

Abstract
Biallelic inactivating mutations in IL21R causes a combined immunodeficiency that is often complicated by cryptosporidium infections. While eight IL-21R-deficient patients have been reported previously, the natural course, immune characteristics of disease, and response to hematopoietic stem cell transplantation (HSCT) remain to be comprehensively examined. In our study, we have collected clinical histories of 13 patients with IL-21R deficiency from eight families across seven centers worldwide, including five novel patients identified by exome or NGS panel sequencing. Eight unique mutations in IL21R were identified in these patients, including two novel mutations. Median age at disease onset was 2.5 years (0.5–7 years). The main clinical manifestations were recurrent bacterial (84.6%), fungal (46.2%), and viral (38.5%) infections; cryptosporidiosis-associated cholangitis (46.2%); and asthma (23.1%). Inflammatory skin diseases (15.3%) and recurrent anaphylaxis (7.9%) constitute novel phenotypes of this combined immunodeficiency. Most patients exhibited hypogammaglobulinemia and reduced proportions of memory B cells, circulating T follicular helper cells, MAIT cells and terminally differentiated NK cells. However, IgE levels were elevated in 50% of IL-21R-deficient patients. Overall survival following HSCT (6 patients, mean follow-up 1.8 year) was 33.3%, with pre-existing organ damage constituting a negative prognostic factor. Mortality of non-transplanted patients (n = 7) was 57.1%. Our detailed analysis of the largest cohort of IL-21R-deficient patients to date provides in-depth clinical, immunological and immunophenotypic features of these patients, thereby establishing critical non-redundant functions of IL-21/IL-21R signaling in lymphocyte differentiation, humoral immunity and host defense against infection, and mechanisms of disease pathogenesis due to IL-21R deficiency. Outcome following HSCT depends on prior chronic infections and organ damage, which should thus be considered as early as possible following molecular diagnosis.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10875-021-01031-5.
Keywords: IL-21/IL-21R signaling, T follicular helper cells, B cell differentiation, STAT3
Introduction
Interleukins (ILs) produced by hematopoietic and non-hematopoietic cells mediate the development, survival, activation, proliferation, and differentation of cells of the immune system [1, 2]. IL-21 is a type I cytokine, produced predominantly by CD4+ T cells, including T follicular helper (Tfh) cells, Th17 and NKT cells [3–6]. On binding its cognate receptor, IL-21 activates JAK/STAT signaling pathways, inducing transcriptional networks that regulate immune cell fate and effector functions [7].
The receptor for IL-21 comprises IL-21R and the “common gamma chain” subunit (γc), which is shared by receptors for IL-2, IL-4, IL-7, IL-9, and IL-15 [8, 9]. Consistent with the broad expression of IL-21R, IL-21 has pleiotropic effects on many immune cell types, including promoting CD8+ T cell activation and proliferation [10], NK cell maturation and cytotoxicity [11], and differentiation of CD4+ T cells, including regulatory T cells [12, 13]. IL-21 appears to exert its most potent effects on B cells, inducing vigorous proliferation, memory B cell formation, isotype switching, and differentiation to plasma cells [14–18]. This parallels the enriched production of IL-21 by Tfh cells within germinal centers (GCs) of secondary lymphoid tissues [3, 19], and strong expression of IL-21R on GC B cells [17].
The non-redundant role of IL-21/IL-21R signaling has been revealed by the identification and characterization of patients with loss-of-function (LOF) biallelic variants in IL21 [20] or IL21R [21–24] who develop an autosomal recessive combined immunodeficiency (CID) with variable presentations. Currently, eight patients from five families with unique homozygous mutations in IL21R have been reported [22–25]. The majority of patients suffered from recurrent respiratory bacterial and gastrointestinal infections, especially cryptosporidial infections associated with chronic cholangitis. Other features of IL-21/IL-21R deficiency are chronic diarrhea, failure to thrive, tinea corporis, herpes labialis, candida and mycobacterial infections, and hypogammaglobulinemia [22–25]. Detailed analysis of immune cells from these individuals revealed impaired T cell proliferation, cytokine production and cytotoxicity [22, 23, 25], reduced Tfh and innate T cell generation [23, 26, 27], impaired B cell differentiation [22–24, 28, 29], and reduced NK cell cytotoxicity [22].
Here, we have identified five novel patients with recessive IL21R mutations, and comprehensively define the clinical and laboratory features of IL-21R deficiency in 13 patients. Furthermore, we expand the clinical and laboratory spectrum of the disease, and report treatment outcomes including those following allogenic hematopoietic stem cell transplant (HSCT).
Material and Methods
Patients
We performed a retrospective analysis of 13 patients with IL-21R deficiency based on a standardized clinical questionnaire. Six patients (P1–P6) were enrolled from three centers in Turkey and an additional five patients came from French (P11), Israeli (P12, P13), and American centers (P9,10). Five patients (P2, P3, P4, P6, P13) were not reported previously. We provide longer-term and/or more detailed clinical follow-up information for six previously reported patients (P1, P5, P9, P10, P11, P12) using the same questionnaire. We additionally included two deceased patients (P7, P8) previously described in the literature [21, 22].
While five of the patients (P1, P4, P5, P6, P12) were identified by a targeted panel of inborn errors of immunity (IEI) genes or whole exome sequencing (WES), variants in P2, P3, P9, P10, P11, and P13 were detected with targeted capillary sequencing.
The study was approved by the respective ethics review boards of the participating institutes, including the Ethics commitees of the Hacettepe University (GO13-228); Marmara University (09.2018.624); Royal Prince Alfred Hospital, Camperdown, Australia (X16-0210/LNR/16/RPAH/257); and Medical University of Vienna, Austria (1796/2018). Written informed consent for genetic investigations was obtained from each family.
Immunophenotyping and Functional Analysis of IL-21R-Deficient Lymphocytes
PBMCs from IL-21R-deficient patients (n = 5–10) and healthy donors (n = 11–40) were analyzed by immunofluorescent staining and flow cytometry [30] to determine proportions of B- (CD20+), T- (CD3+), NK- (CD3−CD56+), and dendritic cells (DCs) (CD19−CD3−CD56− CD235a−CD14−CD20−HLA-DR+). Subsets of T cells (CD4+, CD8+, iNKT (TCR Vα24JαQ+), MAIT (TCR Vα7.2+CD161+), Vγδ (TCR Vγδ+), Vαβ (TCR Vαβ+)) were also determined. Frequencies of naïve (CD45RA+CCR7+), central memory (TCM, CCR7+CD45RA−), effector memory (TEM, CCR7−CD45RA−), and TEMRA (CD45RA+CCR7−) cells within the CD4+ and CD8+ T cell populations, and of Treg (CD25+, CD127−) and Tfh cells (CD45RA−CXCR5+) were enumerated as percentages of total CD4+ or CD8+ T cells. T follicular regulatory (Tfr) cells were defined as the proportion of CXCR5+CD45RA− CD4+ T cells within the Treg population [31]. Th17 and Th1-like phenotypes within memory T cells and Tfh subsets were defined by the differential expression of CXCR3 and CCR6 (CXCR3−CCR6+ and CXCR3+CCR6−, respectively) [26, 31]. Frequencies of transitional (CD27−CD10+), naïve (CD27−CD10−), total memory (CD10−CD27+), class-switched memory (IgD−IgM−IgG+IgA+), and CD21low (CD19+CD21−) B cells were also determined [30, 32]. Lastly, DCs were assessed by quantifying proportions of plasmacytoid DCs (CD123+CD11c−), myeloid DCs (CD11c+CD123+) and mDC subsets cDC1 (CD141+), CD16+ DC (CD16+), cDC2 (CD1c+). Data was acquired on a FACSymphony [30].
Data for B cell and CD4+ T cell function was derived from previously published studies using well-established in vitro assays [26, 32–34]. Specifically, naïve or memory CD4+ T cells sort-purified from PBMCs isolated from healthy donors or IL-21R-deficient patients were cultured (30–40 × 103 cells/well/200 μl) with T cell activation and expansion beads (anti-CD2/CD3/CD28; Miltenyi Biotech) alone (Th0) or under Th17 (2.5 ng/mL TGFβ, 20 ng/mL IL-1 β, 50 ng/mL IL-6, 50 ng/mL IL-21, 100 ng/mL IL-23) or Tfh (50 ng/mL IL-12) polarizing conditions. After 5 days, supernatants were harvested and production of IL-4, IL-5, IL-13, IL-17A, IL-17F, or IL-22 was determined by cytometric bead arrays (Becton Dickinson). To quantify acquisition of expression of IL-21, activated naive CD4+ T cells were re-stimulated with phorbol myristate acetate (PMA, 100 ng/ml)/ionomycin (750 ng/ml) for 6 h, with Brefeldin A (10 μg/ml) added after 2 h. Cells were then fixed, permeabilized, and stained with anti-IL-21 monoclonal antibody. Sort-purified naive B cells were cultured with CD40L alone or together with IL-21 (~5–10 × 103/200 μl/well) for 7 days; immunoglobulin (Ig) secretion was then determined by Ig heavy-chain specific ELISAs.
Genetic Evaluation
Target enriched sequencing was designed to cover 356 IEI genes as previously described [35] and used to diagnose P1 and P6. WES was perfomed for P4, P5, and P12 as described [24, 35]. For Sanger sequencing of IL21R in P2 and P3, genomic DNA was extracted from whole blood (QIamp DNA Blood Kit, Qiagen). Targeted Sanger sequencing of IL21R in P7–P10 has been reported [22, 24]. Primer sequences used to identify IL21R variants in P2, P3, P11, and P13 and validate NGS findings in P1, P4, P5, and P6 are described in Supplementary Table I. Pathogenicity of genetic variants was evaluated based on the allele frequency in public databases including ExAC and gnomAD and prediction tools such as Polyphen-2, SIFT and the combined annotation dependent depletion (CADD) score.
RT-PCR for Qualitative Detection of Rubella Virus
Skin biopsies of P4 were formalin-fixed and paraffin-embedded and RNA-isolated using Maxwell™ RSC DNA FFPE Kit (Promega Corporation) followed by real-time PCR kit for qualitative detection of rubella virus (SACACE Biotechnology).
Statistical Analysis
For single comparisons of independent groups, a Mann-Whitney test was performed. Differences in mean values were considered significant at p < 0.05. All data analysis and figure generation was performed using GraphPad Prism (v. 8.4.2, La Jolla, CA, USA).
Results
Genetic Diagnosis and Mutation Analysis
We identified two novel genetic variants in IL21R in our patient cohort (P4, P6). P4 carries a homozygous missense variant (c.473 T > C, p.L158P) in the extracellular domain, while P6 has a single nucleotide substitution (c.1421C > G), causing a premature termination codon at amino acid p.S474 in the cytoplasmic domain of IL-21R (Fig. 1). Six unique IL21R variants have been described previously: three missense mutations (three families; P7, P8 [R201L]; P11 [W138S]; P12, P13 [R201Q]), a single nucleotide frameshift deletion (1 family: P1, P2, P3 [D179Tfs*51]), a single in-frame codon deletion (1 family; P9, P10 [C81-H82del]), and a splice site mutation (P5 [c.153-1G > T]) (Fig. 1). We performed in silico analysis of the novel and previously reported IL21R variants and found that all were rare alleles (MAF < 0.01), located in highly conserved residues of IL-21R and predicted to be deleterious (SIFT, Polyphen-2, Splicefinder). We also identified a heterozygous variant in CARD14 (Q1001*; CADD score 36) in P6 located at the extreme C-terminus of CARD14 in a region of undefined function. Variants in CARD14 have been identified in individuals with defective skin barrier, such as atopic dermatitis and psoriasis. However, clinical penetrance of several of these variants is incomplete [36]. The impact of this variant on CARD14 remains to be determined.
Fig. 1.
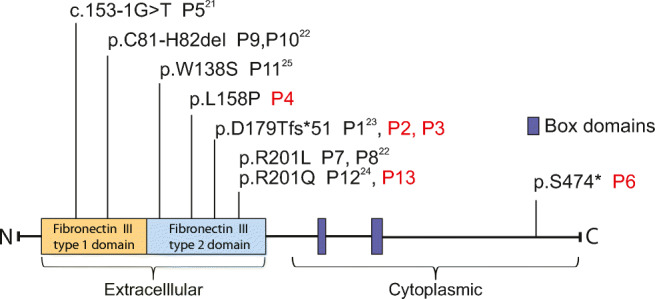
IL-21R domain structure and patient mutations. Domain structure and positions of mutations identified in different patients are indicated. Newly identified patients are marked in red
Impact of IL21R Variants on Protein Structure and Expression
We next assessed the effect of germline variants on the IL-21R structure by identifying the variants’ positions on the crystal structure of the IL-21/IL-21R complex (Fig. 2) [37, 38]:
p.C81-H82del (P9, P10)
Fig. 2.

Pathogenic variants mapped onto the crystal structure of the IL-21/IL-21R complex. IL-21R (orange cartoon) contains two external fibronectin domains, the N-terminal of which (upper) has three internal disulphides (yellow sticks), one involves residue C81 (exploded view). IL-21 (blue cartoon and transparent surface) binds the hinge between the two receptor fibronectin domains. A complex N-linked carbohydrate (gray sticks, extending from N73) bridges the two fibronectin domains and has been postulated to stabilize IL-21R [37, 38]. Residues W138, L158, D179, and R201 (orange sticks) are positioned within the C-terminal fibronectin domain proximal to the extracellular membrane. The side chains of W138 and L158 are buried within the hydrophobic interior of the domain. D179 sits on the exterior surface proximal to, but not directly interfacing, IL-21 while R201 is on an exterior surface distal to IL-21 binding. The side chain of R201 is positioned adjacent to the side chain of W214 (within the WSXWS motif) which is also glycolysylated (dark gray sticks) and may contribute to stability of the N73-projected carbohydrate bridge
This variant removes one of three disulphide linkages within the N-terminal fibronectin domain (each shown in yellow). These deleted amino acids are in close proximity to a protein surface that interfaces with IL-21. Thus, it is likely that this variant affects the fold or interaction surface (i.e., binding), and/or stability of the fibronectin domain, and thus overall expression of IL-21R. Consistent with this latter possibility, we were unable to detect significant expression of IL-21R on EBV-transformed B cells established from P9 and P10 [29], while PBMCs from these patients failed to respond to IL-21, evidenced by a lack of phosphorylation of STAT1, STAT3, and STAT5 [22].
-
b)
W138S (P11), L158P (P4)
The side chains of both W138 and L158 contribute to the hydrophobic core of the C-terminal fibronectin domain fold. In fact, these residues are likely to directly interact with each other. Consequently, the W138S and L158P variants may destabilize the integrity of the fibronectin fold, compromising expression of IL-21R. Indeed, IL-21R could not be detected on the surface of PBMCs from P11 (unpublished observations).
-
c)
R201L/Q (P7, P8, P12, P13)
R201 is spatially adjacent to the first tryptophan residue (W214) in the canonical WSXWS motif present in the extracellular domain of all class I cytokine receptors. W214 undergoes post-translational mannosylation, while its side chain is in close proximity to a complex carbohydrate that bridges across from N73 in the N-terminal fibronectin domain [37, 38]. This carbohydrate bridge may stabilize the IL-21R fold. Notably, of all the possible N-terminal carbohydrate linkages, N73 is critical for IL-21R expression [37]. The R201L and R201Q variants will result in a loss of a positively charged arginine, destabilizing the fibronectin domains of IL-21R, compromising expression and IL-21 binding. Consistent with this possibility, IL-21R was not detected on activated B cells from P12 [R201Q] [24], and IL-21 failed to induce STAT phosphorylation in lymphocytes harboring either the R201L [22] or R201Q [24] variants.
-
d)
D179Tfs*51 (P1, P2, P3)
This variant causes a frameshift at D179, thereby removing the WSXWS motif (aa 214-218) and introducing a premature stop codon 51 amino acids downstream [23]. The carbohydrate bridge formed between N73 and the WSXWS motif is critical for transport of IL-21R from the Golgi to the plasma membrane [38]. Thus, this truncating variant would dramatically reduce, if not abolish, cell surface expression of IL-21R, consistent with a lack of phosphorylation of STAT3 in CD4+ T cells from P1 in response to stimulation with IL-21, but not IL-10 [23].
-
e)
S474* (P6)
This truncating variant deletes 65 amino acids from the C-terminal region of IL-21R. Although there are six conserved tyrosine residues in the cytoplasmic domain of IL-21R, Y510 has been found to be indispensable for IL-21-induced activation of STAT proteins [39]. Thus, while this variant version of IL-21R may be expressed at the cell surface, it is unlikely to be able to initiate IL-21-mediated signaling in immune cells.
Clinical Characteristics
Patient Demographics and Clinical Findings
Median age of disease onset in our cohort was 2.5 years (0.5–7 years) (Supplementary Fig. 1), while the median age at diagnosis was 8.5 (1–19) years. All patients were born to consanguineous parents. Clinical characterics of all patients are shown in Table 1 and Fig. 3, including the family tree of P1 and P2, with a detailed clinical history of each patient provided in the Supplementary results.
Table 1.
Clinical features of patients with IL-21R deficiency
| Patient | Total n (%) | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | P11 | P12 | P13 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| First description | Erman 2015 | Novel | Novel | Novel | Kotlarz 2014 | Novel | Kotlarz 2013 | Kotlarz 2013 | Kotlarz 2013 | Kotlarz 2013 | Ives 2013 | Stepensky 2015 | Novel | |
| Age | 11 | 1 | 8* | 9 | 19* | 19 | 4* | 10* | 18* | 9* | 15* | 15 | 6 | |
| Sex | Male | Female | Male | Male | Male | Female | Male | Male | Male | Male | Female | Female | Female | |
| Age of onset (years) | 0.5 | -*** | 1 | 1.5 | 4 | 2 | 2 | 4 | 5 | 3 | 3 | 2 | -*** | |
| Age at diagnosis (years) | 5 | 1 | 2 | 6 | 19 | 19 | n\a | n\a | 13 | 8 | 13 | 8 | 0.3 | |
| Consanguinity | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Bacterial infections | ||||||||||||||
| Recurrent bacterial respiratory infections | 11 (84.6) | + | + | + | + | + | + | + | + | + | + | + | ||
| Bronchiectasis | 6 (46.2) | + | + | + | + | + | + | |||||||
| Recurrent otitis media | 6 (46.2) | + | + | + | + | + | + | |||||||
| Respiratory mycobacterial infections | 2 (15.4) | M. tuberculosis | M. massilense | |||||||||||
| Fungal infections | ||||||||||||||
| Fungal respratory infections | 5 (38.5) | P. jirovecii | + | P. jirovecii | P. jirovecii | P. jirovecii | ||||||||
| Candidiasis | 5 (38.5) | Esophageal | Hepatosplenic | Esophageal | Esophageal | Systemic | ||||||||
| Parasitic infestations | ||||||||||||||
| Cryptosporidiosis-associated cholangitis | 6 (46.2) | + | + | + | + | + | + | |||||||
| Cryptosporidiosis intestinal inflammation | 5 (38.5) | + | + | + | + | + | ||||||||
| Viral infections | Rubella persistance | CMV penumonia, CMV retinitis, | Chronic norovirus infection | Chronic norovirus infection | CMV infection | |||||||||
| Inflammatory complications | ||||||||||||||
| Liver fibrosis | 2 (18.2) | + | + | |||||||||||
| Failure to thrive | 5 (38.5) | + | + | + | + | + | ||||||||
| Asthma | 3 (23.1) | + | + | + | ||||||||||
| Other | Polydactily, celiac-like findings, giardiasis | Recurrent bronchiolitis, hepatomegaly | Mediastinal and intestinal lymphadenopathy, lupus-like skin inflammation | Food allergies | Recurrent sinusitis, H. pylori associated gastritis, hepatomegaly | Bacteremia. Hematogenous spread of M. massilense | H. pylori gastritis | |||||||
| Treatment | HSCT, Prophylactic antibacterial therapy and IVIG | Prophylactic antibacterial therapy and IVIG | Prophylactic antibacterial therapy and IVIG | Steroids, Prophylactic antibacterial therapy and IVIG | HSCT, prophylactic antibacterial therapy, Voriconazole and IVIG | Omalizumab, Prophylactic antibacterial therapy and IVIG | Liver transplant at age 4 y | HSCT | Prophylactic antibacterial therapy and IVIG | Prophylactic antibacterial therapy and IVIG | HSCT, prophylatic antibacterial therapy, IVIG | HSCT | HSCT | |
| Outcome | Died due to pulmonary aspergillosis day +61 post-HSCT | Alive | Died due to severe diarrhea | Died due to pulmonary complications | Died due to multiorgan failure day +55 post-HSCT | Alive | Died due to bacteremia, systemic CMV infection and multiorgan failure day +542 | Died due to multiorgan failure and graft rejection day +84 | Died due to liver and respiratory failure | Died due to infectious complications with liver and renal failure | Died due to severe digestive GvHD/CMV ARDS day +149 post-HSCT | Alive | Alive | |
n/p not published, n/a not applicable, * age at time of death, ** age at clinical description, ***asymptomatic, CMV cytomegaly virus, HSCT hematopoietic stem cell transplantation, IVIG intravenous immunoglobulin stubstitution, ARDS acute respiratory distress syndrome
Fig. 3.

Clinical features of IL-21R-deficient patients. a Pedigree of the family of P1, P2, and P3. b, c Cutaneous involvement in P4 (face and arm). d H&E stain of a skin biopsy of P4 showing lymphohistiocytic inflammation. Predominant infiltration of CD3+ cells of the superficial dermis invading underlying subcutaneous tissue
Infectious Diseases
Recurrent bacterial infections of the upper and lower respiratory tracts leading to hospitalization were the most common infectious diseases, affecting 11/13 (84.6%) patients and causing bronchiectasis in 6/13 (46.2%) patients. Respiratory bacterial pathogens included Streptococcus pneumoniae and Pseudomonas aeruginosa. Mycobacterium tuberculosis and Mycobacterium massilense were detected in P5 and P10, respectively, suggestive of a significant T cell defect [40]. Viral infections were observed in 5/13 (38.5%) patients. Notably, P5 experienced systemic CMV infection, involving the lung and eyes at age 18 years. P4 suffered granulomatous skin lesions that histologically resemble those reported in ataxia telangiectasia and other CIDs following MMR vaccination (Fig. 3b–d) [41]. In-tissue PCR of biopsied lesions from P4 confirmed long-term persistence of live rubella virus vaccine as the trigger for skin inflammation. Fungal pathogens were detected in 6/13 (46.2%) patients, with four suffering from pulmonary Pneumocystis jirovecii infections and five affected by esophageal, hepatosplenic, or systemic candidiasis.
Inflammatory Diseases
Severe intestinal inflammation was noted for P1 and P3, carrying the same genetic variant, and presented as diarrhea of lethal severity in P3. Cryptosporidiosis-associated cholangitis was reported in 6/13 (46.2%) patients and resulted in hepatic fibrosis in P7 and P8 and lethal liver failure in P9 and P10 (Table 1). Erythematous urticarial plaques on the face and extremities were seen in P5 (Supplementary Fig. 2). A skin punch biopsy revealed basal vacuolar changes, mucin accumulation in the dermis, and superficial and deep perivascular lymphocytic infiltration reminiscent of lupus-like dermatitis. Recurrent attacks of bronchial asthma were a shared feature in P4 and P5, and particularly pronounced in P6 who also suffers from severe food allergy leading to recurrent anaphylactic crises and urticaria (Table 1).
Growth Retardation
Growth retardation affected 5/13 of patients and was particularly severe in P1. Both weight (z-score −5.59) and height (z-score −5.62) were below the third percentile in P1 possibly due to sustained steroid treatment.
Laboratory and Immunological Features of IL-21R Deficiency
Results from laboratory tests for IL-21R-deficient patients are shown in Fig. 4a, b and Supplementary Table II. Serum IgG and IgA levels were reduced in 9/13 (70%) and 7/12 (58%) of patients, respectively. Serum IgM was variable, reduced in 5/12 (42%) but increased in 3/12 (25%) of patients. IgE was elevated in 5/12 (42%) of patients, being particularly high in P6 (up to 40,000 IU/ml) consistent with their asthma and severe allergies. Interestingly, we observed an expansion of CD4−CD8− double negative T cells in 3/12 of patients. B cells, CD4+ T cells, and CD8+ T cells were dysregulated in 45, 55, and 45% of patients, respectively, while NK cell numbers were decreased in 40% of patients.
Fig. 4.

Laboratory features of IL-21R-deficient patients. a Proportions of lymphocyte subsets in IL-21R-deficient patients. One timepoint is displayed per patient. Gray areas indicate healthy donor control ranges [61]. b Laboratory tests of IL-21R-deficient patients. WBC white blood cell count, ALC absolute lymphocyte count
Immunological Profiling of 10 Patients with IL-21R Deficiency
We performed in-depth immunophenotypic analyses on five novel IL-21R-deficient patients (P2–P6). Where possible, this data was combined with data we previously generated from analyses of five other IL-21R-deficient patients (P1, P9, P10, P11, P12) [25–27, 33]. This enabled us to assess the effect of IL-21R deficiency on the development and differentiation of peripheral blood lymphocytes.
Compared with age-matched controls, proportions of T cells were decreased (p < 0.01) in IL-21R-deficient patients (Fig. 5a). However, within the population of total T cells, proportions of TCRαβ, TCRγδ, and iNKT cells were comparable in IL-21R-deficient patients and healthy donors (Fig. 5b–d). MAIT cells were significantly reduced in IL-21R-deficient patients (Fig. 5e). Consistent with CD4+ T cell lymphopenia in one third of patients (Fig. 4b), proportions of CD4+ T cells were also reduced in IL-21R-deficient patients (Fig. 5f). Within the CD4+ T cell lineage, proportions of naïve, TCM, and TEM subsets were comparable in patients and healthy donors. We also quantified memory CD4+ T cell subsets defined by differential expression of CXCR3 and CCR6 [26, 31]. This revealed a trend for fewer Th17-type (CCR6+CXCR3−) cells within the memory T cell compartment of IL-21R-deficient patients compared to healthy donors (p = 0.054, Fig. 5h). This is consistent with a partial reduction in production of Th17 cytokines by IL-21R-deficient memory CD4+ T cells ex vivo compared to memory CD4+ T cells from healthy donors (Fig. 7a), as well as a reduced but not abolished ability of IL-21R-deficient naïve CD4+ T cells to differentiate into Th17-cytokine producing cells in vitro (Fig. 7a) [26, 34]. Other memory CD4+ T cell subsets (e.g., CXCR3+CCR6− Th1-type) were unaffected by abolished IL-21R signaling (Fig. 5h). However, from a functional perspective, production of Th2 cytokines IL-4, IL-5, and IL-13 was greatly enhanced for IL-21R-deficient memory CD4+ T cells ex vivo compared to memory CD4+ T cells from healthy donors (Fig. 7c) [26].
Fig. 5.

IL21R mutations impact Tfh, Th17, MAIT cell formation and NK cell differentiation. PBMC from healthy controls or IL-21R-deficient (n = 5–10) patients were labeled with mAbs against CD3, CD4, CD8, CD56, CD161, TCR Vβ11, TCR Vα7-2, TCR Vα24, CCR7, and CD45RA. a–f Proportions of a total (CD3+) T cells, b αβ T cells, c γδ T cells, d NKT cells, e MAIT cells, and f CD4+ and CD8+ T cells. g–l Within CD4+ T cells, frequencies of g naïve, TCM, or TEM cells; h Th17 (CXCR3−CCR6+), CXCR3+CCR6+, Th1 (CXCR3+CCR6−), or CXCR3−CCR6+; i regulatory T cells; j, k cTfh cells, as well as subsets of l Th17, CXCR3+CCR6+, Th1, or CXCR3−CCR6+, cTfh cells. m, n Proportions of m naïve, TCM, TEM, or TEMRA CD8+ T cells, and n percentages of these subsets expressing CD57. o, p Proportions of o total NK cells within lymphocytes, and of p CD56hi, CD56loCD57−, and CD56loCD57+ NK cell subsets. For all graphs, values represent mean ± SEM, with individual symbols corresponding to one healthy donor or IL-21R-deficient patient. Statistics performed using t tests with Mann-Whitney tests. *p < 0.05, **p < 0.01. FACS data for some of the patients has been published previously [25–27, 33] but is included here to provide an overview of all available patients
Fig. 7.

Functional defects in B and T cells due to inactivating biallelic IL21R variants. a–d Memory (a, c) or naïve (b, d) CD4+ T cells were sort-purified from healthy donors (HD, n = 22–24), or individuals with biallelic variants in IL21R (IL21R, n = 6) or DN variants in STAT3 (STAT3 DN, n = 6–9), and then cultured for 5 days in vitro under non-polarizing Th0 conditions (a, c), Th17 conditions (b) or Tfh-polarizing conditions (d). Secretion of the indicated Th17 (a, b; IL-17A, IL-17F, IL-22) or Th2 (c; IL-4, IL-5, IL-13) cytokines, or expression of the Tfh-cytokine IL-21 (d) was then determined. e, f Naive B cells were sort-purified from healthy donors (HD, n = 35), or individuals with biallelic variants in IL21R (IL21R, n = 6) or STAT3 DN variants (n = 10), and then cultured for 7 days in vitro with CD40L and IL-21. Secretion of IgM (e) or IgG and IgA (f) was then determined. NB: the data depicted in these graphs has been presented in previous publications [26, 33, 34], but is reproduced here to enable comparison between flow cytometric and functional defects, as well as clinical features, resulting from IL-21R deficiency and STAT3 DN mutations
Further analysis of CD4+ T cell subsets revealed normal proportions of regulatory T cells (Treg), as a percentage of all CD4+ T cells, in the patients (Fig. 5i). However, circulating Tfh (cTfh) cells were significantly decreased compared to healthy donors (Fig. 5j). This was also observed when cTfh cells were enumerated as a proportion of memory CD4+ T cells (Fig. 5k), indicating a bona fide cTfh deficit rather than being secondary to a general reduction in memory CD4+ T cells. This is consistent with our previous finding of impaired differentiation of IL-21R-deficient naïve CD4+ T cells into Tfh-type cells in vitro (Fig. 7d) [34]. Th17-type cTfh cells were also reduced, albeit not significantly (p = 0.053), in IL-21R-deficient patients relative to healthy donors (Fig. 5l). Similar to total Tregs, Tfr cells were comparable in IL-21R-deficient patients and healthy donors (data not shown). In contrast to CD4+ T cells, proportions and absolute numbers of total CD8+ T cells (Fig. 5f) as well as naïve, TCM, TEM, and TEMRA subsets (Fig. 5m), and the activation status of CD8+ TCM, TEM, and TEMRA cells [determined by expression of CD57 (Fig. 5n)] were unremarkable in IL-21R-deficient patients.
IL-21 has also been reported to regulate murine NK cell maturation [42]. Thus, we assessed NK cells and subsets. Proportions of total as well as CD56bright and CD56lo NK cell subsets were unaffected by impaired IL-21/IL-21R signaling (Fig. 5o, p). However, the composition of the CD56lo NK cell subset was significantly perturbed by IL-21R-deficiency. Specifically, the CD56lo NK-subset in IL-21R-deficient patients was dominated by CD57− cells, while the corresponding subset in healthy donors is predominantly comprised of CD57+ cells (Fig. 5p). CD56loCD57+ NK cells exhibit greater cytotoxic potential than CD56loCD57− NK cells and thus represent a more mature subset of human NK cells [43].
Proportions of total B cells and naïve B cells were significantly increased, and memory B cells significantly reduced, in IL-21R-deficient patients (Fig. 6a, b). Naive B cells in healthy donors comprise two major populations: IgMloIgDhi (~75%) and IgM−IgDhi (~20–25%) cells. Interestingly, >90% of naïve B cells in IL-21R-deficient patients were IgMloIgDhi cells, resulting in significant reductions in proportions of IgM−IgDhi B cells (Fig. 6c). Within the contracted memory B cell population in IL-21R-deficient patients, proportions of IgG+ or IgA+ switched cells were significantly decreased (Fig. 6d). These flow cytometric data correlated with functional data inasmuch that naïve B cells from IL-21R-deficient patients exhibited no response to the stimulatory effect of IL-21, evidenced by undetectable levels of IgM as well as of the class-switched Ig isotypes IgG and IgA. This contrasts responses of naïve B cells from healthy donors which produce large amounts of IgM, and detectable amounts of IgG and IgA (Fig. 7e, f) [33, 34]. Assessment of myeloid cells revealed comparable proportions of total DCs and plasmacytoid DCs in IL-21R-deficient patients and healthy donors (Fig. 7e, f). However, proportions of myeloid DCs—particularly the CD141+ subset—were lower in IL-21R-deficient patients (Fig. 6f, g).
Fig. 6.

Impaired generation of memory B cells and induction of class switching due to IL-21R deficiency. PBMC from healthy controls or IL-21R-deficient (n = 5–9) patients were labeled with mAbs against CD19, CD20, CD10, CD27, IgG, IgA, CD123, CD11c, CD141, CD16, and CD1c. a–d Proportions of a total B cells within lymphocytes; b transitional, naïve, and memory B cells within the total B cell population; c naive B cells with a IgM−IgD+ or IgMloIgDhi phenotype; and d memory B cells expressing IgG or IgA. e–g Proportions of e DC within all leukocytes, f plasmacytoid (CD123+CD11c−) and myeloid cells (CD11c+CD123+) within the total DC population, and g mDC subsets [cDC1 (CD141+), CD16+ DC (CD16+), cDC2 (CD1c+)]. For all graphs, values represent mean ± SEM, with individual symbols corresponding to one healthy donor or IL-21R-deficient patient. Statistics performed using t tests with Mann-Whitney tests. *P < 0.05, **** P < 0.0001
Collectively, while impaired IL-21R signaling has limited effects on lymphocyte development, further differentiation of these cells is substantially compromised, with significant reductions in CD4+ T cells, MAIT cells, cTfh cells, total and class-switched memory B cells, terminally differentiated NK cells and myeloid-derived DCs, and trends for reductions in Th17 cells.
Therapeutic Approach and Outcome
Prophylactic antibacterial therapy (TMP-SMX) and monthly intravenous immunoglobulins (IVIG) were given to all patients. CMV pneumonitis in P5 required ganciclovir; omalizumab treatment was given to P6 to treat their allergies. P1 and P3 received steroids, azathioprine, and 5-ASA for intestinal inflammation. Although all patients received extensive antimicrobial and supportive therapy, mortality of untransplanted patients was 42.8% (3/7) due to infectious complications. The liver disease in P1 necessitated a differential diagnosis with sclerosing cholangitis and autoimmune hepatitis. The progressive liver disease developed despite clarithromycin being administered to treat cryptosporidium infection.
Five IL-21R-deficient patients in our cohort (P1, P5, P8, P11, P12, P13) underwent HSCT, as outlined in Table 2. All of these patients had a molecular diagnosis of IL-21R deficiency prior to HSCT. We collected HSCT data of these five patients. P8 also underwent transplant, though prior to the initial report of IL-21R-deficiency [22]. Thus, we retrieved transplant information of P8 from the literature [22]. The median age at transplant was 11.5 years. Stem cell source was bone marrow (n = 4) or peripheral blood (n = 1). Fully matched related donors were used for five patients. All patients except for P13, who was asymptomatic, showed lung or GI tract damage prior to HSCT (Table 2). P1, P4, P5, and P11 presented with bronchiectasis, while P5 and P12 had PCR-confirmed CMV viremia pre-transplant. Cholangitis was observed in P1, P8, and P11. This necessitated a liver transplantation in P1 1 month pre-HSCT. P5 also suffered chronic norovirus enteritis at the time of HSCT (Table 2). All patients received pre-HSCT conditioning regimens comprised of either reduced toxicity conditioning (RTC; P1, P11, P12, P13) or reduced intensity conditioning (RIC, P5) to prepare for donor engraftment. Notably P12 and P13, who had the least complications clinically, received RTC consisting of fludarabine (30 mg/m2 × 5 days), treosulfan (14 g/m2 × 3 days), and thiotepa (10 mg/kg). All patients received anti-thymocyte globulin (ATG) for a minimum of 3 days. Cyclosporine A (CSA), methotrexate (MTX), or mycophenolate mofetile (MMF) were administered as GvHD prophylaxis (Table 2).
Table 2.
Details of HSCT and clinical course of transplanted IL-21R-deficient patients
| Patient no. | Age at HSCT | Organ damage prior to HSCT | HSCT conditioning regime | GvHD prophylaxis | HSCT source | Complications | Clinical outcome, follow-up time | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Donor | cell source, cells/kg | Engraftment (neutrophil) | GvHD | Other | ||||||
| 1 | 12 years 11 months | Bronchiectasis, cholangitis, hepatic transplant |
Fludarabine (150 mg/m2) Treosulfan (108 g/m2) |
MTX | 10/10 MSD |
Unknown BMC |
Failed | No | Failure to engraft, severe pulmonary aspergillosis, | Died on day 61 post-HSCT |
| 5 | 20 years 2 months | Bronchiectasis |
Fludarabine (150 mg/m2) Melphalan (100 mg/m2) |
CSA, MTX |
10/10 MFD | 8 × 106 BMC | Failed | No | Failure to engraft, severe bact. Infections, PRES, CMV reactivation, ARDS | Died on day 55 post-HSCT |
| 11 | 15 year 6 months | Bronchiectasis, lung resection, cholangitis |
Fludarabine (180 mg/m2) Busulfan (AUC 12000 micro M.min) |
CSA, MMF |
10/10 MSD | 6.5 × 106 PBSC |
Yes, day +14 |
Acute GvHD, grade IV: skin, GI, liver (D + 40) |
CMV reactivation associated ARDS | Died on day 149 post-HSCT |
| 12 | 9 years 3 months | Bronchiectasis |
Fludarabine (150 mg/m2) Treosulfan (42 g/m2) Thiotepa (10 mg/kg) |
CSA, MMF |
12/12 MSD | 2.5 × 106 BMC |
Yes, day +14 |
No | CMV reactivation | Clinical remission, IVIG not needed |
| 13 | 2 years 6 months | No |
Fludarabine (150 mg/m2) Treosulfan (42 g/m2) Thiotepa (10 mg/kg) |
CSA, MMF |
12/12 MSD | 1.04 × 106 BMC |
Yes, Day +14 |
No | No | Clinical remission, IVIG not needed |
| 8 | 10 years | Cholangitis, liver fibrosis | Not published | Not published | HLA matched | Not published |
1st HSCT: yes 2nd HSCT: n/p |
No | Increased cholangitis, CMV infection, graft rejection, required 2nd HSCT, multiorgan failure | Died on day 84 post-HSCT |
HSCT hematopoietic stem cell transplantation, AUC area under the curve, MTX methotrexate, CSA cyclosporine A, MMF mycofenolate mofetil, MSD matched sibling donor, MFD matched family donor, BMC bone marrow cells, GvHD graft versus host disease, GI gastrointestinal, PRES posterior reversible encephalopathy syndrome, CMV cytomegalovirus, ARDS acute respiratory distress syndrome, IVIG intravenous immunoglobulin substitution
Post-transplant complications occurred in 5 out of 6 patients: Four patients fully engrafted post-HSCT and 1 out of 5 patients experienced GvHD. However, four patients (P1, P5, P8, P11) succumbed post-HSCT. HSCT was complicated in P1 by engraftment failure and invasive pulmonary aspergillosis causing death 61 days post-transplant. P5 failed to engraft and presented with severe systemic bacterial infections, encephalopathy, CMV reactivation, and acute respiratory distress syndrome (ARDS), succumbing 55 days post-transplant. P8 developed increased cholanigits, CMV infection, and required a second HSCT after graft rejection (Table 2). P8 died of multiorgan failure following a second transplant [22]. HSCT was complicated by acute grade 4 GvHD in P11 with skin, GI, and liver involvement starting from day 40 post-transplant, and treated with glucocorticoids, CSA, MMF, sirolimus, infliximab, and ATG. P11 developed CMV reactivation and succumbed to CMV-associated ARDS in the third month post-transplant. In total, four patients suffered from CMV reactivation post-HSCT, which was controlled only in P12 (Table 2).
We report a 33% overall survival rate in our cohort at a mean follow-up of 1.8 years with three patients dying due to infectious complications and one patient succumbing to multiorgan failure of unreported etiology. Both P12 and P13 were successfully transplanted and remain free of IVIG substitution post-HSCT.
Discussion
CIDs are a heterogeneous group of primary immune disorders and exhibit defects in T cell development and function, along with compromised B cell responses [44, 45]. Germline biallelic LOF variants in IL21 or IL21R cause a CID [40]. Recessive IL21R LOF variants were first identified in four patients from two unrelated families from distinct geographical locations (P7–P10) [22]. IL-21R deficiency predisposes to infections associated with CID and primary antibody deficiency (Pneumococci respiratory infections, recurrent otitis media, Cryptosporidium cholangitis), and infections common in patients with T or NK cell deficiencies (invasive/mucocutaneous fungal infections, P. jirovecii lung infections, CMV pneumonia). The newly described infections of giardiasis and systemic candidiasis in our cohort are reminiscent of the infectious phenotype of other CIDs such as CD40L deficiency [46].
Our in-depth dissection of circulating immune cell subsets illustrates obligate roles of IL-21R in differentiation and effector function of human lymphocyte subsets. IL-21/IL-21R signaling unequivocally has a non-redundant role in generating or maintaining Tfh-, memory B-, MAIT-, and CD56dimCD57+ terminally differentiated NK cells, and a likely role in Th17 cells. Our findings confirm, extend, and refine previous studies that delineated specific and unique functions of IL-21 in human and murine lymphocyte development [47]. Our findings also provide insight into mechanisms underlying some clinical features of IL-21R deficiency. Thus, the paucity of memory B cells and Tfh cells, together with an impaired ability of IL-21R-deficient naïve CD4+ T cells to differentiate into IL-21-expressing Tfh cells in vitro and of IL-21R-deficient naïve B cells to differentiate into plasmablasts in response to IL-21 [26, 33, 34], explains hypogammaglobulinemia and impaired humoral immunity. While the in vitro analyses indicated these cellular deficiencies were intrinsic to B cells and T cells, they are also likely to be interdependent, as IL-21 is produced predominantly by Tfh cells, and Tfh-derived IL-21 potently induces human B cell proliferation, class switching, and plasmablast formation, and promotes plasma cell survival [47]. Thus, the B cell intrinsic defect in class switching due to the fundamental inability of IL-21R-deficient naïve B cells to respond to IL-21 could be exacerbated by dysfunctional and/or reduced Tfh cells due to B cell extrinsic defects affecting IL-21R-deficient CD4+ T cells. The partial Th17 deficit in IL-21R-deficient patients likely contributes to frequent fungal infections in some, but not all, of these patients indicating the impact of IL-21R signaling on human Th17 cells.
IL-21 activates STAT1, STAT3, and STAT5 [48]. Prior analysis of individuals with inactivating or dominant negative (DN) variants in STAT1 or STAT3 revealed that the effects of IL-21 on human B and T cell subsets are predominantly, if not exclusively, mediated by STAT3 [25, 26, 29, 32–34]. Consequently, our findings define the contribution of defective IL-21R signaling to disease pathogenesis in autosomal-dominant hyper-IgE syndrome (AD-HIES) due to DN STAT3 mutations, namely recurrent infections, impaired humoral immunity, and candidiasis [2, 9, 22]. Interestingly, recessive mutations in the transcription factor ZNF341 phenocopies the clinical features of STAT3 DN mutations, consistent with ZNF341 regulating IL-21-induced STAT3 activation [49, 50]. The current findings for this larger cohort of IL-21R-deficient patients, together with our observations for STAT3 DN [25, 26, 29, 32–34] and ZNF341 [28] deficiency, establish that IL-21 is a major STAT3-activating cytokine that regulates differentiation of T cells and B cells into key effector subsets (Tfh, Th17, MAIT cells; total and class-switched memory B cells), thereby explaining the overlap in some of the clinical features of individuals with pathogenic variants in IL21R, STAT3, or ZNF341. Interestingly, several clinical, laboratory, and cellular features of STAT3 DN and IL-21R- or ZNF341-deficient individuals also overlap with individuals with DN heterozygous variants in IL6ST [12]. These include reduced Th17, Tfh and memory B cells, Th2-skewed memory CD4+ T cells, hyper-IgE and recurrent fungal infections [26, 28, 32–34, 40, 51]. This highlights the interplay between STAT3/ZNF341-mediated signaling downstream of receptors for IL-6 and IL-21. Importantly, our observation that fungal infections occur in fewer IL-21R-deficient patients (~50%) than STAT3- or ZNF341-deficient patients (>90%) also demonstrates partial redundancy for IL-21R signaling in generating human Th17 cells during host defense against fungi, whereas Th17 deficiency is highly penetrant in these other IEIs [28, 51]. Indeed, our functional analyses demonstrated a partial defect in generating Th17 cells in IL-21R-deficient individuals, but complete abolition of Th17 cell differentiation by STAT3 DN mutations [26, 34, 51, 52]. Our data also establish the Th17 defect is T cell intrinsic, evidenced by impaired differentiation of naïve IL-21R-deficient CD4+ T cells into IL-17A/IL-17F-producing cells [34] (Fig. 6). This highlights important combinatorial contributions of additional STAT3-activating cytokines to Th17-cell generation, such as IL-6 and IL-23, evidenced by partial deficiencies in Th17 cells in individuals with biallelic variants in IL23R [53] or DN heterozygous IL6ST variants [12], and variability in the incidence of fungal infections in these individuals.
IL-21- and IL-21R-deficient patients [20–24], similar to corresponding gene-targeted mice [54], exhibit elevated levels of serum IgE. Increased IgE in some cases of IL-21R-deficiency may contribute to atopic disorders. Consistent with this observation, memory CD4+ T cells from IL-21R-deficient individuals, as well as those with STAT3 DN, IL6ST DN, or homozygous ZNF341 variants, exhibit exaggerated production of Th2 cytokines IL-4, IL-5, and IL-13 (Fig. 6) [12, 26, 28], while IL-21 signaling negatively regulates class switching to IgE and abrogates anaphylaxis in mice [55, 56]. Thus, IL-21 potentially regulates IgE production directly by repressing Ig ε class switching and survival of IgE+ B cells, and indirectly by restraining the generation of Th2 effector cells producing IgE-promoting cytokines such as IL-13. IL-21R-deficiency has not previously been associated with lymphohistiocytic infiltration of the skin. Prolonged persistence of live rubella virus vaccine in granulomatous skin lesions in P4 adds IL-21R deficiency to the growing list of immunodeficiencies associated with rubella-mediated skin inflammation [57]. Furthermore, it highlights that immunity to some viruses is impaired in patients with IL-21R deficiency [58] and suggests cautious consideration of live vaccinations—in particular against rubella—in IL-21R-deficient individuals.
P6 suffered recurrent anaphylaxis and severe atopy in addition to higher IgE levels than other IL-21R-deficient patients (Table 1, Supplementary Table 2). Two scenarios may contribute to these clinical findings. First, the IL21R variant in P6 is predicted to truncate the IL-21R protein at amino acid residue S474 in the cytoplasmic domain. This removes 65 C-terminal amino acids, including Y510 which acts as a phospho-tyrosine docking site for STAT proteins, enabling their activation following receptor engagement by IL-21 (Fig. 1) [39]. Thus, this variant may uniquely impact the structure/function of IL-21R and its interactions with STAT1 and/or STAT3. Second, the heterozygous CARD14 variant identified in P6 may contribute to severe atopy, as has been reported for other individuals with CARD14 variants [36]. These possibilities will be pursued in future studies.
Cryptosporidium-associated liver disease remains the most prominent cause of morbidity in IL-21R deficiency. Correspondingly, P1, P9, and P10 succumbed to cryptosporidiosis-associated sclerosing cholangitis, and P3 suffered severe and subsequently fatal diarrhea triggered by intestinal cryptosporidiosis. P1 and P7 underwent liver transplant after extensive cryptosporidiosis-mediated liver damage. Furthermore, increased cholangitis was observed in P8 as a post-HSCT complication. The cases of P4, P5, and P12 illustrate that even in the absence of cryptosporidiosis, IL-21R-deficient patients may develop severe organ damage necessitating HSCT. Even with antibacterial and supportive treatment, IL-21R deficiency is a potentially lethal disease. Thus, HSCT should ideally be considered prior to occurrence of cryptosporidiosis [59].
HSCT for IL-21R deficiency is difficult and should be considered carefully. Indeed, while some patients have a relatively high quality of life, IL-21R deficiency was associated with high mortality (57%) in non-transplanted patients. Primary and secondary graft failure were observed in 2/6 and 1/6 patients, respectively, ultimately resulting in death. Myeloablative conditioning (MAC) prior to HSCT is generally associated with improved engraftment and reduced risk for graft rejection [60]. Thus, MAC or MAC with RTC may be favorable to RIC protocols in establishing sustained engraftment in IL-21R deficiency. Notably, the two patients who survived HSCT—P12, P13—were the youngest in our cohort and also had the least organ damage. This suggests that early transplant prior to chronic infections is a positive prognostic factor for HSCT of IL-21R-deficient patients. Indeed, P13 was diagnosed prior to disease onset due to previous family history. These observations also suggest that organ damage secondary to opportunistic infections, rather than IL-21R deficiency itself, is responsible for the high mortality of HSCT in these individuals. Even larger patient cohorts of IL-21R-deficient patients need to be studied to provide clear treatment recommendations in the future.
Our comprehensive analysis of IL-21R-deficient patients has enabled us to characterize the critical function of IL-21/IL-21R signaling in human health and disease, and provide mechanisms underlying disease pathogenesis. In conclusion, our report provides additional awareness to IL-21R deficiency as a recently defined IEI, expands the spectrum of disease of affected individuals, and indicates the importance of early diagnosis of IL-21R deficiency to improve survival of the patients by providing early therapeutic options prior to cryptosporidium infection.
Supplementary Information
(PDF 176 kb)
Abbreviations
- AD-HIES
Autosomal-dominant hyper-IgE syndrome
- ARDS
Acute respiratory distress syndrome
- ATG
Anti-thymocyte globuline
- CID
Combined immunodeficiency disorder
- CSA
Cyclosporine A
- cTfh
Circulating T follicular helper
- DCs
Dendritic cells
- DN
Dominant negatve
- GC
Germinal centers
- HSCT
Hematopoietic stem cell transplant
- IEI
Inborn error of immunity
- IL-21R
Interleukin-21 receptor
- Ils
Interleukins
- IVIG
Intravenous immunoglobulins
- LOF
Loss-of-function
- MAC
Myeloablative conditioning
- MMF
Mycophenolate mofetile
- MTX
Methotrexate
- RIC
Reduced intensity conditioning
- RTC
Reduced toxicity conditioning
- Tfh
T follicular helper
- Tfr
T follicular regulatory
- WES
Whole exome sequencing
- γc
Common gamma chain
Authors’ Contribution
DC, DM, SB, KB, and SGT collected and analyzed clinical, laboratory, and/or experimental data; wrote the drafts of the manuscript; and prepared the figures and tables. LW performed immunophenotyping. DBL and DC performed structural analyses of IL21R variants. ESA and RA provided clinical, immunophenotyping data for P1, P2, and P4, and prepared the tables. AM and MT provided patient care, collected samples, and provided clinical, immunophenotyping, and genetic data for P3 and P6. DC, OS, and İT provided patient care, collected samples, and provided clinical and immunophenotyping data for P1, P2, and P4. NK, GK, SHT, EKA, and AO provided patient care, collected samples and clinical data of P5. BK and DUÇ provided HSCT data of P1. AU provided pathological data of P4. SKB provided clinical and technical input and support. JD and RJ-H analyzed NGS data of P1, P4, P5, and P6. FO and A-IS are pathologists who performed RT-PCR for qualitative detection of rubella virus in P4. GU provided clinical, immunophenotyping, and genetic data for P9 and P10. EKD performed and provided immunophenotyping data. BK, KW, and PS provided clinical, laboratory, and genetic data and outcomes following HSCT for P12 and P13. BN and AD provided clinical, laboratory, and genetic data and outcomes following HSCT for P11. CSM performed and provided immunophenotyping data. KB, CSM, and SGT supervised the project. All co-authors contributed to and edited drafts of the original and revised manuscripts and table, and approved the final submitted version.
Funding
E.K.D. is the recipient of a Scientia Award from UNSW Sydney. C.S.M is supported by an Early-Mid Career Research Fellowship from the Department of Health of the New South Wales Government of Australia. S.G.T was a Principal Research Fellow (1042925) of the NHMRC, and is currently a recipient of an NHMRC Leadership 3 Investigator Grant (1176665) and NHMRC program grant (1113904). KB is supported by a European Research Council Consolidator Grant (iDysChart, ERC grant agreement number: 820074). S.B. is supported by the Scientific and Technological Research Council of Turkey (318S202). PS and KW received funding from the Deutsche Forschungsgemeinschaft (DECIDE, DFG WA 1597/4-1).
Compliance with Ethical Standards
Ethics Approval
This work is a review of recently reported genetic variants that represent novel inborn errors of immunity. No human research studies were performed in order to produce this review. Thus, no approvals by appropriate institutional review boards or human research ethics committees were required to undertake the preparation of this report.
Consent to Participate
Not applicable as this is a review of recently reported genetic variants.
Consent to Publish
The authors consent to publish the content of this review. However, as noted above, as this is a review of recently reported genetic variants that represent novel inborn errors of immunity, we did not require consent to publish from participants.
Conflict of Interest
The authors declare that they have no conflict of interest.
Availability of Data and Materials
Not applicable
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Deniz Cagdas, Daniel Mayr and Safa Baris contributed equally to this study as first authors
Kaan Boztug and Stuart G Tangye contributed equally to this study as senior authors
Contributor Information
Deniz Cagdas, Email: deniz.ayvaz@hacettepe.edu.tr.
Kaan Boztug, Email: kaan.boztug@ccri.at.
Stuart G. Tangye, Email: s.tangye@garvan.org.au
References
- 1.Akdis M, Aab A, Altunbulakli C, Azkur K, Costa RA, Crameri R, Duan S, Eiwegger T, Eljaszewicz A, Ferstl R, Frei R, Garbani M, Globinska A, Hess L, Huitema C, Kubo T, Komlosi Z, Konieczna P, Kovacs N, Kucuksezer UC, Meyer N, Morita H, Olzhausen J, O'Mahony L, Pezer M, Prati M, Rebane A, Rhyner C, Rinaldi A, Sokolowska M, Stanic B, Sugita K, Treis A, van de Veen W, Wanke K, Wawrzyniak M, Wawrzyniak P, Wirz OF, Zakzuk JS, Akdis CA. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor β, and TNF-α: receptors, functions, and roles in diseases. J Allergy Clin Immunol. 2016;138(4):984–1010. doi: 10.1016/j.jaci.2016.06.033. [DOI] [PubMed] [Google Scholar]
- 2.Tangye SG, Pelham SJ, Deenick EK, Ma CS. Cytokine-mediated regulation of human lymphocyte development and function: insights from primary immunodeficiencies. J Immunol. 2017;199(6):1949–1958. doi: 10.4049/jimmunol.1700842. [DOI] [PubMed] [Google Scholar]
- 3.Chtanova T, Tangye SG, Newton R, Frank N, Hodge MR, Rolph MS, Mackay CR. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J Immunol. 2004;173(1):68–78. doi: 10.4049/jimmunol.173.1.68. [DOI] [PubMed] [Google Scholar]
- 4.Wei L, Laurence A, Elias KM, O'Shea JJ. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. 2007;282(48):34605–34610. doi: 10.1074/jbc.M705100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spolski R, Leonard WJ. IL-21 and T follicular helper cells. Int Immunol. 2010;22(1):7–12. doi: 10.1093/intimm/dxp112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coquet JM, Kyparissoudis K, Pellicci DG, Besra G, Berzins SP, Smyth MJ, Godfrey DI. IL-21 is produced by NKT cells and modulates NKT cell activation and cytokine production. J Immunol. 2007;178(5):2827–2834. doi: 10.4049/jimmunol.178.5.2827. [DOI] [PubMed] [Google Scholar]
- 7.Brocker C, Thompson D, Matsumoto A, Nebert DW, Vasiliou V. Evolutionary divergence and functions of the human interleukin (IL) gene family. Human Genomics. 2010;5(1):30–55. doi: 10.1186/1479-7364-5-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leonard WJ, Lin JX, O'Shea JJ. The γ(c) family of cytokines: basic biology to therapeutic ramifications. Immunity. 2019;50(4):832–850. doi: 10.1016/j.immuni.2019.03.028. [DOI] [PubMed] [Google Scholar]
- 9.Tangye SG. Advances in IL-21 biology—enhancing our understanding of human disease. Curr Opin Immunol. 2015;34:107–115. doi: 10.1016/j.coi.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 10.Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, Pise-Masison CA, Radonovich MF, Brady JN, Restifo NP, Berzofsky JA, Leonard WJ. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med. 2005;201(1):139–148. doi: 10.1084/jem.20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Z, Yang L, Cui Y, Wang X, Guo C, Huang Z, Kan Q, Liu Z, Liu Y. Il-21 enhances NK cell activation and cytolytic activity and induces Th17 cell differentiation in inflammatory bowel disease. Inflamm Bowel Dis. 2009;15(8):1133–1144. doi: 10.1002/ibd.20923. [DOI] [PubMed] [Google Scholar]
- 12.Beziat V, Tavernier SJ, Chen YH, Ma CS, Materna M, Laurence A, et al. Dominant-negative mutations in human IL6ST underlie hyper-IgE syndrome. J Exp Med. 2020;217(6):e20191804. doi: 10.1084/jem.20191804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmitz I, Schneider C, Frohlich A, Frebel H, Christ D, Leonard WJ, et al. IL-21 restricts virus-driven Treg cell expansion in chronic LCMV infection. PLoS Pathog. 2013;9(5):e1003362. doi: 10.1371/journal.ppat.1003362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avery DT, Bryant VL, Ma CS, de Waal MR, Tangye SG. IL-21-induced isotype switching to IgG and IgA by human naive B cells is differentially regulated by IL-4. J Immunol. 2008;181(3):1767–1779. doi: 10.4049/jimmunol.181.3.1767. [DOI] [PubMed] [Google Scholar]
- 15.Ettinger R, Sims GP, Fairhurst A-M, Robbins R, da Silva YS, Spolski R, Leonard WJ, Lipsky PE. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol. 2005;175(12):7867–7879. doi: 10.4049/jimmunol.175.12.7867. [DOI] [PubMed] [Google Scholar]
- 16.Kuchen S, Robbins R, Sims GP, Sheng C, Phillips TM, Lipsky PE, Ettinger R. Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cell-B cell collaboration. J Immunol. 2007;179(9):5886–5896. doi: 10.4049/jimmunol.179.9.5886. [DOI] [PubMed] [Google Scholar]
- 17.Good KL, Bryant VL, Tangye SG. Kinetics of human B cell behavior and amplification of proliferative responses following stimulation with IL-21. J Immunol. 2006;177(8):5236–5247. doi: 10.4049/jimmunol.177.8.5236. [DOI] [PubMed] [Google Scholar]
- 18.Bryant VL, Ma CS, Avery DT, Li Y, Good KL, Corcoran LM, de Waal Malefyt R, Tangye SG. Cytokine-mediated regulation of human B cell differentiation into Ig-secreting cells: predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J Immunol. 2007;179(12):8180–8190. doi: 10.4049/jimmunol.179.12.8180. [DOI] [PubMed] [Google Scholar]
- 19.Ma CS, Suryani S, Avery DT, Chan A, Nanan R, Santner-Nanan B, Deenick EK, Tangye SG. Early commitment of naive human CD4(+) T cells to the T follicular helper (T(FH)) cell lineage is induced by IL-12. Immunol Cell Biol. 2009;87(8):590–600. doi: 10.1038/icb.2009.64. [DOI] [PubMed] [Google Scholar]
- 20.Salzer E, Kansu A, Sic H, Májek P, Ikincioğullari A, Dogu FE, et al. Early-onset inflammatory bowel disease and common variable immunodeficiency-like disease caused by IL-21 deficiency. J Allergy Clin Immunol. 2014;133(6):1651–9.e12. doi: 10.1016/j.jaci.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 21.Kotlarz D, Zietara N, Milner JD, Klein C. Human IL-21 and IL-21R deficiencies: two novel entities of primary immunodeficiency. Curr Opin Pediatr. 2014;26(6):704–712. doi: 10.1097/MOP.0000000000000160. [DOI] [PubMed] [Google Scholar]
- 22.Kotlarz D, Zietara N, Uzel G, Weidemann T, Braun CJ, Diestelhorst J, et al. Loss-of-function mutations in the IL-21 receptor gene cause a primary immunodeficiency syndrome. J Exp Med. 2013;210(3):433–443. doi: 10.1084/jem.20111229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erman B, Bilic I, Hirschmugl T, Salzer E, Cagdas D, Esenboga S, Akcoren Z, Sanal O, Tezcan I, Boztug K. Combined immunodeficiency with CD4 lymphopenia and sclerosing cholangitis caused by a novel loss-of-function mutation affecting IL21R. Haematologica. 2015;100(6):e216–e219. doi: 10.3324/haematol.2014.120980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stepensky P, Keller B, Abuzaitoun O, Shaag A, Yaacov B, Unger S, Seidl M, Rizzi M, Weintraub M, Elpeleg O, Warnatz K. Extending the clinical and immunological phenotype of human interleukin-21 receptor deficiency. Haematologica. 2015;100(2):e72–e76. doi: 10.3324/haematol.2014.112508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ives ML, Ma CS, Palendira U, Chan A, Bustamante J, Boisson-Dupuis S, Arkwright PD, Engelhard D, Averbuch D, Magdorf K, Roesler J, Peake J, Wong M, Adelstein S, Choo S, Smart JM, French MA, Fulcher DA, Cook MC, Picard C, Durandy A, Tsumura M, Kobayashi M, Uzel G, Casanova JL, Tangye SG, Deenick EK. Signal transducer and activator of transcription 3 (STAT3) mutations underlying autosomal dominant hyper-IgE syndrome impair human CD8(+) T-cell memory formation and function. J Allergy Clin Immunol. 2013;132(2):400–411. doi: 10.1016/j.jaci.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, Bustamante J, Okada S, Stoddard JL, Deenick EK, Pelham SJ, Payne K, Boisson-Dupuis S, Puel A, Kobayashi M, Arkwright PD, Kilic SS, el Baghdadi J, Nonoyama S, Minegishi Y, Mahdaviani SA, Mansouri D, Bousfiha A, Blincoe AK, French MA, Hsu P, Campbell DE, Stormon MO, Wong M, Adelstein S, Smart JM, Fulcher DA, Cook MC, Phan TG, Stepensky P, Boztug K, Kansu A, İkincioğullari A, Baumann U, Beier R, Roscioli T, Ziegler JB, Gray P, Picard C, Grimbacher B, Warnatz K, Holland SM, Casanova JL, Uzel G, Tangye SG. Monogenic mutations differentially affect the quantity and quality of T follicular helper cells in patients with human primary immunodeficiencies. J Allergy Clin Immunol. 2015;136(4):993–1006. doi: 10.1016/j.jaci.2015.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson RP, Ives ML, Rao G, Lau A, Payne K, Kobayashi M, Arkwright PD, Peake J, Wong M, Adelstein S, Smart JM, French MA, Fulcher DA, Picard C, Bustamante J, Boisson-Dupuis S, Gray P, Stepensky P, Warnatz K, Freeman AF, Rossjohn J, McCluskey J, Holland SM, Casanova JL, Uzel G, Ma CS, Tangye SG, Deenick EK. STAT3 is a critical cell-intrinsic regulator of human unconventional T cell numbers and function. J Exp Med. 2015;212(6):855–864. doi: 10.1084/jem.20141992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Béziat V, Li J, Lin JX, Ma CS, Li P, Bousfiha A, et al. A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci Immunol. 2018;3(24). 10.1126/sciimmunol.aat4956. [DOI] [PMC free article] [PubMed]
- 29.Berglund LJ, Avery DT, Ma CS, Moens L, Deenick EK, Bustamante J, Boisson-Dupuis S, Wong M, Adelstein S, Arkwright PD, Bacchetta R, Bezrodnik L, Dadi H, Roifman CM, Fulcher DA, Ziegler JB, Smart JM, Kobayashi M, Picard C, Durandy A, Cook MC, Casanova JL, Uzel G, Tangye SG. IL-21 signalling via STAT3 primes human naive B cells to respond to IL-2 to enhance their differentiation into plasmablasts. Blood. 2013;122(24):3940–3950. doi: 10.1182/blood-2013-06-506865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Payne K, Li W, Salomon R, Ma CS. OMIP-063: 28-color flow cytometry panel for broad human immunophenotyping. Cytometry A. 2020;97(8):777–781. doi: 10.1002/cyto.a.24018. [DOI] [PubMed] [Google Scholar]
- 31.Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, Foucat E, Dullaers M, Oh SK, Sabzghabaei N, Lavecchio EM, Punaro M, Pascual V, Banchereau J, Ueno H. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011;34(1):108–121. doi: 10.1016/j.immuni.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Chew GY, Chan TD, Palendira U, Bustamante J, Boisson-Dupuis S, Choo S, Bleasel KE, Peake J, King C, French MA, Engelhard D, al-Hajjar S, al-Muhsen S, Magdorf K, Roesler J, Arkwright PD, Hissaria P, Riminton DS, Wong M, Brink R, Fulcher DA, Casanova JL, Cook MC, Tangye SG. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med. 2010;207(1):155–171. doi: 10.1084/jem.20091706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deenick EK, Avery DT, Chan A, Berglund LJ, Ives ML, Moens L, Stoddard JL, Bustamante J, Boisson-Dupuis S, Tsumura M, Kobayashi M, Arkwright PD, Averbuch D, Engelhard D, Roesler J, Peake J, Wong M, Adelstein S, Choo S, Smart JM, French MA, Fulcher DA, Cook MC, Picard C, Durandy A, Klein C, Holland SM, Uzel G, Casanova JL, Ma CS, Tangye SG. Naive and memory human B cells have distinct requirements for STAT3 activation to differentiate into antibody-secreting plasma cells. J Exp Med. 2013;210(12):2739–2753. doi: 10.1084/jem.20130323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma CS, Wong N, Rao G, Nguyen A, Avery DT, Payne K, Torpy J, O’Young P, Deenick E, Bustamante J, Puel A, Okada S, Kobayashi M, Martinez-Barricarte R, Elliott M, Sebnem Kilic S, el Baghdadi J, Minegishi Y, Bousfiha A, Robertson N, Hambleton S, Arkwright PD, French M, Blincoe AK, Hsu P, Campbell DE, Stormon MO, Wong M, Adelstein S, Fulcher DA, Cook MC, Stepensky P, Boztug K, Beier R, Ikincioğullari A, Ziegler JB, Gray P, Picard C, Boisson-Dupuis S, Phan TG, Grimbacher B, Warnatz K, Holland SM, Uzel G, Casanova JL, Tangye SG. Unique and shared signaling pathways cooperate to regulate the differentiation of human CD4+ T cells into distinct effector subsets. J Exp Med. 2016;213(8):1589–1608. doi: 10.1084/jem.20151467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willmann KL, Klaver S, Doğu F, Santos-Valente E, Garncarz W, Bilic I, Mace E, Salzer E, Domínguez Conde C, Sic H, Májek P, Banerjee PP, Vladimer GI, Haskoloğlu Ş, Gökalp Bolkent M, Küpesiz A, Condino-Neto A, Colinge J, Superti-Furga G, Pickl WF, van Zelm MC, Eibel H, Orange JS, Ikincioğulları A, Boztuğ K. Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat Commun. 2014;5:5360. doi: 10.1038/ncomms6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Israel L, Mellett M. Clinical and genetic heterogeneity of CARD14 mutations in psoriatic skin disease. Front Immunol. 2018;9:2239. doi: 10.3389/fimmu.2018.02239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamming OJ, Kang L, Svensson A, Karlsen JL, Rahbek-Nielsen H, Paludan SR, Hjorth SA, Bondensgaard K, Hartmann R. Crystal structure of interleukin-21 receptor (IL-21R) bound to IL-21 reveals that sugar chain interacting with WSXWS motif is integral part of IL-21R. J Biol Chem. 2012;287(12):9454–9460. doi: 10.1074/jbc.M111.311084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siupka P, Hamming OT, Kang L, Gad HH, Hartmann R. A conserved sugar bridge connected to the WSXWS motif has an important role for transport of IL-21R to the plasma membrane. Genes Immun. 2015;16(6):405–413. doi: 10.1038/gene.2015.22. [DOI] [PubMed] [Google Scholar]
- 39.Zeng R, Spolski R, Casas E, Zhu W, Levy DE, Leonard WJ. The molecular basis of IL-21-mediated proliferation. Blood. 2007;109(10):4135–4142. doi: 10.1182/blood-2006-10-054973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2020;40(1):24–64. doi: 10.1007/s10875-019-00737-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bodemer C, Sauvage V, Mahlaoui N, Cheval J, Couderc T, Leclerc-Mercier S, Debré M, Pellier I, Gagnieur L, Fraitag S, Fischer A, Blanche S, Lecuit M, Eloit M. Live rubella virus vaccine long-term persistence as an antigenic trigger of cutaneous granulomas in patients with primary immunodeficiency. Clin Microbiol Infect. 2014;20(10):O656–O663. doi: 10.1111/1469-0691.12573. [DOI] [PubMed] [Google Scholar]
- 42.Brady J, Hayakawa Y, Smyth MJ, Nutt SL. IL-21 induces the functional maturation of murine NK cells. J Immunol. 2004;172(4):2048–2058. doi: 10.4049/jimmunol.172.4.2048. [DOI] [PubMed] [Google Scholar]
- 43.Lopez-Vergès S, Milush JM, Pandey S, York VA, Arakawa-Hoyt J, Pircher H, Norris PJ, Nixon DF, Lanier LL. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood. 2010;116(19):3865–3874. doi: 10.1182/blood-2010-04-282301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Casanova J-L, Abel L. Primary immunodeficiencies: a field in its infancy. Science. 2007;317(5838):617–619. doi: 10.1126/science.1142963. [DOI] [PubMed] [Google Scholar]
- 45.Roifman CM, Somech R, Kavadas F, Pires L, Nahum A, Dalal I, Grunebaum E. Defining combined immunodeficiency. J Allergy Clin Immunol. 2012;130(1):177–183. doi: 10.1016/j.jaci.2012.04.029. [DOI] [PubMed] [Google Scholar]
- 46.Winkelstein JA, Marino MC, Ochs H, Fuleihan R, Scholl PR, Geha R, Stiehm ER, Conley ME. The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine (Baltimore) 2003;82(6):373–384. doi: 10.1097/01.md.0000100046.06009.b0. [DOI] [PubMed] [Google Scholar]
- 47.Tangye SG, Ma CS. Regulation of the germinal center and humoral immunity by interleukin-21. J Exp Med. 2020;217(1). 10.1084/jem.20191638. [DOI] [PMC free article] [PubMed]
- 48.Asao H, Okuyama C, Kumaki S, Ishii N, Tsuchiya S, Foster D, Sugamura K. Cutting edge: the common γ-chain is an indispensable subunit of the IL-21 receptor complex. J Immunol. 2001;167(1):1–5. doi: 10.4049/jimmunol.167.1.1. [DOI] [PubMed] [Google Scholar]
- 49.Frey-Jakobs S, Hartberger JM, Fliegauf M, Bossen C, Wehmeyer ML, Neubauer JC, et al. ZNF341 controls STAT3 expression and thereby immunocompetence. Sci Immunol. 2018;3(24). 10.1126/sciimmunol.aat4941. [DOI] [PMC free article] [PubMed]
- 50.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, Metin A, Karasuyama H. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448(7157):1058–1062. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 51.Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature. 2008;455(7214):764–769. doi: 10.1038/nature07345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O’Shea J, Holland SM, Paul WE, Douek DC. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452(7188):773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martínez-Barricarte R, Markle JG, Ma CS, Deenick EK, Ramírez-Alejo N, Mele F, et al. Human IFN-γ immunity to mycobacteria is governed by both IL-12 and IL-23. Sci Immunol. 2018;3(30). 10.1126/sciimmunol.aau6759. [DOI] [PMC free article] [PubMed]
- 54.Ozaki K, Spolski R, Feng CG, Qi C-F, Cheng J, Sher A, Morse HC, III, Liu C, Schwartzberg PL, Leonard WJ. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298(5598):1630–1634. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 55.Kishida T, Hiromura Y, Shin-Ya M, Asada H, Kuriyama H, Sugai M, Shimizu A, Yokota Y, Hama T, Imanishi J, Hisa Y, Mazda O. IL-21 induces inhibitor of differentiation 2 and leads to complete abrogation of anaphylaxis in mice. J Immunol. 2007;179(12):8554–8561. doi: 10.4049/jimmunol.179.12.8554. [DOI] [PubMed] [Google Scholar]
- 56.Tortola L, Pawelski H, Sonar SS, Ampenberger F, Kurrer M, Kopf M. IL-21 promotes allergic airway inflammation by driving apoptosis of FoxP3(+) regulatory T cells. J Allergy Clin Immunol. 2019;143(6):2178–89.e5. doi: 10.1016/j.jaci.2018.11.047. [DOI] [PubMed] [Google Scholar]
- 57.Buchbinder D, Hauck F, Albert MH, Rack A, Bakhtiar S, Shcherbina A, Deripapa E, Sullivan KE, Perelygina L, Eloit M, Neven B, Pérot P, Moshous D, Suarez F, Bodemer C, Bonilla FA, Vaz LE, Krol AL, Klein C, Seppanen M, Nugent DJ, Singh J, Ochs HD. Rubella virus-associated cutaneous granulomatous disease: a unique complication in immune-deficient patients, not limited to DNA repair disorders. J Clin Immunol. 2019;39(1):81–89. doi: 10.1007/s10875-018-0581-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leclerc-Mercier S, Moshous D, Neven B, Mahlaoui N, Martin L, Pellier I, Blanche S, Picard C, Fischer A, Perot P, Eloit M, Fraitag S, Bodemer C. Cutaneous granulomas with primary immunodeficiency in children: a report of 17 new patients and a review of the literature. J Eur Acad Dermatol Venereol. 2019;33(7):1412–1420. doi: 10.1111/jdv.15568. [DOI] [PubMed] [Google Scholar]
- 59.Ferrua F, Galimberti S, Courteille V, Slatter MA, Booth C, Moshous D, Neven B, Blanche S, Cavazzana M, Laberko A, Shcherbina A, Balashov D, Soncini E, Porta F, al-Mousa H, al-Saud B, al-Dhekri H, Arnaout R, Formankova R, Bertrand Y, Lange A, Smart J, Wolska-Kusnierz B, Aquino VM, Dvorak CC, Fasth A, Fouyssac F, Heilmann C, Hoenig M, Schuetz C, Kelečić J, Bredius RGM, Lankester AC, Lindemans CA, Suarez F, Sullivan KE, Albert MH, Kałwak K, Barlogis V, Bhatia M, Bordon V, Czogala W, Alonso L, Dogu F, Gozdzik J, Ikinciogullari A, Kriván G, Ljungman P, Meyts I, Mustillo P, Smith AR, Speckmann C, Sundin M, Keogh SJ, Shaw PJ, Boelens JJ, Schulz AS, Sedlacek P, Veys P, Mahlaoui N, Janda A, Davies EG, Fischer A, Cowan MJ, Gennery AR. Hematopoietic stem cell transplantation for CD40 ligand deficiency: results from an EBMT/ESID-IEWP-SCETIDE-PIDTC study. J Allergy Clin Immunol. 2019;143(6):2238–2253. doi: 10.1016/j.jaci.2018.12.1010. [DOI] [PubMed] [Google Scholar]
- 60.Olsson R, Remberger M, Schaffer M, Berggren DM, Svahn BM, Mattsson J, Ringden O. Graft failure in the modern era of allogeneic hematopoietic SCT. Bone Marrow Transplant. 2013;48(4):537–543. doi: 10.1038/bmt.2012.239. [DOI] [PubMed] [Google Scholar]
- 61.Shearer WT, Rosenblatt HM, Gelman RS, Oyomopito R, Plaeger S, Stiehm ER, Wara DW, Douglas SD, Luzuriaga K, McFarland EJ, Yogev R, Rathore MH, Levy W, Graham BL, Spector SA. Lymphocyte subsets in healthy children from birth through 18 years of age: the pediatric AIDS Clinical Trials Group P1009 study. J Allergy Clin Immunol. 2003;112(5):973–980. doi: 10.1016/j.jaci.2003.07.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 176 kb)
Data Availability Statement
Not applicable