

Abstract
Thousands of genes produce polyadenylated mRNAs that still contain one or more introns. These transcripts are known as retained intron RNAs (RI-RNAs). In the past 10 years, RI-RNAs have been linked to post-transcriptional alternative splicing in a variety of developmental contexts, but they can also be dead-end products fated for RNA decay. Here we discuss the role of intron retention in shaping gene expression programs, as well as recent evidence suggesting that the biogenesis and fate of RI-RNAs is regulated by nuclear organization. We discuss the possibility that proximity of RNA to nuclear speckles – biomolecular condensates that are highly enriched in splicing factors and other RNA binding proteins – is associated with choices ranging from efficient co-transcriptional splicing, export and stability to regulated post-transcriptional splicing and possible vulnerability to decay.
Keywords: Intron retention, splicing, RNA decay, nuclear speckles, transcription
Introduction
The processing of pre-messenger RNAs (pre-mRNAs) into mature mRNA requires 5′-end capping, splicing, 3′-end cleavage, and polyadenylation. A major open question in the field of gene expression is the degree to which pre-mRNAs can be spliced after 3′-end cleavage (post-transcriptionally). Splicing involves the removal of introns and the ligation of neighboring exons by the spliceosome [1,2]. The spliceosome is a multi-subunit molecular machine comprised of five snRNPs (U1, U2, U4, U5, U6) and numerous core and accessory proteins, which must assemble de novo on each intron [1]. In humans, genes contain a median of ~7 introns [3] and are frequently alternatively spliced to produce different transcript and protein isoforms. Sequencing and imaging studies in an array of different organisms has demonstrated that ~75% of splicing is completed co-transcriptionally, meaning that exon-exon ligation occurs before 3′-end cleavage [2,4]. However, post-transcriptional RNAs frequently contain introns [5–8] (Figure 1). For greater than 1/3 of all human and mouse genes in at least one cell type, ~50% of the transcripts they produce contain at least one intron [6]. These incompletely spliced transcripts are known as retained-intron RNAs (RI-RNAs) [5–8]. Many RI-RNAs remain solely in the nucleus and this subclass has also been called detained-intron RNAs [5]. For clarity, we refer to all post-transcriptional intron-containing transcripts as RI-RNAs. In this review, we first examine the potential fates of RI-RNAs including post-transcriptional splicing and RNA decay, and we assess the role of intron-retention in developmental and stress-responsive gene expression programs. Second, we discuss recent evidence that post-transcriptional splicing may be affected by nuclear organization, focusing on biomolecular condensates called nuclear speckles. Nuclear speckles form by liquid-liquid phase separation [9–11] and are highly enriched in both post-transcriptional RNA and RNA processing factors [12–17]. Thus, they are likely candidates to regulate RNA processing efficiency and thereby determine the fate of RI-RNAs.
Figure 1:
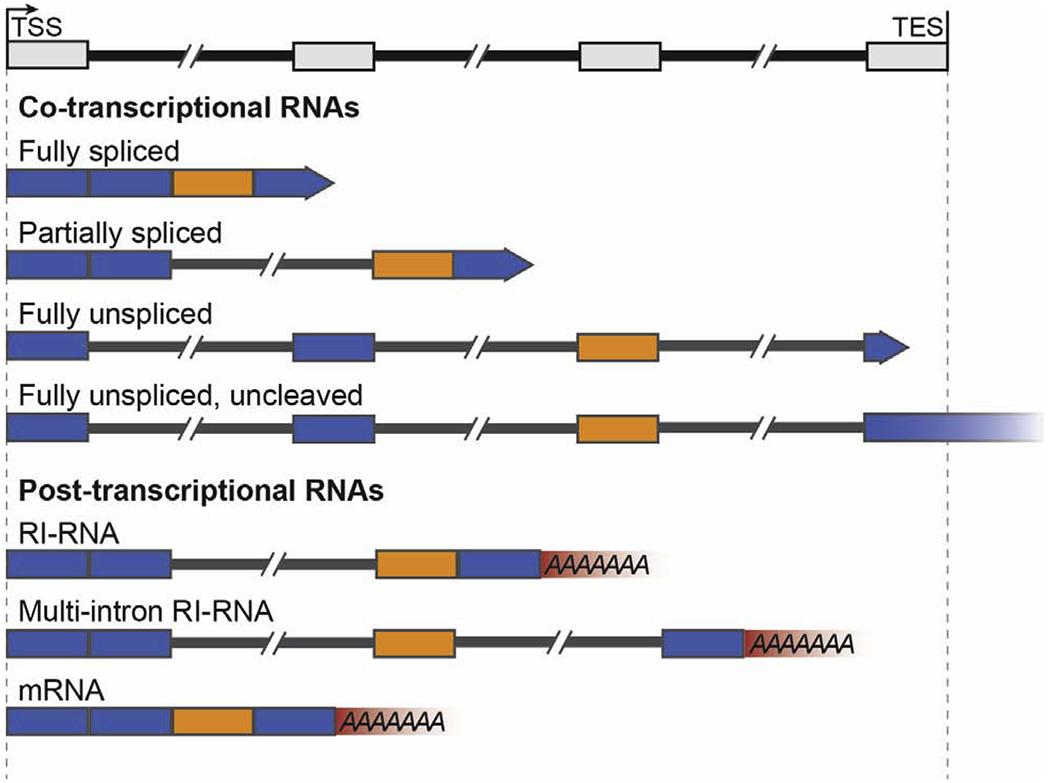
A variety of potential products can be derived from synthesis and processing of a single pre-mRNA, including post-transcriptional RNAs with retained introns. Introns are depicted as thinner black bars spaced between the thicker, colored exons. Co-transcriptional RNAs are elongating, depicted by rightward-facing arrows whereas post-transcriptional RNAs have been cleaved and polyadenylated. Retained introns are often flanked by alternatively spliced exons (orange) compared to constitutively spliced exons (blue).
Unspliced RNAs are fated for post-transcriptional splicing or RNA decay
Single-gene studies have provided several examples of RI-RNAs with two distinct fates: either RNA decay or rapid post-transcriptional splicing in response to signaling. In several tissue types, greater than 50% of transcripts produced from the CLK1 gene, encoding a stress-responsive kinase, have both intron 3 and intron 4 retained post-transcriptionally [18]. CLK1 RI-RNAs remain nuclear and can be spliced post-transcriptionally. In the absence of new transcription, deficiency in Clk1 kinase activity (due to heat shock, osmotic stress, or a Clk1 inhibitor) causes the CLK1 RI-RNA to decrease and the mature mRNA to accumulate. Thus, post-transcriptional splicing of the CLK1 RI-RNA reservoir is auto-regulated by Clk1 kinase activity. Similar studies analyzing precursor-product relationships recapitulated this result with CLK1 RI-RNA; however, two other abundant RI-RNAs, MAT2A and OGT, are not post-transcriptionally spliced into mRNA but are instead dead-end products [19]. Co-transcriptional splicing of MAT2A, encoding an S-Adenosylmethionine (SAM) synthetase, is more efficient in SAM-limiting conditions due to altered adenosine methylation of the MAT2A 3′ UTR [19,20]. Thus, MAT2A uses a feedback loop to produce different ratios of mRNA to RI-RNA depending on environmental conditions [19,20]. These single-gene studies show that RI-RNAs are sometimes fated for decay (or potentially unknown nuclear functions), but in other instances are reservoirs for rapid production of mature mRNA not limited by the rate of transcription activation and elongation (Figure 2a). As these papers illustrate, precursor-product relationships are essential determinants of whether RNAs are bona fide substrates of post-transcriptional splicing [18–20]. It is not sufficient to examine the mRNA to RI-RNA ratio in two cellular conditions, as changes in these conditions can also alter the co-transcriptional splicing efficiency.
Figure 2:
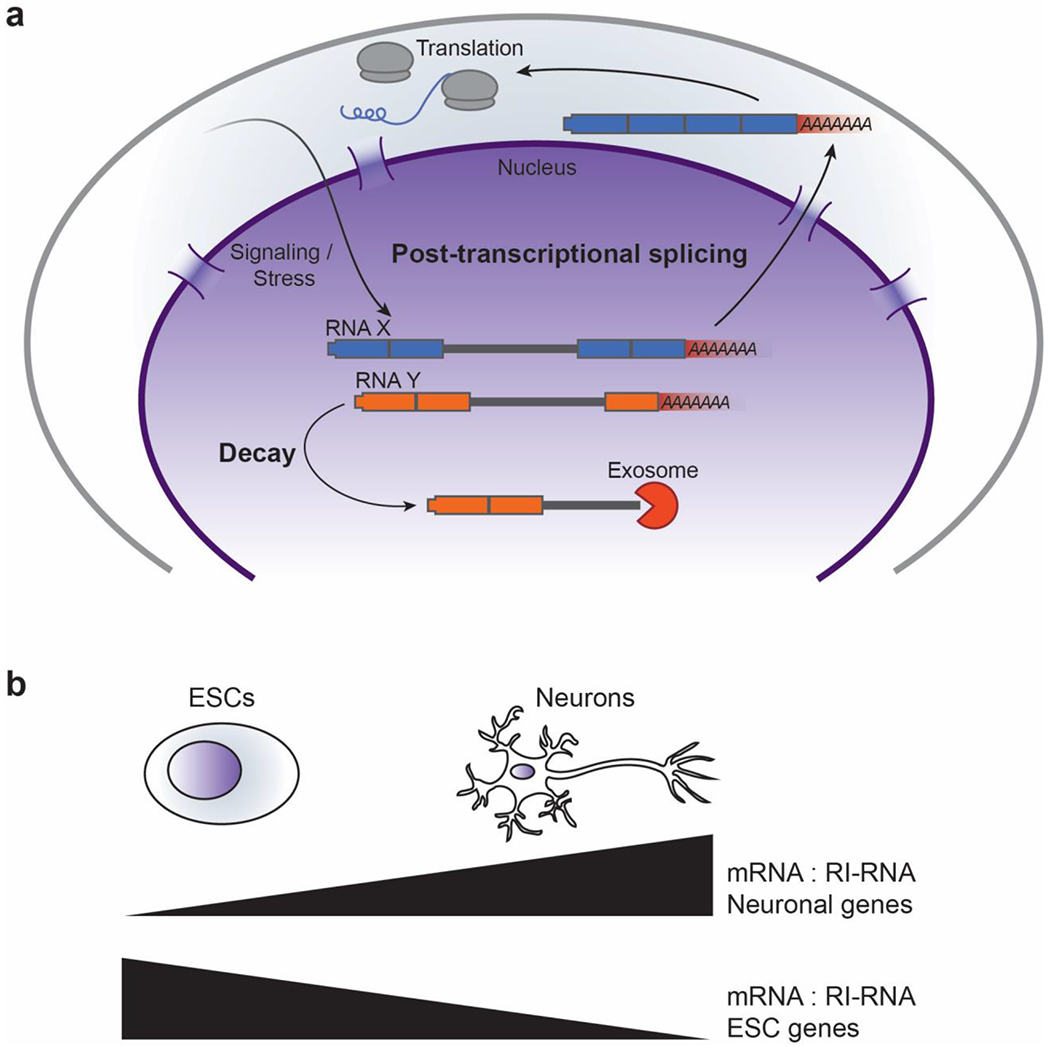
RI-RNAs can be post-transcriptionally spliced or targeted for decay. (a) RI-RNAs can be post-transcriptionally spiced and exported upon cell signaling, developmental transitions, or stress response. In contrast, other RI-RNAs can be degraded through exosome-dependent mechanisms. (b) Intron retention serves as a mechanism to regulate gene expression. The ratio of mRNA to RI-RNA is altered to regulate gene expression in specific cell types.
Recent studies suggest that co-transcriptional splicing of multiple introns within single transcripts is coordinated. Long read sequencing, which sequences whole transcripts from the 5′ to 3′ end, revealed that introns in nascent RNAs that are still being transcribed by RNA polymerase II (Pol II) are spliced in an “all-or-none” fashion [21–23]. Other sequencing methods have also revealed coordination of splicing between introns [24,25]. Analysis of chromatin-bound nascent RNAs through biochemical fractionation in Schizosaccharomyces pombe showed that ~35% of nascent RNAs are fully unspliced and are degraded by the nuclear exosome [21]. Coordinated splicing between neighboring introns was also observed in human K562 and Drosophila S2 cells [22]. Similarly, >75% of co-transcriptional RNAs in Arabidopsis are fully unspliced, and 5-10% of chromatin-bound poly(A) RNAs were also fully unspliced [23]. Whether fully unspliced nascent RNAs are eventually spliced or targeted for decay remains to be determined in different species, cell types and biological contexts. Additionally, whether these RNAs arise from similar mechanisms as those that produce RI-RNAs is unknown.
RI-RNAs shape developmental gene expression programs through alternative splicing and RNA decay
Programmed intron retention is used to control several developmental and stress-induced gene expression programs. In some cases, co-transcriptional splicing is inhibited leading to accumulation of RI-RNAs fated for decay, whereas in other developmental contexts post-transcriptional splicing is used to activate reservoirs of RI-RNAs. In neocortical cells, mimicking synaptic stimulation with a GABAA receptor antagonist resulted in post-transcriptional splicing and export of >200 RI-RNAs [26]. Another striking example of intron retention is seen during mouse spermatogenesis [27]. Here, hundreds of genes enriched for spermatogenesis-related functions are transcribed and produce RI-RNAs. These transcripts are sequestered in the nucleus for several days and eventually post-transcriptionally spliced, exported, and translated. In other situations, production of RI-RNAs is associated with gene repression. This repressive function is particularly prominent when comparing mouse embryonic stem cells before and after differentiation into neurons [6]. Genes important for neuronal function produce mRNAs in neuronal cells but RI-RNAs in mouse ES cells, whereas the converse is true for genes important for ES cells function (Figure 2b) [6]. Specific introns may be retained due to the differential expression of RBPs. For example, genes encoding presynaptic proteins produce RI-RNAs in ES cells due to the activity of Polypyrimidine Tract Binding Protein 1 (PTBP1, also known as hnRNP I) [7]. PTBP1 is repressed in neuronal cells, allowing these genes to produce functional mRNA [7]. Another example wherein dozens of genes are repressed by intron retention is during differentiation of myelocytes into granulocytes. Remarkably, re-expression of only one of these genes (Lmnb1) in the mRNA form instead of RI-RNA form results in alterations to granulocyte nuclear shape, size, and total granulocyte numbers [8]. Specific RI-RNAs are also observed during erythropoiesis, CD4+ T-Cell activation, heat shock, and in a variety of other cell types and conditions [28–32]. Taken together, these data strongly suggest that intron retention is widely used to regulate important genes in several biological contexts.
Bioinformatic and single-molecule approaches indicate that post-transcriptional splicing of RI-RNAs is linked to alternative splicing. Compared to introns that are constitutively spliced, retained introns are more likely than expected by chance to neighbor alternatively spliced exons, including cassette exons, mutually exclusive exons, alternative 5′ splice sites (5′SS) and 3′ splice sites (3′SS) [5,6]. Retained introns also have weaker 5′SS, 3′SS, and polypyrimidine tracts, which are all features associated with alternative splicing [5,6,26,27]. Single-molecule fluorescence in situ hybridization (smFISH) experiments indicate that alternatively spliced exons are post-transcriptionally spliced more frequently than constitutive exons, meaning that transcripts are produced as RI-RNAs and then mature into mRNAs [33]. These data agree with metabolic labeling studies showing that alternative splicing occurs more slowly than constitutive splicing [34], and also with live-cell single-molecule reporters demonstrating that introns with weaker splice sites are removed more slowly than introns with consensus splice sites [35]. The reduced efficiency and rate of alternative splicing likely allows for selection of downstream 5′SS and 3′SS, cassette exons, and mutually exclusive exons. If these exons were spliced quickly, the first transcribed splice sites would be much more likely to be used. Notably, post-transcriptionally retained introns are more evolutionarily conserved than constitutive introns, suggesting that their weaker splicing is functionally important [5].
Mammalian cells effectively prevent protein expression from RI-RNAs by promoting their decay and inhibiting their nuclear export. Cellular fractionation shows that RI-RNAs are primarily restricted to the nucleus in a variety of cell types [5,6,18,27]. RI-RNAs can be targeted for degradation by the nuclear exosome, a 3′-5′ exonuclease complex that operates through several pathways [7,19,36]. The exosome degrades some RI-RNAs by a pathway termed PABPN1 and Poly(A) Polymerase-mediated Decay (PPD) [36]. PPD employs Poly(A) Polymerases α/γ and PABPN1, the nuclear Poly(A) Binding Protein, to synthesize longer than average poly(A) tails, which precedes exosome-dependent decay [36]. PPD-mediated degradation of RI-RNAs may also require ZFC3H1, an exosome-adapter protein, which functions in the same pathway as PABPN1 to degrade other nuclear transcripts [37]. Other RI-RNAs are degraded by the exosome utilizing distinct pathways. For example, neurogenesis-genes produce RI-RNAs in non-neuronal cells that are degraded through a pathway involving the exosome, the nuclear pore protein TPR and PTBP1, but not PPD components [7]. Apart from the exosome, the 5′->3′ exoribonuclease Xrn2 likely prevents the accumulation of unspliced RNAs by promoting co-transcriptional decay of unspliced pre-mRNAs [38]. Accumulation of unspliced RNAs was enriched in Xrn2 knockdown cells upon splicing inhibition. RI-RNAs are also not efficiently exported to the cytoplasm. This effect may be due to the presence of specific cis-acting sequences, such as 5′SS or polypyrimidine tracts, or due to association with splicing factors such as U1 snRNP, PTBP1 and U2AF [7,39,40]. The splicing and export of RI-RNAs may also be influenced by other RBPs such as the SR proteins, a family of proteins with arginine- and serine-rich (RS) domains that function to couple transcription, splicing, and nuclear export [41]. The shuttling competence of some SR proteins has been shown to change during differentiation [42], which may influence which RI-RNAs are exported given that SR proteins bind to retained introns [43] and are known to promote export competence [44,45]. Finally, some RI-RNAs that escape nuclear retention and nuclear decay are repressed by NMD [6,8]. How the exosome-dependent decay pathways differentiate partially spliced RI-RNAs from fully spliced mRNAs is not fully understood [46]. Likely mechanisms may involve RI-RNAs association with spliceosomal snRNPs and intronic RBPs. Moreover, it is unclear whether the nuclear degradation rates of different RI-RNAs impacts their potential for post-transcriptional splicing.
Nuclear speckles are dynamic hubs of poly(A) RNA and RNA binding proteins
A long-standing focus of RNA biology has been to understand how membraneless nuclear compartments impact gene expression. Recently, biomolecular condensation has been identified as a contributing mechanism to the assembly of these compartments [11,47]. In the following section, we discuss nuclear speckles, which are biomolecular condensates that separate from the surrounding nucleoplasm through liquid-liquid phase separation [9,10]. Nuclear speckles are highly enriched in dozens of RBPs that not only include spliceosomal snRNPs, but also other splicing factors such as the SR proteins and SR-like proteins (e.g. SON, SRRM1, SRRM2), transcription factors, cleavage and polyadenylation proteins, and mRNA export proteins (Figure 3a–b) [14–16]. Strikingly, nuclear speckles also contain very high concentrations of poly(A) RNA (Figure 3c) that exchanges rapidly between nuclear speckles and the surrounding nucleoplasm [17,48,49]. In addition to the spliceosomal snRNAs and poly(A) RNA, nuclear speckles contain the metastasis-promoting long non-coding RNA (IncRNA) MALAT1, the levels of which affects speckle size [50,51]. High-resolution imaging revealed that SR proteins form the core of nuclear speckles, with snRNPs and MALAT1 closer to the periphery and poly(A)+ RNA defining a broader region [50]. Nuclear speckles have also been called interchromatin granule clusters in reference to their appearance in transmission electron micrographs and are found in almost all metazoan cell types; interphase nuclei typically contain 20-50 speckles that are each ~0.5 μm in diameter [12,13]. Together, the composition and dynamics of nuclear speckles suggest that they affect mRNA biogenesis, processing, and export. Substantial debate has centered around roles for nuclear speckles as RBP storehouses versus active sites of mRNA biogenesis [12,13]. We favor an active function for nuclear speckles rather than a long-term storage function, because quantitative imaging studies show that poly(A) RNA freely diffuses between speckles and nucleoplasm, and RBPs have similar mobilities in nuclear speckles and at active transcription sites [48,49,52,53].
Figure 3.
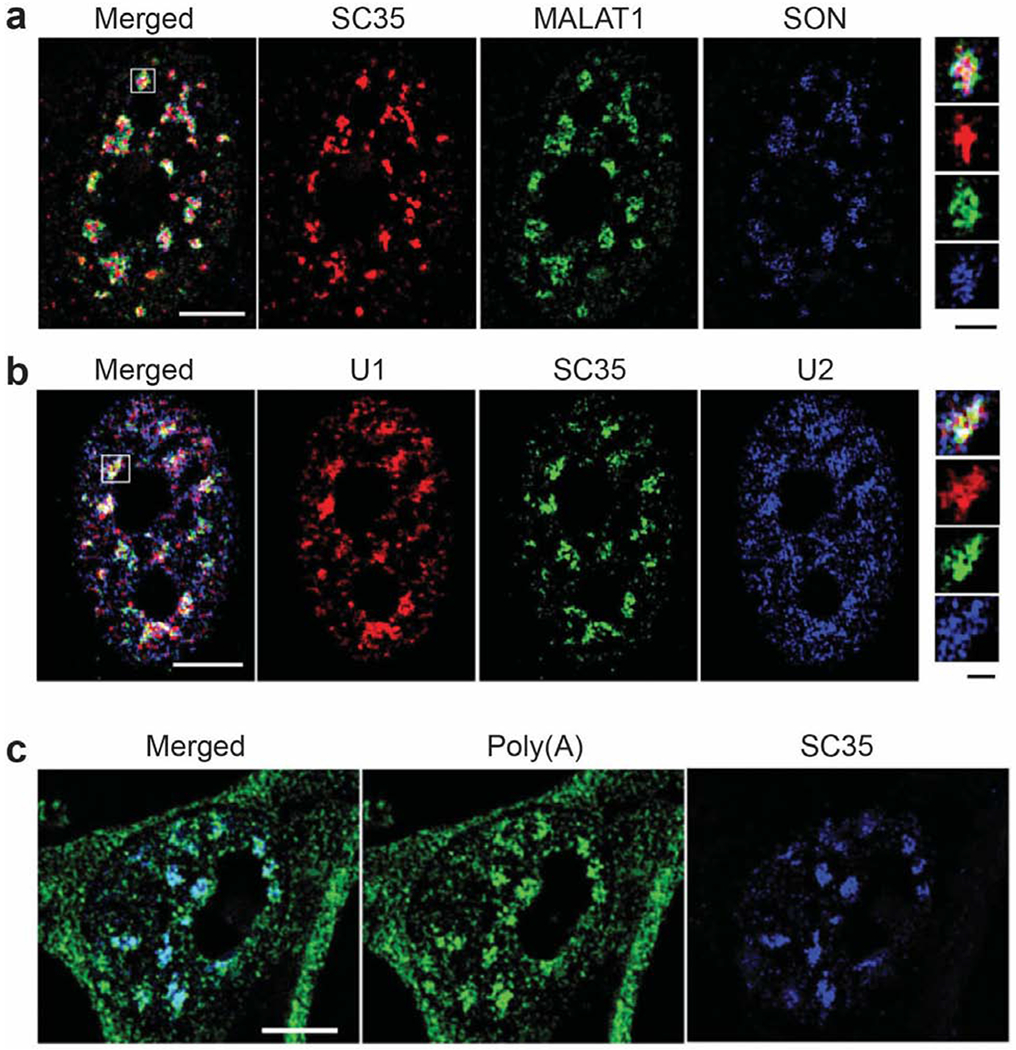
Structured illumination microscopy images of nuclear speckles adapted with permission from the Journal of Cell Science, Fei et al. (2017) [50]. (a) Shown here are nuclear speckle components including the MALAT1 lncRNA, the SR-like protein SON, and many SR proteins detected by the SC35 antibody. SON and SC35 are in the nuclear speckle interior whereas MALAT1 is closer to the periphery. (b) U1 and U2 snRNAs localize to the periphery of nuclear speckles, outside of SC35. (c) Poly(A) RNA is highly enriched in nuclear speckles. White scale bars: 5 μm. Black scalebar of insets: 1 μm.
Evidence that proximity to nuclear speckles increases gene expression through co-transcriptional and post-transcriptional mechanisms
Several studies suggest that proximity to nuclear speckles increases gene expression by promoting transcription, co-transcriptional splicing, and post-transcriptional splicing, all of which can affect the mRNA to RI-RNA ratio (Figure 4). Three different methods for analyzing chromatin compartments genome-wide showed that active genes (compartment A) are highly enriched proximal to nuclear speckles, whereas repressed genes (compartment B) are more distant from speckles [54–56]. These studies have been extensively reviewed elsewhere [57]. Additionally, recent work by Su et al. used multiplexed error-robust FISH (MERFISH) to image hundreds of nascent RNAs, genomic loci, and nuclear speckles in single cells, and found proximity to nuclear speckles correlated with higher transcriptional bursting activity and compartment A genes [58]. Consistent with speckles promoting transcription, intronless genes produce significantly more transcripts when proximal to a nuclear speckle [59,60]. However, nuclear speckles may also directly affect co-transcriptional and post-transcriptional splicing. Using smFISH to analyze splicing efficiency at active transcription sites in single cells, it was shown that an individual gene’s splicing efficiency directly correlated with both transcriptional activity as well as its proximity to nuclear speckles, supporting a role for speckles in co-transcriptional splicing [61]. Other studies using smFISH revealed that post-transcriptionally spliced GFP reporter constructs and Influenza virus MS1 transcripts localize nearby to or within nuclear speckles [33,62]. RNAs are targeted to speckles through several types of cis-acting sequences that affect splicing, including SR protein binding sites and functional 5′SSs and 3′SSs [63,64]. Moreover, global inhibition of splicing results in the localization of poly(A) RNA (which is by definition post-transcriptional) almost exclusively to speckles and away from nucleoplasm [65,66], suggesting that speckles are quality-control centers that detain RI-RNAs until splicing is completed. Further testing this idea will require splicing analyses in mutants that fully or partially eliminate nuclear speckle assembly [67].
Figure 4:
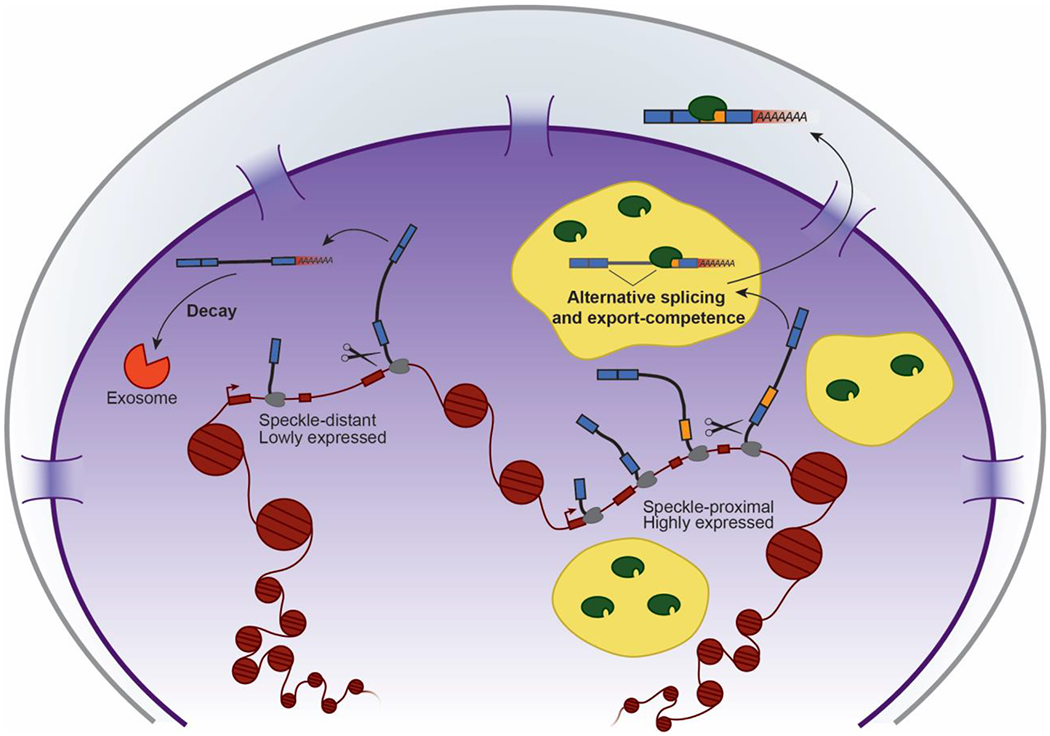
Model of nuclear speckle functions: Proximity to speckles results in increased transcription, splicing, and export-competence. The diagram depicts a model in which nuclear speckle-proximal genes are highly expressed and their RI-RNAs are able to diffuse into speckles for post-transcriptional splicing and export, whereas speckle-distant genes are lowly expressed and their RI-RNAs are fated for decay. Not depicted but discussed in the text is the idea that nuclear speckles also promote co-transcriptional splicing.
Consistent with a global function of speckles in splicing, catalytically active spliceosomes are also highly enriched in nuclear speckles compared to nucleoplasm [68]. Compared to constitutively spliced exons, alternatively spliced exons have increased association with the MALAT1 IncRNA present in nuclear speckles [69], and are also more likely to be spliced post-transcriptionally and associated with intron retention [5,6,33,35]. These observations suggest that proximity to nuclear speckles correlates with increased co-transcriptional and post-transcriptional splicing. Nuclear speckles are also likely associated with assembly of export-competent mRNPs and not RNA decay, because global inhibition of mRNA nuclear export enhances poly(A) RNA localization to nuclear speckles [64]. Intriguingly, nuclear speckles promote export-competence irrespective of splicing. Both microinjected and endogenous intronless mRNAs transit through nuclear speckles to gain export-competence by associating with SR proteins and the TREX complex [63], both of which couple transcription, processing, and export [14,44,64,70]. The exosome is excluded from nuclear speckles, suggesting that speckle-proximal RNAs are less likely to be degraded [50]. Consistent with these ideas, recent work has demonstrated that the elongating form of Pol II directly associates with nuclear speckles in vivo and splicing factor condensates in vitro, whereas the initiation form of Pol II instead associates with transcriptional condensates [9]. Thus, condensate specificity may affect the proteomic environment of mRNA biogenesis, and proximity to nuclear speckles affects the biogenesis and fate of mRNAs and RI-RNAs through co-transcriptional and post-transcriptional mechanisms. In the future, it will be important to determine the molecular mechanisms that govern the biochemical activities proximal to or distal from speckles and how the dynamics of condensates allow for changes in transcription and splicing in response to cell physiology.
Outlook
Intron retention is clearly a significant determinant of gene expression, leading to several outcomes on a per transcript basis. RI-RNAs can either provide reservoirs for rapid mRNA expression or can decrease gene expression by triggering decay. Conversely, the biogenesis and fate of recently discovered fully unspliced RNAs are not well understood. How cells access these regulatory mechanisms for specific or complete intron retention, and how unspliced RNAs are post-transcriptionally regulated by cis-acting sequences and RNA-binding proteins are pressing questions. For example, whether RI-RNAs are bound to preassembled, potentially stalled spliceosomes pending catalysis is unknown. Alternatively, spliceosome assembly would have to be initiated de novo on retained intron-exon junctions entirely post-transcriptionally. Given the influence of the chromatin environment on co-transcriptional splicing [2], post-transcriptional splicing may be subject to different mechanisms of regulation. Although microinjected adenovirus pre-mRNAs form splicing precursors that localize to nuclear speckles [71], it is unclear if this extends to endogenous RI-RNAs. Additionally, it is important to determine quantitatively the fraction of alternative splicing events that occur co- vs. post-transcriptionally, and proximal to or distal from condensates such as nuclear speckles. Advances in single molecule techniques, metabolic-labeling strategies, and long-read sequencing pipelines will better distinguish between co- and post-transcriptional splicing events and help elucidate how cells use intron retention to fine-tune gene expression programs. Recent work in mouse P19 cells has shown that some RI-RNAs are exported to the cytoplasm and can escape NMD [43], and mass spectrometry of melanoma tumor cells showed that RI-RNAs are translated and are a source of neoepitopes presented on MHC type I [72]. Although the biological relevance of these RNAs and neoepitopes is not fully understood, ribosome profiling and genetic screening of intronic sequences could elucidate roles for cytoplasmic RI-RNAs. Likewise, mRNA fate regulation by nuclear speckles is not precisely defined and will require conditional removal of nuclear speckles in otherwise wild-type cells. Although MALAT1, SON, and SR proteins are known structural components of speckles, knockdown of these factors individually only results in speckle disorganization, not disassembly [51,73–75]. A promising direction for this line of research comes from the recent finding that combined knockdown of SON and SRRM2 causes near complete speckle disassembly, as does knockdown of SON along with deletion of SRRM2’s intrinsically disordered domain [67]. It is worth noting that RNA splicing and export in D. melanogaster and C. elegans is promoted by association with nuclear pores, highlighting differences in nuclear architecture between species [76–78]. Furthermore, comparing the relative activities of RNA binding proteins in the nucleoplasm and in nuclear speckles, as well as determining the extent of transcription, splicing, and 3′-end processing within and outside of speckles, may shed light on the role of speckles in coordinating pre-mRNA synthesis and processing.
Highlights.
Intron retention is a widespread feature of mammalian gene expression programs.
Retained introns are associated with post-transcriptional alternative splicing.
Unspliced RNAs are repressed through nuclear retention and RNA decay.
Proximity of genes to nuclear speckles boosts transcription, co-transcriptional splicing, and post-transcriptional splicing.
Acknowledgments
Funding
The authors’ work on this subject is supported by the National Institutes of Health (NIH R01GM112766 to KMN and F32GM134598 to DVP). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflict of interest.
References
- 1.Fica SM, Nagai K: Cryo-electron microscopy snapshots of the spliceosome: structural insights into a dynamic ribonucleoprotein machine. Nat Struct Mol Biol 2017, 24:791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carrocci TJ, Neugebauer KM: Pre-mRNA Splicing in the Nuclear Landscape. Cold Spring Harb Symp Quant Biol 2019, 84:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piovesan A, Caracausi M, Antonaros F, Pelleri MC, Vitale L: GeneBase 1.1: a tool to summarize data from NCBI gene datasets and its application to an update of human gene statistics. Database (Oxford) 2016, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brugiolo M, Herzel L, Neugebauer KM: Counting on co-transcriptional splicing. F1000Prime Rep 2013, 5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boutz PL, Bhutkar A, Sharp PA: Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev 2015, 29:63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos-Pournatzis T, Frey B, Irimia M, Blencowe BJ: Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res 2014, 24:1774–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yap K, Lim ZQ, Khandelia P, Friedman B, Makeyev EV: Coordinated regulation of neuronal mRNA steady-state levels through developmentally controlled intron retention. Genes Dev 2012, 26:1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong JJ, Ritchie W, Ebner OA, Selbach M, Wong JW, Huang Y, Gao D, Pinello N, Gonzalez M, Baidya K, et al. Orchestrated intron retention regulates normal granulocyte differentiation. Cell 2013, 154:583–595. [DOI] [PubMed] [Google Scholar]
- *9.Guo YE, Manteiga JC, Henninger JE, Sabari BR, Dall’Agnese A, Hannett NM, Spille JH, Afeyan LK, Zamudio AV, Shrinivas K, et al. Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 2019, 572:543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that transcriptional condensates and splicing factor condensates, made of proteins present in nuclear speckles, have opposing specificities for Pol II depending on its phosphorylation state. The initiation state Pol II (non-phosphorylated C-terminal domain) is more strongly incorporated into transcriptional condensates with the Mediator complex. In contrast, the elongation state Pol II (phosphorylated CTD by CDK7 or CDK9) is more strongly incorporated into splicing factor condensates.
- 10.Kim J, Han KY, Khanna N, Ha T, Belmont AS: Nuclear speckle fusion via long-range directional motion regulates speckle morphology after transcriptional inhibition. J Cell Sci 2019, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sabari BR, Dall’Agnese A, Young RA: Biomolecular Condensates in the Nucleus. Trends Biochem Sci 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spector DL, Lamond AI: Nuclear speckles. Cold Spring Harb Perspect Biol 2011, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galganski L, Urbanek MO, Krzyzosiak WJ: Nuclear speckles: molecular organization, biological function and role in disease. Nucleic Acids Res 2017, 45:10350–10368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dopie J, Sweredoski MJ, Moradian A, Belmont AS: Tyramide signal amplification mass spectrometry (TSA-MS) ratio identifies nuclear speckle proteins. J Cell Biol 2020, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saitoh N, Spahr CS, Patterson SD, Bubulya P, Neuwald AF, Spector DL: Proteomic analysis of interchromatin granule clusters. Mol Biol Cell 2004, 15:3876–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mintz PJ, Patterson SD, Neuwald AF, Spahr CS, Spector DL: Purification and biochemical characterization of interchromatin granule clusters. EMBO J 1999, 18:4308–4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang S, Deerinck TJ, Ellisman MH, Spector DL: In vivo analysis of the stability and transport of nuclear poly(A)+ RNA. J Cell Biol 1994, 126:877–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ninomiya K, Kataoka N, Hagiwara M: Stress-responsive maturation of Clk1/4 pre-mRNAs promotes phosphorylation of SR splicing factor. J Cell Biol 2011, 195:27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **19.Pendleton KE, Park SK, Hunter OV, Bresson SM, Conrad NK: Balance between MAT2A intron detention and splicing is determined cotranscriptionally. RNA 2018, 24:778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]; In these two papers, the authors demonstrate that RI-RNAs can be dead-end products produced via poor co-transcriptional splicing. In the 2017 paper, the authors show that that the intron retention in MAT2A pre-mRNA, which encodes an S-adenosylmethionine (SAM) synthetase, is regulated by a splicing-dependent feedback loop sensitive to cellular SAM concentrations. In the 2018 paper, it is shown that the MAT2A RI-RNA is a dead-end product regardless of cellular conditions, in contrast to other RI-RNAs that can be spliced post-transcriptionally.
- **20.Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, Conrad NK: The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 2017, 169:824–835 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]; In these two papers, the authors demonstrate that RI-RNAs can be dead-end products produced via poor co-transcriptional splicing. In the 2017 paper, the authors show that that the intron retention in MAT2A pre-mRNA, which encodes an S-adenosylmethionine (SAM) synthetase, is regulated by a splicing-dependent feedback loop sensitive to cellular SAM concentrations. In the 2018 paper, it is shown that the MAT2A RI-RNA is a dead-end product regardless of cellular conditions, in contrast to other RI-RNAs that can be spliced post-transcriptionally.
- **21.Herzel L, Straube K, Neugebauer KM: Long-read sequencing of nascent RNA reveals coupling among RNA processing events. Genome Res 2018, 28:1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]; These papers used long-read sequencing to examine full-length nascent RNAs, demonstrating that introns within single molecules of RNA are spliced in a coordinated manner. Future work using this method to analyze RNA synthesis and isoform usage will likely elucidate more intricate mechanisms of gene expression regulation and coordination among RNA processing events.
- **22.Drexler HL, Choquet K, Churchman LS: Splicing Kinetics and Coordination Revealed by Direct Nascent RNA Sequencing through Nanopores. Mol Cell 2020, 77:985–998 e988. [DOI] [PMC free article] [PubMed] [Google Scholar]; These papers used long-read sequencing to examine full-length nascent RNAs, demonstrating that introns within single molecules of RNA are spliced in a coordinated manner. Future work using this method to analyze RNA synthesis and isoform usage will likely elucidate more intricate mechanisms of gene expression regulation and coordination among RNA processing events.
- **23.Jia J, Long Y, Zhang H, Li Z, Liu Z, Zhao Y, Lu D, Jin X, Deng X, Xia R, et al. Post-transcriptional splicing of nascent RNA contributes to widespread intron retention in plants. Nat Plants 2020. [DOI] [PubMed] [Google Scholar]; These papers used long-read sequencing to examine full-length nascent RNAs, demonstrating that introns within single molecules of RNA are spliced in a coordinated manner. Future work using this method to analyze RNA synthesis and isoform usage will likely elucidate more intricate mechanisms of gene expression regulation and coordination among RNA processing events.
- 24.Tilgner H, Jahanbani F, Gupta I, Collier P, Wei E, Rasmussen M, Snyder M: Microfluidic isoform sequencing shows widespread splicing coordination in the human transcriptome. Genome Res 2018, 28:231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim SW, Taggart AJ, Heintzelman C, Cygan KJ, Hull CG, Wang J, Shrestha B, Fairbrother WG: Widespread intra-dependencies in the removal of introns from human transcripts. Nucleic Acids Res 2017, 45:9503–9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *26.Mauger O, Lemoine F, Scheiffele P: Targeted Intron Retention and Excision for Rapid Gene Regulation in Response to Neuronal Activity. Neuron 2016, 92:1266–1278. [DOI] [PubMed] [Google Scholar]; These papers highlight two distinct contexts in which post-transcriptional splicing of RI-RNAs has a functional role in gene expression regulation. In both cases, cell-type specific Rl-RNAs were found to be spliced post-transcriptionally as either part of a cell signaling response (Mauger et al. 2016) or differentiation programs (Naro et al. 2017).
- *27.Naro C, Jolly A, Di Persio S, Bielli P, Setterblad N, Alberdi AJ, Vicini E, Geremia R, De la Grange P, Sette C: An Orchestrated Intron Retention Program in Meiosis Controls Timely Usage of Transcripts during Germ Cell Differentiation. Dev Cell 2017, 41:82–93 e84. [DOI] [PMC free article] [PubMed] [Google Scholar]; These papers highlight two distinct contexts in which post-transcriptional splicing of RI-RNAs has a functional role in gene expression regulation. In both cases, cell-type specific Rl-RNAs were found to be spliced post-transcriptionally as either part of a cell signaling response (Mauger et al. 2016) or differentiation programs (Naro et al. 2017).
- 28.Shalgi R, Hurt JA, Lindquist S, Burge CB: Widespread inhibition of posttranscriptional splicing shapes the cellular transcriptome following heat shock. Cell Rep 2014, 7:1362–1370. [DOI] [PubMed] [Google Scholar]
- 29.Pimentel H, Parra M, Gee SL, Mohandas N, Pachter L, Conboy JG: A dynamic intron retention program enriched in RNA processing genes regulates gene expression during terminal erythropoiesis. Nucleic Acids Res 2016, 44:838–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ni T, Yang W, Han M, Zhang Y, Shen T, Nie H, Zhou Z, Dai Y, Yang Y, Liu P, et al. Global intron retention mediated gene regulation during CD4+ T cell activation. Nucleic Acids Res 2016, 44:6817–6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monteuuis G, Wong JJL, Bailey CG, Schmitz U, Rasko JEJ: The changing paradigm of intron retention: regulation, ramifications and recipes. Nucleic Acids Res 2019, 47:11497–11513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Braun CJ, Stanciu M, Boutz PL, Patterson JC, Calligaris D, Higuchi F, Neupane R, Fenoglio S, Cahill DP, Wakimoto H, et al. Coordinated Splicing of Regulatory Detained Introns within Oncogenic Transcripts Creates an Exploitable Vulnerability in Malignant Glioma. Cancer Cell 2017, 32:411–426 e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vargas DY, Shah K, Batish M, Levandoski M, Sinha S, Marras SA, Schedl P, Tyagi S: Single-molecule imaging of transcriptionally coupled and uncoupled splicing. Cell 2011, 147:1054–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pai AA, Henriques T, McCue K, Burkholder A, Adelman K, Burge CB: The kinetics of pre-mRNA splicing in the Drosophila genome and the influence of gene architecture. Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin RM, Rino J, Carvalho C, Kirchhausen T, Carmo-Fonseca M: Live-cell visualization of pre-mRNA splicing with single-molecule sensitivity. Cell Rep 2013, 4:1144–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bresson SM, Hunter OV, Hunter AC, Conrad NK: Canonical Poly(A) Polymerase Activity Promotes the Decay of a Wide Variety of Mammalian Nuclear RNAs. PLoS Genet 2015, 11:e1005610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meola N, Domanski M, Karadoulama E, Chen Y, Gentil C, Pultz D, Vitting-Seerup K, Lykke-Andersen S, Andersen JS, Sandelin A, et al. Identification of a Nuclear Exosome Decay Pathway for Processed Transcripts. Mol Cell 2016, 64:520–533. [DOI] [PubMed] [Google Scholar]
- 38.Davidson L, Kerr A, West S: Co-transcriptional degradation of aberrant pre-mRNA by Xrn2. EMBO J 2012, 31:2566–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee ES, Akef A, Mahadevan K, Palazzo AF: The consensus 5′ splice site motif inhibits mRNA nuclear export. PLoS One 2015, 10:e0122743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palazzo AF, Lee ES: Sequence Determinants for Nuclear Retention and Cytoplasmic Export of mRNAs and lncRNAs. Front Genet 2018, 9:440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeong S: SR Proteins: Binders, Regulators, and Connectors of RNA. Mol Cells 2017, 40:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Botti V, McNicoll F, Steiner MC, Richter FM, Solovyeva A, Wegener M, Schwich OD, Poser I, Zarnack K, Wittig I, et al. Cellular differentiation state modulates the mRNA export activity of SR proteins. J Cell Biol 2017, 216:1993–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brugiolo M, Botti V, Liu N, Muller-McNicoll M, Neugebauer KM: Fractionation iCLIP detects persistent SR protein binding to conserved, retained introns in chromatin, nucleoplasm and cytoplasm. Nucleic Acids Res 2017, 45:10452–10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muller-McNicoll M, Botti V, de Jesus Domingues AM, Brandl H, Schwich OD, Steiner MC, Curk T, Poser I, Zarnack K, Neugebauer KM: SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev 2016, 30:553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang Y, Gattoni R, Stévenin J, Steitz JA: SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol Cell 2003, 11:837–843. [DOI] [PubMed] [Google Scholar]
- 46.Wang J, Cheng H: Out or decay: fate determination of nuclear RNAs. Essays Biochem 2020. [DOI] [PubMed] [Google Scholar]
- 47.Courchaine EM, Lu A, Neugebauer KM: Droplet organelles? EMBO J 2016, 35:1603–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Politz JC, Tuft RA, Prasanth KV, Baudendistel N, Fogarty KE, Lifshitz LM, Langowski J, Spector DL, Pederson T: Rapid, diffusional shuttling of poly(A) RNA between nuclear speckles and the nucleoplasm. Mol Biol Cell 2006, 17:1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Molenaar C, Abdulle A, Gena A, Tanke HJ, Dirks RW: Poly(A)+ RNAs roam the cell nucleus and pass through speckle domains in transcriptionally active and inactive cells. J Cell Biol 2004, 165:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fei J, Jadaliha M, Harmon TS, Li ITS, Hua B, Hao Q, Holehouse AS, Reyer M, Sun Q, Freier SM, et al. Quantitative analysis of multilayer organization of proteins and RNA in nuclear speckles at super resolution. J Cell Sci 2017, 130:4180–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 2010, 39:925–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neugebauer KM, Roth MB: Distribution of pre-mRNA splicing factors at sites of RNA polymerase II transcription. Genes Dev 1997, 11:1148–1159. [DOI] [PubMed] [Google Scholar]
- 53.Phair RD, Misteli T: High mobility of proteins in the mammalian cell nucleus. Nature 2000, 404:604–609. [DOI] [PubMed] [Google Scholar]
- 54.Chen Y, Zhang Y, Wang Y, Zhang L, Brinkman EK, Adam SA, Goldman R, van Steensel B, Ma J, Belmont AS: Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J Cell Biol 2018, 217:4025–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, et al. Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 2018, 174:744–757 e724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen W, Yan Z, Li S, Huang N, Huang X, Zhang J, Zhong S: RNAs as Proximity-Labeling Media for Identifying Nuclear Speckle Positions Relative to the Genome. iScience 2018, 4:204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Y, Belmont AS: Genome organization around nuclear speckles. Curr Opin Genet Dev 2019, 55:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *58.Su JH, Zheng P, Kinrot SS, Bintu B, Zhuang X: Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study used an incredibly powerful method, MERFISH, to sequentially image hundreds of nascent RNAs and genomic loci in single cells. MERFISH allows the structure of whole chromosomes or dozens of loci per chromosome to be imaged in situ relative to different nuclear structures, including nuclear speckles and nucleoli. In addition to directly identifying trans-chromosome and long-range chromatin interactions, the authors found that transcriptional activity correlated with compartment A chromatin, and that compartment A chromatin and transcriptional bursting are enriched around nuclear speckles.
- 59.Khanna N, Hu Y, Belmont AS: HSP70 transgene directed motion to nuclear speckles facilitates heat shock activation. Curr Biol 2014, 24:1138–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim J, Venkata NC, Hernandez Gonzalez GA, Khanna N, Belmont AS: Gene expression amplification by nuclear speckle association. J Cell Biol 2020, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **61.Ding F, Elowitz MB: Constitutive splicing and economies of scale in gene expression. Nat Struct Mol Biol 2019, 26:424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]; Several models have been proposed to explain how splicing efficiency is altered between highly and lowly transcribed genes. This paper used smFISH to measure splicing efficiency in the same gene in individual cells. The authors demonstrate that increased splicing efficiency correlated with increased transcription and closer proximity to nuclear speckles, suggesting that splicing and transcription operate on an “economy of scale” that is influenced by nuclear organization.
- 62.Mor A, White A, Zhang K, Thompson M, Esparza M, Munoz-Moreno R, Koide K, Lynch KW, Garcia-Sastre A, Fontoura BM: Influenza virus mRNA trafficking through host nuclear speckles. Nat Microbiol 2016, 1:16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **63.Wang K, Wang L, Wang J, Chen S, Shi M, Cheng H: Intronless mRNAs transit through nuclear speckles to gain export competence. J Cell Biol 2018, 217:3912–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that nuclear speckles promote mRNA export independent of splicing. By analyzing naturally intronless mRNAs as opposed to synthetic cDNA reporters, the authors demonstrate that endogenous mRNAs are targeted to nuclear speckles by their SR protein binding sites and are released for nuclear export by the TREX complex.
- 64.Dias AP, Dufu K, Lei H, Reed R: A role for TREX components in the release of spliced mRNA from nuclear speckle domains. Nat Commun 2010, 1:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol 2007, 3:576–583. [DOI] [PubMed] [Google Scholar]
- 66.Hett A, West S: Inhibition of U4 snRNA in human cells causes the stable retention of polyadenylated pre-mRNA in the nucleus. PLoS One 2014, 9:e96174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ilik İA, Malszycki M, Lübke AK, Schade C, Meierhofer D, Aktaş T: SON and SRRM2 are essential for nuclear speckle formation. Elife 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Girard C, Will CL, Peng J, Makarov EM, Kastner B, Lemm I, Urlaub H, Hartmuth K, Luhrmann R: Post-transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat Commun 2012, 3:994. [DOI] [PubMed] [Google Scholar]
- 69.Engreitz JM, Sirokman K, McDonel P, Shishkin AA, Surka C, Russell P, Grossman SR, Chow AY, Guttman M, Lander ES: RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell 2014, 159:188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Viphakone N, Sudbery I, Griffith L, Heath CG, Sims D, Wilson SA: Co-transcriptional Loading of RNA Export Factors Shapes the Human Transcriptome. Mol Cell 2019, 75:310–323 e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Melcák I, Melcáková S, Kopský V, Vecerová J, Raska I: Prespliceosomal assembly on microinjected precursor mRNA takes place in nuclear speckles. Mol Biol Cell 2001, 12:393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D, Fugmann T, Wong KK, Van Allen EM: Intron retention is a source of neoepitopes in cancer. Nat Biotechnol 2018, 36:1056–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sharma A, Takata H, Shibahara K, Bubulya A, Bubulya PA: Son is essential for nuclear speckle organization and cell cycle progression. Mol Biol Cell 2010, 21:650–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tripathi V, Song DY, Zong X, Shevtsov SP, Hearn S, Fu XD, Dundr M, Prasanth KV: SRSF1 regulates the assembly of pre-mRNA processing factors in nuclear speckles. Mol Biol Cell 2012, 23:3694–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang B, Arun G, Mao YS, Lazar Z, Hung G, Bhattacharjee G, Xiao X, Booth CJ, Wu J, Zhang C, et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep 2012, 2:111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kurshakova MM, Krasnov AN, Kopytova DV, Shidlovskii YV, Nikolenko JV, Nabirochkina EN, Spehner D, Schultz P, Tora L, Georgieva SG: SAGA and a novel Drosophila export complex anchor efficient transcription and mRNA export to NPC. Embo j 2007, 26:4956–4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Texari L, Stutz F: Sumoylation and transcription regulation at nuclear pores. Chromosoma 2015, 124:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rohner S, Kalck V, Wang X, Ikegami K, Lieb JD, Gasser SM, Meister P: Promoter- and RNA polymerase II-dependent hsp-16 gene association with nuclear pores in Caenorhabditis elegans. J Cell Biol 2013, 200:589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]