

Abstract
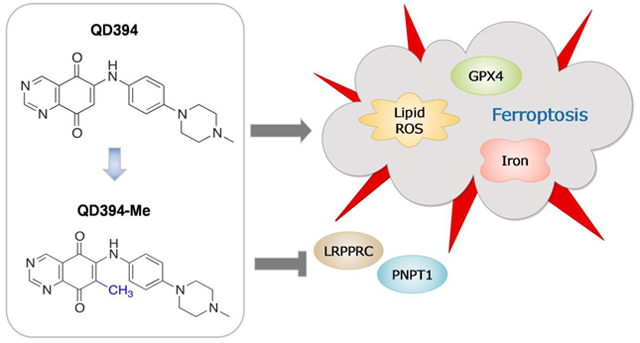
Redox modulators have been developed as an attractive approach to treat cancer. Herein, we report on the synthesis, identification and biological evaluation of a quinazolinedione reactive oxygen species (ROS) inducer, QD394, with significant cytotoxicity in pancreatic cancer cells. QD394 shows a transcriptomic profile remarkably similar to napabucasin, a cancer stemness inhibitor. Both small molecules inhibit STAT3 phosphorylation, increase cellular ROS and decrease the GSH/GSSG ratio. Moreover, QD394 causes an iron- and ROS-dependent, GPX4 mediated cell death, suggesting ferroptosis as a major mechanism. Importantly, QD394 decreases the expression of LRPPRC and PNPT1, two proteins involved in mitochondrial RNA catabolic processes and both negatively correlated with the overall survival of pancreatic cancer patients. Pharmacokinetic-guided lead optimization resulted in the derivative QD394-Me which showed improved plasma stability and reduced toxicity in mice compared to QD394. Overall, QD394 and QD394-Me represent novel ROS-inducing drug-like compounds warranting further development for the treatment of pancreatic cancer.
Graphical Abstract
INTRODUCTION
Pancreatic cancer is the fourth leading cause of cancer death in the United States, and its incidence and mortality rates are continuing to increase worldwide.1, 2 It is predicted to be the second leading cause of cancer death in the USA by 2030.2, 3 Pancreatic cancer is primarily diagnosed at a late stage because early-stage symptoms are non-specific. The 5-year survival rate at the advanced stage is only around 3%.1 The current standard-of-care, FOLFIRINOX (folinic acid, 5-fluorouracil (5-FU), irinotecan, and oxaliplatin), and gemcitabine together with nab-paclitaxel show modest efficacy because of drug resistance and systemic toxicity.2, 4 Therefore, it is imperative to find new improved therapies to treat pancreatic cancer.
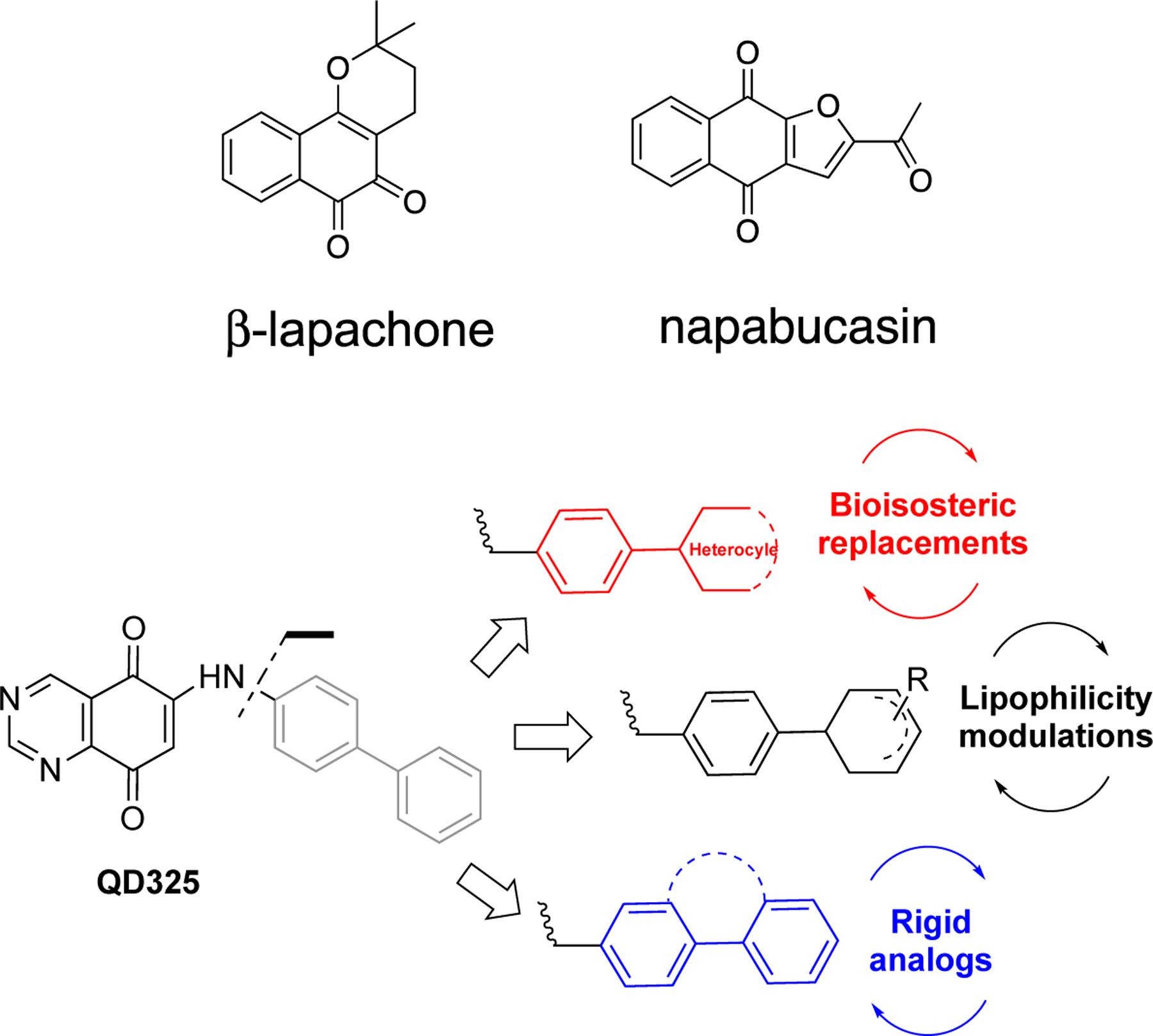
Reprogramming energy metabolism and oxidative stress are two hallmarks of cancer.5–8 Therefore, treating cancer with small molecule reactive oxygen species (ROS) inducers may selectively overwhelm cancer cells. Cellular redox homeostasis is closely related to ROS production and elimination. Cancer cells have higher redox requirements from normal cells due to their increased cell metabolism, dysfunctional mitochondria, scarce nutrients, and oxygen-poor microenvironment.9 Higher intracellular ROS levels force cancer cells to develop robust antioxidant systems to adapt to oxidative stress. At low or moderate levels, ROS serve as signaling molecules to promote cell growth; while at high levels, ROS induce oxidation of lipids, proteins, and DNA, ultimately leading to cell death. Several ROS inducers are in clinical trials to evaluate efficacy and safety profiles in cancer.10–12 ARQ761, a beta-lapachone (Chart 1) prodrug that reacts with NAD(P)H Quinone Dehydrogenase 1 (NQO1) to generate ROS, showed modest efficacy in an open-label, dose-escalation Phase I study.10 Napabucasin (Chart 1) is an orally administered cancer stemness inhibitor with a quinone scaffold and has been tested in Phase III clinical trials in multiple cancers.11, 12 Napabucasin inhibits Signal Transducer and Activator of Transcription 3 (STAT3) signaling and increases ROS levels predominately by NQO1 bioactivation.13 Therefore, inducing cellular ROS is a promising method to treat pancreatic cancer.
Chart 1.
Structures of representative ROS inducers under clinical evaluation, and design of novel quinazolinedione-based ROS modulators.
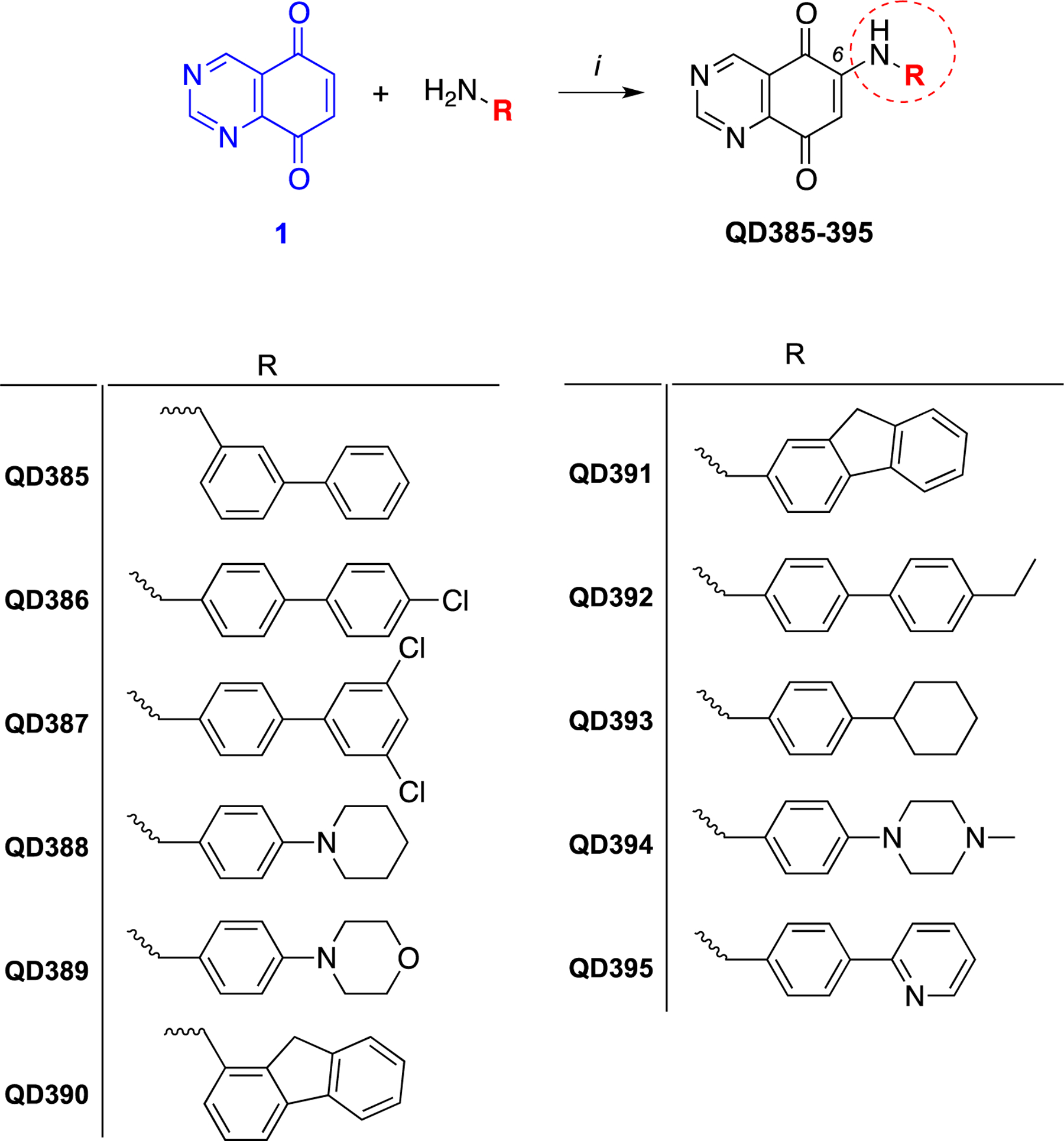
Previously, we developed a series of quinazolinedione (QD)-based ROS inducers that were tested for their utility in pre-clinical models of pancreatic cancer.14 In this study, we have conducted a lead optimization on the most effective compound (i.e. QD325, Chart 1) to select compounds inducing maximum ROS production. A series of structural modifications on arylamino substituent at position 6 of the quinazoline-5,8-dione backbone were made to modulate its physicochemical properties. A set of QD derivatives were synthesized (Scheme 1, Chart 1), and the Me-N-piperazino analogue, i.e. QD394, was selected for mechanistic studies.
Scheme 1.
Preparation of QD385 to QD395a
aReagents and conditions: (i) CeCl3•7H2O, abs EtOH, rt, 1.5 h
Herein, we show for the first time that this new prototype, QD394, displays highly similar transcriptomic profiles with napabucasin, suggesting similar downstream signaling effects. Combination with cell death signaling inhibitors reveals that QD394 induces ferroptosis but not caspase-dependent apoptosis, necroptosis, or autophagy. We further demonstrate that QD394 induces lipid peroxidation, iron-, ROS-, and glutathione peroxidase 4 (GPX4)-mediated cell death, all of which are closely related to ferroptosis. Ferroptosis was first described in 2012 to represent an iron-dependent regulated cell death with increased lipid peroxidation.15 Intrinsic mechanisms of ferroptosis depend on the activity of GPX4 and lipid peroxidation.16, 17 At least four classes of ferroptosis inducers have been developed, including (1) SLC7A11 inhibitor (erastin), (2) GPX4 inhibitor (RSL3), (3) FIN56 (depleting GPX4 protein), and (4) FINO2 (indirectly inhibiting GPX4).16 Ferroptosis inducers show synergistic anti-tumor efficacy with select chemotherapies18 and sensitize cancer cells to radiation.19 We also observed synergy between QD394 and select FDA-approved drugs, suggesting that combination studies with redox modulators as ferroptosis inducers could be a viable approach to treat pancreatic cancer.
Additionally, we performed proteomics analysis and observed a significant decrease in LRPPRC and PNPT1 expression, two proteins involved in mitochondrial RNA catabolic processes.20, 21 Expression of these two genes is negatively correlated with the survival of pancreatic cancer patients in our analysis using TCGA PAAD dataset. Furthermore, a pharmacokinetics (PK)-guided lead optimization campaign was conducted to develop a QD394 analog, QD394-Me, with improved PK properties.
RESULTS
Chemistry.
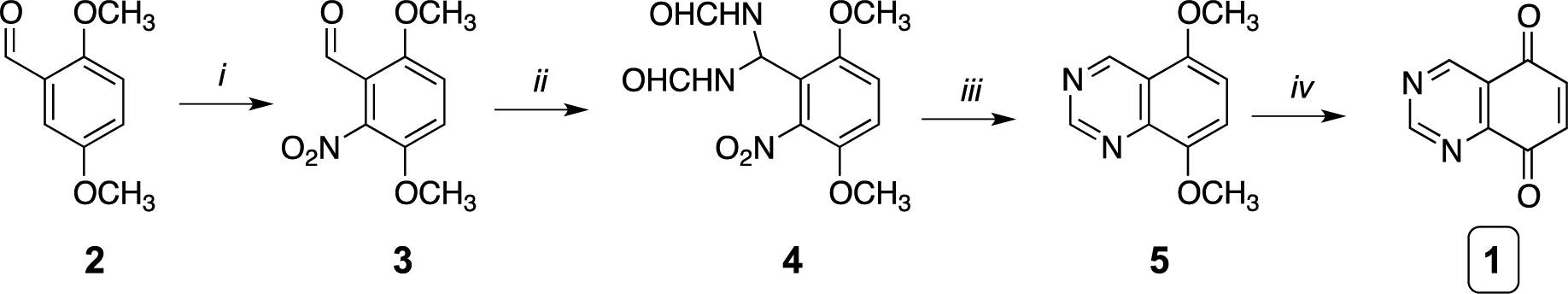
Eleven analogs of QD394 were synthesized to characterize their cytotoxicity in pancreatic cancer cells. The target compounds QD385 to QD395 were synthesized by regioselective coupling of quinazoline-5,8-dione 1 with appropriate amines in the presence of Ce(III) ions (Scheme 1) under mild conditions. The key synthon 1 was prepared by following our previously reported procedure (Scheme 2).14 Briefly, nitration of the dimethoxybenzaldehyde 2 with concentrated nitric acid in the presence of acetic anhydride under simple magnetic stirring afforded the desired 3,6-dimethoxy-2-nitrobenzaldehyde (3) regioisomer, which was isolated in good yield by flash chromatography. Next, 3 was converted to the diformamido-derivative 4 by adding formamide, followed by exposure to gaseous HCl under controlled temperature. Then, compound 4 was treated with zinc powder and acetic acid to obtain the dimethoxyquinazoline 5. Final oxidation of 5 with cerium ammonium nitrate resulted in the production of 1, which was used for final amino conjugation.
Scheme 2.
Synthetic route for the preparation of the synthon 1 and its intermediatesa
aReagents and conditions: (i) conc. HNO3, (CH3CO)2O, 0 °C, 1.5 h; (ii) H2NCHO, HCl(g) from 40 to 80 °C, 1 h; (iii) glacial CH3COOH, Zn, 0 °C for 2 h, rt for 4 h; (iv) (NH4)2Ce(NO3)6, CH3CN/H2O, 0 °C, 20 min. For synthetic details see in ref. 14.
The QD394-Me (namely QD430) was synthesized starting from 2,5-dimethoxy-4-methylbenzaldehyde by adapting the abovementioned synthetic route (Scheme 3), except for the first step. An ipso-nitration on the starting material occurred in the first step reaction, and the 1,4-dimethoxy-2-methyl-5-nitrobenzene (6) was obtained as the main product, while the desired nitrobenzaldehyde 7 was isolated in very low yields (~5%) after chromatographic separation. Scale-up of this reaction was needed to reach the necessary amount of intermediate 7, which resulted in increased consumption of the reagent that dramatically affected the overall yield of this synthetic route. However, the 7-methyl-quinazoline-5,8-dione (10), bearing the methyl at the position 7 on the quinazolinedione ring, was easily obtained after oxidation of 9, which was prepared by cyclizing the diformamido-derivative 8 in the same reaction condition used for the preparation of the nonmethylated analogue.
Scheme 3.
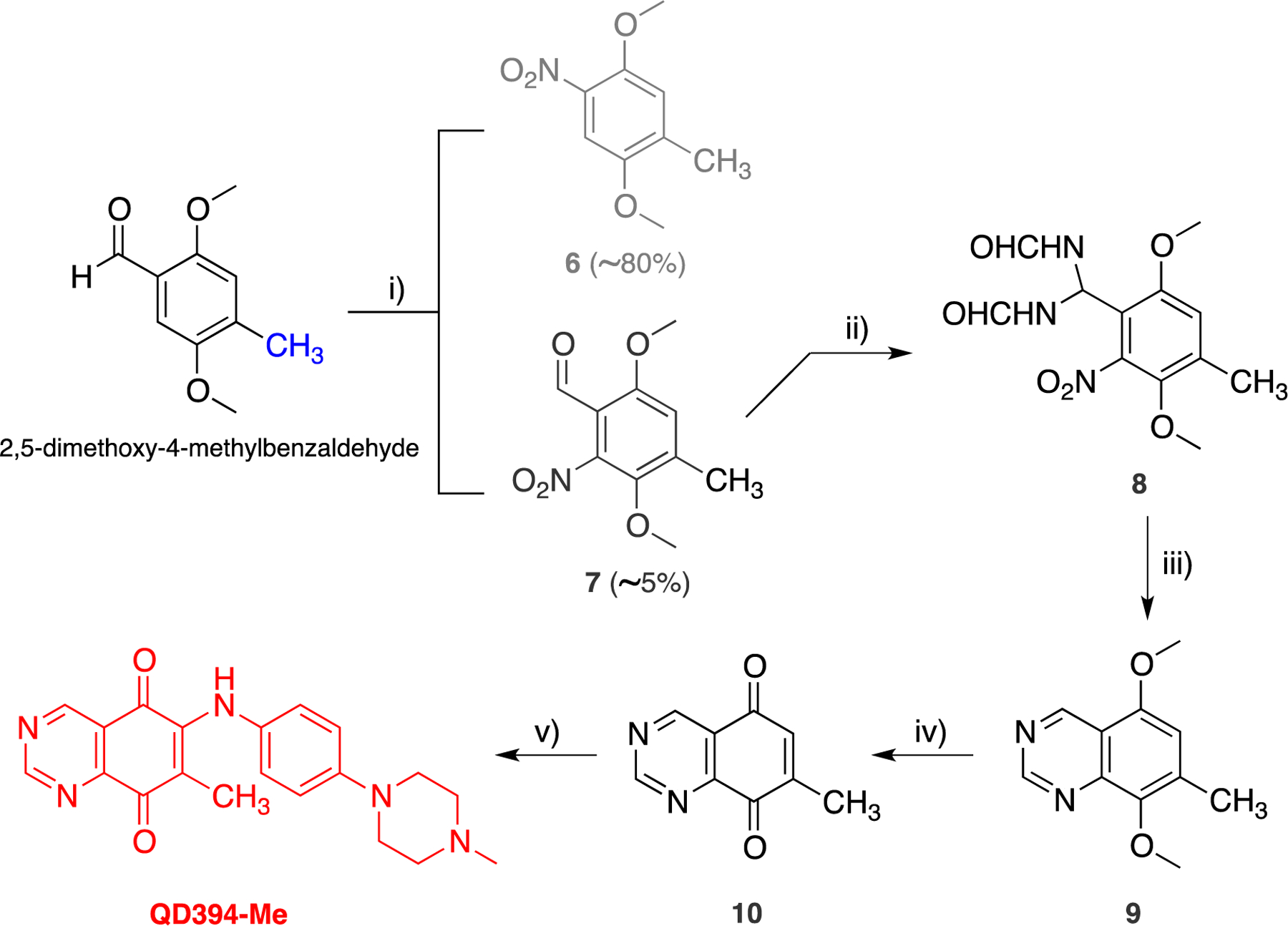
Synthetic Route for the Preparation of QD394-Mea
aReagents and conditions: (i) conc. HNO3, CH2Cl2, rt, 24 h; (ii) H2NCHO, HCl(g) from 40 to 80 °C, 1 h; (iii) glacial CH3COOH, Zn, 0 °C for 2 h, r.t. for 4 h; (iv) (NH4)2Ce(NO3)6, CH3CN/H2O, 0 °C, 20 min.; (v) CeCl3•7H2O, abs EtOH, rt, 1.5 h.
In an attempt to expand the preparation of another QD394-Me isomer, where the methyl substituent was placed at position 6 (Scheme 4), we setup an alternative way to prepare the QD394 with improved overall yield. Specifically, the key methyldiketone 15 was obtained by a two-step ring closure of the N-protected intermediate 12. The latter was prepared by reducing the nitroderivative 6 with ammonium formate and Pd/C to give 11 quantitatively, followed by protection of the amino functionality with ethylcloroacetate in THF. Compound 12 was then reacted with hexamethylenetetramine (HMTA) in trifluoroacetic acid (TFA) to give the dihydroquinazoline 13 after an intramolecular cyclization.22 Finally, oxidative dehydrogenation by using potassium ferricyanide in aqueous ethanolic potassium hydroxide of derivative 13 afforded the synthon 15 in good yield. Instead of the expected QD394-Me isomer, coupling of 15 with 4-(4-methylpiperazin-1-yl)aniline in the presence of cerium ammonium nitrate resulted in the nucleophilic substitution of the methyl group, which led to the isolation of QD394 (Scheme 4), but with about ten-fold lesser overall yields than that obtained from the previous synthetic route.
Scheme 4.
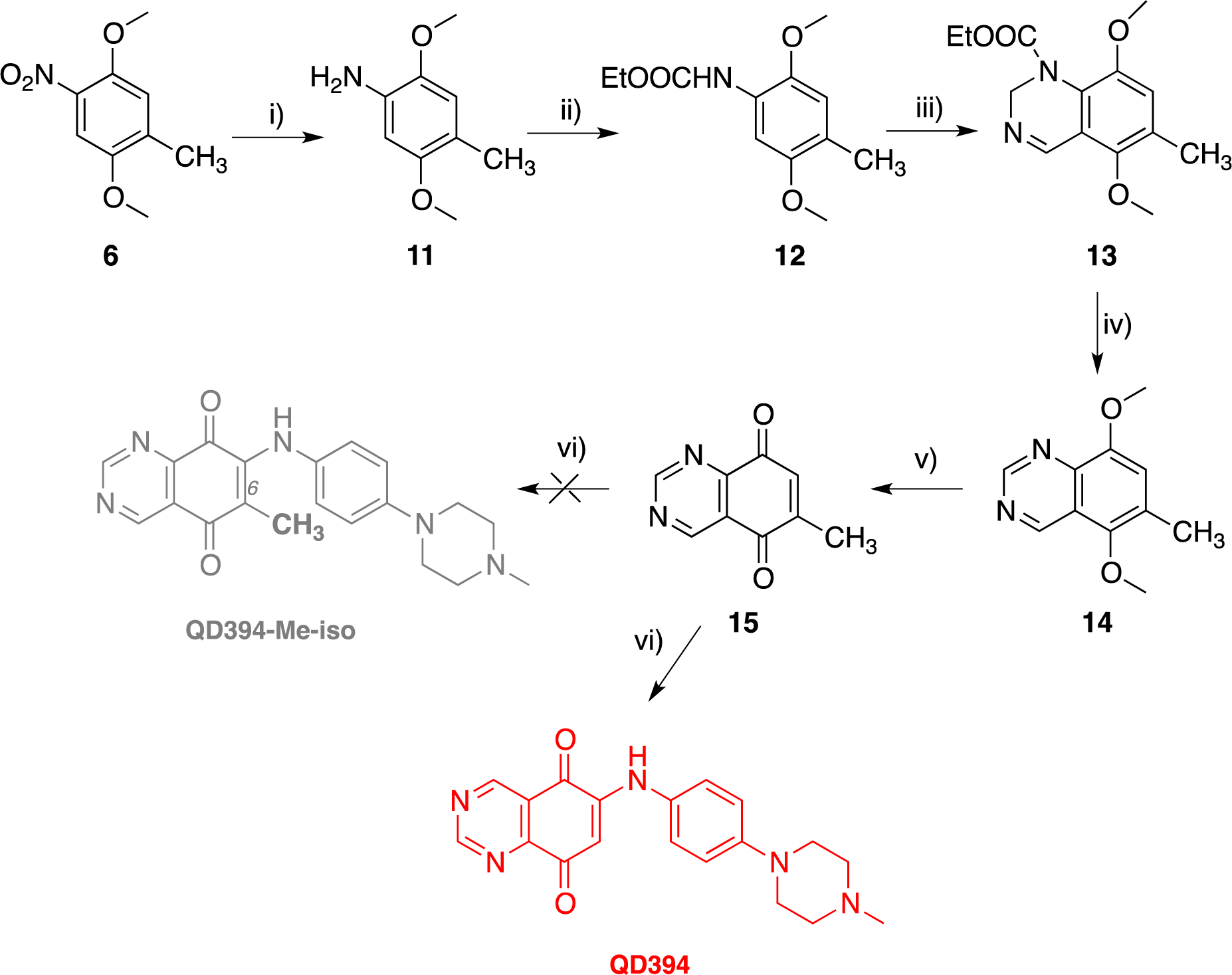
Alternative Synthetic Route for the Preparation of QD394a
aReagents and conditions: Reagents and conditions: (i) HCOONH4; Pd-C; (ii) ClCO2Et, THF; (iii) (1) HMTA, TFA; (iv) KOH, aqueous EtOH, K3Fe(CN)6; (v) (NH4)2Ce(NO3)6,CH3CN/H2O; (vi) 4-(4-methylpiperazin-1-yl)aniline, CeCl3•7H2O, abs EtOH
QD394 shows remarkable similarity to napabucasin and H2O2 in transcriptomic profiles.
Among 11 originally synthesized analogs (Table S1, S2, Scheme 1), QD394 had the highest cytotoxicity in MIA PaCa-2 cells and similar cytotoxicity in PANC-1 and BxPC-3 cells (Table 1). QD394 induced the highest level of intracellular ROS after 1 h compound treatment (Figure S1). QD394 (IC50, 0.64 ± 0. 04 μM) is slightly more cytotoxic and induces more ROS as compared to our previously described QD325 compound (IC50, 0.9 ± 0.2 μM) (Figure S2).14 To better elucidate its mechanism of action, we performed Bru-seq, a nascent RNA-seq technique that uses bromouridine to label newly synthesized RNAs.23 This method detects early changes in the transcriptome caused by drugs and is useful in understanding their mechanisms of action and toxicity. We discovered that the transcriptomic profile of QD394 was highly similar to that of napabucasin and H2O2. The shrunkenLFC (shrunken log2FoldChange) of genes in QD394 Bru-seq samples significantly correlated with that of the napabucasin (r = 0.92) and H2O2 (r = 0.88) samples, indicating similar transcription regulation (Figure 1A). ShrunkenLFC reduces the effect of low-expressing genes that result in over-inflated Log2FoldChange values. Gene Set Enrichment Analysis (GSEA) revealed 82 upregulated and 51 downregulated enriched gene sets in common between QD394 and napabucasin using the C2 curated gene sets from Molecular Signatures (MSigDB) Database v7.0 with FDR below 0.001 (Figure 1B and 1C, Table S3). For the Hallmark and KEGG pathway gene sets shared between QD394 and napabucasin, TNFA signaling, UV response, and hypoxia hallmark gene sets were upregulated, and RNA degradation KEGG gene set was downregulated (Figure 1D and 1E). TNF signaling and ROS are interconnected in the regulation of cell homeostasis, survival, and death in inflammatory diseases and cancers. ROS is closely related to TNF-related downstream signaling, such as NF-κB and JNK activation, and TNF can induce antioxidant NRF2 pathway to protect cells from elevated ROS.24 UV radiation can cause DNA damage and corresponding cell death via oxidative stress.25, 26 Moreover, cellular and mitochondrial ROS activate a series of transcription factors (TFs), which trigger the expression of a number of genes involved in the hypoxic response.27, 28 It is interesting to note that QD394 affects RNA degradation in the KEGG pathway gene set, and this is consistent with our proteomics study (see below).
Table 1.
Cytotoxicity of QD compounds in MIA PaCa-2, PANC-1, and BxPC-3 pancreatic cancer cell lines.
| ID | IC50 (μM) | ||
|---|---|---|---|
| MIA PaCa-2 | PANC-1 | BxPC-3 | |
| QD385 | 2.4 ± 0.1 | 2.3 ± 0.3 | 5.1 ± 1.4 |
| QD386 | 9.7 ± 4.0 | 4.4 ± 1.9 | 10.1 ± 2.8 |
| QD387 | 3.1 ± 0.2 | 1.9 ± 0.2 | 3.3 ± 1.4 |
| QD388 | 2.4 ± 0.6 | 1.4 ± 0.5 | 2.5 ± 0.9 |
| QD389 | 1.5 ± 0.3 | 0.8 ± 0.3 | 2.3 ± 0.4 |
| QD390 | 3.1 ± 1.5 | 4.7 ± 1.5 | 4.5 ± 0.6 |
| QD391 | 2.2 ± 0.7 | 1.6 ± 0.8 | 2.6 ± 0.3 |
| QD392 | 1.5 ± 0.7 | 2.3 ± 0.3 | 1.6 ± 0.6 |
| QD393 | 1.2 ± 0.2 | 0.9 ± 0.4 | 0.5 ± 0.1 |
| QD394 | 0.64 ± 0. 04 | 0.34 ± 0.03 | 0.9 ± 0.2 |
| QD395 | 2.0 ± 0.7 | 0.8 ± 0.2 | 2.7 ± 0.4 |
| QD394-Me | 3.63 ± 0.37 | 0.38 ± 0.01 | 0.87 ± 0.15 |
The IC50 values are shown as mean ± sd. Mean and standard deviation were calculated from three independent experiments.
Figure 1.
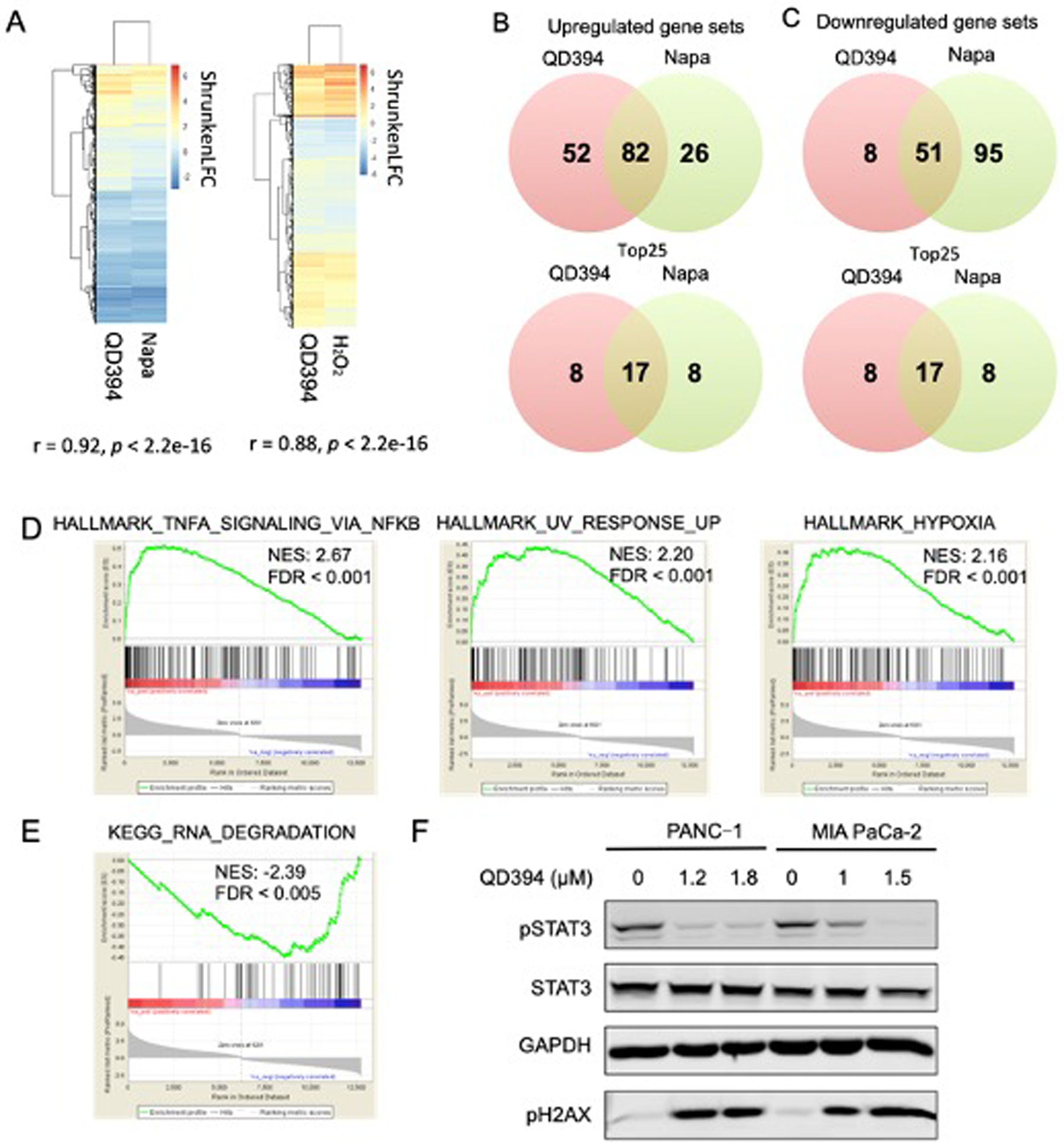
Transcriptomic profile of QD394 is similar to that of napabucasin (napa) and H2O2. (A) Pearson correlation between QD394, napabucasin, and H2O2 samples calculated in R. ShrunkenLFC was used to build heatmaps. (B,C) Upregulated and downregulated gene sets in GSEA from QD394 and napabucasin Bru-seq samples. GSEA plots of upregulated (D) and downregulated (E) enriched gene sets from the QD394 samples that are shared with napabucasin are depicted (FDR < 0.001). NES, normalized enrichment score. (F) Cells were treated with QD394 for 4 h at indicated concentrations and analyzed by immunoblot with STAT3, pSTAT3, GAPDH, and pH2AX antibodies.
Since napabucasin is a STAT3 signaling inhibitor and exhibited a similar transcriptomic profile as QD394, we sought to determine whether QD394 affected STAT3 signaling. Indeed, QD394 inhibited the phosphorylation of STAT3 in MIA PaCa-2 and PANC-1 cells, and it also increased the phosphorylation of H2AX, a DNA damage biomarker (Figure 1F).
QD394 induces ROS-mediated cell death and decreases the GSH/GSSG ratio.
As a redox modulator, QD394 caused time-dependent cellular ROS accumulation within the first 4-hour of treatment in MIA PaCa-2 cells (Figure 2A). The significant increase of ROS level could be reduced in 30 minutes by the antioxidant N-acetylcysteine (NAC) (Figure 2B). Additionally, NAC significantly increased the IC50 of QD394 from 0.53 μM to 1.11 μM (Figure 2C) and rescued colony formation inhibition by QD394 (Figure 2D). Thus, the cytotoxicity of QD394 is partially dependent on ROS induction.
Figure 2.

QD394 increases cellular ROS level and reduces GSH/GSSG ratio in MIA PaCa-2 cells. (A) QD394 increased fluorescence (RFU) indicating cellular ROS generation across treatment time (0 – 4 h). TBHP was used as a positive control. (B) At 30 min, significant increase of ROS was determined in QD394- and TBHP-treated cells, and NAC (3 mM) reduced the elevated ROS levels. ** denotes p < 0.005, *** denotes p < 0.0005. (C) NAC significantly decreased the cytotoxicity of QD394 in the MTT assay. (D) NAC protected the inhibition of colony formation caused by QD394. (E) QD394 reduced cellular GSH/GSSG ratio mainly resulting from GSSG formation. RLU, relative luminescence unit. * denotes p < 0.05, ** denotes p < 0.005, *** denotes p < 0.0005.
Glutathione is a well-known cellular ROS scavenger. In the GSH/GSSG-Glo assay, QD394 significantly decreased the GSH/GSSG ratio at both 2 h and 4 h treatment, similar to napabucasin (Figure 2E). The decrease in ratio is due to reduced GSH level or increased GSSG level or both. The reduction from QD394 treatment was caused by oxidation of GSH to GSSG, rather than the direct decrease of GSH level (Figure S3). Buthionine sulfoximine (BSO) is an inhibitor of GSH synthesis, and both napabucasin and BSO significantly lowered GSH level (Figure 2E and S3).
QD394 causes iron-dependent and GPX4-mediated cell death.
Since we observed that QD394 modulated the GSH/GSSG ratio and its cytotoxicity was dependent on ROS, we hypothesized that QD394 could induce ferroptosis. To determine the cell death mechanism, we combined QD394 with a panel of signaling inhibitors (Table 2). We identified that deferoxamine (DFO) and deferasirox, two iron chelators, prevented cell death caused by QD394, indicating iron is a contributing factor for its cytotoxicity (Figure 3A and S4). Two known ferroptosis inhibitors had different effects on cell death rescue. Liproxstatin-1 modestly rescued cell death caused by QD394, while ferrostatin-1 did not. Z-VAD (a caspase-dependent apoptosis inhibitor) and necrostatin-1 (a necroptosis inhibitor) did not rescue cell death, and chloroquine (an autophagy inhibitor) slightly rescued cell death in the colony formation assay (Figure S4). Interestingly, necrostatin-1 blocked the inhibition of colony formation caused by napabucasin, indicating that cell death by napabucasin may proceed via necroptosis. The link between napabucasin and necroptosis has not been previously reported. Moreover, these results demonstrate that QD394 and napabucasin have different cell death mechanisms.
Table 2.
Select cell death inhibitors that were combined with QD394 in pancreatic cancer cells.
| Cell death | Target | Inhibitor |
|---|---|---|
| Apoptosis | All caspases | Z-VAD-FMK |
| Necroptosis | RIP1 kinase | Necrostatin-1 |
| Selective inhibitor of RIP1 | Necrostatin-1s | |
| Ferroptosis | Iron; iron-dependent prolyl hydroxylases; lysosomal ROS inhibitor by chelating lysosomal redox-active iron | Deferoxamine (DFO), deferasirox |
| Inhibit autooxidation of lipids by trapping peroxyl radicals; not rescue H2O2-induced cell death | Ferrostatin-1 | |
| Inhibit ferroptosis induced by L-buthionine sulphoximine (BSO), erastin and (1S,3R)-RSL3; inhibit autooxidation of lipids by trapping peroxyl radicals | Liproxstatin-1 | |
| Autophagy | Impair autophagosome fusion with lysosomes | Chloroquine |
| Inhibit PI3K activity and the initial phase of the autophagic process | 3-Methyladenine | |
| Parthanatos | PARP inhibitors | Olaparib |
| AIF release inhibitors | N-phenylmaleimide |
Figure 3.
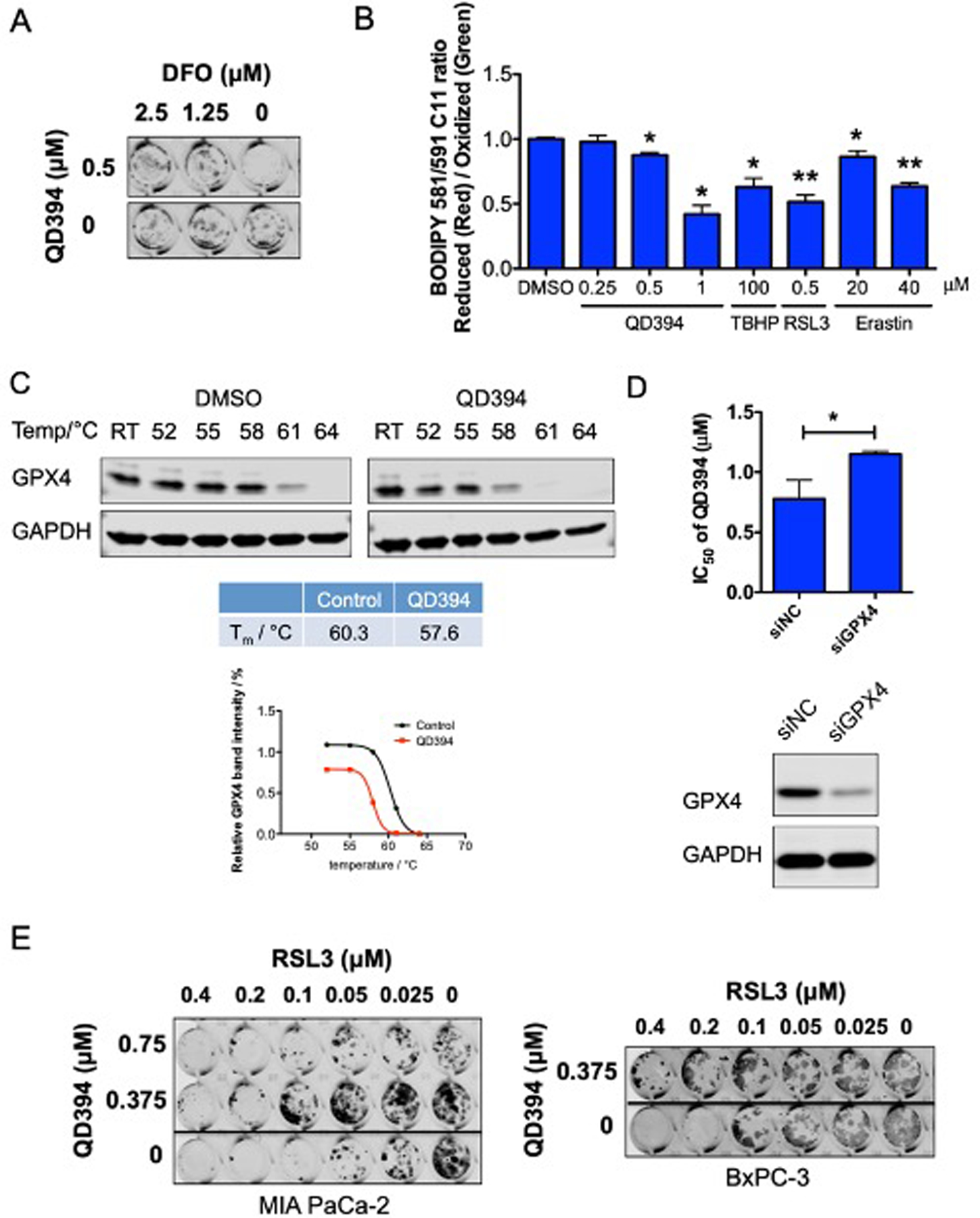
QD394 induces iron-dependent and GPX4-mediated ferroptosis in MIA PaCa-2 cells. (A) DFO decreased the inhibition of colony formation caused by QD394. (B) QD394 significantly induced lipid peroxidation after 24 h treatment, similar to TBHP, RSL3, and erastin. * denotes p < 0.05, ** denotes p < 0.005. (C) The potential interaction between QD394 and GPX4 was determined by cellular thermal shift assay. Detailed procedures are described in the Experimental Section. QD394 decreased the melting temperature of GPX4 protein. Image J and GraphPad Prism were used for quantification. QD394 was tested at 100 μM for 1 h. RT: room temperature, around 21 °C. (D) Knockdown of GPX4 significantly reduced cytotoxicity of QD394. siNC: samples treated with scrambled siRNA; siGPX4: samples treated with GPX4 siRNA. * denotes p < 0.05. Cells were immunoblotted with GPX4 antibody to validate GPX4 knockdown. (E) RSL3 inhibited colony formation in pancreatic cancer cells, and QD394 rescued its effect.
The final step in ferroptosis is an increase in lipid peroxidation.15–18 We measured the lipid ROS level in QD394-treated cells using the BODIPY C11 dye to validate QD394-induced ferroptosis cell death. QD394 significantly increased lipid ROS in a dose-dependent manner (Figure 3B). TBHP (an ROS inducer), RSL3 (a GPX4 inhibitor), and erastin (an SLC7A11 inhibitor) all increased lipid ROS. Beside iron and lipid peroxidation, GPX4 is another central mediator of ferroptosis. GPX4 catalyzes the reduction of hydrogen peroxide, organic hydroperoxides, and lipid hydroperoxides, and thereby protects cells against oxidative damage.15, 29 Therefore, we investigated whether there is a connection between QD394 and GPX4. In the cellular thermal shift assay (CETSA), QD394 destabilized GPX4 protein, indicated by the reduction in its melting temperature (from 60.34 °C to 57.62 °C) in MIA PaCa-2 cells (Figure 3C). Destabilization of GPX4 by QD394 was also observed in PANC-1 cells (Figure S5). Binding to a protein generally stabilizes it, however, QD394 destabilized GPX4 and it may be caused by an alternative binding mode or indirect interaction between them. Furthermore, the cytotoxicity of QD394 was significantly decreased in MIA PaCa-2 cells with GPX4 knocked down using siRNA, further suggesting that QD394 regulates GPX4-mediated cell death (Figure 3D). Additionally, QD394 rescued the cytotoxicity of RSL3 in pancreatic cancer cells, suggesting that QD394 targets GPX4 (Figure 3E). We hypothesized that QD394 and RSL3 may work as competitive inhibitors to bind to the same site of GPX4, thus blocking both their activity when they were combined at certain concentrations. In summary, QD394 induces an iron-dependent and GPX4-mediated cell death, which is closely related to ROS induction.
QD394 interferes with mitochondrial gene expression and RNA catabolic process.
To further investigate the Bru-seq results at the protein level, we performed proteomics and measured the changes caused by QD394. QD394 increased the expression of 84 proteins and decreased the expression of 146 proteins by at least 1.5-fold (FDR < 0.05) after 24 h treatment in MIA PaCa-2 cells (n = 2). There are 17 upregulated and 35 downregulated genes/proteins in common between the proteomics and Bru-seq analysis (Figure 4A). In the STRING analysis,30 the 17 proteins with increased expression were involved in response to endoplasmic reticulum (ER) stress, the unfolded protein response, and the transcription from RNA polymerase II promoter related to oxidative stress (Table S4). Furthermore, the 35 proteins with decreased protein expression had a close connection with mitochondrial genes, mitochondrial translation regulation, and cellular metabolic processes (Table S5, Figure 4B). QD394 reduced expression of proteins involved in mitochondrial RNA catabolic processes, which is consistent with the Bru-seq GSEA analysis showing that QD394 downregulated the KEGG RNA degradation gene set. We observed that LRPPRC and PNPT1 were involved in many of the biological processes downregulated by QD394 (Table S5). LRPPRC (Leucine Rich Pentatricopeptide Repeat Containing) encodes a protein localized primarily to mitochondria, and it may regulate RNA metabolism and transcription in both nuclei and mitochondria. PNPT1 (Polyribonucleotide Nucleotidyltransferase 1) encodes a mitochondrial intermembrane protein in the polynucleotide phosphorylase family comprised of phosphate-dependent 3’-to-5’ exoribonucleases implicated in RNA processing and degradation. More importantly, we discovered that high expression of both genes significantly correlated with reduced overall survival of pancreatic cancer patients in the TCGA PAAD dataset (Figure 4C). In the 24 h proteomics experiment, QD394 reduced the expression of these two proteins (FDR < 0.05); additionally, QD394 significantly decreased their expression at 4 h (Figure 4D). Napabucasin also decreased the protein expression of these two genes at 4 h, while H2O2 did not. We combined NAC and dicoumarol (a NQO1 inhibitor) with QD394 and napabucasin to assess the effects on the expression of LRPPRC and PNPT1 (Figure S6). NAC did not affect the downregulation of these two proteins caused by QD394 and napabucasin; however, dicoumarol blocked the reduction, indicating that NQO1 is a potential regulator in this process. Moreover, in PANC-1 cells, which have little intrinsic NQO1 expression, we did not observe the downregulation of LRPPRC and PNPT1 proteins by QD394 and napabucasin (Figure S6).
Figure 4.
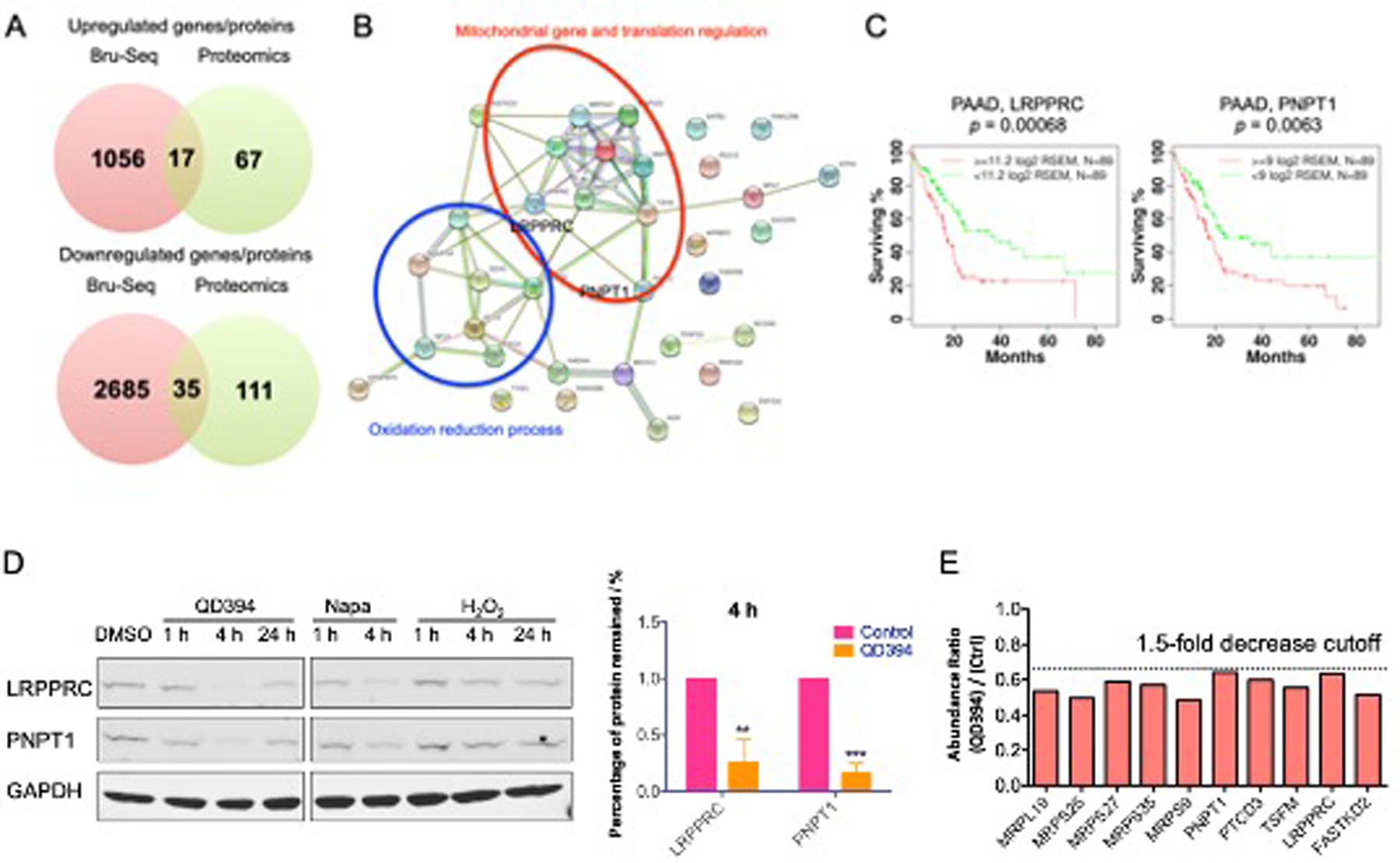
Common genes/proteins between Bru-seq and proteomics analysis reveal that QD394 interferes with mitochondrial gene expression and RNA catabolic process. (A) Venn diagrams of upregulated and downregulated genes/proteins between Bru-seq (fold change over 2) and proteomics (fold change over 1.5) analysis. (B) STRING analysis of 35 downregulated genes/proteins in common between Bru-seq and proteomics studies. Red circle highlights the genes involved in mitochondrial gene and translation regulation. Blue circle indicates the genes associated with the oxidation reduction process. (C) LRPPRC and PNPT1 are significantly related to the overall survival of pancreatic adenocarcinoma patients in TCGA dataset. (D) QD394 decreased the protein expression of LRPPRC and PNPT1 in MIA PaCa-2 cells. QD394 and napabucasin were tested at 3 × IC50 for 1 h and 4 h. QD394 was tested at 2 × IC50 for 24 h. H2O2 was used at 150 μM. * denotes p < 0.05, ** denotes p < 0.005, *** denotes p < 0.0005. (E) The expression levels of mitochondrial proteins decreased in the QD394-treated MIA PaCa-2 cells.
Besides these two mitochondrial proteins related to RNA processing, QD394 also reduced expression of other mitochondrial proteins in the proteomics study, substantiating its possible role in affecting mitochondrial function (Figure 4E). Overall, QD394 downregulated mitochondrial proteins, including LRPPRC and PNPT1 in both Bru-seq and proteomics experiments.
QD394 is synergistic with napabucasin and select FDA-approved drugs.
ROS inducers sensitize cancer cells to standard-of-care chemotherapy and radiation.31 QD394 and napabucasin showed synergism in the colony formation assay in both MIA PaCa-2 and PANC-1 cells (Figure 5A and S7), suggesting the potential development of combination studies between redox modulator QD394 and cancer stemness inhibitors such as napabucasin. We next tested whether QD394 would act in synergy with the ROS inducer arsenic trioxide (As2O3), which is approved to treat acute promyelocytic leukemia. As2O3 has been shown to reduce protein expression of Bcl-2, increase cellular ROS, induce permeability transition pore complex opening, and impair cell proliferation and apoptosis.32 QD394 sensitizes cells to As2O3, supporting synergistic potentials between QD394 and different redox modulators (Figure 5B). Since QD394 causes DNA damage, we assessed the synergy between QD394 and compounds regulating DNA repair. Olaparib, a poly(ADP-ribose) polymerase (PARP) inhibitor, is an FDA-approved drug to treat breast, ovarian, and pancreatic cancer. Combination of QD394 and olaparib resulted in synergistic effects in the colony formation assay (Figure 5C). The full colony formation assay images and their synergism calculation using Highest Single Agent (HSA) and Bliss models in the Combenefit software are shown in the Figure S8. Cumulatively, QD394 is synergistic with cancer stemness inhibitors, other redox modulators, and PARP inhibitors.
Figure 5.
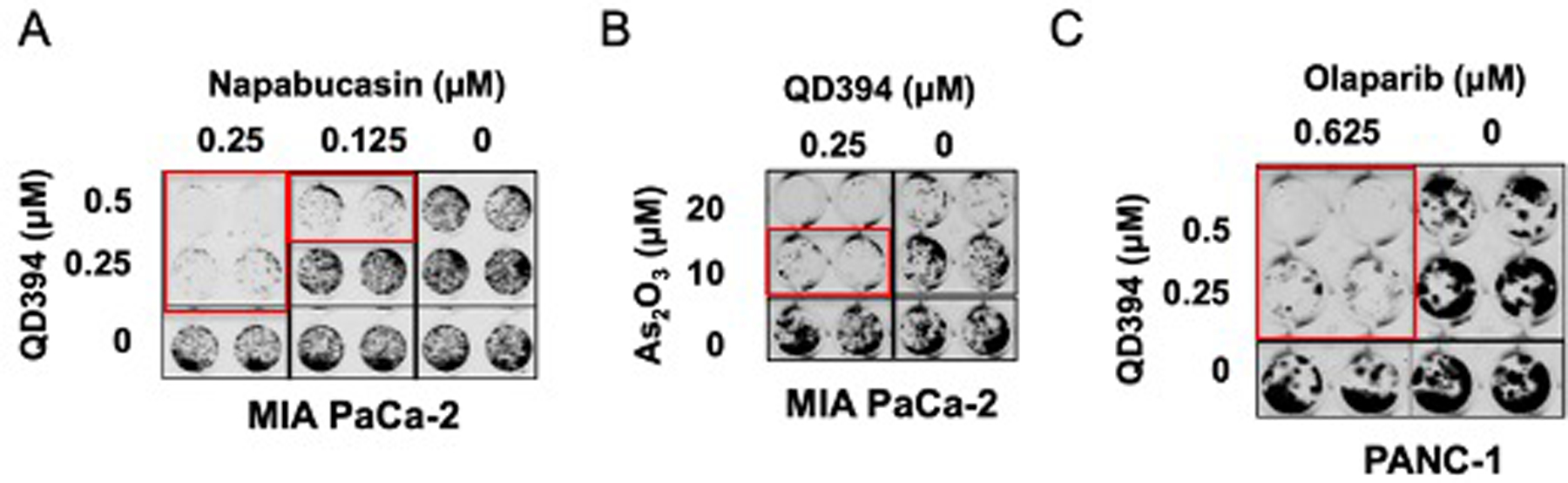
QD394 is synergistic with select FDA-approved drugs in pancreatic cancer cells. Napabucasin (A), As2O3 (B), and olaparib (C) were added 1 h before QD394. Cells were treated with drugs for 24 h. Red rectangles labeled the wells showing synergy.
QD394-Me shows improved plasma stability and reduced toxicity in mice as compared to QD394.
We determined the stability of QD394 in mouse liver microsomes and plasma. The half-life of QD394 is over 60 minutes in mouse liver microsomes, yet only six minutes in mouse plasma, indicating it has good microsomal stability but poor metabolic stability in plasma (Table 3). We then determined its potential metabolites in mouse plasma to better guide the design of more metabolically stable derivatives (Figure 6A). M1 and M2 metabolites have hydroxyl groups on the rings, and M3 and M4 metabolites are produced through a Michael addition. We then synthesized QD394-Me with a methyl group to block Michael addition (Figure 6B). This improved the plasma stability of QD394 from six minutes to over two hours. Next, we calculated the PK parameters of QD394 and QD394-Me in CD-1 mice following intraperitoneal injection (IP, 10 mg/kg), oral administration (PO, 20 mg/kg), or intravenous (IV, 10 mg/kg) injection. Plasma concentration of both QD394 and QD394-Me rapidly decreased within the first two hours (Figure S9). Notably, two out of three mice administered with QD394 via IV injection died likely due to systemic toxicity, while all three mice that received IV injections of QD394-Me survived throughout the study (Table 3). Therefore, QD394-Me appears to be less toxic and more tolerable in mice compared to QD394.
Table 3.
PK optimization of QD394. Microsomal stability, plasma stability, and PK studies were performed to evaluate the PK profiles of QD394 and its analog QD394-Me. Key parameters for each experiment are listed.
| Compound | Microsomal stability T1/2 (min) | Plasma stability T1/2 (min) | Concentration at 7 h in mouse plasma (ng/mL) | |||
|---|---|---|---|---|---|---|
| mouse | mouse | human | IP (n=2) | PO (n=2) | IV (n=3) | |
| QD394 | > 60 | 6 | NA | 5.0, 4.5 | 23.3, 25.8 | 2 out of 3 mice died |
| QD394-Me | 44.6 | > 120 | > 120 | 6.7, 1.1 | 4.2, 3.4 | 3.4 ± 1.2 |
Figure 6.
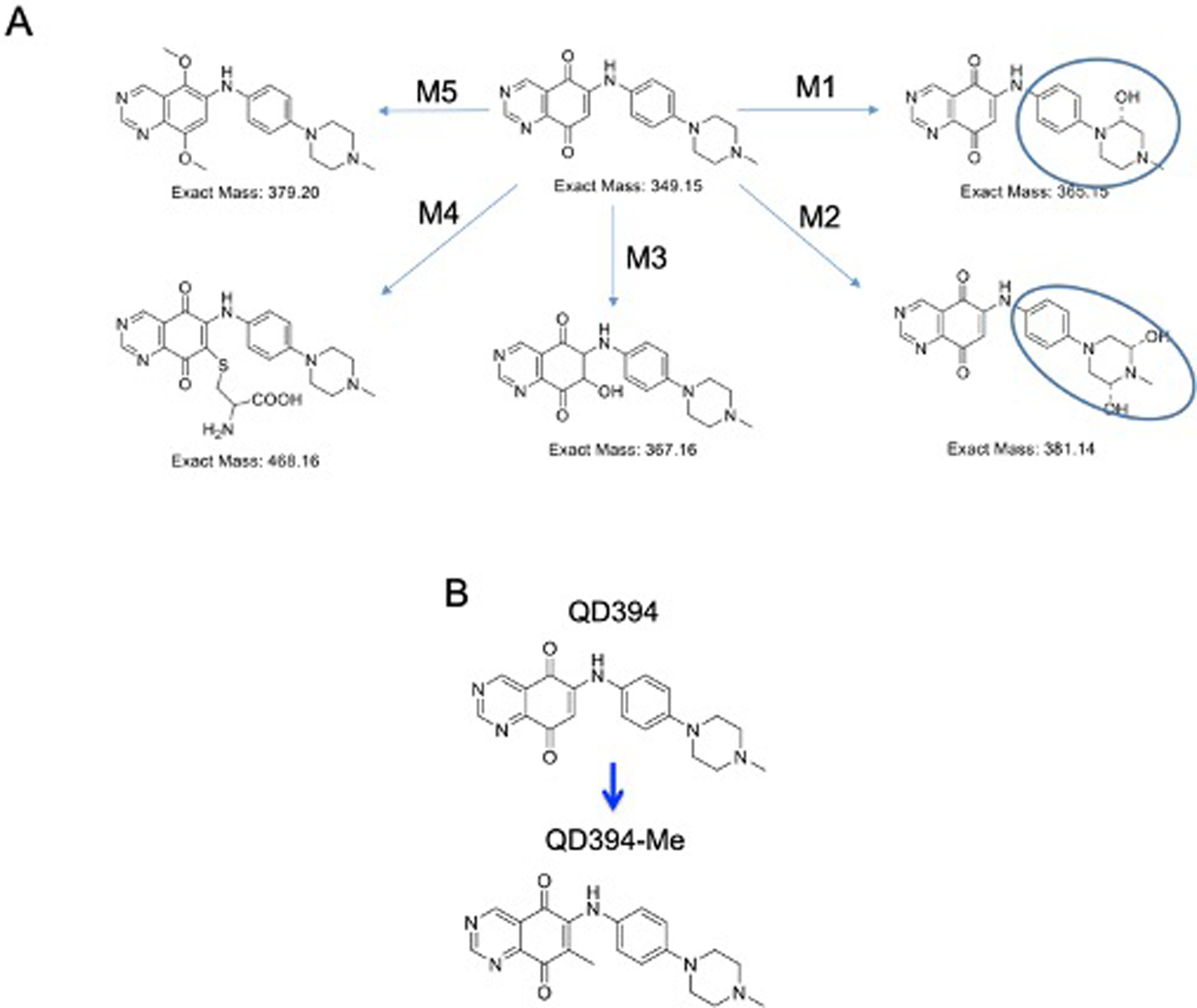
Pharmacokinetics-guided optimization of QD394 led to QD394-Me having better stability in mouse plasma. (A) Potential metabolites of QD394 in mouse plasma. The hydroxyl groups could be on either ring highlighted by circles. (B) Structures of QD394 and QD394-Me.
To understand why the concentration of QD394-Me rapidly decreased in the plasma, we assessed tissue distribution of QD394-Me administered via IV and PO in mice. After one-hour treatment, colon had the highest concentration of QD394-Me, although this was relatively low compared to the dosage (Colon: around 0.3 mg/kg, PO: 20 mg/kg, IV: 10 mg/kg) (Table S6). Besides distribution to organs, we identified potential metabolites of QD394-Me. The major metabolite of QD394-Me contains two hydroxyl groups on the right benzene or piperazine ring (Figure S10). As early as 15 min, we observed the major metabolite in the plasma; however, both the parent compound and metabolite were distributed and excreted quickly over time (Figure S10).
On the basis of tissue distribution results, e.g. where colon tissue had much higher concentration of QD394-Me than pancreas, we conducted a preliminary animal study in immuno-competent Balb/c mice using mouse colon cancer cell line CT-26. Three out of five mice displayed tumor shrinkage in the QD394-Me IV (10 mg/kg) and QD394 IP (20 mg/kg) treatment groups. (Figure S11). A full assessment of in vivo efficacy studies in orthotopic models of colon and pancreatic cancers will be published in due course.
QD394-Me and QD394 share similar cell death mechanisms.
QD394-Me and QD394 show similar cytotoxicity in PANC-1 and BxPC-3 cells, while QD394-Me is less potent than QD394 in MIA PaCa-2 cells (Table 1). QD394-Me also significantly increased cellular ROS in MIA PaCa-2 cells, and NAC blocked the induction of ROS (Figure 7A). Similar to QD394, QD394-Me increased lipid ROS significantly in a dose-dependent manner (Figure 7B). QD394-Me destabilized GPX4 protein in pancreatic cancer cells in the CETSA experiments (Figure 7C and S4), and its cytotoxicity was significantly reduced in the GPX4-knockdown experiments (Figure 7D, Table S7). Moreover, QD394-Me significantly decreased protein expression of LRPPRC and PNPT1 at 4 h, consistent with QD394 in MIA PaCa-2 cells (Figure 7E). Therefore, QD394-Me shows a similar mechanism of action to QD394 in pancreatic cancer cells but has improved plasma stability and less in vivo toxicity.
Figure 7.
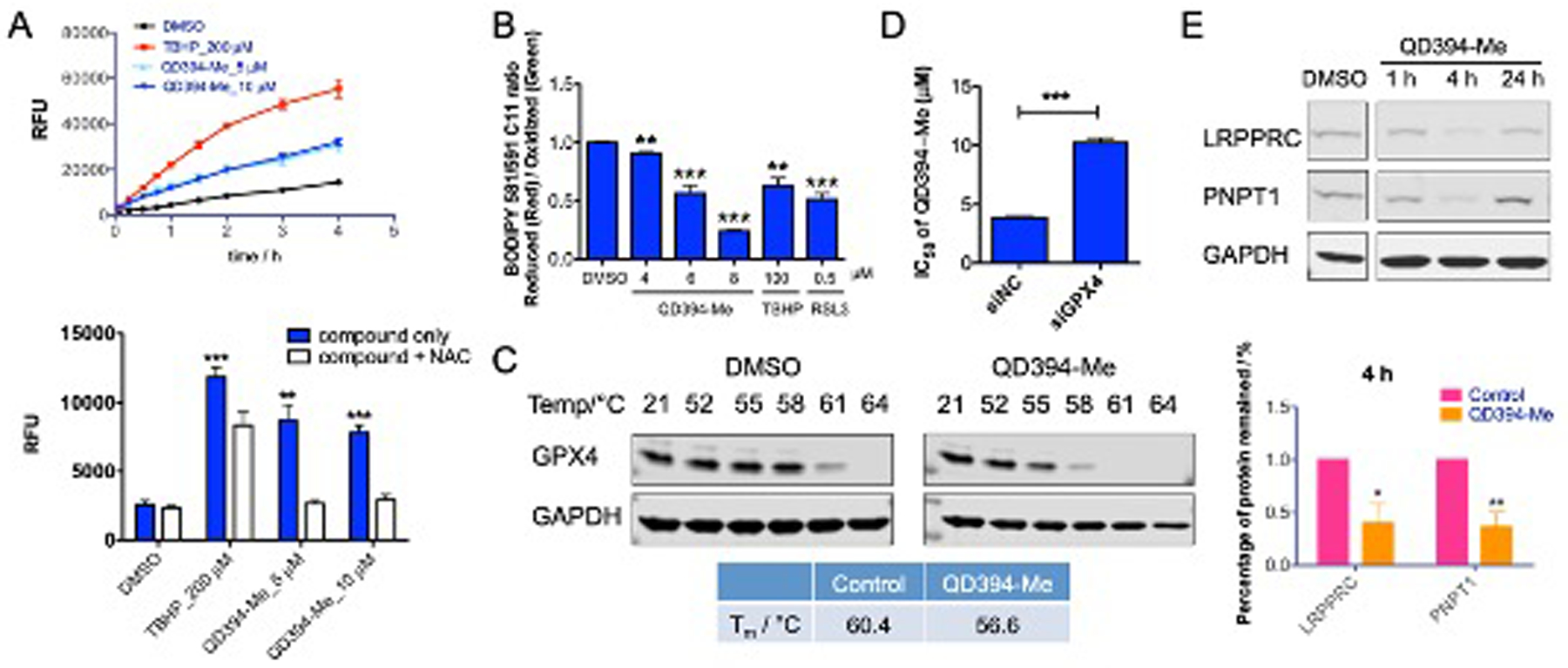
QD394-Me has a similar cellular profile to QD394. (A) QD394-Me increased cellular ROS level, and NAC reduced the induction of ROS at 30 min. ** denotes p < 0.005, *** denotes p < 0.0005. (B) QD394-Me significantly increased lipid peroxidation in MIA PaCa-2 cells. (C) QD394-Me destabilizes GPX4 protein in MIA PaCa-2 cells. QD394-Me was tested at 100 μM. (D) The cytotoxicity of QD394-Me was significantly reduced in the GPX4-knockdown MIA PaCa-2 cells. siNC: samples treated with scrambled siRNA; siGPX4: samples treated with GPX4 siRNA. (E) QD394-Me reduced the protein expression of LRPPRC and PNPT1 significantly at 4 h in MIA PaCa-2 cells. QD394-Me was tested at 3 × IC50 for 1 h and 4 h, and 2 × IC50 for 24 h. * denotes p < 0.05, ** denotes p < 0.005.
Bru-seq analysis of QD394-Me.
We analyzed gene expression profile of QD394-Me in MIA PaCa-2 using Bru-seq. QD394-Me induced hallmark gene sets related to TNF signaling and hypoxia, which is similar to QD394. Additionally, QD394-Me increased unfolded protein response, suggesting potential ER stress (Figure 8, Table S8). QD394-Me also induced ROS pathway and JAK/STAT3 signaling hallmark gene sets, supporting its ROS-inducing capability and STAT3-targeting effects (Table S8). KEGG pathways related to proteasome, ribosome and spliceosome were enriched by QD394-Me, indicating its downstream effects on protein and gene regulations (Figure 8, Table S9). There are no common downregulated enriched gene sets between QD394 and QD394-Me Bru-seq samples.
Figure 8.
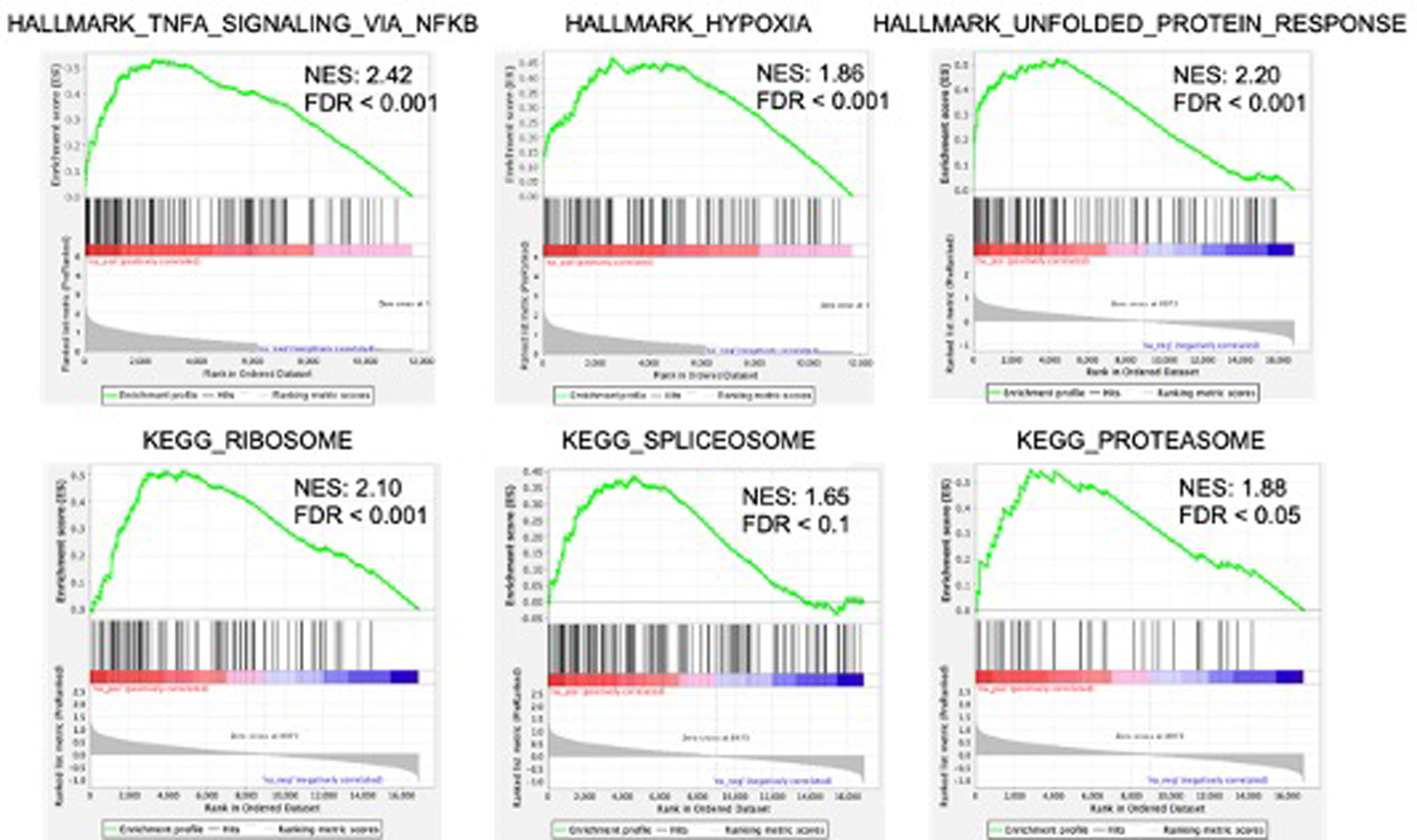
Select enriched gene sets of QD394-Me in MIA PaCa-2 Bru-seq samples.
DISCUSSION
ROS induction alters the activity of multiple transcription factors, including STAT3, NF-κB, and hypoxia-inducible factor-1α (HIF-1A).33 STAT3 signaling is downstream of MAPK, JAK, and Src pathways. Redox modulation can inactivate these kinases leading to either activation or inhibition of STAT3 signaling, depending on cellular context and duration of ROS generation.34 TNFA signaling and hypoxia hallmark gene sets were upregulated by QD394 and napabucasin. TNFA/TNF are important cytokines for maintaining cellular homeostasis and mediating the activation of NF-κB, which in turn regulates downstream prosurvival genes, MAPK signaling, and cell death in cancers and inflammatory diseases.35, 36 The transcription factor HIF-1A is activated by hypoxic conditions and responds to accumulated ROS via NF-κB activation.37, 38 ROS induction by QD394 and napabucasin can trigger these signaling pathways. Napabucasin is a cancer stemness inhibitor that reduces the proliferation, invasion, and stemness of cancer cells.39, 40 Napabucasin is bioactivated by NQO1 and generates ROS to induce DNA damage and cell death.13 In this study, we report that necrostatin-1 rescued napabucasin-mediated cell death, suggesting that napabucasin induces necroptotic cell death in pancreatic cancer cells. However, QD394 did not cause necroptosis, but iron-, ROS-dependent, and GPX4-mediated ferroptosis (Figure 9).
Figure 9.
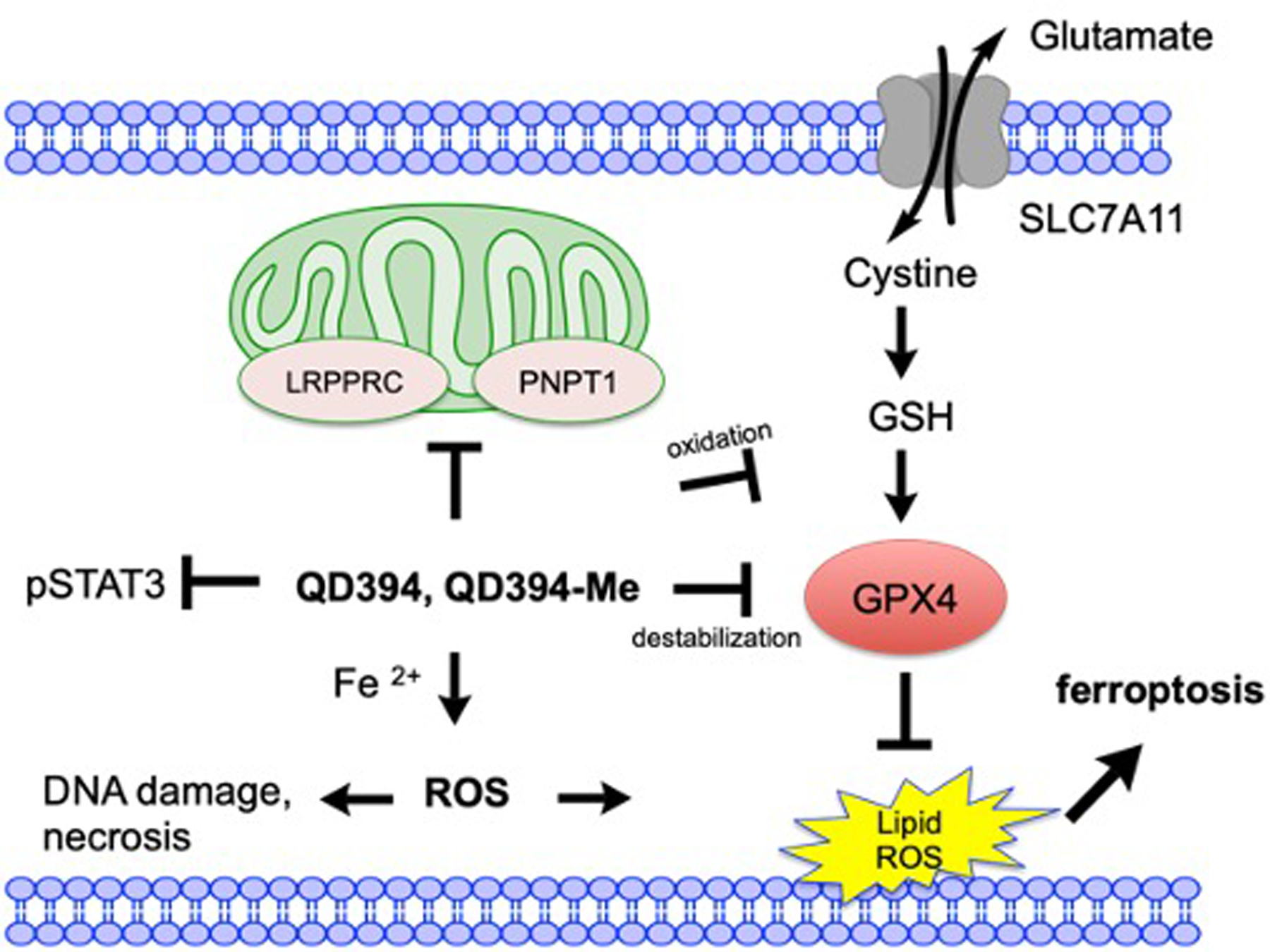
A summary of the working mechanisms of QD394 and QD394-Me in pancreatic cancer cells. QD394 and QD394-Me increase cellular ROS level, induce DNA damage, decrease mitochondrial proteins, and promote accumulation of lipid ROS. They oxidize GSH to GSSG and destabilize GPX4 in the cells. Their cytotoxicity is dependent on iron, ROS, and GPX4 expression. Ferroptosis and necrosis are potentially involved in their cell death mechanisms.
Liproxstatin-1 inhibits ferroptosis induced by erastin, BSO and RSL3 in vitro, and it prevents ferroptosis and ROS accumulation in GPX4 knockout cells both in vitro and in vivo.41, 42 Ferrostatin-1 is a first-generation ferroptosis inhibitor that rescues cell death caused by erastin and RSL3.15, 43 Additionally, both compounds are effective radical-trapping antioxidants in the lipid bilayers.43 However, ferrostatin-1 did not prevent H2O2-induced necrosis, although ferroptosis is defined as a necrotic-like cell death.15 The fact that QD394 induces ROS may explain why ferrostatin-1 did not rescue QD394-induced cell death. Although ferrostatin-1 blocked lipid peroxidation and ferroptosis, QD394 could cause other ROS-involved DNA damage and necrotic cell death (Figure 9). Therefore, we suggest that QD394 and QD394-Me kill cells through a combination of different cell death pathways and ferroptosis is an essential mechanism since GPX4 protein expression regulates the cytotoxicity of QD394 and QD394-Me. Interestingly, we determined potential interactions between QD394, QD394-Me, and GPX4 in the cells, but the detailed binding site requires further investigation.
LRPPRC and PNPT1 are closely related to mitochondrial translation and RNA catabolic processes in different diseases.20, 21, 44, 45 LRPPRC deficiency alters mitochondrial electron transport chain, mitochondrial permeability, and transmembrane ROS diffusion.46 PNPT1 regulates the correct maturation of mitochondrial ND6 transcripts and efficient mitochondrial RNA degradation.21, 45 Both proteins are involved in mitochondrial function and negatively associated with the survival of pancreatic adenocarcinoma patients. Our redox modulators, QD394 and QD394-Me, decreased the expression of these two proteins, which could be used as potential pharmacodynamic biomarkers in pancreatic cancer (Figure 9). Meanwhile, further understanding of the connection between overexpression of these two genes and pancreatic cancer is needed and may lead to novel approaches to treat pancreatic cancer.
CONCLUSIONS
In this study, we developed a novel redox modulator QD394 and comprehensively studied its mechanisms of action in pancreatic cancer cells. QD394 shares a highly similar transcriptomic profile to napabucasin, inhibits STAT3 phosphorylation, increases ROS levels, and induces DNA damage. However, while napabucasin causes necroptosis, QD394 induces an iron-, ROS-dependent, and GPX4-mediated cell death. An essential cell death mechanism of QD394 is ferroptosis, since QD394 increases lipid peroxidation, destabilizes GPX4 protein, and its cytotoxicity is dependent on GPX4. Bru-seq and proteomics analysis reveal that QD394 decreases both nascent transcripts and protein expression of LRPPRC and PNPT1. Furthermore, QD394 acts in synergy with several FDA-approved drugs, suggesting its potential utility to overcome drug resistance. A PK-guided optimization campaign resulted in QD394-Me with improved mouse plasma stability and less systemic toxicity in mice.
EXPERIMENTAL SECTION
Chemistry.
General Methods.
All solvents and other chemicals were obtained from Sigma-Aldrich, Alfa Aesar, Carlo Erba (analytical or anhydrous grade) or Apollo Scientific Limited, Oakwood Chemical, Combi Blocks, Enamine (for amines), and were used without further purification). All reactions involving air- or moisture-sensitive compounds were performed under a nitrogen atmosphere using oven-dried glassware and syringes to transfer solutions. Melting points (Mp) were determined using an electrothermal melting point or a Köfler apparatus and are uncorrected. Nuclear magnetic resonance (H 1H-NMR, 13C-NMR) spectra were determined in CDCl3 or DMSO-d6 and were recorded on a 400 MHz Bruker Avance III. Chemical shifts are reported in parts per million (ppm) downfield from tetramethylsilane (TMS) and used as an internal standard. Splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet; brs, broad singlet; dd, double doublet. The assignment of exchangeable protons (OH and NH) was confirmed by the addition of D2O. Mass spectra were obtained on a Hewlett-Packard 5989 mass engine spectrometer, or a MALDI micro MX (Waters, Micromass) equipped with a reflector analyzer. Analytical thin-layer chromatography (TLC) was carried out on Merck silica gel F-254 plates. Flash chromatography purifications were performed on Merck Silica gel 60 (230–400 mesh ASTM) as a stationary phase. Analytical HPLC for the lead compounds was carried out on an Agilent 1260 Infinity II Quaternary pump (Agilent Technologies, Italia S.p.A.). Elemental analyses for tested compounds were performed on a Perkin-Elmer 2400 spectrometer at Laboratorio di Microanalisi, Dipartimento di Chimica, Università di Sassari (Italy), and were within ± 0.4% of the theoretical values (Table S1), thus confirming ≥95% purity.
General method for the preparation of the compounds QD385 – 395
A solution of quinazoline-5,8-dione (1), cerium (III) chloride heptahydrate (CeCl3•7H2O, 1.1 eq.) and substituted aniline (1.0 eq.) in absolute ethanol was stirred at room temperature for 1.5 h. Next, after removing most of the ethanol under vacuum, the crude residue was diluted with water, extracted with CH2Cl2 and washed with water. The organic layers were dried over sodium sulfate (Na2SO4) and concentrated to dryness. Then, the product was purified by flash chromatography to give the desired compound.
6-(Biphenyl-3-ylamino)quinazoline-5,8-dione [QD 385]
Quinazoline-5,8-dione (1, 0.050 g, 0.312 mmol), cerium (III) chloride heptahydrate (CeCl3•7H2O, 1.1 eq., 0.128 g, 0.343 mmol), 3-aminobiphenyl (1.0 eq., 0.053 g, 0.312 mmol), and absolute ethanol (6 mL). Flash chromatography (dichloromethane-methanol = 9.8:0.2) gave compound QD385 as a violet powder. Yield: 20%. Rf: 0.41 (ether petroleum : ethyl acetate = 3:7). mp: 199–200 °C. 1H-NMR 400 MHz 1H-NMR (DMSO-d6): δ 9.63 (s, 1H), 9.43 (s, 1H), 7.69–7.67 (m, 3H), 7.61–7.59 (d, 2H), 7.49–7.46 (t, 2H), 7.41–7.36 (t, 3H), 6.34 (s, 1H). 13C-NMR (DMSO-d6): δ 163.78, 130.33, 129.03, 127.15, 125.61, 121.74, 105.15. MS: m/z 327 (M+).
6-(4’-Chlorobiphenyl-4-ylamino)quinazoline-5,8-dione [QD 386]
Quinazoline-5,8-dione (1, 0.050 g, 0.312 mmol), cerium (III) chloride heptahydrate (CeCl3•7H2O, 1.1 eq., 0.128 g, 0.343 mmol), 4-amino-4’-chlorobiphenyl (1.0 eq., 0.064 g, 0.312 mmol), and absolute ethanol (6 mL). Flash chromatography (ethyl acetate-petroleum ether = 5:5) gave compound QD386 as a violet powder. Yield: 95%. Rf: 0.20 (ethyl acetate-petroleum ether = 7:3). mp: 292–293 °C. 1H-NMR (DMSO-d6): δ 9.62 (s, 2H), 9.42 (s, 1H), 7.79–7.77 (d, 2H), 7.76–7.74 (d, 2H), 7.55–7.53 (d, 2H), 7.50–7.48 (d, 2H), 6.35 (s, 1H). 13C-NMR (DMSO-d6): δ 180.52, 180.03, 162.59, 155.67, 153.74, 145.89, 138.07, 137.42, 135.77, 132.34, 128.91, 128.26, 127.53, 124.06, 104.06. MS: m/z 361 (M+).
6-(3’,5’-Dichlorobiphenyl-4-ylamino)quinazoline-5,8-dione [QD 387]
Quinazoline-5,8-dione (1, 0.050 g, 0.312 mmol), cerium (III) chloride heptahydrate (CeCl3•7H2O, 1.1 eq., 0.128 g, 0.343 mmol), 4-amino-3’,5’-dichlorobiphenyl (1.0 eq., 0.074 g, 0.312 mmol), and absolute ethanol (6 mL). Flash chromatography (ethyl acetate-petroleum ether = 5:5) gave compound QD387 as a violet powder. Yield: 95%. Rf: 0.50 (ethyl acetate-petroleum ether = 7:3). mp: 260–261 °C. 1H-NMR (DMSO-d6): δ 9.63 (s, 2H), 9.43 (s, 1H), 7.87–7.85 (d, 2H), 7.79 (s, 2H), 7.61 (s, 1H), 7.52–7.50 (d, 2H), 6.38 (s, 1H).13C-NMR (DMSO-d6): δ 180.47, 180.15, 162.60, 155.70, 153.68, 145.75, 142.80, 138.28, 134.68, 133.99, 128.02, 125.20, 123.91, 104.34. MS: m/z 395 (M+).
6-(4-(Piperidin-1-yl)phenylamino)quinazoline-5,8-dione [QD 388]
Quinazoline-5,8-dione (1, 0.050 g, 0.312 mmol), cerium (III) chloride heptahydrate (CeCl3•7H2O, 1.1 eq., 0.128 g, 0.343 mmol), 4-(1-piperidinyl)aniline (1.0 eq., 0.055 g, 0.312 mmol), and absolute ethanol (6 mL). Flash chromatography (ethyl acetate-petroleum ether = 7:3) gave compound QD388 as a violet powder. Yield: 66%. Rf: 0.29 (ethyl acetate-petroleum ether = 7:3). mp: 209 °C. 1H-NMR (DMSO-d6): δ 9.60 (s, 1H), 9.38 (s, 1H), 7.45 (s, 1H), 7.22–7.20 (d, 2H), 7.02–7.00 (d, 2H), 6.13 (s, 1H), 3.16 (s, 4H), 1.63–1.56 (d, 6H). 13C-NMR (DMSO-d6): δ 180.65, 179.30, 162.59, 155.51, 154.08, 149.52, 146.47, 127.71, 124.93, 124.12, 115.91, 102.59, 49.23, 25.09, 23.83. MS: m/z 334 (M+).
6-(4-Morpholinophenylamino)quinazoline-5,8-dione [QD 389]
Quinazoline-5,8-dione (1, 0.050 g, 0.312 mmol), cerium (III) chloride heptahydrate (CeCl3•7H2O, 1.1 eq., 0.128 g, 0.343 mmol), 4-(4-morpholinyl)aniline (1.0 eq., 0.056 g, 0.312 mmol), and absolute ethanol (6 mL). Flash chromatography (ethyl acetate-petroleum ether = 7:3) gave compound QD389 as a violet powder. Yield: 88%. Rf: 0.10 (ethyl acetate-petroleum ether = 7:3). mp: 226–227 °C. 1H-NMR (DMSO-d6): δ 9.60 (s, 1H), 9.47 (s, 1H, H-N), 9.39 (s, 1H), 7.26–7.24 (d, 2H), 7.05–7.03 (d, 2H), 6.14 (s, 1H), 3.76 (s, 4H), 3.15 (s, 4H). 13C-NMR (DMSO-d6): δ 180.62, 179.41, 162.59, 155.53, 149.03, 146.49, 128.56, 124.97, 115.40, 102.70, 66.00, 48.18. MS: m/z 336 (M+).
6-(9H-Fluoren-4-ylamino)quinazoline-5,8-dione [QD390]
Quinazoline-5,8-dione (1, 0.050 g, 0.312 mmol), cerium (III) chloride heptahydrate (CeCl3•7H2O, 1.1 eq., 0.128 g, 0.343 mmol), 1-aminofluorene (1.0 eq., 0.057 g, 0.312 mmol), and absolute ethanol (6 mL). Flash chromatography (ethyl acetate-petroleum ether = 7:3) gave compound QD390 as a violet powder. Yield: 69%. Rf: 0.46 (ethyl acetate-petroleum ether = 7:3). mp: 217 °C. 1H-NMR (CDCl3): δ 9.68 (s, 1H), 9.53 (s, 1H), 7.57–7.55 (d, 2H), 7.52–7.49 (t, 1H), 7.45–7.41 (t, 1H), 7.38–7.35 (t, 1H), 7.31–7.29 (d, 1H), 6.43 (s, 1H), 3.86 (s, 2H). 13C-NMR (CDCl3): δ 163.78, 156.33, 128.79, 127.74, 127.25, 125.18, 121.70, 120.42, 119.04, 105.53. MS: m/z 339 (M+).
6-(9H-Fluoren-2-ylamino)quinazoline-5,8-dione [QD 391]
Quinazoline-5,8-dione (1, 0.050 g, 0.312 mmol), cerium (III) chloride heptahydrate (CeCl3•7H2O, 1.1 eq., 0.128 g, 0.343 mmol), 2-aminofluorene (1.0 eq., 0.057 g, 0.312 mmol), and absolute ethanol (6 mL). Flash chromatography (ethyl acetate-petroleum ether = 5:5) gave compound QD391 as a violet powder. Yield: 92%. Rf: 0.14 (ethyl acetate-petroleum ether = 7:3). mp: 236–237 °C. 1H-NMR (DMSO-d6): δ 9.62 (s, 2H), 9.43 (s, 1H), 8.00–7.97 (d, 1H), 7.92–7.91 (d, 1H), 7.62–7.60 (d, 2H), 7.43–7.39 (t, 2H), 7.35–7.32 (t, 1H), 6.35 (s, 1H), 4.00 (s, 1H). 13C-NMR (DMSO-d6): δ 155.64, 153.82, 146.13, 144.27, 143.19, 140.42, 138.78, 136.25, 126.84, 125.13, 124.14, 122.77, 120.67, 120.54, 119.99, 103.74, 65.70. MS: m/z 339 (M+).
6-(4’-Ethylbiphenyl-4-ylamino)quinazoline-5,8-dione [QD 392]
Quinazoline-5,8-dione (1, 0.050 g, 0.312 mmol), cerium (III) chloride heptahydrate (CeCl3•7H2O, 1.1 eq., 0.128 g, 0.343 mmol), 4-amino-4’-ethylbiphenyl (1.0 eq., 0.061 g, 0.312 mmol), and absolute ethanol (6 mL). Flash chromatography (ethyl acetate-petroleum ether = 5:5) gave compound QD392 as a violet powder. Yield: 77%. Rf: 0.19 (ether petroleum : ethyl acetate = 5:5). mp: 247 °C. 1H-NMR 400 MHz (DMSO-d6): δ 9.63 (s, 2H), 9.43 (s, 1H), 7.76–7.74 (d, 2H), 7.64–7.62 (d, 2H), 7.49–7.47 (d, 2H), 7.33–7.31 (d, 2H), 6.36 (s, 1H), 2.69–2.63 (m, 2H), 1.24–1.21 (t, 3H). 13C-NMR 100MHz (DMSO-d6): δ 185.78, 185.24, 167.73, 161.00, 159.01, 151.19, 148.40, 142.53, 141.91, 133.63, 132.54, 131.68, 129.33, 109.12, 33.02, 20.77. MS: m/z 355 (M+).
6-(4-Cyclohexylphenylamino)quinazoline-5,8-dione [QD 393]
Quinazoline-5,8-dione (1, 0.050 g, 0.312 mmol), cerium (III) chloride heptahydrate (CeCl3•7H2O, 1.1 eq., 0.128 g, 0.343 mmol), 4-cyclohexylaniline (1.0 eq., 0.055 g, 0.312 mmol), and absolute ethanol (6 mL). Flash chromatography (ethyl acetate-petroleum ether = 5:5) gave compound QD393 as a violet powder. Yield 53%. Rf: 0.24 (ether petroleum : ethyl acetate = 5:5). mp: 234 °C. 1H-NMR (DMSO-d6): δ 9.61 (s, 1H), 9.53 (s, 1H), 9.40 (s, 1H), 7.34–7.28 (m, 4H), 6.21 (s, 1H), 1.82–1.80 (m, 5H), 1.73–1.70 (d, 1H), 1.47–1.33 (m, 5H). 13C-NMR (DMSO-d6): δ 180.60, 162.58, 155.59, 153.85, 145.32, 127.53, 123.91, 43.26, 38.87, 26.29, 25.53. MS: m/z 333 (M+).
6-(4-(4-Methylpiperazin-1-yl)phenylamino)quinazoline-5,8-dione [QD 394]
Quinazoline-5,8-dione (1, 0.050 g, 0.312 mmol), cerium (III) chloride heptahydrate (CeCl3•7H2O, 1.1 eq., 0.128 g, 0.343 mmol), 4-(4-methylpiperazin-1-yl)aniline (1.0 eq., 0.060 g, 0.312 mmol), and absolute ethanol (6 mL). Flash chromatography (dicloromethane:methanol = 8:2) gave compound QD394 as a violet powder. Yield: 70%. Rf: 0.30 (dicloromethane:methanol = 8:2). mp: 204–205 °C. 1H-NMR (DMSO-d6): δ 9.60 (s, 1H), 9.46 (s, 1H), 9.39 (s, 1H), 7.24–7.22 (d, 2H), 7.04–7.01 (d, 2H), 6.13 (s, 1H), 3.19–3.17 (m, 4H), 2.47 (s, 4H), 2.23 (s, 3H). MS: m/z 349 (M+).
6-(4-(Pyridin-2-yl)phenylamino)quinazoline-5,8-dione [QD 395]
Quinazoline-5,8-dione (1, 0.075 g, 0.468 mmol), cerium (III) chloride heptahydrate (CeCl3•7H2O, 1.1 eq., 0.192 g, 0.515 mmol), 4-(2-pyridil)aniline (1.0 eq., 0.080 g, 0.468 mmol), and absolute ethanol (9 mL). Flash chromatography (ether petroleum : ethyl acetate = 5:5) gave compound QD395 as a violet powder. Yield: 54%. Rf: 0.16 (ether petroleum : ethyl acetate = 2:8). mp: 239 °C. 1H-NMR (DMSO-d6): δ 9.67(s, 1H), 9.63 (s, 1H), 9.44 (s, 1H), 8.69–8.65 (d, 1H), 8.21–8.18 (d, 2H), 8.01–7.99 (d, 1H), 7.92–7.88 (m, 1H), 7.56–7.53 (dd, 2H), (m, 1H), 6.43 (s, 1H). MS: m/z 328 (M+).
2,5-Dimethoxy-4-methylnitrobenzene [6]
Yield: 80%. Rf = 0.90 (ethyl acetate-petroleum ether 3:7). mp: 116 °C. 1H NMR 400 MHz (CDCl3): δ 10.29 (s, 1H), 6.93 (s, 1H), 3.95 (s, 3H), 3.87 (s, 3H), 2.42 (s, 3H). 13C NMR 400 MHz (CDCl3): δ 150.98, 147.69, 136.77, 135.25, 116.46, 107.05, 57.10, 56.00, 16.83. MS: m/z 198 [M+H]+.
3,6-Dimethoxy-4-methyl-2-nitrobenzaldehyde [7]
To a solution of 3,6-dimethoxy-4-methyl benzaldehyde (1 eq., 4.0 g, 24.07 mmol) in CH2Cl2 (50 mL), conc. HNO3 (4.54 eq., 6 mL) in CH2Cl2 (25 mL) was added at room temperature under stirring. After 24 h, the obtained clear solution was cautiously poured onto aqueous Na2SO4. The organic layer was separated and dried over Na2SO4. The obtained crude product was then purified by flash chromatography on silica gel using ethyl acetate-petroleum ether (2.5:7.5) to give first the major side product, 2,5-dimethoxy-4-methylnitrobenzene (6), and then (by further elution with only ethyl acetate) the desired compound (7). Yield: 5%. Rf = 0.60 (ethyl acetate-petroleum ether 3:7). mp: 85 °C. 1H NMR 400 MHz (CDCl3): δ 10.29 (s, 1H), 6.93 (s, 1H), 3.95 (s, 3H), 3.87 (s, 3H), 2.42 (s, 3H). 13C NMR 400 MHz (CDCl3): δ 185.46, 157.60, 143.47, 142.28, 117.58, 115.54, 114.09, 62.90, 56.63, 17.47. MS: m/z 226 [M+H]+.
N,N′-[(3,6-Dimethoxy-4-methyl-2-nitrophenyl)methanediyl)diformamide [8]
A solution of 3,6-dimethoxy-4-methyl-2-nitrobenzaldehyde (7, 1.1 g, 4.87 mmol) in formamide (66.5 eq., 13 mL, 325 mmol), heated at 40°C, was exposed to dry HCl gas (1 h) until the temperature reached 80°C. Then, the solution was cooled to room temperature, and water/ice was added. A pale brownish yellow colored precipitate was formed, which was filtered, dried and triturated with ethyl acetate and petroleum ether to yield the desired compound. Yield: 70%. Rf = 0.16 (dichloromethane-methanol 9.5:0.5). mp: 238 °C. 1H NMR 400 MHz (DMSO-d6): δ 8.65 (d, 2H), 7.91 (s, 2H), 7.21 (s, 1H), 6.72 (t, 1H), 3.89 (s, 3H), 3.71 (s, 3H), 2.50 (s, 3H). 13C NMR 400 MHz (DMSO- d6): δ 160.14, 152.82, 141.76, 134.61, 119.71, 116.23, 116.09, 62.31, 56.54, 15.89. MS: m/z 298 [M+H]+.
5,8-Dimethoxy-7-methylquinazoline [9]
Zinc powder (15 eq., 1.4 g, 22 mmol,) was added to a suspension of N,N′-[(3,6-Dimethoxy-4-methyl-2-nitrophenyl)methanediyl)diformamide (8, 0.45 g, 1.5 mmol) in triturated ice and glacial acetic acid (2 mL), under constant magnetic stirring. The reaction mixture was stirred for 2 h in an ice bath and for 4 h at room temperature. After filtration of the reaction mixture through filter paper, the resulting solution was dropped on cooled 50% NaOH (5 mL), and the yellow colored suspension thus formed was left without stirring for 1 h. Next, the suspension was filtered and the solid dried (at 30−40 °C) to give a yellow powder. The solid was then solubilized in ethyl acetate, filtered, dried over anhydrous Na2SO4, and concentrated to dryness yielding the desired compound. Yield: 50%. Rf = 0.30 (ethyl acetate-petroleum ether 3:7). mp: 132 °C. 1H NMR 400 MHz (CDCl3): δ 9.65 (s, 1H), 9.31 (s, 1H), 6.72 (s, 1H), 4.02 (s, 3H), 4.00 (s, 3H), 2.51 (s, 3H). 13C NMR 400 MHz (CDCl3): δ 155.68, 155.27, 151.35, 146.12, 144.83, 136.39, 117.04, 108.33, 61.81, 55.86, 17.21. MS: m/z 205 [M+H]+.
7-methylquinazoline-5,8-dione [10]
A solution of 5,8-Dimethoxy-7-methylquinazoline (9, 0.15 g, 0.75 mmol) in (7:3) acetonitrile:water (5 mL) was cooled at 0 °C in an ice bath, and a solution of ceric ammonium nitrate (3 eq., 1.2 g, 2.1 mmol) in (9:1) acetonitrile:water (5 mL) was added dropwise. The reaction mixture was stirred for 20 min, then poured into ice/water and extracted (8−10 times) with dichloromethane. The organic layer was washed (5−6 times) with water, dried over anhydrous Na2SO4 and concentrated to dryness to give a brown powder. Yield: 90%. Rf = 0.52 (ethyl acetate-petroleum ether 3:7). mp: 118 °C. 1H NMR 400 MHz (CDCl3): δ 9.68 (s, 1H), 9.51 (s, 1H), 6.97 (s, 1H), 2.30 (s, 3H). 13C NMR 400 MHz (CDCl3): δ 183.29, 182.88, 162.84, 157.23, 152.51, 149.59, 135.12, 124.24, 16.81. MS: m/z 175 [M+H]+.
7-Methyl-6-((4-(4-methylpiperazin-1-yl)phenyl)amino)quinazoline-5,8-dione [QD394-Me]
A solution of 7-methylquinazoline-5,8-dione (10, 0.050 g, 0.312 mmol), cerium (III) chloride heptahydrate (CeCl3•7H2O, 1.1 eq., 0.128 g, 0.343 mmol) and 4-(4-methylpiperazin-1-yl)aniline (1.0 eq., 0.060 g, 0.312 mmol) in absolute ethanol (6 ml) was stirred at room temperature for 1−2 h. Next, most of the ethanol was removed under vacuum, and water was added, followed by the extraction with dichloromethane. The organic layers were washed with water and brine, dried over anhydrous Na2SO4, and concentrated to dryness. Then, the crude product was purified by flash chromatography to give the expected product. Yield: 90%. Rf = 0.30 (dichloromethane-methanol 9.5:0.5); mp: 142 °C. 1H NMR 400 MHz (DMSO-d6): δ 9.57 (s, 1H), 9.33 (s, 1H), 8.85 (s, 1H), 7.00 (d, 1H), 6.91 (d, 1H), 3.14 (m, 4H), 2.50 (m, 4H), 2.27 (s, 3H), 1.62 (s, 3H). 13C NMR 400 MHz (DMSO-d6): δ 180.92, 180.54, 162.35, 155.21, 153.86, 147.78, 143.71, 131.55, 124.33, 123.77, 117.20, 115.11, 54.40, 47.94, 45.42, 13.22. MS: m/z 364 [M+H]+.
4-methyl-2,5-dimethoxyaniline [11]
To a stirred suspension of 2,5-dimethoxy-4-methylnitrobenzene (6, 1 g, 5.07 mmol) and 10% Pd-C (0.4 g) in dry methanol (20 ml), ammonium formate (4.5 eq., 1.5 g, 23.79 mmol) was added in a single portion. The resulting mixture was stirred at room temperature for 40 min., the catalyst was removed by filtration through a celite pad and washed with dry methanol. The filtrate was evaporated either under reduced or at normal pressure. The resulting residue was extracted with dichloromethane/water. Yield: 70%. Rf = 0.20 (ethyl acetate-petroleum ether 3:7). mp: 255 °C. 1H NMR 400 MHz (DMSO-d6): δ 6.61 (s, 1H), 6.35 (s, 1H), 3.79 (s, 3H), 3.74 (s, 3H), 2.13 (s, 3H).
Ethyl (2,5-dimethoxy-4-methylphenyl)carbamate [12]
To a solution of 4-methyl-2,5-dimethoxyaniline (11, 0.5 g, 2.99 mmol) in tetrahydrofuran (30 mL), ethyl chloroformate (0.65 g, 0.57 mL, 2 eq., 6 mmol) was added. Further trimethylamine (1 mL, 2 eq., 6 mmol) was added into the reaction mixture and stirred at room temperature for 1 hour. Reaction mixture was concentrated and extracted in ethyl acetate/water to obtain the product. Yield: 60%. Rf = 0.10 (ethyl acetate-petroleum ether 3:7). mp: 106 °C. 1H NMR 400 MHz (CDCl3): δ 7.76 (s, 1H), 7.15 (s, 1H), 6.66 (s, 1H), 4.20 (q, 2H), 3.81 (s, 3H), 3.80 (s, 3H), 2.17 (s, 3H), 1.32 (t, 3H).
Ethyl-5,8-dimethoxy-6-methylquinazoline-1-(2H)-carboxylate [13]
A mixture of N-protected N-protected 4-methyl-2,5-dimethoxyaniline (12, 0.2 g, 0.84 mmol) and hexamethylenetetramine (HMTA, 9 eq., 0.84 g, 7.2 mmol) in in trifluoroacetic acid (TFA, 4 mL) was refluxed for 1 hour. After cooling, the mixture was diluted with cold water, neutralized with NaHCO3 and extracted with ethyl acetate. The organic phase was evaporated and residue was treated with cyclohexane. The undissolved solid was filtered off and the solution was evaporated under reduced pressure to give product. Yield: 50%. Rf = 0.45 (ethyl acetate-petroleum ether 3:7); mp: > 320 °C. 1H NMR 400 MHz (CDCl3): 8.58 (s, 1H), 6.83 (d, 2H), 6.56 (s, 1H), 4.14 (q, 2H), 3.94 (s, 3H), 3.84 (s, 3H), 2.30 (s, 3H), 1.28 (t, 3H).
5,8-Dimethoxy-6-methylquinazoline [14]
A solution of 10% KOH in water/EtOH in 1/1 ratio (25 mL), quinazoline carboxylate (13, 0.55 g, 1.97 mmol) was added and refluxed for 1 hour. After cooling, potassium ferricyanide (45 eq., 3.24 g, 89.12 mmol) was added and mixture was refluxed for 4 hour. After cooling, the mixture was diluted with water, extracted with dichloromethane, and the organic phase was evaporated under reduced pressure. The residue was dissolved in the minimum amount of methanol and dilute with water to obtain pure product. Yield: 35%. Rf = 0.30 (ethyl acetate-petroleum ether 3:7). mp: > 320 °C. 1H NMR 400 MHz (CDCl3): δ 9.59 (s, 1H), 9.31 (s, 1H), 7.04 (s, 1H), 4.06 (s, 3H), 3.92 (s, 3H), 2.49 (s, 3H).
6-Methyl quinazoline-5,8-dione [15]
A solution of 5,8-Dimethoxy-6-methylquinazoline (0.15 g, 0.75 mmol) in (7:3) acetonitrile:water (5 mL) was cooled at 0 °C in an ice bath, and a solution of ceric ammonium nitrate (3 eq., 1.2 g, 2.1 mmol) in (9:1) acetonitrile:water (5 mL) was added dropwise. The reaction mixture was stirred for 20 min, then poured into ice/water and extracted (8−10 times) with dichloromethane. The organic layer was washed (5−6 times) with water, dried over anhydrous Na2SO4 and concentrated to dryness to give a brown powder. Yield: 90%. Rf = 0.52 (ethyl acetate-petroleum ether 3:7). mp: > 320 °C. 1H NMR 400 MHz (CDCl3): δ 9.67 (s, 1H), 9.54 (s, 1H), 7.10 (s, 1H), 2.27 (s, 3H). MS: m/z 175 [M+H]+.
Alternative procedure for the preparation of 6-(4-(4-Methylpiperazin-1-yl)phenylamino)quinazoline-5,8-dione [QD 394]
A solution of 6-methylquinazoline-5, 8-dione (15, 0.050 g, 0.312 mmol), cerium (III) chloride heptahydrate (CeCl3•7H2O, 1.1 eq., 0.128 g, 0.343 mmol) and 4-(4-methylpiperazin-1-yl)aniline (1.0 eq., 0.060 g, 0.312 mmol) in absolute ethanol (6 mL) was stirred at room temperature for 1−2 h. Next, most of the ethanol was removed under vacuum, and water was added, followed by the extraction with dichloromethane. The organic layers were washed with water and brine, dried over anhydrous Na2SO4, and concentrated to dryness. Then, the crude product was purified by flash chromatography (dicloromethane:methanol = 8:2) to give the expected product. Yield: 30%. Rf = 0.30 (dichloromethane-methanol 9.5:0.5). mp: > 320 °C. See the previous preparation of QD394 for full characterization.
HPLC analyses.
Analytical HPLC was carried out on an Agilent 1260 Infinity II Quaternary pump HPLC apparatus with a Poroshell 120 EC-C18 reversed column (4.6 × 100 mM, 4 μm) at a flow rate of 0.5 mL/min (injection volume 20 μL) and detected at 254 nm. The mobile phases were mixtures of A = acetonitrile + 0.1% trifluoroacetic acid, and B = H2O + 0.1% trifluoroacetic acid, and are indicated as the ratio A:B. QD 394, HPLC (A:B = 60:40), Rt 1.72 min (100%). QD 394-Me, HPLC (A:B = 60:40), Rt 1.76 min (99.98%).
Chemicals.
Stock solutions of QD compounds and napabucasin were made in dimethylsulfoxide (DMSO) at 10 mM and were stored at −20 °C. Napabucasin was purchased from Medchem Express. N-acetyl cysteine (Sigma) and deferoxamine mesylate salt (Sigma) were dissolved in ultrapure water (Gibco). Z-VAD (MedChemExpress), necrostatin-1 (Selleckchem), RSL3 (Cayman Chemical), deferasirox (Cayman Chemical), liproxstatin-1 (Cayman Chemical), ferrostatin-1 (Cayman Chemical), chloroquine phosphate (LKT LABS), olaparib (MedChemExpress), 3-Methyladenine (Cayman Chemical), and N-phenylmaleimide (Oakwood Chemical) were dissolved in DMSO to provide 10 mM stock solution.
Cell lines and cell culture.
Pancreatic cancer cell lines (MIA PaCa-2, PANC-1, and BxPC-3) were purchased from ATCC. All cell lines were maintained in RPMI 1640 (Gibco) containing 10% FBS (Gibco) at 37 °C in a humidified atmosphere of 5% CO2. All cell lines were in culture under 35 passages and determined to be free of mycoplasma contamination using Plasmo Test (InvivoGen, San Diego, CA).
MTT assay.
Cytotoxicity was determined by a 3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide (MTT) assay. Cells were seeded in 96-well tissue culture plates and allowed to attach overnight. Subsequently, cells were treated with compounds or DMSO for 72 h. MTT solution (0.3 mg/mL) was added to each well for 3 h in the incubator. After removal of the media, DMSO was added, and the plates were shaken for 15 min before measuring the absorbance at a wavelength of 570 nm using a microplate reader (Molecular Devices). IC50 values were calculated using GraphPad Prism.
Colony Formation Assay.
Cells were seeded in 96-well tissue culture plates at 300 cells (MIA PaCa-2) or 600 cells (PANC-1 and BxPC-3) per well. After overnight attachment, cells were treated with compounds continuously. Cell death inhibitors or signaling pathway inhibitors were added 1 h prior to compound treatment. Plates were incubated for 7–9 days until cells in the control wells became 80–90% confluent. Media was removed, and cells were stained with a 0.05% crystal violet solution for 30 min. Cells were then washed with ddH2O to remove excess stain, and the plates were imaged using Odyssey Imaging Systems (LI-COR Biosciences) after overnight drying. Quantification was performed using Image Studio, and synergism was calculated using the HSA and Bliss models in the Combenefit software.
ROS measurement.
Cells were seeded in 96-well flat clear-bottom black tissue-culture plates (Corning #3603) at a density of 1.8 × 104 cells per well. After overnight attachment, cells were stained with CM-H2DCFDA dye (10 μM, Thermo Fisher, C6827) in HBSS for 40 min at 37 °C. Cells were washed with pre-warmed DPBS (Gibco) twice, and 80 μL HBSS was added. Compounds were added in HBSS at designated concentrations. Fluorescent signal was measured using a CLARIO Star plate reader (Ex: 483–15 nm; Em: 530–20 nm; BMG LABTECH).
GSH/GSSG-Glo assay.
GSH/GSSG-Glo assay kit (Promega) was used to determine the GSH/GSSG ratio in cells with drugs or DMSO. 15,000 – 20,000 cells per well were seeded into solid opaque 96-well tissue culture plates (Falcon). After overnight attachment and compound treatment, medium was removed, and cells were lysed with 50 μL of Total or Oxidized Glutathione Reagent per well. The plate was shaken for 5 minutes, and 50 μL fresh Luciferin Generation Reagent was added to each well. After 30-min incubation at room temperature, 100 μL Luciferin Detection Reagent was added. Luminescence was detected using a CLARIOstar plate reader (BMG LABTECH) after 15-min equilibration. GSH/GSSG ratio was calculated with net RLU, using the formula: (net vehicle total glutathione RLU – net vehicle GSSG RLU)/(net vehicle GSSG RLU/2).
Bru-Seq experiments and analysis.
Nascent RNA Bru-seq experiments were performed as previously reported.23 Briefly, 2 × 106 cells were seeded in 10 cm dishes in duplicates. After overnight attachment, cells were treated with compounds (3 × IC50) for 4 h. Cells were added with bromouridine at a final concentration of 2 mM and incubated at 37 °C for the last 30 min of the drug treatments. Cells were then collected and lysed in TRIzol, and total RNA was isolated. The bromouridine-containing RNAs were captured using anti-BrdU antibodies conjugated to magnetic beads and converted to cDNA libraries (Illumina TrueSeq), which were sent for deep sequencing at the University of Michigan Sequencing Core. Sequencing reads were mapped to the hg38 reference genome, and DESeq2 was used to determine differentially expressed genes. The shrunkenLFC (shrunken log2FoldChange) was calculated from DESeq2. We used shurnkenLFC for the heatmap and GSEA analysis. Downstream signaling pathways were identified using GSEA. Heatmaps and statistics were generated using the R programming language.
Immunoblot.
Cells were seeded in 6-well tissue culture plates at 3.5 × 105 cells per well. After compound treatments, cells were washed with DPBS (Gibco) and lysed with RIPA buffer containing 1× protease inhibitor (Sigma) and 1× phosphatase inhibitor (Sigma). Cell lysates were sonicated before centrifugation at 14, 000 rpm for 10 min at 4 °C, and supernatant was collected for BCA protein assay (Thermo Scientific). 25 μg proteins from each sample was resolved on 10% SDS polyacrylamide gels and electrotransferred to PVDF membranes (Trans-Blot® Turbo™ RTA Mini PVDF Transfer Kit, BIO-RAD). After blocking with 5% milk in 1× TBS for 1 h at room temperature, membranes were probed with primary antibodies (1:500–1:1000) in 5% milk in 1× TBST or 5% BSA in 1× TBST overnight at 4 °C. The membranes were then washed with TBST three times and probed with anti-rabbit or anti-mouse secondary antibody (Dylight 800 4 × PEG conjugated; Thermo Scientific; 1:6000) for 1 h at room temperature. Membranes were imaged with Odyssey Imaging Systems (LI-COR Biosciences). Protein expression was quantified with ImageJ and normalized to loading controls.
Lipid ROS measurement.
MIA PaCa-2 cells were treated with compounds for the designated time before 10 μM C11-BODIPY (Thermo Fisher) was added, and cells were incubated for 30 min at 37 °C. Cells were washed with DPBS (Gibco) twice, and 100 μL HBSS was added before measurement using a CLARIOstar plate reader (BMG LABTECH). Plates were read with two filter sets; one at excitation/emission of 581/591 nm (Texas Red) for the reduced dye, and the other at excitation/emission of 488/510 nm (FITC) for the oxidized dye. The ratio of the emission fluorescence intensities using the Texas Red and FITC filters was calculated in Microsoft Excel, and p value was determined using unpaired Student’s t-test.
Cellular Thermal Shift Assay (CETSA).
Cells were seeded at 3 × 106 cells per 10 cm cell culture dish. After overnight attachment, 100 μM QD394, QD394-Me, or DMSO was added to the cells for 1 h. Cells were then trypsinized, washed with DPBS, and suspended in 650 μL DPBS. The suspensions were split into 100 μL aliquots, heated at indicated temperatures for 3 minutes in the Veriti Thermal Cycler (Applied Biosystems), and incubated for 3 minutes at room temperature. Each aliquot was flash-frozen three times and spun at 12,000 rpm for 20 minutes at 4 °C. Supernatants were collected, and 25 μL of each sample was loaded onto a 10% SDS polyacrylamide gel for immunoblot analysis using the protocol detailed above.
siRNA knockdown of GPX4.
GPX4 siRNA (s6110) and negative control siRNA (4390843) were purchased from Thermo Fisher. According to the manufacturer’s instructions, 3.5 × 105 cells were seeded in 6-well tissue culture plates for overnight attachment. The siRNA transfection reagent complex was formed by combining 30 pmol siRNA in 150 μL Opti-MEM medium (Thermo Fisher) with 9 μL Lipofectamine RNAiMAX Reagent (Thermo Fisher) in 150 μL Opti-MEM medium. After 15-min incubation, this complex was added to each well with fresh medium for 48 h. One half of these cells were harvested for immunoblot, and the other half were seeded for the 24 h MTT assay to determine the cytotoxicity of QD394, QD394-Me, and napabucasin.
Proteomics study.
MIA PaCa-2 cells were treated with 1.2 μM QD394 or DMSO control for 24 h before collecting the cell lysates in RIPA buffer. 75 μg proteins of each sample were processed at the Proteomics Core at the University of Michigan in duplicate. TMT10plex Mass Tag Labeling Kit (ThermoFisher) was used to quantify the proteins in each sample, and Abundance Ratio and log2 Abundance Ratio were calculated as treatment over control. STRING analysis was performed using proteins with adjusted p values < 0.05.
Survival analysis of LRPPRC and PNPT1.
Patient sample RNASeq RSEM normalized gene expression values and related survival metadata were sourced from the TCGA GDAC Firehose.47 When multiple samples were available for a given patient, barcodes were sorted alphabetically and the first was selected for analysis. TCGA disease patient samples were evaluated for reduced survivability by comparing survival outcomes for patients with high and low expression of LRPPRC and PNPT1. Thresholding for high and low expression patient populations was evaluated using five different quantile cutoffs: 95%, 90%, 75%, 50%, and 25%. A log-rank test statistic was calculated for each cutoff to compare the survival distributions of high and low expression patient populations with the null hypothesis that there was no difference in survival curves. Survival analysis was performed using the R statistical programming language.
Microsomal stability test.
The 10 mM stock solutions of QD394, QD394-Me, and verapamil (positive control) were prepared in DMSO. Solutions were diluted to 1 mM with acetonitrile and further diluted to 10 μM with 0.1 M phosphate buffer (3.3 mM MgCl2). NADPH (1 mg) was dissolved in 60 μL 0.1 M phosphate buffer (3.3 mM MgCl2). 330 μL 0.1 M phosphate buffer (3.3 mM MgCl2) and 40 μL 10 μM test compounds were added to 10 μL microsomes (20 mg/mL). Data points were collected at 0, 5, 10, 15, 30, 45, and 60 min. Solutions were centrifuged at 3500 g for 10 min to pellet precipitated proteins. The supernatant was used for LC-MS/MS analysis. Natural log peak area ratio (compound peak area / internal standard peak area) was plotted against time, and the gradient of the line was determined to calculate the half-life (t1/2) of the test compound in microsomes.
Plasma stability test.
Pooled mouse plasma was stored at −80 °C prior to use. Compounds were dissolved in DMSO to obtain a concentration of 500 μM. 1 μL compound was added to 500 μL plasma. Samples were collected at time points of 0, 0.17, 0.5, 1, 2, and 4 h. The solution was centrifuged at 3500 g for 10 min to pellet precipitated proteins. The supernatant was used for LC-MS/MS analysis. Natural log peak area ratio (compound peak area / internal standard peak area) was plotted against time, and the gradient of the line was determined to calculate the half-life (t1/2) of the test compound in plasma.
PK study.
QD394 and QD394-Me were administered by IP injection (10 mg/kg), oral PO (20 mg/kg), or IV injection (10 mg/kg) to CD-1 mice. At the given time points (0.5, 2, 4, and 7 h), blood samples were collected using heparinized calibrated pipettes. Samples were centrifuged at 15,000 rpm for 10 min. Subsequently, blood plasma was collected from the upper layer. The plasma was frozen at −80 °C for LC-MS/MS analysis.
Metabolite identification.
QD394 and QD394-Me were incubated with mouse plasma under the same conditions as the plasma stability test. Samples were collected at 0.5 h to determine potential metabolites of QD394 or QD394-Me using LC-MS/MS analysis. Indicated metabolites were also measured in vivo after 1-hour QD394-Me treatment.
Tissue distribution analysis.
Mice were dosed with QD394 at 20 mg/kg PO and QD394-Me at 10 mg/kg IV. After 1 h, indicated organs were collected for later analysis. Dissociation solvent was added at five-fold the tissue weight into each homogenization tube with beads for tissue homogenization, and 30 μL homogenate was isolated for sample preparation. For calibration curve samples, 30 μL blank homogenate was mixed with 30 μL standard solution and 140 μL internal standard solution; for dosed samples, 30 μL homogenate was mixed with 30 μL blank acetonitrile and 140 μL internal standard solution. Samples were vortexed and centrifuged at 3500 g for 10 min, and the supernatant was isolated for LC-MS analysis.
Mice study.
All animal studies were approved by the animal care facility at the University of Michigan-Ann Arbor (Protocol number: PRO00009185) and were handled in accordance with the Institutional Animal Use and Care Committee. Female Balb/c mice were purchased from Envigo. At the time of implantation, all mice were aged 5–6 weeks. Mice were implanted subcutaneously in the right flank with 1 × 106 CT-26 cells in 100 μL DPBS. Seven days after implantation, mice were randomized into groups (n = 5) with mean tumor volumes ranging from 97 to 117 mm3. The negative control group was dosed daily in the intraperitoneal cavity (IP) with the same vehicle used for QD394. QD394 was dosed at 10 mg/kg IP, and QD394-Me was dosed 3 times weekly intravenously (IV) at 20 mg/kg. After 17 days of dosing, four hours post-dose, negative control and treated groups were euthanized by CO2 asphyxiation followed by cervical dislocation. The end of the study was chosen as the first day that any mouse reached humane euthanasia criteria determined by the animal use protocol.
Statistical analysis.
For the Bru-seq analysis, Pearson correlation and p value were calculated using function cor.test() in R (version 3.3.2). Significance levels for assays and immunoblots were calculated using unpaired Student’s t-test in Microsoft Excel. Results are shown as mean ± standard deviation from at least three independent experiments.
Supplementary Material
Acknowledgements
This work was supported by NIH grant R01 CA188252 and a grant from the University of Michigan Forbes Institute for Cancer Discovery. We thank the Fondazione di Sardegna for its partial support. We thank our colleagues Christine Cuthbertson, Andrea Shergalis, and Maha Hanafi for helpful suggestions on manuscript and Zahra Arabzada for technical assistance. We thank Armand Bankhead III for assistance in the survival analysis. We also thank Federico Ladu for his help and assistance, and Vanna Sanna (Nanomater s.r.l.) for the HPLC analyses. We gratefully acknowledge the University of Michigan DNA Sequencing Core and Proteomics Core facilities for their excellent technical assistance.
Abbreviations
- ROS
reactive oxygen species
- STAT3
signal transducer and activator of transcription 3
- GSH
glutathione
- GSSG
glutathione disulfide
- GPX4
glutathione peroxidase 4
- 5-FU
5-fluorouracil
- NQO1
NAD(P)H quinone dehydrogenase 1
- QD
quinazolinedione
- Napa
napabucasin
- HMTA
hexamethylenetetramine
- Bru-seq
bromouridine sequencing
- GSEA
gene set enrichment analysis
- MSigDB
molecular signatures database
- FDR
false discovery rate
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- TF
transcription factor
- NAC
N-acetylcysteine
- BSO
buthionine sulfoximine
- DFO
deferoxamine
- CETSA
cellular thermal shift assay
- ER
endoplasmic reticulum
- LRPPRC
leucine rich pentatricopeptide repeat containing
- PNPT1
polyribonucleotide nucleotidyltransferase 1
- TCGA
The Cancer Genome Atlas
- PAAD
pancreatic adenocarcinoma
- As2O3
arsenic trioxide
- PARP
poly(ADP-ribose) polymerase
- HIF-1A
hypoxia-inducible factor-1α
- H2O2
hydrogen peroxide
- FBS
fetal bovine serum
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide
- DPBS
Dulbecco’s phosphate buffered saline
- HBSS
Hanks’ balanced salt solution
- RLU
relative luminescence units
- RFU
relative fluorescence units
- shrunkenLFC
shrunken log2FoldChange
- LC-MS
liquid chromatography–mass spectrometry
- HPLC
high-performance liquid chromatography
- HSA
highest single agent
Footnotes
Supporting information
Supporting Information Available: This material is available free of charge via the Internet.
ROS assay of QD394 analogs; Colony formation assay of QD394 analogs; GSH level in pancreatic cancer cells treated by QD394, napabucasin and BSO; Colony formation assay of combination studies; CETSA in PANC-1 cells; Combination of NAC, dicoumarol with QD394, QD394-Me, napabucasin and H2O2 in probing LRPPRC and PNPT1 in pancreatic cancer cells; Combination of QD394 and napabucasin in PANC-1 cells; Plasma concentration of QD394 and QD394-Me in CD-1 mice; Major metabolite of QD394-Me in mouse plasma and its abundance profiles in mice; In vivo study with female Balb/c mice implanted subcutaneously with CT-26 cells; HPLC analyses for QD394 and QD394-Me; Elemental analyses for QD394 analogs; Molecular formula strings for all QD394 analogs; Top 25 upregulated and downregulated enriched C2 gene sets in QD394-treated MIA PaCa-2 cells; STRING analysis of 17 upregulated genes/proteins in common between Bru-seq and proteomics; STRING analysis of 35 downregulated genes/proteins in common between Bru-seq and proteomics; Tissue distribution of QD394-Me in mouse; Cytotoxicity of QD394, QD394-Me, and napabucasin in GPX4-knockdown PANC-1 and BxPC-3 cells; Top upregulated and downregulated enriched hallmark gene sets in QD394-Me-treated MIA PaCa-2 cells; Top upregulated enriched KEGG gene sets in QD394-Me-treated MIA PaCa-2 cells
Declarations of interest
The authors declare no competing financial interest.
References
- 1.Siegel RL; Miller KD; Jemal A Cancer statistics, 2020. CA Cancer J Clin 2020, 70, 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Kamisawa T; Wood LD; Itoi T; Takaori K Pancreatic cancer. Lancet 2016, 388, 73–85. [DOI] [PubMed] [Google Scholar]
- 3.Rahib L; Smith BD; Aizenberg R; Rosenzweig AB; Fleshman JM; Matrisian LM Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014, 74, 2913–2921. [DOI] [PubMed] [Google Scholar]
- 4.Mohammad AA Advanced pancreatic cancer: the standard of care and new opportunities. Oncol Rev 2018, 12, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D; Weinberg RA Hallmarks of cancer: the next generation. Cell 2011, 144, 646–74. [DOI] [PubMed] [Google Scholar]
- 6.Fouad YA; Aanei C Revisiting the hallmarks of cancer. Am J Cancer Res 2017, 7, 1016–1036. [PMC free article] [PubMed] [Google Scholar]
- 7.Hornsveld M; Dansen TB The hallmarks of cancer from a redox perspective. Antioxid Redox Signal 2016, 25, 300–25. [DOI] [PubMed] [Google Scholar]
- 8.Pavlova NN; Thompson CB The emerging hallmarks of cancer metabolism. Cell Metab 2016, 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeBerardinis RJ; Chandel NS Fundamentals of cancer metabolism. Sci Adv 2016, 2, e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerber DE; Beg MS; Fattah F; Frankel AE; Fatunde O; Arriaga Y; Dowell JE; Bisen A; Leff RD; Meek CC; Putnam WC; Kallem RR; Subramaniyan I; Dong Y; Bolluyt J; Sarode V; Luo X; Xie Y; Schwartz B; Boothman DA Phase 1 study of ARQ 761, a beta-lapachone analogue that promotes NQO1-mediated programmed cancer cell necrosis. Br J Cancer 2018, 119, 928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sonbol MB; Ahn DH; Goldstein D; Okusaka T; Tabernero J; Macarulla T; Reni M; Li CP; O’Neil B; Van Cutsem E; Bekaii-Saab T CanStem111P trial: a phase III study of napabucasin plus nab-paclitaxel with gemcitabine. Future Oncol 2019, 15, 1295–1302. [DOI] [PubMed] [Google Scholar]
- 12.Grothey A; Shah MA; Yoshino T; Van Cutsem E; Taieb J; Xu RH; Tebbutt NC; Falcone A; Cervantes A; Borodyansky L; Li C CanStem303C trial: A phase III study of napabucasin (BBI-608) in combination with 5-fluorouracil (5-FU), leucovorin, irinotecan (FOLFIRI) in adult patients with previously treated metastatic colorectal cancer (mCRC). J Clin Oncol 2017, 35. [Google Scholar]
- 13.Froeling FEM; Swamynathan MM; Deschenes A; Chio IIC; Brosnan E; Yao MA; Alagesan P; Lucito M; Li J; Chang AY; Trotman LC; Belleau P; Park Y; Rogoff HA; Watson JD; Tuveson DA Bioactivation of napabucasin triggers reactive oxygen species-mediated cancer cell death. Clin Cancer Res 2019, 25, 7162–7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuang Y; Sechi M; Nurra S; Ljungman M; Neamati N Design and synthesis of novel reactive oxygen species inducers for the treatment of pancreatic ductal adenocarcinoma. J Med Chem 2018, 61, 1576–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dixon SJ; Lemberg KM; Lamprecht MR; Skouta R; Zaitsev EM; Gleason CE; Patel DN; Bauer AJ; Cantley AM; Yang WS; Morrison B 3rd; Stockwell BR Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stockwell BR; Friedmann Angeli JP; Bayir H; Bush AI; Conrad M; Dixon SJ; Fulda S; Gascon S; Hatzios SK; Kagan VE; Noel K; Jiang X; Linkermann A; Murphy ME; Overholtzer M; Oyagi A; Pagnussat GC; Park J; Ran Q; Rosenfeld CS; Salnikow K; Tang D; Torti FM; Torti SV; Toyokuni S; Woerpel KA; Zhang DD Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galluzzi L; Vitale I; Aaronson SA; Abrams JM; Adam D; Agostinis P; Alnemri ES; Altucci L; Amelio I; Andrews DW; Annicchiarico-Petruzzelli M; Antonov AV; Arama E; Baehrecke EH; Barlev NA; Bazan NG; Bernassola F; Bertrand MJM; Bianchi K; Blagosklonny MV; Blomgren K; Borner C; Boya P; Brenner C; Campanella M; Candi E; Carmona-Gutierrez D; Cecconi F; Chan FK; Chandel NS; Cheng EH; Chipuk JE; Cidlowski JA; Ciechanover A; Cohen GM; Conrad M; Cubillos-Ruiz JR; Czabotar PE; D’Angiolella V; Dawson TM; Dawson VL; De Laurenzi V; De Maria R; Debatin KM; DeBerardinis RJ; Deshmukh M; Di Daniele N; Di Virgilio F; Dixit VM; Dixon SJ; Duckett CS; Dynlacht BD; El-Deiry WS; Elrod JW; Fimia GM; Fulda S; Garcia-Saez AJ; Garg AD; Garrido C; Gavathiotis E; Golstein P; Gottlieb E; Green DR; Greene LA; Gronemeyer H; Gross A; Hajnoczky G; Hardwick JM; Harris IS; Hengartner MO; Hetz C; Ichijo H; Jaattela M; Joseph B; Jost PJ; Juin PP; Kaiser WJ; Karin M; Kaufmann T; Kepp O; Kimchi A; Kitsis RN; Klionsky DJ; Knight RA; Kumar S; Lee SW; Lemasters JJ; Levine B; Linkermann A; Lipton SA; Lockshin RA; Lopez-Otin C; Lowe SW; Luedde T; Lugli E; MacFarlane M; Madeo F; Malewicz M; Malorni W; Manic G; Marine JC; Martin SJ; Martinou JC; Medema JP; Mehlen P; Meier P; Melino S; Miao EA; Molkentin JD; Moll UM; Munoz-Pinedo C; Nagata S; Nunez G; Oberst A; Oren M; Overholtzer M; Pagano M; Panaretakis T; Pasparakis M; Penninger JM; Pereira DM; Pervaiz S; Peter ME; Piacentini M; Pinton P; Prehn JHM; Puthalakath H; Rabinovich GA; Rehm M; Rizzuto R; Rodrigues CMP; Rubinsztein DC; Rudel T; Ryan KM; Sayan E; Scorrano L; Shao F; Shi Y; Silke J; Simon HU; Sistigu A; Stockwell BR; Strasser A; Szabadkai G; Tait SWG; Tang D; Tavernarakis N; Thorburn A; Tsujimoto Y; Turk B; Vanden Berghe T; Vandenabeele P; Vander Heiden MG; Villunger A; Virgin HW; Vousden KH; Vucic D; Wagner EF; Walczak H; Wallach D; Wang Y; Wells JA; Wood W; Yuan J; Zakeri Z; Zhivotovsky B; Zitvogel L; Melino G; Kroemer G Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018, 25, 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mou Y; Wang J; Wu J; He D; Zhang C; Duan C; Li B Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol 2019, 12, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye LF; Chaudhary KR; Zandkarimi F; Harken AD; Kinslow CJ; Upadhyayula PS; Dovas A; Higgins DM; Tan H; Zhang Y; Buonanno M; Wang TJC; Hei TK; Bruce JN; Canoll PD; Cheng SK; Stockwell BR Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem Biol 2020, 15, 469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui J; Wang L; Ren X; Zhang Y; Zhang H LRPPRC: a multifunctional protein involved in energy metabolism and human disease. Front Physiol 2019, 10, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matilainen S; Carroll CJ; Richter U; Euro L; Pohjanpelto M; Paetau A; Isohanni P; Suomalainen A Defective mitochondrial RNA processing due to PNPT1 variants causes Leigh syndrome. Hum Mol Genet 2017, 26, 3352–3361. [DOI] [PubMed] [Google Scholar]
- 22.Chilin A; Marzaro G; Zanatta S; Barbieri V; Pastorini G; Manzini P; Guiotto A A new access to quinazolines from simple anilines. Tetrahedron 2006, 62, 12351–12356. [Google Scholar]
- 23.Paulsen MT; Veloso A; Prasad J; Bedi K; Ljungman EA; Magnuson B; Wilson TE; Ljungman M Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods 2014, 67, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blaser H; Dostert C; Mak TW; Brenner D TNF and ROS crosstalk in inflammation. Trends Cell Biol 2016, 26, 249–261. [DOI] [PubMed] [Google Scholar]
- 25.Nishigori C; Hattori Y; Toyokuni S Role of reactive oxygen species in skin carcinogenesis. Antioxid Redox Signal 2004, 6, 561–570. [DOI] [PubMed] [Google Scholar]
- 26.Rodust PM; Stockfleth E; Ulrich C; Leverkus M; Eberle J UV-induced squamous cell carcinoma--a role for antiapoptotic signalling pathways. Br J Dermatol 2009, 161 Suppl 3, 107–115. [DOI] [PubMed] [Google Scholar]
- 27.Gorlach A; Dimova EY; Petry A; Martinez-Ruiz A; Hernansanz-Agustin P; Rolo AP; Palmeira CM; Kietzmann T Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol 2015, 6, 372–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tafani M; Sansone L; Limana F; Arcangeli T; De Santis E; Polese M; Fini M; Russo MA The interplay of reactive oxygen species, hypoxia, inflammation, and sirtuins in cancer initiation and progression. Oxid Med Cell Longev 2016, 2016, 3907147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stockwell BR Ferroptosis: death by lipid peroxidation. Free Radical Bio Med 2018, 120, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szklarczyk D; Gable AL; Lyon D; Junge A; Wyder S; Huerta-Cepas J; Simonovic M; Doncheva NT; Morris JH; Bork P; Jensen LJ; Mering CV STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019, 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gupta SC; Hevia D; Patchva S; Park B; Koh W; Aggarwal BB Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid Redox Signal 2012, 16, 1295–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carney DA Arsenic trioxide mechanisms of action--looking beyond acute promyelocytic leukemia. Leuk Lymphoma 2008, 49, 1846–1851. [DOI] [PubMed] [Google Scholar]
- 33.Prasad S; Gupta SC; Tyagi AK Reactive oxygen species (ROS) and cancer: role of antioxidative nutraceuticals. Cancer Lett 2017, 387, 95–105. [DOI] [PubMed] [Google Scholar]
- 34.Fang B Genetic interactions of STAT3 and anticancer drug development. Cancers (Basel) 2014, 6, 494–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Annibaldi A; Meier P Checkpoints in TNF-induced cell death: implications in inflammation and cancer. Trends Mol Med 2018, 24, 49–65. [DOI] [PubMed] [Google Scholar]
- 36.Shen J; Xiao Z; Zhao Q; Li M; Wu X; Zhang L; Hu W; Cho CH Anti-cancer therapy with TNFalpha and IFNgamma: a comprehensive review. Cell Prolif 2018, 51, e12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Semenza GL Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonello S; Zahringer C; BelAiba RS; Djordjevic T; Hess J; Michiels C; Kietzmann T; Gorlach A Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol 2007, 27, 755–761. [DOI] [PubMed] [Google Scholar]
- 39.Han D; Yu T; Dong N; Wang B; Sun F; Jiang D Napabucasin, a novel STAT3 inhibitor suppresses proliferation, invasion and stemness of glioblastoma cells. J Exp Clin Cancer Res 2019, 38, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugarman R; Patel R; Sharma S; Plenker D; Tuveson D; Saif MW Pharmacokinetics and pharmacodynamics of new drugs for pancreatic cancer. Expert Opin Drug Metab Toxicol 2019, 15, 541–552. [DOI] [PubMed] [Google Scholar]
- 41.Friedmann Angeli JP; Schneider M; Proneth B; Tyurina YY; Tyurin VA; Hammond VJ; Herbach N; Aichler M; Walch A; Eggenhofer E; Basavarajappa D; Radmark O; Kobayashi S; Seibt T; Beck H; Neff F; Esposito I; Wanke R; Forster H; Yefremova O; Heinrichmeyer M; Bornkamm GW; Geissler EK; Thomas SB; Stockwell BR; O’Donnell VB; Kagan VE; Schick JA; Conrad M Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014, 16, 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie Y; Hou W; Song X; Yu Y; Huang J; Sun X; Kang R; Tang D Ferroptosis: process and function. Cell Death Differ 2016, 23, 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shah R; Margison K; Pratt DA The potency of diarylamine radical-trapping antioxidants as inhibitors of ferroptosis underscores the role of autoxidation in the mechanism of cell death. ACS Chem Biol 2017, 12, 2538–2545. [DOI] [PubMed] [Google Scholar]
- 44.Zhou W; Sun G; Zhang Z; Zhao L; Xu L; Yuan H; Li S; Dong Z; Song Y; Fang X Proteasome-independent protein knockdown by small-molecule inhibitor for the undruggable lung adenocarcinoma. J Am Chem Soc 2019, 141, 18492–18499. [DOI] [PubMed] [Google Scholar]
- 45.Liu X; Fu R; Pan YD; Meza-Sosa KF; Zhang ZB; Lieberman J PNPT1 release from mitochondria during apoptosis triggers decay of Poly(A) RNAs. Cell 2018, 174, 187–201. [DOI] [PubMed] [Google Scholar]
- 46.Cuillerier A; Honarmand S; Cadete VJJ; Ruiz M; Forest A; Deschenes S; Beauchamp C; Consortium L; Charron G; Rioux JD; Des Rosiers C; Shoubridge EA; Burelle Y Loss of hepatic LRPPRC alters mitochondrial bioenergetics, regulation of permeability transition and trans-membrane ROS diffusion. Hum Mol Genet 2017, 26, 3186–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Broad Institute TCGA Genome Data Analysis Center. Firehose stddata__2016_01_28 run., Broad Institute of MIT Harvard, Cambridge, MA. 2016, DOI: 10.7908/C11G0KM9. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.