

Abstract
As the field of precision medicine progresses, treatments for patients with cancer are starting to be tailored to their molecular as well as their clinical features. The emerging cancer subtypes defined by these molecular features require that dedicated resources be used to assist the discovery of drug candidates for preclinical evaluation. Voluminous gene expression profiles of patients with cancer have been accumulated in public databases, enabling the creation of cancer-specific expression signatures. Meanwhile, large-scale gene expression profiles of cellular responses to chemical compounds have also recently became available. By matching the cancer-specific expression signature to compound-induced gene expression profiles from large drug libraries, researchers can prioritize small molecules that present high potency to reverse expression of signature genes for further experimental testing of their efficacy. This approach has proven to be an efficient and cost-effective way to identify efficacious drug candidates. However, the success of this approach requires multiscale procedures, imposing considerable challenges to many labs. To address this, we developed Open Cancer TherApeutic Discovery (OCTAD; http://octad.org): an open workspace for virtually screening compounds targeting precise groups of patients with cancer using gene expression features. Its database includes 19,127 patient tissue samples covering more than 50 cancer types and expression profiles for 12,442 distinct compounds. The program is used to perform deep-learning-based reference tissue selection, disease gene expression signature creation, drug reversal potency scoring and in silico validation. OCTAD is available as a web portal and a standalone R package to allow experimental and computational scientists to easily navigate the tool.
Introduction
Many cancers are understudied because they are rare or of little public interest, such as Ewing sarcoma, a rare pediatric cancer1, and hepatocellular carcinoma (HCC), a common adult malignancy in Asia but an orphan disease in the United States2. As the field of precision medicine progresses, and we start to tailor treatments for patients with cancer who are classified not only by their clinical features but also by their molecular features (such as MYC amplification and PIK3CA mutation), more cancer subtypes are emerging. The effect of each understudied cancer or cancer subtype in healthcare might be limited, but the cumulative effects of all these diseases could be profound. Ewing sarcoma is one of over 6,000 rare diseases in the United States, affecting ~2.9 per million people1, and all rare diseases combined affect an estimated 25 million people in the United States3. HCC affects fewer than 50,000 people in the United States but is the cause of half a million deaths annually worldwide2. One common research challenge for these diseases is that the resources allocated to them are relatively limited. Compared to common conditions, the large-scale screening of compounds is often challenging, if not impossible, to perform in small labs owing to limited resources.
The decreasing cost of sequencing, however, means that it is now more common to generate gene expression profiles of samples from patients with cancer (e.g., RNA sequencing (RNA-Seq)). Integrating these profiles with the increasing amount of other available open data (such as the effect of chemical compounds on gene expression; Box 1) provides a tremendous opportunity to computationally identify new potential therapeutic candidates.
Box 1 ∣. Public data sources and repositories.
Results from laboratory experiments push scientific knowledge forward, but the raw data generated are also of particular importance. By releasing raw data into an open repository, scientists break their work out of a silo and facilitate further research and reproducibility efforts50,55. For instance, other researchers can re-analyze or combine data from many experiments into meta-analyses that are not possible with each study in isolation. This is especially important for experiments involving rare diseases or uncommonly used cell or tissue types in which data are scarce. Here we detailed a few key open repositories for both disease and drug data.
The Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) from the National Center for Biotechnology Information is a public functional genomics data repository consisting of over 3 million samples from over 110,000 studies as of September 2019.
ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) is another functional genomics dataset that has over 55 TB of data from over 70,000 experiments as of September 2019.
The Immunology Database and Analysis Portal (ImmPort; https://www.immport.org) is a collection of immunology-related studies with genomics and clinical outcome measurements.
The Cancer Genome Atlas (TCGA; https://cancergenome.nih.gov) is a compilation of cancer-related genomics data and outcomes.
The Genomics Data Commons Portal (GDC: https://portal.gdc.cancer.gov/) organizes and harmonizes TCGA and TARGET data and consists of over 350,000 files from ~33,000 cases of 69 primary site cancers (Data Release 13.0).
The Cancer Cell Line Encyclopedia (CCLE; https://portals.broadinstitute.org/ccle) details genetic and pharmacologic properties of human cancer cell line models. As of November 2018, CCLE has data for 1,457 cell lines comprised of over 136,000 datasets.
Met500 is a resource profiling whole-exome and transcriptome data from 500 adult patients with metastatic solid tumors of various lineages and biopsy sites56.
The Treehouse Childhood Cancer Initiative (https://treehousegenomics.soe.ucsc.edu/) is a resource that collects and distributes genomic and clinical data related to childhood cancers and contains over 11,000 tumor samples.
The Genotype-Tissue Expression (GTEx; https://gtexportal.org/home/) contains genotype and expression data for almost 12,000 samples across 53 tissues from over 700 healthy donors (version V7).
For drug-related data, Connectivity Map (CMap; https://www.broadinstitute.org/connectivity-map-cmap and https://clue.io/cmap) from the Broad Institute is a large database of chemical/genetic perturbations on cell lines. Specifically, CMap contains data on transcriptional expression changes due to administration of various chemical compounds on various cell lines.
To scale this project up, CMap evolved into the Library of Network-Based Cellular Signatures (LINCS; http://www.lincsproject.org/) project, where a ‘landmark gene set’ or L1000 of a 978-gene panel has been used to characterize over 1 million profiles covering various types of perturbagens (e.g., compounds, short hairpin RNA and overexpression).
Finding drugs that reverse the disease signature
Like many other investigators4-7, we use a systems-based approach where we analyze gene expression profiles of disease samples and drug-induced gene expression profiles from cancer cell lines to predict new therapeutic candidates. We have used this approach in our studies on HCC8, Ewing sarcoma9 and basal cell carcinoma10.
A disease signature is defined as a list of differentially expressed (DE) genes between disease samples and control samples (i.e., normal tissues). The essential idea is to identify drugs that reverse the gene expression signature of a disease by tamping down overexpressed genes and stimulating weakly expressed ones.
In the Ewing sarcoma study, this systems approach achieved a hit rate of >50% in predicting effective candidates9. This means that, for every ten compounds suggested in the output, more than five turned out to be effective against Ewing sarcoma in vitro.
In the HCC study, we identified deworming pills as therapeutic candidates for HCC and demonstrated that the expression of disease genes was reversed in a clinically relevant mouse model after drug treatment8. The recent pan-cancer analysis demonstrated that the reversal of cancer gene expression correlates to drug efficacy11. Compared to the commonly used target-based drug discovery approach that focuses on interfering with individual targets, this systems approach aims to target a list of critical features of the disease (Supplementary Fig. 1). The previous studies suggested that this efficient and cost-effective approach could be explored to virtually screen novel compounds or existing drugs using existing drug libraries, such as LINCS L100012-14. Looking at a wide selection of existing drugs allows the possibility of repurposing drugs where the safety, toxicology and side effects are already well understood.
We have shown that the success of this approach is made possible by multiscale procedures, such as quality control of tumor samples, selection of appropriate reference normal tissues, evaluation of disease signatures and integration of drug expression profiles from multiple cell lines. For example, the scarcity of adjacent normal tissues for many cancers (e.g., pediatric brain cancers) prevents the creation of disease gene expression signatures using traditional methodologies15. In this study, we estimated that a minimum of ten samples of adjacent normal tissue would be preferred to account for normal tissue heterogeneity. To address this challenge, we developed a deep learning (DL)-based method to select potential reference tissue samples for the selected case samples based on their expression profiles15 and implemented in (Open Cancer TherApeutic Discovery) OCTAD.
Doing this makes a substantial difference; for example, in The Cancer Genome Atlas (TCGA), an adult cancer genomic database, fewer than half of cancers have at least ten adjacent normal tissues. In the pediatric cancer genomic database TARGET, none of cancers has at least ten adjacent normal tissues. Of these tumor tissues, a substantial number of tissues are impure owing to the infiltration of stromal cells and immune cells, leading to a significant bias in the subsequent genomic analysis, including disease signature creation16,17. Some of these impure samples could be detected by correlating their expression profiles with those from cancer cell lines, which are more pure18.
There is a plethora of relevant datasets and analysis modules that are publicly available, but these are isolated in distinct silos (different databases requiring different methods for harmonization). For many labs, it would be tedious or even impossible to collate all these data to implement this approach. In this work, we describe in detail how these resources and data types can be used, as well as challenges with the process. Further, we introduce our publicly available framework and workflow, OCTAD, to streamline the various computational tasks required for the drug discovery.
We have made OCTAD available both as a standalone software package in R for bioinformaticians as well as a web server resource for investigators without a coding background. This protocol provides more detail describing the power of our approach and the novel aspects that enable more refined prediction methods. Since the publication of original key papers, we have substantially improved the protocols in many aspects, including the coverage of samples, the computing performance and the usability. In the ‘Anticipated results’ section, we demonstrate the importance of key parameters, the consistency of the results between the new version and our previously published HCC work15 and the feasibility of using the new version to predict candidates for MYC amplification lung adenocarcinoma and PIK3CA mutation breast cancer.
OCTAD pipeline
Overview of OCTAD
The system includes four main components: OCTAD Dataset, OCTAD Core, OCTAD Desktop and OCTAD Portal (Fig. 1a). OCTAD Dataset stores all sample expressions, processed using the Toil RNA-Seq pipeline, an open-source workflow software that can be used to run scientific workflows on a large scale in cloud or high-performance computing environments19. OCTAD Core includes all R functions needed for all analyses. OCTAD Desktop is an R package that can run on a regular laptop. Its customized functions allow computational biologists to perform more advanced analyses (Fig. 1b). The OCTAD Portal is the web version of the system; it has a front end based on Python Flask and HTML5, supported by the back-end OCTAD Core. We developed a simple four-step strategy to allow scientists without any programming skills to easily perform drug candidate predictions (Fig. 1c). We opted to use Python Flask and HTML5, as the portal uses advanced features, such as sample visualization, job management and parallel computing.
Fig. 1 ∣. Systems description.

(a) System design, (b) workflow for drug prediction and (c) web portal screenshot.
OCTAD Dataset
To minimize the batch effect from multiple studies, we use the same pipeline Toil developed by UCSC to process all raw RNA-Seq profiles. We estimated transcript abundance estimated from STAR (an RNA-Seq sequence aligner)20 and RSEM (for transcript quantitation)21. Because the UCSC Treehouse initiative has already used this pipeline to process samples publicly available, we use their processed samples and extend this pipeline to process new samples. The datasets processed through this pipeline were employed in our recent studies15,22.
Any new samples from the major RNA-Seq repositories, including the Gene Expression Omnibus (GEO), dbGAP and the European Bioinformatics Institute European Genome-phenome Archive (EBI EGA), can be easily processed by our pipeline. We have included samples from TCGA, TARGET, GTEx and Met50, totaling 19,127 samples covering 50 cancer types (Fig. 2 and Table 1) and will continue adding more samples from open RNA-Seq repositories. When possible, we also collected their clinical features (e.g., age, gender and tumor stage) and molecular features (e.g., mutation status, amplification and tumor molecular subtype) that allow the selection of a specific set of disease samples (Supplementary Text). In addition to tissue samples, we compiled 66,612 compound gene expression profiles consisting of 12,442 distinct compounds profiled in 71 cell lines (with 83% of the measurements made primarily in 15 cell lines), using data downloaded from the LINCS consortium. Each profile includes the expression measurement of 978 ‘landmark genes’. The changes in the expression of these landmark genes were computed after compounds were tested in different concentrations (62% of the measurements were made in conditions under 10 μM) for 24 h (49%) or 6 h (ref. 11).
Fig. 2 ∣. OCTAD cancer maps.

Each dot represents one sample colored by sample type. x and y are the first two dimensions of the t-SNE plot. normal: normal tissue samples from GTEx; primary: primary cancer samples mainly from TCGA and TARGET; adjacent: normal tissues adjacent to primary cancer; metastatic: tissue samples from the metastasized site; recurrent: recurrent cancer tissue samples.
Table 1 ∣.
Patient sample statistics
| Database | Tissue type | n | ||||
|---|---|---|---|---|---|---|
| Normal | Adjacent | Primary | Recurrent | Metastatic | ||
| St. Jude HGG | 0 | 0 | 66 | 0 | 0 | 66 |
| GTEx | 7,412 | 0 | 0 | 0 | 0 | 7,412 |
| Met500 | 0 | 0 | 0 | 0 | 387 | 387 |
| TARGET | 0 | 11 | 602 | 120 | 1 | 734 |
| TCGA | 0 | 726 | 9,365 | 44 | 393 | 10,528 |
| Total | 7,412 | 737 | 10,033 | 164 | 781 | 19,127 |
Reference tissue selection
Case samples could be manually defined based on disease clinical and molecular features. Selecting appropriate corresponding normal samples is essential for creating a disease signature. In OCTAD, users can choose adjacent normal tissue samples as control, whereas plenty of cancers, such as brain cancer, have no or insufficient adjacent tissue samples. From Table 1, it is clear that there are not enough data for adjacent normal tissues, but there are a lot of data for normal tissue samples from GTEx, a repository of tissue samples from healthy individuals. Owing to variation of normal tissue samples, only some of them are appropriate to serve as case samples15. We could compute per-gene interquartile range and choose top varying genes to calculate the Spearman correlation between each case sample and all normal samples, although selecting top varying genes might ignore many critical genes. Top features derived from principal component analysis (PCA) could also be used, although PCA might not capture the non-linear relationship between genes. Autoencoder, a type of artificial neural network used to represent input data in an unsupervised manner, can capture non-linear relationships between input features and normalize input data, presenting unique advantages in handling such high-dimensional expression data. Our previous work demonstrated the utility and advantage of autoencoder in control sample selection and the feasibility of adopting highly correlated normal samples taken from the different tissue of origin15. Accordingly, in OCTAD, we use new features encoded by DL autoencoder to select highly correlated normal samples given a set of disease samples.
By default, the top 50 control samples that are mostly correlated to the case samples are selected for further analysis. Control sample selection can be customized in the desktop version. For example, advanced users can compare metastatic cancer samples and primary cancer samples, or TP53 mutation and wild-type samples, although the feasibility of predicting drugs for these types of comparisons needs to be evaluated individually. It is rare, yet possible, that none of normal samples is highly correlated with the case samples; users are encouraged to examine the absolute correlations from the result file.
Disease signature creation
A disease gene expression signature is defined as a list of DE genes between a specific set of case samples and matched reference normal samples. Because case and control samples are often from different studies, raw counts are first normalized through RUVSeq23, where a set of empirical negative control genes (least significantly DE genes based on a first-pass DE analysis) that are assumed to have constant expression across samples are used to adjust for technical effects.
With two groups of samples, standard DE analysis methods (e.g., edgeR24, limma voom25 and DESeq2 (ref. 26)) could be performed, followed by pathway enrichment analysis through the Enrichr API27. Pathway enrichment analysis allows users to examine critical pathways or biological processes associated with dysregulated genes from the disease signature (Box 2). The comparison of the DE methods was performed elsewhere28. EdgeR, one of the most popular and fastest methods for DE analysis, is the default, whereas limma, which uses quantile normalization and is extremely useful for microarray analysis, is another option. Because of the low efficiency, DESeq2 is supported only in the R package. Popular databases, including KEGG and GO, are used as default in the enrichment analysis.
Box 2 ∣. Enrichment analysis.
Pathway enrichment analysis. The process of finding biological pathways that are enriched in a set of genes (i.e., upregulated or downregulated disease genes) more than random chance. OCTAD uses the results of enriched GO terms and KEGG pathways computed from Enrichr. The significance computation in Enrichr is based on a hypergeometric test.
Drug enrichment analysis. The process of finding drug classes that are enriched in the top predicted drugs more than random chance. OCTAD has incorporated the following drug classes: 1,072 drug targets, 2,695 structure clusters and 226 MeSH pharmacological terms. The enrichment score is computed by ssGSEA, and significance is computed by a permutation test.
Reversal of cancer expression
In our earlier studies, we quantified the reversal of disease gene expression as the Reversal Gene Expression Score (RGES)11, a measure modified from the connectivity score developed in other studies4,12. To compute the RGES, we first rank genes based on their expression values in each drug signature. A score for each set of upregulated and downregulated disease genes is computed separately using a Kolmogorov–Smirnov-like statistic, followed by the merge of scores into an RGES from both sides (up/down). The RGES is based on the number of the upregulated (or downregulated) genes enriched at either the top (or bottom) of a drug–gene list ranked by expression change after drug treatment. A negative RGES means upregulated disease genes are downregulated in the drug profile, and/or downregulated disease genes are upregulated in the drug profile. One compound might have multiple available expression profiles owing to having been tested in various cell lines, drug concentrations, treatment durations or even different replicates, resulting in multiple RGESs for one drug–disease prediction. Therefore, we developed a summarization method to mitigate bias and to compute a score representative of the overall reversal potency of a compound to a particular cancer. We refer to this score as sRGES.
sRGES
sRGES is calculated using the following equation:
where N is the number of drug profiles. f(dose(i), time(i)) was estimated by a computational model. Correlation between cell(i) and tumor samples was estimated as the average of correlations between the cell line and individual tumors. The maximum correlation between cell lines and tumor samples can be used to normalize correlation. We set a reference condition (i.e., concentration of 10 μM and treatment duration of 24 h) and used a model to estimate a new RGES if the drug profile under the reference condition was not available. We then weighted the RGES by the degree of correlation between the gene expression profiles of the disease and the cell line in which the compound was tested.
We demonstrated that sRGES is correlated to drug efficacy (measured by log(IC50 nm) or area under the curve (AUC) derived from the dose response curves), and such correlation is retained even when the disease is not represented by cell lines of its own lineage in the drug expression databases. The analyses suggested the feasibility of applying this approach for large-scale screening of compounds for a given disease signature.
! CAUTION At the moment, it is better to calculate sRGES using ranked genes rather than the absolute magnitude of expression changes. Previous attempts to use any form of absolute expression magnitude have resulted in less robust results11. While computing sRGES, restricting the cell lines to the cancer of the same lineage used by LINCS L1000 causes significant loss in the number of drugs evaluated and has not been shown to improve the specificity of results11.
Hit prediction and selection
Previous drug repositioning efforts considered only a couple of thousand Food and Drug Administration (FDA)-approved drugs with more potential to translate into the clinic, leaving over 10,000 compounds in LINCS unused for a broad chemical space for discovery (Fig. 3a; a two-dimensional t-distributed stochastic neighbor embedding (t-SNE) projection of the compounds based on their chemical structural similarity). Three compound structures are displayed to show the structural diversity and complexity of the compound collection. Including those unused compounds might increase the chance of discovering novel compounds. Of these compounds, 14% are commercially available in ZINC29, which is one of the largest collections of commercially available compounds (Fig. 3b). An additional 5% of compounds are structurally similar to ZINC compounds (similarity >0.9), leaving >80% of compounds that are not directly purchasable (Fig. 3b). According to synthetic accessibility scores30, 70% of these inaccessible compounds can be easily synthesized (Fig. 3c). This protocol added several enrichment analyses of drug hits, including enriched Medical Subject Headings (MeSH) terms, protein targets and chemical scaffolds (Box 2). MeSH pharmacological classification and protein targets of LINCS compounds were retrieved from PubChem31 and ChEMBL32, respectively (Fig. 3d,e). Chemical scaffolds of LINCS compounds were created using RDkit (Supplementary Materials and https://www.rdkit.org). We expect such information will facilitate the selection of representative compounds that could be quickly obtained for testing.
Fig. 3 ∣. OCTAD compounds.

a, t-SNE plot of LINCS L1000 compounds colored by synthetic accessibility. x and y axes show t-SNE dimension 1 and 2 projection of samples based on their structural similarity. Each point indicates a compound, and the color shows synthetic accessibility (SA; see the color bar at the bottom). The lower SA score means that it is easier to synthesize the compound. This dataset covers both the traditional drug-like chemical space (purple dots) and novel scaffolds (green dots). Three compounds are highlighted as examples: the one in the middle is benzocaine (SA = 1.45, easy to synthesize); the lower left one is BRD-K00278564 (SA = 3.82, moderate to synthesize); and the upper right one is BRD-K96551169 (SA = 5.41, difficult to synthesize). b, Reagent accessibility (in stock: available in ZINC; similar in stock: structurally similar to the compounds in ZINC; easy to synthesize: synthetic accessibility score <4; hard to synthesize: synthetic accessibility score ≥4). c, Synthetic accessibility score distribution. y shows the density of the number of compounds. It indicates that the compounds in stock have lower synthetic accessibility scores than those for compounds thar are not commercially available. d, MeSH pharmacological classification distribution. x shows the number of compounds per one MeSH term, and y shows the number of MeSH terms. e, Compound target distribution. x shows the number of compounds per one target, and y shows the number of targets. d and e indicate that most MeSH terms and targets are associated with a small set of compounds.
Selection of cell lines and in silico validation
The efficacy of compounds is often first evaluated in cancer cell lines. The transcriptomic comparison of cell lines with disease samples could be employed to select appropriate cell lines to use for the following efficacy validation18,22,33. The emerging large-scale drug sensitivity (or efficacy) data across a variety of cancer cell lines34,35 even enable the validation of predictions using published experimental data without biological experiments. Here we use the transcriptome profiles to select a cell line related to case samples and then leverage published drug sensitivity data of the selected cell line to validate the prediction. We rank transformed gene reads per kilobase of transcript, per million mapped reads (RPKM) values for each CCLE36 cell line and then ranked all the genes according to their rank variation across all CCLE cell lines. The 1,000 most-varied genes were kept as ‘marker genes’. (In addition to 1,000 genes, we explored multiple gene sizes in the early preliminary analysis and did not find the large variation of correlated cell lines, so, in this study, we chose 1,000 most-varied genes.) Given RNA-Seq profiles of a cell line and case samples, we compute the Spearman rank correlation (across the 1,000 marker genes) between the cell line and each case sample. The median value of computed Spearman rank correlation values is defined as the transcriptome similarity between the cell line and the case samples. If the drug sensitivity database Cancer Therapeutics Response Portal (CTRP)35 provides the sensitivity data of the LINCS compounds in the selected cell line, OCTAD allows you to pull out this drug sensitivity data and correlate them with sRGES to evaluate drug predictions. A significant correlation with experimental data would increase the confidence of investigating other drug hits that have not been tested.
Overview of OCTAD Desktop and Portal
To enable users to make use of our pipeline, we release both a freely available and open-source web portal and a workflow in a computational pipeline. The web portal runs many of the OCTAD core functions in the back end but requires no programming expertise. It allows users to perform all key parts of the pipeline, including selecting case and control samples, performing DE analysis to generate a disease signature and generating drug candidates. To make the process as efficient as possible, users can register for the web server, and the various parts of the pipeline can be saved as jobs that will be saved for future visits. The portal assigns each job with a permanent URL and also allows the submission of an anonymous job and the uploading of disease signatures computed from elsewhere. The web server is interactive and produces informative plots and tables that users can interact with and download. The web server also incorporates some, but not all, advanced features of the pipeline, including autoencoder-recommended control sample selection. The full set of features of the web server can be found in both the ‘Procedure’ section and the Supplementary Materials.
The desktop version (R package) can not only perform all of the above components but also provides more flexibility and features. The computational pipeline is built in the R framework and incorporates publicly available Bioconductor and R packages for processing and analyses. Advanced users can perform large-scale drug predictions and explore multiple control selection methods, and it includes in silico validation. We provide a breakdown of the R pipeline in the ‘Procedure’ section and the Supplementary Materials.
Availability
The web portal can be used by anyone without programming expertise or extensive domain knowledge. Users with genetics and molecular biology knowledge will find the results highly interpretable. To use the desktop version, a user will need to have basic skills in R and Bioconductor packages and have knowledge of genomics data. The web portal is available at http://octad.org. The R package.tar.gz, datasets and a tutorial can be accessed at the download page (http://octad.org/download). Alternatively, the files can be accessed here: https://www.synapse.org/#!Synapse:syn22101254/files/.
To reduce the R package to a manageable size, the desktop version, by default, includes expression for 978 genes from the LINCS database to compute sRGES and/or differential gene expression. However, DE analysis using the reduced set might result in bias because count normalization is performed in a smaller gene set. Users are advised to download the entire dataset from the download page (with an h5 format, >2G).
Alternative methods
The related datasets and analysis modules are publicly available but isolated in distinct silos. Genomic data of patients with cancer could be searched and visualized in platforms such as cBioPortal37, Oncoscape38 and TumorMap39. Massive RNA-Seq samples are processed in platforms such as Treehouse19, Rcount40 and ARCHS441. Disease signatures could be created by R packages such as edgeR24 and DESeq2 (ref. 26). A comprehensive enrichment analysis of disease signatures could be performed in Enrichr27 and DAVID42. Given a disease signature, clue.io could predict drug hits using LINCS data13. To be able to predict drugs using public RNA-Seq profiles, researchers have to use different platforms and various tools to accomplish the task. To address this issue, we developed a portal to streamline this process. We provide an agile desktop version that allows computational scientists to customize the code and a web portal version that allows bench scientists and clinicians to easily navigate and predict drug hits.
There are also several existing web resources, tools and applications that can perform somewhat similar procedures and analyses. Rnama (https://rnama.com) is a freely available web application that allows for meta-analysis of publicly available RNA data, seamlessly extracted from the GEO. Comparing case and control groups can be interactively performed, which results in an interactive plot of DE genes. DEBrowser is an R shiny-based package that creates an interactive dashboard to facilitate DE analysis and visualization of RNA count data43. Users can upload their own data, and the application allows for multiple quality control steps and visualization. DrugSig is a web resource that allows for prioritizing potential drug repurposing opportunities and submitting upregulated and downregulated genes from pre-computed disease DE profiles44. RE:fine drugs is an interactive dashboard that pre-calculates potential drug repurposing opportunities, combining information from previously published genome-wide association studies and PheWAS results45. Users can search for a drug name, disease name or gene symbol to see suggestions based on these levels of evidence. DeSigN is an interactive web tool that allows for prediction of drug efficacy against cancer cell lines46. In this application, users can enter a list of upregulated and downregulated genes to be compared against IC50 values to prioritize potential drugs. Drug Gene Budger is a web tool and mobile app to interactively rank drugs to modulate user-specified genes based on transcriptomic profiles47. With this, users can search for a specific gene, and the tool prioritizes drugs to either upregulate or downregulate them using CMAP, LINCS L1000 or CREEDS data. Although the aforementioned tools and applications are indeed useful, none can perform the full gamut of steps necessary for this process. Furthermore, the flexibility of our application, the incorporation and integration of multiple datasets, along with enhancements and incorporation of novel methodologies (e.g., autoencoder reference normal sample identification) makes OCTAD truly a unique and powerful resource.
Many other computational resources and repositories demonstrate the power of drug repurposing. The Drug Repurposing Hub contains an app that allows for dynamic search and exploration of annotated information pertaining to over 8,000 compounds, including targets, mechanism of action and even vendor information14. RepurposeDB48 and repoDB49 collect and curate information about known drug repurposing experiments. These, together with studies from other researchers, demonstrate the feasibility of applying a systems approach to screen drug hits in cancers.
Advantages, limitations and future directions
Our pipeline has several advantages. First, OCTAD covers nearly 20,000 open RNA-Seq samples from multiple sources processed in the same computational pipeline, so that any pipeline effect is minimized. Coupling with the robust control sample selection module makes it possible to predict drugs for cancers or cancer subtypes with no empiric controls. Second, the one-stop drug prediction web portal allows clinicians and bench scientists who might not have sufficient programming expertise to run the various computational tasks necessary to prioritize drug hits for further experimental validation. Third, the flexible desktop-based R package allows advanced users to perform customized drug discovery computations. Fourth, collected molecular and clinical samples enable precise stratification of patient samples and prediction of drug candidates for subsets of patients. Fifth, our unique DL-based models enable appropriate selection of normal samples. Finally, an optimal outcome is generated thanks to the rigorous quality control in each step (e.g., in silico validation of drug hits).
Despite these advantages of OCTAD, a few issues remain to be addressed in future versions. First, the application is limited by the quality and structure of the input data, and some data sources provide more information than others, which restricts search functionality. For instance, TCGA provides a much more comprehensive coverage of clinical and molecular features than many individual studies. Similarly, these various datasets have different nomenclatures in which phenotypes and diseases are characterized. In future iterations, we will perform more intensive harmonization of these labels using common data model ontologies. For now, users will have to curate their selections based on necessity, but the application provides the framework to do so. Additionally, this pipeline is focused only on cancer. We envision that this application could extend easily to other phenotypes. In future iterations of the web application, we will allow for more seamless integration of data from other sources and repositories, such as the GEO, the Sequence Read Archive, the EBI EGA and Treehouse. Furthermore, OCTAD uses only one repurposing methodology (reversal of gene expression) and one molecular datum (gene expression). Finally, the pipeline has been validated only for several cancers; as with all drug repurposing in silico exploration, predictions will have to go through extensive biological and clinical validation experiments to verify utility and efficacy.
Gene expression captures only one aspect of biological systems. With the rapid advances of ‘omics’ technologies, we envision that the system that we developed will greatly facilitate the use of other omics data (proteins, metabolites and single cells) in future therapeutic discovery50. As indicated, identifying therapeutic treatments involves multiple biological systems, and, as such, it is only natural that the drug discovery or repurposing process should involve multiple data types across domains. The types of data that exist to broadly assess therapeutics with phenotype are vast and cross several biological domains. These include, but are not limited to, genome, transcriptome, proteome, metabolome, epigenome and microbiome. In the space of drug discovery and repurposing, it is important to look at these domains not only across different cell types and tissues but also under different time points and conditions, particularly when exposed to drugs. There are also many models in which to perform such experiments, including animals (e.g., rodent and zebrafish), cell lines, organoids, xenografts and tissues.
Materials
For the web server (http://octad.org): developed and tested in Google Chrome
For the desktop version:
R v.4.0.0 or newer (https://www.r-project.org)
RStudio (https://www.rstudio.com/)
Download and install ‘octad’ package with all requisitions from the GitHub directory https://github.com/Bin-Chen-Lab/octad
Whole OCTAD expression can be downloaded from the web portal or directly via https://chenlab-data-public.s3-us-west-2.amazonaws.com/octad/octad.counts.and.tpm.h5
Whole combined tutorial link: https://chenlab-data-public.s3-us-west-2.amazonaws.com/octad/octad_tutorial.pdf
Code with five examples: https://chenlab-data-public.s3-us-west-2.amazonaws.com/octad/octad_example.R
Required hardware for the desktop version:
Computer with ≥8 GB RAM
Hard drive with ≥10 GB free
A stable broadband internet connection
Procedure 1
Desktop version
▲ CRITICAL We illustrate the utility of the desktop pipeline by highlighting a use case for HCC. We provide code and data for investigating DE, pathway enrichment, drug prediction and hit selection and in silico validation using an external dataset. In this workflow, we will select case tissue samples from our compiled TCGA data and compute control tissues from the GTEx data.
Note that our compiled data also contain adjacent normal TCGA HCC samples that can also serve as control tissues. More detailed description of functions and result files is illustrated in Fig. 4.
Fig. 4 ∣.

Key steps of the desktop pipeline.
The links used for setting up are listed in the ‘Materials’ section.
Setup
Install required libraries. Refer to the script to install required R packages that can be found in the ReadMe file from the GitHub directory https://github.com/Bin-Chen-Lab/octad.
Customize the Setup folders. By default, the octad package uses the Small OCTAD dataset containing expression values only for LINCS landmark genes required for sRGES score computation. To download the full expression values, refer to the links to the whole expression dataset. By default, computation results of the pipeline are stored in the working directory.
First-time setup and loading the package
-
3
Before running the pipeline for the first time, install the required packages by typing the following in the R command:
packages=c(‘magrittr’, ‘dplyr’, ‘ggplot2’, ‘doParallel’, ‘foreach’, ‘lme4’, ‘Rfast’) if (length(setdiff(packages, rownames(installed.packages()))) > 0) { install.packages(setdiff(packages, rownames(installed.packages()))) } -
4
Install the required Bioconductor packages:
bioconductor_packages=c(‘edgeR’, ‘RUVSeq’, ‘DESeq2’, ‘limma’, ‘rhd- f5’,‘artMS’) if (length(setdiff(bioconductor_packages, rownames(installed. packages()))) > 0) { if (!requireNamespace(“BiocManager”, quietly = TRUE)) install.packages(“BiocManager”) BiocManager::install(setdiff(bioconductor_packages, rownames (installed.packages()))) -
5
Install the OCTAD version and octad.db that was downloaded. Note that, because the package contains lots of pre-compiled data, it might take ~10 min to install.
install.packages(“https://chenlab-data-public.s3.amazonaws.com/ octad/octad.db_0.99.0.tar.gz%3Fdl%3D0”, method=“libcurl”,repos=NULL,type=“source”) devtools::install_github(‘Bin-Chen-Lab/octad’,build_vignettes = TRUE)
-
6
When the package is installed, load it into R:
library(“octad”)
The package will first check dependable packages and then load necessary packages and datasets. After this step, the pipeline is ready to run.
Case and control samples
▲ CRITICAL Choosing which cases (tumor samples from the phenoDF data.frame) and controls (corresponding samples treated as background samples—e.g., normal tissue, adjacent normal tissue or tumor samples without mutation) to use are the two most important factors in achieving the best results when using this pipeline. Several methods included in the provided code evaluate controls relative to cases, but there are no built-in validation steps that evaluate cases. Each group of cases needs to be evaluated individually for validity by the investigator. Visualization of cases in a t-SNE plot could help understand their relations with other OCTAD samples. Samples sharing similar transcriptomic profiles tend to cluster together in the t-SNE plot. The cases scattering in multiple clusters are not recommended to choose as a group.
▲ CRITICAL The case_id and control_id variables must be simple character vectors containing sample IDs. They are most easily generated by subsetting the metadata matrix phenoDF, but advanced users may assemble them using other means (including querying cBioportal or the NCI GDC portal).
Select case samples
-
7
Phenotype data contains tissue types, such as normal tissue, adjacent normal tissue, primary cancer, recurrent cancer and metastatic cancer. We will select for primary HCC. To list all available samples from the OCTAD database, use the phenoDF data.frame.
To select HCC samples, use the code below:
head(phenoDF) HCC_primary=subset(phenoDF,cancer==‘liver hepatocellular carcino- ma’&sample.type == ‘primary’) #select data case_id=HCC_primary$sample.id #select cases
The sample IDs will be stored in the character vector case_id.
This code can be easily modified to select other cancers or a set of samples based on mutations and copy numbers (e.g., TP53 mutation or MYC amplification). It is also recommended to use the R package cgdsr to select TCGA samples based on more clinical and molecular features.
#choose breast invasive carcinoma samples with PIK3CA mutation BIC_with_PIK3_primary=subset(phenoDF,cancer==‘breast invasive car- cinoma’&sample.type == ‘primary’ &grepl(‘PIK3CA’,mutation_list))
Compute or select control samples
-
8
Use the function computeRefTissue to compute appropriate normal tissues via comparing gene expression features between case samples and normal tissue samples. Users can select adjacent normal tissue samples if available. By default, features from the precomputed autoencoder file are used, but other features, such as top varying genes across samples, can be employed as well. Pairwise Spearman correlation is computed between every case sample and every normal sample using these features. For each normal sample, its median correlation with all case samples is then computed. Top correlated normal samples (defined by control_size) are then selected as control.
#computing top 50 reference tissues control_id=computeRefTissue(case_id,outputFolder=“,output=T,adja- cent=T,source = “octad”,control_size = 50)
-
9
The list of normal IDs is stored into the variable control_id. Use the following code to select adjacent control samples from phenoDF. This code can also be used to select any set of OCTAD samples (including cancer samples) as control.
HCC_adjacent=subset(phenoDF,cancer==‘liver hepatocellular carcino- ma’&sample.type == ‘adjacent’&data.source == ‘TCGA’) control_id=HCC_adjacent$sample.id
-
10
Figure 5a shows the top 50 normal tissues, highlighted in red, that have the highest median correlation with HCC tumors. Check the absolute correlations from this plot or the case_normal_median_cor.csv file to assess the relevance of the control samples. By default, we consider a correlation coefficient >0.285 to be significant according to the background distribution (Supplementary Fig. 2). It is rare, but possible, that none or few control samples in GTEx are highly correlated to case samples. If this happens, users should not proceed.
-
11
The relationships among case, control and other samples are visualized in Fig. 5b. To generate this type of image, use the following code and pre-computed t-SNE matrix. We applied the t-SNE algorithm to the whole OCTAD database for computation of the distance between all samples in the phenoDF for increased visualization timings. t-SNE is a non-linear dimensionality reduction technique that is particularly well suited for the visualization of high-dimensional datasets.
tsne$type <- “others” tsne$type[tsne$sample.id %in% case_id] <- “case” tsne$type[tsne$sample.id %in% control_id] <- “control” #plot (p2 <- ggplot(tsne, aes(X, Y, color = type)) + geom_point(alpha = 0.4) + labs(title = paste (‘TNSE PLOT’), x= ‘TSNE Dim1’, y=‘TSNE Dim2’, caption=“OCTAD”)+ theme_bw())
Fig. 5 ∣. Screen compounds targeting HCC.

a, Correlation between HCC tumor samples and all samples from normal tissues. Highly correlated samples (colored by red) were selected as control. b, Distribution of the samples selected for the HCC study in a cancer map. c, Disease signature visualization. Log TPM value is used. Rows are disease genes; columns are case and control samples. Red and blue show highly and weakly expressed genes, respectively. d, Top compounds that reverse the disease signature. The first column shows a disease signature gene expression; the remaining columns show drug signatures that reverse expression of the corresponding genes. In the first column, red and blue indicate high and low gene expression compared to control samples, respectively. In the remaining columns, red and blue indicate high and low gene expression induced by drug treatment, respectively. e, Correlation between sRGES (predicted score) and drug efficacy data in vitro. f, Enriched drug class. Drugs belonging to the antimetabolite and anti-neoplastic classes are represented as black bars. Lower sRGES means higher potency.
Compute DE genes between case and control samples
-
12
DE can be computed via edgeR, limma + voom or DESeq2. By default, we use edgeR in the analysis. Because the function diffExp computes DE genes between case_id and control_id within the same data matrix, it can be used to find DE genes between any two groups of samples. By default, a small dataset containing only 978 genes shared with the LINCS database is used.
res=diffExp(case_id,control_id,source= ‘octad.small’,output=T)
If you need to use the whole OCTAD dataset as input (see ‘Materials’), make sure that the required h5 file is stored in the R working directory or that the whole path to the file is specified:
res=diffExp(case_id,control_id,source=‘octad.whole’,output=T, n_topGenes=10000,file=‘octad.counts.and.tpm.h5’)
We can also perform DE analysis using an external dataset. Below is an example of how to perform DE analysis between tumor and non-tumor samples using the count data downloaded from the GEO (GSE144269).
data=read.table(‘GSE144269_RSEM_GeneCounts.txt’,header=T,row. names=1) data=as.matrix(data) data=log2(data) #log-convert gene expression for edgeR computation data[is.infinite(data)]=0 #remove infinite numbers samples=colnames(data) #define the case and control cohorts, A samples were obtained from tumors, B samples were obtained from adjacent tissue case_id=samples[grepl(‘A_S’,samples)] control_id=samples[grepl(‘B_S’,samples)] res=diffExp(case_id,control_id,source=‘side’,output=T,n_top- Genes=10000,expSet=data,annotate=F) #compute DE
▲ CRITICAL STEP If you are using a customized dataset, make sure that it is a matrix with rows containing Ensembl gene names. After DE analysis, diffExp() will annotate every expressed gene using Entrez Gene symbols.
Batch normalization
-
13
Perform batch normalization. By default, the normalization and batch correction step is performed (with option normalize_samples = TRUE) that uses RUVSeq to normalize samples so that batch effects between studies can be minimized37. The parameters k and ntop_genes are required in RUVSeq23 if normalize_samples is set to TRUE. These options are used to compute an empirical set of control genes via edgeR (stored in computedEmpGenes.csv). We recommend this step when using samples from different resources (e.g., TCGA cancer and GTEx normal). The disease signature is visualized in a heat map (Fig. 4c).
Compute reverse gene expression scores
-
14
The runsRGES function is used to identify the drugs that potentially reverse the disease signature. Use the code below to choose significant genes; this works by keeping genes that have low adjusted P values and high log-fold changes.
res=subset(res,abs(log2FoldChange)>1&padj<0.001)
-
15
Launch the sRGES computation. It takes a few minutes to compute RGESs. After the job is finished, it will output files all_lincs_score.csv (RGES of individual profiles), sRGES.csv (summarized RGES of individual drugs) and dz_sig_used.csv (signature genes used for drug prediction). Figure 5d is a sample output of drugs that are in clinical trials or are FDA approved. LINCS also provides the imputed expression of the whole transcriptome based on the 978 genes. We will add it in the future when its usage is fully evaluated.
sRGES=runsRGES(res,max_gene_size=100,permutations=10000)
-
16
Identify the hits by choosing those that have sRGESs lower than −0.2.
▲ CRITICAL STEP Using sRGES lower than −0.2 is a method found to produce the best results in a handful of validation cases. There is currently no known superior way to compare hits to aid in lead optimization. Similarly, comparing the magnitude of sRGES results across runs is inappropriate because sRGES is highly dependent on the size of disease genes.
Validate results using published pharmacogenomics data (optional)
-
17
As the pharmacogenomic database CTRPv2 consists of efficacy data of 481 drugs in 860 cancer cell lines35,51, we might leverage this database for further in silico validation of our predictions, even without running any biological experiments. We use the HepG2 cell line to validate the prediction of HCC drugs. In our previous work, we showed that RGESs correlate with drug efficacy, such as AUC or IC50 (ref. 11).
To perform this analysis, run the following chunk:
cell_line_computed=computeCellLine(case_id=case_id,returnDF=T, source=‘octad.whole’,file=‘octad.counts.and.tpm.h5’) topLineEval(topline = c(‘HEPG2’),mysRGES = sRGES)
computeCellLine will produce an object with correlation scores for every cell line and case samples (stored as CellLineCorrelations.csv).
topLineEval will produce CellLineEval*_drug_sensitivity_insilico_results.txt and two .html documents:
*_auc_insilico_validation.html (correlation between drug AUC in the specified cell line and sRGES).
*_ic50_insilico_validation.html (correlation between drug IC50 in the specified cell line and sGRES).
-
18
Validate the predictions using a linear regression model (Fig. 5e). Note that we could use this analysis to optimize the pipeline for the disease of interest by changing various hyperparameters. We expect that a good prediction should be highly correlated with drug sensitivity in a related cell line.
Compute drug enrichment (optional)
-
19
After calculation of sRGES on the L1000 compound dataset, perform drug enrichment analysis to identify interesting drug classes whose member drugs are significantly enriched at the top of the prediction. Example drug classes include anti-inflammatories, EGFR inhibitors and dipines (calcium channel blockers). We combine LINCS drugs into three lists: MeSH, CHEMBL and CHEM_CLUSTER for MeSH term enrichment, target enrichment and chemical structure enrichment, respectively. The enrichment score is calculated using ssGSEA52, and its significance is computed by a permutation test. Figure 5f shows the enrichment of anti-metabolites (drugs that interfere with one or more enzymes or their reactions that are necessary for DNA synthesis) and anti-neoplastics (drugs used to treat cancer) in the top-ranked drugs, suggesting that members of this drug class are more likely drug hits.
octadDrugEnrichment(sRGES = sRGES, target_type = c(‘chembl_tar- gets’,‘mesh’,‘ChemCluster’))
This analysis provides much information for the following candidate selection and experiment design. First, the candidates selected from the same enriched class (i.e., MeSH term and target) are more likely to be true positive than those randomly selected from the top list. Second, when the ideal candidate is not available, it is reasonable to choose an alternative from the same class. Sometimes, it is necessary to choose a new drug for testing (e.g., a second generation of one inhibitor for the same target). Lastly, because many compounds have multiple mechanisms of action (MOAs), this analysis would help interpret the MOA of promising compounds.
Procedure 2
Web server version
Portal landing page/login
At the landing page, create a new account, log into an existing account or go straight to submitting a job (Extended Data Fig. 1). Although having an account is not required to submit the job, we recommend setting one up as it will save all active and past jobs. User account settings can be accessed on the top right of the header. In the main portal, a user’s Job History can be found in the tab on the side menu, where all jobs are listed with information pertaining to disease of interest, status (e.g., Completed and In-Progress), creation time, as well as the access to view and download all output. Users can also delete previous jobs. At the bottom of the screen, the details of the current job can be found by clicking the Summary button; the current job can be saved by clicking the Save button; and navigating forward or backward in the process can be done by clicking the Previous and Next buttons, respectively.
Creating a new job: HCC
▲ CRITICAL Creating a job is separated into four sections: Case, Control, Disease Signature and Drugs.In this procedure, we will go through all steps along with expected output. We will highlight HCC as our featured example.
Case selection
-
2
Upon entering the Case section, all samples available from all included database resources (see ‘Materials’ section; n =19,127) are displayed in a table at the bottom of the page, including cancer type (Cancer), site of derivation (Site), gender, age, cancer subtype, tumor stage, mutation, gain of function (copy number) and loss of function (copy number). Spend some time familiarizing yourself with and exploring this page. The user is able to interact with this table, such as searching in the top right and viewing more information about the samples by clicking the green ‘+’ button under More Info, which shows information such as patient demographics and status for certain mutations. Users can manually select samples to include as well by selecting the check box at the left of the sample row.
▲ CRITICAL STEP For the first time loading, we recommend waiting ~20 s before starting to select case samples, as it takes some time for the browser to download the data from the server. One sign of complete loading is that all samples should be automatically selected in the table during sample selection.
-
3
To begin a job, search for cancers of interest in the search box at the top of the page beside Disease Name; multiple diseases can be selected.
-
4
Refine your search by adding filters (e.g., gender, tissue type and a known mutation status); doing this will automatically update the selected samples below.
-
5
Manually add and remove samples by checking or unchecking rows in the table, respectively.
For the example shown in this protocol, start the search by typing ‘hepatocellular carcinoma’ into the Disease Name search box and choose the corresponding option. After filtering for tissue type, we are left with 369 primary cancer samples.
-
6
Proceed to select Control samples in the next section by clicking the Next button on the bottom of the screen.
Control selection
-
7
Click ‘compute control samples’. The portal will ask whether adjacent samples should be included if available and how many control samples should be chosen. The portal will recommend control samples automatically.
For the example in this protocol, if the DL method is used (which we recommend), we are left with 50 samples. These Control samples are highlighted along with all other normal samples.
-
8
By default, adjacent samples are included for computation. To remove them from the list of potential control samples, uncheck the box ‘adjacent’ before computation. Depending on the user’s goals, this can be a good way to visually verify whether the current selections are adequate.
-
9
Continue to the Disease Signature section by clicking the Next button on the bottom.
Disease signature generation
! CAUTION Sometimes users might want to use a disease signature computed elsewhere. In this case, the signature can be uploaded in the page ‘Upload data’. The expression data must be submitted in a .csv format with the following columns: Symbol containing uppercased HGNC gene symbol and log2FoldChange containing numbers with DE values. An example file is available at the bottom of the page.
-
10
With the Case and Control samples selected, we can now create the disease signature of interest. This section allows for multiple methods to accomplish this, specifically using either edgeR or limma. The portal does not include DESeq2, which takes even longer than edgeR or limma. For this example job, select edgeR and press ‘View signature’ to begin. As indicated by the warning pop-up, this task can take a few minutes, so one benefit of creating an account is that the user can log out while this process is occurring. It is recommended to do this step to quickly evaluate disease signatures (heat map of disease gene expression and pathway enrichment analysis). For example, if case and control samples are not well separated in the heat map, users might want to adjust samples. Users can skip this step and proceed with the job submission by clicking ‘Next’ at the bottom left of the page. In this case, signature creation will be automatically performed before drug prediction.
-
11
Once the signature generation is finalized, this section produces a heat map of DE genes as well as a table below with these data. Explore the data by setting and changing the criteria metrics; two examples of criteria to change are the P value and fold-change cutoffs. Changing these will affect the resulting plots and tables. The heat map and table can be found in the output files for reference.
-
12
This section will provide pathway enrichments (GO terms and KEGG pathways) of both upregulated and downregulated genes in the signature as both table and bar plots.
-
13
Once satisfied with the disease signature that has been generated, proceed to the final stage of the job to calculate the RGES for all available drugs by clicking the Next button on the bottom panel.
Candidate drug selection
-
14
In this section, we can compare the disease signature to all drug signatures obtained from the LINCS resource.
Job submission and tracking
-
15
After submitting the job, an encrypted URL is generated. The pipeline typically takes 10–20 min to run. Use this URL to refresh the page to check the status or revisit the job later. Users can also monitor job status through Job History. The result files are the same as those generated from the R package.
The octad_output_readme.pdf file describes all the output files.
Troubleshooting
Troubleshooting advice for ‘Procedure 1’ section, using the R package, can be found in Table 2.
Table 2 ∣.
Troubleshooting when using the R package (Procedure 1)
| Function | Message | Possible cause | Solution |
|---|---|---|---|
| computeRefTissue | Cannot open file ‘as//…/case_normal_corMatrix.csv’: no such file or directory | Variable outputFolder: name of the output folder is incorrect | Replace it with a valid name |
| Error in apply(expSet_normal, 1, stats::IQR): dim(X) must have a positive length | Variable source: fail to call the expression matrix | Specify the path to the expSet while source is octad.whole | |
| Error in expSet[, normal_id]: incorrect number of dimensions | Variable expSet: input matrix has insufficient samples for computation, or some samples fail to match to the samples provided in the expression matrix | Filter out non-matched samples and/or increase sample size | |
| diffExp | Expression data not sourced; modify expSet option | Variable expSet: did not source the object while using custom expression matrix | Source the object |
| Source case IDs and control IDs vector | Either case or source vector is not sourced | Add case IDs or control IDs | |
| Empty output | If row names of the expSet do not contain Ensembl gene IDs, it will return empty output | If annotate =TRUE, make sure row. names of the custom input contain Ensembl gene IDs | |
| Error in h5checktypeOrOpenLoc (). Cannot open file. File ‘octad.counts. and.tpm.h5’ does not exist | Source | If source = ‘octad.whole’, octad.counts. and.tpm.h5 should be stored in the working directory, or the full path should be sourced through file option | |
| runsRGES | Disease signature input not found | dz_signature | Source disease signature should include columns: Symbol and log2FoldChange |
| Either Symbol or log2FoldChange column in Disease signature is missing | dz_signature | Source disease signature should include columns: Symbol and log2FoldChange | |
| Warning message: in dir.create (outputFolder): cannot create dir; reason ‘Invalid argument’ | outputFolder | Correct output folder | |
| computeCellLine | Case IDs vector input not found | case_id | Case vector is not sourced |
| octadDrugEnrichment | Error in file(file, ifelse(append, “a”, “w”)): cannot open the connection | enrichFolder | Correct output folder |
| sRGES input not found | sRGES | sRGES is not sourced | |
| Error in.local (expr, gset.idx.list,…). No identifiers in the gene sets could be matched to the identifiers in the expression data | sRGES | Make sure column pert_iname is not empty | |
| Either sRGES or pert_iname column in Disease Signature is missing | sRGES | Make sure sRGES input contains both pert_iname and sRGES columns | |
| topLineEval | Error in ‘[.data.frame’(x, r, vars, drop = drop): undefined columns selected | topline | Make sure the cell line vector is valid. You can compare output with computeCellLine output |
Troubleshooting advice relating to Procedure 2, using the web portal version of OCTAD, can be found in Table 3. The details of troubleshooting various issues are updated in the portal (http://octad.org/faq).
Table 3 ∣.
Troubleshooting when using the Web portal (Procedure 2)
| Step | Problem | Cause | Solution |
|---|---|---|---|
| Case | When disease name is clicked, no sample shows up | The system is still loading the data | Wait for ~20 s for samples to be loaded |
| Not all samples are selected in the table | The system is still loading the data; by default, all samples should be selected | Wait for ~20 s for samples to be loaded | |
| Control | After clicking ‘compute control samples’, the page is inactive and could not respond | Depending on the number of case samples, it usually takes within 2 min to finish. It might get slower if many jobs are running | Wait for a few minutes and restart the job |
| Output | Job status is ‘Complete’, but some result files are missing | The system will change the status to Complete when all the key files are created. However, the job is still running to create supplementary files | Wait a few minutes for the system to assemble the output files |
| Upload data | No result file is created | Input file does not conform to the required format | Recreate the input file following the example file format. Column names include at least log2FoldChange and Symbol |
Timing
The test of the web portal was performed on Chrome 80, and the test of the desktop version was performed in Rgui 3.6.3 on a laptop with an Intel Core i7-9700 3-GHz processor and 16G of memory. Code for the desktop example and the timing calculation are available in the GitHub repository (https://github.com/Bin-Chen-Lab/octad; Table 4).
Table 4 ∣.
Benchmarking test performed both for the website and standalone package
| Step Desktop timing |
Description Desktop memory usage |
Web | interface timing | |
|---|---|---|---|---|
| Load library | Load data log-normalized transcripts for computation of case samples | — | 10.35 s | 566.87 Mb |
| Compute reference IDs | Compute top 50 healthy samples that match selected case samples | 10.6 s | 5.88 s | 581.14 Mb |
| DE computation with default subset | Compute DE for case samples (n = 369) versus selected reference samples (n = 50) on LINCS genes (n = 978) | — | 11.38 s | 733.72 Mb |
| DE computation with the whole OCTAD database | Compute DE for case samples (n = 369) versus selected reference samples (n = 50) on the whole OCTAD database with 60,000 transcripts | 4.25 min | 2.62 min | 736.75 Mb |
| sRGES computation | Rank and compute reversed expression scores | 7.25–8.3 min | 1.01 min | 1,347.88 Mb |
Anticipated results
In the procedure for the Desktop version and the related figures (Figs. 4 and 5), we demonstrated the ability of the OCTAD pipeline to select a case of primary cancers from TCGA, compute correlated reference tissues from GTEx to be used as control samples and compute a differential gene expression signature to recommend candidate drugs. This current pipeline is a complete framework with increased functionality compared to our original iteration8. For instance, instead of using DESeq2 to compute DE, we adopted a less time-consuming method: edgeR24. Furthermore, we integrated our methodology to select reference tissue from GTEx data, making possible the prediction of cancers without adjacent normal tissues15. Lastly, we compiled more samples along with their clinical features, enabling prediction of candidates for a subset of patient samples. Here, using HCC as an example, we first demonstrate the consistency of results between the original method and the optimized one in every major step (i.e., disease signature creation, drug prediction and drug enrichment analysis).
To illustrate the potential applications of using OCTAD to screen compounds for identifying putative personalized therapeutics, we predict compounds specifically targeting MYC amplified lung cancer and PIK3CA mutant breast cancer.
Comparison of results obtained using different key parameters
Because the OCTAD pipeline comprises multiple steps, each of which involves multiple parameters, the selection of each parameter might affect the accuracy of the final prediction, and the importance of each parameter is not clear. Although we optimized each step in the original papers11,15, here using three cancers as examples, we systematically investigated eight main parameters (Fig. 6 and Supplementary Fig. 3). For each parameter, we examined a few values commonly used. We enumerated all the combinations and ran the pipeline for each. The final prediction sRGES was compared with the efficacy data compiled in the original paper. In HCC, the correlation in the original paper11 is 0.61, and the average correlation of all combinations here is 0.49 (s.d. = 0.08), suggesting that the selection of parameters does affect the final prediction. Because the performance of each value might be highly confounded by the values from other parameters, we then performed a multiple variant linear regression (cor ~ parameters). The analysis of three cancers revealed that the performance is highly dependent on the DE log2FoldChange threshold and the DE method (Fig. 6 and Supplementary Fig. 3; multivariate regression, P < 0.01). It suggests that using log2FoldChange = 1 and edgeR (or DESeq2) in DE analysis could lead to a consistently better outcome. The effect of other parameters varies, whereas their overall effect on the result is relatively low.
Fig. 6 ∣. Correlation between sRGES and efficacy data under different parameter values in HCC.
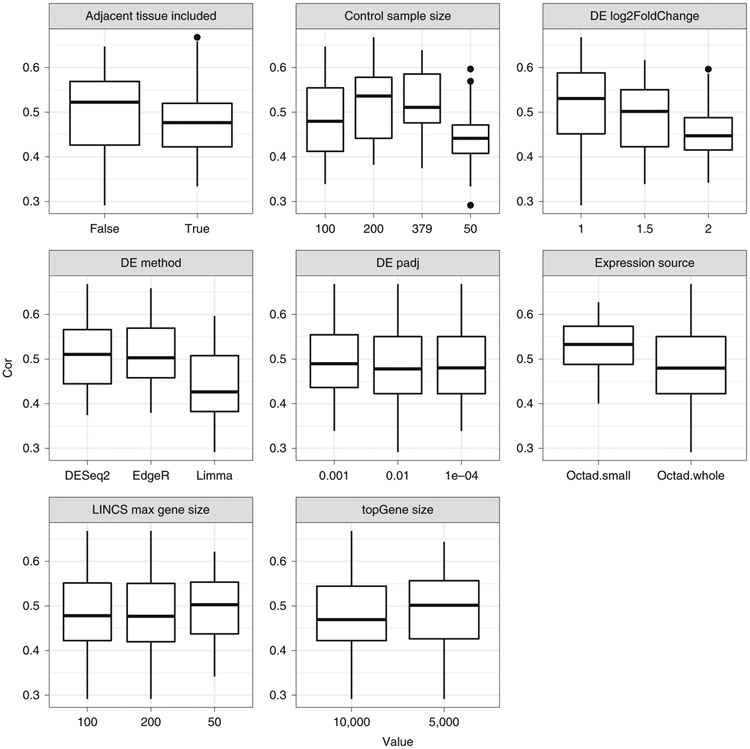
y shows correlation values; x shows the values commonly used. The following parameters were examined: adjacent tissue included, control sample size, DE log2 fold-change threshold, DE method, DE padj threshold, expression source, max gene size in LINCS prediction and topGene size in DE analysis. For each value, we enumerated all the values of other parameters and reported the correlation for each combination. In the box plot, the central line represents the median value, and the bounds represent the 25th and 75th percentiles. The whiskers are 1.5 times the interquartile range plus 25th/75th quartiles.
Comparison between the original method and the current pipeline
We first compared the rank of DE log2 fold change from our original work, which uses DESeq2 (noted as Pub) to the rank of DE in our current pipeline, which uses edgeR (noted as Adj); both use TCGA adjacent normal HCC as a reference control. Next, we compared the original results with our current pipeline using edgeR and computed GTEx normal tissues derived using the top correlated autoencoder method (noted as Ae), generated from the desktop procedure section. Finally, we compared the results from the desktop version with those derived from our online web portal (noted as Online), which also uses edgeR and normal liver tissue from the GTEx database, which was generated from the online procedure section.
The correlation analysis of the DEs from different procedures shows that the workflow using edgeR and adjacent tissues as control had the highest correlation to the original work (Fig. 7a). In addition, both the online and desktop disease signatures are equal to each other, as the parameters for them are the same (Fig. 7a). Both online and desktop results using GTEx as controls also retained high correlation to the original gene expression signatures, indicating that it is feasible to use GTEx controls (Fig. 7a).
Fig. 7 ∣. Evaluation of the results from major steps in HCC prediction.

(a) Consistency of disease signatures, (b) consistency of sRGES, (c) correlation with experimental data (IC50) and (d) consistency of enriched MeSH. adj, using edgeR and adjacent tissue as control; ae, using edgeR and normal tissues selected from the autoencoder approach; online, online web portal; Pub, the published work.
Subsequent drug prediction sRGES is computed using significant differential gene expression. We first observed the small discrepancy of differential gene expression computed using multiple different procedures. We then assessed whether sRGES results are also similar to the original. Unsurprisingly, the workflow using edgeR and adjacent tissues as control had the highest correlation to the original work. Both online and desktop results (Ae) had lower but still significant correlation to the original work (Fig. 7b).
In our original work, we showed that sRGES correlated with drug efficacy data from the CTRP database in which drugs were tested on cancer cell lines. We further compared the correlation of sRGES to the AUC of the corresponding drugs found in CTRP of liver cancer lineage. AUC is computed as the median AUC across all the liver cancer cell lines. The significant correlation between drug efficacy data and sRGES computed from different procedures suggested the utility of using the workflow to in silico predict drug efficacy, even in the absence of adjacent tissue samples (Fig. 7c).
One of the pitfalls of using individual scores such as sRGES is the possibility for false-positive predictions. An additional function of our workflow includes enrichment of drug targets, such as via MeSH terms. This allows for summary of drugs into classes to allow for investigation of groups of drugs rather than individual compounds. A higher score indicates a MeSH drug group to be efficacious against the cancer, whereas a lower score indicates a non-effective MeSH drug group. We further compare the rank correlation of the MeSH scores generated from the different procedures (Fig. 7d). Through drug enrichment analysis, we are able to examine clusters of drugs based on their MOA (e.g., MeSH). Finding novel classes of drugs (e.g., anti-helminths or unconventional chemical structures) allows for the generation of hypotheses that can be prioritized for experimentation. Furthermore, the pipeline also allows us to filter out for drug candidates that might have low value—for example, poor scores were given for anti-hypertensives compared to more conventional classes, such as intercalating agents and HDAC inhibitors.
Screening compounds targeting MYC-amplified lung adenocarcinoma
Targeting MYC, an oncogene amplified in many cancers, including lung adenocarcinoma, is under active research; however, MYC is considered an undruggable target53. One therapeutic strategy is to reverse the gene expression signature of these MYC-amplified tumors. Here, we ran the two sets of TCGA lung adenocarcinoma through our pipeline. In one set, we selected for cancer tissues with MYC amplification, defined as copy numbers of 1 or more. In the second set, we selected for cancer tissues without the MYC amplification, defined as 0 copy numbers. Then we selected drug efficacy data from the CTRP non-small cell lung cancer cell lines with the MYC amplification as a validation set. The Spearman correlation between AUC and sRGES for the MYC run is significant and better than the non-MYC run (rho: −0.415 versus −0.261; Supplementary Fig. 4). This suggests that OCTAD can be used to search for candidates specifically targeting MYC-amplified cancers.
Screening compounds targeting PIK3CA mutation in breast cancer
Likewise, we applied OCTAD to screen compounds targeting tumors harboring PIK3CA mutation. PIK3CA is highly mutated in cancers but currently considered undruggable54. In this case, we ran the two sets of TCGA breast cancers through our pipeline. In one set, we selected for cancer tissues with PIK3CA mutation. In the second set, we selected for cancer tissues without the mutation. Then, we selected drug efficacy data from the CTRP breast cell lines with the PIK3CA mutation as a validation set. Similarly to the previous result, the Spearman correlation between AUC and sRGES for the PIK3CA mutant run was significant (P < 0.05) and better than the non-mutant run (rho: −0.370 versus −0.273; Supplementary Fig. 5). Although more extensive experimental validation of compounds is expected, this exercise demonstrates the feasibility of quickly employing OCTAD to identify candidates for subtypes defined by molecular features. Moreover, OCTAD enables the creation of signatures for various subtypes, from which compounds selectively reversing the signature of one subtype could be fished out.
Summary
The combination of clinical and molecular features in describing patients’ disease could lead to the identification of numerous disease subtypes; for each of these subtypes, the resources available for their study might be very limited. Using open datasets and advanced machine learning methods, OCTAD provides an effective means to screen compounds for one specific cancer subtype for further experimental testing. In this work, we optimized our pipeline and developed OCTAD, which we demonstrate can reproduce results from a previous study that are also consistent when selecting normal tissue data from a different database. Furthermore, the two new cases illustrate the potential of using OCTAD to screen compounds for precisely defined patient groups, although subsequent experimental testing is desired to verify drug candidates. In short, OCTAD provides a useful resource to many wet labs, especially those with limited screening capacity, to prioritize compounds for a specific group of patients, and a powerful platform for computational biologists to run large-scale drug predictions.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data related to this protocol can be found at http://octad.org/download or https://www.synapse.org/#!Synapse:syn22101254. You can also refer to the preprint version of our protocol: https://www.biorxiv.org/content/10.1101/821546v1. This pipeline was verified in our previous research papers.
Software availability
The software is available from http://octad.org/download or https://www.synapse.org/#!Synapse:syn22101254.
Extended Data
Extended Data Fig. 1 ∣. Screenshots of the web portal.

(a) Disease sample selection, (b) control sample selection, (c) drug prediction job submission, (e) job management, (f) predicted drug list and (g) result files.
Supplementary Material
Acknowledgements
The research is supported by R01GM134307, R21 TR001743 and K01 ES028047 and the MSU Global Impact Initiative. Amazon AWS research credits were received to support portal development and hosting. The portal was developed with help from Optra Health and MSU IT. The content is solely the responsibility of the authors and does not necessarily represent the official views of sponsors.
Appendix
Related links
Key references using this protocol
- Chen B et al. Nat. Commun 8, 16022 (2017): 10.1038/s41467-019-10148-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B et al. Gastroenterology 152, 2022–2036 (2017): 10.1053/j.gastro.2017.02.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng WZD, Glicksberg BS, Li Y & Chen B BMC Med. Genomics 12, 21 (2019): 10.1186/s12920-018-0463-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K et al. Nat. Commun 10, 2138 (2019): 10.1038/s41467-019-10148-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Footnotes
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41596-020-00430-z.
Supplementary information is available for this paper at https://doi.org/10.1038/s41596-020-00430-z.
Peer review information Nature Protocols thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Balamuth NJ & Womer RB Ewing’s sarcoma. Lancet Oncol. 11, 184–192 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Torre LA et al. Global cancer statistics, 2012. CA Cancer J. Clin 65, 87–108 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Genetic and Rare Diseases Information Center, National Institutes of Health. FAQs About Rare Diseases. https://rarediseases.info.nih.gov/diseases/pages/31/faqs-about-rare-diseases (2020). [Google Scholar]
- 4.Sirota M et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Science Tranl. Med 3, 96ra77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jahchan NS et al. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov. 3, 1364–1377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Noort V et al. Novel drug candidates for the treatment of metastatic colorectal cancer through global inverse gene-expression profiling. Cancer Res. 74, 5690–5699 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Brum AM et al. Connectivity Map-based discovery of parbendazole reveals targetable human osteogenic pathway. Proc. Natl Acad. Sci. USA 112, 12711–12716 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen B et al. Computational discovery of niclosamide ethanolamine, a repurposed drug candidate that reduces growth of hepatocellular carcinoma cells in vitro and in mice by inhibiting cell division cycle 37 signaling. Gastroenterology 152, 2022–2036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pessetto ZY et al. In silico and in vitro drug screening identifies new therapeutic approaches for Ewing sarcoma. Oncotarget 8, 4079–4095 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mirza AN et al. Combined inhibition of atypical PKC and histone deacetylase 1 is cooperative in basal cell carcinoma treatment. JCI Insight 2, e97071 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen B et al. Reversal of cancer gene expression correlates with drug efficacy and reveals therapeutic targets. Nat. Commun 8, 16022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamb J et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Subramanian A et al. A next generation Connectivity Map: L1000 platform and the first 1,000,000 profiles. Cell 171, 1437–1452 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corsello SM et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat. Med 23, 405–408 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeng WZD, Glicksberg BS, Li Y & Chen B Selecting precise reference normal tissue samples for cancer research using a deep learning approach. BMC Med. Genomics 12, 21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aran D, Sirota M & Butte AJ Systematic pan-cancer analysis of tumour purity. Nat. Commun 6, 8971 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshihara K et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun 4, 2612 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu K et al. Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types. Nat. Commun 10, 3574 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vivian J et al. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol 35, 314–316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu K et al. Evaluating cell lines as models for metastatic breast cancer through integrative analysis of genomic data. Nat. Commun 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Risso D, Ngai J, Speed TP & Dudoit S Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechol 32, 896–902 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Law CW, Chen Y, Shi W & Smyth GK voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen EY et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costa-Silva J, Domingues D & Lopes FM RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 12, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sterling T & Irwin JJ ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model 55, 2324–2337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ertl P & Schuffenhauer A Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform 1, 8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim S et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 47, D1102–D1109 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bento AP et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 42, D1083–D1090 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen B, Sirota M, Fan-Minogue H, Hadley D & Butte AJ Relating hepatocellular carcinoma tumor samples and cell lines using gene expression data in translational research. BMC Med. Genomics 8, S5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iorio F et al. A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seashore-Ludlow B et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 5, 1210–1223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barretina J et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao J et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McFerrin LG et al. Analysis and visualization of linked molecular and clinical cancer data by using Oncoscape. Nat. Genet 50, 1203–1204 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Newton Y et al. TumorMap: exploring the molecular similarities of cancer samples in an interactive portal. Cancer Res. 77, e111–e114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmid MW & Grossniklaus U Rcount: simple and flexible RNA-Seq read counting. Bioinformatics 31, 436–437 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Lachmann A et al. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun 9, 1366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang DW et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 8, R183 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kucukural A, Yukselen O, Ozata DM, Moore MJ & Garber M DEBrowser: interactive differential expression analysis and visualization tool for count data. BMC Genomics 20, 6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu H, Huang J, Zhong Y & Huang Q DrugSig: a resource for computational drug repositioning utilizing gene expression signatures. PLoS ONE 12, e0177743 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moosavinasab S et al. ‘RE:fine drugs’: an interactive dashboard to access drug repurposing opportunities. Database 2016, baw083 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee BKB et al. DeSigN: connecting gene expression with therapeutics for drug repurposing and development. BMC Genomics 18, 934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z et al. Drug Gene Budger (DGB): an application for ranking drugs to modulate a specific gene based on transcriptomic signatures. Bioinformatics 35, 1247–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shameer K et al. Systematic analyses of drugs and disease indications in RepurposeDB reveal pharmacological, biological and epidemiological factors influencing drug repositioning. Brief. Bioinformatics 19, 656–678 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown AS & Patel CJ A standard database for drug repositioning. Sci. Data 4, 170029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen B et al. Harnessing big ‘omics’ data and AI for drug discovery in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol 17, 238–251 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smirnov P et al. PharmacoGx: an R package for analysis of large pharmacogenomic datasets. Bioinformatics 32, 1244–1246 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Hänzelmann S, Castelo R & Guinney J GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dang CV MYC on the path to cancer. Cell 149, 22–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Courtney KD, Corcoran RB & Engelman JA The PI3K pathway as drug target in human cancer. J. Clin. Oncol 28, 1075–1083 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Glicksberg BS, Li L, Chen R, Dudley J & Chen B Leveraging big data to transform drug discovery. Methods Mol. Biol 1939, 91–118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson DR et al. Integrative clinical genomics of metastatic cancer. Nature 548, 297–303 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data related to this protocol can be found at http://octad.org/download or https://www.synapse.org/#!Synapse:syn22101254. You can also refer to the preprint version of our protocol: https://www.biorxiv.org/content/10.1101/821546v1. This pipeline was verified in our previous research papers.