

Abstract
Aberrant aggregation and amyloid formation of tar DNA binding protein (TDP-43) and α-synuclein (αS) underlie frontotemporal dementia (FTD) and Parkinson’s disease (PD), respectively. Amyloid inclusions of TDP-43 and αS are also commonly co-observed in amyotrophic lateral sclerosis (ALS), dementia with Lewy bodies (DLB) and Alzheimer disease (AD). Emerging evidence from cellular and animal models show colocalization of the TDP-43 and αS aggregates, raising the possibility of direct interactions and co-aggregation between the two proteins. In this report, we set out to answer this question by investigating the interactions between αS and prion-like pathogenic C-terminal domain of TDP-43 (TDP-43 PrLD). PrLD is an aggregation-prone fragment generated both by alternative splicing as well as aberrant proteolytic cleavage of full length TDP-43. Our results indicate that two proteins interact in a synergistic manner to augment each other’s aggregation towards hybrid fibrils. While monomers, oligomers and sonicated fibrils of αS seed TDP-43 PrLD monomers, TDP-43 PrLD fibrils failed to seed αS monomers indicating selectivity in interactions. Furthermore, αS modulates liquid droplets formed by TDP-43 PrLD and RNA to promote insoluble amyloid aggregates. Importantly, the cross-seeded hybrid aggregates show greater cytotoxicity as compared to the individual homotypic aggregates suggesting that the interactions between the two proteins have a discernable impact on cellular functions. Together, these results bring forth insights into TDP-43 PrLD – αS interactions that could help explain clinical and pathological presentations in patients with co-morbidities involving the two proteins.
Keywords: α-Synuclein, TDP-43, fibrillization, amyloid, liquid-liquid phase separation, neurodegeneration, amyotrophic lateral sclerosis, frontotemporal dementia, Parkinson’s disease
INTRODUCTION
Protein misfolding and toxic amyloid formation have come to define the pathogenesis of many neurodegenerative disorders including Alzheimer disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) [1, 2]. Each of these pathologies is known to involve aberrant aggregation of a protein that brings to bear cellular abnormalities and toxicities. However, overlapping clinical presentations and comorbidities observed among patients with neurodegenerative diseases have motivated researchers into investigating the possibility of molecular overlaps among the respective amyloidogenic proteins involved. For example, a wealth of evidence accrued over the years indicate that some of these maladies are accompanied by the deposition of common amyloid proteins such as tau and α-synuclein (αS), collectively referred to as tauopathies and synucleinopathies, respectively [3-5]. Abnormal tau inclusions are often observed alongside amyloid-β (Aβ) deposits in AD patients [6] as well as in conditions such as PD [7, 8] and prion disease [9, 10]. Similarly αS, the major protein involved in the formation of pathogenic amyloid inclusions of Lewy bodies (LBs) in PD is also observed in AD [11], dementia with Lewy bodies (DLB), and multiple system atrophy (MSA) [12, 13]. Like tau and αS, insoluble cytoplasmic inclusions of the ribonucleoprotein, tar DNA binding protein (TDP-43) are increasingly observed in many neurodegenerative pathologies such as ALS and FTD [14], as well as in patients with PD, DLB, and AD [15, 16]. There has been a surge in the reports on cross-interactions between amyloidogenic proteins such as the interactions between Aβ/tau, αS/Aβ, αS/tau [8, 17-19], and prion/Aβ [20], which have cemented the hypothesis that cross-interaction mechanisms better define the underlying cause of neurogenerative diseases and related co-pathogenesis. Along the same lines, recently it has come to light that αS and TDP-43 inclusions also co-exist in ALS, FTD and PD [15, 21]. While both αS and TDP-43 are known to independently form cytoplasmic amyloid inclusions [22-25], their co-existence raises the question of whether they interact with one another and if so, what consequence does such an interaction brings to bear. Indeed, recent reports do support this contention; TDP-43 was observed to synergistically interact and enhance αS toxicity in dopaminergic neurons [26], while immunocytochemical and immunoblot analyses on mice models and SH-SY5Y neuroblastoma cells revealed that exogenous αS fibrils promote phosphorylation and aggregation of TDP-43 [19]. Furthermore, co-expression of TDP-43 and αS was shown to induce αS pathology in c.elegans that led to significant neurodegeneration as compared to their individual expressions [27].
αS is a 140 amino acid protein containing three regions; lipid binding N-terminal domain (NTD), an aggregation-prone non-amyloid component (NAC) middle region, and an intrinsically disordered, charged C-terminal domain (CTD). NAC and a part of NTD are the primary regions responsible for aggregation, while CTD seems to play an inhibitory role for this process [28, 29]. On the other hand, TDP-43 is a 414 amino acid containing ribonucloprotein protein with an N-terminal domain, two RNA recognition motifs (RRMs), and a disordered prion-like C-terminal domain (PrLD) containing low complexity sequences [30]. TDP-43 is known to be involved in transcriptional regulation and RNA splicing [31, 32]. In pathology, the protein is translocated into the cytoplasm where it undergoes aberrant proteolytic cleavage that generates different C-terminal fragments (CTFs; C35, C25, and C18), which consequently form toxic insoluble inclusions [33-38]. Furthermore, under stress conditions, cytoplasmic TDP-43 undergoes liquid-liquid phase separation (LLPS) by coacervating with RNA and other proteins to form membraneless stress granules (SGs) [39, 40]. If and how SGs play a role in the formation of cytoplasmic TDP-43 inclusions and cellular toxicity remains unclear. PrLD, a segment of such pathological aggregates, is primarily known to drive the fibrillization process and mediates protein-protein interactions [41, 42]. Despite multiple factors in regulating fibrillization of TDP-43 PrLD, a common biophysical phenomenon, liquid-liquid phase separation (LLPS) is also recently known to modulate the aggregation process via electrostatic interactions [40].
Here, we sought to determine how αS and TDP-43 interact with one another to exacerbate cellular toxicity, by investigating the full-length αS and TDP-43 PrLD as PrLD forms a key part of pathogenic proteolytic products of TDP-43 (Figures 1a and 1b). Our results indicate that αS and TDP-43 PrLD monomers, oligomers or fibrils cross-interact and modulate each other’s aggregation behavior. Moreover, αS also enhance TDP-43 PrLD aggregation by modulating the liquid droplets formed by the phase separation of TDP-43 PrLD in presence of RNA. In all, cross-seeded aggregates show higher cytotoxicity than the individual aggregates suggesting the potential for such mechanisms to present greater pathogenicity.
Figure 1.

1H-15N HMQC spectroscopy of αS and TDP-43 PrLD along with DLS of respective reactions. a-b) Sequence of full-length 1-140 amino acid residues αS (a) and 267-414 amino acid residues TDP-43 PrLD used in the experiment. c) 1H-15N HMQC of 10 μM 15N-labeled αS monomers alone (blue) or in the presence of unlabeled 10 μM TDP-43 PrLD monomers (red) in 20 mM MES buffer pH 6.0 at 37 °C. Some of the residues undergoing chemical shift perturbations (CSPs) are shown in boxes while those with lost peaks are indicated by letters without boxes. d)1H-15N HMQC of 10 μM 15N-labeled TDP-43 PrLD monomers alone (blue) or in presence of unlabeled αS (red). e) CSPs for equimolar incubations of αS and TDP-43 PrLD that were calculated using the equation, from the spectrum in (c). f-g) DLS histograms of 10 μM αS monomer control, TDP-43 PrLD monomer control and co-incubated mixture 1:1 αS and TDP-43 PrLD in the same buffer and temperature conditions taken within 10 minutes of incubation.
RESULTS
αS and TDP-43 PrLD monomers synergistically promote fibrillization.
First, we questioned whether monomers of αS and TDP-43 PrLD are able to interact with one another. To answer this, heteronuclear multiple quantum coherence (HMQC) spectroscopy was performed on a uniformly 15N labeled αS and TDP-43 PrLD individually. Both proteins showed narrow chemical shift dispersions in the amide region (7.8-8.7 ppm for αS and 7.7-8.5 for TDP-43 PrLD in the 1H dimension) confirming the well-known disordered structures for both the proteins (blue; Figures 1c and 1d). To see whether the two proteins interact with one another, HMQC spectra of the 15N-enriched proteins were observed upon incubating with unlabeled, natural isotope-abundant proteins in equimolar concentrations at 37 °C. The spectrum of αS co-incubated with unlabeled TDP-43 PrLD showed the disappearance of several cross-peaks along with significant changes in the chemical shifts (Figure 1c). The disappearance of peaks is largely attributed to the aggregation of proteins which leads to significant line broadening and/or loss of signal intensities [43, 44]. Interestingly, shifts in cross-peaks were largely confined to the N-terminal and C-terminal ends of αS (residues 1-40 and 100-140; boxes, Figure 1c) while the disappearance of cross-peaks corresponded to the central amylodogenic NAC of the αS (residues ~61-95; Figure 1c and 1e). Considering that the N- and C-terminal domains are charged and NAC domain of αS is prone to amyloid formation, it is likely that TDP-43 PrLD induces aggregation of the NAC domain αS. Similarly, a substantial signal loss of cross-peaks was observed for TDP-43 PrLD upon co-incubation with αS indicating potential aggregation of the former (Figure 1d). In order to confirm the NMR observations, aggregation of the co-incubated samples was analyzed by dynamic light scattering (DLS) analysis. The freshly fractionated control monomers of αS and TDP-43 PrLD showed a hydrodynamic diameter of ~ 5 nm individually (Figure 1f and g). One has to bear in mind that even the SEC fractionated monomers may be a mixture containing a small percentage of small oligomers. In contrast, the co-incubated samples showed aggregation with an average diameter of >103 nm within 10 minutes of incubation (Figure 1h). These results confirm the NMR observations that αS and TDP-43 PrLD interact with one another to promote high molecular weight aggregates. Encouraged by these NMR results, a deeper investigation into the dynamics of interactions between the two proteins by means of T2 relaxations and H/D exchange will be carried out and will be reported in the near future, but the data obtained so far unequivocally indicates the interactions between αS and TDP-43 PrLD.
To further understand the mechanism of αS and TDP-43 PrLD interactions, we set forth to investigate the aggregation kinetics using thioflavin T (ThT) fluorescence assay (Figure 2). In one reaction set, αS concentration was held constant at 20 μM while TDP-43 PrLD concentrations were increased from 1 to 20 μM (0.05, 0.25, 0.5 and 1.0 molar equivalents). The reactions were incubated at 37 °C and the kinetics of aggregation was monitored for 48 hours (Figure 2a). All the data points were plotted and fitted using the Boltzmann sigmoidal fit (see Methods). The control αS did not show any aggregation during this period (△; Figure 2a). Similarly, at lowest concentration (1 μM) TDP-43 PrLD control did not show increase in ThT fluorescence (Figure 2a). TDP-43 PrLD control at 5, 10, and 20 μM showed concentration-dependent aggregation with aggregation lag times of 20, 15.4, and 9.6 hours, respectively; (○; Figure 2a), while the co-incubated samples with αS showed significantly decreased lag times (●; Figure 2a) suggesting synergistic augmentation of aggregation between the two proteins. In addition, the co-incubated samples also showed higher ThT intensities, which could either indicate numerically more fibrils or an increased ThT binding vis-à-vis higher fluorescence per fibrils formed by both αS and TDP-43 PrLD. Next, to investigate whether αS can modulate the aggregation behavior of TDP-43 PrLD, TDP-43 PrLD was held constant at 20 μM and αS concentrations were increased from 1 - 40 μM (0.05, 0.5, 1 or 2 molar equivalents) (Figure 2b). TDP-43 PrLD control aggregated with lag time of 9.6 h (○; Figure 2b) while αS did not show aggregation within the 50-hour window (△; Figure 2b). There were small but discernable decreases in TDP-43 PrLD lag time of aggregation with the proportional increase in ThT fluorescence intensities with increasing αS (●; Figure 2b). This effect was most prominent with the 40 μM αS incubation, which led to no detectable lag time in TDP-43 PrLD (●; Figure 2b).
Figure 2.
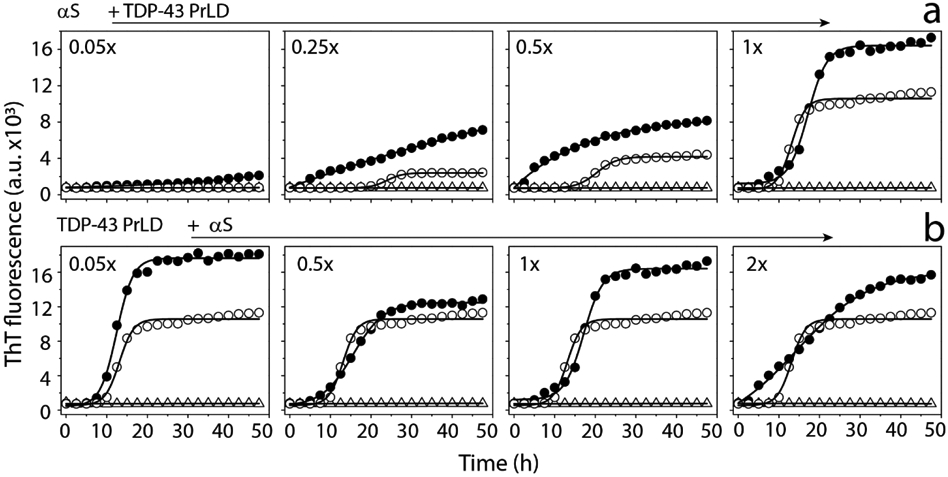
ThT aggregation kinetics of monomeric αS and TDP-43 PrLD in 20 mM MES buffer at pH 6.0 a) ThT fluorescence of 20 μM αS (△) in presence of 0.05 to 1 molar ratio of TDP-43 PrLD to αS (●) and respective TDP-43 PrLD controls (○). b) ThT fluorescence of 20 μM TDP-43 PrLD (○) in presence of 0.05 to 2 molar ratio of αS to TDP-43 PrLD (●) and respective αS controls (△). The data were fit with Boltzmann’s sigmoidal function (see Materials and Methods) to derive lag time information.
In order to monitor the effect of sub-stochiometric co-incubations of the two proteins, higher concentration of αS at 50 μM was incubated in presence of 5 μM (0.1x molar equivalents) and 2 μM (0.04x molar equivalents) TDP-43 PrLD. The reverse incubations with a higher concentration of TDP-43 PrLD were prohibitively difficult due to the increased aggregation propensity of the protein that showed lag time of smaller than three hours for 50 μM TDP-43 PrLD (data not shown). The reactions containing co-incubations of 50 μM αS with sub-stoichiometric TDP-43 PrLD showed significant decreases in lag time of αS fibrillization as compared to either αS or TDP-43 PrLD controls (Figure 3a). The co-incubation with 0.1 and 0.04 molar equivalents showed lag times of 10 and 20 hours, respectively (Figure 3a) suggesting TDP-43 PrLD promotes significant aggregation of αS. After 72 hours of incubation, reaction samples were centrifuged at 18,000xg for 20 minutes to sediment fibrils formed, if any. Both the samples before centrifugation and supernatant of centrifuged samples were then subjected to immunoblot analysis using Syn211 monoclonal αS antibody (Figure 3b). The blot showed the presence of a band corresponding to > 260 kDa that failed to enter the gel in both co-incubated samples (T; Figure 3b) in addition to monomeric, dimeric, and trimeric αS present in all the samples. The corresponding supernatant samples did not contain this high molecular weight band (S; Figure 3b) confirming that the high molecular weight species are sedimentable aggregates or fibrils. To further ascertain the presence of αS fibrils in the co-incubated samples, differential interference contrast (DIC) microscopy analysis of the fibrils was employed. The pellet obtained from the samples centrifuged after 72 hours was resuspended in 20 mM MES buffer pH 6.0 and incubated with 10 μM thioflavin-S (ThS) and imaged after 10 minutes. The images revealed the presence of higher ThS positive fibrils in the reaction compared to TDP-43 PrLD and αS controls (Figure 3c). Further quantitative analysis was carried out to quantitatively assess the proteins present in the fibrils using MALDI-ToF. Relative quantitation was performed using a known amount of cytochrome C (1.42 μmol) as an external standard that was co-analyzed with the samples in the mass spectrometer (Figure S1). Analysis of the data obtained revealed that the relative amounts of αS in the pellet of reactions containing 0.1 and 0.04 molar equivalents of TDP-43 PrLD were higher compared to the individual protein controls (Figure 3d). However, αS amount in the supernatant of the control reaction was significantly higher compared to the reactions (data not shown), indicating proportional increase in αS fibrillization upon co-incubation with TDP-43 PrLD. Similar relative quantification of TDP-43 PrLD to standard in the pellets of reaction showed a higher amount of TDP-43 PrLD than the respective controls indicating enhanced fibrillization in presence of αS (Figure 3e). Since, both αS and TDP-43 PrLD were co-observed within the sedimentable pellets of the cross-seeding reactions, we performed qualitative and quantitative analyses to investigate the presence of hybrid aggregates. For qualitative analysis, 20 μM TDP-43 PrLD was incubated with 2 μM αS at 37 °C for 20 hours and probed using dot blot analysis using both αS and TDP-43 antibodies. We reasoned that since αS does not aggregate within 72 hours (Figure 3a); especially at low concentrations, co-incubation with TDP-43 PrLD will eliminate the possibility of homotypic αS fibrils in 20 hours. Any αS fibrils thus observed will have to be due to co-aggregation with TDP-43 PrLD. As expected, the dot blot analysis of the sample using αS and TDP-43 antibodies showed presence of both αS and TDP-43 PrLD confirming that αS co-aggregates with TDP-43 PrLD during fibrils formation (Figure S2a). Then quantitative analysis of the aggregates were done by examining the stoichiometric ratios of two proteins in the pellets of co-incubated sample containing equimolar (20 μM) αS and TDP-43 PrLD at 37 °C. The volume of the reaction was kept high (500 μL) to recover sufficient amount of fibrils. Fibrils were then isolated by centrifuging the sample after 7.5 hours, the time where the co-incubated reaction had aggregates faster than the individual proteins (Figure S2b inset). Quantitative MALDI-ToF analysis using cytochrome C as standard revealed the presence of both proteins in the fibrils at 1:1.2 ratio of αS to TDP-43 PrLD (Figure S2b). This equimolar distribution of the proteins in the sedimented pellet unequivocally suggests the formation of αS-TDP-43 hybrid fibrils. Taken together, the data indicate that both αS and TDP-43 PrLD monomers interact with one another and promote fibrillization synergistically.
Figure 3.

Interaction of monomeric TDP-43 PrLD and αS in 20 mM MES buffer pH 6.0. a) ThT fluorescence kinetics of 50 μM αS alone (▼) and in presence of 0.04 (■) and 0.1 (●) molar ratio of TDP-43 PrLD to αS, and respective 0.04 (♦) and 0.1 (▲) TDP-43 PrLD controls. b) Western blot of ThT reactions using αS antibodies. Aliquot of sample from the reaction at 72 hours was subjected to western blot as total sample (T), and supernatant (S) after centrifuging at 18,000 xg c) Representative fluorescence microscopic images of Thioflavin S (ThS) stained αS and TDP-43 PrLD aggregation reactions at 72 hours after centrifuging at 18,000 x g (Scale bar = 20 μm). d-e) Relative quantification of αS and TDP-43 PrLD to the Cytochrome C internal standard in pellet of reactions and control at 72 hours after centrifugation at 18,000 xg; 0.1x and 0.04x indicates molar ratio of TDP-43 PrLD to αS.
αS oligomers and sonicated fibrils seed fibrillation of TDP-43 PrLD monomers but αS monomers are innocuous to seeding by TDP-43 PrLD sonicated fibrils.
Having established that monomers of αS and TDP-43 PrLD interact and promote each other’s aggregation, we questioned if their interactions are restricted to only monomers or whether oligomers and sonicated fibrils can also cross-seed respective monomers. To answer these questions, dihydroxyphenyl acetaldehyde (DOPAL), a metabolite of dopamine biogenesis, induced oligomers of αS were used as de facto oligomers along with sonicated fibrils of both αS and TDP-43 PrLD. Isolation of stable oligomers of TDP-43 PrLD was not successful and hence were excluded from the investigation. DOPAL-derived oligomers of αS were generated and isolated by size exclusion chromatography (see Materials and Methods). The αS oligomers showed a monodisperse peak in dynamic light scattering (DLS) around 10 nm diameter (Figure 4a) with a molecular weight centered at ~ 48 kDa (~ 3mer) observed in immunoblot along with two faint bands corresponding to ~ 33 and 64 kDa species (2 and 4mers, respectively) (inset; Figure 4a). Moreover, circular dichroism (CD) spectra of the αS oligomers showed random coiled structure with characteristic minima at 195 nm (Figure 4b) while morphologically they are seen as punctate spheres of ~4-6 nm height by atomic force microscopy (AFM) (inset; Figure 4b). Incubation of these oligomers at 0.5 and 1 μM concentrations with 15 μM of monomeric TDP-43 PrLD in 20 mM MES at pH 6.0 resulted in decrease in lag time of TDP-43 PrLD aggregation to ~ 6h (Figure 4c). Immunoblot analysis of the samples after 12 h showed the formation of high molecular weight fibril bands that is absent in the control sample of TDP-43 PrLD (inset; Figure 4c), clearly indicating that αS oligomers are able to seed TDP-43 PrLD monomers. Seeding of 20 μM monomers of either αS or TDP-43 PrLD with 1, 2 or 4 μM sonicated fibrils of TDP-43 PrLD or αS, respectively showed different behavior. Homotypic seeding of αS monomers by αS sonicated fibrils showed an immediate increase in ThT intensities as expected for seeding by elongation mechanism [45] (Figure 4d). In contrast, the seeding of TDP-43 PrLD monomers by αS sonicated fibrils displayed a more sigmoidal response (Figure 4d) but with a shorter lag time of ThT fluorescence to that of the TDP-43 PrLD control. This suggests that TDP-43 PrLD monomers may undergo conformational alterations before they grow on αS fibrils. Similarly, homotypic seeding of TDP-43 PrLD monomers by TDP-43 sonicated fibrils showed immediate and substantial increases in ThT intensities (Figure 4e). But in stark contrast, seeding of αS monomers by TDP-43 PrLD sonicated fibrils failed to show interactions (Figure 4e). The secondary structure of the cross-seeded reactions from (c-e) was analyzed by FTIR (Figure 4f). The cross-seeded reaction of TDP-43 PrLD monomers with αS sonicated fibrils or DOPAL-derived αS oligomers displayed a β-sheet structure with an absorbance peak centered at 1615 cm−1 (pink and black; Figure 4f) as opposed to the monomeric samples that show predominantly random coil structure at 1643 cm−1 or 1650 cm−1 (blue and red; Figure 4f). In contrast, seeding of αS monomers with TDP-43 PrLD fibrils showed a predominant random coil structure at 1643 cm−1 (green; Figure 4f) suggesting minimal or no seeding. Finally, morphological features of the seeded aggregates were investigated by AFM. Sonicated fibrils of αS and TDP-43 PrLD showed fragmented aggregates as expected (Figures 4g and 4i). TDP-43 PrLD monomers seeded with sonicated αS fibrils after 8h of incubation showed smooth fibrils (Figure 4h). However, αS monomers seeded with TDP-43 PrLD fibrils did not show fibers after 8h of incubation (Figure 4j). These results are in agreement with kinetics data supporting the observation of the difference in the ability to seed. Together, these data suggest that while TDP-43 PrLD monomers seem to be amenable for seeding by different αS species, αS monomers are selective towards TDP-43 PrLD monomers and not sonicated fibrils.
Figure 4.

Cross-seeding of oligomers and sonicated fibrils with monomers. a) DLS analysis of DOPAL-derived αS oligomers isolated from SEC. (inset) SEC fraction containing the oligomer ‘o’ used in the study; ‘m’ refers to control monomer. b) CD spectra and AFM height image (inset) of DOPAL-derived αS oligomers used for cross-seeding reaction (scale bar = 1μm). Cross-seeding reactions with monomers of 15 μM TDP-43 PrLD alone (▲) or in the presence of 0.5 μM (■) or 1 μM (●) DOPAL-derived αS oligomers in the presence of 10 μM ThT. (inset) immunoblot of the reaction after 12 h probed with TDP-43 antibody; ‘c’ refers to TDP-43 PrLD monomer control. d) Seeding of 20 μM αS monomers with 4 μM (○), 2 μM (◇), and 1 μM (△) of αS sonicated fibrils, and 20 μM TDP-43 PrLD monomers seeded with 4 μM (●), 2 μM (♦), 1 μM (▲) of αS sonicated fibrils. e) Seeding of 20 μM TDP-43 PrLD monomers with 4 μM (○), 2 μM (◇), and 1 μM (△) of TDP-43 PrLD sonicated fibrils and seeding of 20 μM αS monomers with 4 μM (●), 2 μM (♦), 1 μM (▲) of TDP-43 PrLD seed. f) FTIR analysis of cross-seeding reactions from (c-e). TDP-43 PrLD monomers cross-seeded with 1 μM DOPAL-derived αS oligomers after 12 h of incubation (—); TDP-43 PrLD monomers seeded with 1 μM αS sonicated fibrils (—); αS monomers seeded with 1 μM TDP-43 PrLD sonicated fibrils (—) along with controls such as αS monomers (—) and TDP-43 PrLD monomers (—) (g-j) AFM height image of αS sonicated fibrils (g), αS sonicated fibrils seeded TDP-43 PrLD monomers (h), TDP-43 PrLD sonicated fibrils (i), and TDP-43 PrLD sonicated fibrils seeded αS monomers (j) (scale bar = 1 μm, inset = 200 nm).
αS modulates LLPS of TDP-43 PrLD and RNA and induces insoluble aggregates.
One of the pivotal roles of TDP-43 in pathophysiology is that under cellular stress conditions, they are known to coacervate with RNA and other proteins to undergo LLPS to form stress granules (SGs) in the cytoplasm [39, 46]. To see whether αS is able to modulate LLPS, TDP-43 PrLD was labeled with Hilyte-647 and mixed with unlabeled TDP-43 PrLD at 1% molar ratio (0.2 μM) to a final total concentration of 20 μM protein. Similarly, αS was labeled with Hilyte-405 and mixed with unlabeled protein in 1:99 ratio, and this sample was used at final concentrations of 5 and 1 μM in 20 mM MES buffer at pH 6.0 at 37 °C. As expected, TDP-43 PrLD spontaneously phase separates in presence of RNA to form liquid droplets (0h; Figure 5a). The addition of αS led to instant changes in the morphology of the droplets to become somewhat distorted, non-spherical structures in both stoichiometric incubations (0h; Figure 5a). The same reactions monitored after 24 hours showed the droplets becoming more distorted and aggregated as opposed to the control, which continued to have well-defined phase-separated liquid droplets (24 h; Figure 5a). To investigate whether the droplet deformity is due to aggregation, the dynamics of liquid droplets were probed by fluorescence recovery after photobleaching (FRAP) analysis. The control TDP-43 PrLD – RNA droplets showed significant FRAP recovery rates both at 0 h and 24 h indicating that the samples retained significant fluid characteristics (Figure 5b and c). However, the FRAP recovery rates of αS co-incubated TDP-43 PrLD droplets reduced significantly at 0 h which remained constant over the period of 24 h with a more dramatic effect with 5 μM than 1 μM αS (Figure 5b and c), which suggest gelation or aggregation samples. To unambiguously confirm if the attenuation of droplet fluidity is due to aggregation, the same reactions were monitored by ThT. The data indicated that the ThT fluorescence did not show much increase in 24 h for the TDP-43 PrLD–RNA control reaction (○; Figure 5d). In contrast, samples co-incubated with αS showed increased ThT fluorescence suggesting aggregation of TDP-43 PrLD as observed in other reactions (● and ●; Figure 5d). Taken together, these data suggest that αS modulate phase separation of TDP-43 PrLD with RNA and enhance its aggregation.
Figure 5.

Modulation of TDP-43 PrLD LLPS by αS. a) Timestamped confocal images of the co-incubations of Hilyte-647 labelled TDP-43 PrLD (20 μM) and RNA (40 μug/mL) in the absence and presence of Hilyte 405 labelled αS (5 or 1 μM) in 20 mM MES buffer pH 6.0 at 37 °C; Bf represents ‘bright field’, b) FRAP analysis on the selected droplets from the reactions before (pre-bleach), during (0 sec), and after photobleaching (45 sec), immediately after incubation (0 h) and after 24 h. c) Normalized kinetics of fluorescence recovery data obtained from FRAP intensity; TDP-43 PrLD and RNA control reaction along with 5 μM and 1 μM αS at 0h (●) and after 24 h (●). The data was fit to a first order exponential growth equation (solid lines). d) Corresponding ThT fluorescence kinetics of the reactions; TDP-43 PrLD and RNA (○) control reaction along with sub-stoichiometric, 1 μM (●) or 5 μM (●) αS incubations.
Cross-seeded heterotypic fibrils are more cytotoxic than the homotypic fibrils of αS or TDP-43 PrLD.
Recently, it was found that TDP-43 enhances the toxicity of αS in dopaminergic neurons resulting in neurodegeneration [26]. Thus, we wanted to investigate and compare the toxicities of heterotypic fibrils formed by cross-seeding and the homotypic ones formed by individual αS or TDP-43 PrLD fibrils in human neuroblastoma SH-SY5Y cells. In order to accomplish this, 20 μM αS monomers were co-incubated with 5 μM TDP-43 PrLD monomers (TDP-43 PrLD: αS = 1:4) in quiescent conditions for 24 hours at 37 °C. Similarly, 20 μM TDP-43 PrLD monomers were co-incubated with 5 μM αS monomers (TDP-43 PrLD: αS = 4:1) under identical conditions. The samples were then diluted five-folds and incubated with freshly cultured SH-SY5Y cells for additional 24 hours. Appropriate control reactions corresponding to homotypic proteins were also set up and incubated under similar conditions. Cell viability of the confluent cells was then determined by XTT assay (see Materials and Methods) [47]. The results indicate that in both stoichiometries, only the co-incubated samples showed higher toxicity than the homotypic aggregates (Figure 6). Among the two, 1:4 reaction of TDP-43 PrLD: αS monomers showed a substantially greater degree of toxicity (~40%) as compared to the 4:1 sample (~28%), suggesting that the effect of sub-stoichiometric addition of TDP-43 PrLD enhances αS aggregation and toxicity. In case of cross-seeding of fibrils to monomers, αS fibrils seeded TDP-43 PrLD aggregation showed a substantial increase in toxicity (~41%) compared to the αS fibril seeds or TDP-43 PrLD monomers (< 20%) (Figure 6). TDP-43 PrLD fibril seeded αS aggregation did not show an increase in toxicity compared to the controls. These results correlate with the kinetics and morphology of the seeding reaction in which only αS fibrils seeded TDP-43 PrLD aggregation showed aggregation (Figure 4). Similarly, the addition of αS monomers to the liquid droplets of TDP-43 PrLD and RNA that promoted insoluble fibrils also showed an increase in toxicity (~30%) as compared to TDP-43 PrLD-RNA control (< 20%). In contrast to these results, only αS oligomer (DOPAL-induced) seeded TDP-43 PrLD fibrils did not show a statistically significant increase in toxicity (Figure 6). One of the main reasons for this result is due to the fact that αS oligomer is DOPAL-induced and not a bonafide aggregation pathway intermediate; clearly, DOPAL-induced αS oligomers showed maximum toxicity (~57%; Figure 6). Therefore, it is possible that fibrils of TDP-43 PrLD seeded by these oligomers do not display any higher degree of toxicity. Together, these data suggest that the synergistic interactions between TDP-43 PrLD and αS lead to the formation of hybrid fibrils which are more toxic than those of their individual proteins.
Figure 6.
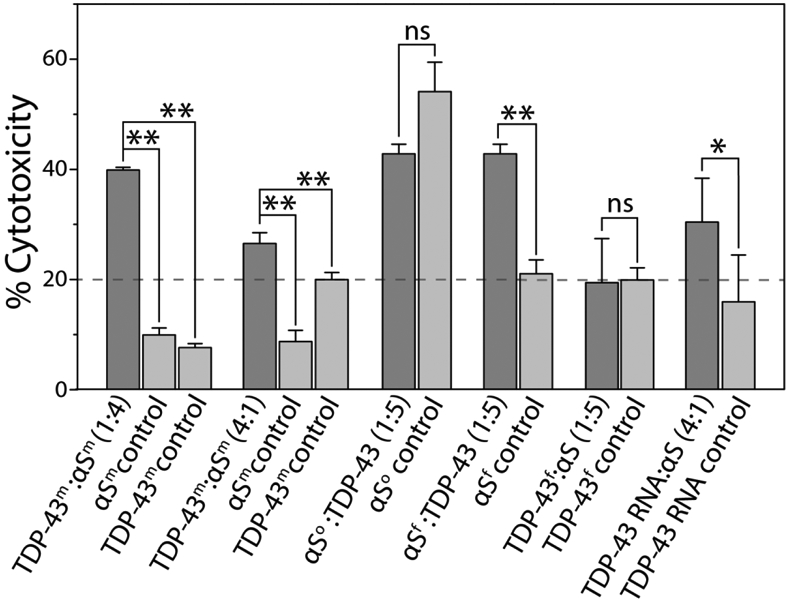
Cytotoxicity of monomeric αS and TDP-43 PrLD species along with homotypic and heterotypic fibrillar species of αS and TDP-43 in SH-SY5Y cells by XTT assay. ‘m’, ‘o’ and ‘f’ in the superscript represents monomer, oligomer, and sonicated fibril respectively. All the data were obtained in triplicates, * represents p<0.1 and **represents p<0.01 based on one-way ANOVA analysis.
DISCUSSION
The work reported here is focused on the effects of TDP-43 PrLD on αS and vice versa. We chose to focus on PrLD instead of the full-length TDP-43 for a variety of reasons; First, the post-mortem ALS and FTD brain tissues show an abundance of TDP-43 CTFs ranging between 35 and 18 kDa (35, 25, 20, and 18 kDa) [48-51], all of which constitute PrLD (17 kDa) a major part. In a more recent study on mass spectrometric analysis of post-mortem brain samples, enhanced levels TDP-43 C-terminal truncation fragment of 266-414 (corresponds to TDP-43 PrLD) was observed in ALS and AD patients [38]. Second, TDP-43 CTFs are primarily involved in the formation of cytoplasmic inclusions generated upon aberrant proteolytic cleavage of the full-length TDP-43 under pathological conditions in which PrLD plays a major role [33, 42, 52]. Third, CTFs ranging between 35 and 18 kDa are also produced by alternative splicing of TDP-43 mRNA and are linked to pathology [51, 53]. Fourth, the PrLD of TDP-43 is known to play a major role in SG formation, aggregation as well as protein-protein interactions [54-56]. Lastly, almost all pathogenic mutations lie within PrLD [42, 57] implying its significance in pathophysiology.
The results presented here demonstrate synergistic interactions between TDP-43 PrLD and αS, which in part, recapitulate the pathobiological observations. Rapid aggregation of the two protein monomers upon co-incubation (Figure 1g), and the concomitant increases in αS or TDP-43 PrLD aggregation rates (Figure 2) suggest a synergistic aggregation possibly during nucleation. In addition, seeding of TDP-43 PrLD by both oligomers and sonicated fibrils of αS also shows cross-interaction between the two proteins. These results collectively suggest potentially a hetero-nucleation mechanism predominating at the initial stages of aggregation and elongation mechanism at the later stages. The results also bring out mechanistic differences in their interactions; the data indicate that the three key αS species such as monomers, oligomers and sonicated fibrils are indiscriminate in interacting with and getting modulated by TDP-43 PrLD monomers (Figure 7). However, only monomeric TDP-43 PrLD, and not the sonicated fibrils, seem to preferentially interact with αS monomers and aggregates (Figure 7). From these results, one may conjecture that early stages of TDP-43 proteinopathies may be susceptible to modulation by the presence of αS aggregates in the form of Lewy bodies. Furthermore, the interaction of TDP-43 PrLD and αS monomers seem to be cooperative with both assisting one another in promoting high molecular weight aggregates expeditiously. We also deduce that co-incubation of monomers is likely to form αS-TDP-43 PrLD hybrid aggregates based on both qualitative and quantitative evidence (Figure S2a and S2b); a) synergistic augmentation in the rate of aggregation of both proteins, and b) the observed molar equivalents of both proteins within the sediments fibrils. However, the data also show that αS monomer, oligomer and fibrils are equally capable of interacting with TDP-43 PrLD monomers (Figure 7) and hence, the possibility of mechanisms such as nucleation-assisted aggregation and protofibril elongations are likely between the two proteins as mentioned earlier. But one also cannot discount other possible mechanisms such as heterologous secondary nucleation, fibril fragmentations and epitaxial growth, which remain to be investigated. Another interesting observation is that the effect of TDP-43 PrLD on αS is far more pronounced than vice versa. This is partly due to αS being relatively slow to aggregate as compared to TDP-43 PrLD; for example, the lag times for αS and TDP-PrLD (20 μM) aggregation at 37 °C and in identical quiescent buffer conditions are > 7 days and 8 hours, respectively (data not shown). Therefore, augmentation of αS aggregation by TDP-43 PrLD is readily comprehensible as the lag time of aggregation is reduced to 20 hours from weeks (Figure 3a). On the other hand, the effect of αS on TDP43 PrLD is discernable but subtle (Figure 2b). This is especially true with DOPAL-derived αS aggregates which augment TDP-43 PrLD aggregation significantly (Figure 4c). Previous studies have shown that the interaction of dopamine with αS results in the formation of structurally distinct oligomers [58, 59]. In particular, small angle X-ray scattering of αS trimers formed in the presence of dopamine has revealed worm-like shape due to laterally associated monomers without end-to-end arrangement [60]. Such trimers are different from those formed in the absence of dopamine exhibiting spherical, chain like or ring like annular structure. Along similar lines, we anticipate that DOPAL-derived αS oligomers will be different both structurally and functionally mainly based on the fact that DOPAL forms covalent, Schiff’s base adduct with αS. αS sonicated fibrils seeding of TDP-43 PrLD monomers show a cooperative mechanism with a delay in elongation, which could indicate conformational reorganization of TDP-43 monomers induced by αS sonicated fibrils. On the other hand, TDP-43 PrLD sonicated fibrils failed to interact with αS monomers suggesting possible incompatibility of TDP-43 PrLD sonicated fibrils structure to seed. Clues about the structural basis of interactions come from the NMR data, which shows that the TDP-43 PrLD seem to preferentially interact with αS on the N-terminal and C-terminal ends of the protein (1-40 and 100-140, respectively) based on the chemical shifts observed, while augmenting aggregation via the amyloidogenic core NAC region (61-95) (Figure 1c). This is readily comprehensible given that the N- and C-terminal ends of αS are hydrophilic and acidic, they could interact electrostatically with highly positively charged TDP-43 PrLD bringing the amyloidogenic NAC region of αS in close proximity to augment aggregation. Unfortunately, similar structural information on TDP-43 PrLD was undecipherable due to rapid aggregation of TDP-43 PrLD and consequent dilution of chemical shifts. Collectively, these data bring forth the synergistic yet selective interactions between broad categories of αS and TDP-43 PrLD species. Yet another significant aspect that highlights the interaction between the two proteins is the ability of αS to modulate LLPS of TDP-43 PrLD and RNA and promote insoluble aggregates. Since these droplets are the main constituents of cytoplasmic SGs, these results demonstrate crucial yet hitherto unseen interactions that may hold significance in neurodegenerative diseases with co-morbidities. Perhaps the most significant of all is the observation that only the hybrid aggregates show far greater cellular toxicity as opposed to the individual aggregates, which suggests that co-morbidities in these pathologies are better defined by the cross-talk between TDP-43 and αS.
Figure 7.

Schematic diagram summarizing the collective results from this work. The square parenthesis indicates theoretical transient oligomers and not used in this study. ‘*’ DOPAL-derived de facto intermediate oligomers but shown along the aggregation pathway for simplicity.
It is well-known that aggregates of αS and TDP-43 individually are observed in many neurodegenerative pathologies including AD, PD, Huntington’s disease, FTD, etc. [11, 61-63]. The term ‘synucleinopathies’ has come to define some of the pathologies in which αS aggregates play a causative or a propagative role(s). In the last decade, aggregates of TDP-43 have also been increasingly observed in as many pathologies in which αS aggregates have been observed [15] invoking a compelling argument to categorize some of these maladies as TDP-43 proteinopathies. However, more interesting is the significant overlap between pathologies in which both aggregates of TDP-43 and αS aggregates are observed. Indeed, many reports have indicated the colocalization of TDP-43 and αS aggregates [27, 64] implicating potential interactions between the two proteins to play a role in these pathologies. The data presented here unequivocally demonstrates the interactions between these two proteins which may underlie the clinical and pathological observations, and open doors to deeper investigations in establishing mechanistic links to co-morbidities in neurodegenerative diseases.
METHODS
Expression and purification of recombinant proteins
Expression and purification of both unlabeled and 15N labeled recombinant TDP-43 PrLD was performed as described previously [40]. TDP-43 PrLD fusion construct (Addgene plasmid #98669) with hexahistidine tag followed by tobacco etch virus (TEV) cleavage site at N-terminus was transformed in BL21 Star™ (DE3) cells, protein expression was induced using 0.5 mM IPTG (Life Technologies) and purified by Ni-NTA affinity chromatography. Briefly, cells were resuspended in lysis buffer (20 mM Tris, 500 mM NaCl, 5 mM imidazole, pH 8.0) and 0.5 mM PMSF was added. Cells were lysed using sonication (Misonix XL-2000) and centrifuged at 20,000 xg for one hour to remove any cellular debris. Supernatant was incubated with Ni-NTA beads for two hours at 4 °C prior to purification and loaded into the column. Impurities were removed by increasing the imidazole concentration in buffer (20 mM Tris pH 8.0, 500 mM NaCl and 6 M urea), 15 mM and 30 mM. Finally, protein was eluted using elution buffer containing 150 mM imidazole followed by buffer exchanged with storage buffer containing 20 mM Tris pH 8.0, 500 mM NaCl, 2 M urea and stored at −80 °C. Concentrated aliquots of protein in storage buffer at −80 °C was thawed on ice and desalted in 20 mM MES buffer pH 6.0 using Zeba Desalting Spin Columns (Thermo) and immediately used for the experiments.
Both unlabeled and 15N labeled recombinant full-length αS was expressed and purified in Rosetta™ 2(DE3) pLysS (EMD Millipore®) cells using IMPACT™ system protocol (New England Biolabs®) as described before [65], with few modifications. Briefly, αS detached from chitin beads using dithiothreitol (DTT) was eluted in elution buffer (20 mM Tris, 100 mM NaCl, 1 mM EDTA at pH 8.0). The eluted fraction was filtered using 30 kDa centrifugal filter units (Thermo) at 7000xg for 20 minutes. The flowthrough was dialyzed against 4L of nanopure water in a 10 kDa dialysis bag to remove DTT and concentrated using a vacufuge. Concentrated αS was directly subjected to reverse phase HPLC purification by the gradient elution of water and acetonitrile (ACN) each containing 0.1% TFA. HPLC fractions containing pure protein were lyophilized and stored at −80 °C. Lyophilized samples were resuspended in 20 mM MES pH 6.0 and subjected to size exclusion chromatography (SEC) to remove any preformed aggregates and thus obtained pure monomeric protein was used as such for studies. Due to better yields, another αS construct containing N-terminus 6-histidine tag and a thrombin cleavage site was also used occasionally in this study. The protein was expressed in BL21(DE3) cells, purified using Ni-NTA affinity chromatography and concentrated using 10 kDa centrifugal filter unit. Concentrated protein was further subjected to SEC and the fractions corresponding to the monomeric protein were used in the experiments. Unused monomer fractions were stored at 4 °C and were used within a week from purification. We confirmed that αS purified using both constructs have similar biophysical characteristics and aggregation kinetics.
Preparation of αS oligomers, αS fibrils and TDP-43 PrLD fibrils
αS oligomers and fibrils were generated similar to previous protocols [66, 67]. Briefly, αS oligomers have been generated by incubating 50 μM αS monomer with 20-fold molar excess of 3,4-Dihydroxyphenylacetaldehyde (DOPAL) at 600 rpm at 37 °C for 24 hours. Oligomers of αS were isolated using superdex-200 size exclusion chromatography (SEC) column in 10 mM Tris pH 8.0. Fibrils of αS were prepared by incubating 5 mg of monomeric protein in presence of 150 mM NaCl at 600 rpm at 37 °C for 7 days. Similarly, TDP-43 PrLD fibrils were generated by incubating 50 μM monomeric TDP-43 in quiescent condition at 37 °C for 7 days. Both fibrils were isolated by centrifuging at 20,000 xg for 20 minutes and stored at −80 °C until use.
Thioflavin-T (ThT) fluorescence
Aggregation kinetics were monitored using BioTek Synergy H1 microplate reader. Samples containing 10 μM ThT were excited at 452 nm and emission was monitored at 485 nm at 37 °C. The data points were plotted as ThT fluorescence versus time and fitted with following Boltzmann sigmoidal function using Origin 8.5.
In this equation, y corresponds to ThT fluorescence intensity, x is time and xo is the time to reach half-maximal ThT fluorescence and A1 and A2 are constants. The lag time of aggregation was calculated as xo - 2dx for each fitted curve.
SDS-PAGE and immnoblotting
Aliquots of the reactions were subjected to SDS-PAGE and immunoblotting using a monoclonal anti-αS antibody, clone Syn211 (Millipore Sigma). Aliquots of the samples were separately mixed in the 4x Laemmli sample buffer and loaded onto SDS-PAGE Biorad Mini-PROTEAN® 4-20% precast gel. Gels were then transferred on to a 0.45 μM Amersham Protran Premium nitrocellulose membrane (GE Life Sciences) and the blot was boiled in 1X PBS for one minute. For dot blot analysis, pellet obtained from αS and TDP-43 co-incubation reaction was treated with formic acid, resuspended in 20 mM MES buffer pH 6.0 and was directly spotted in the nitrocellulose membrane. Blot was then incubated overnight in the blocking buffer (5% non-fat milk, 0.1% Tween®−20 in 1X PBS), followed by primary antibodies against αS or TDP-43 PrLD and horseradish peroxidase-conjugated anti-mouse/anti-rabbit secondary antibodies. Finally, images were obtained by treating with ECL reagent using GelDoc molecular imager (Bio-Rad).
MALDI-ToF mass spectrometry
Quantification of αS and TDP-43 PrLD fibrils was performed on a Bruker Datonics Microflex LT/SH MALDI-ToF system. Aliquots of samples from aggregation reactions after 48 hours were centrifuged at 18,000xg for 20 minutes. The supernatant was discarded and the pellet was washed and resuspended in an equal volume of 20 mM MES buffer pH 6.0. Resuspended pellets were then mixed with an equal volume of formic acid to disaggregate the fibrils. The samples were then mixed with Cytochrome C external standard at 1.42 μM final concentration in 1:1 sinnapinic acid matrix and loaded on to an MSP 96 BC MALDI plate (Bruker Datonics). The instrument was calibrated using Bruker Protein Calibration Standard I (Bruker Datonics) and spectra were collected by adjusting the laser intensity at 70%.
Circular dichroism
Circular dichroism (CD) spectra of 4.25 μM DOPAL-derived αS oligomers in 10 μM Tris buffer pH 8.0 was measured in far UV region (190 to 260 nm) in Jasco J-815 spectrophotometer (Jasco MD) as previously [68].
Fluorescence Microscopy
Fluorescence microscopic images of the reaction with labeled proteins were obtained using Leica SP8 confocal microscope at 40x magnification in a clear glass bottom 96 well black plates (P96-1.5H-N, Cellvis Inc.). Briefly, protein labeling was carried out by incubating three molar excess of fluorescent dyes, Hilyte 405 or Hilyte 647 (AnaSpec Inc) with the proteins for 12 hours at 4°C. Excess dye was removed using PD SpinTrap™ G-25 (Cytiva Life sciences) columns and the labeled and unlabeled protein samples were mixed in 1:99 ratio and used for the experiments. We do not expect the Hilyte dyes to have any discernable effects on our observations as they are comparable to routinely used xanthane dyes. Moreover, to minimize their effects if any, we used only 1% of total protein with labeled ones in our assays. For thioflavin-S (ThS) staining, aliquots of samples from the reactions were centrifuged at 18,000xg for 20 minutes, and pellets were resuspended in 20 mM MES buffer pH 6.0. ThS was added at a final concentration of 10 μM and incubated for 10 minutes prior to imaging.
Fluorescence recovery after photobleaching (FRAP)
Hilyte 647 labeled TDP-43 PrLD and RNA reaction with or without Hilyte 405 labeled αS was monitored using FRAP. The samples were photobleached using 100% laser intensity for 10 seconds with 141 iterations and recovery was monitored for 45 seconds. The FRAP kinetic data was plotted as normalized fluorescence with reference to TDP-43 PrLD and RNA control and was fit using Boltzmann function on Origin 8.5.
NMR spectroscopy
The NMR experiments were carried out using 15N labeled αS and TDP-43 PrLD in 20 mM MES, pH 6.0 with 10% D2O after incubation with and without unlabeled TDP-43 PrLD and αS proteins. For these experiments, N-terminus histidine-tag was removed from TDP-43 PrLD using TEV protease [55]. SOFAST heteronuclear multiple quantum coherence (HMQC) [69] spectra were obtained on the samples at 10 and 37 °C. The data were acquired on a Bruker Advance- III-HD 850 MHz NMR spectrometer equipped with a Bruker TCI cryoprobe at the high-field NMR facility of the University of Alabama, Birmingham as described previously [70, 71]. Briefly, SOFAST HMQC was collected as it allows reduction of the recycle delays to 100 ms while maintaining high sensitivity, and decreasing the overall acquisition time [69]. The protein concentrations were kept at 10 μM for both proteins with 1:1 molar equivalents of unlabeled proteins added for interaction studies. The spectra were then collected with 2048 data points in F2 (1H) with 128 scans coadded for each of the 160 F1 (15N) increments. A 1J(NH) of 90 Hz was used with a 100 ms relaxation delay. The delay interval from F1 to F2 was set at 0.32 ms. The raw HMQC spectra were processed using Bruker TopSpin 3.5 analysis software with standard methods with phase corrections in both dimensions. Cross-peaks of both αS and TDP-43 PrLD were identified as in previous studies [47, 55, 72].
Atomic force microscopy
AFM images were obtained following a previously published method [73]. Briefly, mica was cleaved using tape then attached to a magnet. The mica was then treated with 150 μL of 3-aminopropyltriethoxysilane (APTES) solution (500 μL of 3-aminopropyltriethoxysilane in 50 mL of 1 mM acetic acid) for 30 minutes. The APTES solution was then decanted off and the mica substrate was rinsed three times with nanopure H2O, dried with N2, and stored for an hour. The 1-4 μM reaction samples were diluted to 100-folds and a volume of 150 μL of the sample solution was deposited onto the mica surface and were allowed to absorb for 30 minutes. The sample solution was then decanted from the mica surface and washed with 150 μL of nanopure H2O three times, dried with N2, and stored in a desiccator until imaging. AFM analysis was performed using a Dimension Icon atomic force microscope (Bruker) in PeakForce Tapping mode. AFM scanning was performed using NanoScope 8.15r3sr5 software and the images were analyzed in NanoScope Analysis 1.50 software. Imaging was performed using a sharp silicon nitride cantilever (SNL-C, nominal tip radius of 2 nm; nominal resonance frequency of 56 kHz; nominal spring constant of 0.24 N/m) and a standard probe holder under ambient conditions with 512 × 512 data point resolution. Multiple areas of the mica surface were analyzed, height and phase images were obtained simultaneously, and representative images are reported.
Cell viability XTT assay
Cell viability was measured using 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]- 2H- tetrazolium hydroxide (XTT) assay kit (Biotium) following the previously established protocol with few modifications [74]. Briefly, human neuroblastoma SH-SY5Y cells (ATCC, Manassas, VA) were maintained in a humidified incubator at 37 °C with 5.5% CO2 in 1:1 mixture of DMEM and Ham’s F12K medium with 10% FBS and 1% penicillin/streptomycin. Approximately, 15,000 cells were plated in a clear bottom 96 well black plates (Thermo Scientific) 24 hours prior to sample treatment. All the reaction samples were prepared and purified using autoclaved water and buffer to avoid bacterial contamination. Cell medium from wells was replaced with the reaction samples resuspended in complete growth medium and incubated for 24 hours prior to XTT assay.
Supplementary Material
Highlights.
α-Synuclein and prion-like domain of TDP-43 monomers synergistically promote hybrid fibrils.
Monomers and aggregates of α-Synuclein seed TDP-43 monomers.
TDP-43 fibrils fail to seed α-Synuclein monomers.
Cross-seeded hybrid aggregates are more cytotoxic than homotypic fibrils.
ACKNOWLEDGEMENTS
The authors would like to thank the following agencies for financial support: National Institute of Aging (1R56AG062292-01) and the National Science Foundation (NSF CBET 1802793) to VR. The authors also thank the National Center for Research Resources (5P20RR01647-11) and the National Institute of General Medical Sciences (8 P20 GM103476-11) from the National Institutes of Health for funding through INBRE for the use of their core facilities.
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Soto C & Estrada LD (2008). Protein misfolding and neurodegeneration. Arch. Neurol 65, 184–189. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura T & Lipton SA (2009). Cell death: protein misfolding and neurodegenerative diseases. Apoptosis. 14, 455–468. [DOI] [PubMed] [Google Scholar]
- 3.Sengupta U, Guerrero-Muñoz MJ, Castillo-Carranza DL, Lasagna-Reeves CA, Gerson JE, Paulucci-Holthauzen AA, Krishnamurthy S, Farhed M, Jackson GR & Kayed R (2015). Pathological interface between oligomeric alpha-synuclein and tau in synucleinopathies. Biol. psychiatry 78, 672–683. [DOI] [PubMed] [Google Scholar]
- 4.Galvin JE, Lee VM-Y & Trojanowski JQ (2001). Synucleinopathies: clinical and pathological implications. Arch. Neurol 58, 186–190. [DOI] [PubMed] [Google Scholar]
- 5.Saha P & Sen N (2019). Tauopathy: A common mechanism for neurodegeneration and brain aging. Mech. Ageing Dev 178, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spires-Jones TL & Hyman BT (2014). The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron. 82, 756–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J, Mattison HA, Liu C, Ginghina C, Auinger P, McDermott MP, Stewart T, Kang UJ, Cain KC & Shi M (2013). Longitudinal assessment of tau and amyloid beta in cerebrospinal fluid of Parkinson disease. Acta Neuropathol. 126, 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irwin DJ, Lee VM-Y & Trojanowski JQ (2013). Parkinson's disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci 14, 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vital A, Canron MH, Gil R, Hauw JJ & Vital C (2007). A sporadic case of Creutzfeldt–Jakob disease with beta-amyloid deposits and alpha-synuclein inclusions. Neuropathology. 27, 273–277. [DOI] [PubMed] [Google Scholar]
- 10.Kovacs GG, Rahimi J, Ströbel T, Lutz MI, Regelsberger G, Streichenberger N, Perret-Liaudet A, Höftberger R, Liberski PP & Budka H (2017). Tau pathology in Creutzfeldt-Jakob disease revisited. Brain pathol. 27, 332–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamilton RL (2000). Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using α-synuclein Immunohistochemistry. Brain pathol. 10, 378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burré J (2015). The synaptic function of α-synuclein. J Parkinsons Dis. 5, 699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim WS, Kågedal K & Halliday GM (2014). Alpha-synuclein biology in Lewy body diseases. Alzheimer's Res. Ther 6, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geser F, Lee VMY & Trojanowski JQ (2010). Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: a spectrum of TDP-43 proteinopathies. Neuropathology. 30, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, Arnold SE, Siderowf A, Grossman M & Leverenz JB (2007). Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 114, 221–229. [DOI] [PubMed] [Google Scholar]
- 16.Crews L, Tsigelny I, Hashimoto M & Masliah E (2009). Role of synucleins in Alzheimer's disease. Neurotox. Res 16, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aoki S, Liu AW, Zucca A, Zucca S & Wickens JR (2015). Role of striatal cholinergic interneurons in set-shifting in the rat. J. Neurosci 35, 9424–9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moussaud S, Jones DR, Moussaud-Lamodière EL, Delenclos M, Ross OA & McLean PJ (2014). Alpha-synuclein and tau: teammates in neurodegeneration? Mol. Neurodegener 9, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nonaka T, Masuda-Suzukake M & Hasegawa M (2018). Molecular mechanisms of the co-deposition of multiple pathological proteins in neurodegenerative diseases. Neuropathology. 38, 64–71. [DOI] [PubMed] [Google Scholar]
- 20.Morales R, Estrada LD, Diaz-Espinoza R, Morales-Scheihing D, Jara MC, Castilla J & Soto C (2010). Molecular cross talk between misfolded proteins in animal models of Alzheimer's and prion diseases. J. Neurosci 30, 4528–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K, Togo T, Katsuse O, Uchikado H & Furukawa Y (2007). Concurrence of TDP-43, tau and α-synuclein pathology in brains of Alzheimer's disease and dementia with Lewy bodies. Brain research. 1184, 284–294. [DOI] [PubMed] [Google Scholar]
- 22.Schaser AJ, Osterberg VR, Dent SE, Stackhouse TL, Wakeham CM, Boutros SW, Weston LJ, Owen N, Weissman TA & Luna E (2019). Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep 9, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson RS, Yu L, Trojanowski JQ, Chen E-Y, Boyle PA, Bennett DA & Schneider JA (2013). TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol. 70, 1418–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu WT, Josephs KA, Knopman DS, Boeve BF, Dickson DW, Petersen RC & Parisi JE (2008). Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol. 116, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emamzadeh FN (2016). Alpha-synuclein structure, functions, and interactions. J. Res. Med. Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian T, Huang C, Tong J, Yang M, Zhou H & Xia X-G (2011). TDP-43 potentiates alpha-synuclein toxicity to dopaminergic neurons in transgenic mice. Int J Biol Sci. 7, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen L, Wang C, Chen L, Leung KL, Lo E, Lakso M & Wong G (2020). TDP-1/TDP-43 potentiates human α-Synuclein (HASN) neurodegeneration in Caenorhabditis elegans. Biochim Biophys Acta Mol Basis Dis. 165876. [DOI] [PubMed] [Google Scholar]
- 28.Gallardo J, Escalona-Noguero C & Sot B (2020). Role of α-Synuclein Regions in Nucleation and Elongation of Amyloid Fiber Assembly. ACS Chem. Neurosci 11, 872–879. [DOI] [PubMed] [Google Scholar]
- 29.Stefanis L (2012). α-Synuclein in Parkinson's disease. Cold Spring Harb. Perspect. Med 2, a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.François-Moutal L, Perez-Miller S, Scott DD, Miranda VG, Mollasalehi N & Khanna M (2019). Structural Insights Into TDP-43 and Effects of Post-translational Modifications. Front. Mol. Neurosci 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang G, Yang H, Yan S, Wang C-E, Liu X, Zhao B, Ouyang Z, Yin P, Liu Z & Zhao Y (2015). Cytoplasmic mislocalization of RNA splicing factors and aberrant neuronal gene splicing in TDP-43 transgenic pig brain. Mol. Neurodegener 10, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buratti E & Baralle FE (2008). Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 13, 8. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y-J, Xu Y-F, Cook C, Gendron TF, Roettges P, Link CD, Lin W-L, Tong J, Castanedes-Casey M &Ash P (2009). Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci 106, 7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furukawa Y, Kaneko K & Nukina N (2011). Molecular properties of TAR DNA binding protein-43 fragments are dependent upon its cleavage site. Biochim Biophys Acta Mol Basis Dis. 1812, 1577–1583. [DOI] [PubMed] [Google Scholar]
- 35.Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y, Forman MS, Troost D, Kretzschmar HA & Trojanowski JQ (2009). Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 117, 137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nonaka T, Kametani F, Arai T, Akiyama H & Hasegawa M (2009). Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet 18, 3353–3364. [DOI] [PubMed] [Google Scholar]
- 37.Berning BA & Walker AK (2019). The pathobiology of TDP-43 C-terminal fragments in ALS and FTLD. Front. Neurosci 13, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feneberg E, Charles P, Finelli M, Scott C, Kessler B, Fischer R, Ansorge O, Gray E, Talbot K & Turner M (2020). Detection and quantification of novel C-terminal TDP-43 fragments in ALS-TDP. Brain Pathology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E, Silani V & Ratti A (2009). TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem 111, 1051–1061. [DOI] [PubMed] [Google Scholar]
- 40.Bhopatkar AA, Uversky VN & Rangachari V (2020). Granulins modulate liquid-liquid phase separation and aggregation of prion-like C-terminal domain of the neurodegeneration-associated protein TDP-43. J. Biol. Chem jbc. RA119. 011501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mompean M, Chakrabartty A, Buratti E & Laurents DV (2016). Electrostatic repulsion governs TDP-43 C-terminal domain aggregation. PLoS biol. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berning BA & Walker AK (2019). The pathobiology of TDP-43 C-terminal fragments in ALS and FTLD. Front. Neurosci 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miklos AC, Sumpter M & Zhou H-X (2013). Competitive interactions of ligands and macromolecular crowders with maltose binding protein. PLoS One. 8, e74969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diniz A, Dias JS, Jiménez-Barbero J, Marcelo F & Cabrita EJ (2017). Protein–glycan quinary interactions in crowding environment unveiled by NMR spectroscopy. Chem. Eur. J 23, 13213–13220. [DOI] [PubMed] [Google Scholar]
- 45.Ghosh P, Kumar A, Datta B & Rangachari V (2010). Dynamics of protofibril elongation and association involved in Aβ42 peptide aggregation in Alzheimer’s disease. BMC bioinformatics. 11, S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walker AK, Soo KY, Sundaramoorthy V, Parakh S, Ma Y, Farg MA, Wallace RH, Crouch PJ, Turner BJ & Horne MK (2013). ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PloS one. 8, e81170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dasari AK, Kayed R, Wi S & Lim KH (2019). Tau interacts with the C-terminal region of α-synuclein, promoting formation of toxic aggregates with distinct molecular conformations. Biochemistry. 58, 2814–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL & Grossman M (2008). Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol 173, 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Porta S, Xu Y, Restrepo CR, Kwong LK, Zhang B, Brown HJ, Lee EB, Trojanowski JQ & Lee VM-Y (2018). Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun 9, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feneberg E, Gray E, Ansorge O, Talbot K & Turner MR (2018). Towards a TDP-43-based biomarker for ALS and FTLD. Mol. Neurobiol 55, 7789–7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsuji H, Nonaka T, Yamashita M, Masuda-Suzukake M, Kametani F, Akiyama H, Mann DM, Tamaoka A & Hasegawa M (2012). Epitope mapping of antibodies against TDP-43 and detection of protease-resistant fragments of pathological TDP-43 in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Biochem. Biophys. Res. Commun 417, 116–121. [DOI] [PubMed] [Google Scholar]
- 52.Yang C, Tan W, Whittle C, Qiu L, Cao L, Akbarian S &Xu Z (2010). The C-terminal TDP-43 fragments have a high aggregation propensity and harm neurons by a dominant-negative mechanism. PloS one. 5, e15878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiao S, Sanelli T, Chiang H, Sun Y, Chakrabartty A, Keith J, Rogaeva E, Zinman L & Robertson J (2015). Low molecular weight species of TDP-43 generated by abnormal splicing form inclusions in amyotrophic lateral sclerosis and result in motor neuron death. Acta Neuropathol. 130, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, Rouleau GA &Vande Velde C (2011). TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet 20, 1400–1410. [DOI] [PubMed] [Google Scholar]
- 55.Conicella AE, Zerze GH, Mittal J & Fawzi NL (2016). ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure. 24, 1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T & Taylor JP (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 163, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meyerowitz J, Parker SJ, Vella LJ, Ng DC, Price KA, Liddell JR, Caragounis A, Li Q-X, Masters CL & Nonaka T (2011). C-Jun N-terminal kinase controls TDP-43 accumulation in stress granules induced by oxidative stress. Mol. Neurodegener 6, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cappai R, Leck SL, Tew DJ, Williamson NA, Smith DP, Galatis D, Sharpies RA, Curtain CC, Ali FE & Cherny RA (2005). Dopamine promotes α-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J. 19, 1377–1379. [DOI] [PubMed] [Google Scholar]
- 59.Sengupta U, Puangmalai N, Bhatt N, Garcia S, Zhao Y & Kayed R (2020). Polymorphic α-synuclein strains modified by dopamine and docosahexaenoic acid interact differentially with tau protein. Mol. Neurobiol 57, 2741–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rekas A, Knott RB, Sokolova A, Barnham KJ, Perez KA, Masters CL, Drew SC, Cappai R, Curtain CC & Pham CL (2010). The structure of dopamine induced α-synuclein oligomers. Eur. Biophys. J 39, 1407–1419. [DOI] [PubMed] [Google Scholar]
- 61.Gao J, Wang L, Huntley ML, Perry G & Wang X (2018). Pathomechanisms of TDP-43 in neurodegeneration. J. Neurochem 146, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jo M, Lee S, Jeon Y-M, Kim S, Kwon Y & Kim H-J (2020). The role of TDP-43 propagation in neurodegenerative diseases: integrating insights from clinical and experimental studies. Exp. Mol. Med 52, 1652–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Visanji NP, Lang AE & Kovacs GG (2019). Beyond the synucleinopathies: alpha synuclein as a driving force in neurodegenerative comorbidities. Transl. Neurodegener 8, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koga S, Lin WL, Walton RL, Ross OA & Dickson DW (2018). TDP-43 pathology in multiple system atrophy: colocalization of TDP-43 and α-synuclein in glial cytoplasmic inclusions. Neuropathol Appl Neurobiol. 44, 707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Planchard MS, Exley SE, Morgan SE & Rangachari V (2014). Dopamine-induced α-synuclein oligomers show self-and cross-propagation properties. Protein Sci. 23, 1369–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Plotegher N, Berti G, Ferrari E, Tessari I, Zanetti M, Lunelli L, Greggio E, Bisaglia M, Veronesi M & Girotto S (2017). DOPAL derived alpha-synuclein oligomers impair synaptic vesicles physiological function. Sci. Rep 7, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Strohäker T, Jung BC, Liou S-H, Fernandez CO, Riedel D, Becker S, Halliday GM, Bennati M, Kim WS & Lee S-J (2019). Structural heterogeneity of α-synuclein fibrils amplified from patient brain extracts. Nat. Commun 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dhakal S, Sapkota K, Huang F & Rangachari V (2020). Cloning, expression and purification of the low-complexity region of RanBP9 protein. Protein Expression and Purification. 105630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schanda P (2005). Kupƒçe ƒ, Brutscher B. SOFAST-HMQC Experiments for Recording Two-dimensional Deteronuclear Correlation Spectra of Proteins within a Few Seconds. J. Biomol. NMR 33, 199–211. [DOI] [PubMed] [Google Scholar]
- 70.Bhopatkar AA, Uversky VN & Rangachari V (2020). Granulins modulate liquid–liquid phase separation and aggregation of the prion-like C-terminal domain of the neurodegeneration-associated protein TDP-43. J. Biol. Chem 295, 2506–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ghag G, Holler CJ, Taylor G, Kukar TL, Uversky VN & Rangachari V (2017). Disulfide bonds and disorder in granulin-3: An unusual handshake between structural stability and plasticity. Protein Science. 26, 1759–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu K-P & Baum J (2011). Backbone assignment and dynamics of human α-synuclein in viscous 2 M glucose solution. Biomol. NMR Assign 5, 43–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dean DN, Das PK, Rana P, Burg F, Levites Y, Morgan SE, Ghosh P & Rangachari V (2017). Strain-specific fibril propagation by an Aβ dodecamer. Scientific reports. 7, 40787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Saha J, Dean DN, Dhakal S, Stockmal KA, Morgan SE, Dillon KD, Adamo MF, Levites Y & Rangachari V (2021). Biophysical characteristics of lipid-induced Aβ oligomers correlate to distinctive phenotypes in transgenic mice. FASEB J. 35, e21318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.