2005. March;146(3):1226–35. [DOI] [PubMed] [Google Scholar]
- 34.Mabilleau G, Mieczkowska A, Edmonds ME. Thiazolidinediones induce osteocyte apoptosis and increase sclerostin expression. Diabet. Med 2010. August;27(8):925–32. [DOI] [PubMed] [Google Scholar]
- 35.Kumar S, Hoffman SJ, Samadfam R, Mansell P, Jolette J, Smith SY, Guldberg RE, Fitzpatrick LA. The Effect of Rosiglitazone on Bone Mass and Fragility Is Reversible and Can Be Attenuated With Alendronate. J. Bone Miner. Res 2013. July;28(7):1653–65. [DOI] [PubMed] [Google Scholar]
- 36.Lazarenko OP, Rzonca SO, Hogue WR, Swain FL, Suva LJ, Lecka-Czernik B. Rosiglitazone induces decreases in bone mass and strength that are reminiscent of aged bone. Endocrinology. NIH Public Access; 2007. June;148(6):2669–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ackert-Bicknell CL, Shockley KR, Horton LG, Lecka-Czernik B, Churchill GA, Rosen CJ. Strain-specific effects of rosiglitazone on bone mass, body composition, and serum insulin-like growth factor-I. Endocrinology. 2009;150:1330–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tornvig L, Mosekilde L, Justesen J, Falk E, Kassem M. Troglitazone Treatment Increases Bone Marrow Adipose Tissue Volume but Does not Affect Trabecular Bone Volume in Mice. Calcif. Tissue Int. Springer-Verlag; 2001. July;69(1):46–50. [DOI] [PubMed] [Google Scholar]
- 39.Liu H, He J, Koh SP, Zhong Y, Liu Z, Wang Z, Zhang Y, Li Z, Tam BT, Lin P, Xiao M, Young KH, Amini B, Starbuck MW, Lee HC, Navone NM, Davis RE, Tong Q, Bergsagel PL, Hou J, Yi Q, Orlowski RZ, Gagel RF, Yang J. Reprogrammed marrow adipocytes contribute to myeloma-induced bone disease. Sci. Transl. Med 2019. May 29;11(494):eaau9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reagan MR, Rosen CJ. Navigating the bone marrow niche: translational insights and cancer-driven dysfunction. Nat. Rev. Rheumatol 2016. November 26;12(3):154–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reagan MR, McDonald MM, Terry R, Pettitt J, Le L, Mohanty S, Kramer I, Kneissel M, Brooks DJ, Bouxsein M, Rosen C, Ghobrial I, Croucher PI. Anti-Sclerostin Treatment Prevents Multiple Myeloma Induced Bone Loss and Reduces Tumor Burden. Blood. 2015;126(23):119. [Google Scholar]
- 42.Chandra A, Lin T, Young T, Tong W, Ma X, Tseng W-J, Kramer I, Kneissel M, Levine MA, Zhang Y, Cengel K, Liu XS, Qin L. Suppression of Sclerostin Alleviates Radiation-Induced Bone Loss by Protecting Bone-Forming Cells and Their Progenitors Through Distinct Mechanisms. J. Bone Miner. Res 2017. February;32(2):360–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Motyl KJ, DeMambro VE, Barlow D, Olshan D, Nagano K, Baron R, Rosen CJ, Houseknecht KL. Propranolol Attenuates Risperidone-Induced Trabecular Bone Loss in Female Mice. Endocrinology. The Endocrine Society; 2015. July;156(7):2374–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engblom C, Pfirschke C, Zilionis R, Da Silva Martins J, Bos SA, Courties G, Rickelt S, Severe N, Baryawno N, Faget J, Savova V, Zemmour D, Kline J, Siwicki M, Garris C, Pucci F, Liao H-W, Lin Y-J, Newton A, Yaghi OK, Iwamoto Y, Tricot B, Wojtkiewicz GR, Nahrendorf M, Cortez-Retamozo V, Meylan E, Hynes RO, Demay M, Klein A, Bredella MA, Scadden DT, Weissleder R, Pittet MJ. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF high neutrophils. Science (80-.). 2017. December;358(6367):eaal5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res 2013. January;28(1):2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu P, Ji Y, Yuen T, Rendina-Ruedy E, Demambro VE, Dhawan S, Abu-Amer W, Izadmehr S, Zhou B, Shin A, Latif R, Thangeswaran P, Gupta A, Li J, Shnayder V, Robinson S, Yu YE, Zhang X, Yang F, Lu P, Zhou Y, Zhu L-L, Oberlin DJ, Davies TF, Reagan MR, Brown A, Kumar TR, Epstein S, Iqbal J, New M ari. I, Molina H, Van Klinken JB, Guo EX, Buettner C, Haider S, Bian Z, Sun L, Rosen CJ, Zaidi M. Blocking FSH induces thermogenic adipose tissue and reduces body fat. Nature. 2017;546(7656):107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bornstein S, Moschetta M, Kawano Y, Sacco A, Huynh D, Brooks D, Manier S, Fairfield H, Falank C, Roccaro AM, Nagano K, Baron R, Bouxein M, Vary C, Ghobrial IM, Rosen CJ, Reagan MR. Metformin affects cortical bone mass and marrow adiposity in diet-induced obesity in male mice. Endocrinology. Nature Publishing Group; 2017. December 6;158(10):3369–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res 2010. July;25(7):1468–86. [DOI] [PubMed] [Google Scholar]
- 49.Scheller EL, Troiano N, Vanhoutan JN, Bouxsein MA, Fretz JA, Xi Y, Nelson T, Katz G, Berry R, Church CD, Doucette CR, Rodeheffer MS, Macdougald OA, Rosen CJ, Horowitz MC. Use of osmium tetroxide staining with microcomputerized tomography to visualize and quantify bone marrow adipose tissue in vivo. Methods Enzymol. 2014. January;537:123–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fan Y, Hanai J, Le PT, Bi R, Maridas D, DeMambro V, Figueroa CA, Kir S, Zhou X, Mannstadt M, Baron R, Bronson RT, Horowitz MC, Wu JY, Bilezikian JP, Dempster DW, Rosen CJ, Lanske B. Parathyroid Hormone Directs Bone Marrow Mesenchymal Cell Fate. Cell Metab. 2017;25(3):661–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu W, Wang W, Wang S, Feng Y, Liu K. Rosiglitazone Promotes Bone Marrow Adipogenesis to Impair Myelopoiesis under Stress. PLoS One. Public Library of Science; 2016;11(2):e0149543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colucci S, Brunetti G, Oranger A, Mori G, Sardone F, Specchia G, Rinaldi E, Curci P, Liso V, Passeri G, Zallone A, Rizzi R, Grano M. Myeloma cells suppress osteoblasts through sclerostin secretion. Blood Cancer J. 2011. June;1(6):e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delgado-Calle J, Anderson J, Cregor MD, Condon KW, Kuhstoss SA, Plotkin LI, Bellido T, Roodman GD. Genetic deletion of Sost or pharmacological inhibition of sclerostin prevent multiple myeloma-induced bone disease without affecting tumor growth. Leukemia. 2017. December 6;31(12):2686–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang X-T, He Y-C, Zhou S-Y, Jiang J, Huang Y-M, Liang Y-Z, Lai Y-R. Bone marrow plasma macrophage inflammatory protein protein-1 alpha(MIP-1 alpha) and sclerostin in multiple myeloma: relationship with bone disease and clinical characteristics. Leuk. Res 2014. May;38(5):525–31. [DOI] [PubMed] [Google Scholar]
- 55.Divakaruni AS, Wiley SE, Rogers GW, Andreyev AY, Petrosyan S, Loviscach M, Wall EA, Yadava N, Heuck AP, Ferrick DA, Henry RR, McDonald WG, Colca JR, Simon MI, Ciaraldi TP, Murphy AN. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc. Natl. Acad. Sci. U. S. A 2013. April;110(14):5422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mieczkowska A, Baslé MF, Chappard D, Mabilleau G. Thiazolidinediones induce osteocyte apoptosis by a G protein-coupled receptor 40-dependent mechanism. J. Biol. Chem 2012. July 6;287(28):23517–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao X, Xie L, Wang Z, Wang J, Xu H, Han X, Bai D, Deng P. ZBP1 (DAI/DLM-1) promotes osteogenic differentiation while inhibiting adipogenic differentiation in mesenchymal stem cells through a positive feedback loop of Wnt/β-catenin signaling. Bone Res. Springer Nature; 2020. December;8(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scheller EL, Doucette CR, Learman BS, Cawthorn WP, Khandaker S, Schell B, Wu B, Ding S-Y, Bredella MA, Fazeli PK, Khoury B, Jepsen KJ, Pilch PF, Klibanski A, Rosen CJ, MacDougald OA. Region-specific variation in the properties of skeletal adipocytes reveals regulated and constitutive marrow adipose tissues. Nat. Commun 2015. January;6:7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.