

Abstract
The immunosuppressive tumor microenvironment enables cancer to resist immunotherapies. We have established that intratumoral administration of plant-derived Cowpea mosaic virus (CPMV) nanoparticles as an in situ vaccine overcomes the local immunosuppression and stimulates a potent anti-tumor response in several mouse cancer models and canine patients. CPMV does not infect mammalian cells but acts as a danger signal that leads to the recruitment and activation of innate and subsequently, adaptive immune cells. In the present study we addressed whether other icosahedral viruses or virus-like particles (VLPs) of plant, bacteriophage and mammalian origin can be similarly employed as intratumoral immunotherapy. Our results indicate that CPMV in situ vaccine outperforms Cowpea chlorotic mottle virus (CCMV), Physalis mosaic virus (PhMV), Sesbania mosaic virus (SeMV), bacteriophage Qβ VLPs, or Hepatitis B virus capsids (HBVc). Furthermore, ex vivo and in vitro assays reveal unique features of CPMV that makes it an inherently stronger immune stimulant.
Introduction
Cancer immunotherapies facilitate immune system mediated elimination of transformed cells.1 However, to be effective, such treatment regimen must overcome the immunosuppressive tumor microenvironment (TME) that diminishes the anti-tumor immune response, both quantitatively and qualitatively through a variety of mechanisms.2 Localized intratumoral administration of potent immune modulators can overturn the immunosuppressive TME and instigate an effective anti-tumor immune response from within. 3,4 We have established that plant-derived Cowpea mosaic virus (CPMV) is a highly potent immune modulator of the TME when employed as an in situ vaccine.5 The efficacy of CPMV in situ vaccine has been demonstrated in several syngeneic mouse tumor models and in canine patients with oral melanomas.5–8
While immunotherapy realized by oncolytic viruses is a result of viral infection and propagation in cancer cells resulting in cancer cell lysis and release of tumor antigens, plant viruses do not replicate in or lyse cancer cells; the priming of anti-tumor immunity is triggered by the immunogenic properties of the virus itself leading to modulation of TME, including recruitment of innate immune cells that induce cancer cell lysis. 5,9–11 The CPMV in situ vaccine leads to infiltration and activation of a broad spectrum of innate immune cells in the TME, resulting in an early inflammatory phase characterized by upregulation of pro-inflammatory cytokines (e.g. IFN-γ, IL-6), recruitment of myeloid cell precursors G-MDSCs/M-MDSCs and their conversion to immunostimulatory myeloid cells. Accompanied by reduction of immunosuppressive cytokines levels within TME (e.g. TGF-β and IL-10), these events promote recruitment and repolarization of neutrophils and macrophages to anti-tumor phenotypes (N1, M1) leading to cancer cell killing. Tumor antigens released in the process are picked up by infiltrating antigen-presenting cells (APCs), which then trigger the activation of the adaptive anti-tumor immune response.5,7,8,10,12
The anti-tumor response instigated by the CPMV in situ vaccine is dependent on the recognition of the plant viral capsid protein by the immune system.5,9 The mammalian innate immune system relies on an array of pattern recognizing receptors (PRRs) such as toll-like receptors (TLRs), expressed by immune cells to recognize and respond to invading pathogens.13 Within the TME, tumor-associated macrophages (TAMs) and neutrophils express numerous PRRs that can respond viral antigen.14–16 The utility of CPMV for in situ vaccination raises obvious questions about possible applicability of other (plant) viruses as in situ vaccines. In this study, we specifically asked whether other icosahedral viruses qualify as in situ vaccines for cancer immunotherapy. We employed a library of structurally similar plant viruses; specifically, we evaluated ~30 nm icosahedral plant viruses Cowpea chlorotic mottle virus (CCMV, virus nanoparticles, VNPs), Sesbania mosaic virus (SeMV VNPs) and Physalis mottle virus (PhMV; obtained as genome-free virus-like particle, VLP); in addition we also probed efficacy of bacteriophage Qβ VLPs and Hepatitis B virus capsids (HBVc, a VLP composed of the HBV core protein). Qβ-VLPs have been studied extensively as an adjuvant for vaccines and cancer immunotherapy;17 and HBV capsids have been used as multivalent epitope carriers.18,19 Established tumor models of melanoma, ovarian cancer and colon cancer, as well as ex vivo and in vitro assays were used to probe the immunological properties and potential to elicit anti-tumor immunity of these viruses and their VLPs.
Experimental
Production of plant viruses and virus-like particles (VLPs)
CPMV, CCMV and SeMV viral nanoparticles were produced in V. unguiculata and S. grandiflora plants, respectively using previously described protocols.20–22 PhMV-VLPs were expressed in and purified from ClearColi BL21 (DE3) cells (Lucigen, Middleton, WI) using the synthetic recombinant vector (pR-PhMVCP) using methods described earlier.23–26 CPMV VLPs (eCPMV) were produced using pEAQexpress-VP60–24 K vector and agroinfiltration in N. benthamiana plants using methods described elsewhere.27
Qβ-VLPs were produced using previously disclosed protocols in E. coli BL21 (DE3) cells transformed with Qβ plasmid pET28CP.28 HBVc particles were obtained using previously published protocols through expression of plasmid pET11a-Cp150 in ClearColi BL21(DE3) cells.29
Post purification, VNP/VLP concentrations were determined using Pierce BCA protein Assay Kit (ThermoFisher, Waltham, MA). Particle morphology and integrity was verified by transmission electron microscopy (TEM) performed on the FEI Tecnai Spirit G2 BioTWIN microscope (FEI, Hillsboro, OR). Samples (0.5–1 mg mL−1) were mounted on 400-mesh copper grids bearing Formvar support film and stained with uranyl acetate (2% w/v). Size exclusion chromatography using a Superose6 column on the ÄKTA Explorer chromatography system (GE Healthcare, Marlborough, MA) was also used to validate particle integrity, and determine 260:280 ratios of VNPs and VLPs. Particle charges and presence or absence of RNA was determined by agarose gel electrophoresis. Viral particles (10 μg in 6× loading dye) were subjected to a 100 V-40 minutes electrophoresis on a agarose gels (0.8% w/v) containing 1 μL of GelRed® Nucleic Acid Stain for RNA visualization (GoldBio, St Louis, MO) in Tris borate EDTA (TBE) buffer. Gels were visualized under UV light for nucleic acid and after staining with Coomassie Brilliant Blue (0.25% w/v) (Sigma, St Louis, MO) to visualize the capsids. VNP/VLPs were also characterized using SDS-PAGE gels. Briefly, 10 μg of CPMV, CCMV, SeMV and PhMV-VLP preparations were mixed with SDS running buffer (ThermoFisher Scientific), heated at 100 °C for 5 minutes, and then loaded on pre-cast NuPAGE™ 4–12% bis-tris protein gels (ThermoFisher Scientific); electrophoresis was performed for 40 minutes at 200 V. Gels were then stained using GelCode™ Blue Safe protein stain (ThermoFisher Scientific) to visualize the protein bands corresponding to molecular weight ladders. SDS gel electrophoresis of Qβ-VLPs was performed using 5 μg protein; and PageRuler prestained protein ladder (ThermoFisher Scientific) was used to determine molecular weights. Electrophoresis of HBVc particles was performed on a 6–16% SDS-PAGE at 300 V for 1 hour with the PageRuler prestained protein ladder.
Synthesis of Cy5 labeled plant viruses
CPMV-Cy5, CCMV-Cy5, SeMV-Cy5 and PhMV-Cy5 formulations were synthesized by conjugating N-hydroxysuccinimide-activated esters of Sulfo-Cy5 (NHS-Sulfo-Cy5, Lumiprobe, Hunt Valley, MD) to surface exposed lysine residues. Briefly, Sulfo-Cy5 conjugations were performed on CPMV, CCMV, SeMV and PhMV using dye/lysine ratios of 1, 1/6, 2 and 1/2, respectively. Bioconjugation was carried out overnight in 0.1 M sodium phosphate buffer (KP, pH 7.4) with of 10% (v/v) DMSO and at 2 mg mL−1 protein concentration. Dye-labeled particles were purified by ultracentrifugation (112 000 g, 1 h). The pellet was resuspended in 0.1 M KP buffer and protein concentration was determined using BCA assay. Dye numbers were determined through UV-vis analysis using the molar extinction coeffecient εsuifo-Cy5 = 271 000 L mol−1 cm−1 at 647 nm. Dye conjugation to viral coat proteins was also confirmed using SDS-PAGE as described above and fluorescent bands were visualized on an AlphaImage gel documentation system (protein simple) using a 607 nm excitation.
Cell lines
All cell lines were cultured at 37 °C with 5% CO2. Murine macrophage cell line RAW 264.7 was purchased from ATCC (Manassas, VA) and maintained on DMEM medium (ThermoFisher Scientific) supplemented with 10% (v/v) fetal-bovine serum (FBS) (VWR, Randor, PA) and 1% (v/v) penicillin/streptomycin (Pen/Strep) (ThermoFisher Scientific). The syngeneic ovarian cancer cell line ID8-Defb29/Vegf-a30 was stably transfected with luciferase as previously described31 to enable in vivo tracking and maintained on RPMI 1640 medium (ThermoFisher Scientific) supplemented with 10% (v/v) fetal-bovine serum (FBS) and 1% (v/v) Pen/Strep, 2 mM l-glutamine, 1 mM sodium pyruvate and 0.05 mM 2-mercaptoethanol. B16F10 dermal melanoma cells (ATCC) were cultured in DMEM medium supplemented with 10% (v/v) fetal-bovine serum (FBS) and 1% (v/v) Pen/Strep. CT-26 colon cancer cells (ATCC) were cultured using RPMI-1640 medium supplemented with 10% (v/v) fetal-bovine serum (FBS) and 1% (v/v) Pen/Strep. RAW-Blue™ Cells (Invivogen, San Diego, CA) were maintained in selection media containing Zeocin (Invivogen, San Diego, CA) and Normocin (Invivogen, San Diego, CA) as per instructions by the supplier.
Mice
All animal experiments were carried out in accordance with NIH Guide for the Care and Use of Laboratory Animals in Research (DHEW 78–23), reprinted in 1980 (DHEW 80–23 or succeeding editions), and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California San Diego, California. All mice (six-seven weeks old) were purchased from Jackson Laboratories. Male C57BL6 mice were used for dermal B16F10 melanoma studies comparing CPMV vs. SeMV vs. PhMV in situ vaccines. Female C57BL6 mice were used for all studies involving intraperitoneal ovarian tumors and B16F10 melanoma studies comparing CPMV vs. CCMV and CPMV vs. HBVc in situ vaccinations. Female Balb/c mice were used for comparing CPMV vs. Qβ in situ vaccines against intraperitoneal CT26 tumors.
Tumor inoculations and in situ vaccinations
For in situ vaccine efficacy studies in ovarian cancer, mice (n = 8) were intraperitoneally (i.p.) implanted with 2 × 106 ID8-Defb29/Vegf-A-luc cells in 200 μL PBS. Tumor growth was monitored via bioluminescence imaging on IVIS Spectrum Imaging System (PerkinElmer Ltd, Santa Clara, CA) following luciferin injections (150 mg kg−1, i.p., ThermoScientific). Tumor growth was also monitored by measuring mice weights and circumferences. In situ vaccines were administered through six weekly i.p. injections of viral particles (100 μg in 200 μL PBS) starting at day 7 for plant viral nanoparticles study and day 15 for CPMV vs. HBVc studies (PBS, n = 7, vs. CPMV, n = 5 vs. HBVc, n = 3). For melanoma studies, 200 000 B16F10 cells were injected intradermally into the shaved right flank of C57BL6 mice; tumor volumes were monitored by caliper measurements. VNP/VLPs were injected intratumorally (100 μg in 20 μL PBS) weekly starting at day 7 when comparing plant viruses (PBS, n = 10 vs. CPMV, n = 8 vs. CCMV, n = 9; PBS, n = 4 vs. CPMV, n = 5 vs. SeMV, n = 5 vs. PhMV, n = 5) and every 5th day starting at day 8 in CPMV vs. HBVc study (PBS, n = 6 vs. CPMV, n = 6 vs. HBVc, n = 7). For CT-26 studies, tumors were inoculated by i.p. injections of 5 × 105 CT-26 cells in 200 μL PBS. Tumor burden was monitored by weight measurements. In situ vaccines (CPMV vs. Qβ-VLP, n = 5) were administered intraperitoneally (100 μg in 200 μL PBS) thrice weekly, starting at day 7 from tumor challenge.
Cytokine quantification and flow cytometry
For immune profiling, IP wash was harvested from tumor bearing mice 6 hours after the last injection of in situ vaccines or PBS (n = 5). Cells were separated for staining and flow cytometry analysis whereas supernatant was collected for cytokine quantification using enzyme-linked immunosorbent assays (ELISAs). IP wash supernatant was quantified for IL-10, IFN-γ, IL-6, TNF-α, and IL-12 using ELISA kits (Biolegend) as per manufacturer’s instructions. For flow cytometry analysis, cells were plated at 100 000 cells per well, washed in cold PBS containing 1 mM EDTA and resuspended in staining buffer (PBS with 2% (v/v) FBS, 1 mM EDTA, 0.1% (w/v) sodium azide). Next, Fc receptors were blocked by anti-mouse CD16/CD32 antibody (Biolegend, San Diego, CA) treatment for 15 minutes and then stained with following fluorescently labeled anti-bodies (BioLegend) for 30 minutes at 4 °C: CD45 (30-F11), CD11b (M1/70), CD86 (GL-1), major histocompatibility complex class II (MHCII, M5/114.15.2), Ly6G (1A8), CD11c (N418 A), F4/80 (BM8), Ly6C (HK1.4), NK1.1 (PK136), CD4 (GK1.5), CD3ε (145–2V11 A), CD8α (53–6.7), CD44 (IM7), CD62L (MEL-14), and isotype controls. Flow cytometry was performed on BD LSRII cytometer (BD Biosciences, San Diego, CA) using a gating strategy showed in ESI Fig. S4.† OneComp eBeads (eBioscience, San Diego, CA) were used as compensation controls and data were analyzed using FlowJo software (BD Biosciences).
For ex vivo uptake studies, IP wash was harvested from ovarian tumor bearing mice at day 55 and cells (plated at 2 × 106 cells per well) were incubated in triplicates with 10 μg of Cy5-labeled viral particles for 2 h hours. Cells were washed twice to remove excess particles, and stained with following fluorescent antibodies (BioLegend) for 1 hour at 4 °C: CD45 (30-F11), CD11b (M1/70), CD86 (GL-1), major histocompatibility complex class II (MHCII, M5/114.15.2), Ly6G (1A8), CD11c (N418 A), F4/80 (BM8), Ly6C (HK1.4), and NK1.1 (PK136). Cells were then washed, fixed and flow cytometry was performed as described above.
RAW 264.7 cell uptake and activation studies were performed by incubating 200 000 cells per well with 5 μg of CPMV and CCMV viral particles for 1 hour. Cells were washed off the excess particles, Fc receptors were blocked by anti-mouse CD16/CD32 antibody (BioLegend) treatment for 15 minutes and then stained with fluorescently labeled antibodies (BioLegend) CD86 (GL-1) and major histocompatibility complex class II (MHC-II, M5/114.15.2) for 30 minutes at 4 °C. Cells were then permeabilized with staining solution containing 0.5% (v/v) saponine (10% (w/v) saponine in PBS) and incubated with rabbit α-CPMV and α-CCcMV IgG (Pacific Immunology) at 1: 500 dilutions and then stained with Cy5-conjugated goat anti-rabbit IgG antibody. Cells were then washed, fixed and flow cytometry was performed as described above.
RAW-Blue™ cells/Quanti-Blue SEAP assay
100 000 RAW-Blue™ cells were incubated with 1 μg of CPMV, CCMV, SeMV, PhMV, eCPMV particles, 1× bacterial LPS (eBioscience™), E. coli endotoxin standard control (ThermoScientific, 50 EU mL−1), 0.1 M KP buffer, pH 7.4 and culture media only for 18 hours. The Quanti-Blue assay was performed as per instructions by the supplier, and absorbance was measured at 655 nm using a Tecan microplate reader. In a separate experiment, 5 μg CPMV and HBVc each were similarly incubated with 100 000 RAW-Blue™ cells and probed using the Quanti-Blue assay as described above.
Virus overlay protein binding assay (VOPBA) and dot blots
3 μg recombinant human vimentin (PropSpec) was electrophoresed on a on pre-cast NuPAGE™ 4–12% bis-tris protein gels (ThermoFisher Scientific) for 40 minutes at 200 V. SeeBlue™ Plus2 Pre-stained Protein ladder (ThermoFisher) was used in the gel used for VOPBA of CPMV, CCMV, SeMV and PhMV, and PageRuler™ Prestained Protein Ladder (Thermofisher) was used for gel used in VOPBA comparing CPMV and Qβ. Gels were then blotted on to a nitrocellulose membrane (25 V, 1 hour). The membrane was incubated in denaturing buffer (6 M Guanidine-HCI, 2 mM EDTA, 50 mM DTT, 50 mM Tris/HCl, pH 8.3) for 10 minutes on a rocker. The solution was replaced with fresh denaturing buffer and rocked for another 10 minutes. The membrane was then transferred to 20 mL renaturing buffer (10 mM Tris/HCl, 150 mM NaCl, 2 mM EDTA, 0.1% (v/v) Triton X-100, pH 7.3) with 7.643 g Guanidine-HCl (4 M) at 4 °C for 10 minutes. 6.5 mL, 13.5 mL and 40 mL of renaturing buffer was step-wise added to above solution and rocked for 10 minutes each time at 4 °C. The membrane was then pIaced in fresh renaturing buffer at 4 °C for 10 minutes and blocked with 5% (w/v) dry milk in renaturing buffer overnight. Next day, the membrane was cut into individual strips and each strip was incubated with viral particles (10 μg mL−1 in renaturing buffer containing 1% (w/v) dry milk, 5% (v/v) glycerol) for 2 hours at room temperature. Following this, membranes were washed 4 times with wash buffer (TBS with 0.2% (v/v) Triton X-100) and incubated with primary antibodies (α-VNP/VLP IgGs, Pacific Immunology, Ramona, CA) at 1 : 500 dilutions in 5% (w/v) dry milk in 10 mL wash buffer for 1 hour at room temperature with constant rocking. Mouse monoclonal anti-vimentin antibody v9 (Sigma) was added to one membrane stripe as positive control. After 4× washes, the membrane was incubated with 10 mL of 1: 2000 dilutions secondary antibody (alkaline phosphatase-conjugated goat anti-rabbit IgG for VNPs/VLPs, and HRP-conjugated goat anti-mouse IgG for v9) in wash solution with 5% (w/v) milk, for 1 hour. After 4× washes, 5 minutes each, VNP membranes were developed with 1-Step™ NBT/BCIP substrate (ThermoFisher Scientific), v9 membrane was developed with DAB substrate (Vector Laboratories, Burlingame, CA) as per instuctions by the suppliers.
For dot blots, 10 μg of VNPs/VLPs were spotted on a nitrocellulose membrane (2 μL volume per spot). Following blocking with 5% (w/v) milk at 37 °C for 1 hour, membranes were incubated with vimentin (10 μg mL−1 in PBS, with 5% (w/v) milk) at 4 °C overnight and washed 3 times with wash buffer (PBS with 0.2% (v/v) Triton X-100). Bound vimentin was detected using mouse monoclonal anti-vimentin antibody v9 (1:500) (Sigma) and A647-conjugated anti-mouse IgG secondary antibody (1:2000 dilutions). Developed dots were visualized on an AlphaImager gel documentation system (protein simple) using a 607 nm excitation.
Results and discussion
The viral nanoparticle library
CPMV and CCMV were propagated in black-eyed pea plants (Vigna unguiculata) and SeMV was propagated in Sesbania grandiflora plants using previously described protocols20–22 with yields of 50–100 mg virus/per 100 g of infected leaves. We did not have access to Physalis mottle virus, however had expression constructs to produce VLPs thereof; PhMV-VLPs were purified from ClearColi BL21 (DE3) cells through expression of PhMV-CP by the recombinant vector pR-PhMV-CP using methods described earlier.23–26 Furthermore, we also produced CPMV VLPs (eCPMV) through co-expression of the viral protease and the VP60 precursor protein encoding the small and large coat protein using agro-infiltration method and Nicotiana benthamiana plants, as previously described.27 eCPMV was obtained at yields of 50 mg eCPMV per 100 g of leaves.
We chose CPMV and CCMV because these viruses are similar size (~30 nm in diameter), both have a negative surface charge, and these plant viruses are produced in the same host (V. unguiculata). SeMV also is similar in size with negative surface charge,32,33 but is produced in a different host. SeMV in particular was chosen, because literature indicates that both, SeMV and CPMV interact with the mammalian protein vimentin,33,34 while CCMV is not known to interact with vimentin.35 Lastly, PhMV-VLPs were included because these are also of similar size, yet carry an overall positive surface charge. A further difference is that these are produced as VLPs by heterologous expression in bacteria. Of note is that while all particles are ~30 nm in size, CCMV, SeMV and PhMV exhibit T = 3 symmetry (i.e. their capsids are formed by 180 identical copies of the coat protein unit), while CPMV has a pseudoT or pT = 3 symmetry; CPMV consists of 60 copies of a small (S) and large (L) protein each. The S subunit folds into one jelly roll β-sandwich and clusters around the 5-fold axis, while the L subunit folds into two jelly roll β-sandwich and cluster around the 2-and 3-fold axis.36
VNP/VLP properties are summarized in Fig. 1. Transmission electron microscopy (TEM) and size exclusion chromatography (SEC) was used to verify structural integrity post purification and depict morphological similarities between the four particles (Fig. 1B and C). In TEM, all particles appear as 30 nm-sized particles and SEC revealed simiIar eIution profiles from the Superose6 column. CPMV, CCMV and SeMV showed a characteristic ratio for absorbance at 260 nm and 280 nm (A260:A280) of 1.8 indicating intact capsids with enclosed RNAs, whereas PhMV-VLPs showed a A260:A280 ratio of 0.8 indicating absence of viral RNA. It should be noted though that the PhMV-VLPs package host RNA to a small degree as previously reported;23 this is also observed on native gels stained with GelRed® nucleic acid stain (Fig. 1D). Less stain accumulates in the capsids when comparing PhMV-VLPs vs. the VNPs (CPMV, CCMV and SeMV). The native gels stained for nucleic acid or protein (Fig. 1D) also highlight the differences in surface charge: CPMV, CCMV, and SeMV are negatively charged and migrate toward the anode; PhMV-VLPs are positively charged and migrate toward the cathode. Lastly, analysis of the coat proteins using denaturing SDS-PAGE showed the characteristic coat protein bands: 42 kDa and 24 kDa L and S coat protein for CPMV; and the characteristic coat protein bands of 21 kDa, 29 kDa and 21 kDa for CCMV, SeMV and PhMV, respectively (Fig. 1E).
Fig. 1.
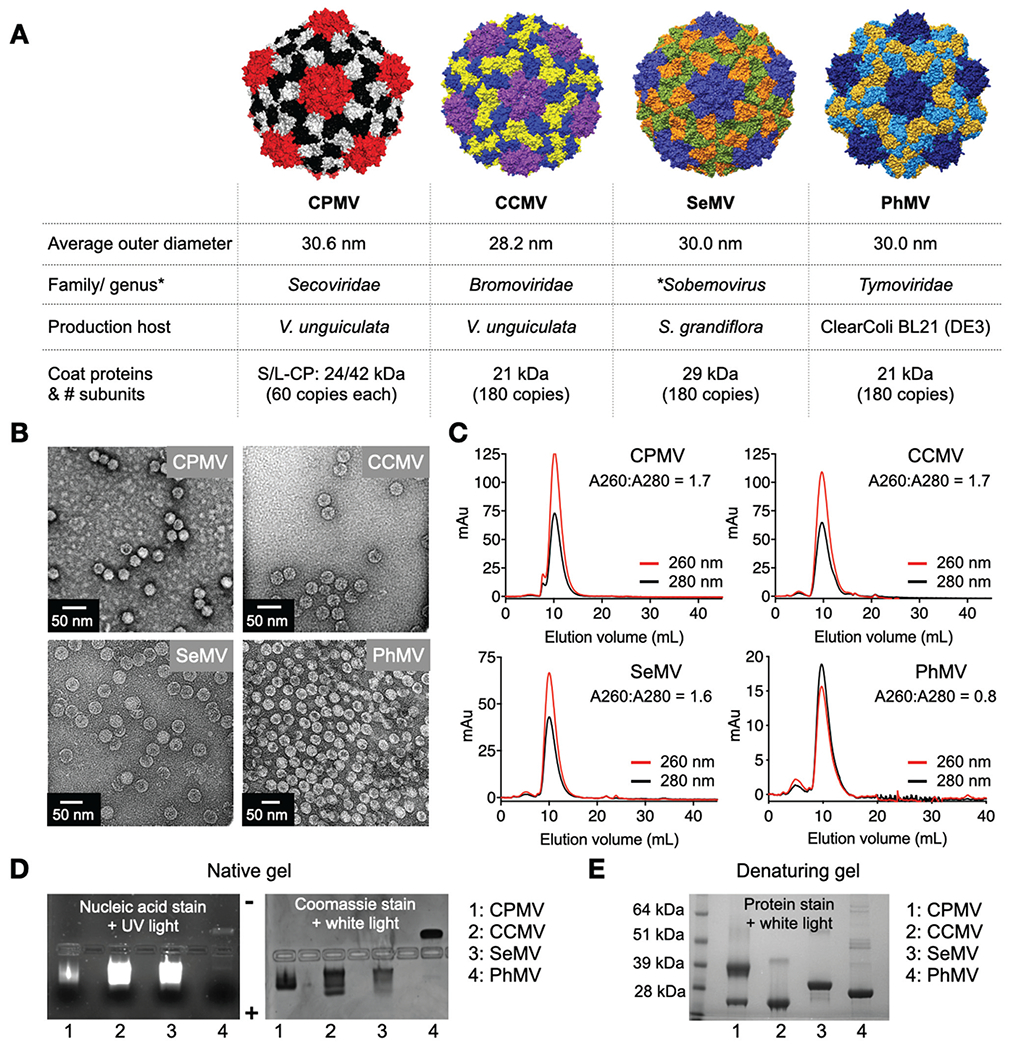
Plant viral nanoparticles. (A) Structure of CPMV, was created using Pymol (v2.3.4) using Protein Data Bank (PDB) entry 1NY7; CCMV, SeMV and PhMV space filling models were created on UCSF Chimera software 1.12 using PDB entries 1CWP, 1SMV and 1E57, respectively. Information on size is as reported on *VIPERdb. (B) TEM images of negatively stained virus nanoparticles. (C) SEC analysis shows characteristics elution profiles from a Superose 6 column; the A260 : A280 ratio indicates presence (~1.8) or absence (0.8) of nucleic acid. (D) Native agarose gel electrophoresis stained with GelRed® nucleic acid stain and Coomassie Brilliant Blue; gels were imaged under UV and white light. (E) Denaturing 4–12% Nu-PAGE gel stained with GelCode™ Blue Safe protein stain shows the coat proteins for the various virus preparations (*VIPERdb).32
The RNA-free VLP of CPMV, eCPMV, was characterized using the same methods (see Fig. S1, ESI†). Size exclusion chromatography confirmed particle integrity through elution profiles similar to those of wild type CPMV, whereas the A260: A280 ratios of ~0.7 indicated absence of encapsidated RNA. Native gel electrophoresis further validated this by the absence of nucleic acid stain. The separation of the slow-migrating fraction representing VLP with the intact S-CP and the fast-migrating fraction representing VLP with C-terminal cleaved S-CP is consistent with previously reported CPMV VLP production.37,38
Furthermore, we considered VLPs from bacteriophage Qβ and mammalian Hepatitis B virus capsids (HBVc). Qβ-VLPs and HBVc particles were produced by heterologous expression using E. coli using previously established methods;28,29 Qβ-VLPs and HBVc particles were obtained at yields of ~16 mg and 3 mg per 100 mL of E. coli culture, respectively. Qβ-VLPs measure 29 nm in size and are composed of an T = 3 icosahedral capsid formed of 180 identical copies of a 14 kDa coat protein. HBVc particles come in two sizes measuring ~36 and 32 nm in diameter and are composed of 240 and 180 copies of the Cp150 variant of the 17 kDa core protein assembly domain, respectively39,40 (Fig. S2, ESI†). The Cp150 protein can form disulfide bonds that reversibly crosslink the capsid.
For particle tracking studies, we prepared fluorescently-labeled VNPs/VLPs by conjugating NHS ester of Sulfo-Cy5 (NHS-Sulfo-Cy5) to the viral particles via the solvent exposed Lys residues (Fig. S3, ESI†). CPMV, CCMV, SeMV and PhMV exhibit 300, 1080, 180 and 720 reactive lysines per capsid, respectively.26,33,41,42 Using dye-to-lysine ratios ranging from 0.5–2, CPMV-Cy5, CCMV-Cy5, SeMV-Cy5 and PhMV-Cy5 particles with 28, 39, 16 and 28 dyes per VNP/VLP were obtained, respectively (the ratios used for each particle are described in detail in the Methods; UV-Vis method was used to determine the degree of labeling). SDS-PAGE analysis confirmed covalent conjugation of Cy5 to the viral coat proteins. Fluorescence intensity of Cy-labeled CPMV, CCMV, SeMV and PhMV-VLPs (at 0.1 mg mL−1, 100 μL total volumes) using 635/655 excitation/emission confirmed that the viral nanoparticles have comparable fluorescent properties: fluorescent intensities of 41 893, 38 845, 46 263 and 42 823 were recorded for CPMV-Cy5, CCMV-Cy5, SeMV-Cy5 and PhMV-VLP-Cy5 (Fig. S3†).
In situ vaccine efficacy
The efficacy of the viral particles as in situ vaccine was evaluated in an intraperitoneal (i.p.) disseminated ovarian tumor model (using ID8-Vegf/Def29b), IP disseminated colon cancer (CT-26) and dermal melanoma (B16F10). First, CPMV, CCMV, SeMV and PhMV-VLPs were used for in situ vaccine treatment using i.p. established ID8-Defb29/Vegf-a ovarian tumors in C57BL6 mice.30 Female C57BL6 mice were inoculated with 2 × 106 luciferase expressing ID8-Defb29/Vegf-a ovarian cancer cells. Starting seven days from tumor inoculation, mice were administered six weekly i.p. injections of CPMV, CCMV, SeMV and PhMV-VLPs (100 μg/200 μL PBS) or 200 μL PBS (Fig. 2A). Tumor progression was monitored using bioluminescence imaging as well as increase in body weight and body circumference resulting from increased tumor burden and developing ascites (Fig. 2B–D). Mice were euthanized upon reaching 30 g weight or a circumference of 10 cm. Untreated mice (PBS group) displayed steep rise in bioluminescence signal intensity starting at day 24 from tumor inoculation with a peak at day 33, consistent with rapid tumor progression. Mice treated with PhMV-VLPs showed a similar trend, indicating ineffectiveness of the treatment. Bioluminescence intensity in SeMV and CCMV treated mice peaked by days 43 and 50, respectively. In contrast, the bioluminescence signal intensity in CPMV treated mice remained lower than all the other treatment groups over the measured 65 days period, highlighting the efficacy of CPMV in situ vaccine in delaying tumor progression. Similarly, body weight and circumference measurements indicated significantly lower tumor burden and slower progression in mice treated with CPMV as compared to untreated or PhMV-VLP, SeMV and CCMV treated mice. This was mirrored in the survival curves (Fig. 2E): PBS-treated mice reached experimental endpoints and were euthanized with a median survival of 55 days. PhMV-VLP in situ vaccination had no therapeutic efficacy as evident from a 55 days median survival matching the PBS-treated mice. SeMV and CCMV in situ vaccines resulted in median survivals of 58 and 61.5 days (p = 0.003 vs. PBS). In contrast, the therapeutic efficacy of CPMV in situ vaccine resulted in a median survival of 81 days (p < 0.0001 vs. PBS or PhMV-VLP and p = 0.001 vs. CCMV or SeMV) (Fig. 2E). These results highlight the unique potency of the CPMV in situ vaccine as an immunotherapy. Other plant virus candidates examined here do not match potency of CPMV.
Fig. 2.
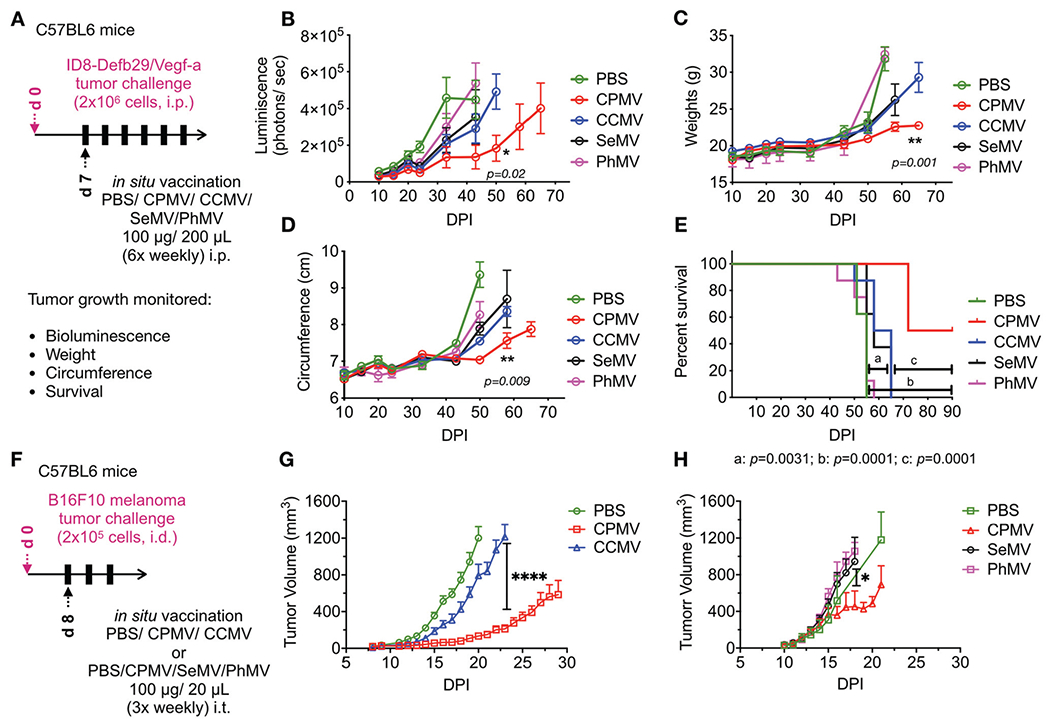
CPMV in situ vaccine has higher therapeutic efficacy over other plant viruses in mouse models of ID8-Defb29/Vegf-a ovarian cancer and B16F10 dermal melanoma. (A) Female C57BL6 mice were inoculated with 2 × 106 luciferase expressing ID8-Defb29/Vegf-a ovarian cancer cells intra-peritoneally. CPMV, CCMV, SeMV and PhMV were administered i.p. 6× weekly (100 μg/200 μL PBS) starting 7 days post-tumor inoculation. Tumor progression was monitored by bioluminescence imaging (B), and by measuring gains in weights (C) and circumference (D) from increasing tumor burden and developing ascites, as well as overall survival (E). Results were compared using unpaired t-test in B-D (with *=p < 0.05, **=p < 0.01) and Log-Rank Mantel-Cox test on survival data (****=p < 0.0001, ***=p < 0.001, **=p < 0.01). (F) C57BL6 mice were challenged with dermal B16F10 tumors by injecting 2 × 105 cells intradermally and treated with 3× weekly intratumoral injections of PBS vs. CPMV vs. CCMV (G) or PBS vs. CPMV vs. SeMV vs. PhMV (H) starting on day 8 from tumor challenge. Treatment efficacy was measured by monitoring tumor volumes. Results were compared using unpaired t-test in G (with ****=p < 0.0001) and one-way ANOVA in H (with *=p < 0.05).
Next, we compared the efficacy of CCMV, SeMV and PhMV-VLP in situ vaccines with CPMV in the B16F10 dermal melanoma model (Fig. 2F–H).12 In the first study, C57BL6 mice were inoculated dermally with B16F10 melanoma cells using 2 × 105 cells per animal. Once the tumors reach 40–60 mm3 (~8 days post tumor challenge), mice were treated with three intratumoral injections CPMV or CCMV (100 μg VNPs/20 μL PBS) every fourth day (20 μL PBS was used as a control). PBS-treated mice showed rapid growth of the dermal melanoma with tumor sizes exceeding 1000 mm3 by day 21 (Fig. 2G). CCMV treated mice followed similar tumor progression indicating no therapeutic effects. In contrast, CPMV treatment significantly reduced tumor burden and slowed disease progression with nearly 6-fold smaller tumor volumes recorded at day 23 from tumor inoculation (p < 0.0001 vs. PBS). In a second study, B16F10 melanoma bearing mice were treated with intratumoral injections using CPMV, SeMV or PhMV-VLPs (Fig. 2H). Again, the CPMV in situ vaccine resulted in slower tumor progression as compared to PBS (negative control), SeMV or PhMV treatments; at day 18, tumors from CPMV-treated animals were 2-fold smaller compared to control and other treatment groups (p = 0.03). SeMV and PhMV treatment showed no significant difference compared to PBS-treated tumors. It was evident from these studies that CCMV, SeMV and PhMV-VLPs did not match the potent efficacy observed with the CPMV in situ vaccine.
Lastly, we also compared the immunotherapeutic efficacy of the CPMV in situ vaccine with icosahedral viral particles of non-plant origin, namely the bacteriophage Qβ-VLPs and HBVc. The effectiveness of CPMV and Qβ-VLP in situ vaccines was tested in an i.p. metastatic CT26 colon carcinoma mouse model (Fig. 3A–C). Balb/c mice were injected with 5 × 105 CT26-Luc cells via i.p. injections and tumor growth was monitored using bioluminescence imaging as well as by measuring circumference and animal’s weight. Tumor bearing mice were treated once a week for three weeks with 100 μg CPMV vs. 100 μg Qβ-VLPs through IP injections starting at day 7 from tumor challenge. Tumor burden monitored as gains in animal weights (from developing ascites) indicated that Qβ-VLP in situ vaccine matched CPMV efficacy in slowing the tumor progression up to day 20. Mice survival analysis showed that Qβ-VLP treatment achieved a median survival of 27 days (p = 0.0235 vs. PBS). However, the CPMV in situ vaccine displayed a significantly higher efficacy over Qβ-VLPs with all mice in the group surviving during a 40-day observation (p = 0.0001 and p = 0.004 vs. PBS and Qβ-VLPs, respectively) (Fig. 3A–C).
Fig. 3.
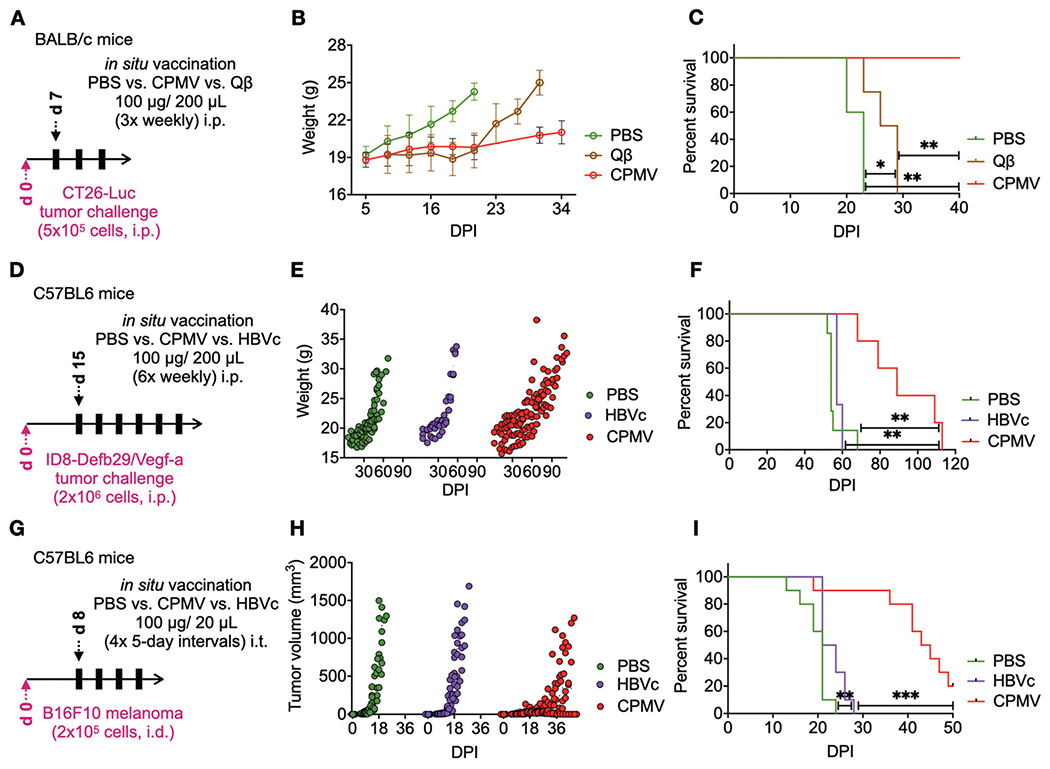
CPMV in situ vaccination outperforms bacteriophage Qβ and mammalian HBVc immunotherapies. (A) Balb/c mice were inoculated with 5 × 105 CT-26 cells intraperitoneally. CPMV (n = 5) and Qβ (n = 5) were administered i.p. 3× weekly (100 μg/200 μL PBS) starting 7 days post-tumor inoculation. Tumor progression was monitored by measuring gains in weights (B) from increasing tumor burden and developing ascites, as well as by overall survival (C). Results were compared using the Log-rank Mantel-Cox test on survival data. (D) Female C57BL6 mice were inoculated intraperitoneally with 2 × 106 ID8-Vegf Defb29-Luc cells to establish ovarian tumors. CPMV (n = 5) and HBVc (n = 3) were administered i.p. 6×, weekly (100 μg/200 μL PBS), starting at day 15 from tumor inoculation. Tumor progression was measured through gains in weight (E) and overall survival (F). Results were compared using the Log-Rank Mantel-Cox test on survival data. (G) Female C57BL6 mice were challenged with 2 × 105 B16F10 melanoma cells intradermally. CPMV (n = 10) or HBVc (n = 10) were administered intratumorally 4× every 5th day (100 μg/20 μL PBS), starting at day 8 post-tumor challenge (n = 10 for all groups). Tumor progression was monitored by measuring tumor volumes (H) and overall survival (I). Results were compared using the Log-Rank Mantel-Cox test on survival data.
The anti-tumor potential of the HBVc particles was assessed using the ID8-Defb29/Vegf-a ovarian tumor model (Fig. 3D–F) as well as the B16F10 dermal melanoma, both established in C57BL6 mice as described above (Fig. 3G–I). Mice bearing ovarian tumors, were treated with 6 weekly i.p. doses of HVBc or CPMV (100 μg viral particles/200 μL PBS) starting at day 15 from tumor inoculation. Disease progression was monitored as previously described through weight and circumference measurements. HBVc treatment showed no advantage over untreated mice (p = 0.31 vs. PBS). In contrast, CPMV treatment significantly slowed tumor progression and prolonged survival (median survival of 89 days vs. 57 days for HBVc and 54 days for PBS treatment with p = 0.0046 and p = 0.002, respectively). Similarly, in the B16F10 dermal melanoma studies, CPMV outperformed HBVc as an in situ vaccine (median survival of 44 days vs. 22.5 days, p = 0.0001) and HBVc showed no significant therapeutic benefit (Fig. 3I). Thus, in all three models, CPMV showed potent anti-tumor efficacy, but the other VNP/VLP formulations did not.
Characterization of immune response
Next, we characterized and compared the immune response induced by the virus-based nanoparticle in situ vaccination. To this end, established ID8-Defb29/Vegf-a tumor bearing mice were treated with i.p. injections of CPMV, CCMV, SeMV and PhMV-VLPs using previously described treatment schedules (see Fig. 2A); 6 hours following the last treatment peritoneal fluid was collected to quantify cytokine levels and assess immune cell profiles (Fig. 4).
Fig. 4.
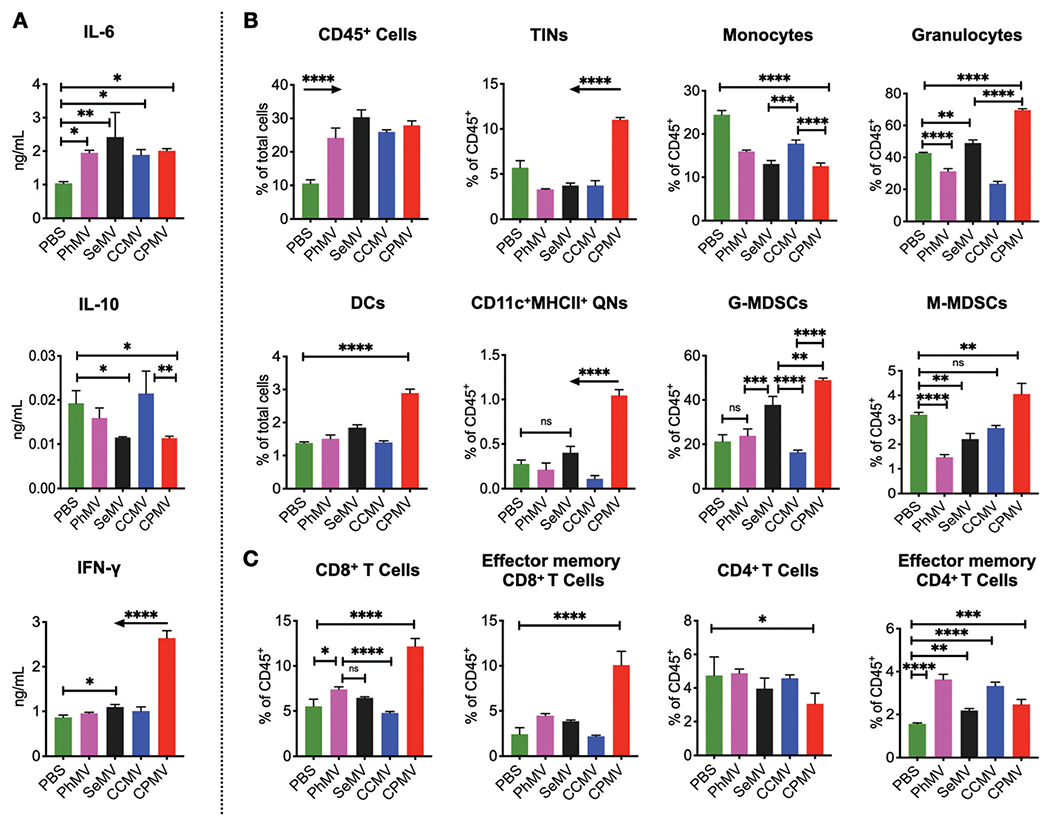
Each plant virus has unique impact on cells and cytokines in the tumor microenvironment of ID8-Defb29/Vegf-a ovarian tumors. IP wash was harvested through peritoneal gavage following six intratumoral administrations of CPMV, CCMV, SeMV and PhMV particles, 6 hours after the last injection for cytokine profile (A), innate immune cell panel (B) and the adaptive immune cell panel (C). ELISA was used to quantify cytokines (A), and flow cytometry was used to assess immune effector cells (B, C) (the gating strategy is shown in Fig. S4†). Results were compared using two-way ANOVA and Tukey’s multiple comparison test with * p < 0.05, *** p < 0.005 and **** p < 0.0001. Arrows indicate significant differences with all other groups.
ELISAs were used to measure the fluctuating levels of selected cytokines in the supernatant separated from the cells (Fig. 4A). As compared to the untreated group, intratumoral administration of the plant viral nanoparticles (CPMV, CCMV, SeMV and PhMV-VLPs) resulted in significantly elevated levels of IL-6, with no significant differences between the treatment arms. Pro-inflammatory IL-6 facilitates recruitment of immune cells into the TME but can also promote tumor progression and metastasis.43 However, in concert with other cytokines and chemokines, IL-6 is also critical for innate to adaptive response transition and development of anti-tumor immunity.44 IL-6 response has also been observed when tobacco mosaic virus (TMV) was used as an in situ vaccine;12 however TMV was less effective at launching anti-tumor immunity compared to CPMV.
Key differences between CPMV and other treatment groups are: only CPMV treatment led to significantly elevated IFN-γ levels (~3 folds higher) as compared to CCMV, SeMV and PhMV-VLPs treatments. IFN-γ is a key cytokine secreted by cytotoxic T cells and helper T cells, which mediate a Th-1 antitumor immune response45 that facilitates anti-tumor immunity. CPMV and SeMV treatment also lowered IL-10 levels in the IP wash significantly as compared to untreated mice, whereas PhMV and CCMV treatment had no influence on IL-10 levels. Tumor cells maintain an immunosuppressive microenvironment to promote disease progression by polarizing macrophages and neutrophils to M2 and N2 phenotypes by secreting cytokines such as IL-10 and TGF-β.46,47 Therefore, reduction in IL-10 levels is an indicator of TME immunomodulation to release immunosuppression. It was evident that CPMV in situ vaccines achieved this most effectively, whereas SeMV had a marginal effect on IL-10; whereas CCMV and PhMV-VLP treatment did not change IL-10 levels.
Flow cytometry analysis focused on innate cell populations indicated that all four plant viral particles CPMV, CCMV, SeMV and PhMV-VLPs induced influx of CD45+ leukocytes into the i.p. space, while reducing the number of monocytes, with no significant differences between the treatments (Fig. 4B, C and Fig. S5, ESI†). However, the phenotype of immune cells varied significantly between the plant viral nanoparticles, both qualitatively and quantitatively. The CPMV in situ vaccine led to significantly higher influx of tumor infiltrating neutrophils (TINs, CD45+CD11b+ Ly6G+MHCII+CD86+), activated quiescent neutrophils (QNs, CD45+CD11b−Ly6G+CD11c+MHCII+), granulocytes (CD45+CD11b+Ly6G+), dendritic cells (DCs, CD45+CD11b+CD11c+), granulocytic myeloid-derived suppressive cells (G-MDSCs, CD45+CD11b+Ly6G+Ly6C-MHCII−CD86−) and monocytic myeloid-derived suppressive cells (M-MDSCs, CD45+CD11b+Ly6G−Ly6C+MHCII−SSClow) over PBS control, CCMV, SeMV and PhMV-VLP in situ administrations. The adaptive immune cell populations were characterized at 24 hours post last treatment; and data indicate a significant increase in the CD8+ T cell population in the IP wash only following CPMV treatment (Fig. 4C). Furthermore, only CPMV treatment led to a higher CD8 effector memory T cells (CD44+ CD62L−). While there were no significant differences in the CD4+ T cell populations amongst the various treatment groups, an increase in CD4+ effector memory T cells was observed in all treatment groups as compared to untreated control. PhMV-VLP and CCMV treatment resulted in a larger influx of CD4 effector memory T cells as compared to SeMV and CPMV treatments (Fig. 4C).
The immune profile from CPMV treated tumors is consistent with our previous reports in metastatic lung B16F10 melanoma,48 dermal B16F10 melanoma12 and i.p. ID8 ovarian tumors.10 In these studies, independent of the tumor models, neutrophils have been identified as primary responders leading to inflammatory response in the tumor microenvironment.46 In addition to mediating cancer cell death, modulating proliferation and cytotoxic activity of NK cells, the N1 polarized neutrophils with elevated expression of MHC-II and CD86 co-stimulation markers also function as antigen presenting cells and thereby serve as key link between the innate and adaptive immune response.10
Under pro-inflammatory conditions, monocytic myeloid precursors M-MDSCs can differentiate into macrophages and DCs whereas granulocytic myeloid precursors G-MDSCs can differentiate into neutrophils and other immune cells.49 DCs are professional APCs that play a critical role in initiating and regulating the adaptive anti-tumor immune response by capturing, processing and cross-presenting tumor-associated antigens to prime T cell responses.50 Again, only the CPMV in situ vaccine promoted significant infiltration of DCs into the tumors; these cell types are expected to capture and process antigen released by the action of neutrophils, NK cells and macrophages.10 Furthermore, the significantly elevated levels of CD8+ T cells and CD8 effector memory T cells within the TME of CPMV-treated tumors also established that CPMV effectively primes the adaptive anti-tumor response.
Overall these observations also matched our previous report in which we compared the efficacy of CPMV in situ vaccine to treat B16F10 dermal melanoma12 with that of plant-based TMV and its structural variants.12 CPMV outperformed TMV in terms of efficacy and this was attributed to distinct immune activation by the two plant viruses: CPMV stimulated an anti-tumor response through recruitment of monocytes into the tumor microenvironment (TME), establishing signaling through the IFN-γ pathway, which also leads to recruitment of tumor infiltrated neutrophils (TINs) and natural killer cells (NKs). In contrast TMV mostly signaled through IL-6 leading to inflammation within the TME that did not activate adaptive anti-tumor immunity. CPMV and TMV also showed differences in IL-6 expression.12 It should be noted that TMV induced expression of IL-6 immediately upon administration with significant levels measured at 24 hours post administration; in contrast significant IL-6 levels were only measured at 4 days post administration using CPMV.
The cytokine and immune cell profile generated by CPMV in situ vaccine shows similarity and differences to previously reported papaya mosaic virus (PapMV) in situ vaccine in B16F10 melanoma tumor model.11 Both plant viruses show increased production of chemokine, pro-inflammatory cytokines and induced influx of CD8+ T cells into the tumor microenvironment. However, while CPMV administration increased the proportions of MDSCs in the tumor microenvironment, PapMV significantly decreased proportions of MDSCs. As noted earlier, pro-inflammatory conditions promote differentiation of MDSCs into macrophages and DCs and therefore these dissimilarities between CPMV and PapMV in situ vaccines could suggest differences in the underlying mechanisms.
Viral nanoparticle-immune cell interactions
Next, we asked which cells the VNPs/VLPs interact with and whether there would be differences in such interactions comparing the various viral nanoparticles. Cy5-labeled, fluorescent VNPs/VLPs of the different plant viruses were inoculated for 2 hours with cells isolated from the day 55 i.p. wash of untreated mice bearing ID8-Defb29/Vegf-a i.p. tumors. Flow cytometry analysis was performed to quantify the uptake of fluorescent Cy5-labeled VNPs/VLPs in CD45+ immune cells including TINs, tumor associated macrophages (TAMs), M-MDSCs, G-MDSCs, natural killer cells (NKs) and DCs (Fig. 5A). Substantial variability in cellular uptake profiles comparing the different VNPs/VLPs was observed. As indicated by the MFI and histograms alike, CCMV and CPMV displayed significantly higher uptake in TINs, TAMs, M-MDSCs, G-MDSCs and NK cells compared to SeMV and PhMV-VLPs. We also assessed cell uptake in vitro using murine macrophages RAW 264.7 cells (Fig. 5B). While all VNPs/VLPs (CPMV, CCMV, SeMV and PhMV-VLPs) demonstrated significant uptake by the macrophages, the trend observed in the ex vivo study was matched in vitro: CPMV and CCMV showed significant uptake; interestingly, CCMV exhibited the highest degree of uptake. However, only CPMV activated the macrophages, as evident from increased expression of macrophage activation markers CD86 and MHC-II following CPMV treatment (Fig. 5C). Despite higher uptake by immune cells in the i.p. wash and in macrophages in vitro, the CCMV in situ vaccine was ineffective; which may be explained by the lack of immune cell stimulation.
Fig. 5.
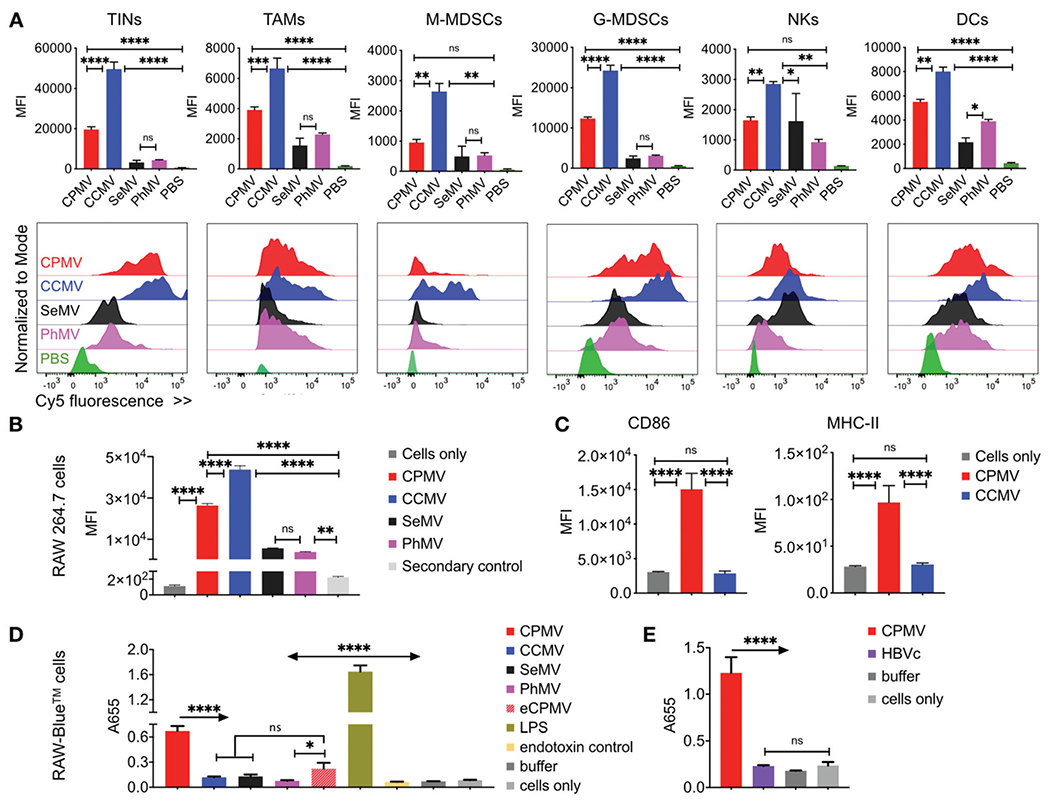
Distinct immune cell uptake and activation displayed by virus-based nanoparticles. (A) Ex vivo uptake of CPMV, CCMV, SeMV and PhMV-VLPs by immune cells harvested by IP gavage was quantified using flow cytometry and Cy5-labeled viral nanoparticles; the VNP/VLPs were incubated with IP wash for 2 hours. Histograms show the relatively shifts in the Cy5 fluorescence between the cellular populations following particle incubations as compared to control cells. (B) In vitro RAW 264.7 cell uptake: murine macrophages RAW 264.7 cells were used to compare the cellular uptake of CPMV, CCMV, SeMV and PhMV-VLPs. (C) Macrophage activation was compared for CCMV and CPMV particles through enhanced expression of activation markers CD86 and MHC-II. (D and E) Immunostimulatory potential of various viral nanoparticles was evaluated using in vitro RAW-Blue™ cell activation through a Quanti-Blue™ assay (1 μg VNPs/VLPs: 100 000 RAW-Blue™ cells in D; 5 μg VNPs/VLPs: 100 000 RAW-Blue™ cells in E). Results were compared using ordinary one-way ANOVA and Tukey’s multiple comparison test with * p < 0.05, *** p < 0.005 and **** p < 0.0001.
Unlike engineered oncolytic viruses that selectively infect, replicate in and lyse cancer cells to debulk tumor and thereby instigate immune response against tumor antigens, non-replicating VLPs of mammalian, plant or bacteriophage origin as well as plant VNPs rely on engaging intratumoral immune cells for modulating the TME; the VNPs/VLPs signals through PRRs. For example, activation of TLR7, which recognizes RNA in viruses, is critical for the PapMV in situ vaccine.11 On the other hand, RNA-free VLPs of CPMV also demonstrate efficacy;48,51 nevertheless data indicate that RNA-Iaden CPMV is more effective vs. CPMV VLP,8 and this is consistent with CPMV signaling through endosomal TLR7 and TLR8 following phagocytic uptake in primary human monocytes.52 The efficacy of PapMV was lost in a TLR7 knockout mouse,11 whereas CPMV activation of human monocytes was partially blocked by a TLR7 antagonist.52 Similarly, TLR-dependent MyD88 signaling pathway was shown to be critical for bacteriophage M13 induced tumor regression, and the anti-tumor effects of M13 were abrogated in MyD88−/− mice.53 In addition to the viral nucleic acids (ssRNA for the plant viruses and ssDNA for the phage M13) that are strong activators of anti-viral immune responses, the highly organized repetitive architecture of viral capsids has been shown to initiate a TLR2 dependent immune response.54 These TLR2 responses are not influenced by capsid shapes, sizes or amino acid composition, but influenced by the multivalent arrangements of the coat protein subunits. Therefore, immune activation through engagement of PRRs is critical for the efficacy of viral particles and quantifying these interactions can compare the potential of VNPs/VLPs to serve as an in situ vaccine.
To further elaborate differences in viral nanoparticle-immune cell interactions and their immune ceII activation potential, we compared activation of RAW-Blue™ cells by the viral nanoparticles using a Quanti-Blue™ assay (Fig. 5D and E).55’56 RAW-Blue™ cells are a mouse macrophage reporter cell line with stably expresses embryonic alkaline phosphatase (SEAP) gene induced by NF-κB and AP-1 transcription factors. These cells express all TLRs (except TLR-5) as well as RIG-1, NOD1 and NOD2 receptors. Presence of respective agonists for these receptors leads to activation of NF-κB and AP-1 and secretion of SEAP, which is detected by Quanti-Blue assay. RAW-Blue™ cells were incubated with CPMV, CCMV, SeMV and PhMV-VLPs as well as eCPMV VLPs (1 μg viral particles/100,000 RAW-Blue™ cells for 18 hours). Bacterial Iipopolysaccharide (LPS, 1x) served as a positive control, whereas untreated cells served as negative control (Fig. 5D). Quanti-Blue assay revealed a 6 folds higher activation of RAW-Blue cells by CPMV over CCMV and SeMV (p < 0.0001), and ~10 folds higher activation over PhMV (p < 0.0001). Furthermore, eCPMV particles also demonstrated ~3 folds higher activation over PhMV (p = 0.0104) and activation comparable to CCMV and SeMV (p = 0.1333 andp = 0.7241, respectively). CPMV also showed ~6 fold higher activation over HBVc VLPs when compared in a separate experiment (Fig. 5E).
Thus, CPMV particles displayed significantly stronger activation of the RAW Blue™ cells as compared to any other VNP/VLPs studied here; these data are consistent with efficacy observed in the tumor mouse models. It was also observed that eCPMV lacking the encapsidated RNA is weaker immune stimulant compared to CPMV. However, eCPMV still triggered stronger activation compared to its counterpart – the PhMV VLPs; eCPMV showed activation of RAW Blue™ cells at levels comparable to both CCMV and SeMV. These results clearly indicate that the CPMV capsid itself (even without the encapsidated nucleic acid) is inherently more potent immunostimulator than PhMV and as potent as RNA-containing CCMV and SeMV. Interestingly, even though LPS showed the strongest stimulation of RAW-Blue™ cells, it does not have the same potency as CPMV in situ vaccine.48 It is likely that LPS mediated inflammatory response triggers a distinct immune pathway not culminating with an effective anti-tumor response. Such divergence has been previously recorded when comparing TMV and CPMV as in situ vaccines in B16F10 melanoma.12 Together, these in vitro results clearly indicate that CPMV is a stronger immune stimulant than other VNPs/VLPs compared in the study and hence a superior in situ vaccine for cancer immunotherapy.
Characterization of the VNP/VLP-vimentin interactions
CPMV is known to interact with cell surface-expressed vimentin on endothelial cells and34 and most importantly, immune cells.57 While vimentin is a cytosolic cytoskeleton protein; cell surface vimentin is present on a broad range of immune cells including neutrophils, T cells and activated macrophages. Whether or not the CPMV-vimentin interaction is critical for the in situ vaccine potency is not known at the present time; however as a first step toward dissecting this, we probed whether any of the other VNP/VLPs would interact with vimentin as well.
Virus overlay protein binding assay (VOPBA)34 and dot blot assays were used to assess the VNP/VLP-vimentin interactions. For VOPBA, vimentin was transferred to nitrocellulose membrane after SDS-PAGE; following denaturing/renaturing treatments and blocking, the membranes were incubated with VNP/VLPs or monoclonal anti-vimentin antibody (v9; positive control). The VNP/VLPs were detected using anti-virus specific rabbit IgGs and alkaline-phosphatase conjugated anti-rabbit IgG secondary antibodies and NBT/BCIP chromogenic substrate. v9 antibody binding was detected using HRP-conjugated anti-mouse IgG secondary and DAB chromogenic substrate. The positive control, v9 antibody detection, shows ~51 kDa band for the vimentin protein (Fig. 6A). As expected, CPMV aIso cIearIy highlights the vimentin band. In contrast SeMV resulted in a faint band, which may suggest a lower degree of SeMV-to-vimentin binding compared to CPMV. CCMV, PhMV-VLPs and Qβ VLPs did not bind to vimentin in VOPBA (Fig. 6A and Fig. S6, ESI†).
Fig. 6.
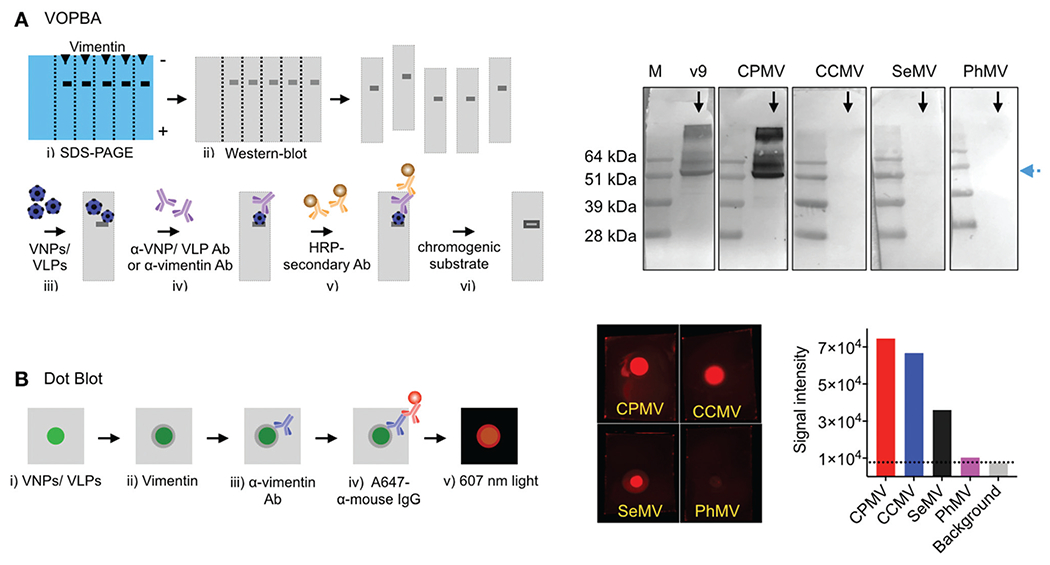
Vimentin interactions: CPMV, CCMV, SeMV and PhMV-VLPs were tested for vimentin binding using a virus overlay protein binding assay (VOPBA) (A) and dot blot (B). For VOPBA, recombinant vimentin protein was blotted to a nitrocellulose membrane following denaturing gel electrophoresis and incubated with the viral nanoparticles. VNP binding was probed using α-VNP antibodies/alkaline phosphatase conjugated secondary antibody. Blots were developed by NBT/BCIP chromogenic substrate. Monoclonal α-vimentin (v9) antibody was used as positive control and was probed using HRP conjugated secondary antibody/DAB chromogenic substrate. For dot blot, 10 μg VNPs (in 2 μL KP buffer) were blotted on the nitrocellulose membrane and incubated with recombinant vimentin protein. Vimentin binding to VNPs was probed using the α-vimentin (v9) antibody/A647-conjugated α-mouse IgG secondary antibody. The fluorescent dots were visualized under 607 nm exposure and signal intensity was measured using the ImageJ software (version 2.0).
To further analyze potential binding to vimentin, dot blots were performed by blotting CPMV, CCMV, SeMV and PhMV-VLPs on the nitrocellulose membrane and incubating the membrane with recombinant vimentin. Vimentin bound to the VNP/VLPs was detected with a mouse anti-vimentin antibody (v9), which was then probed with fluorescent anti-mouse IgG antibody. Dot blot results indicated variable degree of VNP/VLP-vimentin interactions, with signal brightness being strongest for CPMV > CCMV/SeMV » PhMV-VLPs (Fig. 6B). CPMV is strongly positive in both assays and PhMV-VLPs tests negative for vimentin interaction by VOPBA and dot blot; therefore, we conclude that CPMV binds vimentin and PhMV-VLPs does not. SeMV has been reported to test positive for vimentin binding in VOPBA;33 and CCMV has been reported to test negative in VOPBA34 – thus our data are consistent with prior reports. Dot blot assays have not been previously reported; the dot blot and VOPBA are different in a couple of ways, which may explain the different results: first, in VOPBA the VNP/VLPs are detected using antibodies – therefore differences may be explained by differences in staining. The second point is likely the more critical one, i.e. in VOPBA vimentin is denatured/renatured and immobilized on a nitrocellulose membrane; while in the dot blot, native vimentin is added to the VNP/VLPs; therefore, the latter assay may be more sensitive to binding to native vimentin structure.
Cell surface vimentin is recognized as a docking site for several mammalian viruses.58,59 Based on the established CPMV–cell surface vimentin interactions and our results highlighting the differences between the viral nanoparticles, such interactions could be another variable impacting the overall efficacies of in situ vaccines. It is possible that CPMV-vimentin interactions modulate uptake by leukocytes or tumor cells or extend the TME residence time of CPMV, thereby contributing to its enhanced interactions with immune cells as compared to CCMV, SeMV or PhMV-VLP. Such interactions with cell surface vimentin have been recently associated with the modulation of ceIIuIar internalization of Human papillomavirus (HPV). While vimentin is not the receptor for HPV, HPV interacts with cell surface vimentin and vimentin levels regulate HPV cell uptake efficiency.60 These results indicate that the cell surface vimentin may act as a “sticky anchor” for viruses, and therefore could prolong the retention time hence interaction of CPMV in the TME when compared to the other viruses.
Conclusions
In conclusion, this study demonstrates that not all plant viruses (and VLPs thereof), or bacteriophage and mammalian VLPs are effective in situ vaccines for cancer immunotherapy. CPMV outperformed other viral nanoparticle systems studied; data were consistent in vitro and in vivo models highlighting that CPMV is a stronger immune-stimulant priming potent anti-tumor immunity through remodeling of the TME. Comparative immune profiling of the TME revealed that only CPMV treatment led to potent innate immune cell activation, which translated to adaptive anti-tumor immunity. Data indicate that features beyond nanoparticle shape and surface charge govern the effectiveness of plant viral in situ vaccines; whether the key events are extracellular binding, uptake or intracellular signaling are yet to be revealed; vimentin interactions may play a role. It is clear that further research must focus on delineating the underlying mechanism of the CPMV in situ vaccine.
Supplementary Material
Acknowledgements
This work was supported in part by a grant from the NCI Alliance for Nanotechnology in Cancer U01CA218292 and NCI grant R01CA224605 (to N. F. S.). Work focused on the study of the ovarian tumor models are supported in part by the Shaughnessy Family Fund for Nano-ImmunoEngineering (nanoIE) at UCSD.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01219j
Conflicts of interest
Drs Fiering and Steinmetz are co-founders of and have a financial interest in Mosaic ImmunoEngineering Inc. The other authors declare no potential conflict of interest.
References
- 1.Rosenberg SA, Nat. Rev. Clin Oncol, 2014, 11, 630–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joyce JA and Fearon DT, Science, 2015, 348, 74–80. [DOI] [PubMed] [Google Scholar]
- 3.Le QV, Suh J, Choi JJ, Park GT, Lee JW, Shim G and Oh YK, ACS Nano, 2019, 13, 7442–7462. [DOI] [PubMed] [Google Scholar]
- 4.Bommareddy PK and Kaufman HL, J. Clin. Invest, 2018, 128, 1258–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF and Fiering S, Nat. Nanotechnol, 2016, 11, 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoopes PJ, Wagner RJ, Duval K, Kang K, Gladstone DJ, Moodie KL, Crary-Burney M, Ariaspulido H, Veliz FA, Steinmetz NF and Fiering SN, Mol. Pharm, 2018, 15, 3717–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kerstetter-Fogle A, Shukla S, Wang C, Beiss V, Harris PLR, Sloan AE and Steinmetz NF, Cancers (Basel), 2019, 11, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang C, Beiss V and Steinmetz NF, J. Virol, 2019, 93, e00129–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Albakri MM, Veliz FA, Fiering SN, Steinmetz NF and Sieg SF, Immunology, 2020, 159, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang C, Fiering SN and Steinmetz NF, Adv. Ther, 2019, 2, 1900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lebel ME, Chartrand K, Tarrab E, Savard P, Leclerc D and Lamarre A, Nano Lett, 2016, 16, 1826–1832. [DOI] [PubMed] [Google Scholar]
- 12.Murray AA, Wang C, Fiering S and Steinmetz NF, Mol. Pharm, 2018, 15, 3700–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawai T and Akira S, Nat. Immunol, 2010, 11, 373–384. [DOI] [PubMed] [Google Scholar]
- 14.Bellora F, Castriconi R, Dondero A, Pessino A, Nencioni A, Liggieri G, Moretta L, Mantovani A, Moretta A and Bottino C, Eur. J. Immunol, 2014, 44,1814–1822. [DOI] [PubMed] [Google Scholar]
- 15.Chang CY, Tai JA, Li S, Nishikawa T and Kaneda Y, Oncotarget, 2016, 7, 42195–42207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ngambenjawong C, Gustafson HH and Pun SH, Adv. Drug Delivery Rev, 2017, 114, 206–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohsen MO, Speiser DE, Knuth A and Bachmann MF, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12, e1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolb P, Wallich R and Nassal M, PLoS One, 2015, 10, e0136180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nassal M, Skamel C, Vogel M, Kratz PA, Stehle T, Wallich R and Simon MM, Int. J. Med. Microbiol, 2008, 298, 135–142. [DOI] [PubMed] [Google Scholar]
- 20.Ali A and Roossinck MJ, J. Virol. Methods, 2007,141, 84–86. [DOI] [PubMed] [Google Scholar]
- 21.Liu L and Lomonossoff G, J. Virol. Methods, 2002, 105, 343–348. [DOI] [PubMed] [Google Scholar]
- 22.Lokesh GL, Gopinath K, Satheshkumar PS and Savithri HS, Arch. Virol, 2001, 146, 209–223. [DOI] [PubMed] [Google Scholar]
- 23.Sastri M, Kekuda R, Gopinath K, Kumar CT, Jagath JR and Savithri HS, J. Mol. Biol, 1997, 272, 541–552. [DOI] [PubMed] [Google Scholar]
- 24.Sastri M, Reddy DS, Krishna SS, Murthy MR and Savithri HS, J. Mol. Biol, 1999, 289, 905–918. [DOI] [PubMed] [Google Scholar]
- 25.Hu H, Masarapu H, Gu Y, Zhang Y, Yu X and Steinmetz NF, ACS Appl. Mater. Interfaces, 2019, 11, 18213–18223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masarapu H, Patel BK, Chariou PL, Hu H, Gulati NM, Carpenter BL, Ghiladi RA, Shukla S and Steinmetz NF, Biomacromolecules, 2017, 18, 4141–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saunders K, Sainsbury F and Lomonossoff GP, Virology, 2009, 393, 329–337. [DOI] [PubMed] [Google Scholar]
- 28.Isarov SA, Lee PW and Pokorski JK, Biomacromolecules, 2016, 17, 641–648. [DOI] [PubMed] [Google Scholar]
- 29.Stray SJ, Johnson JM, Kopek BG and Zlotnick A, Nat. Biotechnol, 2006, 24, 358–362. [DOI] [PubMed] [Google Scholar]
- 30.Conejo-Garcia JR, Benencia F, Courreges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ, Holtz DO, Jenkins A, Na H, Zhang L, Wagner DS, Katsaros D, Caroll R and Coukos G, Nat. Med, 2004, 10, 950–958. [DOI] [PubMed] [Google Scholar]
- 31.Patel R, Czapar AE, Fiering S, Oleinick NL and Steinmetz NF, ACS Omega, 2018, 3, 3702–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carrillo-Tripp M, Shepherd CM, Borelli IA, Venkataraman S, Lander G, Natarajan P, Johnson JE, Brooks CL 3rd and Reddy VS, Nucleic Acids Res, 2009, 37, D436–D442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vishnu Vardhan GP, Hema M, Sushmitha C, Savithri HS, Natraj U and Murthy MRN, Arch. Virol, 2019, 164, 497–507. [DOI] [PubMed] [Google Scholar]
- 34.Koudelka KJ, Destito G, Plummer EM, Trauger SA, Siuzdak G and Manchester M, PLoS Pathog, 2009, 5, e1000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koudelka KJ, Rae CS, Gonzalez MJ and Manchester M, J. Virol, 2007, 81, 1632–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin T, Chen Z, Usha R, Stauffacher CV, Dai JB, Schmidt T and Johnson JE, Virology, 1999, 265, 20–34. [DOI] [PubMed] [Google Scholar]
- 37.Sainsbury F, Saunders K, Aljabali AA, Evans DJ and Lomonossoff GP, ChemBioChem, 2011, 12, 2435–2440. [DOI] [PubMed] [Google Scholar]
- 38.Steinmetz NF, Evans DJ and Lomonossoff GP, ChemBioChem, 2007, 8, 1131–1136. [DOI] [PubMed] [Google Scholar]
- 39.Lee LS, Brunk N, Haywood DG, Keifer D, Pierson E, Kondylis P, Wang JC, Jacobson SC, Jarrold MF and Zlotnick A, Protein Sci, 2017, 26, 2170–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Venkatakrishnan B and Zlotnick A, Annu. Rev. Virol, 2016, 3, 429–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chatterji A, Ochoa WF, Paine M, Ratna BR, Johnson JE and Lin T, Chem. Biol, 2004, 11, 855–863. [DOI] [PubMed] [Google Scholar]
- 42.Gillitzer E, Willits D, Young M and Douglas T, Chem. Commun, 2002, 2390–2391, DOI: 10.1039/B207853H. [DOI] [PubMed] [Google Scholar]
- 43.Browning L, Patel MR, Horvath EB, Tawara K and Jorcyk CL, Cancer Manage. Res, 2018, 10, 6685–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mauer J, Denson JL and Bruning JC, Trends Immunol, 2015, 36, 92–101. [DOI] [PubMed] [Google Scholar]
- 45.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM and Gajewski TF, J. Exp. Med, 2011, 208, 2005–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS and Albelda SM, Cancer Cell, 2009, 16, 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dennis KL, Blatner NR, Gounari F and Khazaie K, Curr. Opin. Oncol, 2013, 25, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF and Fiering S, Nat. Nanotechnol, 2016, 11, 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Youn JI, Collazo M, Shalova IN, Biswas SK and Gabrilovich DI, J. Leukocyte Biol, 2012, 91, 167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF and Sancho D, Nat. Rev. Immunol, 2020, 20, 7–24. [DOI] [PubMed] [Google Scholar]
- 51.Zheng Y, Lee PW, Wang C, Thomas LD, Stewart PL, Steinmetz NF and Pokorski JK, Nano Lett, 2019, 19, 2099–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Albakri MM, Veliz FA, Fiering SN, Steinmetz NF and Sieg SF, Immunology, 2020, 159, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eriksson F, Tsagozis P, Lundberg K, Parsa R, Mangsbo SM, Persson MA, Harris RA and Pisa P, J. Immunol, 2009, 182, 3105–3111. [DOI] [PubMed] [Google Scholar]
- 54.Shepardson KM, Schwarz B, Larson K, Morton RV, Avera J, McCoy K, Caffrey A, Harmsen A, Douglas T and Rynda-Apple A, mBio, 2017, 8, e01356–17. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Lewis RE, Liao G, Young K, Douglas C and Kontoyiannis DP, Antimicrob. Agents Chemother, 2014, 58, 1738–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Radovic-Moreno AF, Chernyak N, Mader CC, Nallagatla S, Kang RS, Hao L, Walker DA, Halo TL, Merkel TJ, Rische CH, Anantatmula S, Burkhart M, Mirkin CA and Gryaznov SM, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 3892–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gonzalez MJ, Plummer EM, Rae CS and Manchester M, PLoS One, 2009, 4, e7981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Du N, Cong H, Tian H, Zhang H, Zhang W, Song L and Tien P, J. Virol, 2014, 88, 5816–5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liang JJ, Yu CY, Liao CL and Lin YL, Cell. Microbiol, 2011, 13, 1358–1370. [DOI] [PubMed] [Google Scholar]
- 60.Schafer G, Graham LM, Lang DM, Blumenthal MJ, Bergant Marusic M and Katz AA, J. Virol, 2017, 91, e00307–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.