

Abstract
Precise cell targeting is challenging because most mammalian cell types lack a single surface marker that distinguishes them from other cells. A solution would be to target cells based on specific combinations of proteins present on their surfaces. We design colocalization-dependent protein switches (Co-LOCKR) that perform ‘AND’, ‘OR’, and ‘NOT’ Boolean logic operations. These switches activate through a conformational change only when all conditions are met, generating rapid, transcription-independent responses at single-cell resolution within complex cell populations. We implement ‘AND’ gates to redirect T cell specificity against tumor cells expressing two surface antigens while avoiding off-target recognition of single-antigen cells, and 3-input switches that add ‘NOT’ or ‘OR’ logic to avoid or include cells expressing a third antigen. Thus, de novo designed proteins can perform computations on the surface of cells, integrating multiple distinct binding interactions into a single output.
One Sentence Summary:
De novo proteins compute logic on the cell surface by transforming multiple binding events into a single biological output.
Biological systems are complex; therefore, interventions that perturb these systems must achieve specific targeting in mixed populations of closely related cells. Cells displaying a unique surface marker can be targeted with antibodies, but a single marker is rarely sufficient to identify specific cell types. Bispecific antibodies can achieve some selectivity by simultaneously engaging two targets (1, 2), but this strategy requires delicate tuning of the individual binding affinities to reduce interactions with cells expressing just one of the targets. A generalized approach for distinguishing cells based on combinations of surface markers is needed. Towards this end, we sought to develop a modular protein system capable of taking multiple binding events as input, computing combinations of Boolean logic operations (‘AND’, ‘OR’, and ‘NOT’) without requiring cellular machinery for signal integration, and producing a single output (Fig 1a).
Figure 1. A de novo designed protein switch performs AND logic on the cell surface.
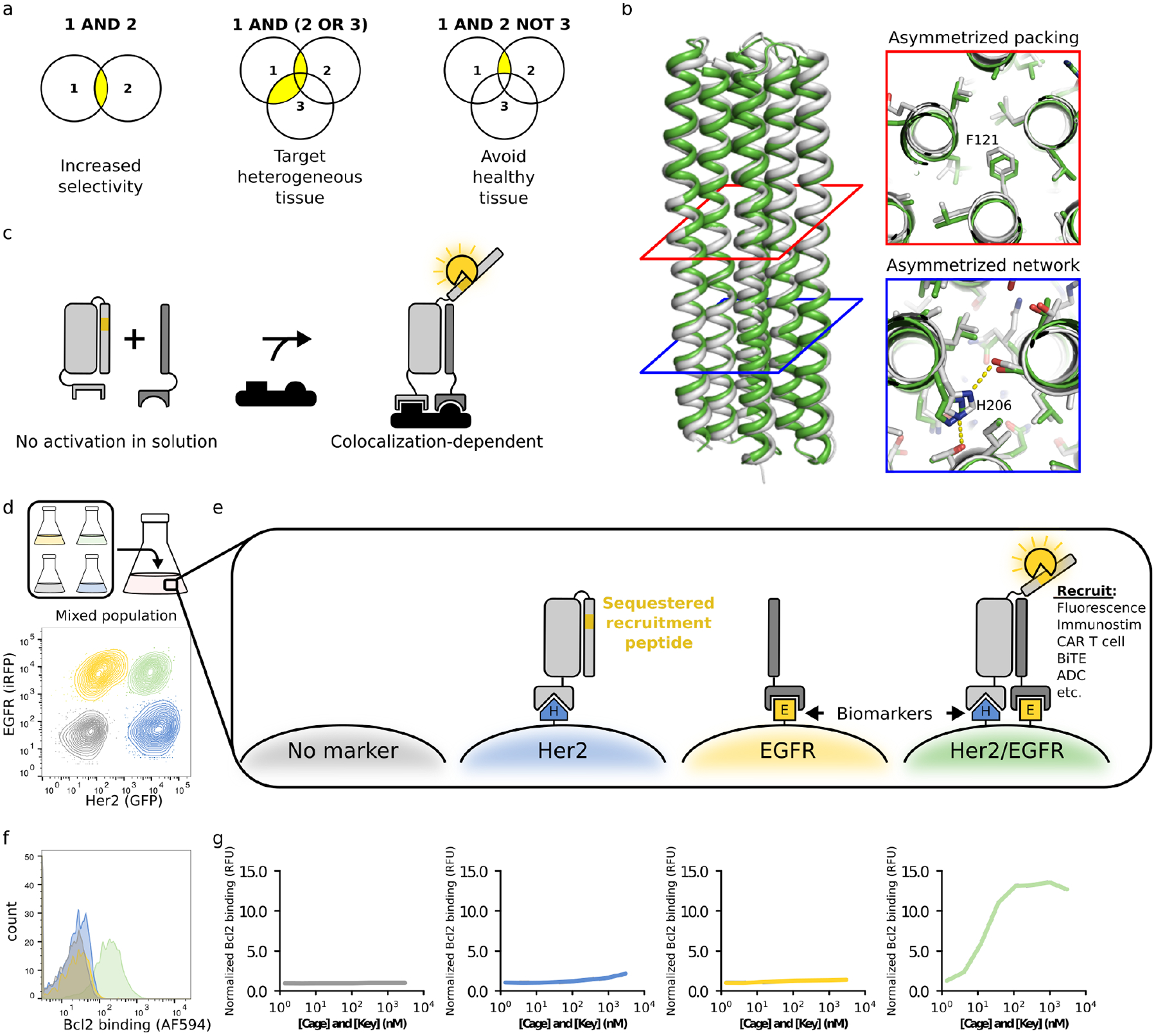
a. The ability to compute logic operations on the surface of cells could increase targeting selectivity, provide flexibility for heterogeneous tissue, and avoid healthy tissue. b. Structure of Cage design used to create Co-LOCKR; the x-ray crystal structure (white, PDB ID: 7JH5) matches the computational design model (green) with 1.1 Å RMSD across all backbone atoms. Cross-sections illustrate asymmetric packing of hydrophobic residues (red square) and an asymmetric hydrogen bond network (blue square). c. Colocalization-dependent protein switches are tuned so that Cage and Key do not interact in solution but strongly interact when colocalized on a surface via targeting domains. d. Flow cytometry discriminates Her2+/EGFR+ cells in a mixed population of K562 cells expressing Her2-eGFP, EGFR-iRFP, both, or neither. e. An Effector protein is recruited only when Cage and Key are colocalized on the surface of the same cell (‘AND’ logic). f. The mixed population of cells from Fig 1d was incubated with 111 nM Her2-targeted Cage, 111 nM EGFR-targeted Key, and 50 nM Bcl2-AF594. Bcl2 binding was only observed for the K562/Her2/EGFR cells. g. The mixed population of cells from Fig 1d was incubated with a dilution series of Her2-targeted Cage and EGFR-targeted Key, washed, and then incubated with 50 nM Bcl2-AF594 Bcl2 binding is reported relative to K562 cells incubated with 3000 nM Her2-targeted Cage, 3000 nM EGFR-targeted Key, and 50 nM Bcl2-AF594.
How does one design a system that activates only on the surface of a cell and not in solution? Given that antigen binding at the cell surface increases the local concentration of the bound protein, such a system potentially could be constructed from an actuator that responds to proximity. The actuation must be modular and independent of target antigen identity to be generally useful. We began from de novo designed protein switches that activate in solution: Latching Orthogonal Cage–Key pRotein (LOCKR) (3) switches consist of a structural “Cage” protein that uses a “Latch” domain to sequester a functional peptide in an inactive conformation until binding of a separate “Key” protein induces a conformational change that permits binding to an “Effector” protein. Cage, Key, and Effector bind in a three-way equilibrium, and the sensitivity of the switch can be tuned by adjusting the relative Cage–Latch and Cage–Key affinities. Previous LOCKR switches functioned in the yeast cytoplasm (3), but were aggregation-prone once purified, likely due to domain swapping of interchangeable symmetric repeats present in the parental homotrimer of a-helical hairpins (4). To alleviate aggregation, we used Rosetta (5) to design a new LOCKR switch with shorter helices, improved hydrophobic packing, and an additional hydrogen bond network to promote interaction specificity among the helices (Fig S1a–d, Computational Protein Design portion of Methods). The new design was nearly 100% monomeric (Fig S2, top), and a 2.1 Å x-ray crystal structure (PDB ID: 7JH5) closely matched the design model (Fig 1b, Table S1) with 1.1 Å root mean squared deviation (RMSD) across all backbone atoms and 0.5 Å RMSD across all sidechain heavy atoms in the newly designed hydrogen bond network (Fig 1b).
To install an output function into Co-LOCKR, we chose the Bim-Bcl2 pair as a model system (6). Bim was encoded into the Latch as a sequestered peptide; Bcl2 was used as the Effector. We added targeting domains that recruit the Co-LOCKR Cage and Key to cells expressing target antigens. While the targeting domains should bind to any cell expressing their target antigens, only cells with both antigens should colocalize Cage and Key (Fig 1c–f). Because Co-LOCKR is thermodynamically controlled, complex formation occurs at much lower concentrations when the components are co-localized on a surface than when they are free in solution (Fig S3a–b); colocalization shifts the binding equilibrium in favor of complex formation (Fig S3c).
To evaluate the ability of Co-LOCKR to target cells co-expressing a precise combination of surface antigens, we developed a mixed population flow cytometry assay by combining four K562 cell lines expressing Her2-eGFP, EGFR-iRFP, both, or neither (Fig 1d). We used Designed Ankyrin Repeat Protein (DARPin) domains (7, 8) to target the Cage and Key to Her2 and EGFR, respectively. If the system functions as designed, only cells co-expressing both Her2 and EGFR should activate Co-LOCKR and bind Bcl2: the Cage contains the sequestered Bim peptide and the Key is required for its exposure. We refer to this Co-LOCKR configuration as CL_CHKE; in this nomenclature “CL” refers to Co-LOCKR, CH indicates that the Cage is targeted to Her2, and KE indicates that the Key is targeted to EGFR (Table S2). When the mixed population of cells was co-incubated with an equimolar dilution series of Cage and Key (3 μM to 1.4 nM) and washed before adding AlexaFluor594-labeled Bcl2 (Bcl2-AF594), the expected sigmoidal binding curve was observed for the Her2/EGFR cells but not for cells expressing either antigen alone (Fig 1g).
We next sought to tune the dynamic range of Co-LOCKR activation to increase colocalization-dependent activation sensitivity and responsiveness. The sensitivity of previous LOCKR switches was tuned by shortening the Latch to produce a ‘toehold’ which allows the key to outcompete the latch (3), but this also promoted aggregation (Fig S2, bottom). We therefore focused on designing mutations rather than toeholds to tune the relative interaction affinities of the Co-LOCKR system to be colocalization-dependent (Fig S4a–c). We mutated large, hydrophobic residues in the Latch (I287A, I287S, I269S) or Cage (L209A) to weaken Cage–Latch affinity (Fig 2a). Biolayer interferometry indicated that increasingly disruptive mutations improved responsiveness (Fig S5a–b), and flow cytometry showed that tuning the Cage–Latch interface enhanced colocalization-dependent signal without compromising specificity (Fig 2b, Fig S5c). Colocalization-dependent activation occurred even at low nanomolar concentrations of CL_CHKE (Fig S5d–e). Very little Effector binding was observed for cells expressing Her2 or EGFR alone, confirming that Co-LOCKR has single-cell targeting resolution in a mixed population. Of the switches tested, I269S exhibited the greatest activation (Fig S6a), the parental Co-LOCKR design exhibited the lowest off-target activation (Fig S6b), and I287A exhibited the highest specificity (on-target signal divided by max off-target signal, Fig S6c).
Figure 2. Tuning Co-LOCKR sensitivity.
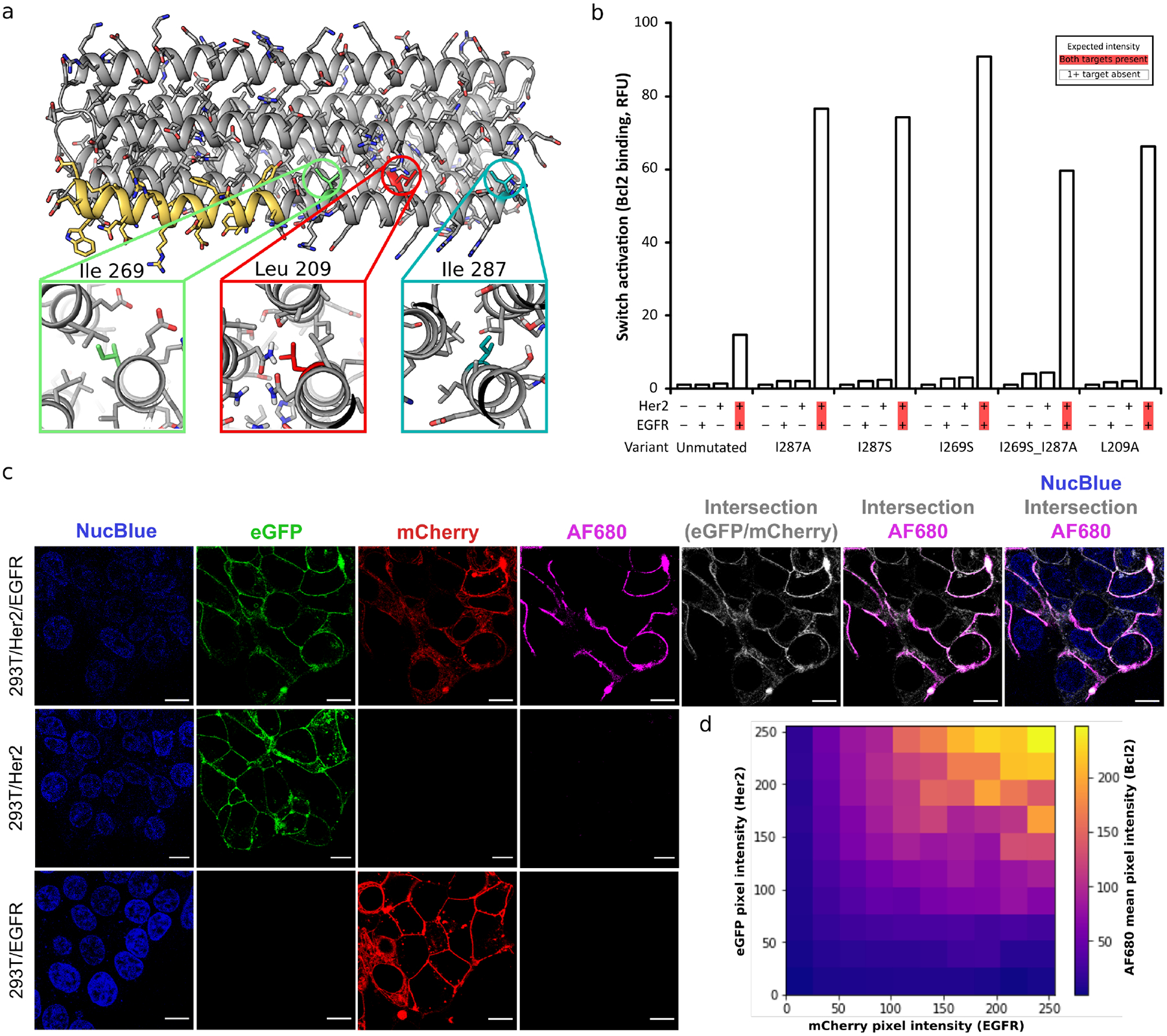
a. Design model of Co-LOCKR with the Bim functional peptide in yellow. Three buried hydrophobic amino acids were mutated to Ala or Ser to weaken the Cage–Latch affinity, thereby favoring Cage–Key binding. b. Tuned Co-LOCKR variants exhibit greater colocalization-dependent activation than the unmutated parental variant. CL_CHKE variants recruiting Bcl2-AF594 were evaluated by flow cytometry using the mixed population of cells from Fig 1d. The data shown represent 12.3 nM CL_CHKE (n = 1), and Fig S5c shows the complete dilution series for each variant. c. Confocal microscopy of HEK293T cell lines shows that Co-LOCKR switches recruit Bcl2-AF680 Effector proteins only where Her2 and EGFR are colocalized. Each cell line was incubated with CL_CHKE (I269S Cage) and Bcl2-AF680 before imaging. NucBlue is a nuclear stain, eGFP indicates Her2 localization, mCherry indicates EGFR localization, AF680 indicates Bcl2 binding in response to Co-LOCKR activation, and white indicates the intersection of Her2-eGFP and EGFR-mCherry signal Scale bars are 10 μm. Uncropped versions of these images are included in Fig S16a–c. d. Heat map showing the intensity of AF680 signal (Co-LOCKR activation) versus eGFP (Her2) and mCherry (EGFR) pixel intensity. Calculations were based on the uncropped 293T/Her2/EGFR image in Fig S16a.
Co-localization dependent activation was observed further at the sub-cellular level by confocal microscopy. CL_CHKE recruited Bcl2-AF680 to the plasma membrane of HEK293T/Her2/EGFR cells but not HEK293T/Her2 or HEK293T/EGFR (Fig 2c). There was a close correspondence between regions of the plasma membrane exhibiting colocalized Her2-eGFP and EGFR-iRFP signal and Co-LOCKR activation (Fig 2d).
To assess the flexibility of Co-LOCKR, we attempted to specifically target alternative pairwise combinations of three cancer-associated antigens (Her2, EGFR, and EpCAM). Each of these antigens are expressed at differing levels by engineered K562 cell lines or human cancer cell lines (Fig S7a, Fig S8a, Table S3). Using the I269S variant to maximize detection of low levels of antigen, (1) Co-LOCKR distinguished the correct pair of antigens in every case, and (2) the magnitude of Bcl2 binding corresponded with the expression level of the lower-expressed of the two target antigens (Fig 3a, Fig S8b–c), consistent with a stoichiometric binding mechanism for colocalization-dependent activation. Taken together, these results demonstrate the modularity of Co-LOCKR to target several antigens expressed at differing levels. While we chose DARPins as targeting domains to allow facile expression of Co-LOCKR variants, single chain variable fragments can also be used (Fig S9).
Figure 3. Co-LOCKR performs 2- and 3-input logic operations in mixed cell populations.
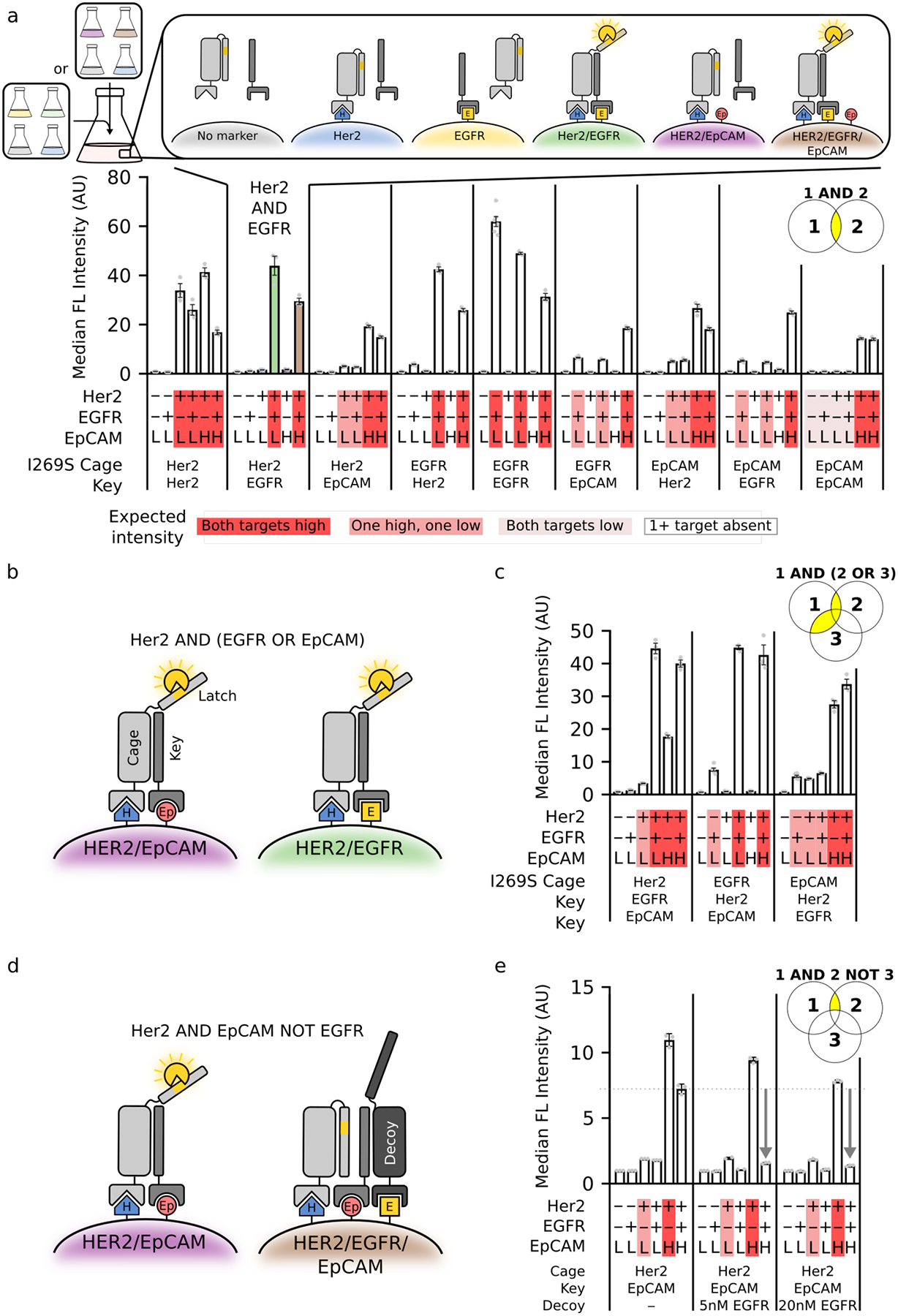
a. Co-LOCKR was used to recruit Bcl2-AF594 for two populations of K562 cells expressing different combinations of Her2, EGFR, and EpCAM. Marker expression for each cell line and identity of the Cage and Key targeting domains are indicated below each bar plot. Red highlighting indicates the expected magnitude of Bcl2-AF594 signal based on relative antigen expression. b. Schematic of [Her2 AND either EGFR OR EpCAM] logic mechanism. c. [Ag1 AND either Ag2 OR Ag3] logic combinations were used to recruit Bcl2-AF594. d. Schematic of [Her2 AND EpCAM NOT EGFR] logic mechanism. The Decoy acts as a sponge to sequester the Key, thereby preventing Cage activation. e. CL_CHKEpDE was used to recruit Bcl2-AF594. Compared to the simple CL_CHKEp ‘AND’ gate (left), recruitment of Decoy to EGFR-expressing cells reduced activation to near background levels. For all panels, population 1 was [K562/EpCAMlo, K562/EGFR/EpCAMlo, K562/EpCAMlo/Her2, and K562/EGFR/EpCAMlo/Her2], and population 2 was [K562/EpCAMlo, K562/EGFR/EpCAMlo, K562/EpCAMhi/Her2, and K562/EGFR/EpCAMhi/Her2]. Error bars represent SEM of 6 independent replicates for K562 and K562/EGFR and 3 independent replicates for all others. Statistics are reported in Table S4.
The colocalization-dependent activation mechanism of Co-LOCKR can in principle be extended to include ‘OR’ logic by adding a second Key fused to a targeting domain against an alternative surface marker (Fig 3b) and ‘NOT’ logic by adding a Decoy protein fused to a targeting domain against a surface marker to be avoided; the Decoy acts as a sponge to sequester the Key, thereby preventing Cage activation (Fig 3d). Using Her2, EGFR, and EpCAM as model antigens (Ag), we first explored [Ag1 AND either Ag2 OR Ag3] logic on the surface of cells (Fig 3b). To assess the programmability of Co-LOCKR targeting, we tested all three combinations: [Her2 AND either EGFR OR EpCAM], [EGFR AND either Her2 OR EpCAM], and [EpCAM AND either Her2 OR EGFR]. In all cases, the correct cell sub-population was targeted at levels consistent with the limiting target antigen (Fig 3c). For example, CL_CEKHKEp targeted cells expressing EGFR/EpCAMlo 10-fold over background, Her2/EGFR/EpCAMlo 59-fold over background, and Her2/EGFR/EpCAMhi 56-fold above background, but exhibited minimal off-target activation on cells missing at least one antigen (middle panel of Fig 3c).
We next explored [Ag1 AND Ag2 NOT Ag3] logic using CL_CHKEpDE (D for Decoy) and the same set of model antigens (Fig 3d). We tuned the Decoy-Key affinity with designed point mutations to maximize the abrogation of activation when the Decoy is targeted, and to minimize the interference of the decoy with activation when it is not targeted (Fig S10). Recruitment of Decoy reduced activation to near background levels on cells with Ag3 on their surface, while reducing activation on cells lacking Ag3 by only 15% (Fig 3e). Consistent with the stoichiometric sequestration mechanism, Ag3 must be expressed at higher levels than Ag2 so that the Decoy can sequester all molecules of the Key (Fig 3d). While bispecific antibodies can be made to approximate [Ag1 AND Ag2] logic by tuning binding affinity, we are not aware of any current approach which can achieve the precise [Ag1 AND Ag2 NOT Ag3] logic of Fig 3e and Fig S11.
As a first step toward a real-world application, we explored the retargeting of primary human T cell effector function against tumor cells in vitro. We designed a Bcl2 CAR that targets Bim peptides displayed on the surface of a target cell; the CAR contains a stabilized variant of human Bcl2, a flexible extracellular spacer domain (9), CD28/CD3ζ signaling domains, and a truncated EGFR (EGFRt) selection marker (10) linked by a T2A ribosomal skipping sequence (Fig S12a). The Bcl2 CAR functions as designed: purified CD8+EGFRt+ Bcl2 CAR T cells efficiently recognized K562 cells stably expressing a surface-exposed Bim-GFP fusion protein (Fig S12b–c).
With Bcl2 CAR T cells in hand, we investigated whether the Co-LOCKR proteins could mediate T cell activation by Raji and K562 cells expressing combinations of Her2, EGFR, and EpCAM. The Raji cells expressed lower levels of transduced antigens than did the K562 cell lines (Fig S7a–b, Table S3), and hence more stringently test Co-LOCKR sensitivity, whereas the K562 cells better assess specificity. CL_CHKEp and CL_CEpKH (using the parental unmutated Cage) promoted IFN-γ release only when co-cultured with Raji/EpCAM/Her2 cells and not Raji/EpCAM or Raji/Her2 (Fig S12d). Titration experiments showed that CAR T effector function could be specifically targeted using between 2.5 nM to 20 nM of Co-LOCKR without causing unintended activation by off-target cells (Fig S13); still lower concentrations would likely be effective using higher affinity binding domains.
Next, we assessed the ability of Co-LOCKR to direct CAR T cell cytotoxicity against specific subsets of cells within a mixed population. Raji, Raji/EpCAM, Raji/Her2, and Raji/EpCAM/Her2 were differentially labeled with fluorescent Cell Trace dyes, mixed together with CAR T cells and CL_CHKEp (Fig S12f), and killing of each of the cell lines was assessed using flow cytometry. After 48 hours, Raji/EpCAM/Her2 cells were preferentially killed, but a fraction of Raji/EpCAM cells were also targeted (Fig S12g), suggesting that even the parental Cage and Key were too leaky for CAR T cell recruitment. We overcame this basal activation by modifying the length of the Key (Fig S12e): the combination of parental Cage and ΔN3 Key (three N-terminal amino acids deleted) selectively targeted Raji/EpCAM/Her2 cells and mitigated unintended killing of Raji/EpCAM and Raji/Her2 cells (Fig S12f–g). A Chromium release assay showed that CL_CHKEp targeted only Raji/EpCAM/Her2 cells and initiated rapid cell killing within 4 hours (Fig S12h). Thus, Co-LOCKR can be used to restrict IFN-γ release and cell killing to only those tumor cells that express a specific pair of antigens.
We next evaluated Co-LOCKR ‘AND’ logic for additional tumor antigen pairs ([Her2 AND EpCAM], [Her2 AND EGFR]) and varying antigen density profiles using K562 cell lines (Fig S7a, Table S3) and solid tumor lines (Fig S8a). Raji cells with low antigen density yielded modest IFN-γ (Fig S14a), K562/Her2/EpCAMlo and SKBR3 breast cancer cells yielded intermediate IFN-γ (Fig 4a, Fig S14b), and both K562/Her2/EpCAMhi and K562/Her2/EGFR cells yielded high IFN-γ release for their respective Co-LOCKRs (Fig 4a, Fig S14c). CL_CEpKH induced IFN-γ release in response to Raji/Her2/EpCAM 3.9-fold above background, SKBR3 4.8-fold above background, K562/Her2/EpCAMlo 16-fold above background, and K562/Her2/EpCAMhi 51-fold above background, with minimal off-target cytokine release. IFN-γ production did not increase appreciably when the target cells expressed high levels of a single antigen. CAR T cells proliferated only upon co-culture with target cells co-expressing the correct pair of antigens, and the degree of proliferation positively correlated with antigen density (Fig 4b, Fig S14d). The flow cytometry-based killing assay confirmed ‘AND’ gate selective cytotoxicity with both CL_CHKEp and CL_CEpKH against Raji/EpCAM/Her2 without depleting single antigen-positive cells (Fig 4c). A similar result was observed for both CL_CHKE and CL_CEKH against Raji/Her2/EGFR (Fig S14e), although killing was less effective, likely due to the lower expression levels of EGFR compared to EpCAM in Raji/Her2/EGFR and Raji/EpCAM/Her2, respectively. We also did not observe fratricide of the EGFRt+ CAR T cells used in the experiment, which could have been targeted by the anti-EGFR DARPin targeting domain of CL_CHKE or CL_CEKH (Fig S14f).
Figure 4. Co-LOCKR directs CAR T cell specificity using 2- and 3-input logic operations.
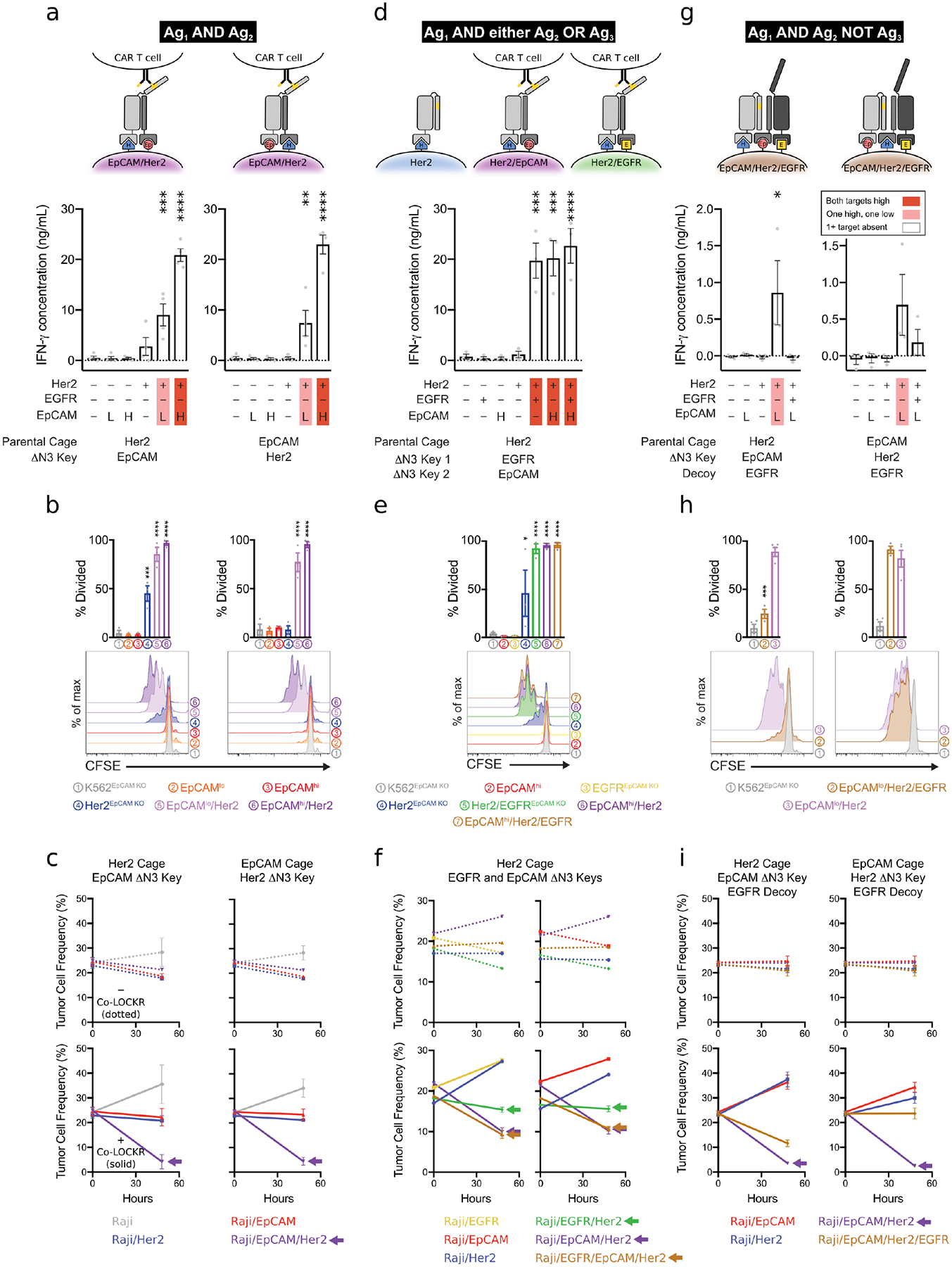
a,d,g. Mean IFN-γ concentration in cell supernatants 24 hours after co-culture of Cage, Key, and K562 cells with CAR T cells. Marker expression for each cell line and identity of the Cage and Key targeting domains are indicated below each bar plot. Red highlighting indicates the expected magnitude of signal based on the target cell’s relative antigen expression. Error bars represent SEM of n = 4 (a) or 3 (d,g) healthy T cell donors. AND/NOT logic is demonstrated with EpCAMlo target K562 cells because T cell effector function was leaky for EpCAMhi target cells (see Fig S15a). b,e,h. CAR T cell proliferation in response to [Her2 AND EpCAM] (c), [Her2 AND EGFR OR EpCAM] (e), or [Her2 AND EpCAM NOT EGFR] (h) logic. Bar plots are the percent of T cells that have undergone at least one cell division by 72 hours after co-culture of CAR T cells, Cage, Key, and target K562 cells. Histograms show flow cytometric analysis of CFSE dye dilution gated on CD8+ lymphocytes. The data are representative of n = 3 biological replicates with healthy T cell donors. c,f,i. CAR T cell cytotoxicity against mixed populations of target Raji cells expressing combinations of Her2, EpCAM, and EGFR. Line graphs show mean frequency of Raji target cells after 0 or 48 hours of co-culture with CAR T cells. n = 4 (c,f) or 3 (i) healthy donors. Arrows indicate cell lines targeted by Co-LOCKR.
Encouraged by robust ‘AND’ logic, we evaluated more complex operations involving combinations of ‘AND’ and either ‘OR’ or ‘NOT’ logic. CAR T cells co-cultured with ‘AND/OR’ Co-LOCKRs (CL_CHKEKEp, CL_CEKHKEp, and CL_CEpKHKE) each carried out [Ag1 AND either Ag2 OR Ag3] logic with respect to IFN-γ production (Fig 4d, Fig S14g) and proliferation (Fig 4e) against K562 cell lines, as well as selective killing in a mixed population of Raji cell lines (Fig 4f, Fig S14h). CAR T cells co-cultured with an ‘AND/NOT’ Co-LOCKR (CL_CHKEpDE) carried out [Her2 AND EpCAM NOT EGFR] logic: IFN-γ production and proliferation were induced in the presence of K562/Her2/EpCAMlo but not K562/EGFR/Her2/EpCAMlo cells (Fig 4g–h). Consistent with the observations above, Ag3 in the ‘NOT’ operation had to be expressed at higher levels than Ag2 (Fig 4g–i, Fig S15a–c). While these data indicate that careful antigen selection and some tuning are necessary for robust control of logical operations, the ability of CL_CEpKHDE to direct CAR T cell mediated killing of Her2/EpCAM cells but not Her2/EpCAM/EGFR cells (right hand panel of Fig 4i) further highlights the power of Co-LOCKR to perform [Ag1 AND Ag2 NOT Ag3] logic for specific cell targeting.
Two previous strategies improved the specificity of CAR T cells by approximating ‘AND’ logic. First, Kloss et al. (11) directly modulated signaling from a sub-optimally-activating first generation CAR (CD3ζ only) using a chimeric costimulatory receptor (CCR, costimulatory domain only) that recognizes a distinct second antigen. Although T cell activation is reduced in the absence of the second antigen, targeting remains leaky because the CAR T cells can lyse both tumor and normal cells expressing the antigen targeted by the CD3ζ CAR, even when the CCR is not engaged. Second, Morsut et al. (12) developed synthetic Notch receptors that modularly induce expression of effector proteins in engineered cells. Roybal et al. (13) applied this technology to enhance CAR T cell specificity using a synthetic Notch receptor that recognizes one antigen to induce expression of a CAR recognizing a second antigen. While this ‘If-Then’ logic strategy has shown promise in pre-clinical models in which the target antigens exist distally, the CAR will kill any nearby cell expressing the target antigen, so off-tumor toxicity can occur when the Notch receptor and CAR targets are expressed in neighboring healthy tissues (14). Co-LOCKR has potential advantages over these approaches as activation requires binding in cis to a precise combination of target antigens before recruiting the cognate CAR T cells, and thus it can direct killing without harming neighboring off-target cells displaying single antigens (Fig 4c, f, i). ‘OR’ (15, 16) and ‘NOT’ (15, 17) logic have also been described for CAR T cells, but not in combination with ‘AND’ logic as we describe here.
Our CAR T cell experiments demonstrate the potential for Co-LOCKR to mediate targeting specificity in vitro; however, several additional challenges will have to be met for Co-LOCKR to be a clinically translatable therapeutic. In vivo studies will be needed to assess and improve the pharmacokinetics of the Co-LOCKR components. Immunogenicity of the designed proteins is also a potential concern, similar to any other system comprising non-human proteins. As Co-LOCKR actuation is thermodynamically controlled, the therapeutic index will depend on the affinity of the targeting domains used to direct the Co-LOCKR proteins to antigens on the target cells: if the affinities are sub-nanomolar, dosing can be far below the 40 nM level where activation starts to occur in solution (Fig S13). CAR T efficacy could also be improved by optimizing the CAR, for example by using alternative signaling molecules (18–20).
The power of Co-LOCKR results from the integration of multiple coherent or competing inputs that determine the magnitude of a single response. The output signal—exposure of the functional peptide on the Latch—is increased by Key binding and countered by Decoy competition. Thus, the proteins can intrinsically perform logic rather than relying on cellular machinery for signal integration. Although our present work has focused on development of the Co-LOCKR system and CAR T cell applications, the Co-LOCKR system should be powerful for engineering biology in any setting that requires proximity-based activation or targeting of specific sub-populations of cells.
Supplementary Material
Acknowledgements:
We would like to thank Andy Scharenberg, Wendell Lim, Rick Klausner, Lance Stewart, Anindya Roy, Zibo Chen, Adrian Briggs, Elizabeth Gray, Daniel Stetson, Marion Pepper, Lauren Carter, Brian Weitzner, Jocelynn Pearl, Howell Moffett, Ian Haydon, Daniel Campbell, Stephen Hauschka, Neil King, and Jesse Zalatan for helpful advice; Donna Prunkard and Han Nguyen for technical assistance with flow cytometry and cell sorting; Alex Kang for setting up crystal trays and looping crystals; Rashmi Ravichandran for protein purification; Stephanie Berger for sharing biotinylated Bcl2, David Hockenbery for sharing SKBR3 cells, and Didier Trono for sharing lentiviral packaging plasmids. We also thank the W.M. Keck Center for Advanced Studies in Neural Signaling (NIH grant S10 OD016240) and the helpful input of Nathaniel Peters and Gary Liu for confocal microscopy experiments.
Funding:
This work was supported by the HHMI (D.B.), the Open Philanthropy Project (D.B.), the NSF (D.B., CHE-1629214), the DTRA (D.B., HDTRA1-18-1-0001), the Nordstrom Barrier IPD Directors Fund (D.B.), the Washington Research Foundation and Translational Research Fund (D.B.), the Audacious Project organized by TED (D.B.), and the NIH (S.R.R., R01 CA114536; J.B., NIGMS T32GM008268). M.J.L. was supported by a Washington Research Foundation Innovation Postdoctoral Fellowship and a Cancer Research Institute Irvington Fellowship from the Cancer Research Institute. S.E.B. was supported by the Burroughs Wellcome Fund Career Award at the Scientific Interface. A.I.S. was supported by the FHCRC interdisciplinary training grant in cancer research and Hearst Foundation. A.O. was supported by NIH NCI grants 1R21CA232430-01 and T32CA080416.
Footnotes
Competing interests:
M.J.L., S.E.B., A.I.S., J.B., R.A.L., M.G., C.E.C., S.R.R., and D.B. are inventors on patents related to this work. M.J.L., S.E.B., A.I.S., R.A.L., M.J.B., S.R.R., and D.B. hold equity in Lyell Immunopharma. D.B. holds equity in Sana Biotechnology. M.J.L., S.E.B., R.A.L., M.J.B., and S.R.R. are employees of Lyell Immunopharma. A.I.S. is a consultant of Lyell Immunopharma.
Data and materials availability:
Coordinates and structure files have been deposited to the Protein Data Bank (PDB) with accession code 7JH5.
Code availability:
The python script used to analyze confocal microscopy images is included in the supplemental material.
Biological material availability:
Expression plasmids and cell lines are available upon request. Primary T cells are not available.
References and Notes
- 1.Sellmann C et al. , Balancing Selectivity and Efficacy of Bispecific Epidermal Growth Factor Receptor (EGFR) × c-MET Antibodies and Antibody-Drug Conjugates. J. Biol. Chem 291, 25106–25119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mazor Y et al. , Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci. Rep 7, 40098 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langan RA et al. , De novo design of bioactive protein switches. Nature 572, 205–210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyken SE et al. , De novo design of protein homo-oligomers with modular hydrogen-bond network-mediated specificity. Science 352, 680–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leaver-Fay A et al. , ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol 487, 545–74 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delgado-Soler L, Pinto M, Tanaka-Gil K, Rubio-Martinez J, Molecular Determinants of Bim(BH3) Peptide Binding to Pro-Survival Proteins. J. Chem. Inf. Model 52, 2107–2118 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Zahnd C et al. , A Designed Ankyrin Repeat Protein Evolved to Picomolar Affinity to Her2. J. Mol. Biol 369, 1015–1028 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Steiner D, Forrer P, Plückthun A, Efficient Selection of DARPins with Sub-nanomolar Affinities using SRP Phage Display. J. Mol. Biol 382, 1211–1227 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Hudecek M et al. , The Nonsignaling Extracellular Spacer Domain of Chimeric Antigen Receptors Is Decisive for In Vivo Antitumor Activity. Cancer Immunol. Res 3, 125–135 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X et al. , A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 118, 1255 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M, Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol 31, 71–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morsut L et al. , Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 164, 780–791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roybal KT et al. , Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 164, 770–779 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srivastava S et al. , Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell 35, 489–503.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho JH, Collins JJ, Wong WW, Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 173, 1426–1438.e11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zah E, Lin M-Y, Silva-Benedict A, Jensen MC, Chen YY, T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res 4, 498–508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fedorov VD, Themeli M, Sadelain M, Sci. Transl. Med, in press, doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tammana S et al. , 4–1BB and CD28 Signaling Plays a Synergistic Role in Redirecting Umbilical Cord Blood T Cells Against B-Cell Malignancies. Hum. Gene Ther 21, 75–86 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kagoya Y et al. , A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat. Med 24, 352–359 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun C et al. , THEMIS-SHP1 Recruitment by 4-1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells. Cancer Cell 37, 216–225.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhlman B, Baker D, Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. U. S. A 97, 10383–8 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fleishman SJ et al. , RosettaScripts: A Scripting Language Interface to the Rosetta Macromolecular Modeling Suite. PLoS One 6, e20161 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Checco JW et al. , α/β-Peptide Foldamers Targeting Intracellular Protein–Protein Interactions with Activity in Living Cells. J. Am. Chem. Soc 137, 11365–11375 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldenzweig A et al. , Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol. Cell 63, 337–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gibson DG, Smith HO, Hutchison CA, Venter JC, Merryman C, Chemical synthesis of the mouse mitochondrial genome. Nat. Methods 7, 901–3 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Stefan N et al. , DARPins Recognizing the Tumor-Associated Antigen EpCAM Selected by Phage and Ribosome Display and Engineered for Multivalency. J. Mol. Biol 413, 826–843 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Kabsch W, IUCr XDS. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCoy AJ et al. , Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams PD et al. , PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terwilliger TC et al. , Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. Sect. D Biol. Crystallogr 64, 61–69 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emsley P, Cowtan K, IUCr, Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Davis IW et al. , MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35, W375–W383 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bandaranayake AD et al. , Daedalus: a robust, turnkey platform for rapid production of decigram quantities of active recombinant proteins in human cell lines using novel lentiviral vectors. Nucleic Acids Res 39, e143–e143 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.