

Abstract
Calsequestrin (CASQ) was discovered in rabbit skeletal muscle tissues in 1971 and has been considered simply a passive Ca2+-buffering protein in the sarcoplasmic reticulum (SR) that provides Ca2+ ions for various Ca2+ signals. For the past three decades, physiologists, biochemists, and structural biologists have examined the roles of the skeletal muscle type of CASQ (CASQ1) in skeletal muscle and revealed that CASQ1 has various important functions as (1) a major Ca2+-buffering protein to maintain the SR with a suitable amount of Ca2+ at each moment, (2) a dynamic Ca2+ sensor in the SR that regulates Ca2+ release from the SR to the cytosol, (3) a structural regulator for the proper formation of terminal cisternae, (4) a reverse-directional regulator of extracellular Ca2+ entries, and (5) a cause of human skeletal muscle diseases. This review is focused on understanding these functions of CASQ1 in the physiological or pathophysiological status of skeletal muscle.
Subject terms: Physiology, Calcium and vitamin D, Diseases
Muscle function: Multiple roles for a muscle modulator
Although previously considered merely a passive regulator of calcium levels, the protein calsequestrin is now known to perform a range of physiological activities essential to skeletal muscle function. The process of muscle contraction depends on the release of calcium ions from an intracellular structure called the sarcoplasmic reticulum (SR). Calsequestrin was originally identified as a “buffering” factor that maintains adequate calcium reserves in the SR, but Eun Hui Lee and colleagues at the Catholic University of Korea review diverse functions that have since been ascribed to this protein. For example, calsequestrin also helps reinforce the structure of the SR, and actively regulates the flux of calcium ions into muscle cells. Perturbations in calsequestrin function also appear to contribute to a number of muscular disorders, including a potential link to Duchenne muscular dystrophy.
Introduction
Generally, the Ca2+ supply in intracellular Ca2+ signals consists of two Ca2+ pools: internal Ca2+ (in the endoplasmic or sarcoplasmic reticulum (ER or SR)) and extracellular Ca2+. To sustain body postures and perform body movements, skeletal muscle contracts or relaxes, and the main key elements governing skeletal muscle contraction and relaxation are well understood1,2. During contraction and relaxation, the movements of contractile proteins in skeletal muscle cells (myotubes in culture conditions and fibers from skeletal muscle tissue) are mainly dependent on the cytosolic level of Ca2+ ions that are released from the SR. Dihydropyridine receptor (DHPR, a voltage-gated Ca2+ channel (i.e., Cav1.1.) on the transverse (t)-tubule membrane) senses the depolarization of the t-tubule membrane (i.e., action potentials) in response to acetylcholine that is released from motor neurons. T-tubules are invaginations of the plasma membrane and have a transverse orientation with respect to the main axis of myofibrils. The conformational changes in DHPR due to the action potential subsequently activate ryanodine receptor type 1 (RyR1, an internal Ca2+ release channel on the SR membrane) via physical interactions (not all RyRs but every other RyR). The interaction allows the release of Ca2+ from the SR to the cytosol through RyR1. The released Ca2+ from the SR turns on contractile proteins and evokes skeletal muscle contraction. In short, a transient elevation in cytosolic Ca2+ levels due to Ca2+ release from the SR couples the action potential and muscle contraction (so-called “excitation–contraction (EC)” coupling). EC coupling is defined as an “orthograde signal” from DHPR in the t-tubule membrane to RyR1 in the SR membrane (to distinguish it from the retrograde signal from RyR1 to DHPR or the reverse-directional signals from proteins in the SR or on the SR membrane to proteins or events in the t-tubule/plasma membrane). Three different genes on chromosomes 19, 1, and 15 encode three distinct isoforms of RyRs (RyR1, RyR2, and RyR3). RyR1 is predominant in skeletal muscle, whereas RyR3 is expressed at much lower amounts than RyR1 in skeletal muscle. In addition to the physical interaction of DHPR and RyR1, the Ca2+ that is released from SR binds and activates nearby RyR1, which does not interact physically with DHPR. This is called Ca2+-induced Ca2+ release, which contributes to rapid intracellular Ca2+ release from the SR and to the amplification of Ca2+-mediated signals3.
Cytosolic [Ca2+] at rest (nM range) rises to the μM range during skeletal muscle contraction4. To establish the Ca2+ increase, in addition to the Ca2+ from the SR, extracellular Ca2+ entry via Ca2+ channels in the t-tubule membrane, such as Orai1 or transient receptor potential canonical type (TRPC), also participates in the cytosolic Ca2+ increase, especially for skeletal muscle contraction during tetanic stimulation or fatigue5–7. The relaxation of skeletal muscle involves the reuptake of Ca2+ from the cytosol to the SR to reset the resting cytosolic Ca2+ level and replenish SR Ca2+. Sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA, a pump in the ER/SR membrane) is mainly responsible for Ca2+ reuptake, and SERCA1a is the major isoform in adult skeletal muscle1,8. In addition to SERCA, the Na+/Ca2+ exchanger (NCX) in the plasma membrane, mNCX in the mitochondrial membrane or Ca2+-ATPase in the plasma membrane (called PMCA) also participate in removing Ca2+ from the cytosol under certain conditions, such as a Ca2+ increase involving store-operated Ca2+ entry (SOCE)9–11. The efficient arrangement of EC coupling-mediating proteins mentioned above along with their regulatory proteins in the triad junction (a specialized membrane structure in skeletal muscle cells, see “SR and triad junction” in this review) is crucial for their proper functions and intermolecular interactions12–14. Therefore, for the successful cycling of the rise and fall in cytosolic Ca2+ level during repetitive skeletal muscle contraction and relaxation during a lifetime, the spatiotemporal distributions of intracellular Ca2+ ions, timely expression of EC coupling-mediating proteins with their regulatory proteins at different levels, and formation of triad junctions should be coordinately prepared with pinpoint accuracy.
SR and triad junction
SR, a skeletal muscle form of ER, is a very dynamic intracellular organelle involved in the reserve, release, and reuptake of Ca2+1,15–18. SR is morphologically very specialized and distinct from the ER16,19,20, which is required for efficient and immediate Ca2+ release from the SR to the cytosol via RyR1. The SR is divided into two functionally distinct but continuous portions: the longitudinal SR (LSR) and the terminal cisternae. LSR is responsible for the largest part of the SR and is the site of Ca2+ reuptake from the cytosol via SERCA21,22. Terminal cisternae can be subdivided into junction-facing SR (JSR) and the remaining part16,23. JSR (also roughly called terminal cisternae in many other articles) is located in close proximity to t-tubules. The membrane structure that comprises two JSRs and a t-tubule between the two JSRs, such as a sandwich, is called the “triad junction”. Close arrangements of the JSR and t-tubule membranes in the triad junction allow physical interaction between DHPR in the t-tubule membrane and RyR1 in the JSR membrane. Therefore, the triad junction is a functional structure for the cytosolic Ca2+ increase during EC coupling.
The formation of the triad junction is primarily mediated by junctophilins (JPHs). Each JPH contains eight repeats of hydrophobic motifs (called the membrane occupation and recognition nexus (MORN)) and a C-terminal transmembrane domain spanning the JSR membrane12. MORN motifs confer the ability to attach to phospholipids in the t-tubule membrane, which causes JPHs to serve as bridges between the t-tubule and SR membranes and subsequently allow close and parallel positioning of the t-tubule and SR membranes in the triad junction12,13. JPH1 and JPH2 are expressed in adult skeletal muscle at different frequencies: JPH1 is found throughout the triad junction when RyR1 is present, and JPH2 is present at a reduced frequency compared to JPH124. In addition to their role as a structural bridge in the triad junction, JPHs also play functional roles by interacting with SR proteins and regulating Ca2+ movements in skeletal muscle. JPH1 can interact with RyR125. Both JPH1 and JPH2 can interact with DHPR26. JPH2, but not JPH1, can interact with TRPC327,28. JPH1 knockout mice show a reduction in the number of triad junctions and structural abnormalities in the SR, such as a vacuolated LSR and swollen triad29. Skeletal muscle fibers from JPH1 knockout mice induce a reduction in the expression of TRPC3, Orai1, and stromal interaction molecule 1 (STIM1), as well as reductions in Orai1-mediated SOCE, cytosolic Ca2+ levels, and Ca2+ amounts in the SR30. For details on SOCE, see “A reverse-directional signal from CASQ1 to SOCE” in this review. Knockdown of both JPH1 and JPH2 in C2C12 myotubes (a skeletal muscle cell line) leads to deformation of the junctional membrane complex (corresponding to the triad junction of skeletal muscle fibers), reductions in SOCE and Ca2+ amounts in the SR, and elevation of the cytosolic Ca2+ level31. S165F, a JPH2 mutant that is found in patients with hypertrophic cardiomyopathy, induces hypertrophic skeletal myotubes along with a reduction in RyR1 activity during EC coupling due to a defect in protein kinase C (PKC)-mediated phosphorylation at Ser165 and the subsequent absence of the interaction between JPH2 and TRPC332. Another JPH2 mutant that is found in other patients with hypertrophic cardiomyopathy, Y141H, also shows hypertrophic skeletal myotubes due to an abnormal junctional membrane complex and increased Orai1-mediated SOCE13. An interaction between TRPC3 and RyR1 has been reported28,33. TRPC3 knockdown in skeletal myotubes results in decreased RyR1 activity and increased expression of JPH1 along with other proteins (TRPC1, CASQ1, and triadin)34. These various reports suggest that all these proteins are physically and functionally related to one another during Ca2+ movements in skeletal muscle.
Mitsugumin 29 (MG29) also participates in the formation of triad junctions during myogenesis or the terminal differentiation of myoblasts (proliferative forms of satellite cells or immature skeletal muscle cells) to myotubes (mature skeletal muscle cells)14. MG29 knockout mice exhibit ultrastructural abnormalities in the triad junction, such as swollen and irregularly directed t-tubules, irregular SR structures, and partial triad junction misformation, along with functional abnormalities such as reduced twitch tension under Ca2+-free conditions. For more details on the formation of triad junctions and the equipment of Ca2+-handling proteins such as DHPRs, RyRs, SERCAs, and CASQs during the development of skeletal muscle, please refer to other reviews and research articles35,36.
In addition to the SR, mitochondria are the second most important intracellular Ca2+ store in skeletal muscle, and they are closely localized in triad junctions to functionally communicate with the SR37–40, which is a very interesting field for further detailed investigation. For details on the relationship between mitochondria and the SR in regulating various Ca2+-mediated cellular events, please refer to another review articles41–43.
Properties of CASQ1 and isoform transition of CASQs during development
CASQ1 is a protein that has been known since its discovery 50 years ago in rabbit skeletal muscle (1971)15,44. The cDNA of CASQ1 was cloned from fast-twitch skeletal muscle from rabbits45, and the cDNA of the cardiac isoform of CASQ (CASQ2) was cloned from the heart and slow-twitch skeletal muscle from canines46. CASQ1 is the major isoform in adult fast-twitch skeletal muscles, while CASQ2 is the only isoform in cardiac muscle and a minor isoform in adult slow-twitch skeletal muscle46,47. Smooth muscle expresses both isoforms15,44,48.
The CASQ1 gene is located on human chromosome 1q2149. CASQ1 presents a high degree of homology to CASQ1 from other species or to CASQ2 in amino acid sequences50: for example, 98% between rabbit and human CASQ1s 86% between dog and human CASQ2s, and 83% between rabbit CASQ1 and dog CASQ2. The crystal structure of CASQ1 is nearly superimposable on that of CASQ251–53. Despite these high homologies, CASQ1 differs from CASQ2 in its C-terminus (a highly extended acidic C-terminus in CASQ2)50. The role of CASQ1 in regulating RyR1 activity differs from that of CASQ2; for example, in single-channel studies in planar lipid bilayers, CASQ2 monomers and/or dimers activate both RyR1 and RyR2, while CASQ1 polymers inhibit RyR1 activity54. The C-termini of CASQs contains one (CASQ1) or two glycosylation sites (CASQ2), as well as one (CASQ1) or three phosphorylation sites (CASQ2) for casein kinase II (CKII)55,56. The phosphorylation of CASQ1 is discussed further in a later section of this review.
CASQ1 and CASQ2 are heavily enriched in the SR, and they are tightly associated with the membrane of JSRs but not LSRs (in the case of rabbit CASQ1, approximately 70% after a purification process)15,18,44,51,54,57–59. CASQs are not evenly distributed in the SR but are concentrated in the vicinity of JSR-bearing RyR1 arrays16,60. For example, CASQ1 is located within 100 nm around RyR1 when it is not anchored to RyR1 or within 20–50 nm when it is anchored to RyR1. Electron microscopy (EM) studies have revealed that CASQ1 (as a form of homopolymer) shows a thin strand-like appearance in the SR and confirms the association of CASQ1 with the SR membrane61,62.
During embryonic development, maturation, or adulthood, the degree of expression of CASQ1 or CASQ2 in fast- and slow-twitch skeletal muscle is dependent on the skeletal muscle type. In a study using a mixture of rabbit fast- and slow-twitch skeletal muscles, both CASQ1 and CASQ2 were present from the embryonic stage, and fast-twitch skeletal muscle displayed more CASQs than slow-twitch skeletal muscle63. The expression of CASQ1 and CASQ2 steeply increased before birth and reached adult values at 4 days after birth. Denervation of postnatal fast- and slow-twitch skeletal muscles decreased the expression levels of CASQ1 and CASQ2 to their levels at the embryonic stage. In addition, during the postnatal development of rabbit skeletal muscle (i.e., during postnatal myogenesis), protein–protein interactions that are involved in EC coupling become more complex and mature for the proper physiological functions of skeletal muscle. The expression of CASQ1 and CASQ2 in the mixture of rabbit fast- and slow-twitch skeletal muscle continued to increase until day 41 and resembled that in adult skeletal muscle36.
Both CASQ1 and CASQ2 are expressed in neonatal rabbit fast-twitch skeletal muscle, and a transition of CASQ isoforms occurs after birth64. The expression of CASQ1 increased up to 1 month, and it reached adult values at approximately 2 months of age. The expression of CASQ2 completely disappeared between 2 and 4 weeks postnatally (i.e., during the maturation of fast-twitch skeletal muscle, downregulation of CASQ2 expression and gradual replacement of CASQ2 with CASQ1). The amount of CASQ1 in the rat fast-twitch extensor digitorum longus (EDL) skeletal muscle fibers was 3.5 times greater than that in slow-twitch soleus skeletal muscle fibers65, in accordance with the higher amount of the released Ca2+ in fast-twitch than in slow-twitch skeletal muscles. Both isoforms are present in rabbit slow-twitch skeletal muscle until adulthood, and CASQ2 accounts for approximately 25% of the total CASQs66.
CASQ1 is a major high-capacity Ca2+-buffering protein and a Ca2+-sensor in the SR
The first role of CASQ1 in skeletal muscle is to buffer Ca2+ in the SR with a low-affinity and high-capacity Ca2+-binding ability (40–50 moles or maximally ~80 moles of Ca2+/1 mole of CASQ1) to prepare rapidly releasable Ca2+ during EC coupling (Fig. 1)15,67,68. Ca2+-binding sites in CASQ1 are very different from those in other Ca2+-binding proteins (i.e., they do not consist of EF-hands). The role of CASQ1 as a high-capacity Ca2+-buffering protein is accomplished by two factors51. First, CASQ1 is an extremely acidic protein. Two-thirds of the C-terminus of CASQ1 is negatively charged, and it serves as the Ca2+-binding site51. Second, CASQ1 self-polymerizes to prepare high-capacity Ca2+-binding at high [Ca2+] in the SR (i.e., Ca2+-dependent homopolymerization of CASQ1 near RyR1, further discussed in the next section of this review)53,69. The polymerization of CASQ1 in skinned fibers from rat skeletal muscle theoretically increases the total [Ca2+]SR up to 20 mM, and simultaneously, it maintains a free [Ca2+]SR at approximately 1 mM50,70, which is important for easier and more efficient Ca2+ reuptake into the SR by SERCA1a during relaxation and for the storage of high [Ca2+] in the SR without harmful osmotic effects. For example, frog skeletal muscle fibers with CASQ1 bear 23 times more Ca2+ than those without CASQ171.
Fig. 1. Summary of the physiological roles of CASQ1 in skeletal muscle.

The physiological roles of CASQ1 in skeletal muscle are summarized in circles (colored blue). Within the circles, corresponding proteins, mechanisms, or events are presented in parentheses. The light purple arrow (in the lower left) indicates the process of CASQ1 polymerization. Proteins that interact with CASQ1 are listed in boxes. Greenish arrows indicate the protein complexes of CASQ1 with CASQ1-interacting proteins along with the effects of the complexes on the functions or protein activities of skeletal muscle. (+), positive or upregulation; (−), negative or downregulation. CASQ1 calsequestrin 1, DHPR dihydropyridine receptor, RyR1 ryanodine receptor 1, STIM1 stromal interaction molecule 1, JP-45 junctional protein 45, SERCA sarcoplasmic/endoplasmic reticulum Ca2+-ATPase, EC coupling excitation-contraction coupling, SOCE store-operated Ca2+ entry, ECCE Ca2+ entry through the DHPR during skeletal EC coupling.
The second role of CASQ1 in skeletal muscle is as a Ca2+ sensor in the SR. CASQ1 senses [Ca2+] in the SR and modulates Ca2+ release from the SR to the cytosol via RyR1 in a conformation (i.e., polymerization)-dependent manner (Fig. 1)67,68,72,73. Lowering the [Ca2+]SR from 1 mM to 100 µM results in conformational changes in CASQ1 (the dissociation of 65–75% of CASQ1 from the JSR membrane). Under this condition, CASQ1 is depolymerized and no longer inhibits RyR1 activity. CASQ1 also protects the SR from deep Ca2+ depletion, which causes massive migration of CASQ1 away from the JSR membrane and remodeling of the SR74,75. Under long-lasting membrane depolarization, such as high-frequency stimuli or early stages of muscle fatigue, Ca2+ in the SR is not fully but partly depleted due to the inactivation of RyR1 at a certain moment by limiting the further depolymerization of CASQ1, which prevents excess Ca2+ release from the SR and catastrophic changes in the SR structure. Therefore, it can also be said that by conformational changes (polymerization or depolymerization) in a Ca2+-dependent manner, CASQ1 senses the degree of Ca2+ depletion from the SR and regulates RyR1 activity. In addition, the regulation of RyR1 activity by CASQ1 requires interactions with other proteins (discussed in ‘Regulation of RyR1 activity by a quaternary complex of CASQ1/RyR1/triadin/junctin’ in this review).
Three-dimensional (3D) structure and polymerization of CASQ1
The 3D structure of CASQ1 was determined at a resolution of 2.4 Å53. The CASQ1 monomer has three almost identical domains (I–III), each with a topology similar to that of thioredoxin from Escherichia coli (i.e., three thioredoxin-like folds occur in CASQ1). Each domain forms a disk-like shape with a fold of α/β structures (a five-stranded β-sheet in the core flanked by four α-helices (two on each side of the β-sheets). The thioredoxin-like fold occurs at ~10 μM [Ca2+]. The roles of the thioredoxin-like domain in CASQ1 have not been studied. Considering that the thioredoxin system participates in the regulation of metabolism, the antioxidant system, cell signaling pathways, and thioredoxin-dependent chaperone actions76, the presence of the three thioredoxin-like domains in CASQ1 suggests that CASQ1 could be directly or indirectly involved in the thioredoxin system.
CASQ1 can exist either as monomers or homopolymers (a wide range of high molecular masses), and polymeric CASQ1 allows various binding to other proteins via the same region, such as the asp-rich/CAS (consecutive aspartate stretch) region. The polymerization of CASQ1 depends on [Ca2+]SR50,53,73,77,78. At 10 μM [Ca2+]SR, CASQ1 exists as a monomer and dissociates from the JSR membrane, which eliminates the inhibitory effect of CASQ1 on RyR1. Two types of dimers facilitate the polymerization of CASQ1. The first type of dimer occurs via a back-to-back interaction. Salt bridges between Glu215-Lys86, Glu215-Lys24, and Glu169-Lys85 from domains I and II stabilize the back-to-back interaction. Two salt bridges between Lys85-Glu223 and between Lys86-Glu215 mediate an interaction between the dihydrobasic sequence (found in several Ca2+-binding proteins79) and a binding site for trifluroperazine (an antipsychotic drug), which also contributes to the back-to-back interaction. In the back-to-back interaction, the C-terminal asp-rich/CAS region from each monomer forms a negatively charged pocket within the dimer, and the negative pocket represents a major Ca2+-binding motif in CASQ180. At low [Ca2+], the asp-rich/CAS region can accommodate 6–8 Ca2+ ions, which neutralizes the acidic property of the asp-rich/CAS region and allows back-to-back stacking. The remaining regions of CASQ1 contain additional Ca2+-binding sites that can progressively support more Ca2+-binding as [Ca2+] increases80. The back-to-back interaction during the polymerization of CASQ1 occurs at 10 μM to 1 mM [Ca2+]SR.
The second type of dimer occurs via a front-to-front interaction. Tetramerization of CASQ1 occurs via a front-to-front interaction between two dimers (via the N-terminus) that are formed by the back-to-back interaction between two monomers (via the C-terminus) when [Ca2+] is further increased. The front-to-front interaction occurs with the insertion of the N-terminal part of one monomer of the dimer into a hydrophobic cleft in the adjacent monomer of another dimer. This process again generates a negatively charged pocket that supports Ca2+-binding. A salt bridge between Glu55–Lys49 contributes to the front-to-front interaction and helps to stabilize CASQ1 polymers. Further polymerization of the CASQ1 tetramers finally forms thin strands (also called ribbon-like linear polymers) that generate more negative pockets, which confer a higher Ca2+-buffering capacity and the ability to bury hydrophobic side chains of amino acids (i.e., losing hydrophobicity) on CASQ1. The highly extended structure of polymeric CASQ1sbecomes more compact by the binding of more Ca2+81. Polymeric CASQ1 is stably anchored to the JSR membrane by binding to other proteins (i.e., triadin and junctin along with RyR1) at ~1 mM [Ca2+]SR and can maximally inhibit RyR1 activity50. The polymerization of CASQ1 is inhibited by increasing [K+]. From these reports, both terms of Ca2+-dependent polymerization of CASQ1 or polymerization-dependent Ca2+-binding of CASQ1 are correct. The Ca2+-buffering and Ca2+-sensing abilities of CASQ1 are not two different roles but are functionally and structurally correlated to each other. In short, the Ca2+-dependent dynamic polymerization and depolymerization of CASQ1 provide the SR with a rapidly releasable Ca2+ reservoir during skeletal muscle contraction and a highly negatively charged surface that easily absorbs Ca2+ at rest or during relaxation. The differences in their abilities in front-to-front and back-to-back interactions and in the composition of the asp-rich/CAS region in CASQ1 and CASQ2 generate isoform-specific Ca2+-binding capacities and different Ca2+-dependent polymerization properties51,73,82.
Ca2+ buffering by CASQ1 in the SR during Ca2+ release from the SR shows a nonlinear pattern51,83,84, which is explained by the polymerization-dependent Ca2+-binding capacity of CASQ1. In the SR fully loaded with Ca2+, CASQ1 is polymerized and bears many Ca2+ to prepare for Ca2+ release from the SR via RyR1. At the beginning of Ca2+ release from the SR, [Ca2+] at the release site of the SR decreases, which causes the depolymerization of CASQ1 at the site and subsequent release of Ca2+ from the depolymerized CASQ1 to the SR lumen via the loss of Ca2+-binding ability (unbuffering of Ca2+ by CASQ1). More Ca2+ release from the SR to the cytosol can occur by the unbuffering of Ca2+ by depolymerized CASQ1. A further cycle of depolymerization and Ca2+ unbuffering of CASQ1 results in more rapid decay of [Ca2+]SR compared with the beginning of Ca2+ release (i.e., a nonlinear pattern). Concurrently, the depolymerized CASQ1 detaches from the JSR membrane, which induces the loss of inhibitory action of CASQ1 on RyR1 activity72,85–87. Further depolymerization of CASQ1 finally terminates Ca2+ release from the SR through RyR1 (i.e., inactivation of RyR1) when Ca2+ depletion from the SR and the detachment of CASQ1 from the JSR membrane progress further74,75. In addition, the binding of CASQ1 to RyR1 is also eliminated by increasing [Ca2+]SR to ≥4 mM (probably mimicking the state of the SR at rest), which consequently eliminates the inhibitory effect of CASQ1 on RyR1 activity54,67,88.
Regulation of RyR1 activity by a quaternary complex of CASQ1/RyR1/triadin/junctin
The acidic asp‐rich/CAS region of CASQ1 in the C-terminus directly interacts with triadin via a KEKE motif in triadin (localized in the SR lumen), which helps CASQ1 anchor to the JSR membrane and allows CASQ1 to bind to Ca2+ in the area near RyR189–91. The same luminal KEKE motif of triadin also interacts with the C-terminal loop of RyR189,92,93. Triadin physically links CASQ1 to RyR1, forming a bridge between CASQ1 and RyR189. It has also been reported that CASQ1 directly interacts with RyR194. The acidic asp‐rich/CAS region of CASQ1 also interacts with junctin via the KEKE motif95,96. All these reports suggest that CASQ1 can participate in generating a protein complex with RyR1, junctin and triadin (i.e., a quaternary complex of CASQ1/RyR1/triadin/junctin).
The possibility has been suggested that the N-terminal residues of CASQ1 may also be responsible for its binding to triadin and junctin, and the CASQ1 polymer may not bind to these proteins53. However, according to EM observations of skeletal muscle and other studies on the binding site of CASQ1 to triadin, as mentioned above61,90,97, the CASQ1 polymer can form a complex with RyR/triadin/junctin. At 1 mM [Ca2+]SR, the CASQ polymer is stable and anchors to the JSR membrane through triadin and junctin, and further increasing [Ca2+]SR to over 10 mM results in the dissociation of CASQ from triadin and junctin50,67,98, which is in accordance with the observation that the CASQ1 polymer at ≥4 mM [Ca2+]SR causes a gradual dissociation of CASQ1 from junctin, triadin and RyR154,67,69,88. Interestingly, both the formation of the quaternary complex and the formation of CASQ1 polymers contribute to the retention of CASQ1 inside the SR in skeletal muscle99.
CASQ1, triadin, and junctin are not essential for survival or skeletal muscle contraction; however, they are related to RyR1 activity and the gain of EC coupling in skeletal muscle. As mentioned in the previous section of this review, CASQ1 acts as a Ca2+ sensor to modulate the activity of RyR1. In early studies, the functional relevance of the interaction between CASQ1 and RyR1 was controversial. CASQ1 was originally described as an activator of RyR1 in isolated heavy SR vesicles100. In a single-channel study with a planar lipid bilayer, the addition of purified CASQ1 to the luminal side enhanced the activity of purified RyR1101. Overexpression of CASQ1 in C2C12 skeletal myotubes enhanced caffeine- and membrane depolarization-induced intracellular Ca2+ release through RyR1102. Overexpression of a deletion mutant of CASQ1 lacking the C-terminal aspartate-rich/CAS region (the triadin- or junctin-binding domain) in C2C12 myotubes resulted in reduced caffeine- and membrane depolarization-induced Ca2+ release90,102. Even a case of no functional relationship between CASQ1 and RyR1 was reported (i.e., no significant functional effect of CASQ1 on RyR1)103. Other groups reported that CASQ1 alone could activate RyR1 activity104, but CASQ1 inhibited RyR1 activity by binding to triadin67,104. Deletion of the C-terminal acidic asp‐rich/CAS region of CASQ1 abolished the inhibitory action of CASQ1 on RyR196. Specifically, in a single-channel study with a planar lipid bilayer, the addition of purified triadin or junctin to the luminal side increased the activity of purified RyR1, and the addition of purified CASQ1 abolished the effect of junctin on RyR1 but not the effect of triadin105. The different compositions of RyR or CASQ isoforms, the different degrees of expression levels of RyR1 or CASQ1 (or the ratio between them), and the different salt concentrations in the various experimental conditions could cause discrepancies in the role of CASQ1 in RyR1 activity. For example, CASQ1 was dissociated from the JSR membrane by increasing the luminal ionic strength (under high-salt conditions), and the inhibitory effect of CASQ1 on RyR1 activity was removed by the dissociation of CASQ167. After all of these reports, it has finally been accepted that at low [Ca2+]SR, CASQ1 functions as a Ca2+ sensor that inhibits RyR1 activity in the presence of triadin at least (considering the quaternary complex, in the presence of both triadin and junctin) to preserve a certain level of Ca2+ in the SR50,51,53,67,72,94. Consistently, triadin knockout mice show skeletal muscle weakness with reduced expression of CASQ1106, and a human patient with a triadin mutation also shows skeletal muscle weakness107. For more details on RyR1 activity along with its partners or regulatory proteins under various pathological conditions, including MH, skeletal muscle myopathies, cardiac arrhythmias, epilepsy, neurodegeneration, and pain, please refer to a review article108.
Overall, the regulation of RyR1 activity by CASQ1 is not a simple one-way process but a very complicated and bidirectional process that is optimized for the fine-tuning of skeletal muscle contraction or relaxation, with dependencies on [Ca2+]SR, the phosphorylation status of CASQ1 (see the next section in this review), the polymerization status of CASQ1, binding to other proteins such as triadin and junctin (i.e., the quaternary complex of CASQ1/RyR1/triadin/junctin), and even RyR1 activity itself (i.e., the nonlinear relationship between Ca2+-buffering and Ca2+ release from the SR, as discussed before).
Phosphorylation of CASQ1
The phosphorylation status of CASQ1 also affects the Ca2+-binding capacity of CASQ1 by regulating the interaction of CASQ1 with other proteins. CKII (which can phosphorylate serine or threonine residues) is present in the SR lumen in rabbit fast-twitch skeletal muscle109. The threonine located at the beginning of the C-terminus of CASQ1 is the only site of CASQ1 phosphorylated by CKII (at 353 in the acidic region, I351NTEDDDDDE-COOH)55. The acidic region partly overlaps with the triadin-binding region (residues 354-36790) and lies near the electronegative pocket formed by the front-to-front interaction during CASQ1 polymerization53. Phosphorylation of CASQ1 at the threonine residue increases the Ca2+-binding capacity of CASQ1 nearly 2-fold110.
Isolated CASQ1 exists in both phosphorylated88,111 and dephosphorylated forms55,112. It is controversial whether the phosphorylation of CASQ1 affects the interactions between CASQ1 and RyR1 or the inhibitory effect of CASQ1 on RyR1. Only dephosphorylated CASQ1 can enhance RyR1 activity without binding to triadin and junctin (approximately fivefold increase in open probability and approximately twofold increase in mean open time at 1 mM of [Ca2+]SR), and only 1% dephosphorylated CASQ1 in the total CASQ1 is sufficient to enhance RyR1 activity113. However, the affinity of phosphorylated CASQ1 to RyR1 is similar to that of dephosphorylated CASQ194. In contrast, both phosphorylated and dephosphorylated CASQ1 have an inhibitory effect on RyR1 activity at 1 mM [Ca2+]SR and only when the quaternary complex of CASQ1/RyR1/triadin/junctin is intact67,88,110. The enhancement of RyR1 activity by dephosphorylated CASQ1 is observed only when CASQ1 is bound to triadin alone110. Considering that functional RyR1 is a very large ion channel (i.e., tetrameric RyR1s with more than 2 MDa), the ambiguity in the relationship between RyR1 activity and CASQ1 could possibly be due to the participation of other proteins that can bind RyR1 and/or exert regulatory effects on RyR1 activity. In addition, interestingly, CASQ1 itself has kinase activity in the presence of Mg-ATP (which is present in the SR) and can autophosphorylate111,114, suggesting that CASQ1 phosphorylates other proteins in the SR, such as EC coupling-mediating proteins, including RyR1. These various reports surely suggest that the regulation of RyR1 activity by CASQ1 depends on the phosphorylation state of CASQ1, [Ca2+]SR, and the binding of triadin and junctin to CASQ1. Overall, the relationship between the phosphorylation status of CASQ1 and the regulation of RyR1 activity by CASQ1 via interaction between RyR1 and CASQ1 should be further investigated.
The Ca2+-binding capacity of dephosphorylated CASQ1 is reduced by more than one-third compared with that of phosphorylated CASQ153,110. Phosphorylation of CASQ1 does not alter CASQ1 polymerization110. Therefore, it is likely that phosphorylated CASQ1 corresponds to polymerized CASQ1.
CASQ1 deficiency
CASQ1 knockout mice are viable, fertile, and have normal muscle performance, although mild atrophy is observed115. Skeletal muscle tissues and fibers from CASQ1 knockout mice show remodeling of the triad junction, such as a narrow lumen of the JSR caused by shrinking, the proliferation of triad-like multilayered junctions (that differ from the typical triad junction, in approximately 70% of EDL fibers, and even in the LSR), an increase in the number and volume of mitochondria, and an increase in the number of RyR1 molecules (even in multilayered junctions) (Fig. 2)115,116. Therefore, CASQ1 is essential for the normal development of the SR and triad junction (which is the third role of CASQ1 in skeletal muscle, Fig. 1), as well as for Ca2+-buffering and Ca2+-sensing in the SR to release appropriate amounts of Ca2+.
Fig. 2. Summary of pathological involvement of CASQ1 in skeletal muscle.

Pathological involvement of CASQ1 in skeletal muscle is summarized in circles (red). In the lower right (deep orange circles), diseases that are related to CASQ1 with no known mechanism are summarized. Corresponding proteins, mechanisms or phenomena are presented in parentheses. Pathological phenomena and tendencies due to the knockout of CASQ1 in mice are listed in the boxes. CASQ calsequestrin, RyR1 ryanodine receptor 1, SR sarcoplasmic reticulum, SOCE store-operated Ca2+ entry, MH malignant hyperthermia, EHS environmental/exertional heat stroke, TAM tubular aggregate myopathy, PAM protein aggregate myopathy, Ab antibody.
The amount of Ca2+ in the SR of fast-twitch EDL skeletal muscle fibers from CASQ1 knockout mice is reduced compared with that in wild-type fibers65,115. The appearance of multilayered junctions with RyR1s could be a compensatory response to the reduced SR Ca2+ storage with the shrinkage of terminal cisternae. There is no compensatory increase in the expression of CASQ2 in either fast- or slow-twitch skeletal muscles115. Fast-twitch EDL skeletal muscle fibers from CASQ1 knockout mice display similar free [Ca2+]cytosol but significantly reduced Ca2+ release from the SR in response to electrical stimulation or caffeine compared with wild-type fibers, while slow-twitch soleus skeletal muscle fibers are not affected115,117. However, tension developed during isometric tetanus for a short duration is not significantly different in the two types of muscle fibers, and the sensitivity to caffeine is also unchanged (similar EC50 values)115,116. Fast-twitch EDL skeletal muscle fibers from CASQ1 knockout show prolonged twitch time parameters (time-to-peak or half-relaxation time, i.e., delayed Ca2+ release or removal)115, which conflicts with the reduced Ca2+ release from the SR in response to electrical stimulation or caffeine in the same EDL fibers. These unexpected results can be induced by an impaired ability of Ca2+ reuptake to the SR or a loss of CASQ1-mediated inhibitory effect on RyR1 activity during EC coupling. The increased number of mitochondria is related to the higher resistance of CASQ1 knockout mice to fatigue than wild-type mice115. EDL muscles from CASQ1 knockout mice also show mitochondrial damage with oxidative stress116,118.
There are no marked differences between mice carrying CASQ1 single- and CASQ1/CASQ2 double-knockouts115,116,119. Fast-twitch FDB skeletal muscle fibers from single- or double-knockout mice do not sustain prolonged muscle activity (such as repetitive electrical stimuli) due to faster SR Ca2+ depletion, a decrease in [Ca2+]SR, impaired Ca2+ reuptake to the SR and finally an inability to sustain [Ca2+]cytosol during contraction119. The proper function of fast-twitch FDB skeletal muscle fibers from double-knockout mice requires external Ca2+ entry (i.e., SOCE)119, suggesting a possible relationship between CASQ1 and SOCE (see the next part of this review). Unlike CASQ1 single-knockout mice, CASQ1/CASQ2 double-knockout mice show a more severely damaged structure of the soleus muscle (alterations in ~30% of soleus fibers but not in EDL fibers), although both single- and double-knockout mice exhibit significant reductions in body weight and grip strength116. However, under tetanic stimulation (2 s and 100 Hz), the soleus was able to sustain contraction, while active tension in the EDL declined by 70–80%. This discordance in the structural damage and function in the slow-twitch soleus skeletal muscle fibers is explained by the nature of the skeletal muscle type: fast-twitch EDL skeletal muscle fibers need the release of large amounts of Ca2+ from the SR during tetanic stimulation; in contrast, slow-twitch soleus fibers require less Ca2+ and have slower cycling than fast-twitch EDL skeletal muscle fibers.
Transient knockdown of CASQ2 alone or both CASQ1 and CASQ2 in C2C12 myotubes induces a reduction in the SR Ca2+ content due to the reduced expression and lower activity of SERCA along with a reduction in the expression of RyR1 compared to wild-type myotubes101.
A reverse-directional signal from CASQ1 to SOCE
In most cell types, the Ca2+ gradient between the cytosol (approximately 100 nM) and extracellular space (mM range) is maintained (approximately 20,000-fold), which suggests that extracellular Ca2+ is an effective source of intracellular Ca2+ signals120. SOCE is one extracellular Ca2+ entry route in various cells121. Generally, SOCE is operated by coordinated interactions between STIMs (STIM1, STIM1L, and STIM2) and Ca2+ release-activated Ca2+ channel proteins (Orai1, Orai2, and Orai3). STIM1 and Orai1 are the major isoforms mediating SOCE. STIM1 in the ER membrane senses the depletion of Ca2+ from the ER via its EF-hand motif, and it activates Orai1 in the plasma membrane via a physical interaction between the cytoplasmic C-terminus of STIM1 and the STIM-Orai1-activating region (SOAR) of Orai1. During this process, sensing Ca2+ by STIM1 (i.e., Ca2+ depletion from the ER) induces the homo-oligomerization of STIM1 and its relocalization to the ER membrane near the plasma membrane where Oria1 is located, which also induces the homo-oligomerization of Orai1. The final hetero-oligomers that are formed by STIM1s and Orai1s are called ‘puncta’.
The presence of SOCE in skeletal muscle was first identified in 2001122. There are several differences between SOCE in skeletal muscle cells and general SOCE in another cell types5–7. First, the SR or t-tubule in skeletal muscle corresponds to the ER or plasma membrane, respectively. Second, without Ca2+ depletion from the SR, puncta are formed during the development of skeletal muscle fibers and the process of terminal differentiation after birth (i.e., prepuncta formation)13,123,124. However, the prepuncta are not functional, and the additional conformational changes in Orai1 and/or STIM1 within the prepuncta are required to evoke SOCE13,123. An advantage of prepuncta formation in skeletal muscle is faster kinetics (rapid Ca2+ entry into the cytosol during skeletal SOCE, in less than 1 s, which is significantly faster than in other types of cells (several seconds)124–126. In skeletal muscle fibers, SOCE is necessary to restore the SR with Ca2+ to the resting level and to sustain Ca2+ in the cytosol for skeletal muscle contraction during tetanic stimulation or fatigue57,124,125,127,128. In addition, it has been found that skeletal SOCE requires both structural and functional interactions with other proteins, such as MG29 (which is required for the formation of the triad junction, mentioned before in this review), RyR1, and RyR3 in the triad junction129.
The fourth role of CASQ1 in skeletal muscle is as a regulator of SOCE via physical interaction between STIM1 and the acidic asp-rich/CAS region of CASQ1 (Fig. 1)102,130,131. Overexpression of CASQ1 in C2C12 myotubes inhibits SOCE102, while SOCE is increased in FDB skeletal muscle fibers from CASQ1 single- or CASQ1/CASQ2 double-knockout mice130,132. Therefore, CASQ1 not only modulates intracellular Ca2+ release but also provides a “reverse-directional signal” to regulate SOCE in skeletal muscle (a signal in the opposite direction of the orthograde signal from DHPR in the t-tubule membrane to RyR1 in the SR membrane during EC coupling, i.e., a signal from CASQ1 in the SR to Orai1 in the t-tubule/plasma membrane)102,133, suggesting that the functional interplay among CASQ1, RyR1 and SOCE could also contribute to pathological Ca2+ overload as well as to physiological Ca2+ signals. Indeed, SOCE is involved in muscle pathophysiology (see the next section on CASQ1-mediated diseases in this review)129,134.
The increased SOCE in FDB skeletal muscle fibers from CASQ1 single- or CASQ1/CASQ2 double-knockout mice130,132 could be one explanation for the sustained high resting [Ca2+]cytosol under reduced Ca2+ in the SR and the increased rate of Ca2+ depletion from the SR in knockout fibers from these mice65,67,116,119,135.
Other CASQ1-binding proteins
Junctional protein 45 (JP-45, a single transmembrane protein) interacts with two EC coupling-mediating proteins: the α1S subunit of DHPR via its cytosolic N-terminus and CASQ1 via its luminal C-terminus136,137. Knockout of JP-45 in young mice results in a loss of skeletal muscle strength due to decreases in the expression and charge movement of DHPR and in membrane depolarization-induced internal Ca2+ release through RyR1 in FDB skeletal muscle fibers138. However, it is controversial whether JP-45 affects the activity of DHPR because both overexpression and knockdown of JP-45 in C2C12 myotubes result in a decrease in the charge movement of DHPR137. Surprisingly, the skeletal muscle phenotype of young JP-45 knockout mice is similar to that of aged mice: reductions in the expression of DHPR and JP-45 and in intracellular Ca2+ release in response to membrane depolarization likewise occur in aged mice139,140.
Ca2+ entry through DHPR during skeletal EC coupling (so-called ECCE) is crucial to maintain muscle force development in the mouse slow-twitch soleus muscle, and ECCE is increased in FDB skeletal muscle fibers from JP-45/CASQ1 double-knockout, JP-45/CASQ2 double-knockout or JP-45/CASQ1/CASQ2 triple-knockout mice141, suggesting another type of ‘reverse-directional signal’ by CASQ1 via complex formation with JP-45. However, it is also controversial whether JP-45 affects ECCE through DHPR. Overexpression of JP-45 in C2C12 myotubes does not induce much change in the voltage-activated inward Ca2+ current (corresponding to ECCE in skeletal muscle fibers or myotubes)142. Therefore, more dedicated examinations are required to clarify the effects of JP-45 on DHPR or ECCE through DHPR in skeletal muscle.
CASQ1 prevents the dimerization of inositol-requiring enzyme 1α (IRE1α, a sensor of ER stress) by interacting with the luminal domain of IRE1α at JSR, which attenuates IRE1α signaling at physiological Ca2+ levels such as during skeletal muscle contraction or relaxation143. In cardiac muscle, IRE1a signaling plays protective roles under cardiac fibrosis or atherosclerosis144,145. These reports suggest the possibility that CASQ1 could participate in the stress-coping responses in skeletal muscle as well as cardiac muscle.
Other Ca2+-buffering proteins in the SR and CASQ-like proteins
Other Ca2+-buffering proteins in the SR have been reported, such as histidine-rich Ca2+-binding protein (HRC, 160 kDa) and sarcalumenin (SLM, 150 kDa)109,146,147. The amounts of HRC and SLM in SR are much lower than the number of CASQ1109. Similar to CASQ1, HRC, and SLM are acidic and act as substrates of CKII. Unlike CASQ1, which is mainly located in the terminal cisternae of the SR, HRC, and SLM are more widely distributed and buffer Ca2+ throughout the SR148. The Ca2+-dependent phosphorylation of SLM and HRC by CKII inhibits the binding affinity of RyR1 to Ca2+147,149. This report suggests that HRC and SLM could regulate RyR1 in a Ca2+- or phosphorylation-dependent manner. In addition to CKII, HRC can be phosphorylated by Ca2+/calmodulin-dependent protein kinase II146. A 53-kDa Ca2+-binding protein is a splice variant of SLM150.
Calreticulin, also known as calregulin, is the ER homolog of CASQ1, and it also exists in skeletal muscle151,152. Calreticulin has no EF-hand consensus sequence; however, the highly acidic C-terminus of calreticulin accounts for its low-affinity and high-capacity Ca2+ binding properties.
CASQ-like proteins have been found in other organisms or tissues, including in smooth muscles of the rat aorta and stomach48, in the urinary bladder of guinea pig153, in Purkinje cells of chicken cerebellum154 and in the liver of rat155, but their functions have not been studied. A CASQ-like protein is found in body wall muscle cells of Caenorhabditis elegans (50% similarity and 30% identity to rabbit CASQ1), but it is not essential for body wall muscle formation and contraction156. CASQ-like proteins that are recognizable by anti-CASQ1 antibodies are found in the eggs of sea urchins (Strongylocentrotus droebachiensis and Arbacia punctulata)157.
Interestingly, CASQ-like proteins have also been found in plants, such as cultured cells of Streptanthus tortuosus or spinach leaves158. The CASQ-like proteins in plants can bind to Ca2+ and are recognized by the anti-CASQ2 antibody. The other plant CASQ-like protein is found in Ca2+-accumulating cells from water lettuce (Pistia stratiotes)159. Various CASQ-like proteins in various types of cells and tissues from diverse species strongly suggest that the functions of CASQ1 could be more diverse than previously known.
CASQ1-related diseases
Tubular aggregate myopathy (TAM) was originally associated with mutations in STIM1 or Orai1, along with the dysregulation of SOCE (higher or lower SOCE than normal)160–162. Patients with heterozygous missense mutations in the CASQ1 gene (D44N, N56Y, G103D, and I385T) show TAM163,164. I385T shows a slight increase in Ca2+-dependent tubular aggregation, but D44N, N56Y, and G103D show a decrease in Ca2+-dependent tubular aggregation. Interestingly, all the mutants reduced the amount of Ca2+ in the SR and D44N and I385T additionally showed a defect in the ability to inhibit SOCE163, again confirming that CASQ1 participates in the modulation of SOCE-mediated Ca2+ homeostasis via a reverse-directional signal.
D244G (a heterozygous missense mutation in the CASQ1 gene) was the first identified CASQ1 mutant in patients showing mild myopathy characterized by muscle weakness, fatigue, and the presence of large vacuoles165–167. D244G induces misfolding and abnormal aggregation of CASQ1 (which is different from the physiological CASQ1 polymers) due to the loss of the electric charge in D244 that is involved in the Ca2+-binding and Ca2+-dependent polymerization of CASQ1. Abnormal CASQ1 aggregates cause sarcoplasmic vacuolar aggregations (which induce protein aggregate myopathy (PAM)) and induce reduced Ca2+ release from the SR.
Under the administration of volatile anesthetics such as isoflurane, sevoflurane, halothane, desflurane, and enflurane or muscle relaxants such as succinylcholine, patients with MH (a pharmacogenetic skeletal muscle disorder) can undergo sudden death by acute life-threatening skeletal muscle contracture with hypermetabolism that is characterized by increased O2 consumption, an extreme elevation in body temperature and rhabdomyolysis168–170. The main cause of the crisis in MH patients is the uncontrolled elevation of [Ca2+]cytosol that is mediated by uncontrolled activation of dominant mutants of RyR1168,169. Hundreds of mutations in the RyR1 gene have been found in MH patients171, and most of these mutations are confined to three regions (called hot spots)172: C35-R614 in the N-terminus, D2129-R2458 in the central region, and I3916-G4942 in the C-terminus. MH is also attributed to mutations in the CACNA1S gene encoding the α1S subunit of DHPR173. In addition to the mutations in RyR1 and DHPR, sustained SOCE may affect the intracellular Ca2+ balance in the skeletal muscles of MH patients174. Until now, the only clinical treatment of MH has been the administration of dantrolene (a muscle relaxant)1,175–178. However, the molecular mechanism of the dantrolene effect has not been clearly demonstrated. What is the direct target of dantrolene? What proteins or pathways are involved in the dantrolene effect?
Interestingly, mutations in CASQ1 in human patients or knockouts of CASQ1 in mice induce symptoms similar to those observed in human patients with MH. Patients with M87T (a missense mutation in the CASQ1 gene) show a mild association with MH167. M87T induces a mild reduction in the Ca2+-binding capacity of CASQ1 due to the shift of the M87-bearing α-helix and the weakening of the back-to-back interaction that is essential for the dimerization of CASQ1. CASQ1 knockout mice clearly show MH-like gain-of-function phenotypes that are very similar to those of RyR1 knock-in mice expressing human MH mutations (Y522S or R163S, gain-of-function mutations in RyR1)135,179–183, such as impaired movement, difficulty breathing, whole-body contractions, an arched back and finally rapid progression to sudden death. Loss of the inhibitory effect of CASQ1 on RyR1 activity and Ca2+ depletion from the SR due to the absence of CASQ1 explain the MH-like phenotypes in CASQ1 knockout mice67,119. A mutation in the CASQ1 gene (1090 G > A in the nucleotide sequence; V364M in the amino acid sequence) has been reported in MH patients, although its pathological mechanism has not yet been studied184. The locations of mutations that are related to patients with MH, TAM, or PAM are indicated in the 3D structure of human CASQ1 (Fig. 3).
Fig. 3. 3D structures of human CASQ1 with indications of residues that are related to human skeletal muscle diseases.
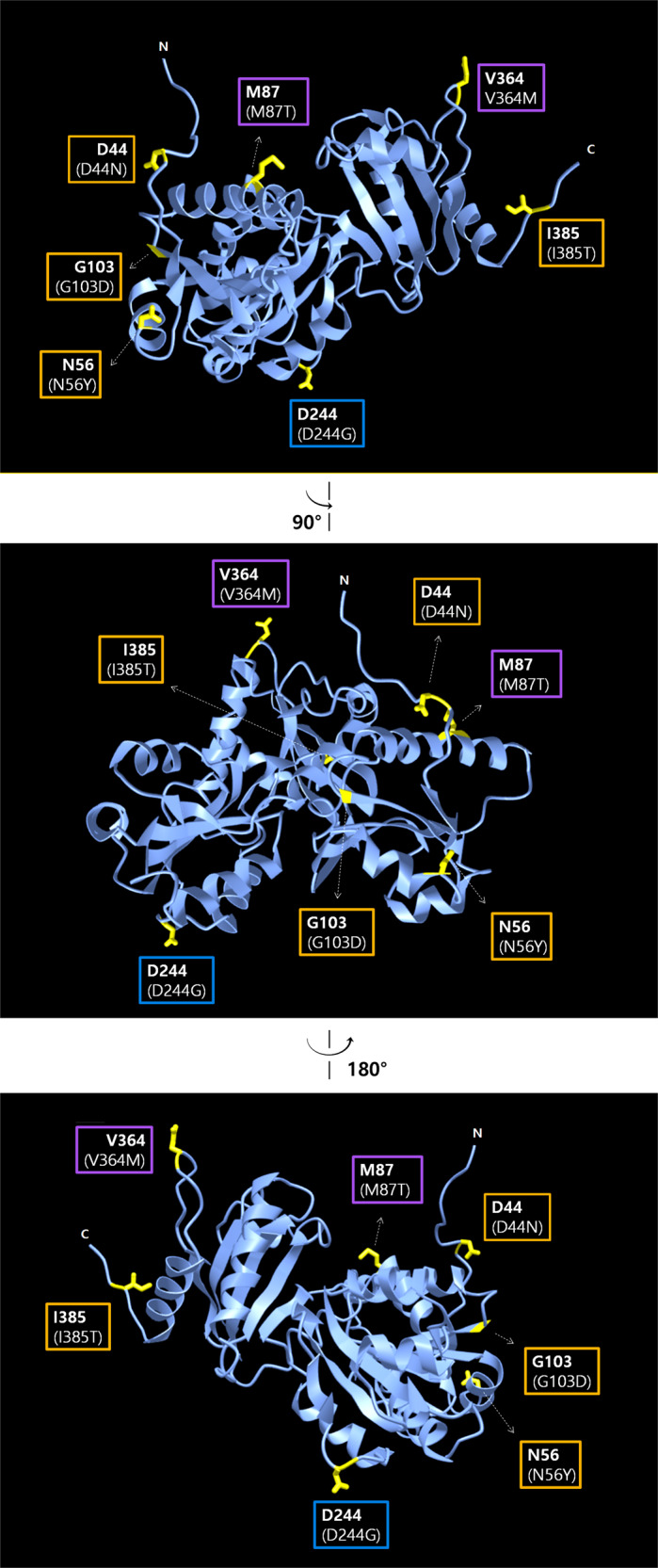
The 3D structure of human CASQ1 (PDB ID: 3UOM) is presented in the top panel as a ribbon diagram using iCn3D (NCBI’s web-based 3D structure viewer202, https://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?Dopt=s&uid=97449). The top image is rotated around the vertical axis (90° or 180° counterclockwise) and presented in the middle or bottom image, respectively. Residues that are involved in human skeletal muscle diseases are presented in the boxes and highlighted in yellow in the 3D structures, and their side chains are presented as sticks. Mutations that are involved in human skeletal muscle diseases are presented in parentheses in the boxes. Numbers indicate the amino acid sequence positions. N or C indicates the N- or C-terminus, respectively. The signal peptide in the N-terminus is not shown. The residues or mutations in the orange boxes are related to TAM (tubular aggregate myopathy); the residues or mutations in the purple boxes are related to MH (malignant hyperthermia); the residue or mutation in the blue box is related to PAM (protein aggregate myopathy).
Unexpectedly, patients with environmental/exertional heat stroke (EHS, triggered either by challenging environmental conditions (such as hot temperatures and high humidity) and/or strenuous exercise) exhibit remarkably similar symptoms to those in patients with MH185, such as lethal hypermetabolic crisis with increases in ATP demand, in body temperature, in contractile sensitivity to temperature, caffeine, and membrane depolarization, and in resting Ca2+, rhabdomyolysis and finally sudden death in response to heat exposure or physical exertion. These EHS symptoms are also found in CASQ1 knockout mice. For example, at low temperature (25 °C), resting free [Ca2+]cytosol and [Ca2+]SR is not significantly different between fast-twitch FDB skeletal muscle fibers from CASQ1 knockout and wild-type mice119. However, at >30 °C, [Ca2+]cytosol is significantly increased in FDB skeletal muscle fibers from CASQ1 knockout mice135. In addition, slow-twitch soleus muscle fibers from CASQ1 knockout mice are not significantly affected by temperature challenge135. These results from CASQ1 knockout mice suggest that the development of increased basal tension in fast-twitch muscle and the impaired control of [Ca2+]cytosol could be the starting point toward the development of contractures during MH or EHS crises (Fig. 2). Therefore, the CASQ1 gene could be a powerful candidate for examining MH and EHS patients who do not have a mutation in the RyR1 or DHPR gene.
Here is “a possible scenario” for the MH- or EHS-like phenomena in CASQ1 single- or CASQ1/CASQ2 double-knockout mice65,67,115,116,119,130,132,135,172,186–188. First, structural remodeling of the triad junction, such as shrinking and abnormal multilayered triad junctions, occurs. Second, CASQ1 knockout induces the loss of inhibitory action of CASQ1 on RyR1 activity. Third, the absence of CASQ1 induces more rapid Ca2+ depletion from the SR due to the absence of Ca2+-buffering ability by CASQ1. Fourth, when exposed to halothane or heat, the knockout fibers show insufficient Ca2+ from the SR due to faster Ca2+ depletion from the SR with impairment of Ca2+ reuptake through SERCA1a into the SR. Fifth, more rapid Ca2+ depletion from the SR lowers the threshold for store overload-induced Ca2+ release (SOICR188). Sixth, the rapid Ca2+ depletion from the SR also increases skeletal SOCE to refill the SR. Seventh, the impaired SERCA1a activity probably increases ATP consumption, thus requiring a greater number of mitochondria (i.e., a hypermetabolic state occurs along with increased metabolic wastes and ionic disturbance115). Eighth, all these abnormal events induce more increases in cytosolic [Ca2+]. Correlations between the abnormal events mentioned above and the causative roles of CASQ1 in MH in humans should be further investigated, although it has been revealed that each abnormal event is related to the characteristic muscle rigidity and rhabdomyolysism showed in patients with MH or EHS and death.
An unexpected property has been observed in CASQ1 knockout mice: a sex dependency. When CASQ1 knockout mice are exposed to halothane (2%, 1 h) or to high environmental temperature (41 °C, 30 min), male CASQ1 knockout mice exhibit a high mortality rate (80%, particularly after 3 months), but female CASQ1 knockout mice show only marginal effects135. This sex-dependent mortality rate has also been found in human patients with MH or EHS189,190. The mechanisms responsible for the higher mortality rate in males have not been studied. A possible explanation for the sex-dependent mortality rate could be hormonal differences between males and females (related to different muscle masses and capacities).
Although the reduction or deficiency of dystrophin is the primary pathological cause of Duchenne muscular dystrophy (DMD, a lethal form of skeletal muscular dystrophy characterized by progressive wasting of the skeletal muscle191), dystrophic skeletal muscle fibers from a murine animal model of DMD (i.e., mdx mice) show reduced expression of CASQ-like proteins of 150–220 kDa (with no change in the expression level of RyR1, DHPR, SERCA1a, and CASQ1)192. The decreased expression of CASQ-like proteins might be related to the altered Ca2+ level in the SR and cytosol and, subsequently, muscle necrosis or apoptosis in DMD patients. In addition, skeletal muscle fibers from another mdx mouse show excessive SOCE due to the increased expression of Orai1193, as shown in FDB muscle fibers from CASQ1 single- or CASQ1/CASQ2 double-knockout mice130,132. These findings suggest a strong possibility that, in addition to the main pathological role of dystrophin in DMD (i.e., muscle damage due to the reduction or deficiency of dystrophin by mutations), abnormal Ca2+-buffering by CASQ1 and/or CASQ-like proteins in the SR, in combination with excessive SOCE, could be related to the pathological mechanism of DMD.
Several other pathological conditions are related to CASQ1, although clear correlations between the pathological phenotypes of patients and CASQ1 have not been defined. CASQ1 is related to diabetes. A diabetic rat model has shown increased expression of CASQ1 with significantly increased Ca2+-buffering capacity194. Polymorphisms in the CASQ1 gene are found in the Caucasian population in North America and in the Amish population with type 2 diabetes, suggesting that sequence variation in the CASQ1 gene could increase the risk for type 2 diabetes195,196. CASQ1 is also related to an abnormal thyroid status. Patients with Graves’ disease (a thyroid disease) show increased mRNA levels of both the CASQ1 and CASQ2 genes in thyroid tissue197. Unlike normal subjects, patients with Graves’ hyperthyroidism without ophthalmopathy and Hashimoto’s thyroiditis with or without ophthalmopathy show significant levels of autoantibodies against CASQ1 in the serum198,199. The acute induction of hyperthyroidism in the rabbit soleus muscle increases the mRNA level of CASQ1200.
Although not discussed in this review, CASQ2 is closely related to human diseases, especially catecholaminergic polymorphic ventricular tachycardia (CPVT, an arrhythmogenic disorder characterized by physical or emotional stress-induced syncopal events and finally sudden cardiac death). Regarding details on the relationship among CASQ2, RyR2, and CPVT in cardiac muscle, please refer to a review article201.
Conclusion and future directions
For the previous three decades, extensive and dedicated studies on CASQ1 in skeletal muscle have been conducted, and the critical roles of CASQ1 have been revealed, which has been a monumental achievement in our understanding of the physiological and path-physiological properties of skeletal muscle. However, considering that CASQ1 is the major Ca2+-buffering protein in the SR of skeletal muscle, it is plausible that the knockout of CASQ1 in skeletal muscle can induce enormous reductions in Ca2+ release from the SR in response to various stimuli and in the amount of Ca2+ available for release from the SR; however, this is not observed (still significant but only relative reductions). In addition, only MH- or EHS-like symptoms, but not other skeletal muscle disease-like symptoms, have been found in CASQ1 single- or CASQ1/CASQ2 double-knockout mice. Muscle fibers from knockout mice still sustain a sufficient degree of Ca2+ release and muscle contraction. These curiosities raise the possibility that either CASQ1 has functions that are not currently understood or other proteins could compensate for the absence of CASQ1 alone or of both CASQ1 and CASQ2.
In addition, no marked difference is observed between CASQ1 single- and CASQ1/CASQ2 double-knockout mice, which raises another important question: what is the physiological relevance of CASQ2 “in skeletal muscle”? It is possible that proteins and/or regulatory mechanisms exist, and further investigations of such proteins and/or mechanisms will help to answer these puzzling questions and improve our understanding of the physiological and pathophysiological properties of skeletal muscle.
In our aging society, the number of patients with skeletal muscle diseases is continuing to increase, and improved understanding of the pathology of skeletal muscle is in great demand. A more detailed understanding of the pathological roles of CASQ1 in skeletal muscle, for example, the roles of CASQ1 in skeletal muscle diseases that are combined with other diseases, could be an excellent way to meet the demand.
Acknowledgements
This work was supported by the Mid-Career Researcher Program through the National Research Foundation of Korea grant (No. NRF-2019R1A2C1086858 to E.H.L.).
Author contributions
E.H.L. designed and arranged the whole process of preparing this review; J.S.W., S.Y.J., J.H.C., J.H.P., and E.H.L. performed the literature searches; E.H.L. wrote the paper and drew Figs. 1 and 2; E.H.L. and J.H.P. drew Fig. 3; J.S.W. and E.H.L. edited or revised the paper. All authors have discussed all content of the paper and reviewed the literature. All authors have read and approved the published version of the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jin Seok Woo, Seung Yeon Jeong
References
- 1.Zucchi R, Ronca-Testoni S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharm. Rev. 1997;49:1–51. [PubMed] [Google Scholar]
- 2.Lee EH. Ca2+ channels and skeletal muscle diseases. Prog. Biophys. Mol. Biol. 2010;103:35–43. doi: 10.1016/j.pbiomolbio.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Endo M. Calcium-induced calcium release in skeletal muscle. Physiol. Rev. 2009;89:1153–1176. doi: 10.1152/physrev.00040.2008. [DOI] [PubMed] [Google Scholar]
- 4.Baylor SM, Hollingworth S. Sarcoplasmic reticulum calcium release compared in slow-twitch and fast-twitch fibres of mouse muscle. J. Physiol. 2003;551:125–138. doi: 10.1113/jphysiol.2003.041608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi, J. H., Jeong, S. Y., Oh, M. R., Allen, P. D. & Lee, E. H. TRPCs: influential mediators in skeletal muscle. Cells9, 850. 10.3390/cells9040850 (2020). [DOI] [PMC free article] [PubMed]
- 6.Cho CH, Lee KJ, Lee EH. With the greatest care, stromal interaction molecule (STIM) proteins verify what skeletal muscle is doing. BMB Rep. 2018;51:378–387. doi: 10.5483/BMBRep.2018.51.8.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho CH, Woo JS, Perez CF, Lee EH. A focus on extracellular Ca2+ entry into skeletal muscle. Exp. Mol. Med. 2017;49:e378. doi: 10.1038/emm.2017.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shamoo AE, MacLennan DH. A Ca++-dependent and -selective ionophore as part of the Ca++ plus Mg++-dependent adenosinetriphosphatase of sarcoplasmic reticulum. Proc. Natl Acad. Sci. USA. 1974;71:3522–3526. doi: 10.1073/pnas.71.9.3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Launikonis BS, Murphy RM, Edwards JN. Toward the roles of store-operated Ca2+ entry in skeletal muscle. Pflug. Arch. 2010;460:813–823. doi: 10.1007/s00424-010-0856-7. [DOI] [PubMed] [Google Scholar]
- 10.Tibbits GF, Thomas MJ. Ca2+ transport across the plasma membrane of striated muscle. Med. Sci. Sports Exerc. 1989;21:399–405. [PubMed] [Google Scholar]
- 11.Calderon JC, Bolanos P, Caputo C. The excitation-contraction coupling mechanism in skeletal muscle. Biophys. Rev. 2014;6:133–160. doi: 10.1007/s12551-013-0135-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol. Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 13.Woo JS, et al. Hypertrophy in skeletal myotubes induced by junctophilin-2 mutant, Y141H, involves an increase in store-operated Ca2+ entry via Orai1. J. Biol. Chem. 2012;287:14336–14348. doi: 10.1074/jbc.M111.304808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishi M, et al. Abnormal features in skeletal muscle from mice lacking mitsugumin29. J. Cell Biol. 1999;147:1473–1480. doi: 10.1083/jcb.147.7.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacLennan DH, Wong PT. Isolation of a calcium-sequestering protein from sarcoplasmic reticulum. Proc. Natl Acad. Sci. USA. 1971;68:1231–1235. doi: 10.1073/pnas.68.6.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saito A, Seiler S, Chu A, Fleischer S. Preparation and morphology of sarcoplasmic reticulum terminal cisternae from rabbit skeletal muscle. J. Cell Biol. 1984;99:875–885. doi: 10.1083/jcb.99.3.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peachey LD. The sarcoplasmic reticulum and transverse tubules of the frog’s sartorius. J. Cell Biol. 1965;25(Suppl):209–231. doi: 10.1083/jcb.25.3.209. [DOI] [PubMed] [Google Scholar]
- 18.Volpe P, Simon BJ. The bulk of Ca2+ released to the myoplasm is free in the sarcoplasmic reticulum and does not unbind from calsequestrin. FEBS Lett. 1991;278:274–278. doi: 10.1016/0014-5793(91)80134-o. [DOI] [PubMed] [Google Scholar]
- 19.Franzini-Armstrong C, Jorgensen AO. Structure and development of E-C coupling units in skeletal muscle. Annu Rev. Physiol. 1994;56:509–534. doi: 10.1146/annurev.ph.56.030194.002453. [DOI] [PubMed] [Google Scholar]
- 20.Koeppen, B. M. & Stanton, B. A. Berne & Levy Physiology (2018).
- 21.Rossi D, Barone V, Giacomello E, Cusimano V, Sorrentino V. The sarcoplasmic reticulum: an organized patchwork of specialized domains. Traffic. 2008;9:1044–1049. doi: 10.1111/j.1600-0854.2008.00717.x. [DOI] [PubMed] [Google Scholar]
- 22.Takekura H, Flucher BE, Franzini-Armstrong C. Sequential docking, molecular differentiation, and positioning of T-Tubule/SR junctions in developing mouse skeletal muscle. Dev. Biol. 2001;239:204–214. doi: 10.1006/dbio.2001.0437. [DOI] [PubMed] [Google Scholar]
- 23.Costello B, et al. Characterization of the junctional face membrane from terminal cisternae of sarcoplasmic reticulum. J. Cell Biol. 1986;103:741–753. doi: 10.1083/jcb.103.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jayasinghe, I. D., Munro, M., Baddeley, D., Launikonis, B. S. & Soeller, C. Observation of the molecular organization of calcium release sites in fast- and slow-twitch skeletal muscle with nanoscale imaging. J. R. Soc. Interface11, 20140570. 10.1098/rsif.2014.0570 (2014). [DOI] [PMC free article] [PubMed]
- 25.Phimister AJ, et al. Conformation-dependent stability of junctophilin 1 (JP1) and ryanodine receptor type 1 (RyR1) channel complex is mediated by their hyper-reactive thiols. J. Biol. Chem. 2007;282:8667–8677. doi: 10.1074/jbc.M609936200. [DOI] [PubMed] [Google Scholar]
- 26.Golini L, et al. Junctophilin 1 and 2 proteins interact with the L-type Ca2+ channel dihydropyridine receptors (DHPRs) in skeletal muscle. J. Biol. Chem. 2011;286:43717–43725. doi: 10.1074/jbc.M111.292755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woo JS, et al. Glutamate at position 227 of junctophilin-2 is involved in binding to TRPC3. Mol. Cell Biochem. 2009;328:25–32. doi: 10.1007/s11010-009-0070-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woo JS, Kim DH, Allen PD, Lee EH. TRPC3-interacting triadic proteins in skeletal muscle. Biochem. J. 2008;411:399–405. doi: 10.1042/bj20071504. [DOI] [PubMed] [Google Scholar]
- 29.Ito K, et al. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J. Cell Biol. 2001;154:1059–1067. doi: 10.1083/jcb.200105040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, et al. Impaired Orai1-mediated resting Ca2+ entry reduces the cytosolic [Ca2+] and sarcoplasmic reticulum Ca2+ loading in quiescent junctophilin 1 knock-out myotubes. J. Biol. Chem. 2010;285:39171–39179. doi: 10.1074/jbc.M110.149690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirata Y, et al. Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys. J. 2006;90:4418–4427. doi: 10.1529/biophysj.105.076570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woo JS, et al. S165F mutation of junctophilin 2 affects Ca2+ signalling in skeletal muscle. Biochem. J. 2010;427:125–134. doi: 10.1042/BJ20091225. [DOI] [PubMed] [Google Scholar]
- 33.Kiselyov KI, et al. Gating of store-operated channels by conformational coupling to ryanodine receptors. Mol. Cell. 2000;6:421–431. doi: 10.1016/s1097-2765(00)00041-1. [DOI] [PubMed] [Google Scholar]
- 34.Lee EH, Cherednichenko G, Pessah IN, Allen PD. Functional coupling between TRPC3 and RyR1 regulates the expressions of key triadic proteins. J. Biol. Chem. 2006;281:10042–10048. doi: 10.1074/jbc.M600981200. [DOI] [PubMed] [Google Scholar]
- 35.Flucher BE, Franzini-Armstrong C. Formation of junctions involved in excitation-contraction coupling in skeletal and cardiac muscle. Proc. Natl Acad. Sci. USA. 1996;93:8101–8106. doi: 10.1073/pnas.93.15.8101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Froemming GR, Ohlendieck K. Oligomerisation of Ca2+-regulatory membrane components involved in the excitation-contraction-relaxation cycle during postnatal development of rabbit skeletal muscle. Biochim. Biophys. Acta. 1998;1387:226–238. doi: 10.1016/s0167-4838(98)00126-5. [DOI] [PubMed] [Google Scholar]
- 37.Boncompagni S, et al. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol. Biol. Cell. 2009;20:1058–1067. doi: 10.1091/mbc.E08-07-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lannergren J, Bruton JD. Mitochondrial Ca2+ in mouse soleus single muscle fibres in response to repeated tetanic contractions. Adv. Exp. Med. Biol. 2003;538:557–562. doi: 10.1007/978-1-4419-9029-7_49. [DOI] [PubMed] [Google Scholar]
- 39.Sembrowich WL, Quintinskie JJ, Li G. Calcium uptake in mitochondria from different skeletal muscle types. J. Appl. Physiol. 1985;59:137–141. doi: 10.1152/jappl.1985.59.1.137. [DOI] [PubMed] [Google Scholar]
- 40.Rossi AE, Boncompagni S, Wei L, Protasi F, Dirksen RT. Differential impact of mitochondrial positioning on mitochondrial Ca2+ uptake and Ca2+ spark suppression in skeletal muscle. Am. J. Physiol. Cell Physiol. 2011;301:C1128–C1139. doi: 10.1152/ajpcell.00194.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eisner V, Csordas G, Hajnoczky G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle - pivotal roles in Ca2+ and reactive oxygen species signaling. J. Cell Sci. 2013;126:2965–2978. doi: 10.1242/jcs.093609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossi AE, Boncompagni S, Dirksen RT. Sarcoplasmic reticulum-mitochondrial symbiosis: bidirectional signaling in skeletal muscle. Exerc. Sport Sci. Rev. 2009;37:29–35. doi: 10.1097/JES.0b013e3181911fa4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 44.Ikemoto N, Bhatnagar GM, Nagy B, Gergely J. Interaction of divalent cations with the 55,000-dalton protein component of the sarcoplasmic reticulum. studies of fluorescence and circular dichroism. J. Biol. Chem. 1972;247:7835–7837. [PubMed] [Google Scholar]
- 45.Zarain-Herzberg A, Fliegel L, MacLennan DH. Structure of the rabbit fast-twitch skeletal muscle calsequestrin gene. J. Biol. Chem. 1988;263:4807–4812. [PubMed] [Google Scholar]
- 46.Scott BT, Simmerman HK, Collins JH, Nadal-Ginard B, Jones LR. Complete amino acid sequence of canine cardiac calsequestrin deduced by cDNA cloning. J. Biol. Chem. 1988;263:8958–8964. [PubMed] [Google Scholar]
- 47.Fliegel L, et al. Amino acid sequence of rabbit fast-twitch skeletal muscle calsequestrin deduced from cDNA and peptide sequencing. Proc. Natl Acad. Sci. USA. 1987;84:1167–1171. doi: 10.1073/pnas.84.5.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Volpe P, Martini A, Furlan S, Meldolesi J. Calsequestrin is a component of smooth muscles: the skeletal- and cardiac-muscle isoforms are both present, although in highly variable amounts and ratios. Biochem. J. 1994;301(Pt 2):465–469. doi: 10.1042/bj3010465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujii J, Willard HF, MacLennan DH. Characterization and localization to human chromosome 1 of human fast-twitch skeletal muscle calsequestrin gene. Somat. Cell Mol. Genet. 1990;16:185–189. doi: 10.1007/BF01233048. [DOI] [PubMed] [Google Scholar]
- 50.Beard NA, Laver DR, Dulhunty AF. Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog. Biophys. Mol. Biol. 2004;85:33–69. doi: 10.1016/j.pbiomolbio.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 51.Park H, et al. Comparing skeletal and cardiac calsequestrin structures and their calcium binding: a proposed mechanism for coupled calcium binding and protein polymerization. J. Biol. Chem. 2004;279:18026–18033. doi: 10.1074/jbc.M311553200. [DOI] [PubMed] [Google Scholar]
- 52.Kim E, et al. Characterization of human cardiac calsequestrin and its deleterious mutants. J. Mol. Biol. 2007;373:1047–1057. doi: 10.1016/j.jmb.2007.08.055. [DOI] [PubMed] [Google Scholar]
- 53.Wang S, et al. Crystal structure of calsequestrin from rabbit skeletal muscle sarcoplasmic reticulum. Nat. Struct. Biol. 1998;5:476–483. doi: 10.1038/nsb0698-476. [DOI] [PubMed] [Google Scholar]
- 54.Wei L, Hanna AD, Beard NA, Dulhunty AF. Unique isoform-specific properties of calsequestrin in the heart and skeletal muscle. Cell Calcium. 2009;45:474–484. doi: 10.1016/j.ceca.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 55.Cala SE, Jones LR. Phosphorylation of cardiac and skeletal muscle calsequestrin isoforms by casein kinase II. Demonstration of a cluster of unique rapidly phosphorylated sites in cardiac calsequestrin. J. Biol. Chem. 1991;266:391–398. [PubMed] [Google Scholar]
- 56.Cala SE, Miles K. Phosphorylation of the cardiac isoform of calsequestrin in cultured rat myotubes and rat skeletal muscle. Biochim. Biophys. Acta. 1992;1118:277–287. doi: 10.1016/0167-4838(92)90285-l. [DOI] [PubMed] [Google Scholar]
- 57.Allen DG, Lamb GD, Westerblad H. Skeletal muscle fatigue: cellular mechanisms. Physiol. Rev. 2008;88:287–332. doi: 10.1152/physrev.00015.2007. [DOI] [PubMed] [Google Scholar]
- 58.Meissner G, Conner GE, Fleischer S. Isolation of sarcoplasmic reticulum by zonal centrifugation and purification of Ca2+-pump and Ca2+-binding proteins. Biochim. Biophys. Acta. 1973;298:246–269. doi: 10.1016/0005-2736(73)90355-6. [DOI] [PubMed] [Google Scholar]
- 59.Jorgensen AO, Shen AC, Campbell KP, MacLennan DH. Ultrastructural localization of calsequestrin in rat skeletal muscle by immunoferritin labeling of ultrathin frozen sections. J. Cell Biol. 1983;97:1573–1581. doi: 10.1083/jcb.97.5.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Campbell KP, MacLennan DH, Jorgensen AO, Mintzer MC. Purification and characterization of calsequestrin from canine cardiac sarcoplasmic reticulum and identification of the 53,000 dalton glycoprotein. J. Biol. Chem. 1983;258:1197–1204. [PubMed] [Google Scholar]
- 61.Franzini-Armstrong C, Kenney LJ, Varriano-Marston E. The structure of calsequestrin in triads of vertebrate skeletal muscle: a deep-etch study. J. Cell Biol. 1987;105:49–56. doi: 10.1083/jcb.105.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wagenknecht T, Hsieh CE, Rath BK, Fleischer S, Marko M. Electron tomography of frozen-hydrated isolated triad junctions. Biophys. J. 2002;83:2491–2501. doi: 10.1016/S0006-3495(02)75260-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leberer E, Seedorf U, Pette D. Neural control of gene expression in skeletal muscle. Calcium-sequestering proteins in developing and chronically stimulated rabbit skeletal muscles. Biochem. J. 1986;239:295–300. doi: 10.1042/bj2390295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sacchetto R, Volpe P, Damiani E, Margreth A. Postnatal development of rabbit fast-twitch skeletal muscle: accumulation, isoform transition and fibre distribution of calsequestrin. J. Muscle Res. Cell Motil. 1993;14:646–653. doi: 10.1007/BF00141561. [DOI] [PubMed] [Google Scholar]
- 65.Murphy RM, Larkins NT, Mollica JP, Beard NA, Lamb GD. Calsequestrin content and SERCA determine normal and maximal Ca2+ storage levels in sarcoplasmic reticulum of fast- and slow-twitch fibres of rat. J. Physiol. 2009;587:443–460. doi: 10.1113/jphysiol.2008.163162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Damiani E, Volpe P, Margreth A. Coexpression of two isoforms of calsequestrin in rabbit slow-twitch muscle. J. Muscle Res. Cell Motil. 1990;11:522–530. doi: 10.1007/BF01745219. [DOI] [PubMed] [Google Scholar]
- 67.Beard NA, Sakowska MM, Dulhunty AF, Laver DR. Calsequestrin is an inhibitor of skeletal muscle ryanodine receptor calcium release channels. Biophys. J. 2002;82:310–320. doi: 10.1016/S0006-3495(02)75396-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ikemoto N, Ronjat M, Meszaros LG, Koshita M. Postulated role of calsequestrin in the regulation of calcium release from sarcoplasmic reticulum. Biochemistry. 1989;28:6764–6771. doi: 10.1021/bi00442a033. [DOI] [PubMed] [Google Scholar]
- 69.He Z, Dunker AK, Wesson CR, Trumble WR. Ca2+-induced folding and aggregation of skeletal muscle sarcoplasmic reticulum calsequestrin. The involvement of the trifluoperazine-binding site. J. Biol. Chem. 1993;268:24635–24641. [PubMed] [Google Scholar]
- 70.Fryer MW, Stephenson DG. Total and sarcoplasmic reticulum calcium contents of skinned fibres from rat skeletal muscle. J. Physiol. 1996;493(Pt 2):357–370. doi: 10.1113/jphysiol.1996.sp021388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pape PC, Fenelon K, Lamboley CR, Stachura D. Role of calsequestrin evaluated from changes in free and total calcium concentrations in the sarcoplasmic reticulum of frog cut skeletal muscle fibres. J. Physiol. 2007;581:319–367. doi: 10.1113/jphysiol.2006.126474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wei L, Varsanyi M, Dulhunty AF, Beard NA. The conformation of calsequestrin determines its ability to regulate skeletal ryanodine receptors. Biophys. J. 2006;91:1288–1301. doi: 10.1529/biophysj.106.082610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Park H, Wu S, Dunker AK, Kang C. Polymerization of calsequestrin. Implications for Ca2+ regulation. J. Biol. Chem. 2003;278:16176–16182. doi: 10.1074/jbc.M300120200. [DOI] [PubMed] [Google Scholar]
- 74.Sztretye M, et al. Measurement of RyR permeability reveals a role of calsequestrin in termination of SR Ca2+ release in skeletal muscle. J. Gen. Physiol. 2011;138:231–247. doi: 10.1085/jgp.201010592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Manno C, et al. Calsequestrin depolymerizes when calcium is depleted in the sarcoplasmic reticulum of working muscle. Proc. Natl Acad. Sci. USA. 2017;114:E638–E647. doi: 10.1073/pnas.1620265114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Balsera M, Buchanan BB. Evolution of the thioredoxin system as a step enabling adaptation to oxidative stress. Free Radic. Biol. Med. 2019;140:28–35. doi: 10.1016/j.freeradbiomed.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 77.Maguire PB, Briggs FN, Lennon NJ, Ohlendieck K. Oligomerization is an intrinsic property of calsequestrin in normal and transformed skeletal muscle. Biochem. Biophys. Res. Commun. 1997;240:721–727. doi: 10.1006/bbrc.1997.7729. [DOI] [PubMed] [Google Scholar]
- 78.Sanchez EJ, Lewis KM, Danna BR, Kang C. High-capacity Ca2+ binding of human skeletal calsequestrin. J. Biol. Chem. 2012;287:11592–11601. doi: 10.1074/jbc.M111.335075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blumenthal DK, et al. Identification of the calmodulin-binding domain of skeletal muscle myosin light chain kinase. Proc. Natl Acad. Sci. USA. 1985;82:3187–3191. doi: 10.1073/pnas.82.10.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kumar A, et al. Identification of calcium binding sites on calsequestrin 1 and their implications for polymerization. Mol. Biosyst. 2013;9:1949–1957. doi: 10.1039/c3mb25588c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cozens B, Reithmeier RA. Size and shape of rabbit skeletal muscle calsequestrin. J. Biol. Chem. 1984;259:6248–6252. [PubMed] [Google Scholar]
- 82.Bal NC, et al. The C-terminal calcium-sensitive disordered motifs regulate isoform-specific polymerization characteristics of calsequestrin. Biopolymers. 2015;103:15–22. doi: 10.1002/bip.22534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Launikonis BS, et al. Confocal imaging of [Ca2+] in cellular organelles by SEER, shifted excitation and emission ratioing of fluorescence. J. Physiol. 2005;567:523–543. doi: 10.1113/jphysiol.2005.087973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Launikonis BS, et al. Depletion “skraps” and dynamic buffering inside the cellular calcium store. Proc. Natl Acad. Sci. USA. 2006;103:2982–2987. doi: 10.1073/pnas.0511252103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pizarro G, Rios E. How source content determines intracellular Ca2+ release kinetics. Simultaneous measurement of [Ca2+] transients and [H+] displacement in skeletal muscle. J. Gen. Physiol. 2004;124:239–258. doi: 10.1085/jgp.200409071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pape PC, Jong DS, Chandler WK. Effects of partial sarcoplasmic reticulum calcium depletion on calcium release in frog cut muscle fibers equilibrated with 20 mM EGTA. J. Gen. Physiol. 1998;112:263–295. doi: 10.1085/jgp.112.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pape PC, Carrier N. Effect of sarcoplasmic reticulum (SR) calcium content on SR calcium release elicited by small voltage-clamp depolarizations in frog cut skeletal muscle fibers equilibrated with 20 mM EGTA. J. Gen. Physiol. 1998;112:161–179. doi: 10.1085/jgp.112.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Beard NA, et al. Regulation of ryanodine receptors by calsequestrin: effect of high luminal Ca2+ and phosphorylation. Biophys. J. 2005;88:3444–3454. doi: 10.1529/biophysj.104.051441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Guo W, Campbell KP. Association of triadin with the ryanodine receptor and calsequestrin in the lumen of the sarcoplasmic reticulum. J. Biol. Chem. 1995;270:9027–9030. doi: 10.1074/jbc.270.16.9027. [DOI] [PubMed] [Google Scholar]
- 90.Shin DW, Ma J, Kim DH. The asp-rich region at the carboxyl-terminus of calsequestrin binds to Ca2+ and interacts with triadin. FEBS Lett. 2000;486:178–182. doi: 10.1016/s0014-5793(00)02246-8. [DOI] [PubMed] [Google Scholar]
- 91.Rossi D, et al. Distinct regions of triadin are required for targeting and retention at the junctional domain of the sarcoplasmic reticulum. Biochem. J. 2014;458:407–417. doi: 10.1042/BJ20130719. [DOI] [PubMed] [Google Scholar]
- 92.Lee JM, et al. Negatively charged amino acids within the intraluminal loop of ryanodine receptor are involved in the interaction with triadin. J. Biol. Chem. 2004;279:6994–7000. doi: 10.1074/jbc.M312446200. [DOI] [PubMed] [Google Scholar]
- 93.Goonasekera SA, et al. Triadin binding to the C-terminal luminal loop of the ryanodine receptor is important for skeletal muscle excitation contraction coupling. J. Gen. Physiol. 2007;130:365–378. doi: 10.1085/jgp.200709790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Herzog A, Szegedi C, Jona I, Herberg FW, Varsanyi M. Surface plasmon resonance studies prove the interaction of skeletal muscle sarcoplasmic reticular Ca2+ release channel/ryanodine receptor with calsequestrin. FEBS Lett. 2000;472:73–77. doi: 10.1016/s0014-5793(00)01431-9. [DOI] [PubMed] [Google Scholar]
- 95.Dulhunty A, Wei L, Beard N. Junctin—the quiet achiever. J. Physiol. 2009;587:3135–3137. doi: 10.1113/jphysiol.2009.171959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Beard NA, Dulhunty AF. C-terminal residues of skeletal muscle calsequestrin are essential for calcium binding and for skeletal ryanodine receptor inhibition. Skelet. Muscle. 2015;5:6. doi: 10.1186/s13395-015-0029-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wagenknecht T, Samso M. Three-dimensional reconstruction of ryanodine receptors. Front. Biosci. 2002;7:d1464–d1474. doi: 10.2741/A853. [DOI] [PubMed] [Google Scholar]
- 98.Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J. Biol. Chem. 1997;272:23389–23397. doi: 10.1074/jbc.272.37.23389. [DOI] [PubMed] [Google Scholar]
- 99.Perni S, Close M, Franzini-Armstrong C. Novel details of calsequestrin gel conformation in situ. J. Biol. Chem. 2013;288:31358–31362. doi: 10.1074/jbc.M113.507749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kawasaki T, Kasai M. Regulation of calcium channel in sarcoplasmic reticulum by calsequestrin. Biochem. Biophys. Res. Commun. 1994;199:1120–1127. doi: 10.1006/bbrc.1994.1347. [DOI] [PubMed] [Google Scholar]
- 101.Wang Y, et al. Knocking down type 2 but not type 1 calsequestrin reduces calcium sequestration and release in C2C12 skeletal muscle myotubes. J. Biol. Chem. 2006;281:15572–15581. doi: 10.1074/jbc.M600090200. [DOI] [PubMed] [Google Scholar]
- 102.Shin DW, et al. A retrograde signal from calsequestrin for the regulation of store-operated Ca2+ entry in skeletal muscle. J. Biol. Chem. 2003;278:3286–3292. doi: 10.1074/jbc.M209045200. [DOI] [PubMed] [Google Scholar]
- 103.Qin J, et al. Ryanodine receptor luminal Ca2+ regulation: swapping calsequestrin and channel isoforms. Biophys. J. 2009;97:1961–1970. doi: 10.1016/j.bpj.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ohkura M, et al. Dual regulation of the skeletal muscle ryanodine receptor by triadin and calsequestrin. Biochemistry. 1998;37:12987–12993. doi: 10.1021/bi972803d. [DOI] [PubMed] [Google Scholar]
- 105.Wei L, Gallant EM, Dulhunty AF, Beard NA. Junctin and triadin each activate skeletal ryanodine receptors but junctin alone mediates functional interactions with calsequestrin. Int. J. Biochem. Cell Biol. 2009;41:2214–2224. doi: 10.1016/j.biocel.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Oddoux S, et al. Triadin deletion induces impaired skeletal muscle function. J. Biol. Chem. 2009;284:34918–34929. doi: 10.1074/jbc.M109.022442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Roux-Buisson N, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum. Mol. Genet. 2012;21:2759–2767. doi: 10.1093/hmg/dds104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mackrill JJ. Ryanodine receptor calcium channels and their partners as drug targets. Biochem. Pharm. 2010;79:1535–1543. doi: 10.1016/j.bcp.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 109.Shoshan-Barmatz V, et al. The identification of the phosphorylated 150/160-kDa proteins of sarcoplasmic reticulum, their kinase and their association with the ryanodine receptor. Biochim. Biophys. Acta. 1996;1283:89–100. doi: 10.1016/0005-2736(96)00079-x. [DOI] [PubMed] [Google Scholar]
- 110.Beard NA, et al. Phosphorylation of skeletal muscle calsequestrin enhances its Ca2+ binding capacity and promotes its association with junctin. Cell Calcium. 2008;44:363–373. doi: 10.1016/j.ceca.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 111.Varsanyi M, Heilmeyer LM., Jr Autocatalytic phosphorylation of calsequestrin. FEBS Lett. 1980;122:227–230. doi: 10.1016/0014-5793(80)80444-3. [DOI] [PubMed] [Google Scholar]
- 112.Nori A, Gola E, Tosato S, Cantini M, Volpe P. Targeting of calsequestrin to sarcoplasmic reticulum after deletions of its acidic carboxy terminus. Am. J. Physiol. 1999;277:C974–C981. doi: 10.1152/ajpcell.1999.277.5.C974. [DOI] [PubMed] [Google Scholar]
- 113.Szegedi C, Sarkozi S, Herzog A, Jona I, Varsanyi M. Calsequestrin: more than ‘only’ a luminal Ca2+ buffer inside the sarcoplasmic reticulum. Biochem. J. 1999;337(Pt 1):19–22. [PMC free article] [PubMed] [Google Scholar]
- 114.Varsanyi M, Heilmeyer LM., Jr. The protein kinase properties of calsequestrin. FEBS Lett. 1979;103:85–88. doi: 10.1016/0014-5793(79)81255-7. [DOI] [PubMed] [Google Scholar]
- 115.Paolini C, et al. Reorganized stores and impaired calcium handling in skeletal muscle of mice lacking calsequestrin-1. J. Physiol. 2007;583:767–784. doi: 10.1113/jphysiol.2007.138024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Paolini C, Quarta M, D’Onofrio L, Reggiani C, Protasi F. Differential effect of calsequestrin ablation on structure and function of fast and slow skeletal muscle fibers. J. Biomed. Biotechnol. 2011;2011:634075. doi: 10.1155/2011/634075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tomasi M, et al. Calsequestrin (CASQ1) rescues function and structure of calcium release units in skeletal muscles of CASQ1-null mice. Am. J. Physiol. Cell Physiol. 2012;302:C575–C586. doi: 10.1152/ajpcell.00119.2011. [DOI] [PubMed] [Google Scholar]
- 118.Paolini C, et al. Oxidative stress, mitochondrial damage, and cores in muscle from calsequestrin-1 knockout mice. Skelet. Muscle. 2015;5:10. doi: 10.1186/s13395-015-0035-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Canato M, et al. Massive alterations of sarcoplasmic reticulum free calcium in skeletal muscle fibers lacking calsequestrin revealed by a genetically encoded probe. Proc. Natl Acad. Sci. USA. 2010;107:22326–22331. doi: 10.1073/pnas.1009168108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 121.Lewis RS. Store-operated calcium channels: from function to structure and back again. Cold Spring Harb. Perspect. Cold Spring Harb. Perspect. Biol. 2020;12:a035055. doi: 10.1101/cshperspect.a035055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kurebayashi N, Ogawa Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J. Physiol. 2001;533:185–199. doi: 10.1111/j.1469-7793.2001.0185b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lee KJ, et al. STIM1 negatively regulates Ca2+ release from the sarcoplasmic reticulum in skeletal myotubes. Biochem. J. 2013;453:187–200. doi: 10.1042/BJ20130178. [DOI] [PubMed] [Google Scholar]
- 124.Stiber J, et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. 2008;10:688–697. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Launikonis BS, Rios E. Store-operated Ca2+ entry during intracellular Ca2+ release in mammalian skeletal muscle. J. Physiol. 2007;583:81–97. doi: 10.1113/jphysiol.2007.135046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Edwards JN, et al. Ultra-rapid activation and deactivation of store-operated Ca2+ entry in skeletal muscle. Cell Calcium. 2010;47:458–467. doi: 10.1016/j.ceca.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 127.Pan Z, Brotto M, Ma J. Store-operated Ca2+ entry in muscle physiology and diseases. BMB Rep. 2014;47:69–79. doi: 10.5483/BMBRep.2014.47.2.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sztretye M, et al. SOCE is important for maintaining sarcoplasmic calcium content and release in skeletal muscle fibers. Biophys. J. 2017;113:2496–2507. doi: 10.1016/j.bpj.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pan Z, et al. Dysfunction of store-operated calcium channel in muscle cells lacking mg29. Nat. Cell Biol. 2002;4:379–383. doi: 10.1038/ncb788. [DOI] [PubMed] [Google Scholar]
- 130.Zhao X, et al. Increased store-operated Ca2+ entry in skeletal muscle with reduced calsequestrin-1 expression. Biophys. J. 2010;99:1556–1564. doi: 10.1016/j.bpj.2010.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhang L, Wang L, Li S, Xue J, Luo D. Calsequestrin-1 regulates store-operated Ca2+ entry by inhibiting STIM1 aggregation. Cell Physiol. Biochem. 2016;38:2183–2193. doi: 10.1159/000445574. [DOI] [PubMed] [Google Scholar]
- 132.Yarotskyy V, Protasi F, Dirksen RT. Accelerated activation of SOCE current in myotubes from two mouse models of anesthetic- and heat-induced sudden death. PLoS ONE. 2013;8:e77633. doi: 10.1371/journal.pone.0077633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ma J, Pan Z. Retrograde activation of store-operated calcium channel. Cell Calcium. 2003;33:375–384. doi: 10.1016/s0143-4160(03)00050-2. [DOI] [PubMed] [Google Scholar]
- 134.Lyfenko AD, Goonasekera SA, Dirksen RT. Dynamic alterations in myoplasmic Ca2+ in malignant hyperthermia and central core disease. Biochem. Biophys. Res. Commun. 2004;322:1256–1266. doi: 10.1016/j.bbrc.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 135.Dainese M, et al. Anesthetic- and heat-induced sudden death in calsequestrin-1-knockout mice. FASEB J. 2009;23:1710–1720. doi: 10.1096/fj.08-121335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Anderson AA, et al. The novel skeletal muscle sarcoplasmic reticulum JP-45 protein. Molecular cloning, tissue distribution, developmental expression, and interaction with alpha 1.1 subunit of the voltage-gated calcium channel. J. Biol. Chem. 2003;278:39987–39992. doi: 10.1074/jbc.M305016200. [DOI] [PubMed] [Google Scholar]
- 137.Anderson AA, et al. The junctional SR protein JP-45 affects the functional expression of the voltage-dependent Ca2+ channel Cav1.1. J. Cell Sci. 2006;119:2145–2155. doi: 10.1242/jcs.02935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Delbono O, et al. Loss of skeletal muscle strength by ablation of the sarcoplasmic reticulum protein JP45. Proc. Natl Acad. Sci. USA. 2007;104:20108–20113. doi: 10.1073/pnas.0707389104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Delbono O, O’Rourke KS, Ettinger WH. Excitation-calcium release uncoupling in aged single human skeletal muscle fibers. J. Membr. Biol. 1995;148:211–222. doi: 10.1007/BF00235039. [DOI] [PubMed] [Google Scholar]
- 140.Renganathan M, Messi ML, Delbono O. Dihydropyridine receptor-ryanodine receptor uncoupling in aged skeletal muscle. J. Membr. Biol. 1997;157:247–253. doi: 10.1007/s002329900233. [DOI] [PubMed] [Google Scholar]
- 141.Mosca B, et al. Role of the JP45-calsequestrin complex on calcium entry in slow twitch skeletal muscles. J. Biol. Chem. 2016;291:14555–14565. doi: 10.1074/jbc.M115.709071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gouadon E, et al. A possible role of the junctional face protein JP-45 in modulating Ca2+ release in skeletal muscle. J. Physiol. 2006;572:269–280. doi: 10.1113/jphysiol.2005.104406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang Q, et al. Two pools of IRE1alpha in cardiac and skeletal muscle cells. FASEB J. 2019;33:8892–8904. doi: 10.1096/fj.201802626R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Groenendyk J, et al. Inhibition of the unfolded protein response mechanism prevents cardiac fibrosis. PLoS ONE. 2016;11:e0159682. doi: 10.1371/journal.pone.0159682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tufanli O, et al. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl Acad. Sci. USA. 2017;114:E1395–E1404. doi: 10.1073/pnas.1621188114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Damiani E, Picello E, Saggin L, Margreth A. Identification of triadin and of histidine-rich Ca2+-binding protein as substrates of 60 kDa calmodulin-dependent protein kinase in junctional terminal cisternae of sarcoplasmic reticulum of rabbit fast muscle. Biochem. Biophys. Res. Commun. 1995;209:457–465. doi: 10.1006/bbrc.1995.1524. [DOI] [PubMed] [Google Scholar]
- 147.Orr I, Shoshan-Barmatz V. Modulation of the skeletal muscle ryanodine receptor by endogenous phosphorylation of 160/150-kDa proteins of the sarcoplasmic reticulum. Biochim. Biophys. Acta. 1996;1283:80–88. doi: 10.1016/0005-2736(96)00078-8. [DOI] [PubMed] [Google Scholar]
- 148.Cala SE, Scott BT, Jones LR. Intralumenal sarcoplasmic reticulum Ca2+-binding proteins. Semin. Cell Biol. 1990;1:265–275. [PubMed] [Google Scholar]
- 149.Orr I, Gechtman Z, Shoshan-Barmatz V. Characterization of Ca2+-dependent endogenous phosphorylation of 160,000- and 150,000-Dalton proteins of sarcoplasmic reticulum. Biochem. J. 1991;276(Pt 1):89–96. doi: 10.1042/bj2760089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Leberer E, et al. Molecular cloning and expression of cDNA encoding the 53,000-dalton glycoprotein of rabbit skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 1989;264:3484–3493. [PubMed] [Google Scholar]
- 151.Fliegel L, Burns K, MacLennan DH, Reithmeier RA, Michalak M. Molecular cloning of the high affinity calcium-binding protein (calreticulin) of skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 1989;264:21522–21528. [PubMed] [Google Scholar]
- 152.Ostwald TJ, MacLennan DH. Isolation of a high affinity calcium-binding protein from sarcoplasmic reticulum. J. Biol. Chem. 1974;249:974–979. [PubMed] [Google Scholar]
- 153.Moore ED, et al. Organization of Ca2+ release units in excitable smooth muscle of the guinea-pig urinary bladder. Biophys. J. 2004;87:1836–1847. doi: 10.1529/biophysj.104.044123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Volpe P, et al. Heterogeneity of microsomal Ca2+ stores in chicken Purkinje neurons. EMBO J. 1991;10:3183–3189. doi: 10.1002/j.1460-2075.1991.tb04880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Damiani E, Spamer C, Heilmann C, Salvatori S, Margreth A. Endoplasmic reticulum of rat liver contains two proteins closely related to skeletal sarcoplasmic reticulum Ca-ATPase and calsequestrin. J. Biol. Chem. 1988;263:340–343. [PubMed] [Google Scholar]
- 156.Cho JH, et al. Calsequestrin, a calcium sequestering protein localized at the sarcoplasmic reticulum, is not essential for body-wall muscle function in Caenorhabditis elegans. J. Cell Sci. 2000;113(Pt 22):3947–3958. doi: 10.1242/jcs.113.22.3947. [DOI] [PubMed] [Google Scholar]
- 157.Oberdorf JA, Lebeche D, Head JF, Kaminer B. Identification of a calsequestrin-like protein from sea urchin eggs. J. Biol. Chem. 1988;263:6806–6809. [PubMed] [Google Scholar]
- 158.Krause KH, Chou M, Thomas MA, Sjolund RD, Campbell KP. Plant cells contain calsequestrin. J. Biol. Chem. 1989;264:4269–4272. [PubMed] [Google Scholar]
- 159.Franceschi VR, Li X, Zhang D, Okita TW. Calsequestrinlike calcium-binding protein is expressed in calcium-accumulating cells of Pistia stratiotes. Proc. Natl Acad. Sci. USA. 1993;90:6986–6990. doi: 10.1073/pnas.90.15.6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bohm J, et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am. J. Hum. Genet. 2013;92:271–278. doi: 10.1016/j.ajhg.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nesin V, et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc. Natl Acad. Sci. USA. 2014;111:4197–4202. doi: 10.1073/pnas.1312520111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Okuma H, et al. Tubular aggregate myopathy caused by a novel mutation in the cytoplasmic domain of STIM1. Neurol. Genet. 2016;2:e50. doi: 10.1212/NXG.0000000000000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Barone V, et al. Identification and characterization of three novel mutations in the CASQ1 gene in four patients with tubular aggregate myopathy. Hum. Mutat. 2017;38:1761–1773. doi: 10.1002/humu.23338. [DOI] [PubMed] [Google Scholar]
- 164.Bohm J, et al. CASQ1 mutations impair calsequestrin polymerization and cause tubular aggregate myopathy. Acta Neuropathol. 2018;135:149–151. doi: 10.1007/s00401-017-1775-x. [DOI] [PubMed] [Google Scholar]
- 165.Rossi D, et al. A mutation in the CASQ1 gene causes a vacuolar myopathy with accumulation of sarcoplasmic reticulum protein aggregates. Hum. Mutat. 2014;35:1163–1170. doi: 10.1002/humu.22631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.D’Adamo MC, et al. A calsequestrin-1 mutation associated with a skeletal muscle disease alters sarcoplasmic Ca2+ release. PLoS ONE. 2016;11:e0155516. doi: 10.1371/journal.pone.0155516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lewis KM, Ronish LA, Rios E, Kang C. Characterization of two human skeletal calsequestrin mutants implicated in malignant hyperthermia and vacuolar aggregate myopathy. J. Biol. Chem. 2015;290:28665–28674. doi: 10.1074/jbc.M115.686261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Nelson TE. Malignant hyperthermia: a pharmacogenetic disease of Ca++ regulating proteins. Curr. Mol. Med. 2002;2:347–369. doi: 10.2174/1566524023362429. [DOI] [PubMed] [Google Scholar]
- 169.MacLennan DH, Phillips MS. Malignant hyperthermia. Science. 1992;256:789–794. doi: 10.1126/science.1589759. [DOI] [PubMed] [Google Scholar]
- 170.Denborough M. Malignant hyperthermia. Lancet. 1998;352:1131–1136. doi: 10.1016/S0140-6736(98)03078-5. [DOI] [PubMed] [Google Scholar]
- 171.Robinson R, Carpenter D, Shaw MA, Halsall J, Hopkins P. Mutations in RYR1 in malignant hyperthermia and central core disease. Hum. Mutat. 2006;27:977–989. doi: 10.1002/humu.20356. [DOI] [PubMed] [Google Scholar]
- 172.Betzenhauser MJ, Marks AR. Ryanodine receptor channelopathies. Pflug. Arch. 2010;460:467–480. doi: 10.1007/s00424-010-0794-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Monnier N, Procaccio V, Stieglitz P, Lunardi J. Malignant-hyperthermia susceptibility is associated with a mutation of the alpha 1-subunit of the human dihydropyridine-sensitive L-type voltage-dependent calcium-channel receptor in skeletal muscle. Am. J. Hum. Genet. 1997;60:1316–1325. doi: 10.1086/515454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Duke AM, Hopkins PM, Calaghan SC, Halsall JP, Steele DS. Store-operated Ca2+ entry in malignant hyperthermia-susceptible human skeletal muscle. J. Biol. Chem. 2010;285:25645–25653. doi: 10.1074/jbc.M110.104976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wedel DJ, Quinlan JG, Iaizzo PA. Clinical effects of intravenously administered dantrolene. Mayo Clin. Proc. 1995;70:241–246. doi: 10.4065/70.3.241. [DOI] [PubMed] [Google Scholar]
- 176.Krause T, Gerbershagen MU, Fiege M, Weisshorn R, Wappler F. Dantrolene-a review of its pharmacology, therapeutic use and new developments. Anaesthesia. 2004;59:364–373. doi: 10.1111/j.1365-2044.2004.03658.x. [DOI] [PubMed] [Google Scholar]
- 177.Bannister RA. Dantrolene-induced inhibition of skeletal L-type Ca2+ current requires RyR1 expression. Biomed. Res. Int. 2013;2013:390493. doi: 10.1155/2013/390493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Oo YW, et al. Essential role of calmodulin in RyR inhibition by dantrolene. Mol. Pharm. 2015;88:57–63. doi: 10.1124/mol.115.097691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Protasi F, Paolini C, Dainese M. Calsequestrin-1: a new candidate gene for malignant hyperthermia and exertional/environmental heat stroke. J. Physiol. 2009;587:3095–3100. doi: 10.1113/jphysiol.2009.171967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Michelucci A, et al. Antioxidants protect calsequestrin-1 knockout mice from halothane- and heat-induced sudden death. Anesthesiology. 2015;123:603–617. doi: 10.1097/ALN.0000000000000748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chelu MG, et al. Heat- and anesthesia-induced malignant hyperthermia in an RyR1 knock-in mouse. FASEB J. 2006;20:329–330. doi: 10.1096/fj.05-4497fje. [DOI] [PubMed] [Google Scholar]
- 182.Yang T, et al. Pharmacologic and functional characterization of malignant hyperthermia in the R163C RyR1 knock-in mouse. Anesthesiology. 2006;105:1164–1175. doi: 10.1097/00000542-200612000-00016. [DOI] [PubMed] [Google Scholar]
- 183.Durham WJ, et al. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133:53–65. doi: 10.1016/j.cell.2008.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bjorksten AR, Gillies RL, Hockey BM, Du Sart D. Sequencing of genes involved in the movement of calcium across human skeletal muscle sarcoplasmic reticulum: continuing the search for genes associated with malignant hyperthermia. Anaesth. Intensive Care. 2016;44:762–768. doi: 10.1177/0310057X1604400625. [DOI] [PubMed] [Google Scholar]
- 185.Bouchama A, Knochel JP. Heat stroke. N. Engl. J. Med. 2002;346:1978–1988. doi: 10.1056/NEJMra011089. [DOI] [PubMed] [Google Scholar]
- 186.Royer L, et al. Paradoxical buffering of calcium by calsequestrin demonstrated for the calcium store of skeletal muscle. J. Gen. Physiol. 2010;136:325–338. doi: 10.1085/jgp.201010454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.MacLennan DH. Ca2+ signalling and muscle disease. Eur. J. Biochem. 2000;267:5291–5297. doi: 10.1046/j.1432-1327.2000.01566.x. [DOI] [PubMed] [Google Scholar]
- 188.MacLennan DH, Chen SR. Store overload-induced Ca2+ release as a triggering mechanism for CPVT and MH episodes caused by mutations in RYR and CASQ genes. J. Physiol. 2009;587:3113–3115. doi: 10.1113/jphysiol.2009.172155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sidman RD, Gallagher EJ. Exertional heat stroke in a young woman: gender differences in response to thermal stress. Acad. Emerg. Med. 1995;2:315–319. doi: 10.1111/j.1553-2712.1995.tb03229.x. [DOI] [PubMed] [Google Scholar]
- 190.Strazis KP, Fox AW. Malignant hyperthermia: a review of published cases. Anesth. Analg. 1993;77:297–304. doi: 10.1213/00000539-199308000-00014. [DOI] [PubMed] [Google Scholar]
- 191.Ervasti JM, Campbell KP. Dystrophin-associated glycoproteins: their possible roles in the pathogenesis of Duchenne muscular dystrophy. Mol. Cell Biol. Hum. Dis. Ser. 1993;3:139–166. doi: 10.1007/978-94-011-1528-5_6. [DOI] [PubMed] [Google Scholar]
- 192.Culligan K, Banville N, Dowling P, Ohlendieck K. Drastic reduction of calsequestrin-like proteins and impaired calcium binding in dystrophic mdx muscle. J. Appl Physiol. (1985) 2002;92:435–445. doi: 10.1152/japplphysiol.00903.2001. [DOI] [PubMed] [Google Scholar]
- 193.Zhao X, Moloughney JG, Zhang S, Komazaki S, Weisleder N. Orai1 mediates exacerbated Ca2+ entry in dystrophic skeletal muscle. PLoS ONE. 2012;7:e49862. doi: 10.1371/journal.pone.0049862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Howarth FC, Glover L, Culligan K, Qureshi MA, Ohlendieck K. Calsequestrin expression and calcium binding is increased in streptozotocin-induced diabetic rat skeletal muscle though not in cardiac muscle. Pflug. Arch. 2002;444:52–58. doi: 10.1007/s00424-002-0784-2. [DOI] [PubMed] [Google Scholar]
- 195.Das SK, Chu W, Zhang Z, Hasstedt SJ, Elbein SC. Calsquestrin 1 (CASQ1) gene polymorphisms under chromosome 1q21 linkage peak are associated with type 2 diabetes in Northern European Caucasians. Diabetes. 2004;53:3300–3306. doi: 10.2337/diabetes.53.12.3300. [DOI] [PubMed] [Google Scholar]
- 196.Fu M, et al. Polymorphism in the calsequestrin 1 (CASQ1) gene on chromosome 1q21 is associated with type 2 diabetes in the old order Amish. Diabetes. 2004;53:3292–3299. doi: 10.2337/diabetes.53.12.3292. [DOI] [PubMed] [Google Scholar]
- 197.Gopinath B, Wescombe L, Nguyen B, Wall JR. Can autoimmunity against calsequestrin explain the eye and eyelid muscle inflammation of thyroid eye disease? Orbit. 2009;28:256–261. [PubMed] [Google Scholar]
- 198.de Haan S, Lahooti H, Morris O, Wall JR. Epitopes, immunoglobulin classes and immunoglobulin G subclasses of calsequestrin antibodies in patients with thyroid eye disease. Autoimmunity. 2010;43:698–703. doi: 10.3109/08916931003774954. [DOI] [PubMed] [Google Scholar]
- 199.Kubota S, Gunji K, Stolarski C, Kennerdell JS, Wall JR. Role of eye muscle antibody measurement in diagnosis of thyroid-associated ophthalmopathy: a laboratory update. Endocr. Pract. 1998;4:127–132. doi: 10.4158/EP.4.3.127. [DOI] [PubMed] [Google Scholar]
- 200.Arai M, Otsu K, MacLennan DH, Alpert NR, Periasamy M. Effect of thyroid hormone on the expression of mRNA encoding sarcoplasmic reticulum proteins. Circ. Res. 1991;69:266–276. doi: 10.1161/01.res.69.2.266. [DOI] [PubMed] [Google Scholar]
- 201.Liu N, Rizzi N, Boveri L, Priori SG. Ryanodine receptor and calsequestrin in arrhythmogenesis: what we have learnt from genetic diseases and transgenic mice. J. Mol. Cell Cardiol. 2009;46:149–159. doi: 10.1016/j.yjmcc.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 202.Madej T, et al. MMDB and VAST+: tracking structural similarities between macromolecular complexes. Nucleic Acids Res. 2014;42:D297–D303. doi: 10.1093/nar/gkt1208. [DOI] [PMC free article] [PubMed] [Google Scholar]