

Abstract
The immunoproteasome (i-20S) has emerged as a therapeutic target for autoimmune and inflammatory disorders and hematological malignancies. Inhibition of the chymotryptic β5i subunit of i-20S inhibits T cell activation, B cell proliferation and dendritic cell differentiation in vitro and suppresses immune responses in animal models of autoimmune disorders and allograft rejection. However, cytotoxicity to immune cells has accompanied the use of covalently reactive β5i inhibitors, whose activity against the constitutive proteasome (c-20S) is cumulative with time of exposure. Herein we report a structure-activity relationship study of a class of noncovalent proteasome inhibitors with picomolar potencies and thousands-fold selectivity for i-20S over c-20S. Furthermore, these inhibitors are specific for β5i over the other 5 active subunits of i-20S and c-20S, providing useful tools to study functions of β5i in immune responses. The potency of these compounds in inhibiting human T cell activation suggests that they may have therapeutic potential.
Keywords: Immunoproteasome, β5i inhibitor, noncovalent, isoform selectivity, T cell activation, immune response
Graphical Abstract
INTRODUCTION
Since the approval of Bortezomib for treatment of once largely untreatable multiple myeloma, components of the ubiquitin proteasome system (UPS) of protein degradation have become attractive drug targets for various malignancies and autoimmune disorders. The UPS degrades cellular proteins via a highly regulated process by which protein substrates are polyubiquitinated through a cascade of reactions catalyzed by a ubiquitin activating enzyme (E1), a ubiquitin conjugating enzyme (E2) and a ubiquitin ligase (E3). Polyubiquitinated proteins are recognized, unfolded and translocated by the proteasome regulatory particle (19S) into the 20S core particle for degradation by three proteolytic β subunits in the 20S core: β1, β2 and β5.1, 2 Through this ordered degradation process, the UPS regulates numerous cellular activities ranging from removal of unfolded or damaged proteins to cell cycle progression, transcription factor activation and immune surveillance by providing antigenic peptides for presentation by MHC class I molecules.2, 3,4 Pan-proteasome inhibitors limit the supply of peptides for MHC class I molecules and thus block antigen presentation.5 Bortezomib is highly toxic to plasma cells6 and has been used to prevent antibody mediated graft rejection of transplanted kidneys, although the effect is modest at the limited doses that are considered tolerable in patients who do not have an incurable malignancy.7, 8 Species selective inhibitors of proteasomes of pathogenic microbes have been developed for treatment of infections by Mycobacterium tuberculosis,9–12 Plasmodia,13–16 Leishmania and Trypanosoma,17, 18 suggesting further potential therapeutic utility of proteasome inhibitors.
To harness the benefit of proteasome inhibitors in the therapy of autoimmune disorders without the toxicity of pan-proteasome inhibition, it is necessary to develop proteasome inhibitors that selectively target one or more of the proteasome subunits that predominate in the immune system rather than those expressed in non-immune cells. There are two major isoforms of proteasome in humans that differ in each of the three proteolytically active subunits: constitutive proteasome (c-20S) and immunoproteasome (i-20S). Recent proteomic studies have found that i-20S predominates in the cells of the human innate and adaptive immune systems.19 The i-20S is also upregulated in other cells at sites of inflammation and upon exposure to interferon-γ.
Selective inhibitors of i-20S β5i over c-20S β5c have been reported, such as the peptide epoxyketone ONX0914, which produced similar immunoregulatory effects in mice as BTZ but with much reduced toxicity.20–26 A recent study reported an improvement on ONX0914 with up to 600-fold selectivity.27 Reports on ONX0914 were followed by reports of other selective i-20S inhibitors (Figure 1); due to space limitations, only reviews and the most recent primary publications are cited.27–33 Most reported inhibitors are peptide epoxyketones or sulfonyl fluorides that act covalently and irreversibly with the active site. Irreversible covalent binding can result in unpredictable liver toxicity, even if off-target binding occurs at a low level,34 and irreversible inhibitors often show diminished selectivity over time.35 Compounds reported in patents also include amino boronates from Merck-Serono and substituted thiazoles and peptide α-keto amides from Roche (Figure 1). These three classes of i-20S inhibitors are either covalent but reversible or noncovalent. They represent alternative directions for developing i-20S selective inhibitors.
Figure 1.
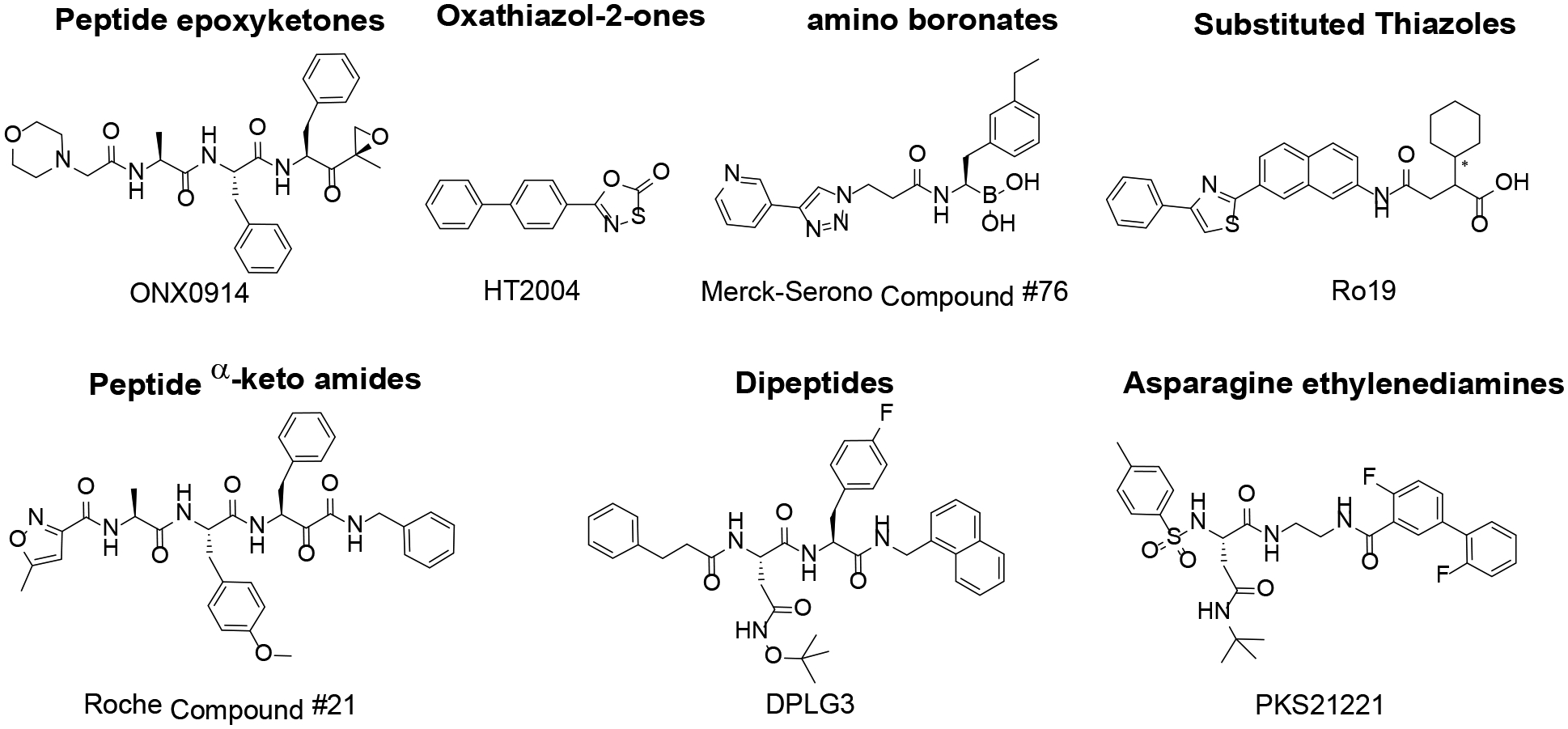
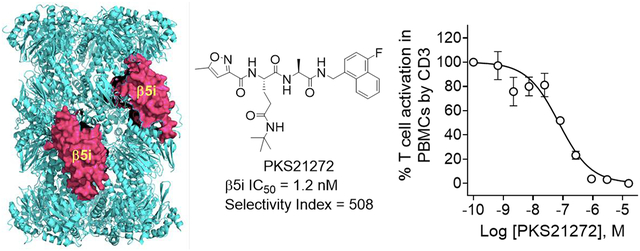
Structures of some reported immunoproteasome inhibitors. ONX0914 was previously named PR957.24 Merck-Serono β5i selective inhibitor 76, Ro19 and Roche Compound 21 were reported in a patent review.17 HT2004, DPLG3 and PKS21221 were recently reported as β5i selective inhibitors.35, 36, 38
Recently, our labs reported three new classes of immunoproteasome inhibitors (IPIs): 1,3,4-oxathiazol-2-ones;36 β-amino acid-based N,C-capped diepeptidomimetics;37 and N,C-capped dipeptides.35 We also discovered asparagine-ethylenediamine IPIs (AsnEDAs) (Figure 1).38 The latter three classes are noncovalent inhibitors and highly β5i- selective. We have demonstrated that compounds from each class inhibit β5i in cells and restrict immunological responses of plasmacytoid dendritic cells, B cells and T cells in vitro.35, 38 We reported that DPLG3 prolonged the survival of MHC-mismatched mouse cardiac allografts in a heterotopic heart transplant model.35 However, DPLG3 lacks drug-like properties. Herein we report our further structure-activity relationship studies of the N,C-capped dipeptides.
RESULTS AND DISCUSSION
Design rationale
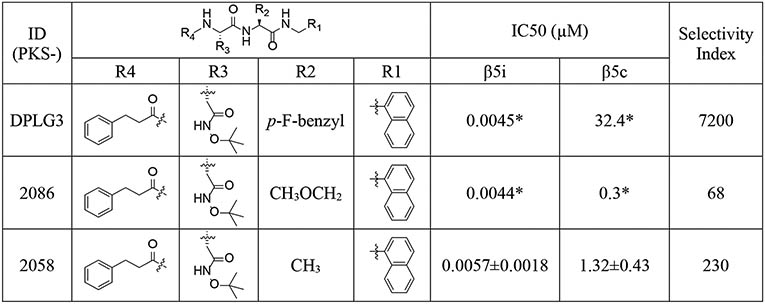
DPLG3 was developed based on DPLG2, which was designed for species selective inhibition of the Mycobacterium tuberculosis proteasome (Mtb20S) over i-20S and c-20S.39 We have shown that i-20S and Mtb20S share similar substrate preferences, especially at the S1 site (Figure 2), which prefers bulky hydrophobic moieties as visualized in the structures of Mtb20S and i-20S.9, 10, 12, 38, 39 We postulated that by anchoring a bulky P1 (N-cap) in the N,C-capped dipeptide, we could explore the P2 (R2), P3 (R3) and N-cap positions to improve potency and in the meantime maintain selectivity and improve pharmacokinetic properties. DPLG3 is a highly selective immunoproteasome β5i inhibitor with single-digit nanomolar potency, but it suffers from poor physicochemical properties and cell permeability.35 We reported that replacing P2 p-F-phenylalanine of DPLG3 with O-methyl-serine (PKS2086) maintained potency, but the isoform selectivity (β5c /β5i) over c-20S was reduced to 68-fold. Replacing P2 O-methyl-serine in PKS2086 with P2 Ala (PKS2058) improved isoform selectivity (β5c/β5i = 230) and maintained comparable inhibition activity. These preliminary structure-activity analyses suggested that even partially exposing S2 to the solvent could be used to tune the physicochemical properties and selectivity profile of N,C-capped dipeptides, inviting further SAR studies (Table 1).
Figure 2.
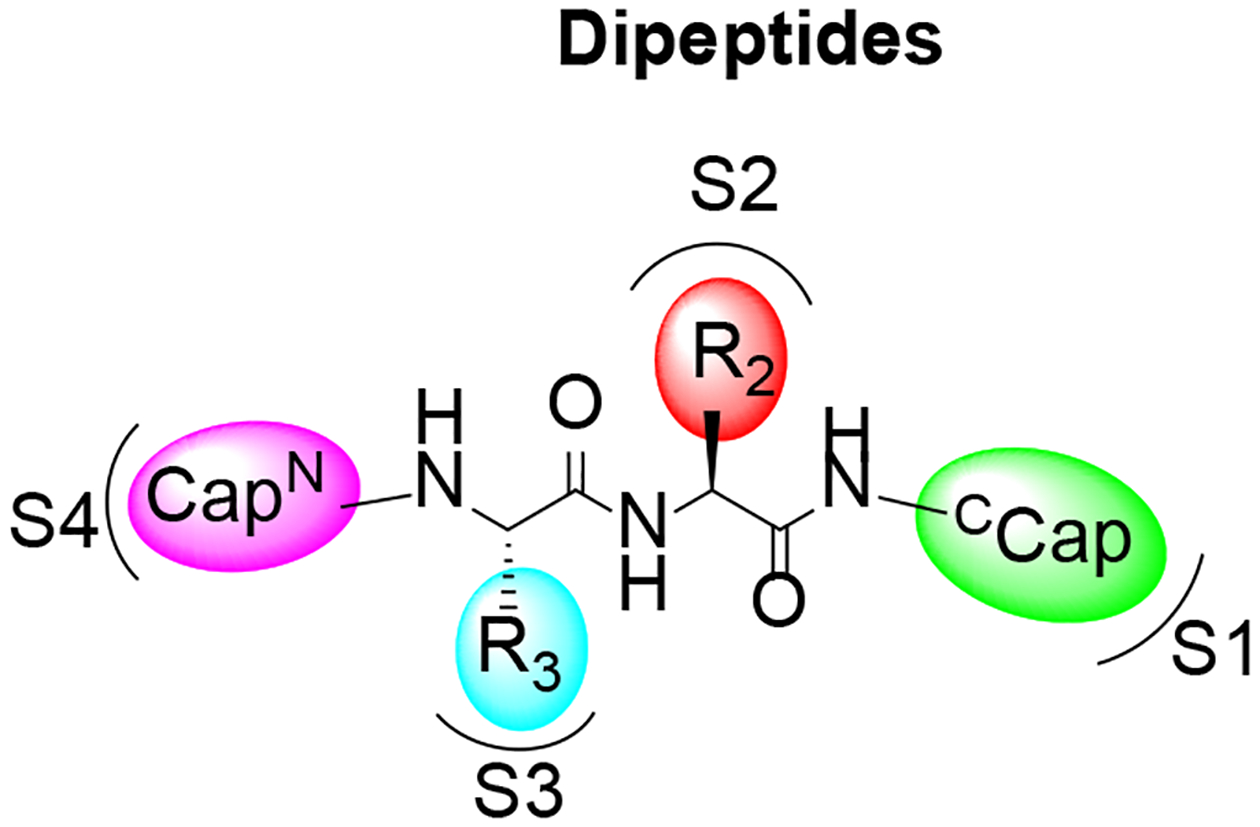
Illustration of N-cap, R2, R3 and C-cap and their binding sites in the β5 active subunits.
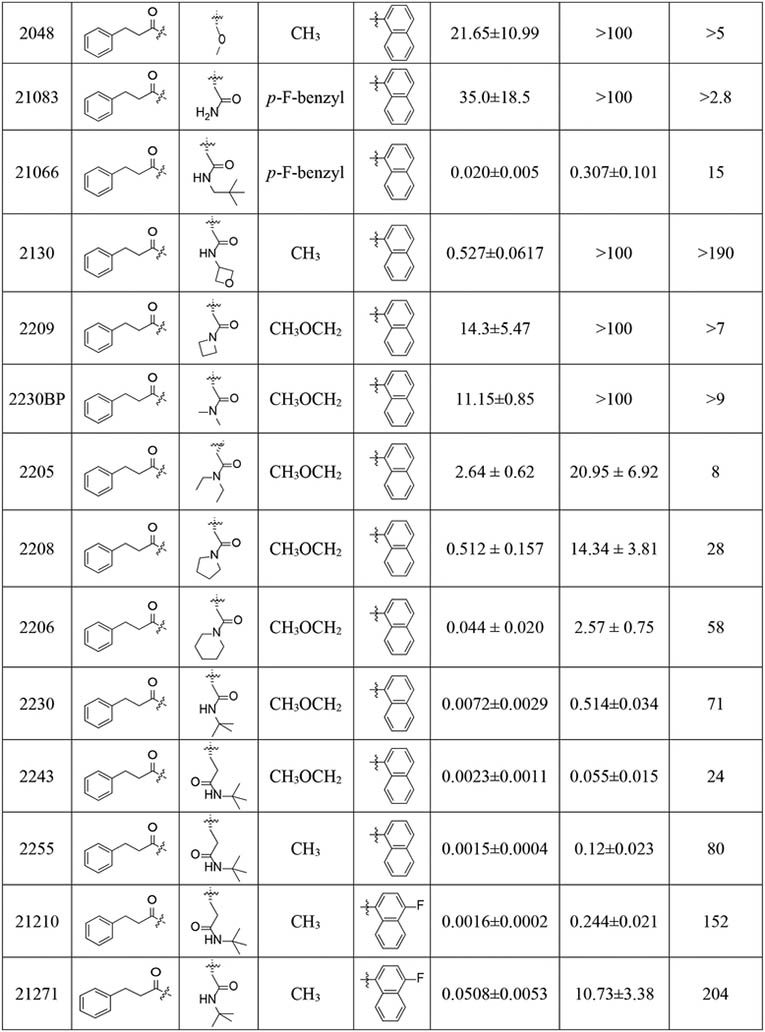
Table 1.
Inhibition IC50 values of compounds with P4 amides against human c-20S β5c and i-20S β5i.
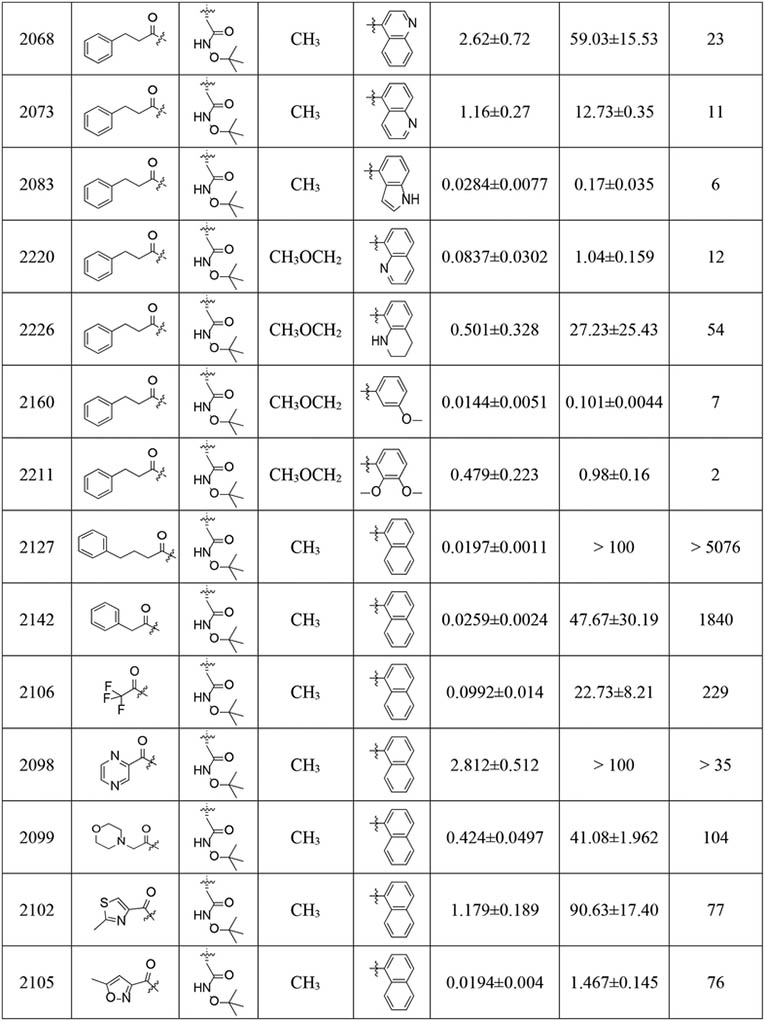
|
C-cap optimization
The C-cap binds to the S1 pocket, which is immediately adjacent to the active site. Structural studies of i-20S and c-20S have revealed that this S1 site dictates the selectivity of an inhibitor between β5i and β5c. We have shown that the P1 naphthyl ring is the selectivity driver, but the naphthyl ring is hydrophobic. To improve solubility and microsomal stability, we replaced the naphthalenemethyl ring in PKS2058 with the following substituents: 4-quinolinmethyl (PKS2068), 5-quinolinmethyl (PKS2073) and 4-indolemethyl (PKS2083). Quinolinyl analogues eliminated inhibitory activity against both β5i and β5c, while the indolyl analog decreased potency against β5i and increased activity against β5c. Replacing 1-naphthalenemethyl on the C-cap of PKS2086 with 8-quinolinmethyl (PKS2220) and 8-tetrahydroquinolinmethyl (PKS2226) led to 19-fold and 114-fold reductions in β5i activities, respectively, further suggesting that a basic P1 moiety is detrimental to its interaction with the hydrophobic S1 pocket of β5i. Using substituted benzyl groups as C-caps, as in PKS2160 and PKS2211, abolished selectivity. Therefore, we kept the naphthalenemethyl moiety as the C-cap for further SAR studies.
N-cap optimization–
Subsequent exploration of the N-cap with small, focused libraries revealed that a hydrophobic N-cap helps maintain selectivity. Extending the phenylpropionate group on the N-cap of PKS2058 yielded PKS2127. Phenylbutanoate-bearing PKS2127 was >5076-fold selective for β5i over β5c and yet with only 3-fold lower potency than PKS2058. Likewise, the abridged analog PKS2142 showed an 1840-fold preference for β5i. Replacement of phenylpropionate in PKS2058 with the smaller trifluoroacetate in PKS2106 resulted in diminished activity with decent selectivity (β5c /β5i = 229). Conversely, polar N-caps were not tolerated, as exemplified by the pyrazine PKS2098, the morpholine PKS2099, the thiazole PKS2102 and the isoxazole PKS2105, all of which suffered a significant reduction in β5i inhibitory potency and compromised selectivity compared to PKS2058.
R3 optimization–
We have detailed the role of the defined S3 pocket in the species-selective binding of N,C-capped dipeptide inhibitors to the Mtb20S28, but we have found no reports exploring interactions with the S3 pocket for improving β5i potency and selectivity for β5i over β5c. Accordingly, we changed R3 from branched chain amino acids to asparagine and glutamine. Replacing the R3 Asn-OtBu with O-methyl-Ser (PKS2048) or Asn (PKS21083) abolished the inhibitory activities against both β5i and β5c, whereas the Asn-neopentyl analogue (PKS21066) was tolerated, suggesting that Asn coupled with a bulky moiety on the side chain would be a potency driver for N,C-capped dipeptide proteasome inhibitors. Replacement of R3 Asn-OtBu with Asn-oxetane (PKS2130) reduced potency against β5i over 100-fold compared to PKS2058, further testifying to the critical role of the bulky OtBu group in inhibition of β5i activity. Replacing the R3 Asn-OtBu (PKS2086) with Asn amide with a fully substituted N atom (PKS2209, 2230BP, 2205 and 2208) resulted in significant loss of affinity for both β5i and β5c. However, the bulky Asn piperidine in PKS2206 did not impose a great loss in potency and retained selectivity. Removal of the O atom from Asn-OtBu of PKS2086 provided PKS2230. While their potencies and selectivity profiles are similar, the physicochemical properties of ‘amide’ PKS2230 would be presumably better than those of the ‘N-alkoxyamide’ PKS2086. Improvement in β5i potency was observed for the R3 Gln-tBu analogue PKS2243 (IC50 = 0.0023 μM), suggesting that Gln-OtBu fits the S3 pocket better than Asn-OtBu, despite their comparable lengths. The trend was further attested by the observation that replacing R3 Asn-OtBu of PKS2058 (IC50 = 0.0057 μM) with Gln-OtBu (PKS2255, IC50 = 0.0015 μM) boosted β5i potency by 3.8-fold. However, potency against β5c was enhanced slightly more, impairing β5i selectivity by 2-to 3-fold (PKS2255 vs. PKS2058, PKS2243 vs. PKS2086). We thus chose Asn-tBu (selectivity) or Gln-tBu (potency) as R3 for further optimization.
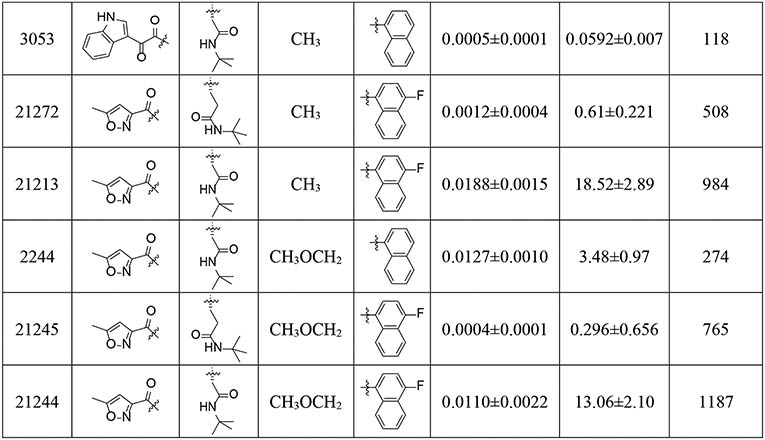
Introduction of a 4-F substituent on the C-cap naphthalenemethyl of PKS2255, which was expected to block metabolism, gave PKS21210, an equipotent β5i inhibitor with about 2-fold improvement in selectivity relative to PKS2255. The isoform selectivity (β5c/β5i = 204) of PKS21271, a hybrid compound incorporating both the 4-F naphthalenemethyl group and the R3 Asn-tBu, was slightly improved relative to that of PKS2255 (β5c /β5i = 152), whereas the potency was compromised by over 30-fold. Because 3-indoleglyoxylate was reported to be an optimal N-cap for dipeptide inhibitors of the human 20S proteasome with a P3 threonine residue,40 we incorporated it into the scaffold of current β5i inhibitor. Combining 3-indoleglyoxylate N-cap with R3 Asn-tBu yielded a β5i inhibitor with pM IC50 and over 100-fold selectivity (PKS3053). Additionally, we revisited the 5-methylisoxazole-3-carboxylate N-cap with an aim to further improve the selectivity profile. Replacing the phenylpropionate group of PKS21210 and PKS21217 with 5-methylisoxazole-3-carboxylate improved selectivity indices from 152- and 204-fold to 508- (PKS21272) and 984-fold (PKS21213), and unexpectedly, also improved β5i inhibition by 1.3- and 2.7- fold. Still better, 765- and 1187- fold isoform selectivities were recorded for two R2 O-methyl-serine analogs, PKS21245 and PKS21244. A 4-F substituent on the C-cap naphthalenemethyl contributed over 4-fold improvement to the noteworthy isoform selectivity of PKS21244 compared to that of PKS2244.
Next, we assessed the influence of a sulfonyl N-cap on asparagine and glutamine derivatives (Table 2). We replaced phenylpropionate with tosyl on the N-cap of PKS2255, yielding PKS2256. IC50s were determined to be 400 pM against β5i and 424 nM against β5c, affording 3 orders of magnitude of selectivity. As in compounds with the selectivity-enhancing 4-F substituents described above, a further 2-fold improvement in selectivity was observed for PKS21209 (β5c /β5i = 2329). However, PKS21209 exhibited a 6-fold reduction in potency against β5i compared to PKS2256. Again, replacing the Gln-tBu of PKS21209 with Asn-tBu (PKS21206) lost 6.8-fold in potency of inhibition of β5i, but improved selectivity to 3274-fold over inhibition of β5c. This pattern of impact on β5i activity was reinforced in the R2 O-methyl-serine analogs PKS21265 and PKS21266, where the Gln-tBu PKS21265 (IC50 = 0.8 nM) exhibited 70-fold greater activity over Asn-tBu PKS21266 (IC50 = 56 nM) (Figure 3).
Table 2.
Inhibition IC50s of compounds with P4 tosyl against human c-20S β5c and i-20S β5i.
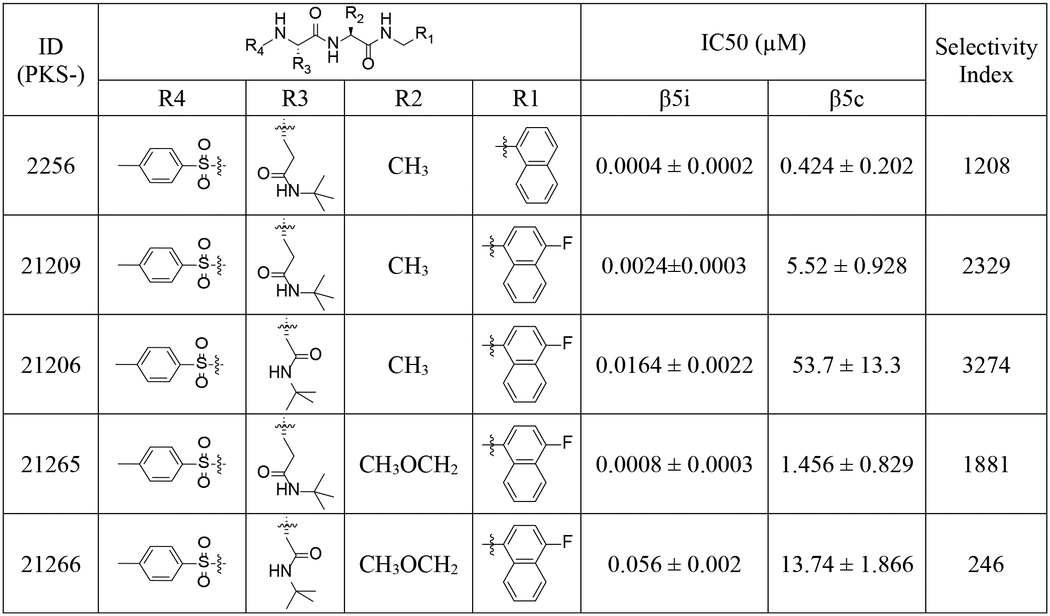
|
Data were taken from reference 35.
Figure 3.
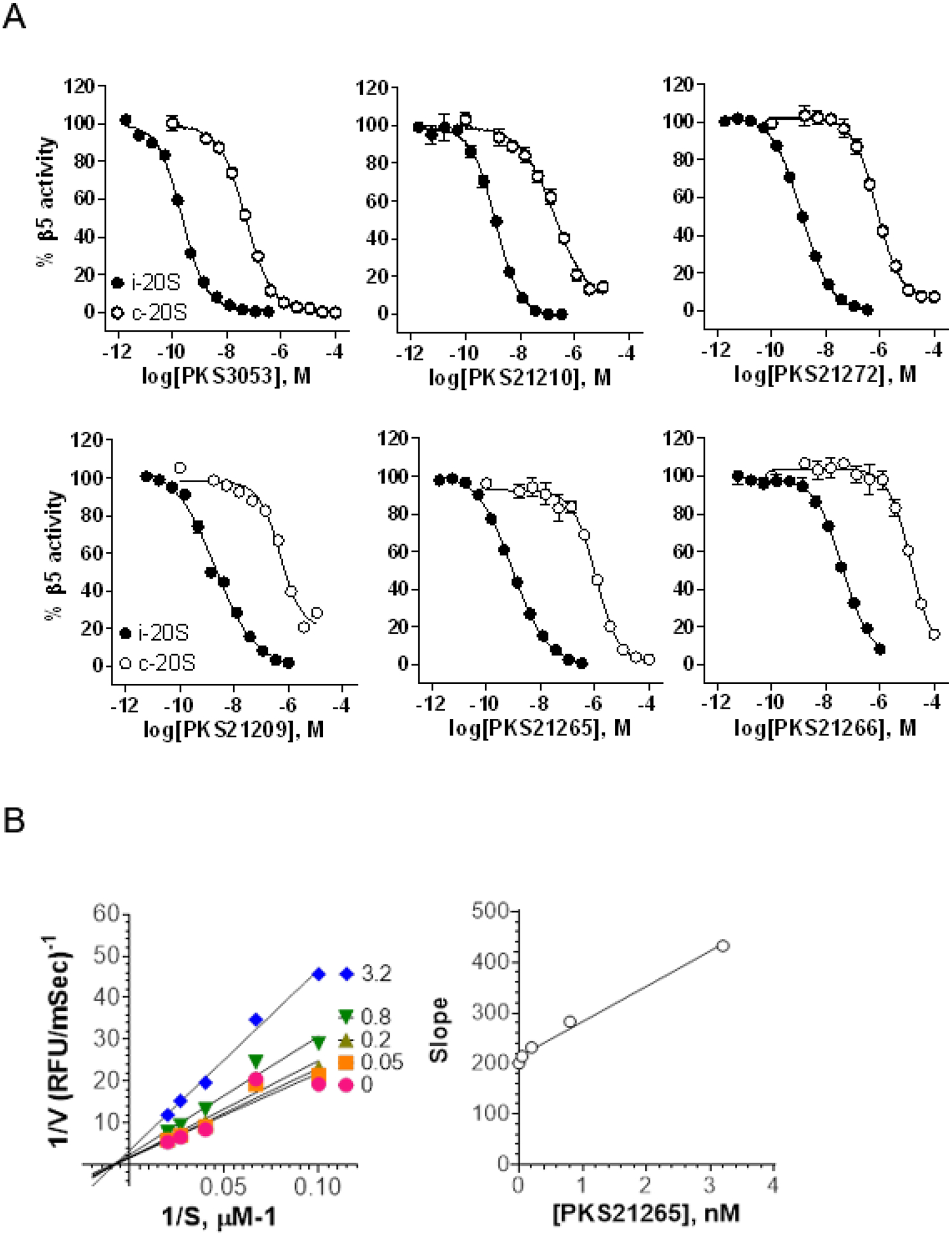
Inhibition of β5i and β5c by select dipeptides. A) Dose-dependent inhibition of β5i and β5c by compounds indicted in the x-axial. B) Ki of PKS21265 was also determined with varying concentrations of PKS21265 and substrate suc-LLVY-AMC. Lineweaver-Burk plot was shown on the left. The plot of slopes against inhibitor concentration yield a Ki 3.1 nM.
We further determined β1 or β2 activities of 6 select compounds in either i-20S or c-20S, and all exhibited <50% inhibition against β1i, β1c, β2i, or β2c at a concentration of 33.3 μM, suggesting they are bona fide β5i specific inhibitors (Table S1).
Blackburn and others have shown that N,C-capped dipeptides are generally specific inhibitors of proteasome over a panel of proteases.40 We next tested these compounds against α-chymotrypsin, cathepsin L and trypsin with the compounds at 100, 33.3 and 11.1 μM. As shown in Table S2, all the compounds were inactive against all three proteases, except for weak inhibition of α-chymotrypsin and cathepsin L by PKS3053. Thus, in agreement with the previous study,40 these compounds were highly specific for proteasome inhibition.
Inhibition kinetics of i-20S β5i
The kinetics of proteasome inhibition can be complicated by the duplication of each active site in different rings of the proteasome..41 We have observed that an active site binding inhibitor can behave like a non-competitive inhibitor.38 We selected PKS21265 for elucidation of the kinetics of inhibition of β5i. The Lineweaver-Burk plot of 1/V versus 1/S in the presence of different concentrations of PKS21265 yielded a series of straight lines that had the same x-intercept, suggesting a typical non-competitive inhibition kinetics (Figure 3B).
Intracellular proteasome inhibition
To investigate if these N,C-capped dipeptides are cell permeable, we selected three compounds with N-cap amides and 3 compounds with N-cap sulfonamides and tested their intracellular proteasome inhibition in Karpas-1106P cells (which express i-20S but not c-20S) and HeLa cells (which express c-20S but not i-20S). These compounds dose-dependently inhibited i-20S β5i in Karpas-1106P and c-20S β5c in Hela cells (Figure 4A), and in situ potencies were correlated well with their activities determined in vitro with purified enzymes (Table 3).
Figure 4.
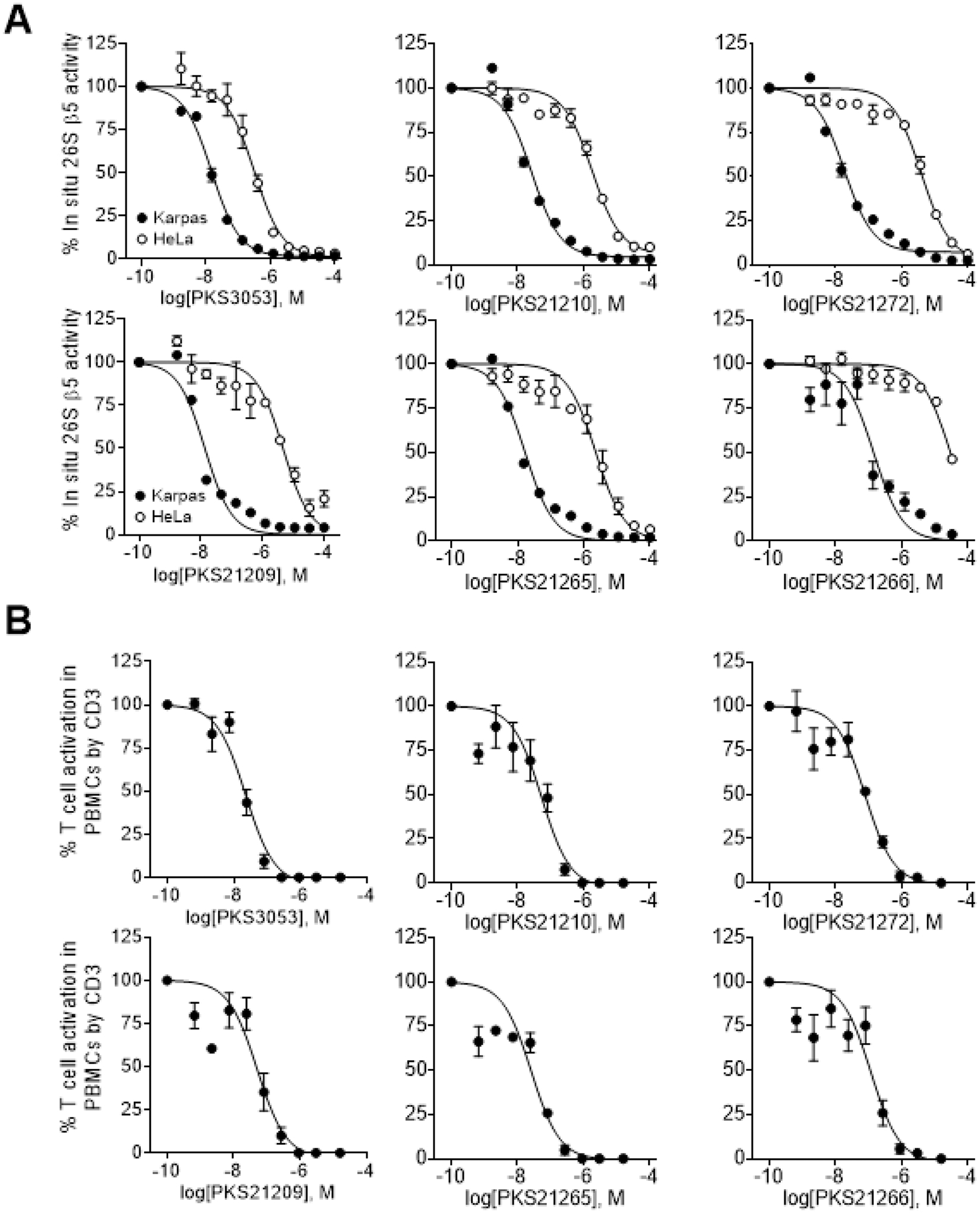
In situ inhibition of i-20S β5i in Karpas-1106P and c-20S β5c in Hela cells and dose-dependent inhibition of T cell activation in peripheral blood mononuclear cells (PBMCs) stimulated by anti-CD3 antibody. All experiments were repeated three times. Representative results are shown and data summarized in Table 3.
Table 3.
Inhibition of i-20S β5i in Karpas-1106P and c-20S β5c in Hela cells by select compounds and their inhibition of T cell proliferation in PBMCs upon stimulation by anti-CD3 antibody.
| Compound | IC50 (μM) | EC50 (μM) | |
|---|---|---|---|
| Karpas-1106P | HELA | ||
| PKS3053 | 0.015 | 0.293 | 0.021 |
| PKS 21210 | 0.039 | 2.031 | 0.044 |
| PKS 21272 | 0.053 | 4.306 | 0.073 |
| PKS 21209 | 0.024 | 3.030 | 0.039 |
| PKS 21265 | 0.028 | 1.861 | 0.018 |
| PKS 21266 | 0.197 | 22.56 | 0.073 |
Inhibition of T cell activation and proliferation
We have shown that β5i-selective β-amino acid based dipeptides inhibit the activation and proliferation of human T cells in peripheral blood mononuclear cells.37 We tested the same 6 compounds as above for their inhibitory activity against primary human T cells stimulated by anti-CD3 antibody (Figure 4B). PKS3053 was two times more active than PKS21210 and > 3 times more active than PKS21272, and the activity tracked with their IC50s against β5i. With its N-cap sulfonamide, PS21265 was the most potent compound, with 2-fold greater potency compared to PKS21209 and 5-fold compared to PKS21266 (Table 3).
CHEMISTRY
The synthesis of dipeptide analogs with different N-caps is shown in Scheme 1. PKS2058 was synthesized by condensation of PKS2105935 with PKS2026.35 The reaction gave poor yield (6%) due to intramolecular cyclization of PKS21059 giving N-tert-butoxysuccinimide byproduct. Other N-cap analogs were synthesized from PKS21054.35 Debenzylation of PKS21054 and coupling with Ala-OBn followed by debenzylation and coupling with 1-methylnaphthylamine gave PKS2095. Finally, BOC-deprotection and coupling with appropriate carboxylic acids gave the N-cap analogs. C-cap analogs were synthesized from PKS21059 (Scheme 2). Coupling of PKS21059 with Ala-OBn and subsequent debenzylation gave PKS2067. Different amines were coupled with PKS2067 to give C-cap analogs. PKS2048 was synthesized from PKS203236 by BOC-deprotection and coupling with 3-phenylpropanoic acid.
Scheme 1.
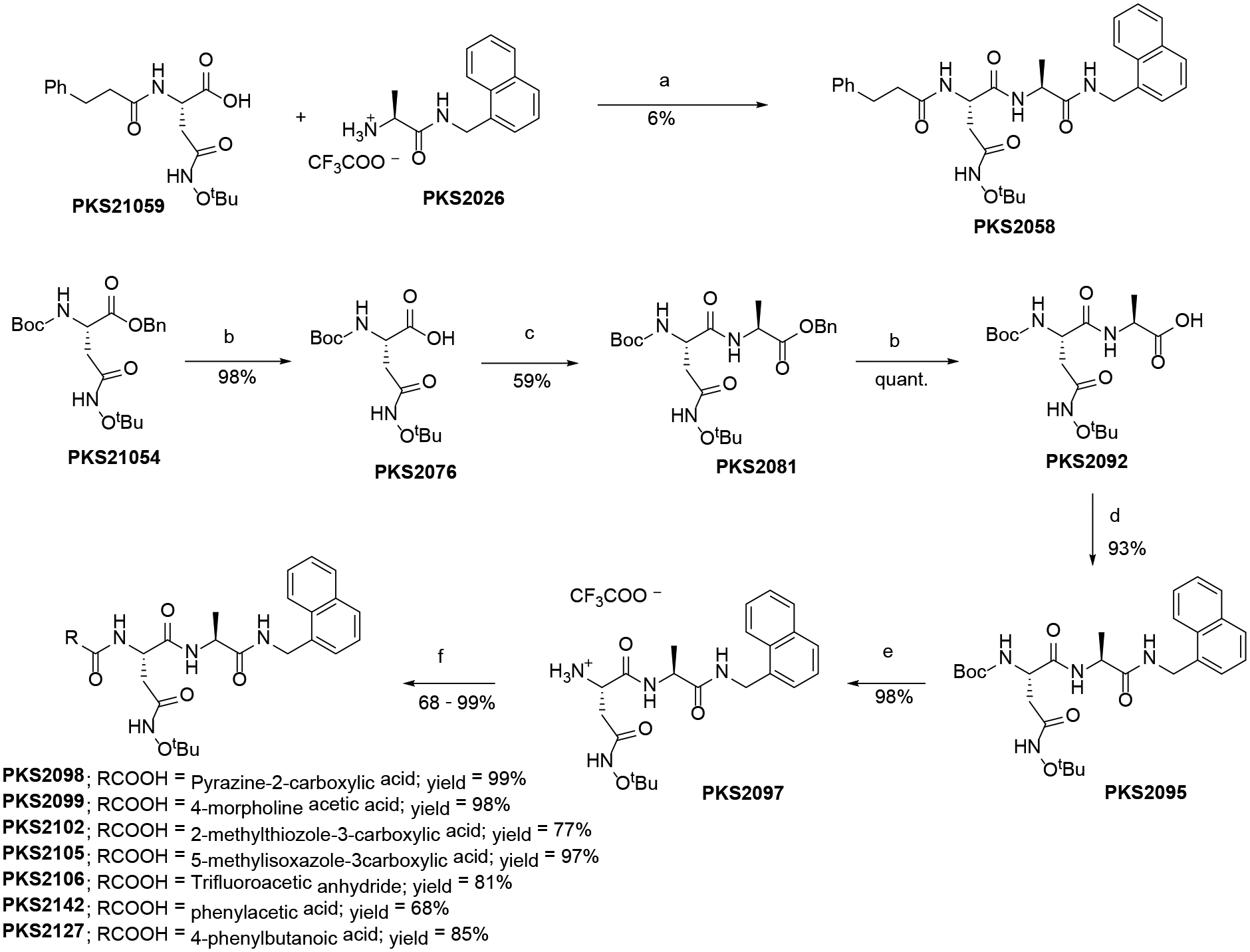
Synthesis of N-Cap analogs.
Reagents and conditions: a) EDCI, HOBt, Hünig’s base; b) 10% Pd on Carbon, Methanol, H2 balloon, 98%; c) O-benzylalanine hydrochloride, HATU, HOAt, Hünig’s base, DMF, 59%; d) 1-naphthylmethylamine, HATU, HOAt, Hünig’s base, DMF; e) 20% TFA in DCM; f) RCOOH, HATU, HOAt, Hünig’s base, DMF
Scheme 2.
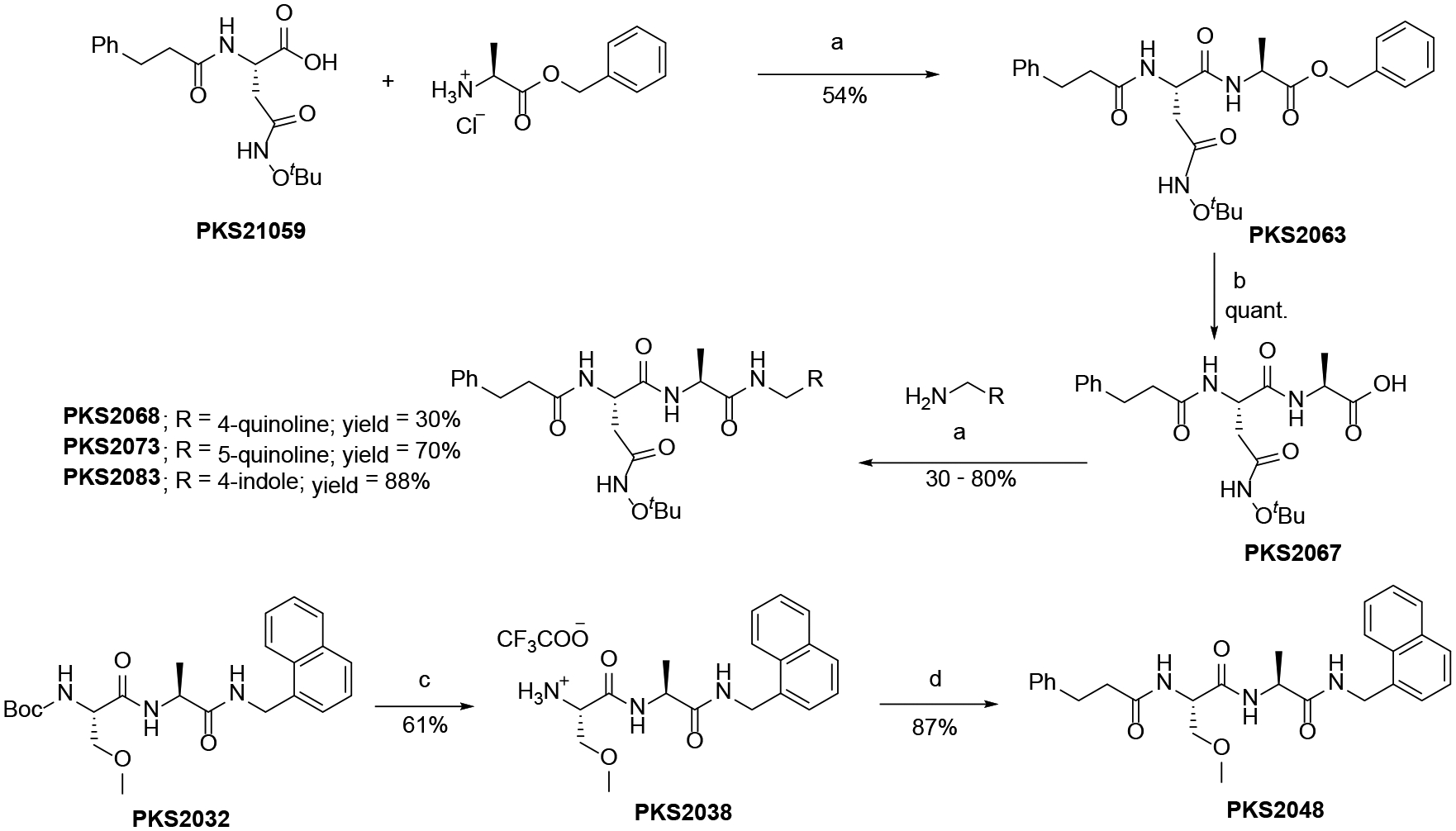
Synthesis of C-cap analogs.
Reagents and conditions: a) HATU, HOAt, Hünig’s base, DMF; b) 10% Pd on Carbon, H2 balloon, EtOH; c) TFA, DCM; d) 3-Phenylpropanoic acid, EDC, HOBt, Hünig’s base, DMF
The synthesis of compounds where P2 is methylserine is shown in Scheme 3. The commercially available O-methylserine was coupled with 3-methoxybenzylamine and subsequent BOC-deprotection gave compound PKS2158. The condensation of PKS21059 with PKS2158 to provide compound PKS2160. Similarly, compound PKS2220 was synthesized starting from O-methylserine and 8-quinolinemethylamine. Compound PKS2211 was synthesized by condensation of O-methylserine with 2,3-dimethoxybenzylamine. The condensation product was subjected to BOC-deprotection and coupled with BOC-Asp(OBn)-OH to give PKS2191. Again BOC-deprotection and coupling with 3-phenylpropanoic acid yielded PKS2200. The debenzylation of PKS2200 and condensation with O-tertbutylhydroxylamine gave the compound PKS2211. Similarly, following the same set of reactions compound PKS2226 was synthesized starting from O-methylserine and 8-quinolinemethylamine. Condensation of PKS212410 with 3-aminooxetane yielded compound PKS2130. Similarly, compound PKS2230 and PKS2209 were synthesized by condensation of PKS220110 with tert-butylamine and azetidine respectively. PKS2244 was synthesized from PKS213110 by converting to tert-butylamide followed by BOC-deprotection and coupling with 5-methylisoxazole-3-carboxylic acid.
Scheme 3.
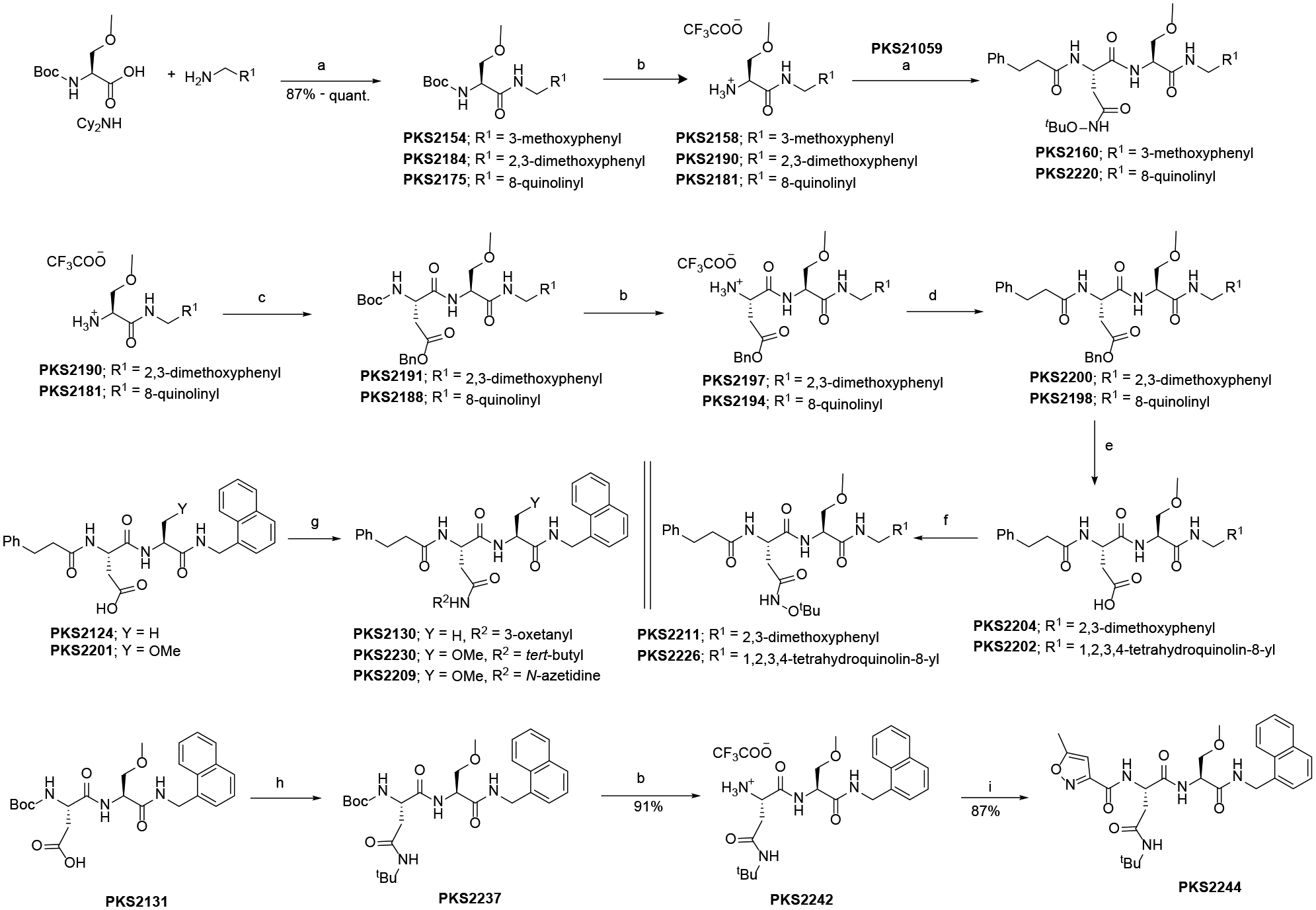
Synthesis of various P2 (methylserine) analogs.
Reagents and conditions: a) HATU, HOAt, Hünig’s base, DMF; b) TFA, DCM; c) BOC-Asp(OBn)-OH, HATU, HOAt, Hünig’s base, DMF; d) 3-phenylpropanoic acid, HATU, HOAt, Hünig’s base, DMF; e) 10% Pd on carbon, methanol; f) O-tert-butylhydroxylamine, HATU, HOAt, Hünig’s base, DMF; g) tert-butylamine or 3-aminooxitane, HATU, HOAt, Hünig’s base, DMF; h) tert-butylamine, HATU, HOAt, Hünig’s base, DMF; i) 5-methylisoxazole-3-carboxylic acid, HATU, HOAt, Hünig’s base, DMF.
Condensation of BOC-Glu-OBn with tert-butylamine followed by BOC-deprotection gave PKS2233. It was then coupled with 3-phenylpropanoic acid and subsequent debenzylation gave PKS2239. PKS21202 was synthesized by condensation of BOC-Ala-OSu with 4-fluoromethyl-1-naphthylmethylamine and subsequent BOC-deprotection. Condensation of PKS2239 with PKS2082,35 PKS2026 and PKS21202 gave compounds PKS2243, PKS2255 and PKS21210 respectively (Scheme 4). PKS305237 was coupled with PKS2026 and PKS2101335 was coupled with PKS21202 to provide compounds PKS3035 and PKS21271 respectively. O-methylserine was coupled with 4-fluoromethyl-1-naphthylmethylamine and subsequently BOC was deprotected to give compound PKS21238. PKS2117837 was coupled with PKS21202 and PKS21238 to give compounds PKS21213 and PKS21244 respectively. 5-methylisoxazole-3-carboxylic acid was subjected to condensation with PKS2233 and subsequent debenzylation furnished the compound PKS21233. PKS21233 was coupled with PKS21202 and PKS21238 to give compounds PKS21272 and PKS21245 respectively. Finally, compounds with N-sulfonyl cap were synthesized by HATU mediated amide formation of corresponding acids and amines (Scheme 6)
Scheme 4.
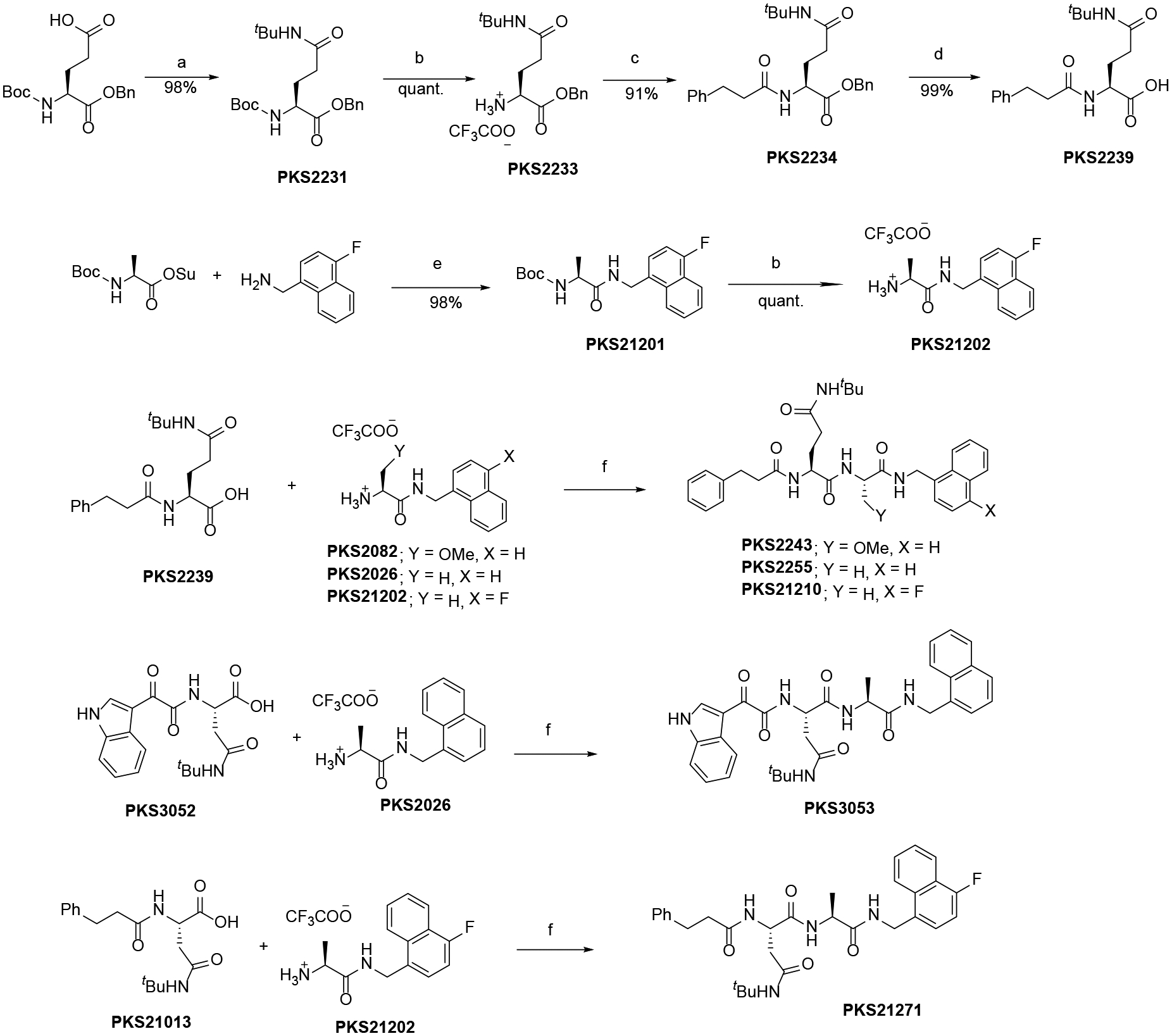
Reagents and conditions: a) tert-butylamine, EDC, HOBt, Hünig’s base; b) TFA, DCM; c) 3-phenylpropanoic acid, HATU, HOAt, Hünig’s base, DMF; d) 10% Pd on carbon, methanol, hydrogen balloon; e) triethylamine, DCM; f) HATU, HOAt, Hünig’s base, DMF
Scheme 6.
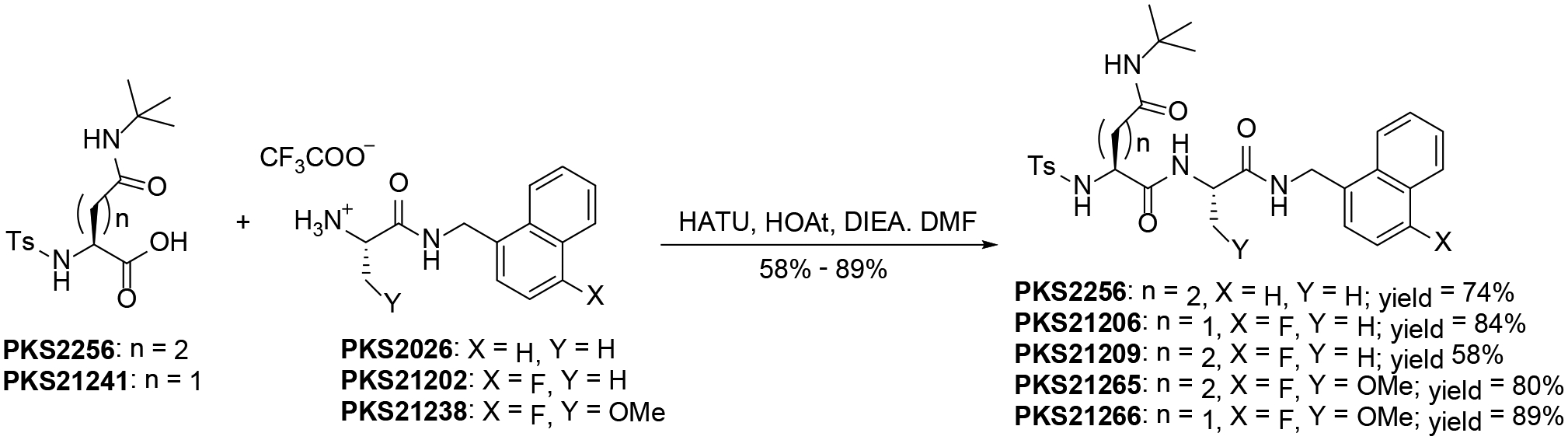
Synthesis of compounds with N-sulfonyl cap
CONCLUSIONS
We found that several factors significantly contribute to potency and selectivity of N,C-capped dipeptides: 1) for inhibition, neither β5i nor β5c prefers benzopyridinyl at P1, but an indole moiety is acceptable, suggesting the hydrophobic nature of the S1 pocket and H-bond acceptor presented; 2) a bulky P1 (C-cap) is the major determinant of selective inhibition of β5i over β5c; 2) P3 glutamine with t-butyl amide side chain generally improved potency by 3- to 50-fold for β5i and by 30- to 40-fold for β5c compared to P3 asparagine, without affecting the selectivity; 4) a hydrophobic N-cap is generally preferred; 5) these inhibitors act only on the chymotryptic β5 subunits.
Recent studies of β5i selective inhibitors have presented several observations: 1) inhibition of both β5i and β5c without inhibition of β2i/c and β1i/c can kill cancer cells;42 however, whether inhibition of only β5i or β5c can induce cell death is still understudied; 2) selective inhibition of β5i by peptide boronates does not lead to cell death of plasma cells and pDCs;43 3) inhibition of both β5 and β2 was synergistic for death of breast cancer cell line cells and inhibition of both β5 and β1 was additive.44, 45 In contrast, non-cytotoxic inhibition of β5i with compounds that showed no inhibitory activity against β2c, β2i, β1c or β1i sufficed to inhibit human T cell proliferation and might afford partial inhibition of the function of many types of immune cells with the cumulative effect of therapeutic efficacy in chronic autoimmune states.
EXPERIMENTAL SECTION
Suc-LLVY-AMC, Ac-ANW-AMC, Z-VLR-AMC, Ac-LLE-AMC, Ac-PAL-AMC, human c-20S (RBC) and i-20S (PBMC) were from Boston Biochem (Cambridge, MA). Enzymatic assays were recorded on a Molecular Devices SpectraMax M5 plate readers. The human blood samples were sourced ethically and their research use was in accord with the terms of the informed consent. Purity of all final compounds were > 95%.
Synthetic Methods
Chemicals and Spectroscopy:
All commercially available materials were purchased from Bachem, Aldrich, P3 BioSystems, or other vendors and were used as received unless stated otherwise. All non-aqueous reactions were performed under argon in oven-dried glasswares. Reactions were monitored using Waters Acquity Ultra Performance Liquid Chromatography (UPLC). All HPLC purifications were done by Varian PrepStar HPLC system or Waters Autopure (mass directed purification system) using Prep C18 5μm OBD (19 X 150 mm) column. 1H- and 13C- NMR spectra were acquired on a Bruker DRX-500 spectrometer. Chemical shifts δ are expressed in parts per million, with the solvent resonance as an internal standard (chloroform-d, 1H: 7.26; 13C: 77.16 ppm; DMSO-d6, 1H: 2.50 ppm; 13C: 39.52 ppm). Hexafluorobenzene was used as internal standard for 19F NMR. NMR data are reported as following: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, b = broad), coupling constant, and integration. We followed our reported procedure for HATU or EDC mediated amide bond formation, boc-deprotection and O-debenzylation.37,38 All final compounds are > 95% pure.
PKS2058:
Synthesized by following the general procedure for EDC mediated amide bond formation. Yield = 6%. 1H NMR (500 MHz, DMSO-d6) δ 10.40 (s, 1H), 8.46 (t, J = 5.8 Hz, 1H), 8.20 – 8.16 (m, 1H), 8.15 (d, J = 7.2Hz, 1H), 8.10 – 8.02 (m, 1H), 7.96 – 7.90 (m, 1H), 7.86 – 7.80 (m, 1H), 7.56 – 7.50 (m, 2H), 7.47 – 7.41 (m, 2H), 7.28 – 7.24 (m, 2H), 7.20 – 7.13 (m, 3H), 4.70 – 4.67 (m, 2H), 4.62 – 4.56 (m, 1H), 4.29 – 4.22 (m, 1H), 2.82 – 2.75 (m, 2H), 2.55 – 2.48 (m, 1H), 2.44 – 2.38 (m, 2H), 2.36 – 2.30 (m, 1H), 1.24 (d, J = 7.2 Hz, 3H), 1.22 & 1.10 (s, rotamers, 9H). LCMS calc. for C31H39N4O5 [M+H]+ : 547.3. Found: 547.4.
PKS2076:
Synthesized by following the general procedure for O-debenzylation of PKS21054 (592 mg, 1.5 mmol). After completion of reaction (5 h), the mixture was filtered through celite and filtrate was evaporated to give product (450 mg, 98%). 1H NMR (500 MHz, Chloroform-d) δ 9.28 & 8.98 (s, rotamers, 1H), 6.92 & 5.76 (b, rotamers, 1H), 4.59 – 4.47 (m, 1H), 3.22 – 2.69 (m, 2H), 1.44 (s, 9H), 1.27 & 1.24 (s, rotamers, 9H). LCMS calc. for C13H23N2O6 [M−H]−: 303.2. Found: 303.5.
PKS2081:
Synthesized by following the general procedure for HATU mediated coupling of PKS2076 (304 mg, 1 mmol) and O-benzylalanine hydrochloride (237 mg, 1.1 mmol). Isolated crude product was purified by recrystallization with ethanol-water to give pure product (276 mg, 59%). 1H NMR (500 MHz, Chloroform-d) δ 8.20 (s, 1H), 7.51 – 7.34 (m, 6H), 6.13 & 5.89 (b, rotamers, 1H), 5.20 (d, J = 12.4 Hz, 1H), 5.15 (d, J = 12.4 Hz, 1H), 4.60 – 4.45 (m, 2H), 2.79 – 2.66 (m, 1H), 2.49 – 2.45 (m, 1H), 1.46 (s, 9H), 1.41 (d, J = 7.2 Hz, 3H), 1.26 (s, 9H). LCMS calc. for C23H36N3O7 [M+H]+ : 466.3. Found: 466.5.
PKS2092:
Synthesized by following the general procedure for O-debenzylation of PKS2081 (150 mg, 0.32 mmol). After completion of reaction, mixture was filtered through celite and evaporated to give product (120 mg, quant.). 1H NMR (500 MHz, DMSO-d6) δ 12.96 (b, 1H), 10.37 (s, 1H), 7.94 (d, J = 6.7 Hz, 1H), 7.01 (d, J = 6.6 Hz, 1H), 4.28 – 4.23 (m, 1H), 4.10 – 4.04 (m, 1H), 2.39 (dd, J = 14.4, 4.4 Hz, 1H), 2.27 (dd, J = 14.4, 9.9 Hz, 1H), 1.37 (s, 9H), 1.23 (d, J = 7.2 Hz, 3H), 1.14 (s, 9H).
PKS2095:
Synthesized following the general procedure for HATU mediated coupling of PKS2092 (120 mg, 0.32 mmol) and 1-naphthylmethylamine (56 μl, 0.38 mmol). After completion of reaction (6 h), the mixture was precipitated with water. The precipitate was filtered and dried to give product (153 mg, 93%). 1H NMR (500 MHz, DMSO-d6) δ 10.26 (s, 1H), 8.47 (t, J = 5.7 Hz, 1H), 8.06 – 8.04 (m, 1H), 8.01 (d, J = 7.4 Hz, 1H), 7.96 – 7.94 (m, 1H), 7.86 – 7.84 (m, 1H), 7.57 – 7.52 (m, 2H), 7.48 – 7.43 (m, 2H), 6.94 (d, J = 8.1 Hz, 1H), 4.74 (d, J = 5.8 Hz, 2H), 4.32 – 4.26 (m, 2H), 2.46 (dd, J = 14.6, 5.5 Hz, 1H), 2.29 (dd, J = 14.6, 8.5 Hz, 1H), 1.37 (s, 9H), 1.24 (d, J = 7.0 Hz, 3H), 1.13 (s, 9H).
PKS2097:
Synthesized by following the general procedure for boc-deprotection of PKS2095 (118 mg, 0.23 mmol). Isolated crude was triturated with diethyl ether to give product (120 mg, 98%). 1H NMR (500 MHz, DMSO-d6) δ 10.68 (s, 1H), 8.71 (d, J = 7.3 Hz, 1H), 8.58 (t, J = 5.7 Hz, 1H), 8.16 (s, 3H), 8.07 – 8.05 (m, 1H), 7.97 – 7.95 (m, 1H), 7.87 – 7.85 (m, 1H), 7.58 – 7.53 (m, 2H), 7.49 – 7.44 (m, 2H), 4.79 – 4.71 (m, 2H), 4.39 – 4.34 (m, 1H), 4.13 (m, 1H), 2.70 (dd, J = 16.4, 4.9 Hz, 1H), 2.56 (dd, J = 16.4, 8.1 Hz, 1H), 1.28 (d, J = 7.0 Hz, 3H), 1.15 (s, 9H). LCMS calc. for C22H31N4O4 [M+H]+ : 415.2. Found: 415.5.
PKS2098:
Synthesized by following the general procedure for HATU mediated coupling of pyrazine-2-carboxylic acid (2.5 mg, 0.02 mmol) and PKS2097 (10.6 mg, 0.02 mmol). The crude was purified by HPLC to give 10.3 mg (99%) of product. 1H NMR (500 MHz, DMSO-d6) δ10.45 (s, 1H), 9.19 (d, J = 1.4 Hz, 1H), 8.94 (d, J = 8.1 Hz, 1H), 8.92 (d, J = 2.5 Hz, 1H), 8.78 (t, J = 2.0 Hz, 1H), 8.50 (t, J = 5.8 Hz, 1H), 8.37 (d, J = 7.3 Hz, 1H), 8.07 – 8.05 (m, 1H), 7.95 – 7.93 (m, 1H), 7.85 – 7.83 (m, 1H), 7.56 – 7.52 (m, 2H), 7.48 – 7.44 (m, 2H), 4.85 – 4.81 (m, 1H), 4.75 (d, J = 5.8 Hz, 2H), 4.36 – 4.30 (m, 1H), 2.68 (dd, J = 14.7, 7.2 Hz, 1H), 2.63 (dd, J = 14.7, 5.2 Hz, 1H), 1.27 (d, J = 7.1 Hz, 3H), 1.06 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 172.3, 170.2, 168.0, 162.8, 148.3, 144.5, 143.9, 134.9, 133.7, 131.2, 128.9, 127.9, 126.6, 126.2, 125.8, 125.6, 123.8, 81.1, 50.5, 49.2, 40.6, 35.4, 26.6, 18.4. LCMS calc. for C27H33N6O5 [M+H]+ : 521.3. Found: 521.5.
PKS2099:
Synthesized by following the general procedure for HATU mediated coupling of morpholine-4-acetic acid (3.0 mg, 0.02 mmol) and PKS2097 (10.6 mg, 0.02 mmol). The crude was purified by HPLC to give 10.6 mg (98%) of product. 1H NMR (500 MHz, DMSO-d6) δ 10.38 (s, 1H), 8.50 (t, J = 5.8 Hz, 1H), 8.20 (d, J = 7.3 Hz, 1H), 8.08 – 8.06 (m, 1H), 7.98 – 8.02 (m, 1H), 7.96 – 7.94 (m, 1H), 7.85 (dd, J = 7.0, 2.6 Hz, 1H), 7.57 – 7.52 (m, 2H), 7.48 – 7.44 (m, 2H), 4.73 (d, J = 5.7 Hz, 2H), 4.62 – 4.57 (m, 1H), 4.31 – 4.27 (m, 1H), 3.61 (t, J = 4.6 Hz, 4H), 2.94 (s, 2H), 2.50 – 2.43 (m, 6H), 1.25 (d, J = 7.2 Hz, 3H), 1.11 (s, 9H). LCMS calc. for C28H40N5O6 [M+H]+: 542.3. Found: 542.5.
PKS2102:
Synthesized by following the general procedure for HATU mediated coupling of 2-methylthiazole-3-carboxylic acid (2.9 mg, 0.02 mmol) and PKS2097 (10.6 mg, 0.02 mmol). The crude was purified by HPLC to give 8.4 mg (77%) of product. 1H NMR (500 MHz, DMSO-d6) δ 10.44 (s, 1H), 8.52 (t, J = 5.8 Hz, 1H), 8.32 (d, J = 7.3 Hz, 1H), 8.29 (d, J = 8.1 Hz, 1H), 8.12 (s, 1H), 8.08 – 8.06 (m, 1H), 7.96 – 7.94 (m, 1H), 7.84 (dd, J = 7.0, 2.5 Hz, 1H), 7.56 – 7.52 (m, 2H), 7.48 – 7.44 (m, 2H), 4.78 – 4.74 (m, 3H), 4.35 – 4.29 (m, 1H), 2.71 (s, 3H), 2.63 (dd, J = 14.6, 7.3 Hz, 1H), 2.58 (dd, J = 14.6, 5.3 Hz, 1H), 1.26 (d, J = 7.1 Hz, 3H), 1.07 (s, 9H). 13C NMR (125 MHz, DMSO-d6) δ 172.4, 170.4, 168.0, 166.7, 160.4, 149.2, 134.9, 133.7, 131.3, 128.9, 127.9, 126.6, 126.2, 125.9, 125.6, 124.7, 123.8, 81.1, 50.3, 49.1, 40.6, 35.6, 26.6, 19.2, 18.5. LCMS calc. for C27H34N5O5S [M+H]+: 540.2. Found: 540.5.
PKS2105:
Synthesized by following the general procedure for HATU mediated coupling of 5-methylisoxazole-3-carboxylic acid (2.5 mg, 0.02 mmol) and PKS2097 (10.6 mg, 0.02 mmol). The crude was purified by HPLC to give 10.2 mg (97%) of product. 1H NMR (500 MHz, DMSO-d6) δ 10.37 (s, 1H), 8.62 (d, J = 8.0 Hz, 1H), 8.48 (t, J = 5.8 Hz, 1H), 8.27 (d, J = 7.4 Hz, 1H), 8.06 – 8.04 (m, 1H), 7.96 – 7.94 (m, 1H), 7.85 (dd, J = 7.4, 1.9 Hz, 1H), 7.56 – 7.52 (m, 2H), 7.48 – 7.43 (m, 2H), 6.54 (s, 1H), 4.79 – 4.76 (m, 1H), 4.74 (d, J = 5.8 Hz, 2H), 4.34 – 4.28 (m, 1H), 2.60 (dd, J = 14.7, 5.5 Hz, 1H), 2.55 (dd, J = 14.7, 8.0 Hz, 1H), 2.47 (s, 3H), 1.25 (d, J = 7.1 Hz, 3H), 1.09 (s, 9H). 13C NMR (125 MHz, DMSO-d6) δ 171.9, 171.4, 169.7, 167.5, 158.4, 158.4, 134.4, 133.2, 130.8, 128.5, 127.5, 126.2, 125.8, 125.4, 125.2, 123.4, 101.3, 80.6, 50.0, 48.6, 40.1, 34.6, 26.2, 18.1, 11.8. LCMS calc. for C27H34N5O6 [M+H]+: 524.3. Found: 524.5.
PKS2106:
Substrate (PKS2097) (10.6 mg, 0.02 mmol) was dissolved in 1 mL tetrahydrofuran and the solution was cooled to 0 °C. The mixture was basified with N-methylmorpholine (4.4 mL, 0.04 mmol). Trifluoroacetic anhydride (2.8 mL, 0.02 mmol) was added and mixture was stirred for one hour at 0 °C. Solvent was evaporated and crude was purified by HPLC to give pure product (8.3 mg, 81%). 1H NMR (500 MHz, DMSO-d6) δ 10.38 (s, 1H), 9.67 (b, 1H), 8.48 (t, J = 5.7 Hz, 1H), 8.41 (d, J = 7.3 Hz, 1H), 8.06 – 8.04 (m, 1H), 7.96 – 7.94 (m, 1H), 7.85 (d, J = 7.6 Hz, 1H), 7.57 – 7.53 (m, 2H), 7.48 – 7.43 (m, 2H), 4.74 – 4.70 (m, 3H), 4.33 – 4.27 (m, 1H), 2.63 (dd, J = 15.2, 5.0 Hz, 1H), 2.54 – 2.49 (m, 1H), 1.26 (d, J = 7.1 Hz, 3H), 1.12 (s, 9H). 13C NMR (125 MHz, DMSO-d6) δ 172.4, 169.5, 167.4, 134.9, 133.7, 131.2, 128.9, 127.9, 126.6, 126.2, 125.9, 125.6, 123.8, 81.1, 50.7, 49.2, 40.6, 34.6, 26.6, 18.5. 19F NMR (471 MHz, DMSO, C6F6 external reference) δ −71.81. LCMS calc. for C24H30F3N4O5 [M+H]+: 511.2. Found: 511.4.
PKS2127:
Synthesized by following the general procedure for HATU mediated coupling of 4-phenylbutanoic acid (5.4 mg, 0.033 mmol) and PKS2097 (17.3 mg, 0.033 mmol). The crude was purified by HPLC to give 15.7 mg (85%) of product. 1H NMR (500 MHz, DMSO-d6) δ 10.34 (s, 1H), 8.46 (t, J = 5.9 Hz, 1H), 8.11 – 8.06 (m, 3H), 7.95 – 7.93 (m, 1H), 7.85 – 7.83 (m, 1H), 7.56 – 7.52 (m, 2H), 7.45 (d, J = 5.0 Hz, 2H), 7.28 – 7.25 (m, 2H), 7.19 – 7.16 (m, 3H), 4.75 (dd, J = 15.3, 5.9 Hz, 1H), 4.70 (dd, J = 15.3, 5.7 Hz, 1H), 4.60 – 4.56 (m, 1H), 4.30 – 4.24 (m, 1H), 2.56 – 2.50 (m, 3H), 2.34 (dd, J = 14.8, 7.8 Hz, 1H), 2.13 (t, J = 7.4 Hz, 2H), 1.80 – 1.74 (m, 2H), 1.24 (d, J = 7.0 Hz, 3H), 1.10 (s, 9H). 13C NMR (125 MHz, DMSO-d6) δ 172.4, 172.4, 171.0, 168.1, 142.3, 134.9, 133.7, 131.3, 128.9, 128.8, 128.7, 127.9, 126.6, 126.2, 126.2, 125.8, 125.7, 123.8, 81.00, 50.2, 49.0, 35.2, 35.1, 35.0, 27.5, 26.7, 18.5. LCMS calc. for C32H41N4O5 [M+H]+: 561.3. Found: 561.5.
PKS2142:
Synthesized by following the general procedure for HATU mediated coupling of phenylacetic acid (4.9 mg, 0.036 mmol) and PKS2097 (17.3 mg, 0.033 mmol). The crude was purified by HPLC to give 12.1 mg (68%) of product. 1H NMR (500 MHz, DMSO-d6) δ 10.38 (s, 1H), 8.47 (t, J = 5.9 Hz, 1H), 8.36 (d, J = 7.9 Hz, 1H), 8.18 (d, J = 7.4 Hz, 1H), 8.09 – 8.07 (m, 1H), 7.95 – 7.93 (m, 1H), 7.85 – 7.83 (m, 1H), 7.56 – 7.52 (m, 2H), 7.45 (d, J = 5.0 Hz, 2H), 7.29 – 7.19 (m, 5H), 4.76 (dd, J = 15.3, 6.0 Hz, 1H), 4.69 (dd, J = 15.3, 5.8 Hz, 1H), 4.60 – 4.56 (m, 1H), 4.29 – 4.23 (m, 1H), 3.46 (s, 2H), 2.56 – 2.51 (m, 1H), 2.37 (dd, J = 14.9, 7.3 Hz, 1H), 1.22 (d, J = 7.1 Hz, 3H), 1.10 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 172.4, 170.9, 170.5, 168.1, 136.6, 134.9, 133.7, 131.3, 129.5, 128.9, 128.6, 127.9, 126.8, 126.6, 126.2, 125.8, 125.7, 123.9, 81.0, 50.2, 49.1, 42.4, 40.6, 35.3, 26.7, 18.4. LCMS calc. for C30H37N4O5 [M+H]+: 533.3. Found: 533.5.
PKS2063:
Synthesized by following the general procedure for HATU mediated coupling of PKS21059 (33.6 mg, 0.1 mmol) and H-Ala-OBn.HCl (21.5 mg, 0.1 mmol). After completion of reaction, the mixture was precipitated by the addition of cold water. The precipitate was filtered and dried to give the product (27 mg, 54%). 1H NMR (500 MHz, DMSO-d6) δ 10.20 (s, 1H), 8.31 (d, J = 7.0 Hz, 1H), 8.07 (d, J = 8.2 Hz, 1H), 7.40 – 7.32 (m, 5H), 7.28 – 7.25 (m, 2H), 7.20 – 7.15 (m, 3H), 5.11 (s, 2H), 4.66 (td, J = 9.0, 4.9 Hz, 1H), 4.33 – 4.27 (m, 1H), 2.80 – 2.76 (m, 2H), 2.41 – 2.37 (m, 3H), 2.26 (dd, J = 14.8, 9.5 Hz, 1H), 1.29 (d, J = 7.2 Hz, 3H), 1.14 (s, 9H).
PKS2067:
Synthesized by following the general procedure for O-debenzylation of PKS2063 (25.0 mg, 0.05 mmol). After completion of reaction (5 h), the mixture was filtered through celite. The filtrate was evaporated and dried under vacuum to give the product (20 mg, quant.). 1H NMR (500 MHz, DMSO-d6) δ 12.35 (b, 1H), 9.75 (b, 1H), 7.80 (b, 1H), 7.20 – 7.17 (m, 2H), 7.12 – 7.09 (m, 1H), 7.01 (d, J = 7.4 Hz, 2H), 4.32 – 4.28 (m, 1H), 3.67 – 3.61 (m, 1H), 2.67 – 2.58 (m, 2H), 2.43 – 2.37 (m, 1H), 2.34 – 2.28 (m, 1H), 2.23 – 2.21 (m, 2H), 1.15 (d, J = 6.8 Hz, 3H), 1.12 (s, 9H).
PKS2068:
Synthesized by following the general procedure for HATU mediated coupling of PKS2067 (5.0 mg, 0.0123 mmol) and quinolin-4-ylmethylamine dihydrochloride (2.8 mg, 0.0123 mmol). The crude was purified by HPLC to give 2.0 mg (30%) of product. 1H NMR (500 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.84 (d, J = 4.5 Hz, 1H), 8.60 (t, J = 6.0 Hz, 1H), 8.29 (d, J = 7.1 Hz, 1H), 8.19 – 8.15 (m, 2H), 8.05 (dd, J = 8.5, 1.3 Hz, 1H), 7.80 – 7.76 (m, 1H), 7.66 – 7.63 (m, 1H), 7.40 (d, J = 4.4 Hz, 1H), 7.27 – 7.24 (m, 2H), 7.20 – 7.15 (m, 3H), 4.80 – 4.78 (m, 2H), 4.64 – 4.59 (m, 1H), 4.30 – 4.27 (m, 1H), 2.80 – 2.77 (m, 2H), 2.56 – 2.51 (m, 1H), 2.44 – 2.40 (m, 2H), 2.35 (dd, J = 14.9, 7.1 Hz, 1H), 1.29 (d, J = 7.1 Hz, 3H), 1.06 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 172.4, 171.4, 170.7, 167.7, 150.1, 147.3, 144.6, 141.2, 129.4, 129.3, 128.3, 128.1, 126.6, 125.9, 125.8, 123.5, 118.9, 80.5, 64.9, 49.6, 48.8, 36.7, 34.8, 30.9, 26.2, 17.7. HRMS calc. for C30H38N5O5 [M+H]+: 548.2873. Found: 548.2857.
PKS2073:
The title compound was prepared by following the general procedure for HATU mediated coupling of PKS2067 (5.0 mg, 0.0123 mmol) and quinolin-5-ylmethylamine (2.0 mg, 0.0123 mmol). The crude was purified by HPLC to give 4.7 mg (70%) of product. 1H NMR (500 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.92 – 8.91 (m, 1H), 8.55 (d, J = 8.6 Hz, 1H), 8.51 (t, J = 6.0 Hz, 1H), 8.18 – 8.15 (m, 2H), 7.94 (d, J = 8.4 Hz, 1H), 7.70 (dd, J = 8.5, 7.0 Hz, 1H), 7.57 – 7.54 (m, 2H), 7.27 – 7.24 (m, 2H), 7.19 – 7.15 (m, 3H), 4.79 (dd, J = 15.4, 6.1 Hz, 1H), 4.70 (dd, J = 15.4, 5.7 Hz, 1H), 4.61 – 4.56 (m, 1H), 4.27 – 4.21 (m, 1H), 2.80 – 2.76 (m, 2H), 2.53 – 2.48 (m, 1H), 2.43 – 2.39 (m, 2H), 2.33 (dd, J = 14.8, 7.2 Hz, 1H), 1.23 (d, J = 7.1 Hz, 3H), 1.10 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 172.4, 171.9, 171.0, 168.1, 150.6, 148.4, 141.7, 136.0, 132.5, 129.4, 128.9, 128.7, 128.6, 126.4, 126.3, 126.2, 121.7, 81.0, 70.2, 50.0, 49.1, 37.2, 35.3, 31.4, 26.7, 18.3. HRMS calc. for C30H38N5O5 [M+H]+: 548.2867. Found: 548.2879.
PKS2083:
Synthesized by following the general procedure for HATU mediated coupling of PKS2067 (5.0 mg, 0.0123 mmol) and 4-(aminomethyl)indole (1.8 mg, 0.0123 mmol). The crude was purified by HPLC to give 5.8 mg (88%) of product. 1H NMR (500 MHz, DMSO-d6) δ 11.10 (s, 1H), 10.35 (s, 1H), 8.36 (t, J = 5.9 Hz, 1H), 8.16 (d, J = 8.0 Hz, 1H), 8.05 (d, J = 7.4 Hz, 1H), 7.32 – 7.25 (m, 4H), 7.20 – 7.16 (m, 3H), 7.01 (t, J = 7.6 Hz, 1H), 6.89 (d, J = 7.1 Hz, 1H), 6.52 – 6.51 (m, 1H), 4.61 – 4.48 (m, 3H), 4.28 – 4.24 (m, 1H), 2.81 – 2.77 (m, 2H), 2.53 – 2.49 (m, 1H), 2.46 – 2.38 (m, 2H), 2.33 (dd, J = 14.8, 7.6 Hz, 1H), 1.23 (d, J = 7.1 Hz, 3H), 1.13 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 172.1, 171.8, 170.8, 168.1, 141.7, 136.2, 130.5, 128.7, 128.6, 128.6, 126.6, 126.3, 125.3, 121.1, 117.8, 110.8, 81.0, 50.1, 49.0, 41.0, 37.2, 35.3, 31.4, 26.7, 18.7. HRMS calc. for C29H37N5O5Na [M+Na]+: 558.2687. Found: 558.2698.
PKS2038:
Synthesized by following the general procedure for boc-deprotection of PKS2032 (95 mg, 0.22 mmol). After completion of reaction, dichloromethane and excess trifluoroacetic acid were evaporated and crude was triturated with diethyl ether. The mixture was filtered to give product (60 mg, 61%) as white powder. 1H NMR (500 MHz, DMSO-d6) δ 8.73 (d, J = 7.6 Hz, 1H), 8.51 (t, J = 5.7 Hz, 1H), 8.18 (b, 3H), 8.05 – 8.03 (m, 1H), 7.97 – 7.95 (m, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.58 – 7.54 (m, 2H), 7.49 – 7.43 (m, 2H), 4.80 – 4.72 (m, 2H), 4.45 – 4.39 (m, 1H), 4.03 (m, 1H), 3.65 (dd, J = 10.7, 3.8 Hz, 1H), 3.56 (dd, J = 10.7, 7.2 Hz, 1H), 3.25 (s, 3H), 1.28 (d, J = 7.0 Hz, 3H).
PKS2048:
Synthesized following the general procedure for EDC coupling of 3-phenylpropanoic acid (16.5 mg, 0.11 mmol) with PKS2038 (44.3 mg, 0.1 mmol). The crude was purified by silica gel column chromatography to give 44.1 mg (87%) of product. 1H NMR (500 MHz, DMSO-d6) δ 8.24 – 8.18 (m, 2H), 8.15 (d, J = 7.8 Hz, 1H), 8.04 – 8.02 (m, 1H), 7.96 – 7.94 (m, 1H), 7.86 – 7.84 (m, 1H), 7.56 – 7.53 (m, 2H), 7.45 – 7.40 (m, 2H), 7.27 – 7.24 (m, 2H), 7.20 – 7.16 (m, 3H), 4.75 (d, J = 5.7 Hz, 2H), 4.50 (dt, J = 7.9, 6.0 Hz, 1H), 4.36 – 4.30 (m, 1H), 3.45 – 3.41 (m, 2H), 3.12 (s, 3H), 2.79 (t, J = 7.9 Hz, 2H), 2.47 – 2.44 (m, 2H), 1.26 (d, J = 7.1 Hz, 3H). LCMS calc. for C27H32N3O4 [M+H]+: 462.2. Found: 462.3.
PKS2154:
Synthesized following the general procedure of HATU mediated coupling of boc-β-methoxyalanine dicyclohexylamine (100 mg, 0.25 mmol) and 3-methoxybenzyl amine (36 mL, 0.275 mmol). The product was isolated by ethyl acetate extraction and purified by silica-gel column chromatography (yield 74 mg, 87%). 1H NMR (500 MHz, Chloroform-d) δ 7.26 – 7.22 (m, 1H), 6.86– 6.84 (m, 1H), 6.82 – 6.80 (m, 2H), 6.75 (t, J = 6.2 Hz, 1H), 5.42 (b, 1H), 4.47 – 4.46 (m, 2H), 4.28 (m, 1H), 3.85 (dd, J = 9.2, 3.8 Hz, 1H), 3.80 (s, 3H), 3.51 (dd, J = 9.2, 6.2 Hz, 1H), 3.38 (s, 3H), 1.44 (s, 9H).
PKS2158:
Synthesized by following the general procedure for boc-deprotection of PKS2154 (74 mg, 0.219 mmol). Isolated crude was dried under vacuum to give product (70 mg, 91%). 1H NMR (500 MHz, Chloroform-d) δ 9.13 (b, 1H), 8.01 (b, 2H), 7.56 (t, J = 5.8 Hz, 1H), 7.22 (t, J = 7.9 Hz, 1H), 6.80 (dd, J = 8.3, 2.5 Hz, 1H), 6.75 (d, J = 7.6 Hz, 1H), 6.73 – 6.72 (m, 1H), 4.41 – 4.35 (m, 2H), 4.29 (dd, J = 15.0, 5.5 Hz, 1H), 3.75 (s, 3H), 3.73 (dd, J = 10.4, 4.7 Hz, 1H), 3.67 (dd, J = 10.4, 5.3 Hz, 1H), 3.32 (s, 3H).
PKS2160:
Synthesized following the general procedure for HATU mediated coupling of PKS21059 (15.8 mg, 0.047 mmol) and PKS2158 (18.4 mg, 0.052 mmol). The product was purified by HPLC (yield 16.2 mg, 62%).1H NMR (500 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.51 (t, J = 6.1 Hz, 1H), 8.20 (d, J = 7.9 Hz, 1H), 8.09 (d, J = 7.7 Hz, 1H), 7.27 – 7.25 (m, 2H), 7.21 – 7.15 (m, 4H), 6.82 – 6.81 (m, 2H), 6.77 (dd, J = 8.1, 2.4 Hz, 1H), 4.67 – 4.63 (m, 1H), 4.40 – 4.37 (m, 1H), 4.30 – 4.21 (m, 2H), 3.72 (s, 3H), 3.61 (dd, J = 9.8, 5.9 Hz, 1H), 3.50 (dd, J = 9.8, 4.6 Hz, 1H), 3.25 (s, 3H), 2.80 – 2.76 (m, 2H), 2.53 – 2.48 (m, 1H), 2.43 – 2.39 (m, 2H), 2.34 (dd, J = 14.8, 7.6 Hz, 1H), 1.11 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 172.0, 171.4, 169.7, 168.1, 159.8, 141.7, 141.3, 129.7, 128.8, 128.6, 126.4, 119.6, 112.9, 112.6, 81.1, 72.3, 58.8, 55.4, 53.7, 50.1, 42.5, 37.3, 35.3, 31.5, 26.7. HRMS calc. for C29H40N4O7Na [M+Na]+: 579.2789. Found: 579.2774.
PKS2184:
Synthesized following the general procedure of HATU mediated coupling of boc-β-methoxyalanine dicyclohexylamine (200 mg, 0.5 mmol) and 2,3-dimethoxybenzyl amine (83 μL, 0.55 mmol). The product was isolated by ethyl acetate extraction and purified by silica-gel column chromatography (yield = 184 mg, quant.). 1H NMR (500 MHz, DMSO-d6) δ 8.24 (t, J = 5.9 Hz, 1H), 7.00 – 6.97 (m, 1H), 6.93 (dd, J = 8.3, 1.7 Hz, 1H), 6.86 (d, J = 8.3 Hz, 1H), 6.81 (dd, J = 7.6, 1.7 Hz, 1H), 4.31 – 4.23 (m, 2H), 4.20 – 4.15 (m, 1H), 3.79 (s, 3H), 3.72 (s, 3H), 3.48 – 3.46 (m, 2H), 3.24 (s, 3H), 1.38 (s, 9H).
PKS2190:
Synthesized by following the general procedure for boc-deprotection of PKS2184 (180 mg, 0.49 mmol). Crude product was dried under vacuum to give viscous paste, which upon standing turned solid. 1H NMR (500 MHz, DMSO-d6) δ 8.78 (t, J = 5.7 Hz, 1H), 8.21 (b, 3H), 7.05 – 7.02 (m, 1H), 6.97 (dd, J = 8.2, 1.6 Hz, 1H), 6.83 (dd, J = 7.6, 1.6 Hz, 1H), 4.36 (dd, J = 15.1, 5.7 Hz, 1H), 4.31 (dd, J = 15.1, 5.6 Hz, 1H), 4.04 – 4.01 (m, 1H), 3.80 (s, 3H), 3.75 (s, 3H), 3.66 (d, J = 5.1 Hz, 2H), 3.30 (s, 3H).
PKS2191:
Synthesized following the general procedure of HATU mediated coupling of N-(tert-butoxycarbonyl)-L-aspartic acid 4-benzyl ester (159 mg, 0.49 mmol) and PKS2190 (0.49 mmol, from previous step). The product was isolated by ethyl acetate extraction and recrystallized with ethanol-water (yield = 245 mg, 87% for 2 steps). 1H NMR (500 MHz, DMSO-d6) δ 8.29 (t, J = 5.8 Hz, 1H), 7.86 (d, J = 7.9 Hz, 1H), 7.38 – 7.31 (m, 5H), 7.28 (d, J = 8.0 Hz, 1H), 7.00 – 6.97 (m, 1H), 6.92 (dd, J = 8.2, 1.6 Hz, 1H), 6.78 (dd, J = 7.7, 1.6 Hz, 1H), 5.08 (d, J = 12.6 Hz, 1H), 5.05 (d, J = 12.6 Hz, 1H), 4.45 – 4.37 (m, 2H), 4.31 – 4.23 (m, 2H), 3.78 (s, 3H), 3.71 (s, 3H), 3.56 (dd, J = 9.7, 5.6 Hz, 1H), 3.46 (dd, J = 9.7, 5.1 Hz, 1H), 3.24 (s, 3H), 2.82 – 2.78 (m, 1H), 2.61 (dd, J = 16.3, 8.9 Hz, 1 H), 1.37 (s, 9H).
PKS2197:
Synthesized by following the general procedure for boc-deprotection of PKS2191 (115 mg, 0.2 mmol). The crude was used in next step without further purification. 1H NMR (500 MHz, DMSO-d6) δ 8.81 (d, J = 7.8 Hz, 1H), 8.37 (t, J = 5.9 Hz, 1H), 8.26 (b, 3H), 7.39 – 7.34 (m, 5H), 6.99 (t, J = 7.9 Hz, 1H), 6.93 (dd, J = 8.3, 1.6 Hz, 1H), 6.78 (dd, J = 7.7, 1.6 Hz, 1H), 5.14 (d, J = 12.4 Hz, 1H), 5.11 (d, J = 12.4 Hz, 1H), 4.55 – 4.51 (m, 1H), 4.33 – 4.23 (m, 3H), 3.78 (s, 3H), 3.71 (s, 3H), 3.59 (dd, J = 9.9, 6.2 Hz, 1H), 3.52 (dd, J = 9.9, 4.8 Hz, 1H), 3.27 (s, 3H), 3.02 (dd, J = 17.5, 4.1 Hz, 1H), 2.82 (dd, J = 17.5, 8.8 Hz, 1H).
PKS2200:
Synthesized following the general procedure of HATU mediated coupling of 3-phenylpropanoic acid (33 mg, 0.22 mmol) and PKS2197 (0.2 mmol, from previous step). The reaction mixture was precipitated with water and the precipitate was filtered and dried to give product (110 mg, 91% for 2 steps). 1H NMR (500 MHz, DMSO-d6) δ 8.30 (d, J = 7.9 Hz, 1H), 8.24 (t, J = 5.9 Hz, 1H), 8.04 (d, J = 7.9 Hz, 1H), 7.38 – 7.32 (m, 5H), 7.28 – 7.25 (m, 2H), 7.18 – 7.15 (m, 3H), 6.99 (t, J = 7.9 Hz, 1H), 6.92 (dd, J = 8.0, 1.6 Hz, 1H), 6.79 (dd, J = 7.5, 1.6 Hz, 1H), 5.04 (s, 2H), 4.74 (td, J = 8.1, 5.7 Hz, 1H), 4.42 (dt, J = 7.8, 5.4 Hz, 1H), 4.31 – 4.24 (m, 2H), 3.78 (s, 3H), 3.72 (s, 3H), 3.56 (dd, J = 9.8, 5.8 Hz, 1H), 3.47 (dd, J = 9.8, 5.0 Hz, 1H), 3.24 (s, 3H), 2.85 – 2.77 (m, 3H), 2.59 (dd, J = 16.2, 8.3 Hz, 1H), 2.41 – 2.38 (m, 2H).
PKS2204:
Synthesized by following general procedure for O-debenzylation. The reaction was not complete and isolated crude was purified by HPLC to give 40 mg product (50% bsrm; 16.0 mg starting material was recovered). 1H NMR (500 MHz, DMSO-d6) δ 12.34 (s, 1H), 8.27 – 8.24 (m, 2H), 7.95 (d, J = 7.9 Hz, 1H), 7.28 – 7.25 (m, 2H), 7.20 – 7.15 (m, 3H), 6.99 (t, J = 7.9 Hz, 1H), 6.93 (dd, J = 8.2, 1.5 Hz, 1H), 6.80 (dd, J = 7.7, 1.5 Hz, 1H), 4.66 – 4.60 (m, 1H), 4.43 – 4. 38 (m, 1H), 4.28 (d, J = 5.9 Hz, 2H), 3.78 (s, 3H), 3.72 (s, 3H), 3.56 (dd, J = 9.7, 5.8 Hz, 1H), 3.47 (dd, J = 9.7, 5.0 Hz, 1H), 3.24 (s, 3H), 2.79 (t, J = 7.9 Hz, 2H), 2.68 (dd, J = 16.6, 5.9 Hz, 1H), 2.48 – 2.39 (m, 3H).
PKS2211:
Synthesized following the general procedure for HATU mediated coupling of O-tert-butyl hydroxylamine hydrochloride (6.6 mg, 0.0525 mmol) and PKS2204 (18.0 mg, 0.035 mmol). After completion of reaction (1 h), mixture was diluted with water and extracted with ethyl acetate. The organic layer was evaporated and purified by HPLC to give the product (11.0 mg, 54%). 1H NMR (500 MHz, DMSO-d6) δ 10.37 (s, 1H), 8.37 (t, J = 6.0 Hz, 1H), 8.20 (d, J = 7.9 Hz, 1H), 8.05 (d, J = 7.7 Hz, 1H), 7.28 – 7.25 (m, 2H), 7.19 – 7.15 (m, 3H), 6.98 (t, J = 7.9 Hz, 1H), 6.92 (dd, J = 8.1, 1.6 Hz, 1H), 6.82 (d, J = 7.7 Hz, 1H), 4.67 – 4.63 (m, 1H), 4.40 (dt, J = 7.6, 5.3 Hz, 1H), 4.31 – 4.24 (m, 2H), 3.78 (s, 3H), 3.72 (s, 3H), 3.59 (dd, J = 9.8, 6.0 Hz, 1H), 3.49 (dd, J = 9.8, 4.6 Hz, 1H), 3.24 (s, 3H), 2.80 – 2.76 (m, 2H), 2.52 – 2.48 (m, 1H), 2.43 – 2.39 (m, 2H), 2.33 (dd, J = 14.9, 7.8 Hz, 1H), 1.11 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 172.0, 171.3, 169.7, 168.0, 152.6, 146.5, 141.7, 132.8, 128.8, 128.6, 126.3, 124.1, 120.3, 112.0, 81.0, 72.3, 60.4, 58.7, 56.1, 53.6, 50.1, 37.4, 37.2, 35.3, 31.4, 26.7. HRMS calc. for C30H42N4O8Na [M+Na]+: 609.2895. Found: 609.2897.
PKS2175:
Synthesized following the general procedure for HATU coupling of boc-β-methoxyalanine dicyclohexylamine (80 mg, 0.02 mmol) and quinolin-8-ylmethylamine dihydrochloride (46 mg, 0.2 mmol) in 2 mL dimethylformamide. (Note: reaction mixture was not soluble in dimethylformamide) After completion of reaction (3 h), water was added to reaction mixture (reaction mixture turned transparent) and extracted twice with chloroform. The combined organic layer was washed with water followed by brine, dried over anhydrous sodium sulfate and evaporated. The crude product was purified by HPLC to give 68.5 mg (95%) of pure product. 1H NMR (500 MHz, DMSO-d6) δ 8.96 – 8.94 (m, 1H), 8.46 (t, J = 6.2 Hz, 1H), 8.39 (dd, J = 8.1, 1.6 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.64 (d, J = 7.1 Hz, 1H), 7.58 (dd, J = 8.3, 4.2 Hz, 1H), 7.54 (t, J = 7.6 Hz, 1H), 6.96 (d, J = 8.1 Hz, 1H), 4.96 – 4.87 (m, 2H), 4.26 – 4.22 (m, 1H), 3.54 – 3.53 (m, 2H), 3.27 (s, 3H), 1.39 (s, 9H).
PKS2181:
Synthesized by following the general procedure for boc-deprotection of PKS2175 (68.5 mg, 0.19 mmol). The crude was used in next step. 1H NMR (500 MHz, DMSO-d6) δ 8.98 (dd, J = 4.2, 1.8 Hz, 1H), 8.95 (t, J = 5.9 Hz, 1H), 8.42 (dd, J = 8.2, 1.8 Hz, 1H), 8.22 (b, 3H), 7.93 (dd, J = 8.1, 1.5 Hz, 1H), 7.67 (dd, J = 7.1, 1.5 Hz, 1H), 7.62 – 7.58 (m, 2H), 5.02 (dd, J = 15.9, 5.9 Hz, 1H), 4.96 (dd, J = 15.9, 5.7 Hz, 1H), 4.13 – 4.10 (m, 1H), 3.75 – 3.69 (m, 2H), 3.32 (s, 3H).
PKS2188:
Synthesized following the general procedure for HATU mediated coupling of N-(tert-butoxycarbonyl)-L-aspartic acid 4-benzyl ester (61.4 mg, 0.19 mmol) and PKS2181 (0.19 mmol, from previous step). After completion of reaction (1 h), reaction mixture was diluted with water and extracted twice with ethyl acetate. Ethyl acetate layer was dried over anhydrous Na2SO4 and evaporated. The crude was dried under high vacuum and used in next step without further purification. 1H NMR (500 MHz, DMSO-d6) δ 8.96 (dd, J = 4.3, 1.8 Hz, 1H), 8.49 (t, J = 5.9 Hz, 1H), 8.43 (dd, J = 8.3, 1.8 Hz, 1H), 7.95 (d, J = 7.8 Hz, 1H), 7.90 (dd, J = 8.0, 1.5 Hz, 1H), 7.63 (dd, J = 7.1, 1.5 Hz, 1H), 7.61 – 7.55 (m, 2H), 7.37 – 7.30 (m, 6H), 5.09 (d, J = 12.7 Hz, 1H), 5.05 (d, J = 12.7 Hz, 1H), 4.96 (dd, J = 16.4, 6.0 Hz, 1H), 4.89 (dd, J = 16.4, 5.9 Hz, 1H), 4.51 – 4.48 (m, 1H), 4.43 (td, J = 8.5, 5.0 Hz, 1H), 3.63 (dd, J = 9.7, 5.5 Hz, 1H), 3.52 (dd, J = 9.7, 5.1 Hz, 1H), 3.28 (s, 3H), 2.85 – 2.81 (m, 1H), 2.62 (dd, J = 16.2, 8.9 Hz, 1H), 1.38 (s, 9H).
PKS2194:
Synthesized by following the general procedure for boc-deprotection of PKS2188 (0.19 mmol, from previous step). After completion of reaction (3 h), excess trfluoroacetic acid and dichloromethane were evaporated. Crude paste was washed twice with diethyl ether to give product as off-white solid (yield = 111 mg, 84% for 3 steps). 1H NMR (500 MHz, DMSO-d6) δ 8.94 (dd, J = 4.2, 1.8 Hz, 1H), 8.87 (d, J = 7.8 Hz, 1H), 8.53 (t, J = 6.0 Hz, 1H), 8.39 (dd, J = 8.4, 1.8 Hz, 1H), 8.27 (b, 3H), 7.88 (dd, J = 8.1, 1.5 Hz, 1H), 7.61 (dd, J = 7.2, 1.5 Hz, 1H), 7.58 – 7.54 (m, 2H), 7.40 – 7.31 (m, 5H), 5.13 (J = 12.5 Hz, 1H), 5.10 (d, J = 12.5 Hz, 1H), 4.97 (dd, J = 16.4, 6.1 Hz, 1H), 4.90 (dd, J = 16.4, 5.9 Hz, 1H), 4.62 – 4.58 (m, 1H), 4.27 (m, 1H), 3.66 (dd, J = 9.8, 6.0 Hz, 1H), 3.57 (dd, J = 9.8, 4.8 Hz, 1H), 3.30 (s, 3H), 3.04 (dd, J = 17.5, 3.9 Hz, 1H), 2.85 – 2.80 (m, 1H). LCMS calc. for C25H28N4O5 [M+H]+: 464.2. Found: 464.3.
PKS2198:
Synthesized following the general procedure for HATU mediated coupling of 3-phenylproapnoic acid (26.4 mg, 0.176 mmol) and PKS2194 (111 mg, 0.16 mmol). After completion of reaction (3 h), the mixture was precipitated with water. The precipitate was filtered and dried to give the product (77 mg, 81%). 1H NMR (500 MHz, DMSO-d6) δ 8.94 (dd, J = 4.2, 1.8 Hz, 1H), 8.43 (t, J = 6.1 Hz, 1H), 8.38 (dd, J = 8.3, 1.8 Hz, 1H), 8.32 (d, J = 8.0 Hz, 1H), 8.12 (d, J = 7.8 Hz, 1H), 7.87 (dd, J = 8.1, 1.5 Hz, 1H), 7.61 (dd, J = 7.2, 1.5 Hz, 1H), 7.57 – 7.53 (m, 2H), 7.35 – 7.30 (m, 5H), 7.27 – 7.24 (m, 2H), 7.19 – 7.15 (m, 3H), 5.03 (s, 2H), 4.95 (dd, J = 16.4, 6.1 Hz, 1H), 4.90 (dd, J = 16.4, 5.9 Hz, 1H), 4.77 (td, J = 8.2, 5.7 Hz, 1H), 4.49 (dt, J = 7.8, 5.3 Hz, 1H), 3.62 (dd, J = 9.7, 5.8 Hz, 1H), 3.52 (dd, J = 9.7, 5.0 Hz, 1H), 3.28 (s, 3H), 2.85 – 2.77 (m, 3H), 2.59 (dd, J = 16.2, 8.4 Hz, 1H), 2.42 – 2.38 (m, 2H).
PKS2202:
Synthesized by following general procedure for O-debenzylation of PKS2198 (73 mg, 0.122 mmol). The mixture was filtered through celite, evaporated and purified by HPLC to give the product (19 mg, 30%). 1H NMR (500 MHz, DMSO-d6) δ 12.34 (s, 1H), 8.26 – 8.23 (m, 2H), 7.99 (d, J = 7.8 Hz, 1H), 7.28 – 7.25 (m, 2H), 7.21 – 7.15 (m, 3H), 6.81 (dd, J = 7.5, 1.5 Hz, 1H), 6.76 (dd, J = 7.5, 1.5 Hz, 1H), 6.40 (t, J = 7.4 Hz, 1H), 4.63 (td, J = 7.8, 5.8 Hz, 1H), 4.40 (dt, J = 7.7, 5.4 Hz, 1H), 4.08 (dd, J = 15.3, 6.1 Hz, 1H), 4.03 (dd, J = 15.3, 5.9 Hz, 1H), 3.56 (dd, J = 9.8, 5.8 Hz, 1H), 3.47 (dd, J = 9.8, 5.0 Hz, 1H), 3.24 (s, 3H), 3.22 (t, J = 5.5 Hz, 2H), 2.80 (t, J = 7.9 Hz, 2H), 2.70 – 2.65 (m, 3H), 2.48 – 2.39 (m, 3H), 1.79 – 1.74 (m, 2H).
PKS2226:
Synthesized following the general procedure for HATU mediated coupling of PKS2202 (19.0 mg, 0.037 mmol) and O-tert-butylhydroxylamine hydrochloride (5.1 mg, 0.041 mmol). The product was purified by HPLC (17.9 mg, 83%) 1H NMR (500 MHz, DMSO-d6) δ 10.40 (s, 1H), 8.41 (t, J = 6.1 Hz, 1H), 8.18 (d, J = 7.9 Hz, 1H), 8.10 (d, J = 7.8 Hz, 1H), 7.28 – 7.25 (m, 2H), 7.20 – 7.15 (m, 3H), 6.84 (d, J = 7.5 Hz, 1H), 6.75 (d, J = 7.5 Hz, 1H), 6.38 (t, J = 7.4 Hz, 1H), 5.24 – 5.20 (m, 1H), 4.67 – 4.62 (m, 1H), 4.40 – 4.37 (m, 1H), 4.10 (dd, J = 15.1, 6.3 Hz, 1H), 4.00 (dd, J = 15.1, 5.8 Hz, 1H), 3.60 (dd, J = 9.8, 5.9 Hz, 1H), 3.49 (dd, J = 9.8, 4.5 Hz, 1H), 3.24 (s, 3H), 3.22 – 3.21 (m, 2H), 2.80 – 2.76 (m, 2H), 2.66 (t, J = 6.4 Hz, 2H), 2.52 – 2.48 (m, 1H), 2.42 – 2.39 (m, 2H), 2.35 (dd, J = 14.8, 7.5 Hz, 1H), 1.79 – 1.74 (m, 2H), 1.14 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 171.5, 170.9, 169.5, 167.6, 142.5, 141.2, 128.3, 128.1, 128.0, 126.5, 125.8, 121.3, 120.0, 114.6, 80.6, 71.7, 58.3, 53.1, 49.6, 41.3, 36.8, 34.8, 31.00, 27.2, 26.3, 21.3. HRMS calc. for C31H43N5O6Na [M+Na]+: 604.3106. Found: 604.3105.
PKS2220:
Synthesized following the general procedure for HATU mediated coupling of PKS21059 (20.2 mg, 0.06 mmol) and PKS2181 (32.2 mg, 0.066 mmol). The product was purified be HPLC (yield = 21.6 mg, 62%). 1H NMR (500 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.95 (dd, J = 4.2, 1.8 Hz, 1H), 8.57 (t, J = 6.1 Hz, 1H), 8.38 (dd, J = 8.3, 1.8 Hz, 1H), 8.22 (d, J = 8.0 Hz, 1H), 8.15 (d, J = 7.8 Hz, 1H), 7.87 (dd, J = 8.3, 1.5 Hz 1H), 7.63 (dd, J = 7.4, 1.5 Hz, 1H), 7.59 – 7.54 (m, 2H), 7.28 – 7.25 (m, 2H), 7.20 – 7.17 (m, 3H), 4.98 – 4.89 (m, 2H), 4.71 – 4.67 (m, 1H), 4.50 – 4.46 (m, 1H), 3.66 (dd, J = 9.7, 5.8 Hz, 1H), 3.55 (dd, J = 9.7, 4.7 Hz, 1H), 3.29 (s, 3H), 2.80 – 2.77 (m, 2H), 2.55 – 2.51 (m, 1H), 2.43 – 2.40 (m, 2H), 2.35 (dd, J = 14.7, 7.7 Hz, 1H), 1.06 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 172.0, 171.5, 170.0, 168.0, 150.1, 145.8, 141.7, 136.9, 136.8, 128.8, 128.6, 128.1, 127.1, 126.9, 126.7, 126.3, 121.9, 81.0, 72.3, 58.8, 53.7, 50.1, 39.2, 37.2, 35.3, 31.5, 26.7. HRMS calc. for C31H39N5O6Na [M+Na]+: 600.2793. Found: 600.2783.
PKS2130:
Synthesized following the general procedure for HATU mediated coupling of PKS2124 (12.4 mg, 0.026 mmol) and 3-aminoxetane (2.2 μL, 0.031 mmol). The mixture was purified by HPLC to give product (12.0 mg, 87%). 1H NMR (500 MHz, DMSO-d6) δ 8.71 (d, J = 6.4 Hz, 1H), 8.44 (t, J = 5.8 Hz, 1H), 8.21 (d, J = 7.3 Hz, 1H), 8.15 (d, J = 7.9 Hz, 1H), 8.07 – 8.05 (m, 1H), 7.95 – 7.94 (m, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.57 – 7.52 (m, 2H), 7.47 – 7.42 (m, 2H), 7.27 – 7.24 (m, 2H), 7.19 – 7.15 (m, 3H), 4.76 (dd, J = 15.4, 5.9 Hz, 1H), 4.70 (dd, J = 15.4, 5.8 Hz, 1H), 4.64 – 4.51 (m, 4H), 4.34 – 4.22 (m, 3H), 2.78 (t, J = 7.8 Hz, 2H), 2.58 (dd, J = 15.2, 7.5 Hz, 1H), 2.44 – 2.40 (m, 3H), 1.25 (d, J = 7.2 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 172.4, 172.0, 171.0, 169.8, 141.7, 134.8, 133.7, 131.2, 128.9, 128.7, 128.6, 127.9, 126.6, 126.3, 126.2, 125.8, 125.6, 123.8, 77.4, 77.3, 50.0, 49.1, 44.3, 40.6, 37.8, 37.1, 31.4, 18.3. LCMS calc. for C30H35N4O5 [M+H]+: 531.3. Found: 531.3.
PKS2209:
Synthesized following the general procedure for HATU mediated coupling of PKS2201 (15.2 mg, 0.03 mmol) and azetidine hydrochloride (4.2 mg, 0.045 mmol). The mixture was purified by HPLC to give pure product (10.5 mg, 64%). 1H NMR (500 MHz, DMSO-d6) δ 8.51 (t, J = 5.9 Hz, 1H), 8.29 (d, J = 7.7 Hz, 1H), 8.16 (d, J = 7.9 Hz, 1H), 8.08 – 8.06 (m, 1H), 7.95 – 7.93 (m, 1H), 7.84 – 7.82 (m, 1H), 7.55 – 7.52 (m, 2H), 7.46 – 7.41 (m, 2H), 7.28 – 7.25 (m, 2H), 7.20 – 7.15 (m, 3H), 4.80 – 4.62 (m, 3H), 4.43 – 4.39 (m, 1H), 4.07 (td, J = 8.7, 6.3 Hz, 1H), 4.00 (td, J = 8.7, 6.3 Hz, 1H), 3.72 – 3.56 (m, 3H), 3.53 (dd, J = 9.8, 4.3 Hz, 1H), 3.24 (s, 3H), 2.79 (t, J = 7.8 Hz, 2H), 2.47 – 2.36 (m, 3H), 2.29 (dd, J = 15.4, 5.8 Hz, 1H), 2.12 – 2.03 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 171.4, 171.0, 169.3, 169.2, 141.2, 134.2, 133.2, 130.7, 128.4, 128.2, 127.3, 126.1, 125.8, 125.7, 125.3, 124.9, 124.8, 123.4, 71.7, 58.2, 53.5, 49.6, 49.1, 47.3, 40.2, 36.6, 33.1, 31.0, 14.5. LCMS calc. for C31H37N4O5 [M+H]+: 545.3. Found: 545.2.
PKS2230:
Synthesized following the general procedure for HATU mediated coupling of PKS2201 (15.2 mg, 0.03 mmol) and tert-butylamine (9.5 μL, 0.09 mmol). The product was purified by HPLC to give pure product (8.7 mg, 52%). 1H NMR (500 MHz, DMSO-d6) δ 8.66 (t, J = 5.9 Hz, 1H), 8.19 (d, J = 7.7 Hz, 1H), 8.12 (d, J = 8.0 Hz, 1H), 8.07 – 8.05 (m, 1H), 7.94 – 7.92 (m, 1H), 7.83 – 7.81 (m, 1H), 7.55 – 7.50 (m, 3H), 7.46 – 7.40 (m, 2H), 7.28 – 7.25 (m, 2H), 7.20 – 7.15 (m, 3H), 4.79 (dd, J = 15.6, 6.0 Hz, 1H), 4.70 (dd, J = 15.6, 5.8 Hz, 1H), 4.63 – 4.59 (m, 1H), 4.45 – 4.41 (m, 1H), 3.63 (dd, J = 9.8, 6.1 Hz, 1H), 3.54 (dd, J = 9.8, 4.4 Hz, 1H), 3.24 (s, 3H), 2.78 (t, J = 7.9 Hz, 2H), 2.55 – 2.51 (m, 1H), 2.43 – 2.34 (m, 3H), 1.14 (s, 9H).13C NMR (126 MHz, DMSO-d6) δ 171.3, 171.2, 169.3, 169.1, 141.3, 134.2, 133.2, 130.7, 128.4, 128.3, 128.1, 127.3, 126.1, 125.8, 125.7, 125.3, 124.9, 123.3, 71.8, 58.2, 53.3, 50.1, 49.7, 40.2, 38.5, 36.7, 31.0, 28.4. HRMS calc. for C32H40N4O5Na [M+Na]+: 583.2891. Found: 583.2877.
PKS2230BP:
Isolated as a byproduct in the above reaction. 1H NMR (500 MHz, DMSO-d6) δ 8.44 (t, J = 6.0 Hz, 1H), 8.32 (d, J = 7.7 Hz, 1H), 8.15 (d, J = 8.0 Hz, 1H), 8.09 – 8.04 (m, 1H), 7.96 – 7.91 (m, 1H), 7.84 – 7.80 (m, 1H), 7.55 – 7.50 (m, 2H), 7.45 – 7.39 (m, 2H), 7.28 – 7.23 (m, 2H), 7.20 – 7.15 (m, 3H), 4.77 (dd, J = 15.6, 5.8 Hz, 1H), 4.74 – 4.64 (m, 2H), 4.42 – 4.37 (m, 1H), 3.66 (dd, J = 9.7, 6.3 Hz, 1H), 3.55 (dd, J = 9.7, 4.1 Hz, 1H), 3.24 (s, 3H), 2.87 (s, 3H), 2.79 (t, J = 7.8 Hz, 2H), 2.67 (dd, J = 16.2, 8.4 Hz, 1H), 2.54 – 2.60 (m, 1H), 2.57 (s, 3H), 2.46 – 2.37 (m, 2H). LCMS calc. for C30H37N4O5 [M+H]+: 533.3. Found: 533.5.
PKS2237:
The title compound was prepared following the general procedure for HATU mediated coupling of PKS2131 (118 mg, 0.25 mmol) and tert-butylamine (32 mL, 0.3 mmol). The product was isolated by ethylacetate extraction and purified by HPLC (yield = 37.0 mg). 1H NMR (500 MHz, DMSO-d6) δ 8.59 (t, J = 5.8 Hz, 1H), 8.05 – 8.03 (m, 1H), 7.98 – 7.93 (m, 2H), 7.84 – 7.82 (m, 1H), 7.55 – 7.51 (m, 2H), 7.46 – 7.43 (m, 3H), 6.93 (d, J = 8.1 Hz, 1H), 4.79 – 4.70 (m, 2H), 4.47 – 4.43 (m, 1H), 4.31 – 4.26 (m, 1H), 3.61 (dd, J = 9.7, 5.7 Hz, 1H), 3.51 (dd, J = 9.7, 4.8 Hz, 1H), 3.24 (s, 3H), 2.50 – 2.45 (m, 1H), 2.33 (dd, J = 14.9, 7.8 Hz, 1H), 1.36 (s, 9H), 1.18 (s, 9H). PKS2242: Synthesized by following the general procedure for boc-deprotection of PKS2237 (34.4 mg, 0.065 mmol). Yield = 32 mg, 91%. 1H NMR (500 MHz, DMSO-d6) δ 8.76 (d, J = 7.7 Hz, 1H), 8.67 (t, J = 5.8 Hz, 1H), 8.14 (b, 3H), 8.04 – 8.02 (m, 1H), 7.95 – 7.93 (m, 1H), 7.88 (s, 1H), 7.85 – 7.83 (m, 1H), 7.56 – 7.52 (m, 2H), 7.47 – 7.43 (m, 2H), 4.80 – 4.72 (m, 2H), 4.53 (ddd, J = 7.7, 5.9, 4.6 Hz, 1H), 4.16 – 4.13 (m, 1H), 3.63 (dd, J = 9.8, 5.9 Hz, 1H), 3.53 (dd, J = 9.8, 4.6 Hz, 1H), 3.26 (s, 3H), 2.73 (dd, J = 16.7, 5.2 Hz, 1H), 2.60 – 2.55 (m, 1H), 1.21 (s, 9H). LCMS calc. for C23H33N4O4 [M+H]+: 429.2. Found 429.5.
PKS2244:
Synthesized following the general procedure for HATU mediated coupling of 5-methylisoxazole-3-carboxylic acid (5.6 mg, 0.044 mmol) and PKS2242 (21.7 mg, 0.04 mmol). Isolated crude was purified by HPLC to give pure product (18.6 mg, 87%). 1H NMR (500 MHz, DMSO-d6) δ 8.62 (t, J = 5.8 Hz, 1H), 8.56 (d, J = 8.0 Hz, 1H), 8.25 (d, J = 7.8 Hz, 1H), 8.05 – 8.03 (m, 1H), 7.95 – 7.93 (m, 1H), 7.85 – 7.82 (m, 1H), 7.54 – 7.51 (m, 3H), 7.45 – 7.43 (m, 2H), 6.54 (s, 1H), 4.80 – 4.74 (m, 3H), 4.50 – 4.46 (m, 1H), 3.60 (dd, J = 9.8, 5.9 Hz, 1H), 3.53 (dd, J = 9.8, 4.9 Hz, 1H), 3.23 (s, 3H), 2.58 (d, J = 6.7 Hz, 2H), 2.47 (s, 3H), 1.16 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 171.4, 170.4, 169.2, 168.9, 158.5, 158.3, 134.2, 133.2, 130.7, 128.4, 127.4, 126.1, 125.7, 125.4, 125.0, 123.3, 101.3, 71.8, 58.2, 53.1, 50.2, 50.1, 40.3, 38.1, 28.3, 11.8. HRMS calc. for C28H35N5O6Na [M+Na]+: 560.2480. Found: 560.2478.
PKS2231:
Synthesized by following the general procedure of EDC mediated coupling of Boc-Glu-OBn (5.06 g, 15.0 mmol) with tert-butylamine (2.37 mL, 22.5 mmol). After completion of reaction, water was added to the mixture. Mixture was extracted twice with ethyl acetate. Combined organic layer was washed with aq. NaHCO3, water, 1N HCl, water followed by saturated brine solution. Ethyl acetate layer was dried over anhydrous Na2SO4 and evaporated to give product (5.78 g, 98%) as white solid. The product was used in next step without further purification. 1H NMR (500 MHz, Chloroform-d) δ 7.43 – 7.29 (m, 5H), 5.58 (s, 1H), 5.27 (d, J = 8.3 Hz, 1H), 5.20 (d, J = 12.3 Hz, 1H), 5.13 (d, J = 12.3 Hz, 1H), 4.36 – 4.23 (m, 1H), 2.22 – 2.06 (m, 3H), 2.02 – 1.87 (m, 1H), 1.42 (s, 9H), 1.32 (s, 9H).
PKS2233:
Synthesized by following the general procedure for boc-deprotection of PKS2231 (3.68 g, 9.38 mmol). Crude was dried under high vacuum to give product (3.81 g, quant.) as colorless paste. Product was used in next step without further purification. 1H NMR (500 MHz, Chloroform-d) δ 8.44 (s, 3H), 7.42 – 7.29 (m, 5H), 6.07 (b, 1H), 5.27 (d, J = 11.9 Hz, 1H), 5.20 (d, J = 11.9 Hz, 1H), 4.24 – 4.16 (m, 1H), 2.53 – 2.44 (m, 1H), 2.43 – 2.34 (m, 1H), 2.34 – 2.24 (m, 1H), 2.24 – 2.13 (m, 1H), 1.30 (d, J = 2.1 Hz, 9H).
PKS2234:
Synthesized by following the general procedure for HATU mediated coupling of 3-phenylpropanoic acid (82.6 mg, 0.55 mmol) and PKS2233 (203.2 mg, 0.5 mmol). Crude was purified by column chromatography to give product (193 mg, 91%) as white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.28 (d, J = 7.4 Hz, 1H), 7.41 – 7.30 (m, 5H), 7.30 – 7.21 (m, 3H), 7.21 – 7.13 (m, 3H), 5.13 (d, J = 12.6 Hz, 1H), 5.10 (d, J = 12.6 Hz,1H), 4.31 – 4.20 (m, 1H), 2.80 – 2.76 (m, 2H), 2.45 – 2.38 (m, 2H), 2.07 (t, J = 7.7 Hz, 2H), 1.97 – 1.84 (m, 1H), 1.79 – 1.67 (m, 1H), 1.22 (s, 9H).
PKS2239:
Synthesized by following the general procedure for O-debenzylation of PKS2234 (180 mg, 0.34 mmol). The product was isolated as a white solid (140 mg, 99%). 1H NMR (500 MHz, DMSO-d6) δ 12.58 (s, 1H), 8.10 (d, J = 7.7 Hz, 1H), 7.37 (s, 1H), 7.27 – 7.13 (m, 5H), 4.19 – 4.11 (m, 1H), 2.81 (t, J = 7.9 Hz, 2H), 2.49 – 2.36 (m, 2H), 2.10 – 1.98 (m, 2H), 1.96 – 1.85 (m, 1H), 1.76 – 1.63 (m, 1H), 1.23 (s, 9H). LCMS calc. for C18H25N2O4 [M−H]+: 333.2. Found: 333.4.
PKS2243:
Synthesized by following the general procedure for HATU mediated coupling of PKS2239 (20.1 mg, 0.06 mmol) and PKS2082 (22.3 mg, 0.06 mmol). The crude was purified by HPLC to give product (18.8 mg, 54%) as white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.55 (t, J = 5.8 Hz, 1H), 8.10 – 8.00 (m, 3H), 7.96 – 7.92 (m, 1H), 7.83 (dd, J = 6.1, 3.4 Hz, 1H), 7.56 – 7.50 (m, 2H), 7.47 – 7.43 (m, 2H), 7.35 (s, 1H), 7.28 – 7.23 (m, 2H), 7.21 – 7.13 (m, 3H), 4.75 (d, J = 5.7 Hz, 2H), 4.51 (dt, J = 7.7, 5.6 Hz, 1H), 4.27 (td, J = 8.3, 5.3 Hz, 1H), 3.55 (dd, J = 9.7, 6.0 Hz, 1H), 3.50 (dd, J = 9.7, 5.4 Hz, 1H), 3.24 (s, 3H), 2.79 (t, J = 7.9 Hz, 2H), 2.48 – 2.37 (m, 2H), 2.03 (t, J = 8.0 Hz, 2H), 1.92 – 1.78 (m, 1H), 1.77 – 1.60 (m, 1H), 1.22 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 171.6, 171.5, 171.2, 169.4, 141.3, 134.1, 133.2, 130.7, 128.4, 128.2, 128.1, 127.4, 126.1, 125.8, 125.7, 125.4, 124.9, 123.4, 71.9, 58.2, 52.6, 52.3, 49.8, 40.3, 36.8, 32.6, 31.0, 28.5, 28.3. HRMS calc. for C32H42N4O5Na [M+Na]+: 597.3047. Found: 597.3027.
PKS2255:
Synthesized by following the general procedure for HATU mediated coupling of PKS2239 (18.4 mg, 0.055 mmol) and PKS2026 (17.1 mg, 0.05 mmol). The crude was purified by HPLC to give product (20.2 mg, 74%) as white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.45 (t, J = 5.7 Hz, 1H), 8.10 – 8.01 (m, 3H), 7.98 – 7.90 (m, 1H), 7.84 (dd, J = 7.5, 1.9 Hz, 1H), 7.58 – 7.50 (m, 2H), 7.50 – 7.41 (m, 2H), 7.35 (s, 1H), 7.29 – 7.22 (m, 2H), 7.22 – 7.11 (m, 3H), 4.76 (dd, J = 14.2, 4.6 Hz, 1H), 4.73 (dd, J = 14.2, 4.5 Hz, 1H), 4.37 – 4.27 (m, 1H), 4.27 – 4.18 (m, 1H), 2.79 (t, J = 7.9 Hz, 2H), 2.47 – 2.36 (m, 2H), 2.09 – 2.00 (m, 2H), 1.90 – 1.79 (m, 1H), 1.75 – 1.63 (m, 1H), 1.24 (d, J = 7.1 Hz, 3H), 1.22 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 172.1, 171.6, 171.2, 171.2, 141.3, 134.4, 133.2, 130.8, 128.5, 128.2, 128.2, 127.5, 126.2, 125.8, 125.8, 125.4, 125.1, 123.4, 52.3, 49.8, 48.3, 40.2, 36.7, 32.6, 31.0, 28.5, 28.3, 18.2. HRMS calc. for C32H40N4O4Na [M+Na]+: 567.2942. Found: 567.2937.
PKS21201:
Boc-Ala-OSu (143 mg, 0.5 mmol) was dissolved in dichloromethane (5 mL) and (4-fluoro-1-naphthyl)methanamine (88 mg, 0.5 mmol) was added. The solution was cooled to 0 °C and triethylamine (51 mg, 0.5 mmol) was added. The reaction mixture was allowed to warm to room temperature and stirred at room temperature for 2 hours. After completion of reaction, dichloromethane was evaporated and water was added. Mixture was extracted with ethyl acetate twice. Combined organic layer was washed with 1N HCl followed by brine and dried over anhydrous sodium sulfate. Organic layer was evaporated to give product (170 mg, 98%) as white solid. Product was used in next step without further purification. 1H NMR (500 MHz, Chloroform-d) δ 8.16 – 8.09 (m, 1H), 7.96 (d, J = 7.8 Hz, 1H), 7.62 – 7.52 (m, 2H), 7.35 (dd, J = 7.8, 5.4 Hz, 1H), 7.07 (dd, J = 10.2, 7.9 Hz, 1H), 6.48 (b, 1H), 4.95 (b, 1H), 4.88 – 4.76 (m, 2H), 4.21 – 4.06 (m, 1H), 1.36 (d, J = 7.0 Hz, 3H), 1.33 (s, 9H). LCMS calc. for C19H24FN2O3 [M+H]+: 347.2. Found 347.1.
PKS21202:
Synthesized by following general procedure for boc-deprotection of PKS21201. Crude was dried under vacuum to give colorless paste (which turned off-white solid upon standing overnight at 0 °C). Product was used in next step without further purification. 1H NMR (500 MHz, DMSO-d6) δ 8.89 (t, J = 5.6 Hz, 1H), 8.16 – 8.06 (m, 5H), 7.72 – 7.64 (m, 2H), 7.47 (dd, J = 7.9, 5.5 Hz, 1H), 7.32 (dd, J = 10.6, 7.9 Hz, 1H), 4.80 (dd, J = 15.1, 5.6 Hz, 1H), 4.75 (dd, J = 15.1, 5.5 Hz, 1H), 3.92 – 3.80 (m, 1H), 1.35 (d, J = 6.9 Hz, 3H). LCMS calc. for C14H16FN2O [M+H]+: 247.1. Found 247.2.
PKS21210:
Synthesized by following the general procedure for HATU mediated coupling of PKS2239 (16.7 mg, 0.05 mmol) and PKS21202 (18.0 mg, 0.05 mmol). Isolated mixture was purified by HPLC to give product (19.6 mg, 70%) as white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.44 (t, J = 5.6 Hz, 1H), 8.12 – 8.05 (m, 3H), 8.03 (d, J = 7.7 Hz, 1H), 7.68 – 7.62 (m, 2H), 7.42 (dd, J = 7.8, 5.6 Hz, 1H), 7.35 (s, 1H), 7.30 – 7.23 (m, 3H), 7.21 – 7.13 (m, 3H), 4.71 (d, J = 5.6 Hz, 2H), 4.34 – 4.26 (m, 1H), 4.25 – 4.17 (m, 1H), 2.78 (t, J = 7.8 Hz, 2H), 2.48 – 2.36 (m, 2H), 2.03 (t, J = 8.0 Hz, 2H), 1.89 – 1.78 (m, 1H), 1.74 – 1.62 (m, 1H), 1.28 – 1.20 (m, 12H). 13C NMR (126 MHz, DMSO- d6) δ 172.2, 171.6, 171.2, 171.2, 157.4 (d, J = 248.8 Hz), 141.3, 132.0 (d, J = 3.7 Hz), 130.8 (d, J = 3.2 Hz), 128.2, 128.1, 127.3, 126.6, 125.8, 125.2 (d, J = 8.9 Hz), 123.8, 122.9 (d, J = 16.3 Hz), 120.4 (d, J = 5.3 Hz), 108.9 (d, J = 19.8 Hz), 52.3, 49.8, 48.3, 39.8, 36.7, 32.6, 31.0, 28.5, 28.3, 18.2. HRMS calc. for C32H39FN4O4Na [M+Na]+: 585.2848. Found: 585.2843.
PKS3053:
Synthesized by following the general procedure for HATU mediated coupling of PKS3052 (7.2 mg, 0.02 mmol) and PKS2026 (7.5 mg, 0.022 mmol). The mixture was purified by HPLC to give product (6.4 mg, 56%) as white solid. 1H NMR (500 MHz, DMSO-d6) δ 12.26 (d, J = 3.4 Hz, 1H), 8.77 (d, J = 3.2 Hz, 1H), 8.71 (d, J = 8.1 Hz, 1H), 8.52 (t, J = 5.8 Hz, 1H), 8.30 – 8.21 (m, 2H), 8.08 – 8.02 (m, 1H), 7.97 – 7.91 (m, 1H), 7.86 – 7.80 (m, 1H), 7.57 (s, 1H), 7.56 – 7.49 (m, 3H), 7.47 – 7.41 (m, 2H), 7.31 – 7.24 (m, 2H), 4.75 (d, J = 5.8 Hz, 2H), 4.70 – 4.60 (m, 1H), 4.38 – 4.25 (m, 1H), 2.66 – 2.54 (m, 2H), 1.27 (d, J = 7.1 Hz, 3H), 1.17 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 181.1, 171.9, 170.0, 169.0, 162.8, 138.6, 136.2, 134.4, 133.2, 130.8, 128.5, 127.5, 126.1, 125.7, 125.4, 125.1, 123.5, 123.3, 122.6, 121.3, 112.6, 112.1, 50.2, 50.1, 48.7, 40.2, 38.1, 28.4, 18.1. HRMS calc. for C32H35N5O5Na [M+Na]+: 592.2530. Found: 592.2508.
PKS21271:
Synthesized by following the general procedure for HATU mediated coupling of PKS21013 (16.0 mg, 0.05 mmol) and PKS21202 (18.0 mg, 0.05 mmol). Isolated mixture was purified by HPLC to give product (21.6 mg, 79%) as white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.57 (t, J = 6.1 Hz, 1H), 8.26 (d, J = 7.4 Hz, 1H), 8.16 – 8.04 (m, 3H), 7.69 – 7.61 (m, 2H), 7.53 (s, 1H), 7.46 – 7.39 (m, 1H), 7.29 – 7.22 (m, 3H), 7.21 – 7.12 (m, 3H), 4.76 (dd, J = 15.6, 5.9 Hz, 1H), 4.64 (dd, J = 15.6, 5.5 Hz, 1H), 4.55 – 4.47 (m, 1H), 4.29 – 4.18 (m, 1H), 2.78 (t, J = 8.0 Hz, 2H), 2.56 – 2.50 (m, 1H), 2.41 (t, J = 8.0 Hz, 2H), 2.35 (dd, J = 15.3, 6.1 Hz, 1H), 1.25 (d, J = 6.7 Hz, 3H), 1.12 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 172.0, 171.3, 170.8, 169.3, 157.3 (d, J = 249.2 Hz), 141.3, 132.0 (d, J = 5.0 Hz), 130.8 (d, J = 5.3 Hz), 128.3, 128.2, 127.3, 126.5, 125.9, 125.1 (d, J = 8.7 Hz), 123.8, 122.9 (d, J = 16.3 Hz), 120.4 (d, J = 5.4 Hz), 108.8 (d, J = 19.2 Hz), 50.1, 49.8, 48.7, 39.8, 38.4, 36.7, 31.0, 28.3, 17.8. HRMS calc. for C31H37FN4O4Na [M+Na]+: 571.2691. Found: 571.2689.
PKS21237:
Synthesized by following general procedure for HATU mediated amide bond formation of boc-β-methoxyalanine dicyclohexylamine (401 mg, 1 mmol) with (4-fluoro-1-naphthyl)methanamine (175 mg, 1 mmol). Crude was isolated by ethyl acetate extraction and purified by combi-flash to give product (337 mg, 90%) as off-white solid. 1H NMR (500 MHz, Chloroform-d) δ 8.16 – 8.08 (m, 1H), 7.95 (d, J = 7.8 Hz, 1H), 7.61 – 7.51 (m, 2H), 7.35 (dd, J = 7.9, 5.3 Hz, 1H), 7.11 – 7.01 (m, 1H), 6.71 (t, J = 5.6 Hz, 1H), 5.47 – 5.25 (m, 1H), 4.94 – 4.76 (m, 2H), 4.34 – 4.18 (m, 1H), 3.81 (dd, J = 9.3, 3.8 Hz, 1H), 3.51 – 3.43 (m, 1H), 3.27 (s, 3H), 1.37 (s, 9H).
PKS21238:
Synthesized by following general procedure for boc-deprotection of PKS21237 (335 mg, 0.9 mmol). Isolated crude was dried under vacuum to give product (347 mg, quant.) as off-white solid. Product was used in next step without further purification. 1H NMR (500 MHz, DMSO- d6) δ 8.95 (b, 1H), 8.23 (b, 3H), 8.14 – 8.06 (m, 2H), 7.72 – 7.64 (m, 2H), 7.51 – 7.44 (m, 1H), 7.32 (dd, J = 10.3, 8.2 Hz, 1H), 4.82 (dd, J = 15.2, 5.7 Hz, 1H), 4.75 (dd, J = 15.2, 5.4 Hz, 1H), 4.09 – 3.98 (m, 1H), 3.72 – 3.59 (m, 2H), 3.27 (s, 3H). LCMS calc. for C15H18FN2O2 [M+H]+: 277.1. Found 277.7.
PKS21218:
Synthesized by following the general procedure for HATU mediated coupling of 5-methylisoxazole-3-carboxylic acid (720 mg, 3.00 mmol) and PKS2233 (880 mg, 3.00 mmol). After completion of reaction (2 h), water was added and mixture was stirred for 30 minutes. A white precipitate appeared. Precipitate was filtered, washed with water and dried in air to give product (1.03 g, 86%) as white solid.1H NMR (500 MHz, Chloroform-d) δ 7.58 (d, J = 7.9 Hz, 1H), 7.39 – 7.30 (m, 5H), 6.41 (s, 1H), 5.47 (s, 1H), 5.20 (s, 2H), 4.80 – 4.72 (m, 1H), 2.47 (s, 3H), 2.35 – 2.25 (m, 1H), 2.23 – 2.04 (m, 3H), 1.31 (s, 9H). LCMS calc. for C21H28N3O5 [M+H]+: 402.2. Found 402.3.
PKS21233:
Synthesized by following the general procedure for O-debenzylation of PKS21218 (1.02 g, 2.54 mmol). The product (780 mg, 99%) was isolated as white solid. 1H NMR (500 MHz, DMSO- d6) δ 12.71 (s, 1H), 8.78 (d, J = 7.6 Hz, 1H), 7.41 (s, 1H), 6.55 (s, 1H), 4.35 – 4.27 (m, 1H), 2.47 (s, 3H), 2.16 – 2.10 (m, 2H), 2.10 – 2.00 (m, 1H), 1.97 – 1.86 (m, 1H), 1.21 (s, 9H). LCMS calc. for C14H20N3O5 [M−H]+: 310.1. Found: 310.4.
PKS21213:
Synthesized following general procedure for HATU mediated amide bond formation of PKS21178 (14.9 mg, 0.05 mmol) with PKS21202 (18.0 mg, 0.05 mmol). After completion of reaction (1 h), mixture was purified by HPLC to give product (18.6 mg, 71%) as white solid. 1H NMR (500 MHz, DMSO- d6) δ 8.54 (d, J = 7.9 Hz, 1H), 8.50 (t, J = 5.8 Hz, 1H), 8.27 (d, J = 7.3 Hz, 1H), 8.16 – 8.05 (m, 2H), 7.69 – 7.59 (m, 2H), 7.54 (s, 1H), 7.42 (dd, J = 7.8, 5.7 Hz, 1H), 7.27 (dd, J = 10.5, 8.0 Hz, 1H), 6.53 (s, 1H), 4.75 – 4.63 (m, 3H), 4.34 – 4.23 (m, 1H), 2.57 (d, J = 6.7 Hz, 2H), 2.47 (s, 3H), 1.25 (d, J = 7.1 Hz, 3H), 1.15 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 171.9, 171.3, 170.0, 169.0, 158.5, 158.3, 157.3 (d, J = 249.3 Hz), 132.0 (d, J = 5.0 Hz), 130.8 (d, J = 5.2 Hz), 127.3, 126.5, 125.2 (d, J = 8.8 Hz), 123.8, 122.9 (d, J = 16.0 Hz), 120.4 (d, J = 5.9 Hz), 108.9 (d, J = 19.9 Hz), 101.3,50.2, 50.1, 48.7, 39.8, 38.0, 28.3, 18.0, 11.8. HRMS calc. for C27H32FN5O5Na [M+Na]+: 548.2280. Found: 548.2267.
PKS21272:
Synthesized following general procedure for HATU mediated amide bond formation of PKS21233 (15.6 mg, 0.05 mmol) with PKS21202 (18.0 mg, 0.05 mmol). After completion of reaction (1 h), mixture was purified by HPLC to give product (20.7 mg, 77%) as white solid. 1H NMR (500 MHz, DMSO- d6) δ 8.54 (d, J = 7.7 Hz, 1H), 8.50 (t, J = 6.0 Hz, 1H), 8.26 (d, J = 7.3 Hz, 1H), 8.12 – 8.04 (m, 2H), 7.69 – 7.60 (m, 2H), 7.45 – 7.37 (m, 2H), 7.32 – 7.24 (m, 1H), 6.54 (s, 1H), 4.76 – 4.65 (m, 2H), 4.45 – 4.38 (m, 1H), 4.38 – 4.29 (m, 1H), 2.46 (s, 3H), 2.18 – 2.05 (m, 2H), 2.03 – 1.93 (m, 1H), 1.92 – 1.81 (m, 1H), 1.24 (d, J = 7.1 Hz, 3H), 1.21 (s, 9H). 13C NMR (126 MHz, DMSO- d6) δ 172.0, 171.2, 171.2, 170.4, 158.5, 158.5, 157.4 (d, J = 248.8 Hz), 132.0 (d, J = 5.3 Hz), 130.8 (d, J = 5.3 Hz), 127.4, 126.6, 125.2 (d, J = 10.2 Hz), 123.8, 122.9 (d, J = 16.3 Hz), 120.4 (d, J = 5.4 Hz), 108.9 (d, J = 19.5 Hz), 101.3, 52.7, 49.9, 48.3, 39.8, 32.6, 28.5, 27.9, 18.3, 11.8. HRMS calc. for C28H34FN5O5Na [M+Na]+: 562.2436. Found: 562.2433.
PKS21244:
Synthesized following general procedure for HATU mediated amide bond formation of PKS21178 (14.9 mg, 0.05 mmol) with PKS21238 (19.5 mg, 0.05 mmol). After completion of reaction (1 h), mixture was purified by preparative LCMS to give product (19.7 mg, 71%) as white solid. 1H NMR (500 MHz, DMSO- d6) δ 8.62 (t, J = 5.9 Hz, 1H), 8.55 (d, J = 7.9 Hz, 1H), 8.25 (d, J = 7.7 Hz, 1H), 8.13 – 8.04 (m, 2H), 7.68 – 7.60 (m, 2H), 7.53 (s, 1H), 7.46 – 7.38 (m, 1H), 7.31 – 7.22 (m, 1H), 6.53 (s, 1H), 4.81 – 4.74 (m, 1H), 4.74 – 4.66 (m, 2H), 4.50 – 4.41 (m, 1H), 3.63 – 3.55 (m, 1H), 3.52 (dd, J = 10.2, 4.7 Hz, 1H), 3.22 (s, 3H), 2.57 (d, J = 6.7 Hz, 2H), 2.47 (s, 3H), 1.15 (s, 9H). 13C NMR (126 MHz, DMSO- d6) δ 171.4, 170.4, 169.2, 168.9, 158.5, 158.3, 157.3 (d, J = 249.3 Hz), 131.9 (d, J = 4.4 Hz), 130.6 (d, J = 3.1 Hz), 127.3, 126.5, 125.1 (d, J = 8.6 Hz), 123.8, 122.9 (d, J = 16.4 Hz), 120.4 (d, J = 5.4 Hz), 108.8 (d, J = 19.7 Hz), 101.3, 71.8, 58.3, 53.2, 50.2, 50.1, 40.0, 38.1, 28.3, 11.8. HRMS calc. for C28H34FN5O6Na [M+Na]+: 578.2385. Found: 578.2377.
PKS21245:
Synthesized following general procedure for HATU mediated amide bond formation of PKS21233 (15.6 mg, 0.05 mmol) with PKS21238 (19.5 mg, 0.05 mmol). After completion of reaction (1 h), mixture was purified by preparative LCMS to give product (22.5 g, 79%) as white solid. 1H NMR (500 MHz, DMSO- d6) δ 8.63 – 8.57 (m, 1H), 8.56 (d, J = 7.8 Hz, 1H), 8.25 (d, J = 7.8 Hz, 1H), 8.11 – 8.03 (m, 2H), 7.69 – 7.60 (m, 2H), 7.45 – 7.34 (m, 2H), 7.28 (dd, J = 10.6, 8.1 Hz, 1H), 6.54 (s, 1H), 4.72 (d, J = 5.6 Hz, 2H), 4.56 – 4.49 (m, 1H), 4.49 – 4.40 (m, 1H), 3.57 – 3.45 (m, 2H), 3.23 (s, 3H), 2.46 (s, 3H), 2.17 – 2.03 (m, 2H), 2.02 – 1.91 (m, 1H), 1.91 – 1.77 (m, 1H), 1.21 (s, 9H). 13C NMR (126 MHz, DMSO- d6) δ 171.2, 171.1, 170.8, 169.3, 158.5, 158.5, 157.4 (d, J = 248.9 Hz), 131.9, 130.5, 127.3, 126.5, 125.1 (d, J = 8.6 Hz), 123.8, 122.9 (d, J = 16.1 Hz), 120.4, 108.9 (d, J = 19.1 Hz), 101.3, 71.9, 58.2, 52.8, 52.6, 49.9, 39.9, 32.6, 28.5, 27.9, 11.8. HRMS calc. for C29H36FN5O6Na [M+Na]+: 592.2542. Found: 592.2537.
PKS2238:
PKS2233 (203.0 mg, 0.5 mmol) was dissolved in dichloromethane (6 mL) and triethylamine (140 μL, 1.0 mmol) was added. After stirring the mixture for 10 minutes at room temperature, 4-toluenesulfonyl chloride (143.0 mg, 0.75 mmol) was added. The reaction mixture was stirred 2 hours at room temperature. Dichloromethane was evaporated and the crude was dissolved in ethyl acetate. The solution was washed with water, 1N HCl followed by brine. The product was purified by column chromatography to give product (177.0 mg, 79%) as white solid. 1H NMR (500 MHz, Chloroform-d) δ 7.66 (d, J = 8.5 Hz, 2H), 7.35 – 7.29 (m, 3H), 7.20 (d, J = 8.0 Hz, 2H), 7.18 – 7.13 (m, 2H), 5.52 (s, 1H), 5.47 (d, J = 9.1 Hz, 1H), 4.91 (d, J = 12.2 Hz, 1H), 4.87 (d, J = 12.2 Hz, 1H), 3.93 – 3.84 (m, 1H), 2.38 (s, 3H), 2.33 – 2.11 (m, 3H), 1.88 – 1.77 (m, 1H), 1.35 (s, 9H). LCMS calc. for C23H32N2O5S [M+H]+: 447.2. Found 447.3.
PKS2254:
Synthesized by following the general procedure for O-debenzylation of PKS2238 (170 mg, 0.38 mmol). The product (135 mg, quant.) was isolated as white solid. 1H NMR (500 MHz, Chloroform-d) δ 7.69 (d, J = 7.9 Hz, 2H), 7.26 (d, J = 7.9 Hz, 2H), 6.15 – 5.91 (m, 2H), 3.82 – 3.69 (m, 1H), 2.39 (s, 3H), 2.35 – 2.27 (m, 1H), 2.21 – 2.12 (m, 1H), 2.11 – 2.02 (m, 1H), 1.99 – 1.88 (m, 1H), 1.31 (s, 9H). LCMS calc. for C16H23N2O5S [M−H]+: 355.1. Found: 355.0.
PKS2256:
Synthesized by following the general procedure for HATU mediated coupling of PKS2254 (29.0 mg, 0.08 mmol) and PKS2026 (27.4 mg, 0.08 mmol). The crude was purified by HPLC to give product (32.8 mg, 74%) as white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.43 (t, J = 5.7 Hz, 1H), 8.09 (d, J = 7.3 Hz, 1H), 8.04 – 7.98 (m, 1H), 7.97 – 7.90 (m, 1H), 7.86 (b, 1H), 7.84 (d, J = 8.1 Hz, 1H), 7.63 (d, J = 8.3 Hz, 2H), 7.58 – 7.49 (m, 2H), 7.49 – 7.42 (m, 1H), 7.42 – 7.39 (m, 1H), 7.38 (s, 1H), 7.30 (d, J = 7.9 Hz, 2H), 4.75 (dd, J = 15.4, 5.8 Hz, 1H), 4.69 (dd, J = 15.4, 5.6 Hz, 1H), 4.11 – 4.01 (m, 1H), 3.74 – 3.67 (m, 1H), 2.34 (s, 3H), 2.11 – 1.93 (m, 2H), 1.77 – 1.66 (m, 1H), 1.66 – 1.56 (m, 1H), 1.21 (s, 9H), 1.07 (d, J = 7.0 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 171.8, 171.0, 169.9, 142.4, 138.0, 134.3, 133.2, 130.8, 129.2, 128.5, 127.5, 126.7, 126.2, 125.8, 125.4, 125.1, 123.4, 55.8, 49.8, 48.1, 40.1, 32.3, 29.2, 28.5, 20.9, 18.1. LCMS calc. for C30H39N4O5S [M+H]+: 567.3. Found: 567.1.
PKS21206:
Synthesized following general procedure for HATU mediated amide bond formation of PKS21241 (17.1 mg, 0.05 mmol) with PKS21202 (18.0 mg, 0.05 mmol). After completion of reaction (1 h), mixture was purified by HPLC to give product (24.0 mg, 84%) as white solid. 1H NMR (500 MHz, DMSO- d6) δ 8.46 (t, J = 6.0 Hz, 1H), 8.21 (d, J = 7.2 Hz, 1H), 8.12 – 8.03 (m, 2H), 8.03 – 7.93 (m, 1H), 7.68 – 7.60 (m, 4H), 7.54 (s, 1H), 7.36 (dd, J = 7.9, 5.6 Hz, 1H), 7.32 (d, J = 7.9 Hz, 2H), 7.23 (dd, J = 10.6, 7.9 Hz, 1H), 4.72 (dd, J = 15.5, 6.1 Hz, 1H), 4.59 (dd, J = 15.5, 5.6 Hz, 1H), 4.05 – 3.95 (m, 1H), 3.91 – 3.81 (m, 1H), 2.49 – 2.45 (m, 1H), 2.35 (s, 3H), 2.29 (dd, J = 15.2, 5.5 Hz, 1H), 1.11 – 1.01 (m, 12H). 13C NMR (126 MHz, DMSO- d6) δ 171.8, 169.7, 168.7, 157.3 (d, J = 249.5 Hz), 142.4, 138.3, 131.9 (d, J = 5.2 Hz), 130.7 (d, J = 3.3 Hz), 129.2, 127.3, 126.6, 126.5, 125.0 (d, J = 8.0 Hz), 123.7, 122.9 (d, J = 16.4 Hz), 120.4 (d, J = 6.7 Hz), 108.8 (d, J = 18.4 Hz), 53.0, 50.1, 48.6, 39.7, 39.2, 28.2, 20.9, 17.4. HRMS calc. for C29H35FN4O5SNa [M+Na]+: 593.2204. Found: 593.2206.
PKS21209:
Synthesized following general procedure for HATU mediated amide bond formation of PKS2254 (17.8 mg, 0.05 mmol) with PKS21202 (18.0 mg, 0.05 mmol). After completion of reaction (1 h), mixture was purified by HPLC to give product (17.0 mg, 58%) as white solid. 1H NMR (500 MHz, DMSO- d6) δ 8.42 (t, J = 5.7 Hz, 1H), 8.12 – 8.02 (m, 3H), 7.90 – 7.78 (m, 1H), 7.70 – 7.57 (m, 4H), 7.42 – 7.33 (m, 2H), 7.32 – 7.21 (m, 3H), 4.74 – 4.62 (m, 2H), 4.08 – 3.97 (m, 1H), 3.74 – 3.63 (m, 1H), 2.34 (s, 3H), 2.09 – 1.91 (m, 2H), 1.76 – 1.65 (m, 1H), 1.65 – 1.51 (m, 1H), 1.20 (s, 9H), 1.05 (d, J = 7.0 Hz, 3H). 13C NMR (126 MHz, DMSO- d6) δ 171.9, 171.1, 170.0, 157.4 (d, J = 248.9 Hz), 142.4, 138.0, 132.0 (d, J = 5.2 Hz), 130.7 (d, J = 5.1 Hz), 129.2, 127.4, 126.7, 126.5, 125.2 (d, J = 9.0 Hz), 123.8, 122.9 (d, J = 16.3 Hz), 120.4 (d, J = 5.4 Hz), 108.9 (d, J = 18.4 Hz), 55.8, 49.8, 48.1, 39.8, 32.3, 29.2, 28.5, 20.9, 18.1. HRMS calc. for C30H37FN4O5SNa [M+Na]+: 607.2361. Found: 607.2349.
PKS21265:
Synthesized following general procedure for HATU mediated amide bond formation of PKS2254 (17.8 mg, 0.05 mmol) with PKS21238 (19.5 mg, 0.05 mmol). After completion of reaction (1 h), mixture was purified by preparative LCMS to give product (24.5 g, 80%) as white solid. 1H NMR (500 MHz, DMSO- d6) δ 8.59 – 8.48 (m, 1H), 8.11 (d, J = 7.8 Hz, 1H), 8.09 – 8.02 (m, 2H), 7.89 (s, 1H), 7.69 – 7.59 (m, 4H), 7.43 – 7.35 (m, 2H), 7.32 (d, J = 7.9 Hz, 2H), 7.30 – 7.22 (m, 1H), 4.75 – 4.64 (m, 2H), 4.31 – 4.21 (m, 1H), 3.81 – 3.70 (m, 1H), 3.31 – 3.23 (m, 2H), 3.19 (s, 3H), 2.35 (s, 3H), 2.10 – 2.00 (m, 1H), 2.00 – 1.90 (m, 1H), 1.75 – 1.64 (m, 1H), 1.64 – 1.50 (m, 1H), 1.20 (s, 9H). 13C NMR (126 MHz, DMSO- d6) δ 171.1, 170.5, 169.2, 157.4 (d, J = 248.9 Hz), 142.4, 138.0, 131.9 (d, J = 5.2 Hz), 130.5 (d, J = 4.7 Hz), 129.3, 127.4, 126.7, 126.6, 125.1 (d, J = 8.8 Hz), 123.8, 122.9 (d, J = 16.3 Hz), 120.4 (d, J = 6.8 Hz), 108.9 (d, J = 20.0 Hz), 71.7, 58.3, 55.8, 52.3, 49.9, 39.9, 32.4, 29.3, 28.5, 21.0. HRMS calc. for C31H39FN4O6SNa [M+Na]+: 637.2467. Found: 637.2464.
PKS21266:
Synthesized following general procedure for HATU mediated amide bond formation of PKS21241 (17.1 mg, 0.05 mmol) with PKS21238 (19.5 mg, 0.05 mmol). After completion of reaction (1 h), mixture was purified by preparative LCMS to give product (26.8 mg, 89%) as white solid. 1H NMR (500 MHz, DMSO- d6) δ 8.57 (t, J = 6.0 Hz, 1H), 8.20 (d, J = 7.6 Hz, 1H), 8.12 – 8.03 (m, 2H), 7.93 (d, J = 8.6 Hz, 1H), 7.68 – 7.59 (m, 4H), 7.48 (s, 1H), 7.41 – 7.36 (m, 1H), 7.32 (d, J = 7.9 Hz, 2H), 7.23 (dd, J = 10.8, 8.2 Hz, 1H), 4.73 (dd, J = 15.7, 5.9 Hz, 1H), 4.63 (dd, J = 15.7, 5.5 Hz, 1H), 4.20 – 4.07 (m, 2H), 3.53 – 3.46 (m, 1H), 3.29 – 3.23 (m, 1H), 3.20 (s, 3H), 2.44 (dd, J = 15.6, 8.2 Hz, 1H), 2.36 (s, 3H), 2.26 (dd, J = 15.6, 6.2 Hz, 1H), 1.08 (s, 9H). 13C NMR (126 MHz, DMSO- d6) δ 170.3, 169.2, 168.5, 157.3 (d, J = 248.8 Hz), 142.4, 138.3, 131.9 (d, J = 3.8 Hz), 130.6 (d, J = 3.3 Hz), 129.3, 127.2, 126.6, 126.5, 125.0 (d, J = 7.5 Hz), 123.8, 122.8 (d, J = 16.2 Hz), 120.3 (d, J = 5.3 Hz), 108.8 (d, J = 19.5 Hz), 71.5, 58.3, 53.3, 52.9, 50.1, 39.9, 39.4, 28.3, 21.0 . HRMS calc. for C30H37FN4O6SNa [M+Na]+: 623.2310. Found: 623.2307.
PKS21066:
Synthesized following general procedure for HATU mediated amide bond formation of PKS2141 (22.8 mg, 0.04 mmol) with neo-pentylamine (9.4 μL, 0.08 mmol). After completion of reaction (1 h), mixture was purified by HPLC to give product (17.8 mg, 70%) as white solid. 1H NMR (500 MHz, DMSO- d6) δ 8.59 (t, J = 5.9 Hz, 1H), 8.21 (d, J = 8.2 Hz, 1H), 8.15 (d, J = 8.0 Hz, 1H), 8.07 (d, J = 7.9 Hz, 1H), 7.94 (d, J = 7.4 Hz, 1H), 7.89 – 7.81 (m, 2H), 7.57 – 7.51 (m, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.36 (d, J = 6.9 Hz, 1H), 7.30 – 7.13 (m, 7H), 7.04 – 6.95 (m, 2H), 4.78 (dd, J = 15.3, 5.9 Hz, 1H), 4.68 (dd, J = 15.3, 5.6 Hz, 1H), 4.59 – 4.52 (m, 1H), 4.50 – 4.42 (m, 1H), 3.09 (dd, J = 13.9, 4.7 Hz, 1H), 2.87 – 2.79 (m, 2H), 2.77 – 2.70 (m, 3H), 2.55 (dd, J = 15.0, 7.1 Hz, 1H), 2.41 (dd, J = 15.0, 7.2 Hz, 1H), 2.35 (t, J = 8.0 Hz, 2H), 0.76 (s, 9H). 13C NMR (126 MHz, DMSO- d6) δ 171.3, 171.0, 170.5, 169.7, 160.9 (d, J = 240.3 Hz), 141.3, 134.2, 134.0, 133.2, 131.0 (d, J = 7.7 Hz), 130.8, 128.5, 128.3, 128.1, 127.5, 126.2, 125.8, 125.7, 125.4, 125.3, 123.4, 114.7 (d, J = 20.2 Hz), 54.3, 49.9, 49.7, 40.3, 37.5, 36.8, 36.2, 31.8, 31.0, 27.2. HRMS calc. for C38H43FN4O4Na [M+Na]+: 661.3161. Found: 661.3154.
PKS21076:
Synthesized by following general procedure for HATU coupling of Boc-Asn-OH (130 mg, 0.56 mmol) and PKS2123 (245 mg, 0.56 mmol). After completion of reaction (1 h), water was added. A white precipitate was formed. Precipitate was filtered, washed with water and dried to give product (298 mg, 99%) as white solid. Product was next step without further purification. 1H NMR (500 MHz, DMSO- d6) δ 8.55 (d, J = 5.9 Hz, 1H), 8.08 – 7.98 (m, 2H), 7.98 – 7.92 (m, 1H), 7.84 (d, J = 8.1 Hz, 1H), 7.60 – 7.49 (m, 2H), 7.47 – 7.40 (m, 1H), 7.40 – 7.28 (m, 2H), 7.28 – 7.15 (m, 2H), 7.11 – 6.97 (m, 2H), 6.97 – 6.87 (m, 2H), 4.72 (d, J = 5.6 Hz, 2H), 4.58 – 4.43 (m, 1H), 4.27 – 4.10 (m, 1H), 3.02 (dd, J = 13.9, 5.0 Hz, 1H), 2.85 (dd, J = 13.9, 8.9 Hz, 1H), 2.43 (dd, J = 15.3, 5.9 Hz, 1H), 2.32 (dd, J = 15.3, 7.9 Hz, 1H), 1.35 (s, 9H). LCMS calc. for C29H34FN4O5 [M+H]+: 537.2. Found: 537.4.
PKS21078:
Synthesized by following general procedure for boc-deprotection of PKS21076 (298 mg, 0.56 mmol). Isolated crude was triturated with diethyl ether to give a white solid. Diethyl ether was decanted and white solid was dried under high vacuum to give product (300 mg, 98%). Product was used in next step without further purification. 1H NMR (500 MHz, DMSO- d6) δ 8.74 (d, J = 8.0 Hz, 1H), 8.72 – 8.66 (m, 1H), 8.15 – 7.98 (m, 4H), 7.96 (d, J = 6.7 Hz, 1H), 7.86 (d, J = 8.2 Hz, 1H), 7.72 (s, 1H), 7.59 – 7.52 (m, 2H), 7.47 – 7.41 (m, 1H), 7.33 – 7.28 (m, 2H), 7.28 – 7.22 (m, 2H), 7.09 – 7.02 (m, 2H), 4.77 (dd, J = 15.5, 5.7 Hz, 1H), 4.69 (dd, J = 15.5, 5.4 Hz, 1H), 4.60 – 4.52 (m, 1H), 4.09 – 3.98 (m, 1H), 3.00 (dd, J = 14.1, 5.1 Hz, 1H), 2.85 – 2.72 (m, 2H), 2.58 (dd, J = 17.3, 9.0 Hz, 1H). LCMS calc. for C24H26FN4O3 [M+H]+: 437.2. Found: 437.4.
PKS21083:
Synthesized by following general procedure for HATU mediated amide bond formation of 3-phenylpropanoic acid (8.3 mg, 0.055 mmol) and PKS21078 (27.5 mg. 0.05 mmol). After completion of reaction (1 h), the mixture was purified by HPLC to give product (21.6 mg, 76%) as white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.61 – 8.49 (m, 1H), 8.16 (d, J = 8.2 Hz, 1H), 8.07 (d, J = 7.9 Hz, 2H), 7.94 (d, J = 7.5 Hz, 1H), 7.84 (d, J = 8.2 Hz, 1H), 7.61 – 7.50 (m, 2H), 7.44 (t, J = 7.6 Hz, 1H), 7.40 (s, 1H), 7.36 (d, J = 7.2 Hz, 1H), 7.29 – 7.22 (m, 2H), 7.24 – 7.13 (m, 5H), 7.03 – 6.95 (m, 2H), 6.94 (s, 1H), 4.76 (dd, J = 15.6, 5.7 Hz, 1H), 4.70 (dd, J = 15.6, 5.7 Hz, 1H), 4.61 – 4.51 (m, 1H), 4.50 – 4.41 (m, 1H), 3.08 (dd, J = 14.3, 4.5 Hz, 1H), 2.83 (dd, J = 13.9, 9.7 Hz, 1H), 2.78 – 2.70 (m, 2H), 2.57 – 2.49 (m, 1H), 2.42 – 2.29 (m, 3H). 13C NMR (126 MHz, DMSO- d6) δ 171.8, 171.3, 170.9, 170.5, 160.9 (d, J = 240.0 Hz), 141.3, 134.2, 134.0, 133.2, 130.9 (d, J = 6.3 Hz), 130.7, 128.5, 128.3, 128.1, 127.4, 126.2, 125.8, 125.7, 125.4, 125.0, 123.4, 114.7 (d, J = 21.6 Hz), 54.3, 49.5, 40.2, 37.0, 36.7, 36.2, 31.0.19F NMR (471 MHz, DMSO-d6) δ −119.31 (m). HRMS calc. for C33H33FN4O4Na [M+Na]+: 591.2378. Found: 591.2374.
IC50 Determination.
IC50 values of all compounds against hu i-20S and c-20S were performed using a 96-well format assay, as described.37
Cell-based proteasome β5 activity assay.
Karpas-1106P (22,000 cells per well) or Hela (3,000 cells per well) were plated in 384-well plate and incubated with compound at indicated concentrations for 2 h at 37 °C. The activity of the overall β5 activity including β5i and β5c in each was measured in situ after compound removal with Proteasome-Glo assay kit according to manufacturer’s instructions. Luminescence was recorded on a SpectraMax M5 plate reader. Relative percentage of RLU was used to calculate the IC50s.
Human PBMC Proliferation Assay.
IC50 values of select compounds against T cell proliferation in PBMCs were determined as reported.37
Kinetic assay of PKS21265 vs hu i-20S.
The assay was performed on a SpectraMax Gemini plate-reader from Molecular Devices (Sunnyvale, CA). 1 μL of PKS21265 at 100x indicated concentrations in DMSO were spotted at the bottom of the wells of a black wall/clear bottom 96-well plate. 99 μL of assay mixture containing 0.4 nM human i-20S and the indicated concentration of Suc-LLVY-AMC in HEPES buffer (25 mM HEPES, 0.5 mM EDTA, 0.02% Sodium dodecyl sulfate, pH 7.4) were dispensed into the designated wells. The reaction progress of each well was immediately recorded by monitoring fluorescence at Ex 360nm and Em 460 nm for 1.2 h at 37°C. Ki value for PKS21225 was derived by fitting data to equation V = (Vmax / (1 + I / (α*Ki))) * S / (KM * (1+ I / Ki) / (1 + I / (α*Ki)) + S) in Prism (GraphPad Software, Inc. La Jolla, CA).
Supplementary Material
Scheme 5.
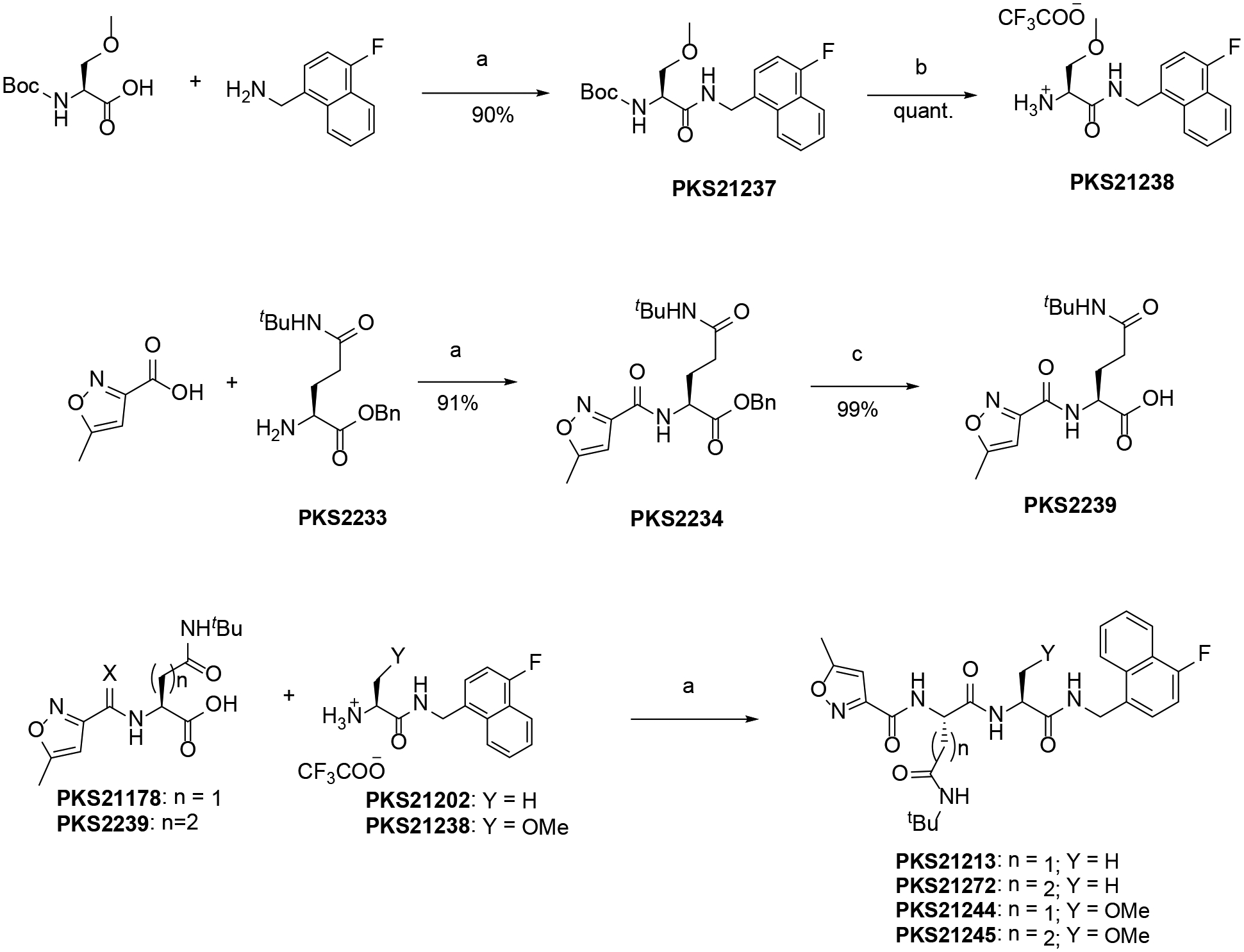
Reagents and conditions: a) HATU, HOAt, Hünig’s base, DMF; b) TFA, DCM; c) 10% Pd on carbon, methanol, hydrogen balloon
Funding Sources
This work was supported by R21 AI101393 (GL), R01AI143714 (GL), R01AI134842 (Jamil Azzi at Brigham and Women’s Hospital, G.L. subaward), Alliance for Lupus Research (Award 255848, GL), the Daedalus Fund for Innovation (GL) of Weill Cornell Medicine, the Milstein Program in Translational Medicine and Chemical Biology (CN). The Department of Microbiology and Immunology is supported by the William Randolph Hearst Foundation. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748.
ABBREVIATIONS
- i-20S
immunoproteasome
- c-20S
constitutive proteasome
Footnotes
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website at http://pubs.acs.org.
Molecular formula strings (CSV)
Synthesis and compound characterization of all new compounds and Table S1.
The authors declare the following competing financial interest(s): Cornell University has obtained a United States patent on these immunoproteasome inhibitors, patent number 9,988,421, as well as corresponding foreign patents. G. Lin, C. Nathan and X. Ma are listed as inventors on these patents.
References
- 1.Baumeister W; Walz J; Zuhl F; Seemuller E, The proteasome: paradigm of a self-compartmentalizing protease. Cell 1998, 92, 367–380. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg AL, Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans 2007, 35, 12–17. [DOI] [PubMed] [Google Scholar]
- 3.Rock KL; Gramm C; Rothstein L; Clark K; Stein R; Dick L; Hwang D; Goldberg AL, Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 1994, 78, 761–771. [DOI] [PubMed] [Google Scholar]
- 4.Liepe J; Marino F; Sidney J; Jeko A; Bunting DE; Sette A; Kloetzel PM; Stumpf MP; Heck AJ; Mishto M, A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 2016, 354, 354–358. [DOI] [PubMed] [Google Scholar]
- 5.Rock KL; York IA; Saric T; Goldberg AL, Protein degradation and the generation of MHC class I-presented peptides. Adv Immunol 2002, 80, 1–70. [DOI] [PubMed] [Google Scholar]
- 6.Neubert K; Meister S; Moser K; Weisel F; Maseda D; Amann K; Wiethe C; Winkler TH; Kalden JR; Manz RA; Voll RE, The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med 2008, 14, 748–755. [DOI] [PubMed] [Google Scholar]
- 7.Sureshkumar KK; Hussain SM; Marcus RJ; Ko TY; Khan AS; Tom K; Vivas CA; Parris G; Jasnosz KM; Thai NL, Proteasome inhibition with bortezomib: an effective therapy for severe antibody mediated rejection after renal transplantation. Clin Nephrol 2012, 77, 246–253. [DOI] [PubMed] [Google Scholar]
- 8.Moreno Gonzales MA; Gandhi MJ; Schinstock CA; Moore NA; Smith BH; Braaten NY; Stegall MD, 32 Doses of Bortezomib for Desensitization Is Not Well Tolerated and Is Associated With Only Modest Reductions in Anti-HLA Antibody. Transplantation 2017, 101, 1222–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin G; Li D; Chidawanyika T; Nathan C; Li H, Fellutamide B is a potent inhibitor of the Mycobacterium tuberculosis proteasome. Arch Biochem Biophys 2010, 501, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu HC; Singh PK; Fan H; Wang R; Sukenick G; Nathan C; Lin G; Li H, Structural Basis for the Species-Selective Binding of N,C-Capped Dipeptides to the Mycobacterium tuberculosis Proteasome. Biochemistry 2017, 56, 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin G; Tsu C; Dick L; Zhou XK; Nathan C, Distinct specificities of Mycobacterium tuberculosis and mammalian proteasomes for N-acetyl tripeptide substrates. J Biol Chem 2008, 283, 34423–34431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin G; Li D; de Carvalho LP; Deng H; Tao H; Vogt G; Wu K; Schneider J; Chidawanyika T; Warren JD; Li H; Nathan C, Inhibitors selective for mycobacterial versus human proteasomes. Nature 2009, 461, 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhan W; Visone J; Ouellette T; Harris JC; Wang R; Zhang H; Singh PK; Ginn J; Sukenick G; Wong TT; Okoro JI; Scales RM; Tumwebaze PK; Rosenthal PJ; Kafsack BFC; Cooper RA; Meinke PT; Kirkman LA; Lin G, Improvement of asparagine ethylenediamines as anti-malarial Plasmodium-selective proteasome inhibitors. J Med Chem 2019, 62, 6137–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoo E; Stokes BH; de Jong H; Vanaerschot M; Kumar T; Lawrence N; Njoroge M; Garcia A; Van der Westhuyzen R; Momper JD; Ng CL; Fidock DA; Bogyo M, Defining the determinants of specificity of Plasmodium proteasome inhibitors. J Am Chem Soc 2018, 140, 11424–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirkman LA; Zhan W; Visone J; Dziedziech A; Singh PK; Fan H; Tong X; Bruzual I; Hara R; Kawasaki M; Imaeda T; Okamoto R; Sato K; Michino M; Alvaro EF; Guiang LF; Sanz L; Mota DJ; Govindasamy K; Wang R; Ling Y; Tumwebaze PK; Sukenick G; Shi L; Vendome J; Bhanot P; Rosenthal PJ; Aso K; Foley MA; Cooper RA; Kafsack B; Doggett JS; Nathan CF; Lin G, Antimalarial proteasome inhibitor reveals collateral sensitivity from intersubunit interactions and fitness cost of resistance. Proc Natl Acad Sci U S A 2018, 115, E6863–E6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H; O’Donoghue AJ; van der Linden WA; Xie SC; Yoo E; Foe IT; Tilley L; Craik CS; da Fonseca PC; Bogyo M, Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 2016, 530, 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wyllie S; Brand S; Thomas M; De Rycker M; Chung CW; Pena I; Bingham RP; Bueren-Calabuig JA; Cantizani J; Cebrian D; Craggs PD; Ferguson L; Goswami P; Hobrath J; Howe J; Jeacock L; Ko EJ; Korczynska J; MacLean L; Manthri S; Martinez MS; Mata-Cantero L; Moniz S; Nuhs A; Osuna-Cabello M; Pinto E; Riley J; Robinson S; Rowland P; Simeons FRC; Shishikura Y; Spinks D; Stojanovski L; Thomas J; Thompson S; Viayna Gaza E; Wall RJ; Zuccotto F; Horn D; Ferguson MAJ; Fairlamb AH; Fiandor JM; Martin J; Gray DW; Miles TJ; Gilbert IH; Read KD; Marco M; Wyatt PG, Preclinical candidate for the treatment of visceral leishmaniasis that acts through proteasome inhibition. Proc Natl Acad Sci U S A 2019, 116, 9318–9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khare S; Nagle AS; Biggart A; Lai YH; Liang F; Davis LC; Barnes SW; Mathison CJ; Myburgh E; Gao MY; Gillespie JR; Liu X; Tan JL; Stinson M; Rivera IC; Ballard J; Yeh V; Groessl T; Federe G; Koh HX; Venable JD; Bursulaya B; Shapiro M; Mishra PK; Spraggon G; Brock A; Mottram JC; Buckner FS; Rao SP; Wen BG; Walker JR; Tuntland T; Molteni V; Glynne RJ; Supek F, Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim MS; Pinto SM; Getnet D; Nirujogi RS; Manda SS; Chaerkady R; Madugundu AK; Kelkar DS; Isserlin R; Jain S; Thomas JK; Muthusamy B; Leal-Rojas P; Kumar P; Sahasrabuddhe NA; Balakrishnan L; Advani J; George B; Renuse S; Selvan LD; Patil AH; Nanjappa V; Radhakrishnan A; Prasad S; Subbannayya T; Raju R; Kumar M; Sreenivasamurthy SK; Marimuthu A; Sathe GJ; Chavan S; Datta KK; Subbannayya Y; Sahu A; Yelamanchi SD; Jayaram S; Rajagopalan P; Sharma J; Murthy KR; Syed N; Goel R; Khan AA; Ahmad S; Dey G; Mudgal K; Chatterjee A; Huang TC; Zhong J; Wu X; Shaw PG; Freed D; Zahari MS; Mukherjee KK; Shankar S; Mahadevan A; Lam H; Mitchell CJ; Shankar SK; Satishchandra P; Schroeder JT; Sirdeshmukh R; Maitra A; Leach SD; Drake CG; Halushka MK; Prasad TS; Hruban RH; Kerr CL; Bader GD; Iacobuzio-Donahue CA; Gowda H; Pandey A, A draft map of the human proteome. Nature 2014, 509, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basler M; Lauer C; Groettrup M, Prevention of autoimmune diabetes by a selective inhibitor of the immunoproteasome. Wien Klin Wochenschr 2008, 120, 165–165. [Google Scholar]
- 21.Basler M; Mundt S; Muchamuel T; Moll C; Jiang J; Groettrup M; Kirk CJ, Inhibition of the immunoproteasome ameliorates experimental autoimmune encephalomyelitis. EMBO Mol Med 2014, 6, 226–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ichikawa HT; Conley T; Muchamuel T; Jiang J; Lee S; Owen T; Barnard J; Nevarez S; Goldman BI; Kirk CJ; Looney RJ; Anolik JH, Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum 2012, 64, 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalim KW; Basler M; Kirk CJ; Groettrup M, Immunoproteasome subunit LMP7 deficiency and inhibition suppresses Th1 and Th17 but enhances regulatory T cell differentiation. J Immunol 2012, 189, 4182–4193. [DOI] [PubMed] [Google Scholar]
- 24.Muchamuel T; Basler M; Aujay MA; Suzuki E; Kalim KW; Lauer C; Sylvain C; Ring ER; Shields J; Jiang J; Shwonek P; Parlati F; Demo SD; Bennett MK; Kirk CJ; Groettrup M, A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med 2009, 15, 781–787. [DOI] [PubMed] [Google Scholar]
- 25.Muchamuel T; Jiang J; Lee SJ; Kirk CJ, ONX-0914, A selective inhibitor of the immunoproteasome, protects against experimental autoimmune encephalomyelitis and suppresses the TH17 Response. Neurology 2011, 76, A459–A460. [Google Scholar]
- 26.Vachharajani N; Joeris T; Luu M; Hartmann S; Pautz S; Jenike E; Pantazis G; Prinz I; Hofer MJ; Steinhoff U; Visekruna A, Prevention of colitis-associated cancer by selective targeting of immunoproteasome subunit LMP7. Oncotarget 2017, 8, 50447–50459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kasam V; Lee NR; Kim KB; Zhan CG, Selective immunoproteasome inhibitors with non-peptide scaffolds identified from structure-based virtual screening. Bioorg Med Chem Lett 2014, 24, 3614–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kisselev AF; Groettrup M, Subunit specific inhibitors of proteasomes and their potential for immunomodulation. Curr Opin Chem Biol 2014, 23C, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee W; Kim KB, The Immunoproteasome: An emerging therapeutic target. Curr Top Med Chem 2011, 11, 2923–2930. [DOI] [PubMed] [Google Scholar]
- 30.Basler M; Groettrup M, Immunoproteasome-specific inhibitors and their application. Methods Mol Biol 2012, 832, 391–401. [DOI] [PubMed] [Google Scholar]
- 31.Dubiella C; Cui H; Gersch M; Brouwer AJ; Sieber SA; Kruger A; Liskamp RM; Groll M, Selective inhibition of the immunoproteasome by ligand-induced crosslinking of the active site. Angew Chem Int Ed Engl 2014, 53, 11969–11973. [DOI] [PubMed] [Google Scholar]
- 32.Sosic I; Gobec M; Brus B; Knez D; Zivec M; Konc J; Lesnik S; Ogrizek M; Obreza A; Zigon D; Janezic D; Mlinaric-Rascan I; Gobec S, Nonpeptidic selective inhibitors of the chymotrypsin-like (beta5 i) subunit of the immunoproteasome. Angew Chem Int Ed Engl 2016, 55, 5745–5748. [DOI] [PubMed] [Google Scholar]
- 33.Cui H; Baur R; Le Chapelain C; Dubiella C; Heinemeyer W; Huber EM; Groll M, Structural elucidation of a non-peptidic inhibitor specific for the human immunoproteasome. Chembiochem 2017, 18, 523–526 [DOI] [PubMed] [Google Scholar]
- 34.Evans DC; Watt AP; Nicoll-Griffith DA; Baillie TA, Drug-protein adducts: an industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chem Res Toxicol 2004, 17, 3–16. [DOI] [PubMed] [Google Scholar]
- 35.Sula Karreci E; Fan H; Uehara M; Mihali AB; Singh PK; Kurdi AT; Solhjou Z; Riella LV; Ghobrial I; Laragione T; Routray S; Assaker JP; Wang R; Sukenick G; Shi L; Barrat FJ; Nathan CF; Lin G; Azzi J, Brief treatment with a highly selective immunoproteasome inhibitor promotes long-term cardiac allograft acceptance in mice. Proc Natl Acad Sci U S A 2016, 113, E8425–E8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan H; Angelo NG; Warren JD; Nathan CF; Lin G, Oxathiazolones selectively inhibit the human immunoproteasome over the constitutive proteasome. ACS Med Chem Lett 2014, 5, 405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh PK; Fan H; Jiang X; Shi L; Nathan CF; Lin G, Immunoproteasome beta5i-selective dipeptidomimetic inhibitors. ChemMedChem 2016, 11, 2127–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santos RLA; Bai L; Singh PK; Murakami N; Fan H; Zhan W; Zhu Y; Jiang X; Zhang K; Assker JP; Nathan CF; Li H; Azzi J; Lin G, Structure of human immunoproteasome with a reversible and noncompetitive inhibitor that selectively inhibits activated lymphocytes. Nat Commun 2017, 8, 1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin G; Chidawanyika T; Tsu C; Warrier T; Vaubourgeix J; Blackburn C; Gigstad K; Sintchak M; Dick L; Nathan C, N,C-Capped dipeptides with selectivity for mycobacterial proteasome over human proteasomes: role of S3 and S1 binding pockets. J Am Chem Soc 2013, 135, 9968–9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blackburn C; Gigstad KM; Hales P; Garcia K; Jones M; Bruzzese FJ; Barrett C; Liu JX; Soucy TA; Sappal DS; Bump N; Olhava EJ; Fleming P; Dick LR; Tsu C; Sintchak MD; Blank JL, Characterization of a new series of noncovalent proteasome inhibitors with exquisite potency and selectivity for the 20S beta5-subunit. Biochem J 2010, 430, 461–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stein RL; Melandri F; Dick L, Kinetic characterization of the chymotryptic activity of the 20S proteasome. Biochemistry 1996, 35, 3899–3908. [DOI] [PubMed] [Google Scholar]
- 42.Roccaro AM; Sacco A; Aujay M; Ngo HT; Azab AK; Azab F; Quang P; Maiso P; Runnels J; Anderson KC; Demo S; Ghobrial IM, Selective inhibition of chymotrypsin-like activity of the immunoproteasome and constitutive proteasome in Waldenstrom macroglobulinemia. Blood 2010, 115, 4051–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ladi E; Everett C; Stivala CE; Daniels BE; Durk MR; Harris SF; Huestis MP; Purkey HE; Staben ST; Augustin M; Blaesse M; Steinbacher S; Eidenschenk C; Pappu R; Siu M, Design and evaluation of highly selective human immunoproteasome inhibitors reveal a compensatory process that preserves immune cell viability. J Med Chem 2019, 62, 7032–7041. [DOI] [PubMed] [Google Scholar]
- 44.Weyburne ES; Wilkins OM; Sha Z; Williams DA; Pletnev AA; de Bruin G; Overkleeft HS; Goldberg AL; Cole MD; Kisselev AF, Inhibition of the proteasome beta2 site sensitizes triple-negative breast cancer cells to beta5 inhibitors and suppresses nrf1 activation. Cell Chem Biol 2017, 24, 218–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Besse A; Besse L; Kraus M; Mendez-Lopez M; Bader J; Xin BT; de Bruin G; Maurits E; Overkleeft HS; Driessen C, Proteasome inhibition in multiple myeloma: head-to-head comparison of currently available proteasome inhibitors. Cell Chem Biol 2019, 26, 340–351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.