

Abstract
Pneumonia-induced lung injury and acute respiratory distress syndrome can develop because of an inappropriate inflammatory response to acute infections, leading to a compromised alveolar barrier. Recent work suggests that hospitalized patients with allergies/asthma are less likely to die of pulmonary infections and that there is a correlation between survival from acute respiratory distress syndrome and higher eosinophil counts; thus, we hypothesized that eosinophils associated with a type 2 immune response may protect against pneumonia-induced acute lung injury. To test this hypothesis, mice were treated with the type 2–initiating cytokine IL-33 intratracheally 3 days before induction of pneumonia with airway administration of a lethal dose of Staphylococcus aureus. Interestingly, IL-33 pretreatment promoted survival by inhibiting acute lung injury: amount of BAL fluid proinflammatory cytokines and pulmonary edema were both reduced, with an associated increase in oxygen saturation. Pulmonary neutrophilia was also reduced, whereas eosinophilia was strongly increased. This eosinophilia was key to protection; eosinophil reduction eliminated both IL-33–mediated protection against mortality and inhibition of neutrophilia and pulmonary edema. Together, these data reveal a novel role for eosinophils in protection against lung injury and suggest that modulation of pulmonary type 2 immunity may represent a novel therapeutic strategy.
Keywords: IL-33, eosinophilia, lung injury, neutrophils, Staphylococcus aureus
Pathogen-induced pneumonia develops in critically ill patients after lower respiratory infection or infection at extrapulmonary sites, resulting in hypoxemic respiratory failure (1). Approximately 1 million adult Americans are hospitalized annually with pneumonia, with high mortality rates at 30% to 40% (2–4). Pneumonia is a leading risk factor for developing acute respiratory distress syndrome (ARDS) and may be caused by a variety of infections, including bacterial, viral, and fungal infections, with Staphylococcus aureus being among the most common causes (5).
During pneumonia, invading pathogens elicit a robust proinflammatory response from the host in an effort to clear the pathogen, promoting release of proinflammatory cytokines/chemokines, including TNF-α, IL-6, G-CSF, and KC (6). Leukocytes, particularly neutrophils, infiltrate the lung and migrate into the airway in association with increased lung permeability and fluid, flooding the alveoli (7). This compromised alveolar barrier is a sine qua non of acute lung injury (ALI) and is associated with hypoxia and dyspnea. In addition, pathogens themselves can cause direct lung injury, as in the case of S. aureus releasing α-hemolysin toxin, resulting in alveolar damage and pulmonary edema (8). Although neutrophils are one of the first leukocytes to respond to pathogen-induced inflammation, they can cause severe tissue damage upon accumulation in the lungs. Once activated, neutrophils will produce reactive oxygen species and extracellular traps in an effort to promote pathogen clearance (9). All of these processes can result in respiratory failure and hypoxemia, leading to multisystem organ failure in the most severe cases.
Several studies have suggested that antiinflammatory responses may counterregulate tissue damage induced by an overabundant proinflammatory response, and disruption in this balance of pro- and antiinflammatory responses may result in mortality (10). Studies from our laboratory, however, have focused on an alternative possibility: we postulate that type 2 (allergic) immunity may provide a beneficial response that acts to counterbalance the overabundant inflammation that leads to sepsis and ARDS.
In support of this hypothesis, we and others recently demonstrated that patients with type 2 diseases such as asthma or allergies were 50% less likely to die in the hospital when admitted for acute pulmonary infections (11, 12). Furthermore, eosinopenia among patients with ARDS was first recognized nearly 40 years ago and may be a risk factor for mortality, implying that eosinophils play an important beneficial role in ARDS (13–17). To test this hypothesis, we used a mouse model of S. aureus pneumonia to investigate the putative protection by type 2 immunity during ALI. We find that prior activation of the pulmonary type 2 response using intratracheal treatment with IL-33 protects mice from lethal S. aureus–induced pneumonia. Specifically, the eosinophilia of the type 2 response inhibited pulmonary inflammation and lung injury, leading to a significant reduction in hypoxemia. This work suggests that modulation of type 2 immunity during acute pulmonary infections may represent a novel therapeutic adjunct to reduce the development of ARDS and pneumonia-associated mortality.
Methods
Mouse Studies
C57BL/6J mice and IL-5−/− mice on a C57BL/6J background were purchased from Jackson Laboratories and maintained in house. iPHIL mice on a C57BL/6J background were a generous gift from Drs. Nancy and James Lee (Mayo Clinic). For all experiments, mice were 6- to 9-week-old females. All animals were housed in specific pathogen–free conditions, and animal procedures were approved by the University of Chicago Institutional Animal Care and Use Committee as outlined in the levels “Guide for the Care and Use of Laboratory Animals,” published by the National Academy Press.
Murine Model for S. aureus–induced Pneumonia
Bacteria were cultured according to previously published methods (18, 19). An overnight 3-ml culture was grown in tryptic soy broth to a stationary phase. The following day, a 1:100 dilution of the overnight culture was used to subculture bacteria to a log phase with an optical density reading between 0.45 and 0.65. Bacteria were washed and resuspended in sterile PBS. Mice were infected with 2 × 108 USA300 cfu/50 μl intratracheally. Mice were treated intratracheally with either PBS or 100 ng of murine IL-33 (BioLegend) on Days −3 to −1 and were infected on Day 0. For survival analysis, mice were weighed and monitored every day for 7 days. For all other experiments, noninfected control animals received either PBS or 100 ng/d of IL-33 for 3 days.
Bacterial Burden Quantification
Mice were killed and lungs were harvested. Lungs were homogenized in 1 ml of sterile PBS on ice and plated using serial dilutions on tryptic soy agar plates for incubation overnight at 37°C. Colonies were quantified and normalized to tissue weight.
Flow Cytometry Analysis
Upon euthanasia, BAL fluid was harvested at 6, 18, and 36 hours post-infection (p.i.) Noninfected control animals were killed on the same day. For BAL fluid collection, 800 μl of cold, sterile PBS was gently lavaged and aspirated from the airway via a tracheal cannula. For flow cytometry analysis, this was repeated for a total of four times, and all four fractions were pooled together. Cells were resuspended in single-cell suspension (500,000 cells) in 100 μl of PBS containing 0.1% sodium azide and 0.2% BSA. Cells were incubated with anti-CD16/CD32 (2.4G2) to block against nonspecific antibody binding. For quantification of granulocytes and red blood cells (RBCs), the following antibodies were used for cell-surface staining: CD45.2 APC-Cy7 (clone 30-F11), Ly6G BrilliantViolet 711 (clone 1A8), CD11b-BrilliantViolet 510 (clone M1/70), CD11c PE-Cy7 (clone N418), F4/80 APC (clone BM8), Ter-119 FITC (clone 116206; all from Biolegend), and Siglec F PE (clone E50-2440; BD Biosciences). We selected for live cells by gating on single-cell, DAPI (Sigma-Aldrich)–negative, CD45-positive cells. Flow cytometry data were collected using a BD Biosciences LSRFortessa flow cytometer and were analyzed with FlowJo software (Tree Star, Inc.). All instruments are maintained by the University of Chicago Flow Cytometry and Antibody Technology Core Facility.
Quantification of BAL Fluid Albumin and Cytokines
Upon euthanasia, BAL fluid was harvested as previously stated. Albumin and cytokine measurements were performed on the supernatant of the first wash. BAL fluid albumin was quantified using an ELISA as per the manufacturer’s protocol (Bethyl Laboratories, Inc.) Cytokine analysis was performed on BAL fluid using a multiplex assay according to the manufacturer’s instructions (R&D Systems).
FITC–Dextran Assay
Lung permeability was assessed using pulmonary leakage of a fluorescent dextran (FITC–dextran; Sigma-Aldrich) according to previously published methods (20, 21). Briefly, mice were intratracheally administered 10 mg/kg of FITC–dextran (Sigma-Aldrich) in a 50-μl volume. After 1 hour, blood was collected through the retroorbital sinus and serum was isolated. The fluorescence intensity of FITC in the serum was measured using a fluorescent plate reader.
Histological Analysis
Lungs were perfused with PBS and the mouse left lobe was fixed in 10% formalin, which was followed by staining with hematoxylin and eosin. For all histology scoring, lungs were taken from mice in which BAL fluid isolation was not performed, the scorer was blinded to the mouse treatment, and slides were presented in a random order. Images were taken and analyzed with a Pannoramic Viewer (3DHISTECH Ltd.) (https://www.3dhistech.com/research/software/software-downloads/). Lung edema was scored as previously described (18). The edema scoring schematic was as follows: 0 = no RBCs present, 1 = small patch of airway with RBCs infiltrating, 2 = one-eighth of the lung lobe contains RBCs, 3 = one-fourth of the lung lobe contains RBCs, 4 = one-half of the lung lobe contains RBCs, and 5 = the entire lobe shows RBCs in airways. Inflammation was scored as follows: 0 = few to almost no infiltrating leukocytes, 1 = small patch of leukocytes signifying inflammation, 2 = one-eighth of the lung lobe contains inflammation, 3 = one-fourth of the lung lobe contains inflammation, 4 = one-half of the lung lobe contains inflammation, and 5 = the entire lobe is inflamed (18, 21).
Measurement of Mouse Oximetry
The cardiopulmonary health status of mice was assessed by measuring mouse oximetry in live, anesthetized mice using the MouseOx Plus system (Starr Life Sciences) in accordance with the manufacturer’s instructions and previously published methods (22). Mice were anesthetized with ketamine/xylazine, and hair around the neck was removed by shaving and with Nair (Church and Dwight Co., Inc.) hair removal cream. Oxygen saturation was measured for 10 minutes per mouse. The average reading for 10 minutes for each mouse was reported.
Eosinophil Reduction
Eosinophil counts were reduced on the basis of modification of previously published methods (18, 23). Briefly, iPHIL mice were treated with intratracheal PBS or IL-33 on Days −3 to −1. On Day −1, eosinophils were depleted using intraperitoneal injection of diphtheria toxin (DT) (45 ng/g of body weight; Sigma-Aldrich). Mice were infected with 2 × 108 cfu intratracheally on Day 0. For survival analysis, mice were treated with DT every other day to maintain decreased eosinophil counts and were monitored for survival for 7 days p.i. For flow cytometry analysis, all groups were killed at 18 hours p.i.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism software, and P values less than 0.05 were considered to indicate significance. For comparisons of two groups, an unpaired Student’s two-tailed t test (parametric) or Mann-Whitney test (nonparametric) was performed. Mann-Whitney tests were used when variances were not normally distributed, as determined using an F test. For comparisons of three or more groups, a one-way ANOVA with Sidak post hoc multiple comparison was conducted. For comparisons of two or more groups at multiple time points, a two-way ANOVA was used with a Tukey’s multiple comparison test. Survival analysis was performed using a log-rank (Mantel-Cox) test. Error bars represent the SEM.
Results
IL-33 Pretreatment Protects against Mortality During S. aureus–induced Pneumonia
To determine whether the type 2 response may be protective against S. aureus–induced pneumonia, we used IL-33 to activate a pulmonary, innate, type 2 inflammatory response, which was followed by lethal S. aureus lung infection (18, 24). C57BL/6 mice were treated intratracheally with IL-33 or PBS for 3 days before infection. On Day 0, both groups were intratracheally infected with a lethal dose of S. aureus USA300, a community-associated methicillin-resistant strain, and monitored for survival for 7 days (Figure 1A). Remarkably, none of the IL-33–treated mice died, whereas 50% of PBS-treated mice died (Figure 1B).
Figure 1.

IL-33 pretreatment protects against Staphylococcus aureus–induced pneumonia. C57BL/6 mice were treated intratracheal (i.t.) for 3 days with IL-33 or PBS. On Day 0, both groups were infected i.t. with 2 × 108 S. aureus cfu. Mice were monitored for survival. (A) Diagram demonstrating the experimental setup. (B) Kaplan-Meier survival curve. Statistical significance was determined using a log-rank (Mantel-Cox) test (n = 7–13 mice per group). (C) Bacterial burden in the lung at 6 and 18 hours post-infection (p.i.). The bacterial burden was measured in C57BL/6 mice treated with PBS or IL-33. Bacteria was quantified using serial dilution of colony-forming units (n = 4–9 mice per group). Significance was determined using an unpaired t test. *P < 0.05. cfu = colony-forming unit; S. aureus = Staphylococcus aureus.
To determine whether IL-33 pretreatment induced protection by promoting bacterial clearance, mice were treated with intratracheal IL-33 or PBS, and bacterial counts in the lung were quantified by serial dilution at 6 and 18 hours p.i. IL-33 pretreatment did not influence bacterial clearance, as no difference was found in lung colony-forming units between PBS and IL-33 treatment (Figure 1C). Given these results, we concluded that IL-33–mediated protection is independent of bacterial clearance and instead depends on modulating pathogen-induced inflammation early during infection.
IL-33 Pretreatment Protects against Bacteria-induced ALI
We investigated the impact of IL-33 pretreatment on ALI during S. aureus–induced pneumonia using several methods. As expected, S. aureus infection caused ALI, with increased BAL fluid albumin, lung permeability, and pulmonary edema compared with PBS-treated control animals (Figures 2A–2D). In comparison, IL-33 pretreatment attenuated ALI, as seen by lower BAL fluid albumin, lung permeability, and pulmonary edema than those seen in mice treated with PBS + S. aureus alone.
Figure 2.
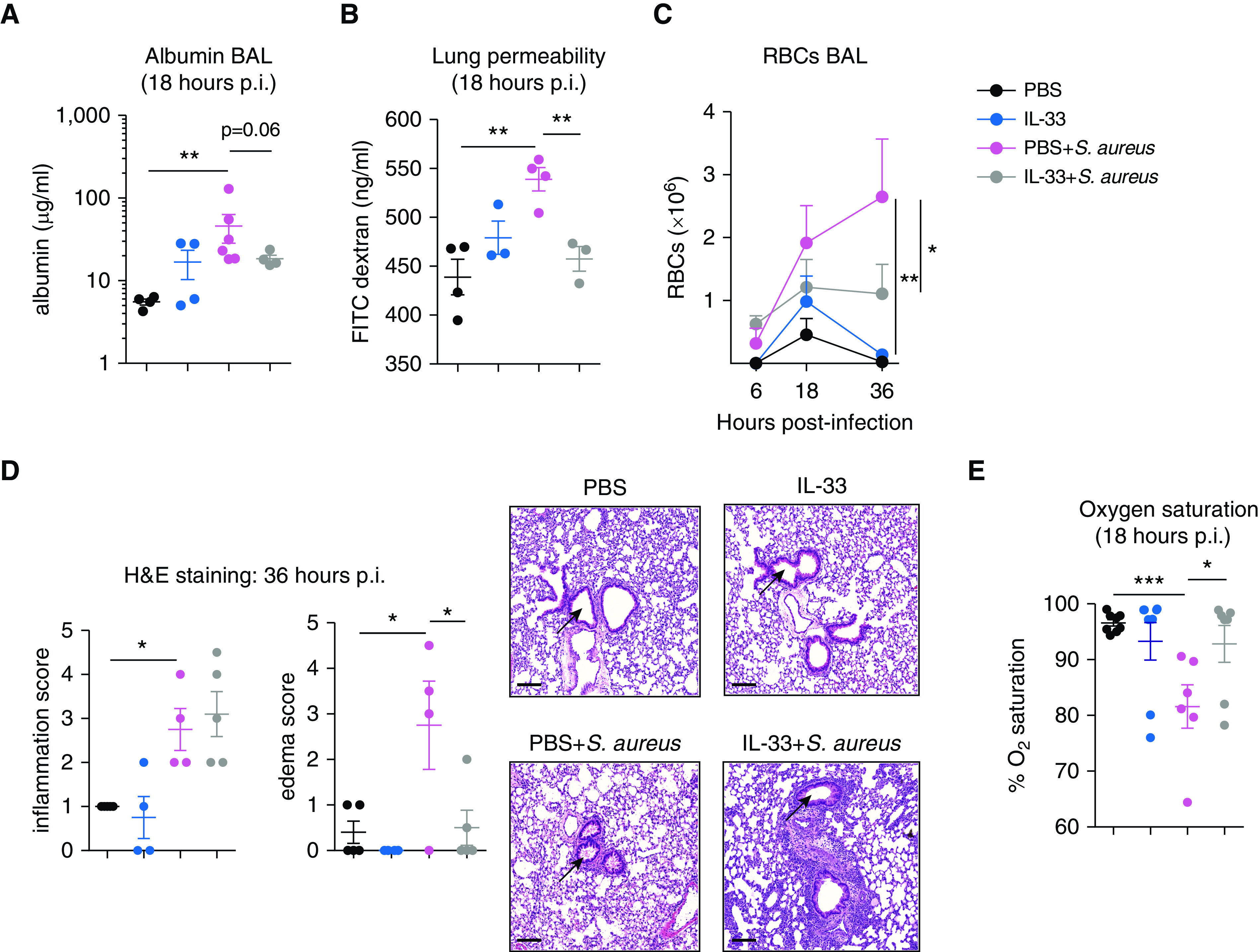
IL-33 pretreatment reduces S. aureus–induced lung injury. C57BL/6 mice were treated i.t. with PBS, IL-33, PBS + S. aureus, or IL-33 + S. aureus. (A) BAL fluid albumin was measured at 18 hours p.i. (B) Quantification of lung permeability using FITC–dextran leakage from alveoli into serum at 18 hours p.i. Pulmonary edema was quantified using (C) flow cytometry measurement of BAL fluid red blood cell (RBC) counts or (D) H&E staining of lung sections with arrows indicating the area assessed for edema within the airways. RBCs were measured in the BAL fluid at 6, 18 and 36 hours p.i. and defined as single cells, live CD45−Ter119+FSClo. (E) Oxygen saturation was measured in live, anesthetized mice at 18 hours p.i. for 10 minutes using the MouseOx Plus system. For A–C, representative graphs of two experiments with sample sizes of three to six per group are shown. For E, two experiments were pooled, with total sample sizes between six and eight per group. For C, statistical significance was determined using a two-way ANOVA with a Tukey post hoc test, whereas for A, B, D, and E, significance was determined using an unpaired t test. Scale bars, 100 μm. *P < 0.05, **P < 0.01, and ***P < 0.001. H&E = hematoxylin and eosin.
Mortality from ARDS is caused, in part, by the inability to adequately oxygenate the blood because of the overwhelming pulmonary edema. The resultant hypoxemia can lead to tissue injury and multisystem organ failure throughout the body. We therefore tested the effect of IL-33 pretreatment on S. aureus–induced hypoxemia by measuring pulse oximetry in live, anesthetized mice. Remarkably, infected mice treated with IL-33 maintained normal oxygen saturation, rescuing them from hypoxemia during pulmonary infection, in contrast to those infected with S. aureus alone (Figure 2E). Altogether, these findings demonstrate that prior induction of the innate type 2 inflammatory response inhibits lung injury and rescues mice from hypoxemia during pneumonia, thereby resulting in reduced mortality.
IL-33 Promotes Airway Eosinophilia while Inhibiting ALI Early during Infection
During pneumonia and ARDS, proinflammatory cytokines are released into the airway as part of the innate immune response to promote pathogen clearance (6). However, this inflammation also promotes destruction of the alveolar barrier. Leukocytes, cytokines, and RBCs flood the alveoli, inducing edema, respiratory distress, and hypoxemia. To further analyze the influence of IL-33 pretreatment on the proinflammatory response induced by S. aureus, we measured secreted inflammatory factors in the BAL fluid. As expected, S. aureus promoted secretion of several inflammatory factors, including G-CSF, IFN-γ, and IL-1α, with G-CSF being important for neutrophil development (Figure 3) (25). IL-33 pretreatment significantly inhibited G-CSF, IFN-γ, and IL-1α production and was associated with nonsignificant decreases in KC/CXCL1, TNF-α, and IL-6. IL-33 promoted IL-5 secretion, a known growth factor for eosinophils, irrespective of infection. Although S. aureus induced changes in IL-12, MCP-2, or MMP-8 levels, no difference was found between infected groups, indicating that IL-33 pretreatment does not suppress their secretion.
Figure 3.
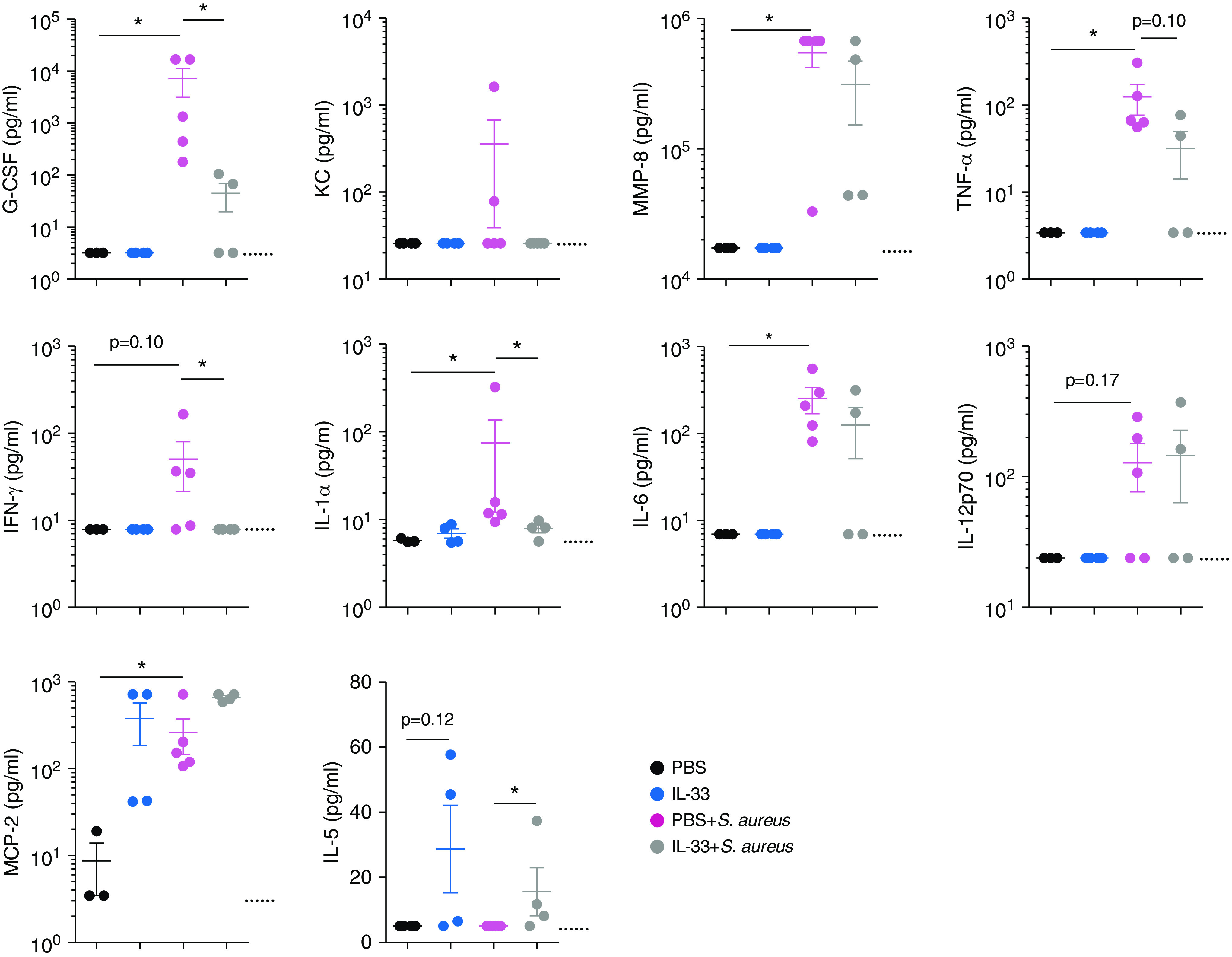
IL-33 pretreatment inhibits induction of early inflammatory factors after S. aureus–induced pneumonia. C57BL/6 mice were treated i.t. with PBS, IL-33, PBS + S. aureus, or IL-33 + S. aureus. All groups were killed at 18 hours p.i. BAL fluid cytokine and chemokine levels were quantified with a multiplex assay. Dotted lines indicate the limit of detection. Statistical significance was determined using a Mann-Whitney test. Sample sizes were three to five per group. *P < 0.05. G-CSF = granulocyte colony-stimulating factor; KC = keratinocyte-derived chemokine; MCP-2 = monocyte chemotactic protein-2; MMP-8 = matrix metalloproteinase-8.
To investigate the mechanism of how IL-33 inhibited ALI, airway cell infiltration was quantified at multiple time points during the course of infection. We measured counts of neutrophils (which are essential for bacterial clearance but are also associated with lung injury), eosinophils (which are activated during the IL-33–dependent type 2 response), and various lymphocyte populations. Although airway neutrophilia in the S. aureus–infected mice rapidly increased throughout the course of infection, IL-33 pretreatment suppressed this neutrophilia (Figure 4A). At 36 hours p.i., airway neutrophil counts peaked in S. aureus–infected mice, whereas counts in mice treated with IL-33 before infection remained significantly lower. Similar results were found in the lung digest, with neutrophil counts in IL-33–pretreated mice infected with S. aureus remaining at baseline (see Figure E1A in the data supplement). IL-33 treatment promoted eosinophilia in the airway and lung digest compared with PBS treatment, regardless of whether or not the mice were infected with S. aureus (Figures 4B and E1B). Interestingly, eosinophil counts were lower in S. aureus–infected mice treated with IL-33 than in uninfected mice treated with IL-33. No differences were found among macrophage, T-cell, or B-cell populations in the BAL fluid or lung digest among infected groups (data not shown). These data suggest that IL-33–mediated eosinophilia inhibited the neutrophilic response induced by bacterial pneumonia.
Figure 4.

S. aureus–induced neutrophilia during pneumonia is suppressed by IL-33 treatment in association with an increase in eosinophils. C57BL/6 mice were treated i.t. with PBS, IL-33, PBS + S. aureus, or IL-33 + S. aureus. All groups were killed, and flow cytometry was performed on BAL fluid. Shown are graphs depicting (A) neutrophil counts or (B) eosinophil counts at 6, 18, or 36 hours p.i. Neutrophils were defined as singlet, live, CD11b+Ly6Ghi F4/80−. Eosinophils were defined as singlet, live, CD45+Ly6GloCD11c−SiglecF+SSChi. Significance was determined using a two-way ANOVA with a Tukey post hoc test. Each time point is a representative experiment of two independent experiments, with sample sizes of three to six per group. *P < 0.05, **P < 0.001, and ****P < 0.0001.
IL-5–Dependent Eosinophilia Is Required for the Protective Effect of IL-33 during S. aureus–induced ALI
We have previously demonstrated that IL-33 promotes eosinophilia via the release of IL-5 from type 2 innate lymphoid cells (ILC2s) in the lung (26). We therefore hypothesized that IL-33–mediated protection would be abrogated in IL-5−/− mice because of the inability to recruit eosinophils to the lungs in response to IL-33. IL-5−/− mice or heterozygote IL-5+/− littermate control animals were intratracheally treated with IL-33 or PBS for 3 days, which was followed by intratracheal infection with S. aureus. As shown in Figure 5, IL-33 was unable to rescue IL-5−/− mice from lethal S. aureus pulmonary infection, suggesting that activation of the innate type 2 response is important for protection.
Figure 5.

IL-5 expression is required for IL-33–mediated survival of S. aureus–induced pneumonia. Shown is a Kaplan-Meier survival curve for IL-5+/− or IL-5−/− mice. Mice were treated i.t. with PBS or IL-33 for 3 days, which was followed by i.t. infection with S. aureus on Day 0. All groups were monitored for survival for 7 days. Statistical significance was determined using a log-rank (Mantel-Cox) test. The graph is pooled from two experiments (n = 5–9 mice per group). *P < 0.05.
To determine whether eosinophils are required for IL-33–mediated protection against ALI, we used the iPHIL mouse model to allow for specific reduction of eosinophils during S. aureus pulmonary infection. iPHIL mice express the DT receptor under the control of the eosinophil peroxidase promoter, which leads to eosinophil cell death upon systemic administration of DT (23). iPHIL mice were intratracheally treated with IL-33 or PBS, followed by intratracheal infection with S. aureus, with or without administration of DT (Figure 6A). Eosinophil reduction (shown in Figure E2) abrogated IL-33–mediated protection, with reduced survival noted among iPHIL mice treated with IL-33 + DT + S. aureus (Figure 6B). iPHIL mice infected with S. aureus were treated with DT as a control, and no difference was found in mortality between this group and the PBS + S. aureus group, indicating that DT administration did not influence mortality (Figure 6B). In addition, eosinophil reduction resulted in an increase in both pulmonary neutrophilia and pulmonary edema associated with S. aureus–induced ALI, as measured by flow cytometry (Figures 6C and 6D). Taken together, these data demonstrate that IL-5–dependent eosinophilia protects against S. aureus–dependent mortality by preventing ALI (Figure 7).
Figure 6.
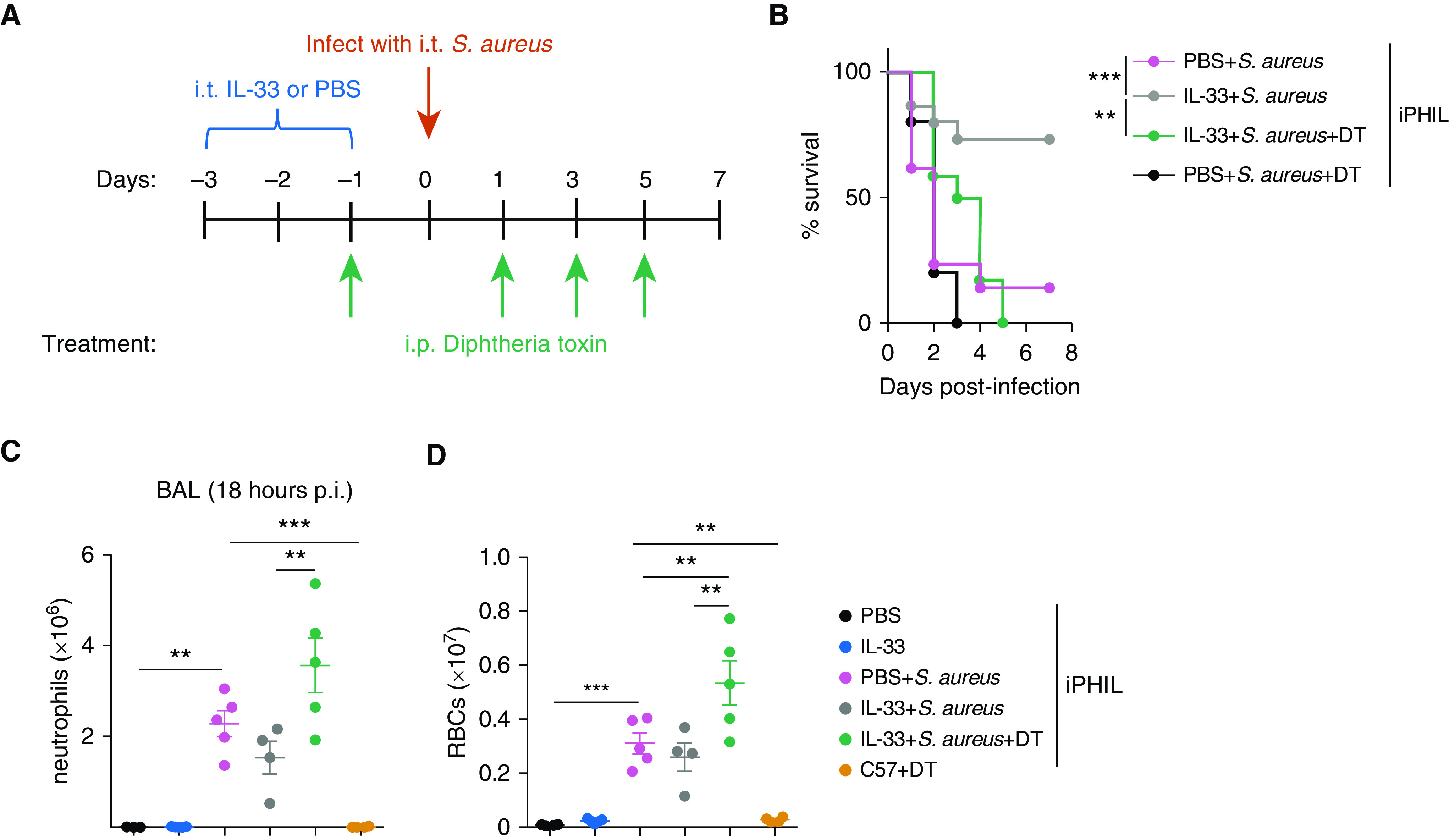
Eosinophils are required for survival and inhibition of S. aureus–induced neutrophilia and RBC infiltration during pneumonia. (A) Diagram of the experimental setup for the survival curve. On Days −3 to −1, iPHIL mice were treated i.t. with PBS or IL-33. S. aureus + diphtheria toxin (DT) mice received DT intraperitoneally on Day −1 and every other day through Day 5. All groups were infected i.t. with S. aureus on Day 0. All groups were monitored for survival for 7 days. (B) Kaplan-Meier survival curve. Statistical significance was determined using a log-rank (Mantel-Cox) test. The graph is pooled from a sample size of four experiments, with n = 10–21 per group. (C) BAL fluid neutrophil and (D) RBC counts were measured in iPHIL mice depleted of eosinophils. The treatment schedule was similar to the setup in A, with the exception of all groups being killed at 18 hours p.i. for flow cytometry analysis of BAL fluid. As a negative control, C57BL/6 mice were treated with DT alone. Statistical analysis was performed with a one-way ANOVA. Graphs are representative of two independent experiments, with n = 3–5 per group. **P < 0.01 and ***P < 0.001. iPHIL = inducible eosinophil-deficient.
Figure 7.
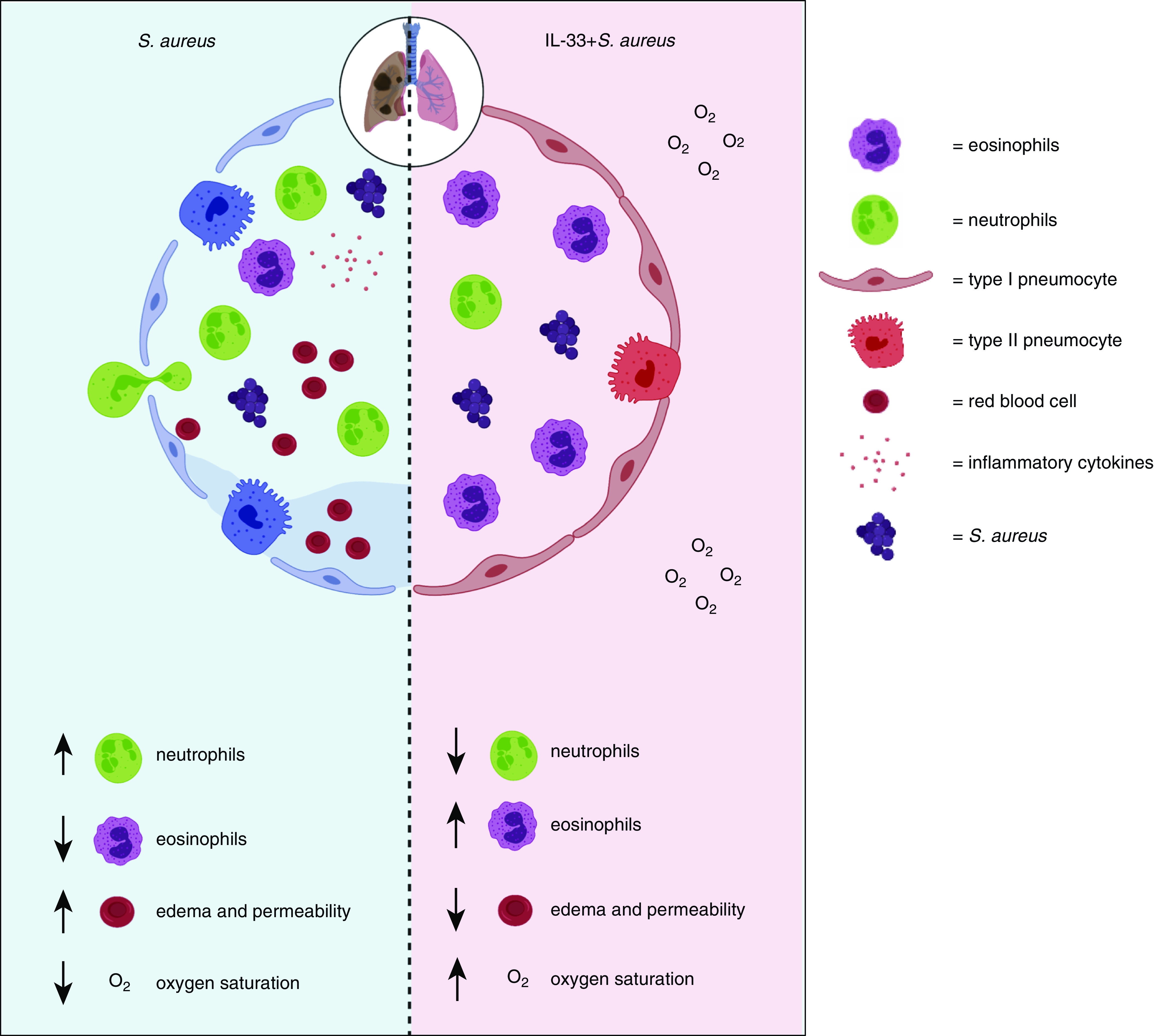
IL-33–mediated eosinophilia protects against lethal S. aureus pneumonia by inhibiting lung injury, with up arrows indicating an increase and down arrows indicating a decrease of the indicated feature. During S. aureus–induced pneumonia, neutrophils extravasate from the blood and interstitium into alveoli early during the immune response to promote bacterial clearance. However, neutrophils can also cause tissue damage, resulting in destruction of epithelial cells lining the alveoli. This causes infiltration of inflammatory cytokines, increased pulmonary edema, and finally hypoxemia. Our study demonstrates that IL-33–mediated eosinophilia inhibited induction of airway inflammatory cytokines, neutrophilia, pulmonary edema, lung permeability, and hypoxemia.
Discussion
Bacterial pneumonia and subsequent ARDS is a serious respiratory disorder with high mortality rates and few treatments currently available beyond antibiotics and supportive care. In this study, we demonstrated that prior activation of the type 2 inflammatory response with IL-33 promoted survival in S. aureus–induced pneumonia by inhibiting pulmonary inflammation, ALI, and hypoxemia. Importantly, IL-33 induction of eosinophilia was critical for the protection against S. aureus–induced mortality, demonstrating a novel and potentially beneficial role for these cells.
Our observation that activation of the pulmonary type 2 inflammatory response protects mice against development of ALI due to lethal S. aureus pneumonia is consistent with several recent clinical studies. In an analysis of several national inpatient data sets, Zein and colleagues showed that patients with asthma hospitalized for pneumonia were much less likely to develop sepsis or die while in the hospital (11). Furthermore, in our own analysis of hospitalizations for pulmonary infections at the University of Chicago, we found that the mortality rate of patients with any type 2–mediated immune disease (defined as allergic rhinitis, atopy, food allergies, or asthma) was only 4.5%, compared with a 10.4% mortality rate for patients without type 2–mediated immune diseases (12). We have also demonstrated that patients who survive hospitalization with S. aureus infections have a higher number of circulating T-helper cell type 2 (Th2) lymphocytes, and lower numbers of Th17 cells and neutrophils, compared with nonsurvivors (18, 27). In terms of the recent coronavirus disease (COVID-19) pandemic, several studies indicate that low eosinophil counts are associated with COVID-19 mortality, whereas recovery from COVID-19 is associated with a restoration of eosinophil counts (13–15, 28). Finally, two recent studies indicate that patients with allergies/asthma are underrepresented among those with diagnosed COVID-19, suggesting a protective effect of allergic responses on acute viral lung injury (13, 29).
In exploring possible mechanisms for this protective effect, we observed that several proinflammatory cytokines and proteins, including G-CSF, IFN-γ, TNF-α, MMP-8, IL-6, KC/CXCL1, and IL-1α, were elevated during S. aureus lung infection and ALI. However, levels of many of these were decreased when mice had an active pulmonary type 2 immune response before infection, indicating that stimulation of type 2 immunity markedly impacts the host response to a lethal pathogen. The reduction of these cytokines was associated with a significant reduction in pulmonary neutrophils; given that neutrophils and neutrophil extracellular traps can cause significant pulmonary injury, the reduction in neutrophils may contribute to the reduced lung injury observed in these mice (9, 30, 31). Notably, these results agree with those obtained by Dulek and colleagues, who used a model of ovalbumin sensitization/challenge, followed by Klebsiella pneumoniae pulmonary infection, and observed similar decreases in cytokines and neutrophil counts (32). However, they used neutrophil depletion to suggest that the type 2 response recruits “highly activated neutrophils” to the airways; we would posit instead, on the basis of the current work and our prior studies, that removal of neutrophils completely compromises the host, regardless of the type 2 response (18). As such, both an inadequate and an overwhelming neutrophil response can be detrimental during infection, and in this work, we show that the type 2 response can partially attenuate the neutrophilic response and mobilize tissue repair responses, resulting in protection against lethality. It remains unclear, however, how the type 2 response (and specifically IL-33 pretreatment) leads to a reduction in neutrophil-inducing proinflammatory cytokines.
IL-33 is known to activate ILC2s, which, in turn, secrete the canonical type 2 cytokines IL-5 and IL-13, leading to eosinophil recruitment, smooth muscle hyperplasia, and the promotion of mucus secretion from goblet cells (26). Mucus secretion may also contribute to the protection against S. aureus infection, given that mucus acts as a barrier to trap pathogens; indeed, mice lacking the mucus protein Muc5b die of overwhelming S. aureus pneumonia (33, 34). IL-33 can also promote lung repair in a non–eosinophil-dependent manner through induction of amphiregulin from ILC2s (35). Other elements of the type 2 immune response, such as IL-5, have been shown to protect against lung injury and promote survival in sepsis (18, 21, 36). We confirmed that IL-5 was important in our model as well, observing that IL-33 was unable to protect IL-5−/− mice from lethal S. aureus pulmonary infection. We therefore focused our attention on the potential beneficial role of eosinophils, given recent clinical studies demonstrating their association with survival among patients with ARDS and acute pulmonary infections as well as our own studies suggesting a protective role for eosinophils during sepsis (12–15, 37).
Eosinophils produce several growth factors, including TGF-α and TGF-β, that induce tissue repair via epithelial, vascular, and endothelial cell regeneration as well as fibroblast and smooth muscle hyperplasia (38, 39). In addition, Willetts and colleagues found that surviving patients with ALI had higher lung eosinophil counts and increased eosinophil degranulation compared with nonsurviving patients (17). These data are consistent with our observation that eosinophil reduction exacerbates neutrophil influx and ALI during S. aureus pulmonary infection. Eosinophils also produce granules with antibacterial and antiviral properties, including major basic protein-1, eosinophil peroxidase, and eosinophil-derived neurotoxin (40, 41). Furthermore, a critical role for type 2 immune responses and eosinophils in maintenance of the alveolar–capillary barrier is strongly suggested by the observation that mice treated with IL-33 do not develop hypoxemia when challenged with pulmonary infection. Indeed, this may partly explain why the mice do not die; because they do not become hypoxemic, they are able to maintain adequate organ perfusion, allowing them to clear the infection and avoid multiorgan system failure. Future studies will focus on the mechanisms by which eosinophils mediate protection, such as through direct interactions with neutrophils, sequestration of pathogens, and/or maintenance of the alveolar barrier.
Although we have demonstrated a critical role for eosinophils in mediating protection, our work does not exclude the possibility of additional protective mechanisms that are either associated with the type 2 immune response or function through type 2–independent effects of IL-33. For example, IL-33 has been shown to protect against S. aureus pulmonary superinfection in the context of prior influenza infection in a non–type 2–dependent manner via the recruitment of neutrophils; however, this effect may reflect the altered immune milieu induced by the STAT2-dependent viral response, which can impact both macrophage and neutrophil recruitment and function (42, 43). Furthermore, the importance of type 2 immunity in protection against lung injury may depend on both the pathogen and the degree of activation of the type 2 immune response: both Clement and colleagues and Sanfilippo and colleagues used a model of Streptococcus pneumoniae lung infection 3–4 days after activation of the Th2 response (using OVA sensitization and challenge) and failed to identify a robust protective effect, which was possibly due to different virulence factors in pneumococcus (44, 45). However, Th2 activation 10 days before S. pneumoniae infection resulted in complete protection against lethal infection in a macrophage-dependent manner (44). In both of these contexts, eosinophilia is likely minimal compared with our model, and the existence of protection 10 days after Th2 stimulation likely reflects a distinct mechanism.
In summary, our study adds to the growing body of literature indicating that the type 2 immune response may counteract pathogen-induced inflammation during acute infections, suggesting that augmenting type 2 immunity may serve as a beneficial way to supplement antibiotic treatment in patients with pneumonia (12, 18, 24, 27, 32, 46, 47). Understanding the importance of type 2 immunity in this context can help explain the marked variability in outcomes due to infections but may also inform the development of novel immune-modulating therapeutic strategies to serve as an adjunct to supportive care for patients with ARDS, particularly during the current COVID-19 pandemic.
Supplementary Material
Acknowledgments
Acknowledgment
The graphical abstract was created with Biorender.com using an academic license. The authors thank Kelly Blaine for excellent technical assistance.
Footnotes
Supported by U.S. National Institutes of Health grants K08 HL132109 (P.A.V.), UL1 TR000430 (P.A.V.), R01 AI125644 (A.I.S.), and R01 HL118758 (A.I.S.); American Heart Association grant 16POST31200033 (P.A.K.); and American Heart Association and the Circle of Service Foundation grant 18POST33990075 (P.A.K.).
Author Contributions: P.A.K., A.I.S., and P.A.V. conceived and designed the study and also drafted and revised the manuscript. P.A.K., M.K.H., T.G.K., T.S.D., T.J.L., and C.L.H. performed the experiments and analyzed the data that were acquired. A.I.S. and P.A.V. also provided scientific guidance throughout the course of the study. All authors approved the final version to be published.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0166OC on February 11, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Englert JA, Bobba C, Baron RM. Integrating molecular pathogenesis and clinical translation in sepsis-induced acute respiratory distress syndrome. JCI Insight. 2019;4:e124061. doi: 10.1172/jci.insight.124061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, et al. CDC Prevention Epicenter Program. Incidence and trends of sepsis in US hospitals using clinical vs claims data, 2009-2014. JAMA. 2017;318:1241–1249. doi: 10.1001/jama.2017.13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5:18. doi: 10.1038/s41572-019-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wuerth BA, Bonnewell JP, Wiemken TL, Arnold FW. Trends in pneumonia mortality rates and hospitalizations by organism, United States, 2002-2011. Emerg Infect Dis. 2016;22:1624–1627. doi: 10.3201/eid2209.150680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomer L, Schneewind O, Missiakas D. Pathogenesis of Staphylococcus aureus bloodstream infections. Annu Rev Pathol. 2016;11:343–364. doi: 10.1146/annurev-pathol-012615-044351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ventura CL, Higdon R, Hohmann L, Martin D, Kolker E, Liggitt HD, et al. Staphylococcus aureus elicits marked alterations in the airway proteome during early pneumonia. Infect Immun. 2008;76:5862–5872. doi: 10.1128/IAI.00865-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Feng G, Guo Q, Wardenburg JB, Lin S, Inoshima I, et al. Transcriptional events during the recovery from MRSA lung infection: a mouse pneumonia model. PLoS One. 2013;8:e70176. doi: 10.1371/journal.pone.0070176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hook JL, Islam MN, Parker D, Prince AS, Bhattacharya S, Bhattacharya J. Disruption of staphylococcal aggregation protects against lethal lung injury. J Clin Invest. 2018;128:1074–1086. doi: 10.1172/JCI95823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spaan AN, Surewaard BGJ, Nijland R, van Strijp JAG. Neutrophils versus Staphylococcus aureus: a biological tug of war. Annu Rev Microbiol. 2013;67:629–650. doi: 10.1146/annurev-micro-092412-155746. [DOI] [PubMed] [Google Scholar]
- 10.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13:862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zein JG, Love TE, Erzurum SC. Asthma is associated with a lower risk of sepsis and sepsis-related mortality. Am J Respir Crit Care Med. 2017;196:787–790. doi: 10.1164/rccm.201608-1583LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verhoef PA, Bhavani SV, Carey KA, Churpek MM. Allergic immune diseases and the risk of mortality among patients hospitalized for acute infection. Crit Care Med. 2019;47:1735–1742. doi: 10.1097/CCM.0000000000004020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J-J, Dong X, Cao Y-Y, Yuan Y-D, Yang Y-B, Yan Y-Q, et al. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy. 2020;75:1730–1741. doi: 10.1111/all.14238. [DOI] [PubMed] [Google Scholar]
- 14.Liu F, Xu A, Zhang Y, Xuan W, Yan T, Pan K, et al. Patients of COVID-19 may benefit from sustained Lopinavir-combined regimen and the increase of eosinophil may predict the outcome of COVID-19 progression. Int J Infect Dis. 2020;95:183–191. doi: 10.1016/j.ijid.2020.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du Y, Tu L, Zhu P, Mu M, Wang R, Yang P, et al. Clinical features of 85 fatal cases of COVID-19 from Wuhan: a retrospective observational study. Am J Respir Crit Care Med. 2020;201:1372–1379. doi: 10.1164/rccm.202003-0543OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hällgren R, Borg T, Venge P, Modig J. Signs of neutrophil and eosinophil activation in adult respiratory distress syndrome. Crit Care Med. 1984;12:14–18. doi: 10.1097/00003246-198401000-00004. [DOI] [PubMed] [Google Scholar]
- 17.Willetts L, Parker K, Wesselius LJ, Protheroe CA, Jaben E, Graziano P, et al. Immunodetection of occult eosinophils in lung tissue biopsies may help predict survival in acute lung injury. Respir Res. 2011;12:116. doi: 10.1186/1465-9921-12-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krishack PA, Louviere TJ, Decker TS, Kuzel TG, Greenberg JA, Camacho DF, et al. Protection against Staphylococcus aureus bacteremia-induced mortality depends on ILC2s and eosinophils. JCI Insight. 2019;4:e124168. doi: 10.1172/jci.insight.124168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frank KM, Zhou T, Moreno-Vinasco L, Hollett B, Garcia JG, Bubeck Wardenburg J. Host response signature to Staphylococcus aureus alpha-hemolysin implicates pulmonary Th17 response. Infect Immun. 2012;80:3161–3169. doi: 10.1128/IAI.00191-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen H, Wu S, Lu R, Zhang YG, Zheng Y, Sun J. Pulmonary permeability assessed by fluorescent-labeled dextran instilled intranasally into mice with LPS-induced acute lung injury. PLoS One. 2014;9:e101925. doi: 10.1371/journal.pone.0101925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hrusch CL, Manns ST, Bryazka D, Casaos J, Bonham CA, Jaffery MR, et al. ICOS protects against mortality from acute lung injury through activation of IL-5+ ILC2s. Mucosal Immunol. 2018;11:61–70. doi: 10.1038/mi.2017.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lax S, Wilson MR, Takata M, Thickett DR. Using a non-invasive assessment of lung injury in a murine model of acute lung injury. BMJ Open Resp Res. 2014;1:e000014. doi: 10.1136/bmjresp-2013-000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobsen EA, Lesuer WE, Willetts L, Zellner KR, Mazzolini K, Antonios N, et al. Eosinophil activities modulate the immune/inflammatory character of allergic respiratory responses in mice. Allergy. 2014;69:315–327. doi: 10.1111/all.12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krishack PA, Wang K, Rzhetsky A, Solway J, Sperling AI, Verhoef PA. Preexisting type 2 immune activation protects against the development of sepsis. Am J Respir Cell Mol Biol. 2017;57:628–630. doi: 10.1165/rcmb.2017-0277LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang H, Jiang W, Cheng J, Lu Y, Liu A, Kan L, et al. Balancing innate immunity and inflammatory state via modulation of neutrophil function: a novel strategy to fight sepsis. J Immunol Res. 2015;2015:187048. doi: 10.1155/2015/187048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verhoef PA, Constantinides MG, McDonald BD, Urban JF, Jr, Sperling AI, Bendelac A. Intrinsic functional defects of type 2 innate lymphoid cells impair innate allergic inflammation in promyelocytic leukemia zinc finger (PLZF)-deficient mice. J Allergy Clin Immunol. 2016;137:591–600, e1. doi: 10.1016/j.jaci.2015.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenberg JA, Hrusch CL, Jaffery MR, David MZ, Daum RS, Hall JB, et al. Distinct T-helper cell responses to Staphylococcus aureus bacteremia reflect immunologic comorbidities and correlate with mortality. Crit Care. 2018;22:107. doi: 10.1186/s13054-018-2025-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanni F, Akker E, Zaman MM, Figueroa N, Tharian B, Hupart KH. Eosinopenia and COVID-19. J Am Osteopath Assoc. 2020;120:504–508. doi: 10.7556/jaoa.2020.091. [DOI] [PubMed] [Google Scholar]
- 29.Green I, Merzon E, Vinker S, Golan-Cohen A, Magen E. COVID-19 susceptibility in bronchial asthma. J Allergy Clin Immunol Pract. 2021;9:684–692, e1. doi: 10.1016/j.jaip.2020.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cassatella MA, Östberg NK, Tamassia N, Soehnlein O. Biological roles of neutrophil-derived granule proteins and cytokines. Trends Immunol. 2019;40:648–664. doi: 10.1016/j.it.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 31.Pechous RD. With friends like these: the complex role of neutrophils in the progression of severe pneumonia. Front Cell Infect Microbiol. 2017;7:160. doi: 10.3389/fcimb.2017.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dulek DE, Newcomb DC, Goleniewska K, Cephus J, Zhou W, Reiss S, et al. Allergic airway inflammation decreases lung bacterial burden following acute Klebsiella pneumoniae infection in a neutrophil- and CCL8-dependent manner. Infect Immun. 2014;82:3723–3739. doi: 10.1128/IAI.00035-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, et al. Muc5b is required for airway defence. Nature. 2014;505:412–416. doi: 10.1038/nature12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zanin M, Baviskar P, Webster R, Webby R. The interaction between respiratory pathogens and mucus. Cell Host Microbe. 2016;19:159–168. doi: 10.1016/j.chom.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaiss DMW, Gause WC, Osborne LC, Artis D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity. 2015;42:216–226. doi: 10.1016/j.immuni.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linch SN, Danielson ET, Kelly AM, Tamakawa RA, Lee JJ, Gold JA. Interleukin 5 is protective during sepsis in an eosinophil-independent manner. Am J Respir Crit Care Med. 2012;186:246–254. doi: 10.1164/rccm.201201-0134OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grasselli G, Zangrillo A, Zanella A, Antonelli M, Cabrini L, Castelli A, et al. COVID-19 Lombardy ICU Network. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy region, Italy. JAMA. 2020;323:1574–1581. doi: 10.1001/jama.2020.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shah K, Ignacio A, McCoy KD, Harris NL. The emerging roles of eosinophils in mucosal homeostasis. Mucosal Immunol. 2020;13:574–583. doi: 10.1038/s41385-020-0281-y. [DOI] [PubMed] [Google Scholar]
- 39.Wen T, Rothenberg ME. The regulatory function of eosinophils. Microbiol Spectr. 2016;4:MCHD-0020-2015. doi: 10.1128/microbiolspec.MCHD-0020-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Acharya KR, Ackerman SJ. Eosinophil granule proteins: form and function. J Biol Chem. 2014;289:17406–17415. doi: 10.1074/jbc.R113.546218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Linch SN, Kelly AM, Danielson ET, Pero R, Lee JJ, Gold JA. Mouse eosinophils possess potent antibacterial properties in vivo. Infect Immun. 2009;77:4976–4982. doi: 10.1128/IAI.00306-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gopal R, Lee B, McHugh KJ, Rich HE, Ramanan K, Mandalapu S, et al. STAT2 signaling regulates macrophage phenotype during influenza and bacterial super-infection. Front Immunol. 2018;9:2151. doi: 10.3389/fimmu.2018.02151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson KM, Ramanan K, Clay ME, McHugh KJ, Rich HE, Alcorn JF. Novel protective mechanism for interleukin-33 at the mucosal barrier during influenza-associated bacterial superinfection. Mucosal Immunol. 2018;11:199–208. doi: 10.1038/mi.2017.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanfilippo AM, Furuya Y, Roberts S, Salmon SL, Metzger DW. Allergic lung inflammation reduces tissue invasion and enhances survival from pulmonary pneumococcal infection in mice, which correlates with increased expression of transforming growth factor β1 and SiglecFlow alveolar macrophages. Infect Immun. 2015;83:2976–2983. doi: 10.1128/IAI.00142-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clement CG, Tuvim MJ, Evans CM, Tuvin DM, Dickey BF, Evans SE. Allergic lung inflammation alters neither susceptibility to Streptococcus pneumoniae infection nor inducibility of innate resistance in mice. Respir Res. 2009;10:70. doi: 10.1186/1465-9921-10-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allen JE, Wynn TA. Evolution of Th2 immunity: a rapid repair response to tissue destructive pathogens. PLoS Pathog. 2011;7:e1002003. doi: 10.1371/journal.ppat.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Q, Dwyer GK, Zhao Y, Li H, Mathews LR, Chakka AB, et al. IL-33-mediated IL-13 secretion by ST2+ Tregs controls inflammation after lung injury. JCI Insight. 2019;4:e123919. doi: 10.1172/jci.insight.123919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.