

Abstract
Bacterial pneumonia is a significant cause of morbidity, mortality, and health care expenditures. Optimum neutrophil recruitment and their function are critical defense mechanisms against respiratory pathogens. The nucleotide-binding oligomerization domain-like receptor (NLRP) 6 controls gut microbiota and immune response to systemic and enteric infections. However, the importance of NLRP6 in neutrophil homeostasis following lung infection remains elusive. To investigate the role of NLRs in neutrophil homeostasis, we used Nlrp6 gene-deficient (Nlrp6−/−) mice in a model of Klebsiella pneumoniae-induced pneumonia-derived sepsis. We demonstrated that NLRP6 is critical for host survival, bacterial clearance, neutrophil influx, and CXC-chemokine production. Kp-infected Nlrp6−/− mice have reduced numbers of hematopoietic stem cells and granulocyte-monocyte progenitors but increased retention of matured neutrophils in bone marrow. Neutrophil extracellular trap (NET) formation and NET-mediated bacterial killing were also impaired in Nlrp6−/− neutrophils in vitro. Furthermore, recombinant CXCL1 rescued the impaired host defense, granulopoietic response, and NETosis in Kp-infected Nlrp6−/− mice. Using A/J background mice and co-housing experiments, our findings revealed that the susceptible phenotype of Nlrp6−/− mice is not strain-specific and gut microbiota-dependent. Taken together, these data unveil NLRP6 as a central regulator of neutrophil recruitment, generation and function during bacterial pneumonia followed by sepsis.
Keywords: Pneumonia, Sepsis, Innate Immunity, Neutrophil, Granulopoiesis, NETosis, Mobilization
INTRODUCTION
Bacterial lung infection is a significant cause of morbidity and mortality1, 2. The growing number of antibiotic-resistant bacteria, termed ESKAPE pathogens, has become a serious public health threat, as they are frequently associated with life-threatening infections3, 4. Klebsiella pneumoniae (Kp), an ESKAPE pathogen, induces necrotizing pneumonia, abscesses in various organs, and septicemia5–7. In recent years, the rapid spread of multi-drug resistant bacteria, including Carbapenem-resistant Kp strains, has caused a greater than 50% increase in lung-infection induced mortality worldwide8, 9.
Nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) are implicated in the recognition of pathogens or danger signals, modulation of inflammation, and maintenance of homeostatic processes1, 10, 11. In particular, NOD-like receptor pyrin domain-6 (NLRP6) has been extensively studied in the context of intestinal homeostasis and tumorigenesis12, 13. As compared to wild-type (WT) mice, those deficient in NLRP6 (Nlrp6−/−) develop severe and transferable colitis, which is associated with pro-colitogenic gut microbiota14. Further, NLRP6 deficiency imposes microbiota alterations that lead to the development of hepatic steatosis and obesity15. However, the finding that NLRP6 controls gut microbiota dysbiosis has been challenged by recent studies16.
NLRP6 has been shown to interact with Dhx15 to regulate MAVS-dependent responses, thereby controlling enteric encephalomyocarditis during viral infections17. Recently multiple reports suggested that NLRP6 recognizes bacterial components and modulates host defense mechanisms1, 13, 18, 19 Importantly, Nlrp6−/− mice were shown to be resistant to bacterial infection due to increased inflammation and augmented bacterial clearance following infection with Listeria monocytogenes, Salmonella typhimurium, Escherichia coli, and Staphylococcus aureus13, 18, 19. Similarly, our previous work demonstrated that Nlrp6−/− mice exhibit higher neutrophil recruitment and augmented bacterial clearance in response to pulmonary S. aureus infection18. On the other hand, Nlrp6−/− mice were unable to clear enteric Citrobacter rodentium infection, which was associated with compromised intestinal barrier function and mucus secretion20. However, results obtained with gram-positive and enteric bacteria cannot be extrapolated to Gram-negative pathogens in the lung because every bacterium has unique virulence factors and induces specific pathogenesis in different organs. In case of Kp-induced pneumonia, several NLRs, including NLRP3, NLRP12 and NLRC4, have been shown to regulate neutrophil-dependent host defense21–23. However, whether NLRP6 modulates host protection, neutrophil homeostasis, and neutrophil function during pulmonary infection with Kp remains unclear.
The goal of this study was to define the role of NLRP6 in neutrophil homeostasis following Kp infection and identify the downstream molecules that contribute to host response in order to gain more in-depth insight into immunological mechanisms in Kp-induced pneumonia-derived sepsis.
RESULTS
NLRP6 expression is upregulated in mouse lungs and human cells following pulmonary Kp infection.
Since NLRP6 expression is enhanced in human pneumonic lung sections18, we investigated whether NLRP6 is upregulated in Kp-infected mouse lungs using immunohistochemistry. We observed increased expression of NLRP6 in Ly6G+ neutrophils, F4/80+ macrophages, and cytokeratin-5+ epithelial cells in Kp-infected mouse lungs (Fig. 1a). Immunoblotting confirmed that Kp-infected mouse lungs had increased NLRP6 expression at 6, 24, and 48 h post-infection (Fig. 1b). Increased NLRP6 expression was further confirmed by immunoblotting Kp-infected bone-marrow derived neutrophils (BMDN) and macrophages (BMDM) at 2 and 4 h post-infection (Fig. 1c, d). As anticipated, no NLRP6 staining was found in the lung tissue of gene-deficient mice (data not shown).
Figure 1. NLRP6 expression in mouse lungs following Kp infection.
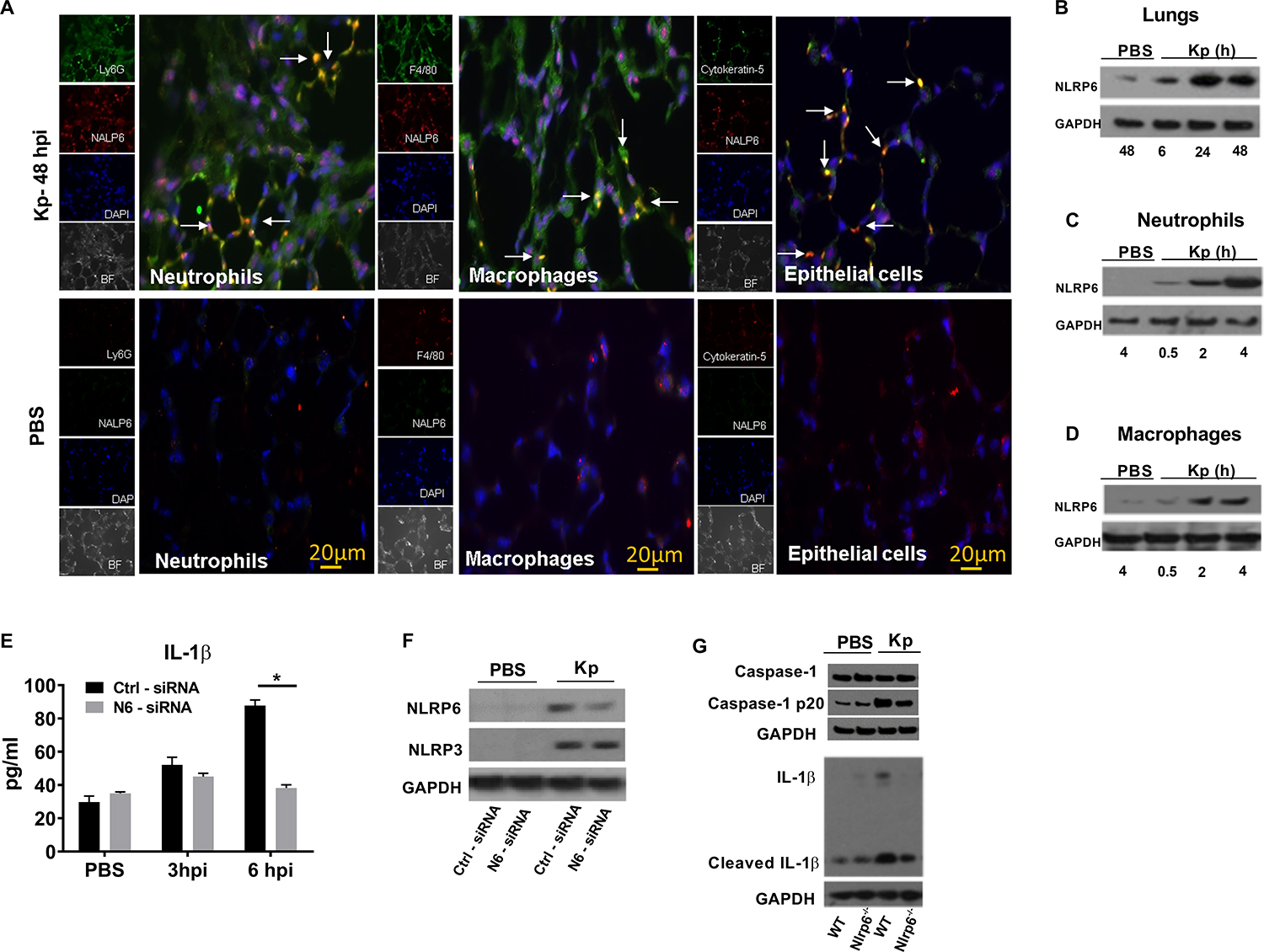
(A) NLRP6 expression at 48 h in lung sections from WT mice following Kp (103 CFU/mouse) or PBS inoculation. In infected group, red staining indicates NALP6 expression. Neutrophils (Ly6G+), macrophages (F4/80+), and epithelial cells (Cytokeratin-5+) are stained green. In PBS group, Green staining shows NLRP6 expression. Ly6G+ neutrophils, F4/80+ macrophages, and cytokeratin-5+ epithelial cells are stained red. This is a representative image of five lung sections with similar results. Original magnification, X 40. Arrows show co-localization. (B) Immunoblotting of NLRP6 in mouse lungs following Kp (103 CFU/mouse) infection at designated time points (hpi). (C–D) Expression of NLRP6 in BMDN and BMDMs at different time points following infection. (E–F) siRNA knockdown of NLRP6 in THP-1 (human macrophage) cells followed by infection with Kp (MOI 1) at indicated time points. THP1 cells (0.5×106) were transfected with NLRP6 or scrambled control siRNA for 48 hours and infected with Kp (MOI 1) for 6 hours. (E) Level of human IL-1β in culture supernatants using triplicate wells. Statistical significance was determined by unpaired t-test. (F) Expression of NLRP6 and NLRP3 proteins in cell lysates after siRNA knockdowns. (G) Immunoblotting of pro-form and cleaved Caspase-1 (p20), and IL-1β in mouse lungs following Kp (103 CFU/mouse) at 48 hpi. Western blots are representative of 3 separate experiments. PBS served as a control. *, p<0.05. BF: bright field, DAPI: 4’, 6-diamidino-2-phenylindole, hpi: hour post-infection, CFU: colony forming units, MOI: multiplicity of infection.
Recent studies have shown that NLRP6 can form an inflammasome complex which results in maturation of caspase-1 and IL-1β in response to Gram-positive bacterial infection13, 18. To show the translational relevance of the experimental model, we utilized a siRNA knockdown approach to suppress NLRP6 expression in human monocytes (THP1 cells) and measured production of the inflammasome-dependent cytokine, IL-1β. Our results illustrate that Nlrp6 siRNA-transfected THP1 cells secrete a reduced level of IL-1β compared to the control siRNA-transfected cells (Fig. 1e, f). As a control, Nlrp6 siRNA-transfected THP1 cells display no difference in NLRP3 protein expression as compared with scrambled siRNA-transfected cells (Fig. 1F). Additionally, lung lysates from Nlrp6−/− mice show attenuated levels of cleaved Caspase 1 and IL-β compared to Kp-infected WT mice (Fig. 1g).
Nlrp6−/− mice have impaired neutrophil-dependent host defense against Kp infection.
To assess the importance of NLRP6 in host defense against a Gram-negative Kp infection, WT and Nlrp6−/− mice were infected intratracheally (i.t.) with a high (104 CFU/mouse) or low (103 CFU/mouse) inoculum of Kp and survival was monitored for 15 days. Unlike the resistance of Nlrp6−/− mice to infection with Gram-positive bacteria13, 18, Nlrp6−/− mice displayed increased mortality following infection with Kp at either dose (Fig. 2a). Importantly, the reduced survival of Nlrp6−/− mice was associated with impaired bacterial clearance in the lungs and bacterial dissemination to the spleen at 48 h post-infection (Fig. 2b, c). Previously, we reported that accumulation of neutrophils in the lungs is a critical step for clearance of Kp24. Thus, total white blood cells and neutrophils were enumerated in BALF of mice following Kp infection. As expected, Nlrp6−/− mice recruited reduced numbers of total white blood cells and neutrophils at both 24 and 48 h post-infection compared to Kp-infected WT mice (Fig. 2d, e). Additionally, myeloperoxidase (MPO) levels in the lungs of Nlrp6−/− mice were reduced at 24 and 48 h post-infection compared to WT mice (Fig. 2f). Based on the % of section infiltrated by inflammatory leukocytes, semi-quantitative lung histology revealed that Nlrp6−/− mice had reduced inflammation scores (cellular recruitment) compared to WT mice at 24 h post-infection (Fig, 2g, h). Neutrophil migration is primarily regulated by pro-inflammatory cytokines and chemokines in the lungs24. As anticipated, CXC chemokines (CXCL1, CXCL2, and CXCL5) were decreased in KP-infected Nlrp6−/− mice (Fig. 2I–K). Furthermore, pro-inflammatory cytokines (TNF-α, IL-6, G-CSF, IL-1β) were reduced in Kp-infected Nlrp6−/− mice (Supplemental Fig. 1A–F).
Figure 2. Impact of NLRP6 on survival, bacterial clearance, cellular recruitment, and cytokine/chemokine production during Kp infection.
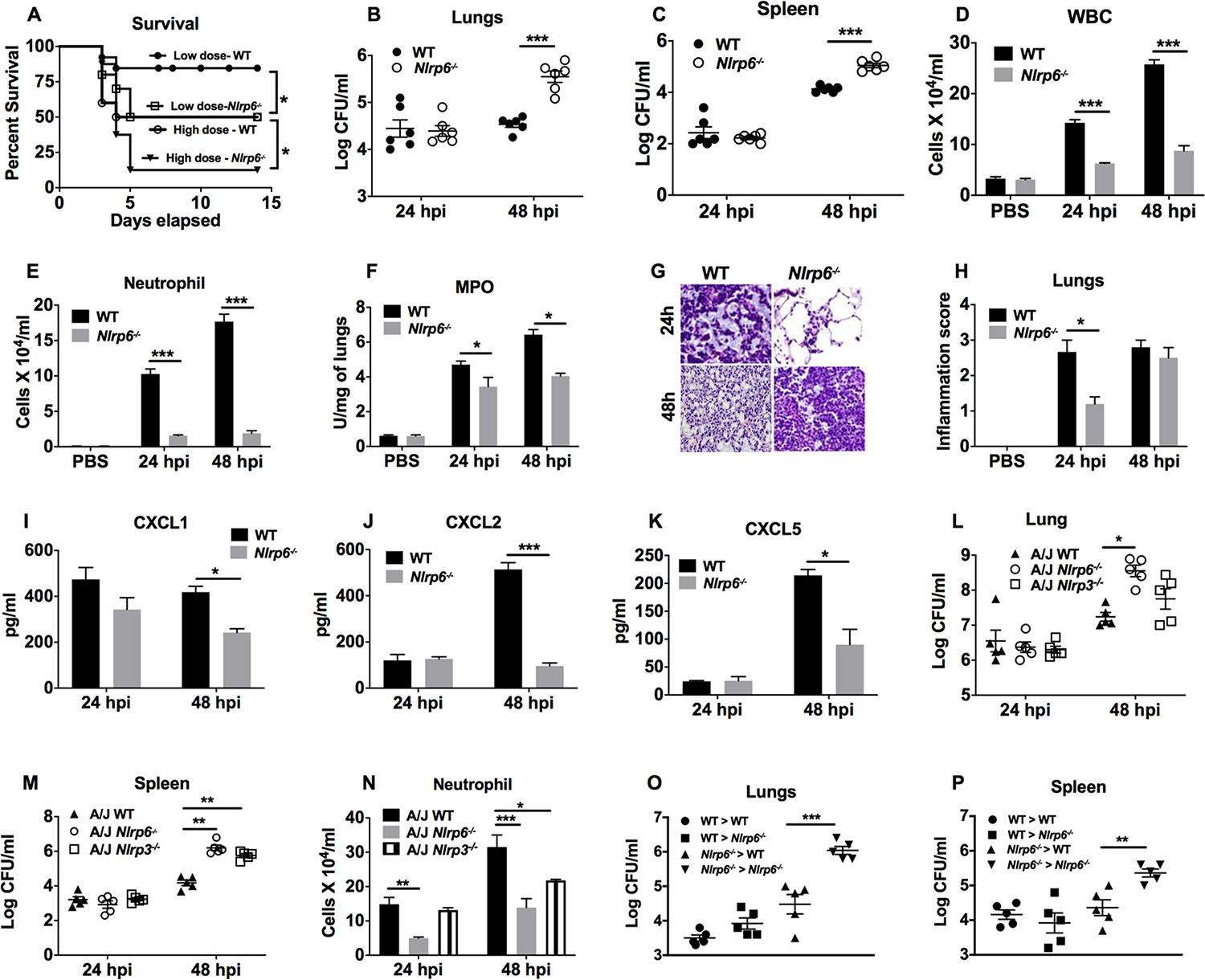
(A) Nlrp6−/− and WT mice were infected with 103 or 104 CFU of Kp and survival was recorded for 15 days. A Kaplan-Meier plot is used to show survival of mice from each group. (n=20/group). Statistical significance was determined by log-rank. (B–K) Nlrp6−/− and WT mice were infected with 103 CFU of Kp and BALF and organs were harvested at 24 and 48 h post-infection. Bacterial burden in lungs (B) and spleen (C) was assessed at the indicated time points (n=6/group). Total number of white blood cells (D), neutrophils (E) in BALF, and MPO activity in lung homogenates (F) were measured at 24 and 48 h post-infection (n=3/group). (G–H) Lungs from Kp-infected mice were perfused and processed for histology, stained with H&E, and inflammatory changes in histological sections were scored. (G) Representative histological mouse lung sections are presented. (H) Inflammation score obtained by semiquantitative histology are shown at 24 and 48 h post-infection (n=5/group). Original magnification, 40X. (I–K) Levels of neutrophil-attracting chemokines CXCL1 (I), CXCL2 (J), and CXCL5 (K) in BALF were measured at 24 and 48 h post-infection (n=3/group). (L–N) Nlrp6−/− or Nlrp3−/− on the A/J background mice were inoculated with Kp (103 CFU/mouse) and BALF and organs were harvested at 24 and 48 h post-infection. Bacterial burden in lungs (L), spleens (M), and neutrophils (N) in BALF were enumerated at 24 and 48 h post-infection (n=5/group). (O, P) Bone marrow chimeric mice were generated and infected with Kp (103 CFU/mouse). At 48 h post-infection, the bacterial burden was assessed in the lungs (O) and spleens (P). (n = 4–6/infection group). Unpaired t-test (B–F, H–K), and ANOVA (followed by Tukey’s multiple comparisons) (L–P). *, p<0.05; **, p<0.01; ***, p<0.001. WBC: White blood cells, MPO: Myeloperoxidase.
In order to show whether genetic background of the mouse strain dictate the phenotype of the immune response to infection25, we also infected WT and Nlrp6−/− mice on an A/J background and assessed bacterial clearance and neutrophil influx in the lungs. Similar to Nlrp6−/− mice on a C57BL6/J background, Nlrp6−/− mice on the A/J background displayed defective bacterial clearance in the lungs and spleen, which were associated with reduced neutrophil influx in BALF (Fig. 2l–n). We used another inflammasome (NLRP3) gene-deficient mice on the A/J background to demonstrate the role of NLRP3 in Kp infection because mice deficient in Nlrp3 on the C57Bl/6 background results in decreased survival and attenuated lung inflammation.23. Here, we found that Nlrp3−/− mice on the A/J background23 also displayed defective bacterial clearance in the spleen and reduced neutrophil recruitment to BALF (Fig. 2l–n). Because both myeloid and stromal cells display NLRP6 upregulation following Kp infection (Fig. 1a, b), we next performed bone marrow transplantation to study the relative importance of NLRP6 expression in hematopoietic (neutrophil and macrophage) and non-hematopoietic (epithelial) compartments. The bacterial burden was higher in the lung and spleen of the Nlrp6−/−→Nlrp6−/− chimeric group compared to the WT→WT group (Fig. 2o, p). Moreover, compared with the Nlrp6−/− →Nlrp6−/− chimeric mice, the bacterial burden was reduced in the lungs and spleens of WT→Nlrp6−/− and Nlrp6−/−→WT chimeric mice (Fig. 2o, p). However, the bacterial burden in the lung and spleen were similar between chimeric WT→Nlrp6−/−, Nlrp6−/− →WT mice, and WT→WT mice, suggesting NLRP6 signaling in both compartments is critical for host defense (Fig. 2o, p).
Defective neutrophil recruitment and bacterial clearance in Nlrp6−/− mice is not dependent of gut microbiota composition.
It has been reported that alterations in the gut microbiota regulate susceptibility to many human diseases26. Although there is some debate, microbial dysbiosis resulting from genetic deficiencies of NLRP6 and ASC-linked inflammasomes has been shown to worsen colitis and obesity, both of which are transferable14–16. To this end, we carried out 16S rDNA-based sequencing to investigate the potential dysbiosis of the gut microbiota in WT and Nlrp6−/− mice. Regarding alpha diversity, the bacterial community richness (Chao1) and diversity (Shannon) did not differ between WT and Nlrp6−/− mice following Kp infection (Fig. 3a, b). Moreover, a PCoA analysis of microbial OTUs revealed a differential clustering of microbial communities between WT and Nlrp6−/− mice (Fig. 3c). Consistent with a previous report14, taxonomic analysis showed Nlrp6−/− mice had increased abundance of Prevotellaceae and Paraprevotellaceae compared to WT mice (Fig. 3d–f). To determine if the gut microbiota contributes to host susceptibility in Nlrp6−/− mice, we cohoused WT and Nlrp6−/− mice for four weeks prior to infection to equalize differences in the microbiota of mice14, 18. Similar to separately housed Nlrp6−/− mice, the co-housed Nlrp6−/− mice also showed increased bacterial burdens in the lungs and spleens, which were associated with decreased influx of total white blood cells and neutrophils in BALF (Fig. 3g–j). However, we have not examined the gut composition after co-housing to show equalization of differences in microbiota.
Figure 3. Importance of the gut microbiota in Nlrp6−/− mice for bacterial clearance and neutrophil influx following Kp infection.
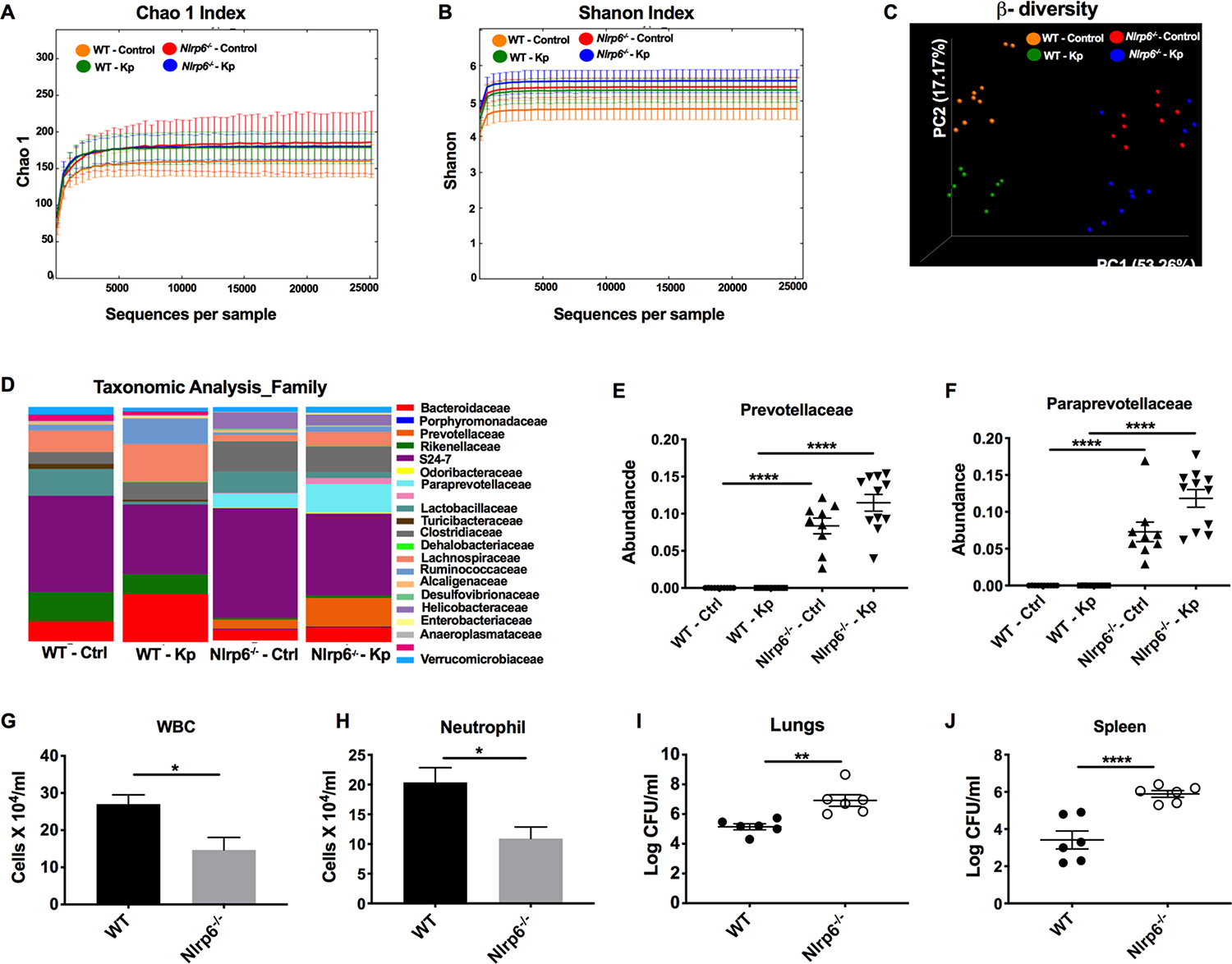
(A–F) WT and Nlrp6−/− mice were infected with Kp (103 CFU/mouse) or PBS. At 48 h post-infection, fecal samples were collected and the microbiota was analyzed using 16S rDNA-based phylogenetics method. Rarefaction curves calculated for Chao1 (A) and Shannon index (B) are shown. (C) PCoA plots based on the weighted Unifrac distance matrix are presented. (D) Heat maps showing taxonomic composition of microbial communities at the family level. ANOSIM, R= 0.786, P=0.001. Relative abundance of prevotellaceae (E) and pararevotellaceae (F) families is presented. (n=10 mice/group). #, ## denotes orders with unknown families. (G–J) WT and Nlrp6−/− mice were co-housed for 4 weeks and inoculated with Kp (103 CFU/mouse). Total number of white blood cells (G), neutrophils (H) in BALF and bacterial burden in lungs (I) and spleen (J) were enumerated at 48 h post-infection. (n = 5–6/group). Statistical significance was determined by ANOVA (followed by Tukey’s multiple comparisons) (E, F) and unpaired t-test (G–J). *, p<0.05; **, p<0.001; ****, p<0.0001.
Bone marrow of Nlrp6−/− mice has a defect in granulopoiesis and neutrophil release during pulmonary Kp infection.
Numerous studies have demonstrated that neutrophils are continuously generated in the bone marrow and rapidly mobilize to sites of infection during inflammatory episodes through a well-orchestrated process called emergency granulopoiesis27. Since Nlrp6−/− mice showed reduced levels of neutrophil recruitment and CXC-chemokines and granulocytic cytokines (Fig. 2e, f and Supplemental Fig. 1A–F), we hypothesized that emergency granulopoiesis may be altered in Nlrp6−/− mice during Kp-pneumonia. In this context, we investigated the granulopoietic compartments of Kp-infected Nlrp6−/− mice using flow cytometry, as previously described28, 29. During inflammatory episodes, immature neutrophils gradually lose c-Kit and acquire the maturation marker, Ly6G. In Kp-infected bone marrow undergoing granulopoiesis, subpopulations #1 (c-KithighLy6Gneg), #2 (c-KitintLy6Gneg), and #3 (c-KitintLy6Gint) represent early c-Kit+ immature granulocyte precursors; whereas subpopulation #4 (c-KitnegLy6Gint) and #5 (c-KitnegLy6Ghigh) are immature and mature neutrophils respectively (Fig. 4a). At 48 h post-infection, WT mice displayed an increase in subpopulation #1 (early granulocyte precursors) and a decrease in subpopulation #5 (mature neutrophils to be released) in the granulopoietic compartments compared to their steady state (Fig. 4b–d). In contrast, Kp-infected Nlrp6−/− mice exhibited impairment in the increase of subpopulation #1 and a decrease in subpopulation #5 in the granulopoietic compartments compared to WT mice (Fig. 4b–d). However, the distributions of granulopoietic subpopulations #1–5 were identical in both uninfected WT and Nlrp6−/− mice (Fig. 4b–d). Furthermore, we investigated early granulocytic compartments, particularly hematopoietic stem cells (HSC) and myeloid progenitors, in Nlrp6−/− mice after Kp infection, as described previously28, 29. Both WT and Nlrp6−/− mice showed an increase in HSCs (c-Kit+Sca-1+Lin−) after Kp infection at steady state, although the numbers of HSCs were higher in WT mice than in Nlrp6−/− mice (Fig. 4e, f). At 48 h post-infection, the population of granulocyte-monocyte progenitor (GMP) cells increased within the c-Kit+Sca-1− Lin− HSC subpopulation in WT mice compared to Nlrp6−/− mice (Fig. 4g, h). Furthermore, blood neutrophil counts reduced in Nlrp6−/−mice as compared to their WT counterparts at 48 h post-infection with Kp (Fig. 4I).
Figure 4. Role of NLRP6 in emergency granulopoiesis and neutrophil release during Kp-induced lung infection.
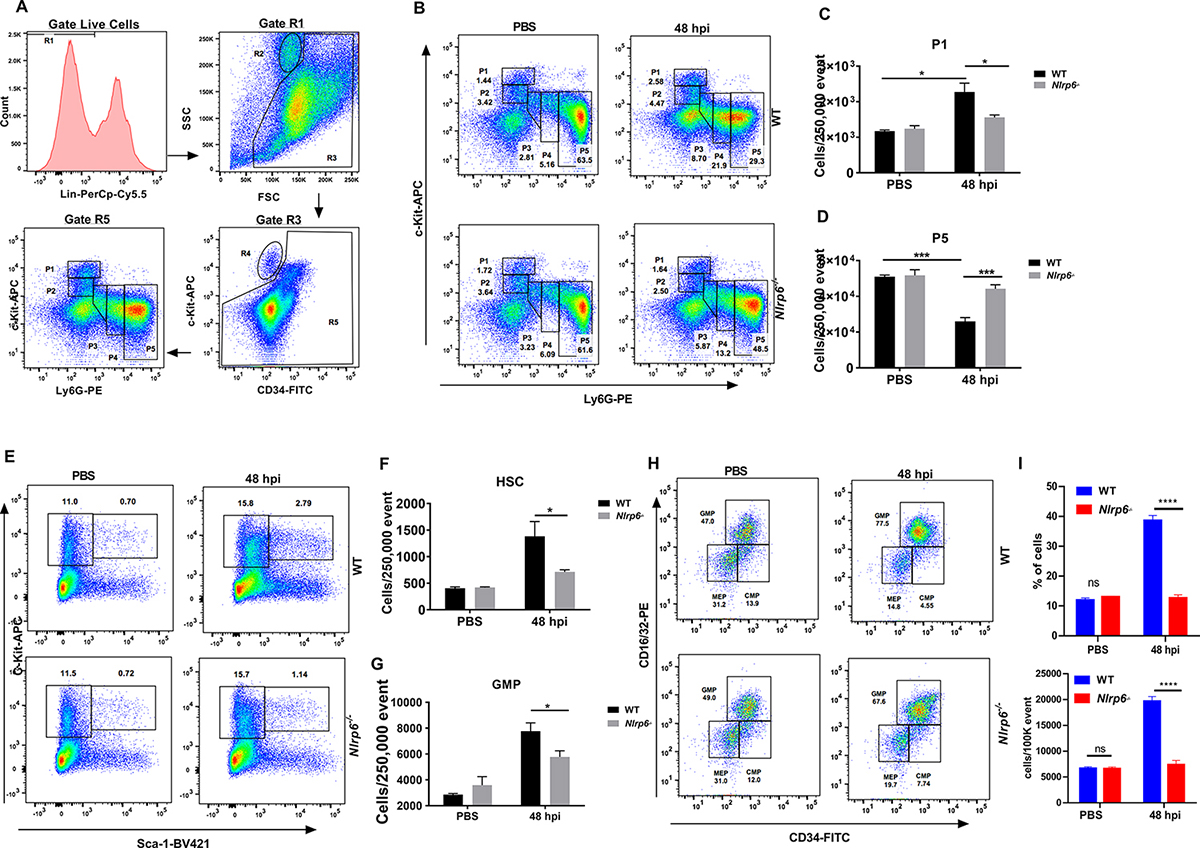
(A–H) WT and Nlrp6−/− mice were infected with Kp (103 CFU/mouse) and lungs were harvested at 48 h post-infection. (A) Flow cytometric analysis of granulopoiesis. First, BM cells that are lineage positive for B, T and erythroid cells were excluded as they have lost the potential to give rise to granulocytes. The remainder, gate R5, was then plotted against expression of c-Kit and Ly-6G. Populations R2 and R4 represent eosinophilic and megakaryocyte-erythroid progenitors, respectively. FACS dot plot (B), percentage of subpopulation #1 (C), and subpopulation #5 (D) within the granulopoietic compartment are presented. FACS analysis plot (E) and number (F) of hematopoietic stem cells (HSC)(c-Kit+Sca-1+Lin−) and FACS dot plot (G) number (H) of granulocyte-monocyte progenitor (GMP) cells (within c-Kit+Sca-1−Lin−) at 48 h post-infection. (n= 4–6 mice/infection group, n=3 mice/control group). (I) FACS analysis of blood neutrophils at 48 h post-infection with Kp (n=6 mice/group). The % and number of cells were determined after PBS instillation or infection with Kp. Statistical significance was determined by unpaired t-test (C, D, F, G). *p<0.05; ***, p<0.001. CMP: Common myeloid progenitors, MEP: Megakaryocyte-erythroid progenitors.
Nlrp6−/− mice display decreased neutrophil extracellular trap (NET) formation and NET-mediated bacterial killing.
In addition to an accelerated granulopoietic response, a successful immune response also requires effective neutrophil function to clear bacteria24. To investigate the role of NLRP6 in neutrophil function, we depleted neutrophils from Nlrp6−/− mice as described previously30 and replenished mice with either WT or Nlrp6−/−-BMDNs. Interestingly, the bacterial burden in the lungs of Nlrp6−/− mice was higher when Nlrp6−/−-BMDNs were used compared to WT-BMDNs (Fig. 5a). The formation of neutrophil extracellular traps (NETs), a process is known as NETosis, has emerged as a critical mechanism used by neutrophils to kill extracellular pathogens31. NETs are composed of DNA studded with antimicrobial proteins, such as citrullinated histones, MPO, neutrophil elastase, and α-defensins31, 32. Compared to WT-BMDNs, Nlrp6−/−-BMDNs showed impaired NETosis at different time points (Fig. 5b). Utilizing immunofluorescence microscopy, we demonstrate that Kp-infected Nlrp6−/−-BMDNs exhibit reduced NET forming cells compared to WT-BMDNs (Fig. 5c, d). Regarding the NET structure, Brinkmann et al. described NETs as long string-like extracellular threads and globular domains, as seen under scanning electron microscopy (SEM)31. Utilizing SEM, we show long string-like extracellular threads entangling bacteria in Kp-infected WT-BMDNs, but not in Nlrp6−/−-BMDNs (Fig. 5e). To investigate if NLRP6-dependent NETosis is important for Kp clearance, we infected cytochalasin pre-treated BMDNs from Nlrp6−/− or WT mice in the presence or absence of DNase with Kp and measured Kp burden in culture supernatants. As expected, the culture supernatant of WT-BMDNs had a lower bacterial burden compared to Nlrp6−/−-BMDNs following Kp infection (Fig. 5f). Importantly, when treated with DNase, the bacterial burden in the supernatant of WT-BMDNs increased, highlighting the importance of NET-mediated killing (Fig. 5f). However, DNase-treated and untreated Nlrp6−/−-BMDNs exhibited comparable bacterial burdens in the culture supernatant, suggesting NLRP6 is upstream and regulates NET formation (Fig. 5f). Furthermore, BMDNs show comparable intracellular killing of bacteria between Nlrp6−/− and WT mice at 30 and 60 min after infection (Fig. 5G). In lung tissue homogenates after Kp infection, reduced expression of citrullinated histone 3 (Cit-H3), a marker of NET formation, is noted in Nlrp6−/− mice (Fig. 5H).
Figure 5. The effect of NLRP6 on NETosis and NET-mediated bacterial killing.
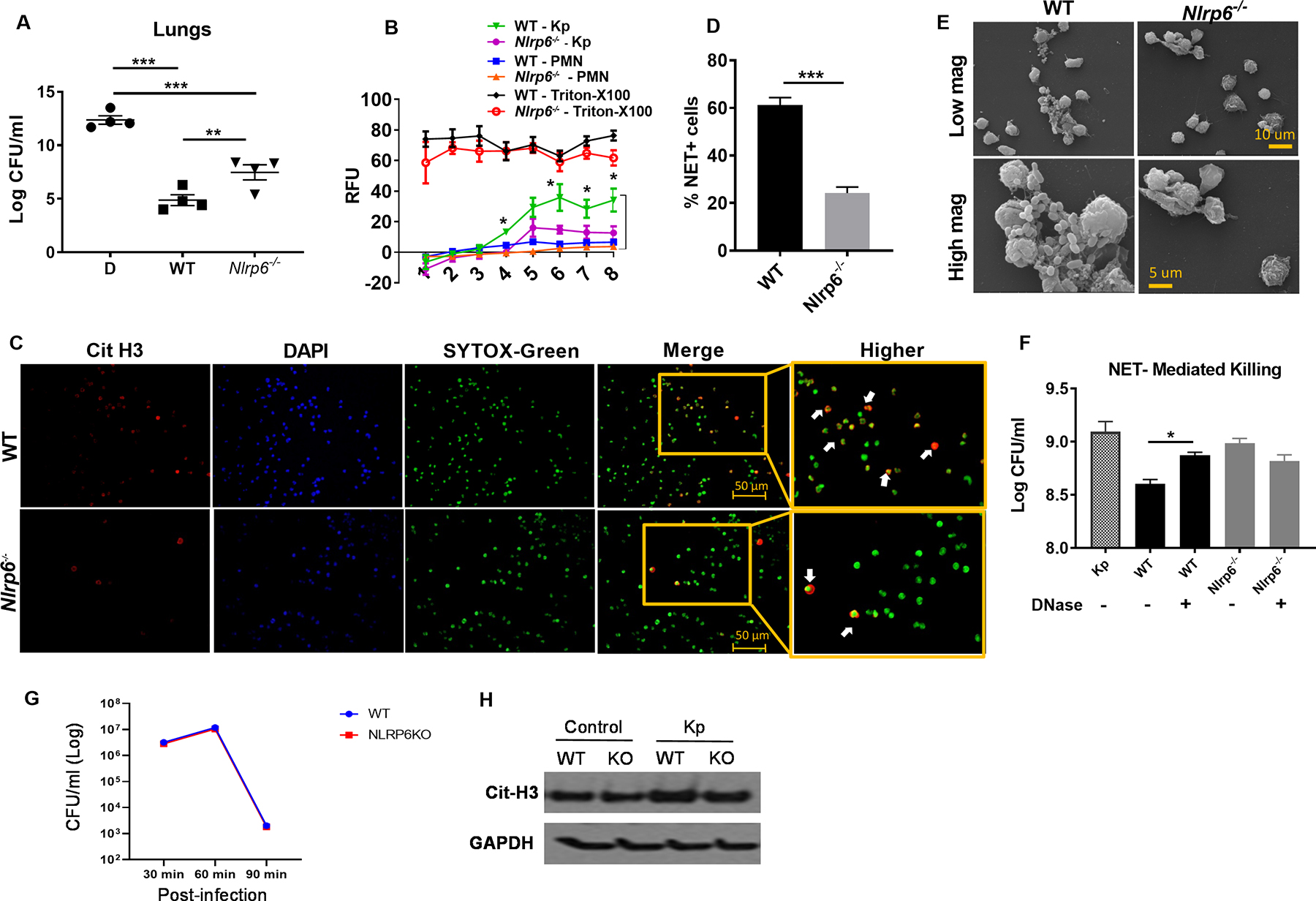
(A) In Nlrp6−/− mice, neutrophils were depleted by administration of anti-Ly6G mAb intraperitoneally and replenished with freshly isolated BMDNs (2 ×106 cells/mouse) from WT or Nlrp6−/− mice i.t. 30 min prior to Kp infection. At 48 h post-infection, bacterial burden was assessed in lungs (n=4/group). (B) BMDNs from WT and Nlrp6−/− mice were seeded in 96-well plates and infected with Kp. SYTOX green (5 μM) was added to the plates and they were monitored every hour to assess extracellular DNA release. The relative fluorescence intensity (RFU) was recorded to evaluate NETosis each hour up to 8 h post-infection. (C–D) WT and Nlrp6−/−-BMDNs were seeded and infected, SYTOX was added and cells were incubated for 8 hours and then fixed. Double positive cells for citrullinated H3 and SYTOX green (extracellular DNA) were counted (indicated by white arrowheads) as NETosis. Representative immunofluorescence images (C) and percentage of NET-forming (double positive) cells (D) are presented. Original magnification, ×20. (E) Morphological features of NETosis in Kp-infected BMDNs were analyzed by scanning electron microscopy. Presence of long thread-like structures is evidence of NETosis. (F) NET-mediated killings of WT and Nlrp6−/− mice. BMDNs was determined by assessing extracellular bacterial burden in supernatants following Kp (MOI 1) infection at 6 h post-infection in absence or presence of DNase (100 U/ml). BDMNs were pretreated with cytochalasin D (10 μg/ml) to inhibit phagocytosis. (G) Intracellular bacterial killing by neutrophils. Kp clearance from bone marrow-derived neutrophils of Nlrp6−/− and WT mice. Bone marrow neutrophils were infected with Kp at an MOI of 10 and determined for bacterial killing capacity by estimating intracellular CFU at 30, 60 and 90 min after infection using 5 wells/group. (H) Expression of citrullinated-H3 protein level in the lungs from WT and Nlrp6−/− mice following Kp infection at 48 hpi. Western blots are representative of 3 separate experiments. Statistical significance was determined by ANOVA (followed by Bonferroni’s post hoc comparisons) (A, F) and unpaired t-test (B, C). *p<0.05; **p<0.01; ***, p<0.001.
Recombinant CXCL1 rescues impaired host defense, neutrophil homeostasis and NETosis in Nlrp6−/− mice after Kp infection.
We have previously shown that CXCL1 regulates granulopoiesis, neutrophil migration, and neutrophil function in numerous disease models33, 34. The lack of a significant neutrophil homeostasis in Nlrp6−/− mice to Kp infection may be due to a decrease in production of CXCL1. Our results demonstrate that Nlrp6−/− mice have reduced CXCL1 in BALF, lungs, and blood (Fig. 2i and Supplemental Fig. 2A, B). Kp-infected IL-1β−/− mice exhibit reduced CXCL1 in BALF (Supplemental Fig. 3A). CXCL1 is produced by both myeloid and non-myeloid cells and is essential for host protection during Kp-induced pneumonia33. To determine if recombinant CXCL1 (rCXCL1) restores host defense against Kp-pneumonia in Nlrp6−/− mice, we administered rCXCL1, rIL-17A, or BSA i.t. to Nlrp6−/− mice 1 h post-infection. Since the level of IL-17A remains unaltered in Nlrp6−/− mice (Supplemental Fig. 2), rIL-17A was used as the control cytokine despite its granulopoietic activity via G-CSF35. To our surprise, administration of rCXCL1, but not rIL-17A, rescued host survival (Fig. 6a), although both rCXCL1 and rIL-17A led to a reduced bacterial burden in Nlrp6−/− mice as compared to BSA-treated mice following Kp infection (Fig. 6b–d). Furthermore, administration of rCXCL1, but not rIL-17A, augmented the recruitment of total white blood cells, neutrophil influx in the BALF, and MPO activity in lung homogenates (Fig. 6e–g). Additionally, Nlrp6−/− mice treated with rCXCL1, but not rIL-17A, had increased levels of cytokines (IL-6, TNF-α, IL-1β) and the CXCL5 chemokine in BALF following Kp infection (Supplemental Fig. 4A–F). Since impaired host protection in Nlrp6−/− mice is associated with defective granulopoietic responses and NETosis, we next investigated whether administration of rCXCL1 can rescue these defects in Kp-infected Nlrp6−/− mice. Moreover, we have demonstrated previously that CXCL1 rescues NETosis in neutrophils isolated from alcohol-challenged septic mice34. Pretreatment of Nlrp6−/−-BMDNs with rCXCL1 enhanced NETosis (Fig. 6h). We have previously shown a critical role of CXCL1 in emergency granulopoiesis during pneumococcal infection29. Thus, to investigate if administration of rCXCL1 can rescue emergency granulopoiesis, we administered rCXCL1 to Nlrp6−/− mice at the time of Kp infection and examined granulopoietic subpopulations #1–5 and GMPs. Nlrp6−/− mice administered rCXCL1 exhibit a decrease in subpopulation #5 following Kp infection, indicating enhanced release (Fig. 6i, j). Additionally, rCXCL1-treated Nlrp6−/− mice also display increased GMPs compared to BSA-treated mice (Fig. 6k, l).
Figure 6. Effects of recombinant CXCL1 on host defense, emergency granulopoiesis, and NETosis in Nlrp6−/− mice following Kp infection.
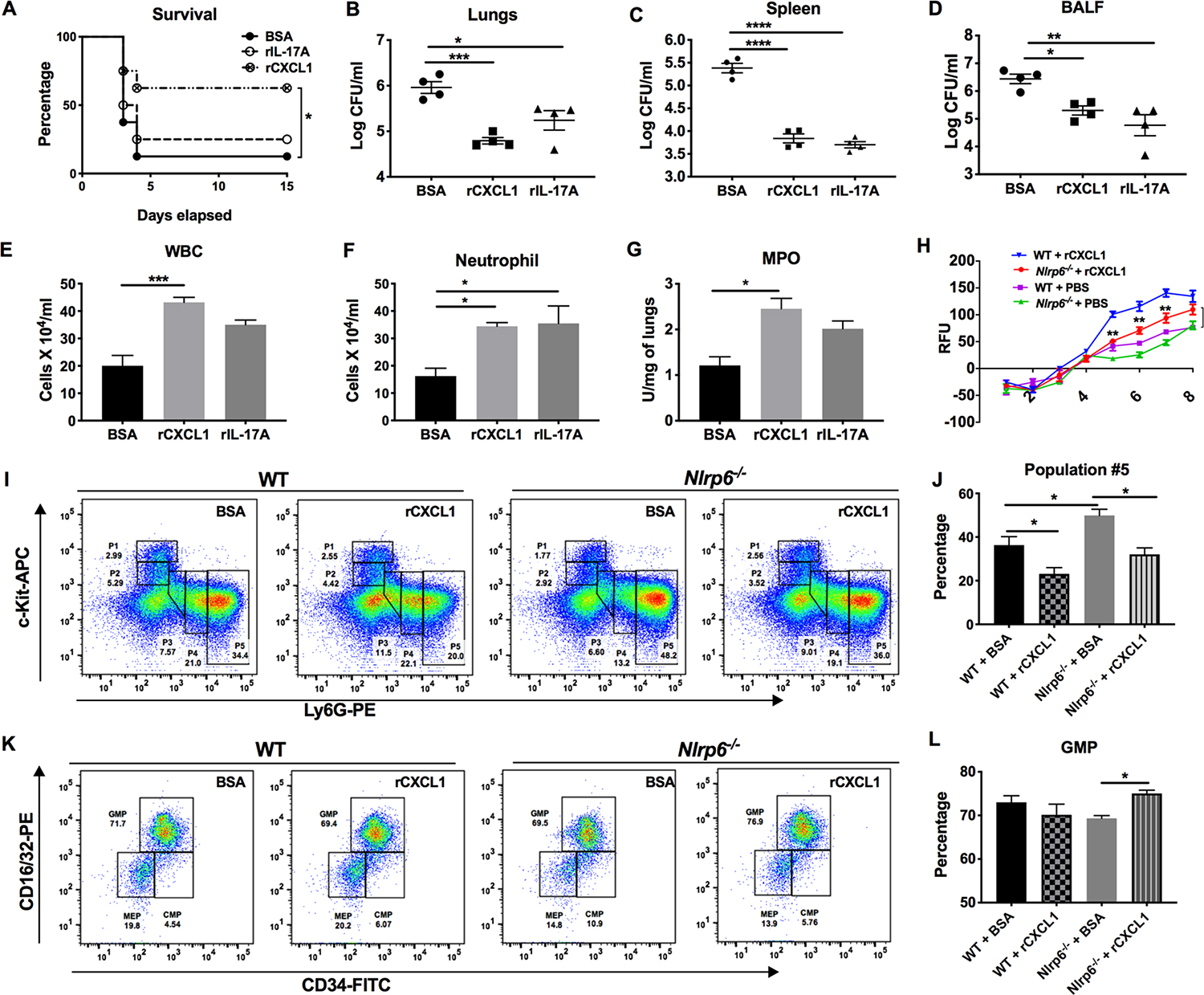
(A–G) Nlrp6−/− mice were infected with Kp (103 CFU/mouse) with or without administration of rCXCL1 or rIL-17A or BSA at 1 h post-infection. (A) Survival was monitored for 15 days. Statistical significance was determined by log-rank (n = 8 mice/group). (B–D) The bacterial burden was assessed in lungs, spleen, and BALF at 48 h post-infection. (E–G) Total numbers of white blood cells, neutrophils in BALF, and MPO activity in lung homogenates were measured. (n=4 mice/group). (H) Nlrp6−/− BMDNs were seeded, pre-treated in the presence or absence of rCXCL1 (5nM) for an hour, infected, then SYTOX was added and incubated for 8 hours. Relative fluorescence intensity (RFU) was recorded to evaluate NETosis each hour up to 8 h post-infection. (H-L) Nlrp6−/− mice were infected with Kp (103 CFU/mouse) with or without administration of rCXCL1 or BSA at 1 h post-infection. Bone marrow was harvested at 48 h post infection. The FACs plot (I) and percentage of subpopulations #5 (J) and FACs plot (K) and percentage of GMPs (L) within the granulopoietic compartment are presented. (n = 4–6 mice/infection group, n = 3 mice/control group). In vitro experiments had at least four technical replicates. ANOVA (followed by Bonferroni’s post hoc comparisons) (B–G, I, K), and unpaired t-test (H). *, p<0.05; **, p<0.001; ***, p<0.001****, p<0.0001.
DISCUSSION
In this report, we demonstrate the protective role of NLRP6 in Gram-negative bacterial infection in the lungs followed by sepsis. Kp-challenged Nlrp6−/− mice show augmented mortality, higher bacterial burden in the lung and spleen, and impaired neutrophil influx in the lungs. Because Kp causes sepsis following dissemination, we also explored granulopoiesis following Kp infection and found it to be impaired in Nlrp6−/− mice. In addition to attenuated neutrophil influx, NET formation and NET-mediated bacterial killing are also attenuated in Nlrp6−/− neutrophils. Administration of rCXCL1 rescued impaired neutrophil accumulation, granulopoiesis, and NET formation in Nlrp6−/− mice. While Nlrp6−/− mice display increased abundance of Prevotellaceae and Paraprevotellaceae families in the gut than their WT counterparts, the composition of the microbiota was not found to affect susceptibility of Nlrp6−/− mice to Kp infection because co-housing of mice did not appear to alter the phenotypes. A limitation in this study is that we did not perform gut microbiota analysis to demonstrate equalization of microbiota after co-housing. Finally, utilizing bone marrow chimeras, we illustrate that NLRP6 expression in both hematopoietic and resident cells is essential for bacterial clearance in the lungs and limiting sepsis.
NLRP6 has initially been shown to be a negative regulator of inflammation against bacterial infections13, 18, 19. For example, Nlrp6−/− mice were shown to be protected from systemic bacterial infections with Listeria monocytogenes, Salmonella typhimurium and Escherichia coli due to increased numbers of inflammatory cells. These studies have shown that the expression of NLRP6 in both hematopoietic and resident cells was found to be detrimental to host defense19. Recently, the protective phenotype of Nlrp6−/− mice during L. monocytogenes was further defined, as the NLRP6/Caspase11 axis seems to exacerbate Gram-positive pathogen (L. monocytogenes) infection through IL-18 production13. We also reported that Nlrp6−/− mice are protected against S. aureus-pneumonia18, and that S. aureus-infected Nlrp6−/− mice have increased neutrophil accumulation and higher IFN-γ levels leading to bacterial clearance18. In contrast, recent reports demonstrate NLRP6 as a positive regulator of host defense against enteric bacterial and viral pathogens. For example, Nlrp6−/− mice are unable to clear Citrobacter rodentium due to defective mucus exocytosis and autophagy of goblet cells20. Furthermore, NLRP6 protects against enteric encephalomyocarditis virus infection as it interacts with Dhx15 to regulate MAVS-dependent anti-viral responses17. Although not related to infection, Nlrp6−/− mice are vulnerable to colitis and colitis-induced carcinogenesis12, 14, 36. We also found NLRP6 as a positive regulator because Nlrp6−/− mice are susceptible to Kp infection-induced pneumonia-derived sepsis. While activation of NLRP6 with systemic L. monocytogenes, E. coli, and S. typhimurium infection and pulmonary S. aureus (Gram-positive) result in overt pathology if uncontrolled18, 19, it is likely that NLRP6 is critical for the amplification of inflammatory responses during Gram-negative lung and viral infections where pathogens do not induce robust inflammatory or pathological response.
NLRP6 is primarily studied in intestinal inflammation models and is abundantly expressed in the apical surface of gut epithelium12. It is initially established that NLRP6 is essential for intestinal homeostasis through gut microbiota. During dextran sulfate sodium (DSS)-induced colitis, Nlrp6−/− mice develop transferable and pro-colitogenic microbiota14. A recent report, however, indicated that loss of NLRP6 and the ASC-dependent inflammasome do not shape the composition of gut microbiota in mice16. Our findings are in agreement with the concept that NLRP6-regulated gut microbiota composition does not have a significant role in neutrophil-dependent host defense against Kp-induced pneumonia-derived sepsis. Although molecular mechanisms that regulate neutrophil function can be mediated by extrinsic pathways induced by extracellular messengers and intrinsic pathways induced by intracellular signals, controlling bacterial infection by neutrophils via NLRP6 is primarily extrinsic.
Emergency granulopoiesis is a mechanism of neutrophil generation in the bone marrow in response to systematic infection, which is well-orchestrated by numerous mediators and transcription factors27. Hematopoietic progenitor stem cells are known to express TLR4, which initiates LPS-induced emergency granulopoiesis37. Our work extends this notion by demonstrating a new role for NLRP6 against Gram-negative pneumonia-derived sepsis since Nlrp6−/− mice exhibit defective emergency granulopoiesis and granulocyte release and attenuated blood neutrophil numbers following Kp infection. Kp-infected Nlrp6−/− mice also showed reduced levels of CXC-chemokines as well as G-CSF. Therefore, we speculate that defective granulopoiesis in Nlrp6−/− mice may be due to reduced CXC-chemokines and/or G-CSF, thereby resulting in decreased accumulation of neutrophils in the lungs. We also showed that Kp-induced granulopoiesis is dependent on CXCL1, as its administration to Nlrp6−/− mice leads to an increase in HSCs and GMPs. However, we cannot rule out the possibility that NLRP6 controls expression of known regulators of emergency granulopoiesis, such as STAT3, STAT5A/5B, and C/EBPβ27.
Successful neutrophil recruitment and efficient neutrophil function at the site of infection are critical steps for efficient clearance of bacteria. NETosis is a unique form of programmed pathogen killing by neutrophils, where decondensed chromatin and antimicrobial granules are released into the extracellular space to trap and kill extracellular pathogens31. It is likely that NLRP6-dependent NETosis may be regulated through ROS production, as NETosis is linked to NADPH-dependent ROS production38. Here, we show that NLRP6 regulates NETosis in neutrophils and the mechanism underlying this can be rescued by CXCL1 treatment of Nlrp6−/− neutrophils. In this regard, we previously reported that CXCL1 regulates NET formation in a ROS-dependent manner34. As peptidylarginine deaminase 4 (PAD4) is required for NET formation39, future studies are needed to determine whether PAD4 participates in NLPR6-dependent NETosis. Although our findings indicate that NETosis in neutrophils is an essential mechanism for bacterial killing in vitro, this study does not rule out whether NETosis is an important mechanism for bacterial clearance in the lung. Moreover, our results ruled out the possibility that NLRP6 plays an important role in mediating neutrophil-dependent intracellular killing of Kp.
As for acute bacterial infections in the lung, we speculate that resident (alveolar) macrophages and alveolar epithelial cells first interact with pathogens through the NLRP6-dependent manner, including Kp in order to recruit PMNs to the lung from the bloodstream1–2. In this context, NLRP6 is expressed in both myeloid and resident cells18, 19 and our results are consistent with previous reports. Moreover, expression of NLRP6 by both cell types is essential for Kp clearance from the lungs and extrapulmonary organs. Since both myeloid and resident cell-derived CXCL1 is also important for pulmonary Kp clearance33, our findings suggest that NLRP6-IL-1β axis is upstream of CXCL1 production in both cell types. Regarding inflammasome complex formation, NLRP6 has been well characterized in non-myeloid cells, such as enterocytes40. NLRP6 forms an inflammasome complex, which processes caspase-1 and induces IL-1β expression in myeloid cells in response to S. aureus and L. monocytogenes infections13, 18. We demonstrate that NLRP6 mediates activation of caspase-1 and maturation of IL-1β following Kp infection likely through the formation of inflammasome. Since lipoteichoic acid and hemolysin of Gram-positive bacteria are implicated in sensing by NLRP613, 18, future mechanistic studies to investigate if NLRP6 senses LPS, other components of Kp, or possibly even endogenous ligands are of significant interest. With regard to Kp infection, NLRP3, NLRC4, and NLRP12 inflammasomes participate in protective host immunity although partial protection has been observed in these studies33. Therefore, cooperative interactions of multiple inflammasomes may be important for complete host protection against pulmonary Kp infection, a mechanism proposed in the context of intestinal health and homeostasis41.
In summary, the data presented here demonstrate NLRP6 is essential for neutrophil-dependent host defense, emergency granulopoiesis, and neutrophil function, including NETosis during Kp-pneumonia-derived sepsis through CXCL1 production. Notably, NLRP6-dependent host responses to Kp infection are independent of gut microbiota composition. Clearly, future studies using double- or triple-inflammasome knockout mice are still required to address the precise role of multiple inflammasome interaction in host protection. These findings also suggest modulation of NLRP6 activity is an attractive, druggable target for host protection against pneumonia-derived sepsis exclusively caused by Gram-negative bacterial pathogens.
METHODS
Animals.
NLRP6 gene-deficient (Nlrp6−/−) mice were obtained from Millennium Pharmaceuticals18. Mouse strains were backcrossed at least 10 times onto either C57BL/6J or A/J backgrounds. Eight to ten-week-old female WT controls were purchased from Jackson Laboratory because male mice are more resistant to intratracheal Kp infection as reported in previous publications22, 42, 43. The Institutional Animal Care and Use Committee at Louisiana State University approved our protocols.
Pneumonia-induced sepsis.
Pneumonia-derived sepsis was induced by intratracheal inoculation of Kp serotype 2 strain (ATCC 43816) as described previously43. Following the anesthesia, mice were inoculated with 103CFU/mouse for all experiments except survival (104 CFU/mouse) in 50 μL saline. The collection of lungs, spleen, and bronchoalveolar lavage fluid (BALF) was performed as described previously30, 43, 44. To assess the bacterial burden, 20 μL of organ homogenates and BALF were serially diluted, plated onto MacConkey Agar plates, and incubated overnight. In some experiments, 1 μg of rCXCL1 or rIL-17A or BSA (R&D systems) was intratracheally administered at 1 h post-infection29, 30.
Human macrophages and NLRP6 SiRNA.
Human macrophages (THP-1 cells; an acute monocytic leukemia cell line) were obtained from Invivogen. THP-1 cells were cultured in the presence of RPMI 1640 medium (Gibco) containing 10% FBS at 37°C in a 5% CO2 humidified incubator. For knockdowns, a pre-validated pool of siRNA for human Nlrp6 or control siRNA (Santa Cruz Biotechnology) was used. THP1 cells (0.5×106) were transfected with either 40 nM siRNA or a control siRNA using Lipofectamine 2000 (Thermofischer Scientific) for 48 hours according to manufactures guidelines. The transfected cells were infected with Kp (MOI 1) for 6 hours. Culture supernatants were used for ELISA and cell lysates were used for immunoblotting.
Immunoblotting and cytokine measurement.
Western blotting for lung homogenates and cell lysates was performed as described in earlier publications22, 30. Antibodies to mCaspase-1 (Adipogen), mIl-1β (Abcam), mNLRP6 (Sigma), hNLRP6 (Abgent), hNLRP3 (Cell Signaling), citrullinated histone H3 (Abcam) and GAPDH (Cell signaling) were used for immunoblotting. Human IL-1β and IL-18 and mouse TNF-α, MCP-1, IL-1β, IL-6, G-CSF, and IL-17A (eBioscience), and mouse CXCL1, CXCL2, and CXCL5 (R&D systems) were quantified according to the manufacturers’ protocols. To perform ELISA in mouse lung homogenates, snap frozen lung lobes were thawed, homogenized in 2 mL PBS containing 0.1% triton X-100 supplemented with complete protease/phosphatase inhibitor (1 tablet/50 mL) (Roche), centrifuged, and filtered prior to use for ELISA. Readings were normalized to total protein of lung homogenates and expressed as pg/mg.
Myeloperoxidase (MPO) assay.
MPO activity in the mouse lungs was measured as described previously33, 43. In brief, snap frozen lungs were thawed, weighed, and homogenized in 0.5% Hexadecyl Trimethyl-Ammonium Bromide (HTAB) in 50mM potassium phosphate buffer. The lung homogenates were centrifuged for 5 minutes and 7μl of supernatant was transferred into a flat bottom 96-well plate. Immediately after the addition of 200 μl of O-dianisidine hydrochloride solution, an increment in the absorbance between 0 sec and 90 sec were recorded at 460 nm using a spectrophotometer. MPO activity was as determined as U/mg of lung tissue.
Isolation of macrophages and neutrophils.
For bone marrow derived macrophages (BMDMs), femur/tibia were harvested, flushed with PBS to collect bone marrow cells. After RBC lysis, cells were centrifuged, washed, and set up in T75 flask with complete DMEM (supplemented with 10% FBS and Penicillin/Streptomycin (100U/ml)). Murine-CSF (final concentration of 50 ng/ml) was added at days 0, 2, 4, 6 for a week until they differentiate to macrophages. For bone marrow derived neutrophils (BMDNs), RBC-lysed bone marrow cells were passed through enriched neutrophil isolation cocktail EasySep neutrophil isolation kit (STEMCELL Technologies) using a magnetic negative selection.
Lung histology.
Lungs were perfused with saline, excised, and fixed in 4% phosphate-buffered formalin for 24 hours. After fixing, lungs were embedded in paraffin, and 5-μm thick sections were cut and stained with H&E. A veterinary pathologist examined the stained sections and scored them semiquantitatively in a blinded manner using previously described method. In brief, following scored system was used; 1, <5% of section is infiltrated by inflammatory leukocytes; 2, 5–10% of section is infiltrated by inflammatory leukocytes; and 3, >10% of section is infiltrated by inflammatory leukocytes.
Immunofluorescence microscopy.
Immunofluorescence staining of tissue slides was done as described previously18, 30, 43. Primary antibodies were used; anti-NLRP6 Ab (Abgent), anti-Ly6G Ab for PMNs (BioLegend), anti-cytokeratin-5 Ab for epithelial cells (BioLegend), and the anti-F4/80 Ab for macrophages (BioLegend). Appropriate Alexa-conjugated secondary antibodies were used (Invitrogen).
Chimeras.
Bone marrow chimeric mice were generated as described previously30, 43. Briefly, Recipient WT or Nlrp6−/− mice were irradiated from a cesium source in two 525-rad doses at 3 hours apart and freshly isolated BM cells (8 × 106) from donor naïve WT or Nlrp6−/− mice were injected through tail vein intravenously. BM recipient mice were then rested for two weeks with 0.2% neomycin sulfate in water. Two months after BM reconstitution, pneumonia was induced and bacterial burdens were enumerated. In these experiments, we found > 80% of blood leukocytes were derived from donor marrow as confirmed by GFP expressing cells.
Co-housing.
Co-housing experiments were performed as described previously18, 19. In brief, age- and sex-matched WT and Nlrp6−/− mice along with their cage contents were mixed together in 1:1 ratio and co-housed for 4 weeks prior to infection.
16S rDNA-based phylogenetics.
Stool samples were collected for the sequencing of 16S rDNA and phylogenetic analysis. The V4 hypervariable region was amplified using 515F: GTGCCAGCMGCCGCGGTAA and 805R: GGACTACHVGGGTWTCTAA 16S primers along with standard Illumina sequencing adapters. Amplicons were indexed, pooled, and sequenced on an Illumina MiSeq using a V2 2×250 paired end (500 Cycle) sequencing kit. Raw sequencing reads were processed through the DADA2 pipeline45 using trimLeft = 20 to remove 16S primers and truncLen = 210 for forward reads and 170 for reverse reads to discard low sequencing quality tails. Inferred sequence variants were merged and placed into a sequence variant table. Diversity metrics and taxonomic classification were performed using the QIIMEv2 pipeline46 and Ribosomal Database Project Classifier47.
Granulopoiesis.
Granulopoiesis was studied using flow cytometry as described previously28, 29. In brief, tibias/femur were harvested, flushed with PBS (supplemented with EDTA), RBCs were lysed, and cells were stained with the following antibodies: anti-Ly6G (IA8), c-Kit (2B8), CD34 (RAM34, eBioscience), and the cocktail of (CD3ε (145–2C11), CD4 (RM4.5), CD19 (6D5), CD8α (53–6.7), B220 (RA3–6B2), and TER119 (Ter119)). For gating granulopoietic sub-populations, bone marrow cells that had lost the potential to give rise to granulocytes were removed from the target population (Fig. 5a). The remaining cells were then analyzed for expression of c-Kit and Ly-6G (Fig. 5a). First, cells expressing lineage marker for T cells (CD3, CD4, CD8), B cells (B220), and erythroid cells (Ter119) are gated out along with dead cells (R1 gate). Second, the population of SSChighFSCint cells in R2 gate representing eosinophils is removed too from further analysis. Next, within R3 gate, when analyzed for the expression of CD34 and c-Kit, population of cells in R4 gate representing megakaryocyte–erythroid progenitors (MEP) (c-KithighCD34low cells) are also removed from further analysis. Finally, the remaining cells (R5) were then analyzed for the expression of c-Kit and Ly6G and divided into five cell subpopulations (#1–#5). The subpopulations #1 (c-KithighLy6Gneg), #2 (c-KitintLy6Gneg), #3 (c-KitintLy6Gint) represent early granulocytic compartments, whereas subpopulation #4 (c-KitnegLy6Gint) and #5 (c-KitnegLy6Ghigh) are immature to mature neutrophils ready to be released. For hematopoietic stem cells (HSCs) and myeloid progenitors28, 29, 48, BM cells were stained with anti-CD34 (RAM34, eBioscience), FCRγIII/II (93), Sca-1 (D7), c-Kit (2B8), and lineage cocktail (Lin-1). Appropriate autofluorescence and isotype controls were used. Cells were acquired either on a FACS Calibur or LSRFortessaX20 (BD Biosciences). FlowJo 10 (Treestar, Sanford, CA) was used to analyze data.
Blood neutrophil estimation.
For blood neutrophil count, blood was collected from both uninfected and infected WT and NLRP6 KO mice at 48 h post-infection. Briefly, 100 μl of blood was added to a 5 ml Polystyrene Round Bottom tube and RBCs were lysed using RBC lysis buffer (Gibco, USA). Cells were incubated for 20 min at 4°C in cell staining buffer (Biolegend, USA) containing anti-mouse Fc receptor blocking reagent (Miltenyi Biotec). Afterwards, cells were stained with Ly6G (1A8) and CD11b (M1/70) antibodies (eBioscience) for 30 min at 4°C. Appropriate Isotype-matched control antibodies were used in a similar manner. Unstained samples were included in the analyses for the compensation. The cells were fixed with 2% paraformaldehyde and analyzed using either FACS caliber or LSRFortessaX20 (BD). Data was analyzed using FlowJo v10.0 software.
NETosis and NET-mediated killing.
NETs were quantified using fluorimetry as described previously31, 34. Briefly, BMDNs (1×105 cells/well) were seeded in 96-well plates coated with poly L-lysine and infected with Kp. SYTOX green (5 μM; a non-cell-permanent DNA binding dye) was added to the plates and monitored to assess extracellular DNA release every hour up to 8 hours. Immunofluorescent detection of NETosis in Kp-infected BMDNs was performed as described previously34. Briefly, BMDNs were seeded, infected with Kp, and scanned for double-positive cells using DNA dye-SYTOX Green and H3-Cit staining. DAPI was used to stain cell nuclei. SEM images of NETosis in Kp-infected BMDNs were obtained as described previously34, 49. For NET-mediated killings, BMDNs were pretreated with cytochalasin-D (10 μg/ml) (to inhibit phagocytosis), infected with Kp (MOI 1) in the absence or presence of DNase (100 U/ml), and the bacterial burden in the supernatant was determined.
Intracellular killing assay.
The BMDN-dependent intracellular killing was performed as reported previously50. BMDN isolation from WT and NLRP6 KO mice were performed using the EasySep™ Mouse Neutrophil Enrichment Kit (Stemcell™ Technologies) as indicated in manufacturer’s instructions. Next, 0.25 ×106 neutrophils were suspended in RPMI 1640 with 10% v/v FBS and infected with K. pneumoniae (MOI 10) for the designated time points, such as 30 min, 60 min and 90 min. For the 90 min time point only, cells were treated with Gentamicin (250ug/ml) after 1 hour (to kill extracellular bacteria) and incubated for the additional 30 min prior to washing. At each time point, cells were washed numerous times with PBS to eliminate extracellular bacteria and gentamicin. Finally, cells were lysed with 120 μL of 0.1% triton-X to liberate intracellular bacteria. To count intracellular bacteria, the lysates were serially diluted with PBS and 20 ul of sample was plated on MacConkey and Tryptic Soy Agar and incubated 37°C overnight for bacterial enumeration.
Statistical analysis.
Data are expressed as mean ± SEM. Statistical analysis was performed in Prism 4.0 software (GraphPad Software Inc.). Unpaired t-test or ANOVA (followed by Bonferroni’s post hoc analysis) was performed as appropriate. Survival studies were analyzed using a Kaplan-Meier plot and compared using log-rank tests. The data are from 2–3 independent experiments. A p-value of *<0.05, **p<0.01, and ***p<0.001.
Supplementary Material
ACKNOWLEDGMENTS
We thank Marilyn Dietrich for assistance with flow cytometry. We thank previous Lung Biology laboratory members for their contributions, helpful discussions and critical reading of the manuscript. This work was supported by grants from NIH/NHLBI (R01HL133336 to S.C; R01HL091958 to S.J), grants from NIH/NIAID (R01AI113720, and R21AI133681), NIH/NIGMS (P20GM130555) (SJ), a predoctoral fellowship from NIH/NHLBI (F31HL137287) (S.P.), and a postdoctoral fellowship from the American Heart Association (L.J.).
Footnotes
Conflict-of-interest disclosure
The authors declare no competing financial interests.
REFERENCES
- 1.Ravi Kumar S, Paudel S, Ghimire L, Bergeron S, Cai S, Zemans RL et al. Emerging Roles of Inflammasomes in Acute Pneumonia. Am J Respir Crit Care Med 2018; 197(2): 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rider AC, Frazee BW. Community-Acquired Pneumonia. Emerg Med Clin North Am 2018; 36(4): 665–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rice LB. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis 2008; 197(8): 1079–1081. [DOI] [PubMed] [Google Scholar]
- 4.Rice LB. Progress and challenges in implementing the research on ESKAPE pathogens. Infect Control Hosp Epidemiol 2010; 31 Suppl 1: S7–10. [DOI] [PubMed] [Google Scholar]
- 5.Li B, Zhao Y, Liu C, Chen Z, Zhou D. Molecular pathogenesis of Klebsiella pneumoniae. Future Microbiol 2014; 9(9): 1071–1081. [DOI] [PubMed] [Google Scholar]
- 6.Siu LK, Yeh KM, Lin JC, Fung CP, Chang FY. Klebsiella pneumoniae liver abscess: a new invasive syndrome. Lancet Infect Dis 2012; 12(11): 881–887. [DOI] [PubMed] [Google Scholar]
- 7.Bengoechea JA, Sa Pessoa J. Klebsiella pneumoniae infection biology: living to counteract host defences. FEMS Microbiol Rev 2019; 43(2): 123–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Endimiani A, Depasquale JM, Forero S, Perez F, Hujer AM, Roberts-Pollack D et al. Emergence of blaKPC-containing Klebsiella pneumoniae in a long-term acute care hospital: a new challenge to our healthcare system. J Antimicrob Chemother 2009; 64(5): 1102–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mataseje LF, Boyd DA, Willey BM, Prayitno N, Kreiswirth N, Gelosia A et al. Plasmid comparison and molecular analysis of Klebsiella pneumoniae harbouring bla(KPC) from New York City and Toronto. J Antimicrob Chemother 2011; 66(6): 1273–1277. [DOI] [PubMed] [Google Scholar]
- 10.Chen GY. Role of Nlrp6 and Nlrp12 in the maintenance of intestinal homeostasis. Eur J Immunol 2014; 44(2): 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pedra JH, Cassel SL, Sutterwala FS. Sensing pathogens and danger signals by the inflammasome. Curr Opin Immunol 2009; 21(1): 10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen GY, Liu M, Wang F, Bertin J, Nunez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol 2011; 186(12): 7187–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hara H, Seregin SS, Yang D, Fukase K, Chamaillard M, Alnemri ES et al. The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell 2018; 175(6): 1651–1664 e1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011; 145(5): 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012; 482(7384): 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mamantopoulos M, Ronchi F, Van Hauwermeiren F, Vieira-Silva S, Yilmaz B, Martens L et al. Nlrp6- and ASC-Dependent Inflammasomes Do Not Shape the Commensal Gut Microbiota Composition. Immunity 2017; 47(2): 339–348 e334. [DOI] [PubMed] [Google Scholar]
- 17.Wang P, Zhu S, Yang L, Cui S, Pan W, Jackson R et al. Nlrp6 regulates intestinal antiviral innate immunity. Science 2015; 350(6262): 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghimire L, Paudel S, Jin L, Baral P, Cai S, Jeyaseelan S. NLRP6 negatively regulates pulmonary host defense in Gram-positive bacterial infection through modulating neutrophil recruitment and function. PLoS Pathog 2018; 14(9): e1007308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 2012; 488(7411): 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014; 156(5): 1045–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allen IC, McElvania-TeKippe E, Wilson JE, Lich JD, Arthur JC, Sullivan JT et al. Characterization of NLRP12 during the in vivo host immune response to Klebsiella pneumoniae and Mycobacterium tuberculosis. PloS one 2013; 8(4): e60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai S, Batra S, Wakamatsu N, Pacher P, Jeyaseelan S. NLRC4 Inflammasome-Mediated Production of IL-1beta Modulates Mucosal Immunity in the Lung against Gram-Negative Bacterial Infection. J Immunol 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ et al. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol 2009; 183(3): 2008–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Craig A, Mai J, Cai S, Jeyaseelan S. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect Immun 2009; 77(2): 568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sellers RS, Clifford CB, Treuting PM, Brayton C. Immunological variation between inbred laboratory mouse strains: points to consider in phenotyping genetically immunomodified mice. Vet Pathol 2012; 49(1): 32–43. [DOI] [PubMed] [Google Scholar]
- 26.Gilbert JA, Quinn RA, Debelius J, Xu ZZ, Morton J, Garg N et al. Microbiome-wide association studies link dynamic microbial consortia to disease. Nature 2016; 535(7610): 94–103. [DOI] [PubMed] [Google Scholar]
- 27.Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol 2014; 14(5): 302–314. [DOI] [PubMed] [Google Scholar]
- 28.Satake S, Hirai H, Hayashi Y, Shime N, Tamura A, Yao H et al. C/EBPbeta is involved in the amplification of early granulocyte precursors during candidemia-induced “emergency” granulopoiesis. J Immunol 2012; 189(9): 4546–4555. [DOI] [PubMed] [Google Scholar]
- 29.Paudel S, Baral P, Ghimire L, Bergeron S, Jin L, DeCorte JA et al. CXCL1 regulates neutrophil homeostasis in pneumonia-derived sepsis caused by Streptococcus pneumoniae serotype 3. Blood 2019; 133(12): 1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paudel S, Ghimire L, Jin L, Baral P, Cai S, Jeyaseelan S. NLRC4 suppresses IL-17A-mediated neutrophil-dependent host defense through upregulation of IL-18 and induction of necroptosis during Gram-positive pneumonia. Mucosal Immunol 2019; 12(1): 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS et al. Neutrophil extracellular traps kill bacteria. Science 2004; 303(5663): 1532–1535. [DOI] [PubMed] [Google Scholar]
- 32.Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012; 12(1): 109–116. [DOI] [PubMed] [Google Scholar]
- 33.Cai S, Batra S, Lira SA, Kolls JK, Jeyaseelan S. CXCL1 regulates pulmonary host defense to Klebsiella Infection via CXCL2, CXCL5, NF-kappaB, and MAPKs. J Immunol 2010; 185(10): 6214–6225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin L, Batra S, Jeyaseelan S. Diminished neutrophil extracellular trap (NET) formation is a novel innate immune deficiency induced by acute ethanol exposure in polymicrobial sepsis, which can be rescued by CXCL1. PLoS Pathog 2017; 13(9): e1006637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 2001; 194(4): 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci U S A 2011; 108(23): 9601–9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boettcher S, Ziegler P, Schmid MA, Takizawa H, van Rooijen N, Kopf M et al. Cutting edge: LPS-induced emergency myelopoiesis depends on TLR4-expressing nonhematopoietic cells. J Immunol 2012; 188(12): 5824–5828. [DOI] [PubMed] [Google Scholar]
- 38.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 2007; 176(2): 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 2010; 207(9): 1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levy M, Thaiss CA, Zeevi D, Dohnalova L, Zilberman-Schapira G, Mahdi JA et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015; 163(6): 1428–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zmora N, Levy M, Pevsner-Fishcer M, Elinav E. Inflammasomes and intestinal inflammation. Mucosal Immunol 2017; 10(4): 865–883. [DOI] [PubMed] [Google Scholar]
- 42.Batra S, Cai S, Balamayooran G, Jeyaseelan S. Intrapulmonary administration of leukotriene B4 augments neutrophil accumulation and responses in the lung to Klebsiella infection in CXCL1 knockout mice. The Journal of Immunology 2012; 188(7): 3458–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai S, Batra S, Del Piero F, Jeyaseelan S. NLRP12 modulates host defense through IL-17A-CXCL1 axis. Mucosal Immunol 2016; 9(2): 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kulkarni R, Caskey J, Singh SK, Paudel S, Baral P, Schexnayder M et al. Cigarette Smoke Extract-Exposed Methicillin-Resistant Staphylococcus aureus Regulates Leukocyte Function for Pulmonary Persistence. Am J Respir Cell Mol Biol 2016; 55(4): 586–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 2016; 13(7): 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7(5): 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 2007; 73(16): 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J et al. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat Immunol 2006; 7(7): 732–739. [DOI] [PubMed] [Google Scholar]
- 49.Douda DN, Jackson R, Grasemann H, Palaniyar N. Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. J Immunol 2011; 187(4): 1856–1865. [DOI] [PubMed] [Google Scholar]
- 50.Jin L, Batra S, Jeyaseelan S. Deletion of Nlrp3 augments survival during polymicrobial sepsis by decreasing autophagy and enhancing phagocytosis. The Journal of Immunology 2017; 198(3): 1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.