

Abstract
Pinin (PNN), a desmosome associated protein, was demonstrated to be over-expressed and act as a tumor-promoting factor in ovarian cancer, hepatocellular carcinoma and colorectal cancer. However, the precise role of PNN in prostate cancer is still unknown. In the study, we reported that PNN was upregulated in prostate cancer tissues and PNN expression was positively associated with Gleason score, tumor stage and tumor metastasis. PNN promoted cell growth and tumorigenicity in vitro and in vivo, and modulated cell growth through driving G1/S transition via CDK6, CDK2, and Cyclin D1 in prostate cancer cells. Furthermore, PNN accelerated cell invasion, migration and EMT processes of prostate cancer cells, accompanied with the up-regulation of MMP-2, MMP-9, N-cadherin, Vimentin and down-regulation of E-cadherin. Mechanism study demonstrated that the proliferation- and motility-promoting effects of PNN on prostate cancer cells dependent on the activation of CREB, which was reversed by CREB inhibition. More important, PNN activated CREB via PI3K/AKT and ERK/MAPK pathway. Collectively, these findings indicated that PNN plays important roles in prostate cancer tumorigenesis and progression and it is a potential therapeutic target for prostate cancer treatment.
Keywords: PNN, prostate cancer, tumor progression, CREB, PI3K/AKT, ERK/MAPK
Introduction
Prostate cancer (PCa) is the most common malignancy of the male urogenital system [1]. In the United States, the incidence of prostate cancer ranks first with accounting for more than 1 in 5 new diagnoses, and its mortality ranks second among all male malignant tumors in 2020 [2]. Although the incidence and mortality of prostate cancer is lower in China, it increases rapidly in the past 20 years [3,4]. When the cancerous cells are still confined within the prostate capsule, patients usually receive prostatectomy or radiation therapy and the outcome is well with the 5-year survival rate almost 99%. However, many patients are diagnosed after the cancer has spread and the 5-year survival rate of metastatic PCa is only 28% [5]. Although metastatic cancer has been focused of PCa, the mechanisms of PCa malignant progression are not fully understood.
Pinin (PNN), a desmosome associated protein, was found to locate at the cytoplasmic surface of the desmosome structure in corneal epithelia for the first time [6,7]. The presence of PNN within the desmosome was correlated to highly organized, perpendicular bundles of keratin filaments, and primarily stabilized the intermediate fiber-desmosome composite structure and reinforcing epithelial cell to cell adhesion [8,9]. Furthermore, PNN was found to widely distribute in the nuclear plaque region, which is involved in the transcription regulation of genes and the selective splicing of pre-mRNA and lncRNA [10-13]. Recent studies have found that PNN played important roles in various tumors. Down-regulation of PNN induced apoptosis in breast cancer MCF-7 cell [14]. PNN was over-expressed in ovarian cancer and PNN knockdown reduced the cell viability and tumorigenicity [15,16]. Over-expression of PNN was found to promote cell survival, proliferation and migration via activation of ERK pathway in hepatocellular carcinoma and colorectal cancer [17,18]. These results indicated that PNN may be a potential proto-oncogene. However, the role of PNN in prostate cancer is still unknown.
In this study, we revealed that up-regulated PNN was positively correlated with prostate cancer malignant progression. PNN over-expression promoted prostate cancer cell proliferation, invasion and migration via activating CREB through PI3K/AKT and ERK/MAPK pathway. These findings demonstrated that PNN plays an important role in prostate cancer and it is a valuable therapeutic target for prostate cancer.
Materials and methods
Tissue samples
In the study, a total of 81 prostate cancer samples and 22 normal prostate samples were obtained from patients who underwent prostate biopsy in Department of Urology, Ningbo First Hospital (Ningbo, China) between 2012 and 2017. These samples were then formalin-fixed and paraffin-embedded followed by pathological diagnosis and immunohistochemical staining. The study protocol was approved by the Ethics Committee of Ningbo First Hospital and the information written consent was obtained from all the subjects prior to their participation in the study. The clinicopathological characteristics of patient samples were obtained from medical records within Ningbo First Hospital and were summarized in Table 1.
Table 1.
Clinicopathological characteristics of patient samples and expression of PNN in prostate cancer
| Characteristics | Number of Patients | Percentage (%) |
|---|---|---|
| PSA Level (ng/ml) | ||
| < 10 | 11 | 13.6 |
| 10-50 | 34 | 42.0 |
| > 50 | 36 | 44.4 |
| Gleason Score | ||
| 6 | 13 | 16.0 |
| 7 | 21 | 25.9 |
| 8 | 25 | 30.9 |
| 9 | 18 | 22.2 |
| 10 | 4 | 4.9 |
| Tumor Stage | ||
| I | 2 | 2.5 |
| II | 52 | 64.2 |
| III | 17 | 21.0 |
| IV | 9 | 11.1 |
| Unknown | 1 | 1.2 |
| Tumor Metastasis | ||
| Yes | 45 | 55.6 |
| No | 35 | 43.2 |
| Unknown | 1 | 1.2 |
| Biochemical recurrence | ||
| Yes | 20 | 24.7 |
| No | 61 | 75.3 |
| PNN Expression in Tumors | ||
| Negative | 5 | 6.2 |
| Low | 38 | 46.9 |
| Moderate | 33 | 40.7 |
| High | 5 | 6.2 |
| PNN Expression in Normal samples | ||
| Negative | 15 | 68.2 |
| Low | 7 | 31.8 |
Immunohistochemical staining
Immunohistochemical staining (IHC) was performed on paraffin-embedded tissue sections to determine PNN levels in prostate cancer tissues and normal prostate samples using a staining kit (absin, Shanghai, China). Briefly, sections were deparaffinized with xylene and rehydrated with ethanol-aqueous solutions, then antigen retrieval was done by heating the slides for 15 min in a microwave oven in 10 mM citrate buffer (pH 6.0). After eliminating endogenous peroxidase activity using 3% H2O2 and blocking with 5% BSA, the sections were incubated with anti-PNN antibody (1:50, HPA001378, Sigma-Aldrich, Merck KGaA, Germany) overnight at 4°C and followed with secondary antibody. The sections were then incubated with Diaminobenzidine (DAB) and counterstained with Mayer’s hematoxylin.
TCGA data analysis
The gene expression matrix and clinical phenotype of prostate cancer patients from TCGA were downloaded from UCSC (https://xenabrowser.net/datapages/), then PNN expression in prostate cancer was analyzed by GraphPad Prism 8.0. Kaplan-Meier survival curves, applied to analyze the effect of PNN expression on PCa patients’ recurrence and survival, were analyzed with GEPIA (Gene Expression Profiling Interactive Analysis) [19].
Cell culture and cell transfection
Human prostate cancer cells DU145, 22Rv1, LNCaP clone FGC and PC-3, and human embryonic kidney cells 293T used in the study were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and were cultured in MEM, RPMI 1640, F-12K or DMEM medium (Gibco, Thermo Fisher SCIENTIFIC, USA) containing 10% fetal bovine serum (PAN, Germany). All cell cultures were carried out in a humidified chamber at 37°C with an atmosphere of 5% CO2.
PNN expression vector pcDNA3.1-3xFlag-C-PNN (pcDNA3.1-PNN) and primer vector pcDNA3.1-3xFlag-C (pcDNA3.1) used in the study was purchased from YouBio (Changsha, Hunan, China). Lipofectamine™ 3000 reagent (Invitrogen, Thermo Fisher SCIENTIFIC, USA) was used to cell transfection following the manufacturer’s instructions.
Lentivirus production and generation of stably PNN-knockdown PC-3 cells
The shRNAs targeted human PNN were obtained from GPP Web Portal (https://portals.broadinstitute.org/gpp/public/gene/search). The shRNAs sequences used in the study included shPNN #1 (5’-CCGACAGAAAGAGGTCTATAT-3’), shPNN #2 (5’-GAAGGTAGACGCATCGAATTT-3’), shPNN #3 (5’-GGTAGAGGACGTGGTAGTTTA-3’) and a scramble shRNA (5’-CCTAAGGTTAAGTCGCCCTCG-3’). We cloned the shRNAs into pLKO.1-puro (Plasmid #8453, Addgene) at Age I and EcoR I to construct pLKO.1-shPNN or pLKO.1-shSCR vectors. Three plasmids pCMV-dR8.2 dvpr (Plasmid #8455, Addgene), pCMV-VSV-G (Plasmid #8454, Addgene) and pLKO.1-shPNN or pLKO.1-shSCR were co-transfected into 293T cells and cultured for 72 h. Lentivirus-containing supernatants were then harvested using 0.45 μm sterilizing filter. Lentiviruses were then used to infect PC-3 cells and the infected cells were then selected by complete medium containing 2 ug/ml puromycin to acquire stably PNN-knockdown PC-3 cells.
Western blotting
Tumor tissues and cells were lysed and protein was harvested with RIPA buffer (Solarbio, Beijing, China) supplemented with 1% protease inhibitor mix (Cell Signaling Technology, USA) and 1% phosphatase inhibitor mix (Sangon Biotech, Shanghai, China). Proteins were separated on SDS-PAGE gel, then transferred onto PVDF membranes (BIO-RAD, USA) followed with blocking by 5% non-fat milk. The membranes were incubated with the primary antibody at 4°C overnight. The following primary antibodies were used: E-Cadherin, N-Cadherin, Vimentin, Fibronectin, GAPDH, MMP-2, MMP-9, CDK4, CDK6, CDK2, CyclinD1, CyclinE1, PI3K p110α, PI3K p110β, PI3K p110γ, PI3K p85, p-PI3K p85 (Tyr458), AKT, p-AKT (Ser473), CREB and p-CREB (Ser133) were purchased from Cell Signaling Technology (USA), PNN antibody was obtained from Sigma-Aldrich (Merck KGaA, Germany). Then, membranes were incubated with HRP (horseradish peroxidase)-labeled secondary antibody (Boster, Wuhan, China) and detected by chemiluminescence.
RNA extraction and quantitative real time-PCR (qRT-PCR)
Total RNAs were extracted from prostate cancer cells using TRIzol Reagent (Invitrogen, Thermo Fisher SCIENTIFIC, USA). The RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher SCIENTIFIC, USA) was used to synthesize cDNA. The cDNAs were amplified by qRT-PCR using SYBR Green PCR Master Mix (Roche, US) on a LightCycler480 system, and fold changes were calculated by relative quantification (2-ΔΔCt). The PCR primers sequences were as follows: Forward primer: 5’-ACCCACTCCTCCACCTTTGAC-3’ and Reverse primer: 5’-TGTTGCTGTAGCCAAATTCGTT-3’ for GAPDH, Forward primer: 5’-CCTGTAAAGCAGTCTCAAGCC-3’ and Reverse primer: 5’-CGAATGTTCTCATCCACGTTCT-3’ for PNN.
Transwell assay and wound healing assay
The transwell assay was performed to determine the ability of cellular invasion and migration using 8 μm Transwell® Permeable Supports (Costar, Corning, Bedford, MA, USA). For cell migration assay, a suspension of cells (1 × 105 cells/well for PC-3 and 5 × 104 cells/well for DU145) in serum free basal medium was seeded to the upper chambers. The lower chambers were filled with growth media containing 10% FBS. After incubation at 37°C (24 h for DU145 cell and 48 h for PC-3 cell), the non-invaded cells in upper chamber were removed and the invaded cells were fixed with 90% methyl alcohol, then stained with 0.1% crystal violet (Sigma-Aldrich, Merck KGaA, Germany). The invasion assay was performed using the same procedure with the following modifications: (i) the seeding density was 2 × 105 cells/well for PC-3 and 1 × 105 cells/well for DU145, (ii) the upper chambers were pre-coated with Matrigel® Matrix (Corning, Bedford, MA, USA), and (iii) PC-3 and DU145 cells were all incubated for 48 h at 37°C.
The wound healing assay was performed to determine the cell motility. When the seeded cells reached 95% confluence in 24-well plate, a linear wound was created with a micropipette tip across the diameter of the well, and then PBS was used to rinse the non-adherent cells. The medium containing 0.5% FBS was added to allow cells to move into the gap without the influence of cell growth. Three different equidistant points of the scratched area were photographically measured and imaged by an inverted phase contrast microscope (Olympus, Japan) at 0 h and 24 h. Migration rate was calculated as the proportion of initial scratch distant of each sample using the mean distance between both borderline that remain cell-free after cell migration.
MTS assay
MTS assay was performed to determined cell proliferation using Cell Titer 96® Aqueous One Solution Reagent (MTS, Promega, Madison, USA) according to the manufacturer’s protocol. In brief, a suspension of cells (2000 cells/well for DU145 and 4000 cells/well for PC-3) in 100 μl of growth media was seeded to 96-well plate, Following incubation for 4, 24, 48, 72 and 96 h, 20 μl of MTS reagent was added to each well. The absorbance (OD value) was measured at 490 nm after incubation for another 2 h using iMarkTM Microplate Reader (Bio-Rad, US).
Cell cycle and apoptosis assay
Cells were harvested by trypsinization and washed with PBS, then the cells were stained followed the Cell Cycle Staining Kit (MultiSciences, Hangzhou, China). After staining, cell cycle analysis was performed using a flow cytometer (Beckman Coulter, Fullerton, CA, USA).
Cell apoptosis was assessed using Annexin V-FITC/PI apoptosis kit (MultiSciences, Hangzhou, China). In brief, cells were harvested and washed with pre-cold PBS, then cells were stained with fluorescein isothiocyanate (FITC)-conjugated Annexin V and PI followed with apoptosis analysis by Beckman flow cytometer.
Tumorigenesis assay of tumor cells
Colony formation assays and Xenograft model in nude mice were performed to detect the tumorigenicity of prostate cancer cells. For colony formation assays, PC-3 cells were counted and seeded in 6-well plate at a density of 1000 cells per well. After incubation for 14 days, the cells were fixed with 90% methyl alcohol for 15 min and then stained with 0.1% crystal violet (Sigma-Aldrich, Merck KGaA, Germany) for 15 min. The number of colonies consisting of more than 50 cells was counted.
Five-week-old male nude mice (BALB/C) (Shanghai laboratory animal center, China) were used as xenograft model with a protocol approved by the Institutional Animal Ethics Committee of Ningbo University. 4 × 106 stably PNN-knockdown PC-3 cells or PC-3/Control cells were injected subcutaneously into the flank of mouse (5 for each group) and tumor formation was monitored. On day 32 after inoculation, the nude mice were sacrificed to collect the tumors. The tumor volume was calculated with the formula V = L * W2/2 (V, volume; L, length; W, width).
Statistical analysis
Statistical analyses were performed with SPSS software (SPSS Inc., Chicago, IL, USA). The correlation between PNN expression and the clinicopathological features was analyzed with chi-square test. Bivariate correlations between study variables were calculated using the Spearman’s rank correlation coeffcient. Variance among the control and tested groups were analyzed using one-way ANOVA analysis followed by Dunnett post hoc test. Variance between two groups was assessed using student t test. The data were showed as Mean ± standard deviation (SD). P < 0.05 was considered statistically significant in all tests.
Results
Over-expressed PNN correlates with malignant progression of prostate cancer
Previous studies have demonstrated that PNN is up-regulated in ovarian cancer, colorectal cancer and hepatocellular carcinoma promoting tumor cell growth and metastasis and inhibiting apoptosis [15-18]. Here, we firstly analyzed PNN expression using immunohistochemical staining (IHC) in a cohort of 81 prostate cancer tissues and 22 normal prostate samples obtained by prostate biopsy. The results indicated that PNN was only expressed in 31.8% (7/22) normal prostate tissues at low levels (Tables 1, 2; Figure 1A). However, PNN was positively expressed in 93.8% (76/81) prostate cancer samples, and a moderate to strong staining was observed in 39/81 tumor samples (Tables 1, 2; Figure 1A). In addition, the results of 20 paired tumor-normal samples in the cohort showed that PNN is significantly up-regulated in tumors compared to their paired normal samples (P < 0.001) (Table 2).
Table 2.
Over-expressed PNN in prostate cancer samples
| Characteristics | PNN Expression | χ2 | P | |
|---|---|---|---|---|
|
| ||||
| Negative (%) | Positive (%) | |||
| PNN expression in all samples | ||||
| Nomal vs Tumor | 42.515 | < 0.001 | ||
| Normal | 15 (68.2) | 7 (31.8) | ||
| Tumor | 5 (6.2) | 76 (93.8) | ||
| PNN expression in pair-matched samples | ||||
| Nomal vs Tumor | 18.027 | < 0.001 | ||
| Normal | 14 (70.0) | 6 (30.0) | ||
| Tumor | 1 (5.0) | 19 (95.0) | ||
Figure 1.
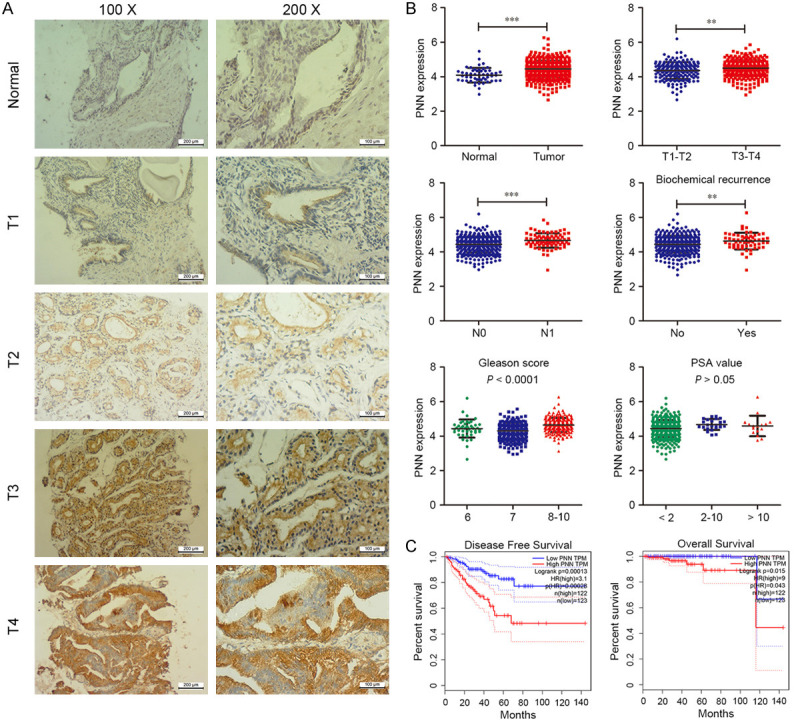
PNN is over-expressed in human prostate cancer. A. PNN is up-regulated in tumor samples. Representative images of immunohistochemical staining with PNN in a cohort of 81 prostate cancer tissues and 22 normal prostate samples. The scale bar = 200 μm (left), 100 μm (right). B. TCGA data analysis of prostate cancer. PNN is over-expressed in tumors, and its over-expression correlates with tumor stage, lymph nodal metastasis, biochemical recurrence and Gleason score. C. The survival analysis based on the TCGA dateset. PNN over-expression indicates poor survival of prostate cancer patients. **P < 0.01, ***P < 0.001.
To better understand the significance of PNN expression in prostate cancer, a correlation analysis was performed between PNN expression and clinicopathological parameters. As shown in Tables 3 and 4, PNN expression was positively correlated with Gleason score (P < 0.01), tumor stage (P < 0.05) and tumor metastasis (P < 0.05), but not PSA level and biochemical recurrence.
Table 3.
Correlation between PNN expression and clinicopathologic characteristics in prostate cancer patients
| Characteristics | PNN Expression | χ2 | P | |
|---|---|---|---|---|
|
| ||||
| Negative and Low (%) | Moderate and High (%) | |||
| PSA Level | 2.021 | 0.364 | ||
| < 10 | 7 (63.6) | 4 (36.4) | ||
| 10-50 | 20 (58.8) | 14 (41.2) | ||
| > 50 | 16 (44.4) | 20 (55.6) | ||
| Gleason Score | 7.546 | 0.023 | ||
| < 7 | 10 (76.9) | 3 (23.1) | ||
| = 7 | 14 (66.7) | 7 (33.3) | ||
| > 7 | 19 (40.4) | 28 (59.6) | ||
| Tumor Stage | 4.941 | 0.026 | ||
| I-II | 33 (61.1) | 21 (38.9) | ||
| III-IV | 9 (34.6) | 17 (65.4) | ||
| Tumor Metastasis | 4.357 | 0.037 | ||
| No | 23 (65.7) | 12 (34.3) | ||
| Yes | 19 (42.2) | 26 (57.8) | ||
| Biochemical Recurrence | 0.102 | 0.750 | ||
| No | 33 (54.1) | 28 (45.9) | ||
| Yes | 10 (50.0) | 10 (50.0) | ||
Table 4.
Spearman analysis of correlation between PNN and clinicopathological features
| Variables | PNN Expression | |
|---|---|---|
|
| ||
| Spearman Correlation | P | |
| PSA Level | 0.156 | 0.164 |
| Gleason Score | 0.305 | 0.006 |
| Tumor Stage | 0.249 | 0.026 |
| Tumor Metastasis | 0.233 | 0.037 |
| Biochemical Recurrence | -0.035 | 0.754 |
Meanwhile, we downloaded the gene expression matrix and clinical data of prostate cancer from TCGA database, and then analyzed PNN expression stratified by the clinicopathological parameters. As Figure 1B shown, PNN was up-regulated in tumors (P < 0.001), and its over-expression was correlated with tumor stage (P < 0.01), lymph nodal metastasis (P < 0.001), biochemical recurrence (P < 0.01) and Gleason score (P < 0.001). These results were basically consistent with our immunohistochemical staining results. Furthermore, to explore the relationship between PNN expression and the clinical prognosis of patients with prostate cancer, we determined the prognostic significance of PNN in TCGA cohort. The result of Kaplan-Meier analysis demonstrated that high PNN expression could predict significantly unfavorable PFS (progression-free survival) and OS (overall survival) (Figure 1C).
Collectively, these findings indicated that PNN is up-regulated in human prostate cancers and its over-expression correlates with prostate cancer malignant progression and poor prognosis.
PNN has positive effects on cell growth and cell cycle in prostate cancer cells
To address the biological importance of PNN, the expression of PNN in prostate cancer cells was detected. The results showed that a higher level of PNN in PC-3 cells than the other cells both in protein and mRNA levels, however, LNCaP clone FGC (LNCaP) and DU145 cells showed lower PNN expression (Figure 2A, 2B). We overexpressed or stably depleted PNN in DU145 or PC-3 cells respectively (Figure 2C). As determined by MTS assay, PNN-depleted PC-3 cells displayed a slower growth rate than the controls (Figure 2D), whereas overexpression of PNN significantly promoted cell proliferation (Figure 2E). Similarly, colony formation assay showed that the ability of tumorigenicity was reduced in PNN-depleted PC-3 cells (Figure 2F).
Figure 2.
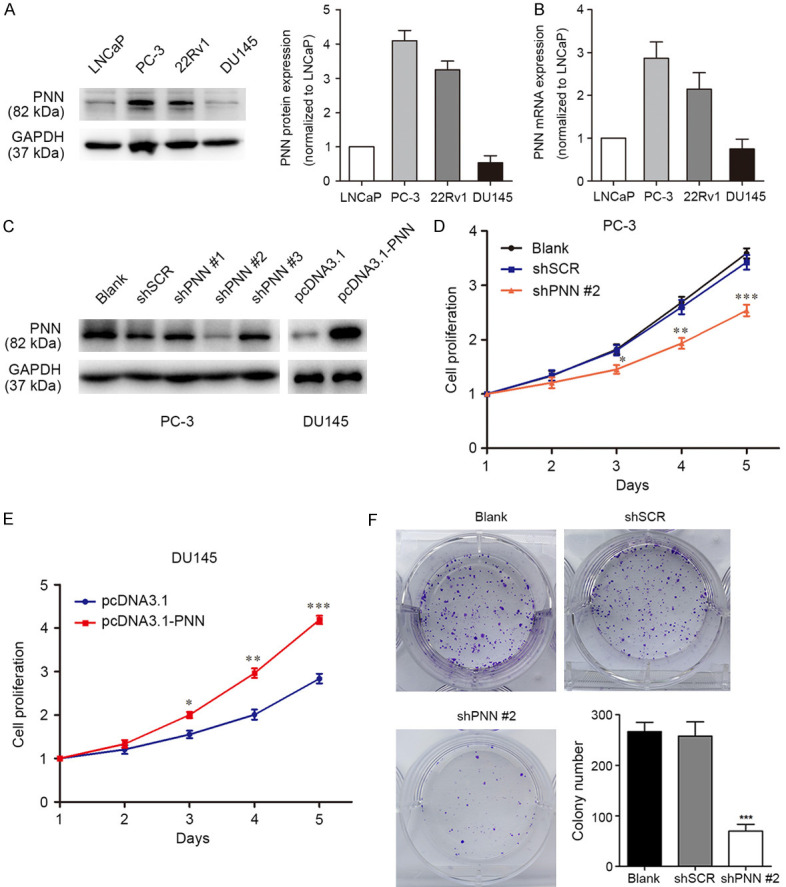
PNN promotes PCa cell proliferation in vitro. A. PNN protein levels in prostate cancer cells. Western blotting was performed to detect PNN expression, GAPDH was measured as control. B. RT-qPCR was performed to analyse PNN mRNA expression in prostate cancer cells. GAPDH was measured as control. The fold changes were calculated by relative quantification (2-ΔΔCt). C. Verification of PNN expression by western blotting after prostate cancer cells infecting PNN-shRNA-lentivirus or transfecting with PNN over-expression vector. D. Down-regulation of PNN repressed cell growth in PC-3 cells. Cell growth was detected by MTS assay. E. PNN up-regulation showed proliferation-promoting effect in DU145 cells. F. The tumorigenesis ability was reduced in PNN-silenced PC-3 cells. PC-3 cell tumorigenesis was detected by colony formation assays with incubation for 14 days followed by crystal violet staining. *P < 0.05, **P < 0.01, ***P < 0.001.
Furthermore, we examined whether cell cycle and some important checkpoint molecules were also mediated by PNN in prostate cancer cells. Flow cytometry analysis showed that a marked elevation in the percentage of G0/G1 phase was observed in PC-3 cells with PNN depletion, which also displayed a significant reduction of G1/S checkpoint molecules expression, including CDK6, CDK2, and Cyclin D1 (Figure 3A, 3B). Apoptosis assay was also performed and the results showed that PNN expression had no significant effect on PC-3 cell apoptosis (shPNN #2 vs shSCR: 3.64% vs 3.58%, P > 0.05, Figure 3C). Meanwhile, PNN-depletion also didn’t trigger the activation of apoptotic effectors Caspase-3 and PARP (Figure 3D). Taken together, these findings demonstrate that PNN might modulate cell growth through G1/S transition via CDK6, CDK2, and Cyclin D1 in prostate cancer cells.
Figure 3.
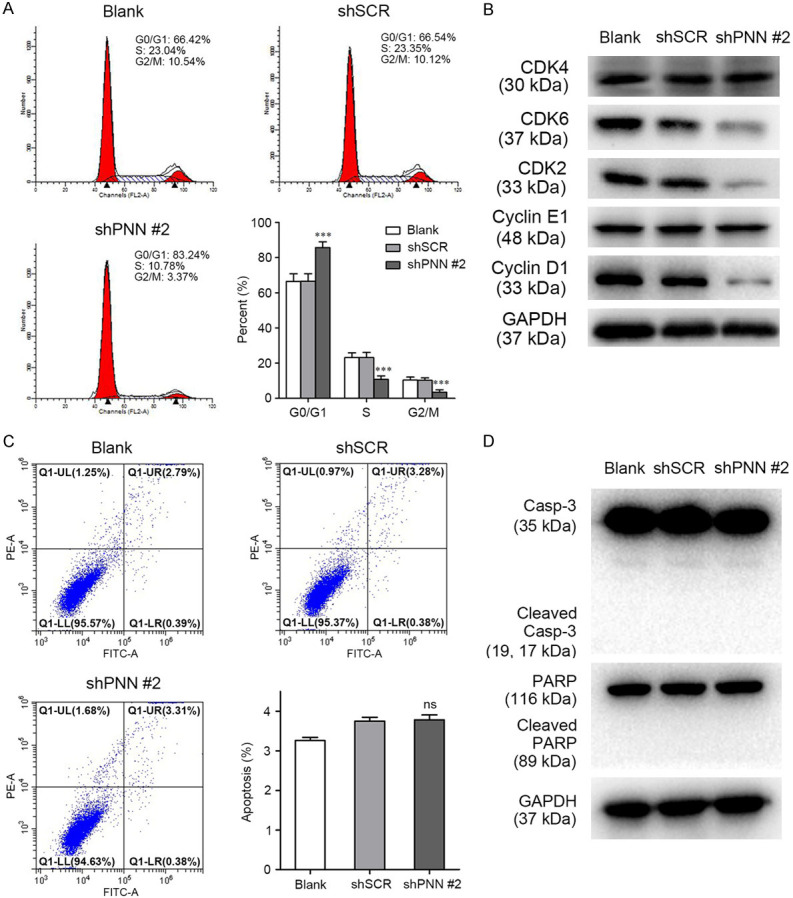
PNN modulates PCa cell growth through cell cycle control. A. PNN knockdown induced PC-3 cell arrest at G0/G1 phase. Flow cytometry was employed to analyse cell cycle in PC-3 cell. B. Down-regulation of PNN decreased the expression of CDK2, CDK6 and Cyclin D1 in PC-3 cells. The expression of proteins control the cell fate in G0/G1 phase and G1 to S transformation was detected by western blotting. C. PNN showed less effect on PCa cell apoptosis. Cells were stained with Annexin V-FITC and PI followed with flow cytometry assay. D. PNN-depletion didn’t trigger the activation of apoptotic effectors Caspase-3 and PARP. ***P < 0.001.
PNN promotes the tumorigenesis of prostate cancer cell in vivo
Our data suggested that PNN positively regulates cell growth in vitro and drove us to further explore whether it plays an important role in the modulation of prostate cancer tumorigenesis and progression in vivo. To confirm the impact of PNN in prostate cancer, we performed an in vivo experiments with subcutaneous xeno-transplanted tumor models based on BALB/C nude mice. As shown in Figure 4A-E, mice bearing PNN-depleted PC-3 cells showed a drastic regression of tumor growth compared with the control mice. Furthermore, western blotting assay revealed the reduction of expression of PNN and MCM2, a marker of proliferation, in tumors derived from PNN-depleted PC-3 cells (Figure 4F). Taken together, these findings demonstrated that PNN plays an important role in the tumorigenesis and progression of prostate cancer cells in vivo.
Figure 4.
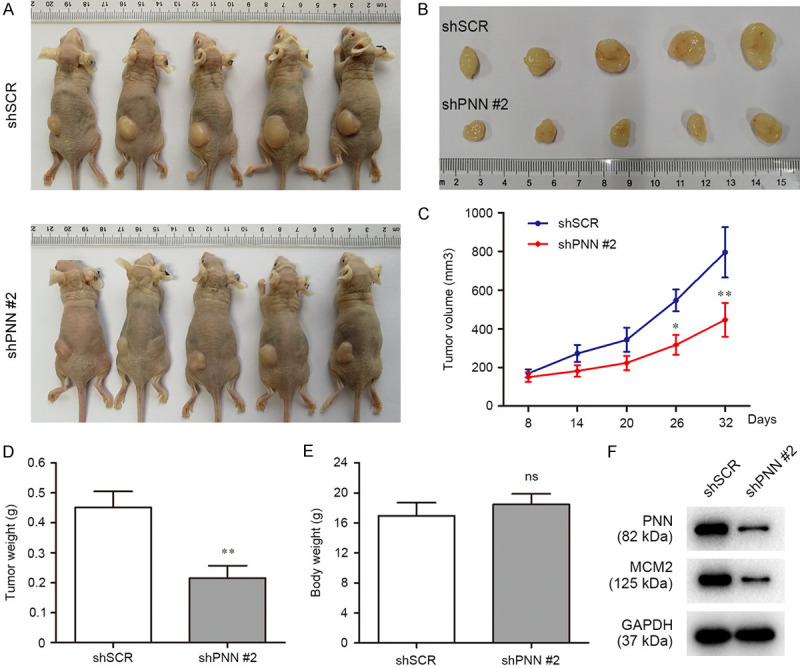
PNN modulates the tumorigenesis of prostate cancer cell in vivo. A. Images of nude mice. Thirty-two days after tumor cells were injected subcutaneously, the nude mice were sacrificed to collect the tumors. B. Images of excised tumors from nude mice. C. Tumor growth curves. On day 8 after injection of transfected PC3 cells, the tumor formation was monitored and measured intermittently. D and E. Average weight of xenograft tumor (D) and mice body (E). F. PNN and MCM2 expression in tumors. Reduction of the marker of proliferation MCM2 was indicated in xeno-transplanted tumors with PNN-depleted PC-3 cells. *P < 0.05, **P < 0.01.
PNN accelerates prostate cancer cell invasion and migration in vitro
Previous efforts reported that PNN facilitated metastasis of colorectal cancer and hepatocellular carcinoma [17,18], it led to us to further explore the functions of PNN on cell invasion and migration in prostate cancer. As shown in Figure 5A, PNN depletion suppressed invasion of PC-3 cell (Figure 5A). Wound healing and transwell assays indicated that depletion of PNN also suppressed prostate cancer cell migration (Figure 5B, 5C). On the contrary, we found that up-regulation of PNN in DU145 cells accelerated cell migration and invasion (Figure 5D, 5E). Collectively, these findings reveal that PNN accelerates prostate cancer cell invasion and migration in vitro.
Figure 5.
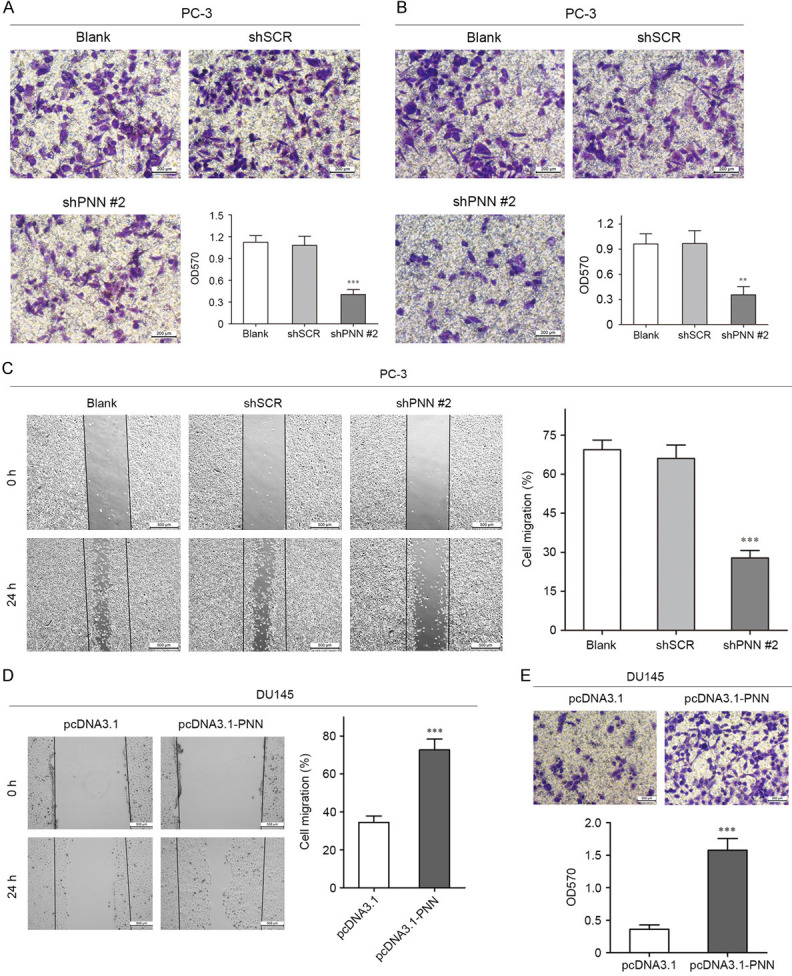
PNN accelerates prostate cancer cell invasion and migration in vitro. A. Transwell assay covered with matrigel revealed that PNN down-regulation reduced the invasive ability of PC-3 cells. The scale bar = 200 μm. B and C. Down-regulation of PNN significantly suppressed prostate cancer cell migration in Wound healing assay and Transwell assay. The scale bar = 200 μm (B), 500 μm (C). D and E. Wound healing assay and Transwell assay demonstrated that up-regulation of PNN accelerated cell migration and invasion. The scale bar = 500 μm (D), 200 μm (E). **P < 0.01, ***P < 0.001.
PNN over-expression induces prostate cancer cell EMT
Tumor cell invasion and migration are multistep processes which are finely regulated [20-22]. The epithelial-to-mesenchymal transition (EMT) has been identified as an important process of tumor cell invasion and migration [23-25]. Therefore, we determined the effects of PNN on EMT in prostate cancer cell. As shown in Figure 6A, the expression of epithelial cell marker E-cadherin was elevated, whereas expression of mesenchymal cell markers N-cadherin and Vimentin, as well as matrix metalloproteinase MMP-2 and MMP-9 were reduced in PNN-depleted PC-3 cell (Figure 6A). However, the opposite results were found in DU145 cells when PNN was overexpressed (Figure 6B). Moreover, we detected the expression of EMT-related proteins in tumors derived from control and PNN-depleted xenografts, similar results were observed (Figure 6C). During EMT, polarized epithelial cells rearrange cytoskeleton, dissolve the cell-cell junctions and convert into non-polarized mesenchymal cells [26-28]. Here we found that depletion of PNN changed the morphology from long, polygonal form to spindle appearances in PC-3 cells (Figure 6D). These results revealed that PNN has a positive effect on EMT process.
Figure 6.
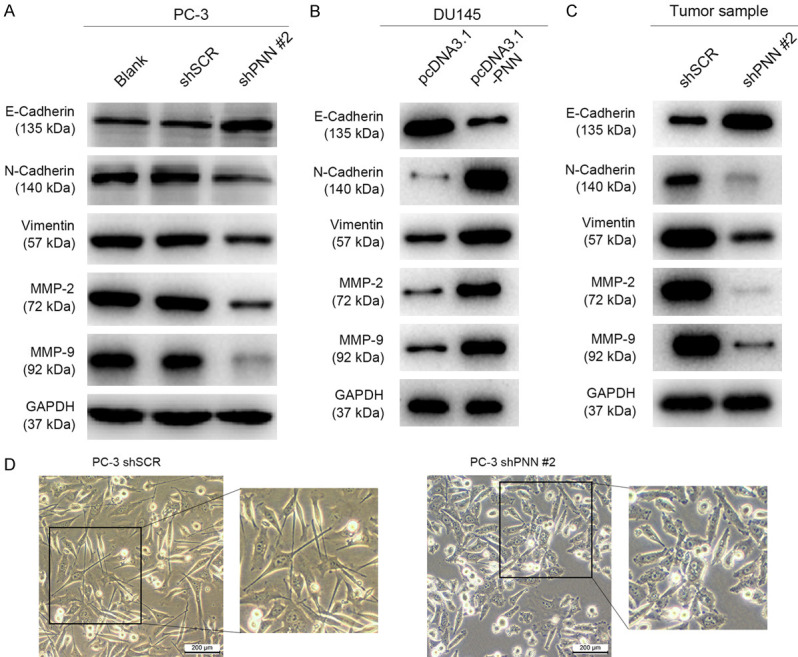
PNN over-expression induces prostate cancer cell EMT. A and B. The expression levels of epithelial-to-mesenchymal transition (EMT) related proteins in prostate cancer cells after altering PNN expression. PNN up-regulation triggers tumor cell epithelial-to-mesenchymal transition. C. EMT-related proteins level in xenografts. D. Down-regulation of PNN induced PC-3 cell morphological changes from long, polygonal form to spindle appearances. The scale bar = 200 μm.
PNN regulates prostate cancer cell proliferation and migration via activating CREB
Cyclic-AMP response element binding protein (CREB) is a proto-oncogenic transcription factor, which generally regulates various cell functions by enhancing the expression of target genes [29,30]. Our previous studies indicated that the over-expression and abnormal activation of CREB (phosphorylation at Ser133) promote tumorigenesis and tumor migration [31,32]. Herein, we observed that phosphorylated CREB at Ser133 was positively correlated with PNN levels (Figure 7A). Therefore, we postulated that overexpression of PNN promotes prostate cancer progression through accelerating CREB phosphorylation and activation.
Figure 7.
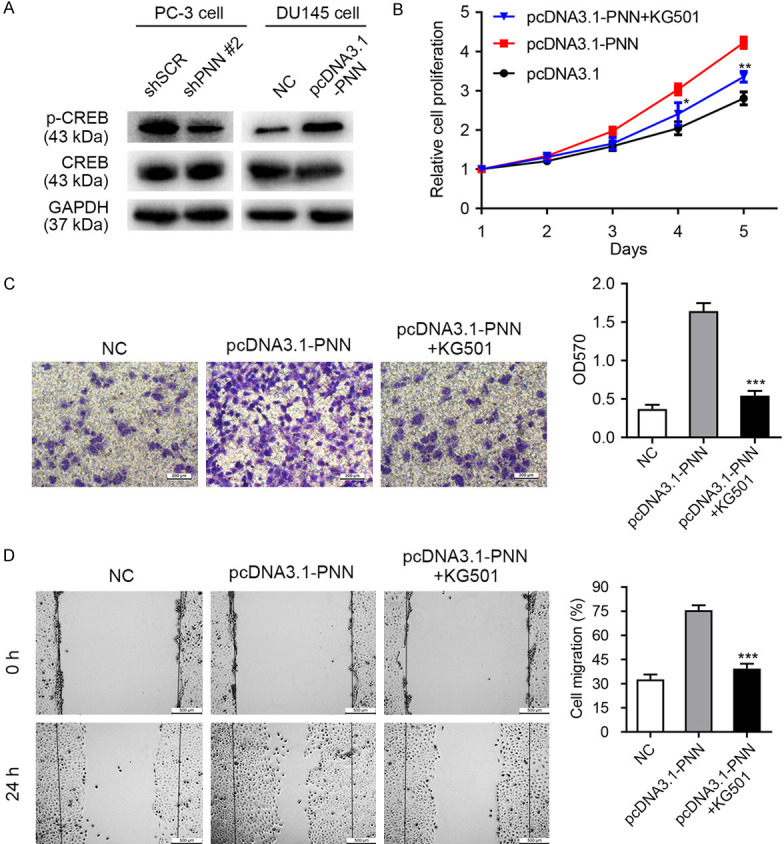
PNN regulates prostate cancer cell proliferation and migration via activating CREB. A. PNN over-expression accelerates CREB phosphorylation (Ser133) and activation. B-D. Phosphorylated CREB involved in PNN-mediated cell proliferation, invasion and migration. The MTS, transwell, and wound healing assays demonstrated that the PNN overexpression-caused cell phenotype changes could be reversed by CREB inhibitor KG501. The scale bar = 200 μm (C), 500 μm (D). *P < 0.05, **P < 0.01, ***P < 0.001.
To verify this hypothesis, small molecule inhibitor KG501 targeting CREB was utilized. The MTS, transwell and wound healing assays demonstrated that the PNN overexpression-caused cell phenotype changes could be reversed by CREB inhibitor KG501 (Figure 7B-D). Furthermore, these results were re-identified by siRNA-mediated CREB knockdown (Figure S1). Taken together, these results indicated that PNN modulates prostate cancer progression dependent on the activation of CREB.
PNN activates CREB via PI3K/AKT and ERK/MAPK signaling
CREB belongs to a large family of basic leucine zipper (bZIP)-containing transcription factors [33,34], a salient feature of this transcription factor is that its transcriptional activity is induced upon phosphorylation at Ser133 by many different serine/threonine (Ser/Thr) protein kinases to yield phosphorylated CREB (p-CREB) [35,36].
AKT/protein kinase B (PKB), a known kinase that phosphorylates CREB, is one of the most important proto-oncogene which regulates cell survival, proliferation, metabolism as well as cytoskeletal reorganization in human cancers including prostate cancer [37-39]. In the study, we demonstrated that PNN activated PI3K/AKT signaling as the levels of PI3K p110β, phosphorylated p85 (p-p85) and phosphorylated AKT (p-AKT) were in accordance with PNN expression (Figure 8A). According to these results, we considered that PNN activates CREB may via PI3K/AKT signaling.
Figure 8.
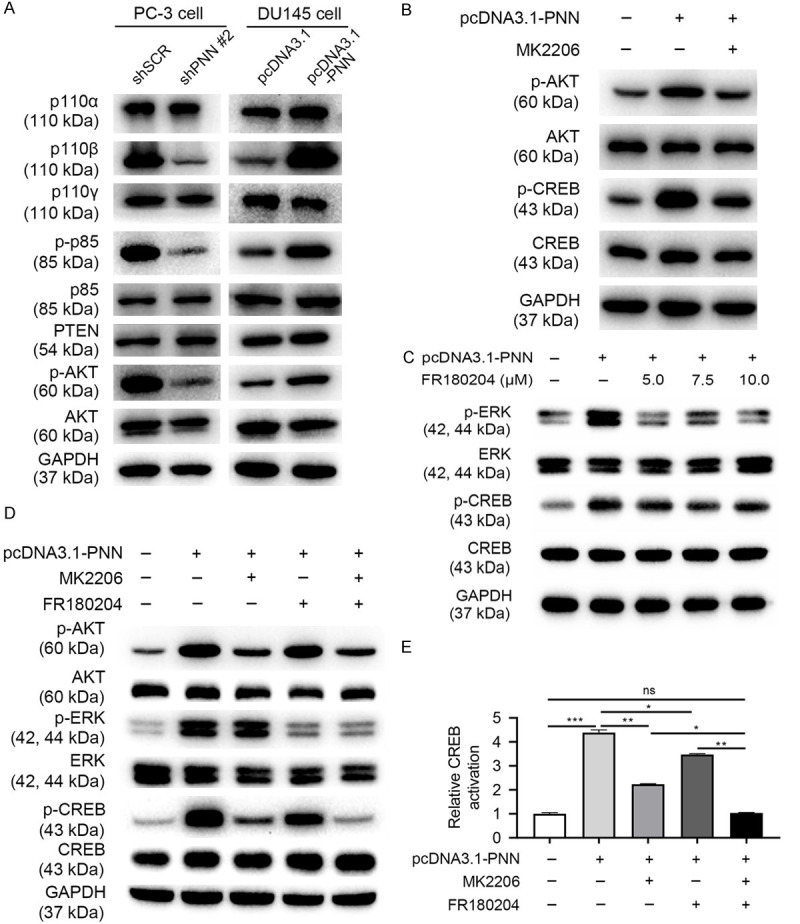
PNN activates CREB via PI3K/AKT and ERK/MAPK signaling. A. The activity and protein levels of PI3K/AKT pathway in cells after altering PNN expression. PNN activates PI3K/AKT signaling. B. PNN regulates CREB activation through PI3K/AKT signaling. C. ERK partly involves in PNN phosphorylating and activating CREB. D, E. PNN activates CREB via PI3K/AKT and ERK/MAPK signaling, and PI3K/AKT pathway play the major role. E. CREB activation is generated from pCREB/CREB normalized to negative control (pcDNA3.1-PNN (-), MK2206 (-) and FR180204 (-)).
To verify this hypothesis, we used MK2206 inhibiting AKT and determined p-CREB level. The western blotting assay showed that PNN overexpression-caused expression change of p-CREB partly rescued by MK2206 (Figure 8B). These results indicated that PI3K/AKT signaling take part in PNN regulating CREB activation. Meanwhile, these results also indicated that PNN may regulate CREB through other pathways.
ERK/MAPK is a serine/threonine protein kinase involved in many cellular programs, such as cell proliferation, differentiation, motility, and death [40,41], and research investigators consider it an important target in the diagnosis and treatment of cancer [42]. Furthermore, ERK/MAPK is also a known kinase that phosphorylates CREB [43,44]. In the study, we demonstrated that PNN activates ERK/MAPK signaling (Figure 8C). In addition, FR180204, a selective ERK inhibitor, could partly rescue PNN caused CREB activation (Figure 8C).
Additionally, we used MK2206 combined with FR180204 and then detected the level of p-CREB in PNN up-regulated cells. As shown in Figure 8D and 8E, combination of MK2206 and FR180204 could rescue PNN overexpression-caused CREB activation. Furthermore, PI3K/AKT pathway plays the major role in PNN regulating CREB activation when compare with ERK/MAPK signaling (Figure 8D, 8E).
Taken together, these results indicated that PNN activates CREB via PI3K/AKT and ERK/MAPK signaling, and PI3K/AKT pathway plays the major role.
Discussion
In the present study, we demonstrated that PNN was over-expressed in prostate cancer tissues and its over-expression was positively correlated with Gleason score, tumor stage and tumor metastasis (Figure 1; Tables 2, 3 and 4). These findings suggest that PNN may play an important role in progression and metastasis in prostate cancer.
Furthermore, we found ectopic expression of PNN promoted prostate cancer cell proliferation in vitro, while knockdown of PNN inhibited proliferation in vitro and tumorigenesis in vivo (Figures 2, 4). Cell cycle and apoptosis are the main events that affect cell growth. The experimental data displayed that PNN-silenced cell arrested at G0/G1 phase, and PNN knockdown distinctly repressed the expression of Cyclin D1, CDK2 and CDK6, which control the cell fate in G0/G1 phase and G1 to S transformation (Figure 3). However, the effect of PNN expression on cell apoptosis was not significant in the study (Figure 3). These results suggest that proliferation-promoting effect of PNN was through cell cycle control in prostate cancer.
Moreover, we found that over-expressed PNN accelerated prostate cancer cell invasion and migration in vitro (Figure 5). Tumor cell invasion and migration, the major events in tumor progression, are multistep processes [45,46]. It is well known that EMT had been demonstrated to be an important process of tumor cell invasion and migration. After EMT, tumor cells lose the polarity and gain motility, secrete matrix metalloproteinases to degrade extracellular matrix, then invade surrounding tissues and transfer to other organs [47,48]. In the study, we also found that PC-3 cell with down-regulation of PNN changed the morphology from long, polygonal form to spindle appearances (Figure 6). Furthermore, up-regulation of PNN induced mesenchymal cell markers N-cadherin, Vimentin, Fibronectin and matrix metalloproteinase MMP-2 and MMP-9 expression and suppressed epithelial cell marker E-cadherin. In addition, the opposite results were found in PNN silenced PC-3 cells (Figure 6). These results revealed that altered PNN expression induced both morphological and molecular biological EMT. Therefore, we believed that PNN accelerated invasion and migration via inducing tumor cell EMT in prostate cancer.
CREB belongs to a large family of basic leucine zipper-containing transcription factors [33,34], its over-expression and abnormal activation could promote tumorigenesis and tumor migration [31,32]. Herein, we found that PNN activates CREB and modulates prostate cancer progression via CREB (Figure 7). But how dose PNN regulate CREB activation? A salient feature of CREB is that its activation is induced upon phosphorylation at Ser133 by many different Ser/Thr protein kinases [35,36]. AKT and ERK are the important kinases that phosphorylate CREB [43,49].
PI3K/AKT signaling pathway is one of the most important pathways in human cancers [37-39] and has emerged as a noteworthy goal for cancer treatment, such as PI3K inhibitors Zydelig® (Idelalisib), Aliqopa® (Copanlisib) and Piqray® (Alpelisib) have been approved by FDA [50-55]. There are also lots of drugs are now in clinical trials including AKT phosphorylation inhibitors Ipatasertib (GDC-0068) and Capivasertib (AZD5363) [56-59].
In the study, we demonstrated that over-expression of PNN activated both PI3K/AKT and ERK/MAPK pathway. Moreover, inhibition of AKT and ERK activity completely reversed the effects of PNN on CREB activation (Figure 8). These findings suggested PNN activates CREB via PI3K/AKT and ERK/MAPK signaling. More important, PI3K/AKT pathway plays the major role in PNN regulating CREB activation.
Collectively, PNN was over-expressed in prostate cancer tissues and cells, and its over-expression indicated malignant progression. PNN promoted prostate cancer cell proliferation, invasion and migration via activating CREB through PI3K/AKT and ERK/MAPK pathway. In conclusion, PNN is a potential proto-oncogene and a potential therapeutic target for prostate cancer treatment.
Acknowledgements
We thank Dr. Yu-Qi Wang for English editing on our manuscript. The comments from Dr. Yu-qi Wang (School of Life Sciences, Fudan University) in modifying the manuscript were greatly appreciated. This study was supported by Natural Science Foundation of Zhejiang Province (Grant No. LY20H050002 to Qi Ma, Grant No. LQ20H160008 to Jun-feng Chen and Grant No. LY18H050003 to Jun-Hui Jiang), Ningbo Natural Science Foundation (Grant No. 2018A610297 to Qi Ma and Grant No. 202003N4259 to Ke-jie Wang), Ningbo Science and Technology Planning Project (Grant No. 202002N3192 to Qi Ma). and the Fund of Ningbo Clinical Research Center for Urological Disease (2019A21001).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 3.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 4.Zheng RS, Sun KX, Zhang SW, Zeng HM, Zou XN, Chen R, Gu XY, Wei WW, He J. Report of cancer epidemiology in China, 2015. Zhonghua Zhong Liu Za Zhi. 2019;41:19–28. doi: 10.3760/cma.j.issn.0253-3766.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Nowak DG, Katsenelson KC, Watrud KE, Chen M, Mathew G, D’Andrea VD, Lee MF, Swamynathan MM, Casanova-Salas I, Jibilian MC, Buckholtz CL, Ambrico AJ, Pan CH, Wilkinson JE, Newton AC, Trotman LC. The PHLPP2 phosphatase is a druggable driver of prostate cancer progression. J Cell Biol. 2019;218:1943–1957. doi: 10.1083/jcb.201902048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ouyang P, Sugrue SP. Identification of an epithelial protein related to the desmosome and intermediate filament network. J Cell Biol. 1992;118:1477–1488. doi: 10.1083/jcb.118.6.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ouyang P, Sugrue SP. Characterization of pinin, a novel protein associated with the desmosome-intermediate filament complex. J Cell Biol. 1996;135:1027–1042. doi: 10.1083/jcb.135.4.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi Y, Tabesh M, Sugrue SP. Role of cell adhesion-associated protein, pinin (DRS/memA), in corneal epithelial migration. Invest Ophthalmol Vis Sci. 2000;41:1337–1345. [PubMed] [Google Scholar]
- 9.Joo JH, Alpatov R, Munguba GC, Jackson MR, Hunt ME, Sugrue SP. Reduction of Pnn by RNAi induces loss of cell-cell adhesion between human corneal epithelial cells. Mol Vis. 2005;11:133–142. [PubMed] [Google Scholar]
- 10.Brandner JM, Reidenbach S, Franke WW. Evidence that “pinin”, reportedly a differentiation-specific desmosomal protein, is actually a widespread nuclear protein. Differentiation. 1997;62:119–127. doi: 10.1046/j.1432-0436.1997.6230119.x. [DOI] [PubMed] [Google Scholar]
- 11.Akin D, Newman JR, McIntyre LM, Sugrue SP. RNA-seq analysis of impact of PNN on gene expression and alternative splicing in corneal epithelial cells. Mol Vis. 2016;22:40–60. [PMC free article] [PubMed] [Google Scholar]
- 12.Joo JH, Correia GP, Li JL, Lopez MC, Baker HV, Sugrue SP. Transcriptomic analysis of PNN- and ESRP1-regulated alternative pre-mRNA splicing in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2013;54:697–707. doi: 10.1167/iovs.12-10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joo JH, Ryu D, Peng Q, Sugrue SP. Role of Pnn in alternative splicing of a specific subset of lncRNAs of the corneal epithelium. Mol Vis. 2014;20:1629–1642. [PMC free article] [PubMed] [Google Scholar]
- 14.Leu S, Lin YM, Wu CH, Ouyang P. Loss of Pnn expression results in mouse early embryonic lethality and cellular apoptosis through SRSF1-mediated alternative expression of Bcl-xS and ICAD. J Cell Sci. 2012;125:3164–3172. doi: 10.1242/jcs.100859. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Kwok JS, Choi PW, Liu M, Yang J, Singh M, Ng SK, Welch WR, Muto MG, Tsui SK, Sugrue SP, Berkowitz RS, Ng SW. Pinin interacts with C-terminal binding proteins for RNA alternative splicing and epithelial cell identity of human ovarian cancer cells. Oncotarget. 2016;7:11397–11411. doi: 10.18632/oncotarget.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Kwok JS, Choi PW, Liu M, Yang J, Singh M, Ng SK, Welch WR, Muto MG, Tsui SK, Sugrue SP, Berkowitz RS, Ng SW. Correction: pinin interacts with C-terminal binding proteins for RNA alternative splicing and epithelial cell identity of human ovarian cancer cells. Oncotarget. 2017;8:12533. doi: 10.18632/oncotarget.15321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei Z, Ma W, Qi X, Zhu X, Wang Y, Xu Z, Luo J, Wang D, Guo W, Li X, Xin S, Yu J, Li G. Pinin facilitated proliferation and metastasis of colorectal cancer through activating EGFR/ERK signaling pathway. Oncotarget. 2016;7:29429–29439. doi: 10.18632/oncotarget.8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang X, Sun D, Dong C, Tian Y, Gao Z, Wang L. Pinin associates with prognosis of hepatocellular carcinoma through promoting cell proliferation and suppressing glucose deprivation-induced apoptosis. Oncotarget. 2016;7:39694–39704. doi: 10.18632/oncotarget.9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, Psaila B, Kaplan RN, Bromberg JF, Kang Y, Bissell MJ, Cox TR, Giaccia AJ, Erler JT, Hiratsuka S, Ghajar CM, Lyden D. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. 2017;17:302–317. doi: 10.1038/nrc.2017.6. [DOI] [PubMed] [Google Scholar]
- 21.Zhang C, Xie C, Wang X, Huang Y, Gao S, Lu J, Lu Y, Zhang S. Aberrant USP11 expression regulates NF90 to promote proliferation and metastasis in hepatocellular carcinoma. Am J Cancer Res. 2020;10:1416–1428. [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng Y, Wang C, Song A, Jiang F, Zhou J, Li G, Zhang W, Ye J, Ding X, Zhang W, Du Y, Zhang H, Wu H, Song X, Wu Y. CMTM6 promotes cell proliferation and invasion in oral squamous cell carcinoma by interacting with NRP1. Am J Cancer Res. 2020;10:1691–1709. [PMC free article] [PubMed] [Google Scholar]
- 23.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 24.Sheng W, Shi X, Lin Y, Tang J, Jia C, Cao R, Sun J, Wang G, Zhou L, Dong M. Musashi2 promotes EGF-induced EMT in pancreatic cancer via ZEB1-ERK/MAPK signaling. J Exp Clin Cancer Res. 2020;39:16. doi: 10.1186/s13046-020-1521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang R, Shi H, Ren F, Feng W, Cao Y, Li G, Liu Z, Ji P, Zhang M. MicroRNA-338-3p suppresses ovarian cancer cells growth and metastasis: implication of Wnt/catenin beta and MEK/ERK signaling pathways. J Exp Clin Cancer Res. 2019;38:494. doi: 10.1186/s13046-019-1494-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, Van Keymeulen A, Brown D, Moers V, Lemaire S, De Clercq S, Minguijon E, Balsat C, Sokolow Y, Dubois C, De Cock F, Scozzaro S, Sopena F, Lanas A, D’Haene N, Salmon I, Marine JC, Voet T, Sotiropoulou PA, Blanpain C. Identification of the tumour transition states occurring during EMT. Nature. 2018;556:463–468. doi: 10.1038/s41586-018-0040-3. [DOI] [PubMed] [Google Scholar]
- 27.Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20:69–84. doi: 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- 28.Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, Campbell K, Cano A, Casanova J, Christofori G, Dedhar S, Derynck R, Ford HL, Fuxe J, García de Herreros A, Goodall GJ, Hadjantonakis AK, Huang RJY, Kalcheim C, Kalluri R, Kang Y, Khew-Goodall Y, Levine H, Liu J, Longmore GD, Mani SA, Massagué J, Mayor R, McClay D, Mostov KE, Newgreen DF, Nieto MA, Puisieux A, Runyan R, Savagner P, Stanger B, Stemmler MP, Takahashi Y, Takeichi M, Theveneau E, Thiery JP, Thompson EW, Weinberg RA, Williams ED, Xing J, Zhou BP, Sheng G EMT International Association (TEMTIA) Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2020;21:341–352. doi: 10.1038/s41580-020-0237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 30.Sandoval S, Kraus C, Cho EC, Cho M, Bies J, Manara E, Accordi B, Landaw EM, Wolff L, Pigazzi M, Sakamoto KM. Sox4 cooperates with CREB in myeloid transformation. Blood. 2012;120:155–165. doi: 10.1182/blood-2011-05-357418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhuang H, Meng X, Li Y, Wang X, Huang S, Liu K, Hehir M, Fang R, Jiang L, Zhou JX, Wang P, Ren Y. Cyclic AMP responsive element-binding protein promotes renal cell carcinoma proliferation probably via the expression of spindle and kinetochore-associated protein 2. Oncotarget. 2016;7:16325–16337. doi: 10.18632/oncotarget.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Ren Y, Zhuang H, Meng X, Huang S, Li Y, Hehir M, Wang P. Decrease of phosphorylated proto-oncogene CREB at Ser 133 site inhibits growth and metastatic activity of renal cell cancer. Expert Opin Ther Targets. 2015;19:985–995. doi: 10.1517/14728222.2015.1053208. [DOI] [PubMed] [Google Scholar]
- 33.Montminy MR, Bilezikjian LM. Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature. 1987;328:175–178. doi: 10.1038/328175a0. [DOI] [PubMed] [Google Scholar]
- 34.Montminy MR, Sevarino KA, Wagner JA, Mandel G, Goodman RH. Identification of a cyclic-AMP-responsive element within the rat somatostatin gene. Proc Natl Acad Sci U S A. 1986;83:6682–6686. doi: 10.1073/pnas.83.18.6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 36.Vinson CR, Sigler PB, McKnight SL. Scissors-grip model for DNA recognition by a family of leucine zipper proteins. Science. 1989;246:911–916. doi: 10.1126/science.2683088. [DOI] [PubMed] [Google Scholar]
- 37.Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170:605–635. doi: 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20:74–88. doi: 10.1038/s41568-019-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu X, Yu Y, Zong K, Lv P, Gu Y. Up-regulation of IGF2BP2 by multiple mechanisms in pancreatic cancer promotes cancer proliferation by activating the PI3K/Akt signaling pathway. J Exp Clin Cancer Res. 2019;38:497. doi: 10.1186/s13046-019-1470-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–3239. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 41.Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 43.Ginty DD, Bonni A, Greenberg ME. Nerve growth factor activates a Ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation of CREB. Cell. 1994;77:713–725. doi: 10.1016/0092-8674(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 44.Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb MJ. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 1996;15:4629–4642. [PMC free article] [PubMed] [Google Scholar]
- 45.Mierke CT. The matrix environmental and cell mechanical properties regulate cell migration and contribute to the invasive phenotype of cancer cells. Rep Prog Phys. 2019;82:064602. doi: 10.1088/1361-6633/ab1628. [DOI] [PubMed] [Google Scholar]
- 46.Polacheck WJ, Zervantonakis IK, Kamm RD. Tumor cell migration in complex microenvironments. Cell Mol Life Sci. 2013;70:1335–1356. doi: 10.1007/s00018-012-1115-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams ED, Gao D, Redfern A, Thompson EW. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat Rev Cancer. 2019;19:716–732. doi: 10.1038/s41568-019-0213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marcucci F, Stassi G, De Maria R. Epithelial-mesenchymal transition: a new target in anticancer drug discovery. Nat Rev Drug Discov. 2016;15:311–325. doi: 10.1038/nrd.2015.13. [DOI] [PubMed] [Google Scholar]
- 49.Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273:32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- 50.André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, Iwata H, Conte P, Mayer IA, Kaufman B, Yamashita T, Lu YS, Inoue K, Takahashi M, Pápai Z, Longin AS, Mills D, Wilke C, Hirawat S, Juric D SOLAR-1 Study Group. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med. 2019;380:1929–1940. doi: 10.1056/NEJMoa1813904. [DOI] [PubMed] [Google Scholar]
- 51.Juric D, Janku F, Rodon J, Burris HA, Mayer IA, Schuler M, Seggewiss-Bernhardt R, Gil-Martin M, Middleton MR, Baselga J, Bootle D, Demanse D, Blumenstein L, Schumacher K, Huang A, Quadt C, Rugo HS. Alpelisib plus fulvestrant in PIK3CA-altered and PIK3CA-wild-type estrogen receptor-positive advanced breast cancer: a phase 1b clinical trial. JAMA Oncol. 2019;5:e184475. doi: 10.1001/jamaoncol.2018.4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, Ghia P, Eradat H, Ervin T, Lamanna N, Coiffier B, Pettitt AR, Ma S, Stilgenbauer S, Cramer P, Aiello M, Johnson DM, Miller LL, Li D, Jahn TM, Dansey RD, Hallek M, O’Brien SM. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370:997–1007. doi: 10.1056/NEJMoa1315226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dreyling M, Morschhauser F, Bouabdallah K, Bron D, Cunningham D, Assouline SE, Verhoef G, Linton K, Thieblemont C, Vitolo U, Hiemeyer F, Giurescu M, Garcia-Vargas J, Gorbatchevsky I, Liu L, Koechert K, Pena C, Neves M, Childs BH, Zinzani PL. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann Oncol. 2017;28:2169–2178. doi: 10.1093/annonc/mdx289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dreyling M, Santoro A, Mollica L, Leppa S, Follows GA, Lenz G, Kim WS, Nagler A, Panayiotidis P, Demeter J, Ozcan M, Kosinova M, Bouabdallah K, Morschhauser F, Stevens DA, Trevarthen D, Giurescu M, Cupit L, Liu L, Kochert K, Seidel H, Pena C, Yin S, Hiemeyer F, Garcia-Vargas J, Childs BH, Zinzani PL. Phosphatidylinositol 3-kinase inhibition by copanlisib in relapsed or refractory indolent lymphoma. J. Clin. Oncol. 2017;35:3898–3905. doi: 10.1200/JCO.2017.75.4648. [DOI] [PubMed] [Google Scholar]
- 55.Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, Flinn IW, Flowers CR, Martin P, Viardot A, Blum KA, Goy AH, Davies AJ, Zinzani PL, Dreyling M, Johnson D, Miller LL, Holes L, Li D, Dansey RD, Godfrey WR, Salles GA. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370:1008–1018. doi: 10.1056/NEJMoa1314583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim SB, Dent R, Im SA, Espié M, Blau S, Tan AR, Isakoff SJ, Oliveira M, Saura C, Wongchenko MJ, Kapp AV, Chan WY, Singel SM, Maslyar DJ, Baselga J LOTUS investigators. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18:1360–1372. doi: 10.1016/S1470-2045(17)30450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oliveira M, Saura C, Nuciforo P, Calvo I, Andersen J, Passos-Coelho JL, Gil Gil M, Bermejo B, Patt DA, Ciruelos E, de la Peña L, Xu N, Wongchenko M, Shi Z, Singel SM, Isakoff SJ. FAIRLANE, a double-blind placebo-controlled randomized phase II trial of neoadjuvant ipatasertib plus paclitaxel for early triple-negative breast cancer. Ann Oncol. 2019;30:1289–1297. doi: 10.1093/annonc/mdz177. [DOI] [PubMed] [Google Scholar]
- 58.Jones RH, Casbard A, Carucci M, Cox C, Butler R, Alchami F, Madden TA, Bale C, Bezecny P, Joffe J, Moon S, Twelves C, Venkitaraman R, Waters S, Foxley A, Howell SJ. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2020;21:345–357. doi: 10.1016/S1470-2045(19)30817-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kolinsky MP, Rescigno P, Bianchini D, Zafeiriou Z, Mehra N, Mateo J, Michalarea V, Riisnaes R, Crespo M, Figueiredo I, Miranda S, Nava Rodrigues D, Flohr P, Tunariu N, Banerji U, Ruddle R, Sharp A, Welti J, Lambros M, Carreira S, Raynaud FI, Swales KE, Plymate S, Luo J, Tovey H, Porta N, Slade R, Leonard L, Hall E, de Bono JS. A phase I dose-escalation study of enzalutamide in combination with the AKT inhibitor AZD5363 (capivasertib) in patients with metastatic castration-resistant prostate cancer. Ann Oncol. 2020;31:619–625. doi: 10.1016/j.annonc.2020.01.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.