

Abstract
Cigarette smoke (CS) contains many toxins that collectively harm nearly every organ in the body, and smoking is a key risk factor for many chronic diseases. Aside from its toxic actions, CS may alter expression of the drug- and steroid-binding pregnane X receptor (PXR), which when activated upregulates expression of cytochrome P450 (CYP) enzymes, glutathione transferases (GSTs), and multidrug resistance protein 1 (MDR1), an adaptive metabolic array that mediates clearance of CS component toxins. We sought to identify new PXR agonists that may be useful for restoring PXR activity in conditions wherein it is suppressed, and their mechanisms of PXR binding and activation. PXR has a uniquely larger, hydrophobic, and highly flexible ligand-binding domain (LBD) vs. other nuclear receptors, enabling it to interact with structurally diverse molecules. We tested certain calcium channel blockers (CCBs) as a pharmacological subset of potential PXR ligands, analyzing by molecular docking methods, and identified a putative active site in the PXR LBD, along with the relevant bonds and bonding energies. We analyzed felodipine binding and agonist activity in detail, as it showed the lowest binding energy among CCBs tested. We found felodipine was a potent PXR agonist as measured by luciferase reporter assay, whereas CCBs with higher binding energies were less potent (amlodipine) or nearly inactive (manidipine), and it induced CYP3A4 expression in HepG2 cells, a known target of PXR agonism. Felodipine also both induced PXR mRNA in HepG2 hepatocytes and reduced CS extract-induced diminution of PXR levels, indicating it modulates PXR expression. The results illuminate mechanisms of ligand-induced PXR activation and identify felodipine as a novel PXR agonist.
Keywords: Molecular modeling, Calcium channel blocker, Cytochrome p450, HepG2, Cigarette smoke, Nuclear receptor
1. Introduction
Cigarette smoke (CS) contains hundreds of toxic chemicals that harm nearly every organ in the body [1], and smoking is a significant risk factor for many diseases, including chronic obstructive lung disease, atherosclerosis, stroke, and cancers. Smoking rates have stabilized in some countries, but have continued to rise in others. More than 1.1 billion people worldwide smoke tobacco, causing substantial public health problems [2]. Besides inducing pathogenic changes in organs including lung and liver, CS also may also alter expression of the pregnane X receptor (PXR; also known as SXR [steroid and xenobiotic receptor] or PAR [pregnane activated receptor]), a drug- and steroid-binding receptor. When activated, PXR upregulates expression of cytochrome P450 (CYP) enzymes, glutathione transferases (GSTs), and multidrug resistance protein 1 (MDR1) [3] – a potent and adaptive metabolic array that mediates clearance of CS' component toxins [4,5]. This has motivated us to seek to develop novel PXR ligands that can restore PXR activation, as a potential new means to prevent or mitigate the harmful effects of CS.
PXR is a member of the nuclear receptor (NR) superfamily of ligand-activated transcription factors, encoded by NR1I2 [6]. Like other NRs, PXR contains three distinct domains: a ligand-binding domain (LBD), a DNA-binding domain, and a transactivation domain that contains activation functions-1 [AF-1] and −2 [AF-2]) [7]. Nevertheless, PXR is distinct from classical NRs in harboring a larger, hydrophobic, and highly flexible LBD [8] – a unique feature that renders PXR promiscuous, enabling interaction with structurally diverse molecules including steroids [6], dietary supplements [9], antibiotics [10], antifungals [11], and environmental pollutants [12]. When activated by agonist binding, PXR interacts with coactivators, such as SRC-1 and TIF2, and binds to PXR-responsive elements (PXRE) or xenobiotic DNA response elements [13] to induce expression of CYP, GST, and MDR1. Such upregulation by PXR can thus broadly impact both drug metabolism and disease conditions, yet its roles in these domains have remained largely unexplored [14,15].
We therefore aimed to identify novel PXR agonists and determine their abilities to mitigate noxious actions of CS. In silico computational approaches such as ligand-based and protein structure-based drug design methods provide potent means for rational drug development, and facilitate progress in drug discovery [16]. Experimentally-derived PXR structures have informed and enabled computational studies to create visualizable PXR-ligand interactions such as ligand-based models [17,18], pharmacophore models [19,20], structure-activity relationship (SAR) models [21], and machine learning approaches [22,23]. Many of these studies relied primarily on computational methods, but evaluating ligand properties and activities are critical to understanding the mechanism and determinants of the relevant molecular interactions.
Therefore, as a means to identify new PXR agonists and the determinants of their activities and potential utility, in this study we first modeled a putative ligand-binding site by analyzing 12 crystallographic ligand-PXR complexes. Because certain calcium channel blockers (CCBs) are safe, clinically approved medications, we focused on CCBs as a pharmacological subset of potential ligands, and screened some of these as new potential PXR-ligands, using molecular docking methods. We identified several novel PXR activators that interact with critical amino acid residues of the PXR LBD, and evaluated their abilities to induce PXR transcriptional activity and to ameliorate the toxic effects of CS.
2. Materials and methods
2.1. Chemical structures
The 3-dimensional (3-D) structures of selective CCBs: amlodipine, diltiazem, felodipine, isradipine, manidipine, and verapamil (Supplemental Table S1) were downloaded from PubChem (the U.S. National Library of Medicine; https://pubchem.ncbi.nlm.nih.gov/). The structures were energy minimized using Chemistry at Harvard Macromolecular Mechanics (CHARMM36) force fields [24]. The geometry optimized structures were used as ligands in docking studies.
2.2. Molecular modelling and docking
Twelve-crystallographic ligand-PXR complexes (Protein Data Bank entries 1ilh, 1m13, 1nrl, 1skx, 2o9i, 2qnv, 3r8d, 4ny9, 4xif, 4xhd, 5x0r, 6hty) were obtained from the RCSB Protein Data Bank, and the ligand-PXR interactions were analyzed to determine the volume occupancy and to determine the active site amino acids interacting with the ligands (Supplemental Table S2). The X-ray structure of 1ilg [8] with a 2.52 Å resolution (the complete structure with the highest resolution among those selected) was chosen to generate an active site using Discovery Studio (BIOVIA, San Diego, CA, USA). Water and other small molecules that co-crystallized were removed, and the missing residues were added using the Build and Edit Protein tools. CHARMM was added to the receptor molecule and energy minimized with constrained heavy atoms. After simulation, the binding site was created using the ligand-volume occupancy determined, as noted earlier. This molecule was used in the docking studies. Molecular docking was performed using Discovery Studio to determine the interactions of selected CCBs with the PXR LBD. The energy minimized 3-D ligands, and an active site defined-receptor molecule was used for docking. The receptor-ligand complexes with the lowest binding energy ΔG (kcal/mol) were further analyzed to determine the interactions of the ligands, as noted earlier.
2.3. PXR reporter assay
The efficacy of selective CCBs to activate PXR was determined using a PXR driven luciferase reporter assay (cat. no. IB07001; Indigo Biosciences, State College, PA, USA) following the manufacturer’s instructions. Briefly, PXR reporter cells cultured in Cell Recovery Medium were treated with 0.0412 −30 μM of indicated compounds or rifampicin (positive control) for 24 hours. After the incubation, the medium was removed by suction, and luciferase detection reagent was added and incubated for 5 minutes at room temperature. The produced luminescence was quantified using a luminometer.
2.4. Cell culture
HepG2 cells were obtained from American Type Culture Collection (cat. no. HB-8065; Manassas, VA, USA) and maintained as subconfluent monolayers in DMEM with 10% fetal bovine serum (Hyclone, Logan, UT, USA) and 100 units/ml penicillin plus 100 μg/ml streptomycin at 37°C with 5% CO2. Serum starved cells at ~90% confluence were exposed to CS extract (CSE) followed by treatment with vehicle (DMSO) or felodipine (Cayman Chemical, Ann Arbor, MI, USA) at the indicated concentrations. After treatment, whole-cell lysates were prepared. Protein concentrations were determined using the BCA Protein Assay kit (Pierce, Rockford, IL, USA).
2.5. Cigarette smoke extract preparation
CS from research-grade cigarettes (3R4F; Kentucky Tobacco Research and Development Center, University of Kentucky, Lexington, KY, USA) was puffed (one puff per minute) into DMEM medium according to the FTC guidelines in an Erlenmeyer flask with a sidearm. The solution was sterilized using a sterile syringe filter and defined as 100% CSE. The optical density of the solution was measured using a NanoDrop spectrophotometer at a wavelength of 320 nm. Freshly prepared CSE was diluted to indicated concentrations and used immediately.
2.6. Western blotting
Protein extracts were mixed with 4x Laemmli sample buffer (cat. no. 1610747; Bio-Rad Laboratories, Hercules, CA, USA) then separated on Mini-PROTEAN TGX Precast Gels (cat. no. 4561036; Bio-Rad) and electroblotted onto a nitrocellulose membrane (cat. no. 1620112; Bio-Rad). The membrane was blocked in blocking buffer (Intercept, LI-COR Biosciences, Lincoln, NE, USA) for 1 hour at room temperature. Blots were then incubated overnight at 4°C with the primary antibody against the target protein. PXR (1:1000; cat. no. sc-25381) and GAPDH (1:2500; cat. no. sc-20357) primary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). IRDye secondary antibodies were obtained from LI-COR Biosciences. The infrared signal was detected with an Odyssey Infrared Imager (LI-COR Biosciences).
2.7. Real-time PCR
The RNeasy Plus Kit (Qiagen, Valencia, CA, USA) was used to isolate RNA from cells. RNA concentration was quantified using a NanoDrop spectrophotometer at a wavelength of 260 nm. The cDNA was synthesized from RNA using the cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). The expression of PXR and CYP3A4 were quantitated by performing real-time PCR with specific primers (Supplemental Table S3) and SYBR Green Master Mix (Applied Biosystems). The relative expression normalized to GAPDH is shown.
2.8. Statistical analysis
The data are represented as mean ± standard deviation. We used GraphPad Prism (GraphPad Software, La Jolla, CA, USA) for statistical analyses; the differences among different treatment conditions were determined by ANOVA followed by a Bonferroni correction for multiple comparisons. The EC50 was determined from dose-response curves with non-linear regression fitting. P values < 0.05 were considered statistically significant.
3. Results
3.1. Molecular interactions of selective calcium channel blockers with the PXR LBD
As an initial strategy toward identifying CCBs as potential PXR ligands, we used in silico modeling. To determine key amino acids, we compiled a list of amino acids within the PXR LBD that interacted with 12 known ligands in crystallographic ligand-PXR complexes (1ilh, 1m13, 1nrl, 1skx, 2o9i, 2qnv, 3r8d, 4ny9, 4xif, 4xhd, 5x0r, 6hty; Supplemental Table 2). We used this information to generate a putative active site in the PXR LBD (Fig. 1) to use in docking analyses. The CCB ligands tested, their structures (Supplemental Table 1), and their associated chemical information and binding energies, ΔG (kcal/mol), are summarized in Table 1. For each such complex, we analyzed the most optimal pose with low binding energy to determine the relevant ligand-receptor interactions, and generated two-dimensional (2-D) ligand plots. The interactions of felodipine, amlodipine, diltiazem, isradipine, verapamil, and manidipine with the PXR LBD are shown in Supplemental Fig. 1A-F.
Figure 1. PXR LBD.

(A) The putative ligand-binding site (red sphere) in the N-terminal region of PXR LBD was generated by analyzing twelve-crystallographic ligand-PXR complexes and (B) with the hydrogen (H)-bond donor and acceptor surface. Figures were generated using Discovery Studio Visualizer.
Table 1.
Calcium channel blockers (CCBs) tested along with their associated chemical information and binding energies.
| S. No | Compound Name | Molecular Formula | Molecular Weight g/mol |
Lowest Binding Energy ΔG (kcal/mol) |
|---|---|---|---|---|
| 1 | Felodipine | C18H19Cl2NO4 | 384.2 | −10.82 |
| 2 | Amlodipine | C20H25ClN2O5 | 408.9 | −9.70 |
| 3 | Diltiazem | C22H26N2O4S | 414.5 | −9.80 |
| 4 | Isradipine | C19H21N3O5 | 371.4 | −9.27 |
| 5 | Verapamil | C27H38N2O4 | 454.6 | −9.76 |
| 6 | Manidipine | C35H38N4O6 | 601.7 | −8.32 |
3.2. Molecular interactions in the felodipine-PXR LBD complex
Because felodipine-PXR LBD (Supplemental Fig. 1A) yielded the highest-ranking complex among those tested, we analyzed the molecular interactions involved. The felodipine-PXR LBD complex (Fig. 2A) was similar to the experimentally-observed position of SR12813, as described earlier [8]. The oxygen atoms of the methyl carboxylate group formed a short hydrogen bond (2.75 Å) with Trp299 and pi-alkyl interactions with Gln285, Met323, and His327, and that of the ethyl carboxylate group formed a much shorter hydrogen bond (2.06 Å) with Ser247. The chlorine (position 3) in the (2,3)-dichlorophenyl group formed several pi-alkyl interactions with Leu209, Met243, Phe251, Phe281, and Cys284. We also detected a pi-sulfur interaction between the Met323 sulfur atom and the (2,3)-dichlorophenyl ring (Fig. 2B-C).
Figure 2. Molecular docking of the felodipine-PXR complex.

(A) Molecular model of felodipine bound to PXR LBD. (B) Stereo views of the PXR-felodipine complex encompassing felodipine (element colored stick model) and PXR amino acid residues (colored line models) that interact and stabilize felodipine in the selected pose. PXR is shown as a secondary structure colored ribbon. (C) 2-D-ligand interaction plot of the complex. Felodipine is shown as an element-colored stick model with interacting amino acids represented as balls and sticks. Hydrogen bond interactions are indicated by green arrows; alkyl and pi-alkyl interactions are indicated by pink arrows; pi-sulfur interaction is indicated by a yellow arrowhead. Figures were generated using Discovery Studio Visualizer.
3.3. Felodipine is a potent PXR agonist
We further validated the in silico PXR-ligand docking findings using in vitro PXR luciferase reporter assays to test PXR transactivation by felodipine, amlodipine, and manidipine. We used the known potent PXR activator rifampicin as a positive control. We selected felodipine and amlodipine for testing because our in silico analyses yielded the lowest binding energies for them, suggesting potential to serve as potent PXR agonists. In contrast, we predicted manidipine would be a weak PXR agonist based on its in silico docking results. The EC50, a measurement of the effective ligand concentration required to reach a 50% response, is a well-established measurement of potency. Thus, a more potent ligand has a lower EC50 value. As predicted, rifampicin yielded a robust response in the reporter assay, with an EC50 of ~3.8 μM. As predicted from in silico computations, felodipine yielded a more potent EC50 of ~1.0 μM, followed by amlodipine at ~2.5 μM, and manidipine was least potent (~6.5 μM) (Fig. 3). These in vitro findings correlate very closely with the in silico computational analyses, and suggest that felodipine is a potent PXR agonist.
Figure 3. Felodipine activates PXR.

PXR reporter cells were treated with indicated compounds at 0.0412–30 μM for 24 hours. PXR transcriptional activity was then measured by luciferase activity. Dose-response curve fitting was performed by non-linear regression analysis. Data are expressed as mean ± SD; n = 3.
3.4. Felodipine reverses CS extract-induced PXR degradation and induces CYP3A4
To further test the validity of felodipine as a bona fide PXR agonist, we tested its functional effects on PXR activation. Prior reports suggested that PXR is regulated by environmental toxins, such as CS [5]. To test if CS altered PXR expression and function, we used CS extract (CSE) as in vitro stimulus in HepG2 hepatocytes, a well-characterized cell line. We first assessed the concentration-response relationship for CSE on PXR protein expression, by western blotting (Fig. 4A), and found that CSE markedly and concentration-dependently reduced PXR protein levels. We determined the time course of these effects using a submaximal CSE concentration, which revealed increasing degradation of PXR with time, which was maximal at 6 hours (Fig. 4B). Based on these findings, we tested if felodipine treatment similarly influenced PXR in CSE-treated HepG2 hepatocytes. In resting conditions, felodipine elevated PXR protein and mRNA expression vs. their baseline levels. Also, felodipine treatment reversed CSE-induced reduction of PXR levels, returning PXR protein and mRNA expression to baseline levels (Fig. 4C-D).
Figure 4. Felodipine reverses CSE-induced PXR degradation and induces CYP3A4.
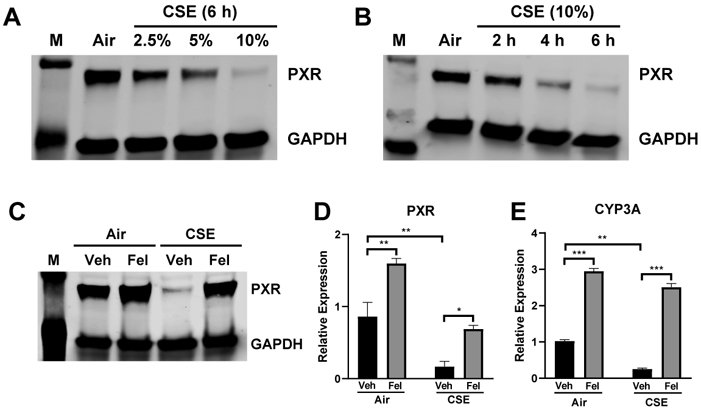
HepG2 cells exposed to filtered air-treated medium (Air) or CSE medium for indicated (A) dose and (B) time. PXR expression was determined by western blotting. Next, HepG2 cells were exposed to Air or 10% CSE medium for 6 hours. Felodipine was added 1 hour after CSE treatment. (C) PXR expression, determined by western blotting, with GAPDH as loading control. M indicates the protein marker. (D) PXR and (E) CYP3A4 mRNA expression were determined by real-time PCR. Fel: felodipine, Veh: vehicle (DMSO). Data are expressed as mean ± SD; n = 3, *P < 0.05, **P < 0.01, ***P < 0.001.
Because CYP genes are direct targets of activated PXR, we also tested if felodipine induces CYP3A4 expression. Treating HepG2 cells with felodipine (10 μM) for 6 hours significantly elevated CYP3A4 mRNA levels (Fig. 4E). Overall, the results demonstrate that felodipine activates biological responses via PXR.
4. Discussion
Our findings addressed our study's aims of 1) discovering novel PXR modulators/agonists and 2) evaluating their ability to activate PXR and potential roles as modulators of cellular defense mechanisms against environmental insults such as CS. We exploited the ability of CCBs to interact with and activate PXR to analyze the molecular interactions involved. Molecular docking analysis revealed that some CCBs form one or more hydrogen bond acceptor/donor potentials, which anchor the ligand to the PXR LBD. We further found that hydrogen bonding was essential to high-affinity interactions between CCBs and PXR. PXR has been described as a chemical “sensor” that contains a flexible LBD [8], and our findings suggest that PXR also adapts a flexible conformation that accommodates CCBs to fit inside the binding cavity.
In silico methods are useful to identify novel PXR ligands [20,23], but are too limited to enable predicting a ligand’s potential to activate PXR given the diverse chemical compounds in multiple orientations that can bind to the large and flexible PXR LBD. We therefore confirmed the in silico-predicted ability of CCBs to bind to PXR, by using a PXR-driven luciferase reporter assay which validated the efficacy of felodipine and amlodipine (lowest binding energies) and manidipine (high binding energy). We found that both felodipine and amlodipine, strong in silico candidates, dose-dependently induced PXR activity. Manidipine, which was predicted to be a weak ligand by in silico methods, accordingly failed to yield any significant effect. CCBs of the dihydropyridine family have been found to activate PXR and induce CYP3A4 expression [25], and our present findings expand the range of CCBs displaying such PXR activation, to include felodipine and amlodipine.
Prior findings suggested that PXR expression was reduced by environmental toxins such as CS [5]. We found that CS dose- and time-dependently decreased PXR expression, yielding near-complete abrogation with longer exposure periods and higher doses. Such reduction of PXR expression inhibits CYP transcription, and later its expression [26]. In contrast, PXR activation induces multiple xenobiotic- and toxin-metabolizing enzymes, including CYPs [27]. CS was found earlier to markedly decrease CYP-encoding transcript levels in alveolar macrophages [4] and in livers of rats exposed to whole-body CS [5]. Presently, we expanded upon these insights by finding that CS significantly reduced CYP3A4 expression in human liver cells. Furthermore, we found that felodipine is a true PXR agonist, as its binding causes PXR activation and thus increases CYP3A4 transcription.
Together, our findings revealed felodipine is a novel and potent PXR agonist, and we anticipate that exploring further scaffolds will yield development of highly potent ligands that favor hydrogen bond interactions with critical amino acid residues in the PXR LBD. We also found that felodipine restored CS-suppressed PXR expression and thus CYP3A4 production. Because CS is a potent air pollutant containing hundreds of toxic chemicals, identifying the precise molecular mechanisms by which felodipine-PXR responds to CS warrants further investigation, beyond our present scope. These findings significantly advance understanding of CCB-PXR interactions, by identifying both novel PXR agonists and mechanisms involved. Further research in in vitro and in vivo models may yield translationally useful agents for protection against PXR-dysregulating agents.
Supplementary Material
Highlights.
Analyzed calcium channel blocker (CCB) interactions with pregnane X receptor (PXR)
Identified and characterized selective CCBs as novel PXR agonists
Cigarette smoke (CS) downregulates PXR mRNA and protein levels
CCB felodipine reverses such CS-induced downregulation
Acknowledgments
Funding
This work was supported by the Merit Review Award from the US Department of Veterans Affairs (CX001048 and CX000105), National Institute of Health (HL149719), and AHA transformational grant to TN (19TPA34830061).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Kleinstreuer C, Feng Y, Lung deposition analyses of inhaled toxic aerosols in conventional and less harmful cigarette smoke: a review, International journal of environmental research and public health 10 (2013) 4454–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ahluwalia IB, Smith T, Arrazola RA, Palipudi KM, Garcia de Quevedo I, Prasad VM, Commar A, Schotte K, Garwood PD, Armour BS, Current Tobacco Smoking, Quit Attempts, and Knowledge About Smoking Risks Among Persons Aged >/=15 Years - Global Adult Tobacco Survey, 28 Countries, 2008-2016, MMWR Morb Mortal Wkly Rep 67 (2018) 1072–1076. 10.15585/mmwr.mm6738a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kliewer SA, The nuclear pregnane X receptor regulates xenobiotic detoxification, The Journal of nutrition 133 (2003) 2444S–2447S. [DOI] [PubMed] [Google Scholar]
- [4].Hukkanen J, Väisänen T, Lassila A, Piipari R, Anttila S, Pelkonen O, Raunio H, Hakkola J, Regulation of CYP3A5 by glucocorticoids and cigarette smoke in human lung-derived cells, Journal of Pharmacology and Experimental Therapeutics 304 (2003) 745–752. [DOI] [PubMed] [Google Scholar]
- [5].Li X, Yan Z, Wu Q, Sun X, Li F, Zhang S, Li K, Li L, Wu J, Xu L, Glucocorticoid receptor contributes to the altered expression of hepatic cytochrome P450 upon cigarette smoking, Molecular Medicine Reports 14 (2016) 5271–5280. [DOI] [PubMed] [Google Scholar]
- [6].Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterström RH, An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway, Cell 92 (1998) 73–82. [DOI] [PubMed] [Google Scholar]
- [7].Khorasanizadeh S, Rastinejad F, Visualizing the architectures and interactions of nuclear receptors, Endocrinology 157 (2016) 4212–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR, The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity, Science 292 (2001) 2329–2333. [DOI] [PubMed] [Google Scholar]
- [9].Staudinger JL, Ding X, Lichti K, Pregnane X receptor and natural products: beyond drug–drug interactions, Expert opinion on drug metabolism & toxicology 2 (2006) 847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yasuda K, Ranade A, Venkataramanan R, Strom S, Chupka J, Ekins S, Schuetz E, Bachmann K, A comprehensive in vitro and in silico analysis of antibiotics that activate pregnane X receptor and induce CYP3A4 in liver and intestine, Drug Metabolism and Disposition 36 (2008) 1689–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Svecova L, Vrzal R, Burysek L, Anzenbacherova E, Cerveny L, Grim J, Trejtnar F, Kunes J, Pour M, Staud F, Azole antimycotics differentially affect rifampicin-induced pregnane X receptor-mediated CYP3A4 gene expression, Drug metabolism and disposition 36 (2008) 339–348. [DOI] [PubMed] [Google Scholar]
- [12].Wyde ME, Bartolucci E, Ueda A, Zhang H, Yan B, Negishi M, You L, The environmental pollutant 1, 1-dichloro-2, 2-bis (p-chlorophenyl) ethylene induces rat hepatic cytochrome P450 2B and 3A expression through the constitutive androstane receptor and pregnane X receptor, Molecular pharmacology 64 (2003) 474–481. [DOI] [PubMed] [Google Scholar]
- [13].Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA, The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions, The Journal of clinical investigation 102 (1998) 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Banerjee M, Robbins D, Chen T, Targeting xenobiotic receptors PXR and CAR in human diseases, Drug discovery today 20 (2015) 618–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang Y-M, Ong SS, Chai SC, Chen T, Role of CAR and PXR in xenobiotic sensing and metabolism, Expert opinion on drug metabolism & toxicology 8 (2012) 803–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Macalino SJY, Gosu V, Hong S, Choi S, Role of computer-aided drug design in modern drug discovery, Archives of pharmacal research 38 (2015) 1686–1701. [DOI] [PubMed] [Google Scholar]
- [17].Ekins S, Chang C, Mani S, Krasowski MD, Reschly EJ, Iyer M, Kholodovych V, Ai N, Welsh WJ, Sinz M, Human pregnane X receptor antagonists and agonists define molecular requirements for different binding sites, Molecular pharmacology 72 (2007) 592–603. [DOI] [PubMed] [Google Scholar]
- [18].Ekins S, Kortagere S, Iyer M, Reschly EJ, Lill MA, Redinbo MR, Krasowski MD, Challenges predicting ligand-receptor interactions of promiscuous proteins: the nuclear receptor PXR, PLoS Comput Biol 5 (2009) e1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ekins S, Erickson JA, A pharmacophore for human pregnane X receptor ligands, Drug Metabolism and Disposition 30 (2002) 96–99. [DOI] [PubMed] [Google Scholar]
- [20].Ekins S, Kholodovych V, Ai N, Sinz M, Gal J, Gera L, Welsh WJ, Bachmann K, Mani S, Computational discovery of novel low micromolar human pregnane X receptor antagonists, Molecular pharmacology 74 (2008) 662–672. [DOI] [PubMed] [Google Scholar]
- [21].Dring AM, Anderson LE, Qamar S, Stoner MA, Rational quantitative structure–activity relationship (RQSAR) screen for PXR and CAR isoform-specific nuclear receptor ligands, Chemico-biological interactions 188 (2010) 512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Khandelwal A, Krasowski MD, Reschly EJ, Sinz MW, Swaan PW, Ekins S, Machine learning methods and docking for predicting human pregnane X receptor activation, Chemical research in toxicology 21 (2008) 1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ung CY, Li H, Yap CW, Chen YZ, In silico prediction of pregnane x receptor activators by machine learning approache, Molecular pharmacology 71 (2007) 158–168. [DOI] [PubMed] [Google Scholar]
- [24].Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field, Journal of chemical theory and computation 12 (2016) 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Drocourt L, Pascussi J-M, Assenat E, Fabre J-M, Maurel P, Vilarem M-J, Calcium channel modulators of the dihydropyridine family are human pregnane X receptor activators and inducers of CYP3A, CYP2B, and CYP2C in human hepatocytes, Drug Metabolism and Disposition 29 (2001) 1325–1331. [PubMed] [Google Scholar]
- [26].Beigneux AP, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR, Reduction in cytochrome P-450 enzyme expression is associated with repression of CAR (constitutive androstane receptor) and PXR (pregnane X receptor) in mouse liver during the acute phase response, Biochemical and biophysical research communications 293 (2002) 145–149. [DOI] [PubMed] [Google Scholar]
- [27].Kliewer SA, Goodwin B, Willson TM, The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism, Endocrine reviews 23 (2002) 687–702. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.