

Abstract
Protein kinase C family members are multi-domain proteins whose function is exquisitely tuned by inter-domain interactions that control the spatiotemporal dynamics of their signaling. Despite extensive mechanistic studies on this family of enzymes, no structure of a full-length enzyme that includes all domains has been solved. Here we take into account the biochemical mechanisms that control autoinhibition, the properties of each individual domain, and previous structural studies to propose a unifying model for the general architecture of protein kinase C family members. This model shows how the C2 domains of conventional and novel protein kinase C isozymes, which have different topologies and different positions in the primary structure, can occupy the same position in the tertiary structure of the kinase. This common architecture of conventional and novel protein kinase C isozymes provides a framework for understanding how disease-associated mutations impair PKC function.
Keywords: protein kinase C, C1 domain, C2 domain
Text:
Protein kinase C is a multi-module Ser/Thr protein kinase that transduces the abundance of signals resulting in phospholipid hydrolysis (1). Binding of diacylglycerol (DG) allosterically and reversibly activates these enzymes to effect their major role in maintaining cellular homeostasis. This allosteric regulation is precisely controlled by multiple mechanisms that ensure that the enzyme is only active for specific times at specific locations in response to specific signals. Deregulation of any of these mechanisms result in pathophysiologies, with loss-of-function associated with cancer and enhanced function associated with degenerative disease (2). Maintaining autoinhibition is critical to the function of PKC.
DG-sensing PKC isozymes fall into one of two classes: conventional PKCs (cPKC: α, β, γ) have a low-affinity DG-sensing C1B domain and require a Ca2+ sensor (C2 domain) for activation, whereas novel PKCs (nPKC: δ, ε, θ, η) have a high-affinity DG sensing C1B domain and are not regulated by Ca2+. Both subtypes share a common architecture of an N-terminal regulatory moiety containing allosteric sensors that constrain the catalytic activity of a C-terminal kinase domain (Figure 1A). This is achieved by an autoinhibitory pseudosubstrate segment that occupies the substrate-binding cavity in the inactive conformation. The pseudosubstrate is immediately followed by tandem C1 domains (C1A and C1B) which serve as DG sensors, although only the C1B binds DG in the context of the ‘mature’ protein (3). cPKCs have a Ca2+-sensing C2 domain which targets these PKCs to the plasma membrane via a recognition motif for phosphatidylinositol-4,5-bisphosphate (PIP2), a lipid localized to the plasma membrane. The nPKCs also have a C2 domain but, curiously, it has a different topology (4), lacks residues that coordinate Ca2+, and precedes the pseudosubstrate-C1A-C1B segment. The kinase domain is followed by a C-Tail that is a hallmark regulatory region of AGC kinases and serves as docking site for regulatory proteins (5). Understanding the mechanism of autoinhibition of PKC would open avenues for allosteric modulators to treat deregulated signaling in disease.
Figure 1.
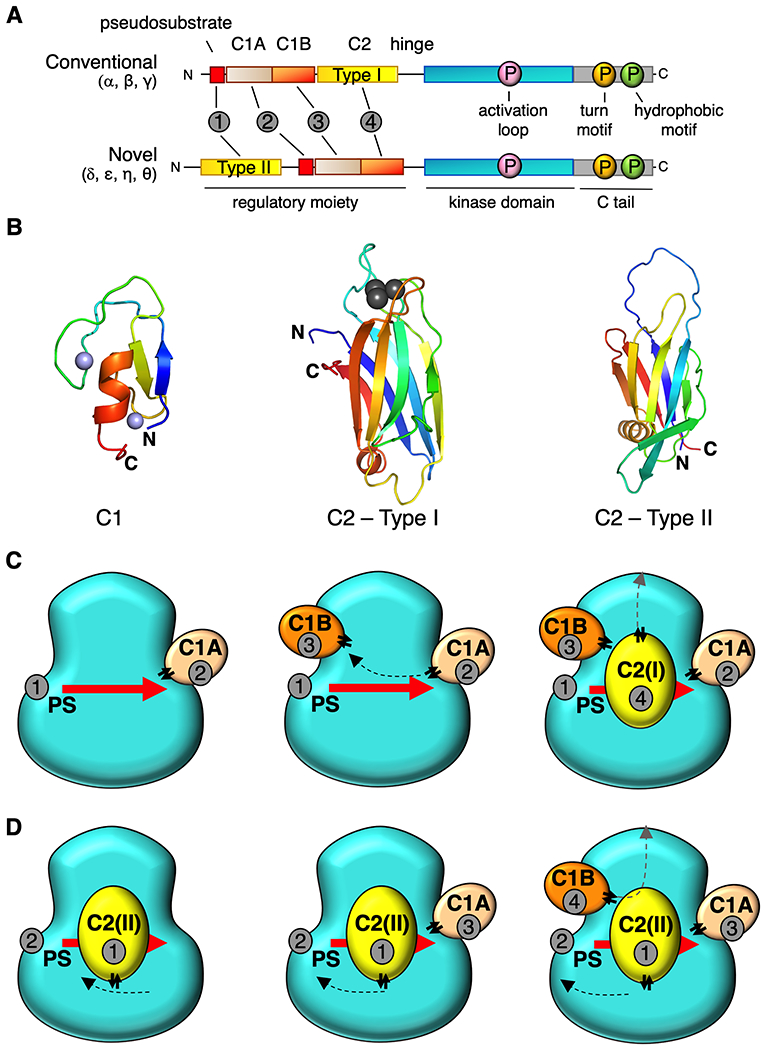
Proposed architecture for conventional and novel PKC isozymes. (A) Primary structure and domain composition of conventional and novel PKC isoforms showing pseudosubstrate (PS, red), C1A domain (sand), C1B domain (orange), C2 domain (yellow), kinase (cyan), and C-tail (grey). Three processing phosphorylations indicated in magenta, orange, and green for the activation loop, turn motif, and hydrophobic motif, respectively. Order of domains indicated by grey circles. (B) Structure of C1 domain (PKCδ, PDB ID code 2YUU)) and two types of C2 domain: Type1 C2 (PKCβ PDB ID code 1A25) in conventional PKCs and Type 2 C2 (PKCθ PDB ID code 2ENJ) in novel PKCs highlighting different topology but same overall architecture. Notably, the N and C termini are on opposite ends of each the two types of C2. Zn2+ ions (C1 domains) in purple and Ca2+ ions (Type I C2 domain) in grey (C) Proposed positions of each domain on the kinase module for conventional PKCs and novel PKCs (D) showing common tertiary structure. Domains are numbered in the order they appear in the primary structure in (A). Double arrows indicate positions of the “plugs”.
The key role of the pseudosubstrate as an autoinhibitory segment that binds the substrate-binding cavity of the kinase domain was identified by Kemp and colleagues over three decades ago (6). While this segment is necessary for the autoinhibition of all PKC family members, numerous studies in the ensuing years have revealed it is not sufficient (7–10). Rather, all domains in the regulatory module participate in a network of interactions to maintain autoinhibition. Of these, the C1A domain is the most critical: the binding affinity of the isolated regulatory module (see Figure 1A) for the isolated kinase domain is reduced by three orders of magnitude upon deletion of the C1A domain, but only about 20-fold upon deletion of the C1B or C2 domains (11). Thus, whereas all modules participate in a network of interactions to maintain autoinhibition, the pseudosubstate-C1A is the dominant autoinhibitory module. But what is this network of interactions?
Unraveling the structure of full-length PKC and demonstrating how these domains communicate with the kinase core has been challenging, likely because these molecules are intrinsically highly dynamic. While the structures of each of the domains of PKC from multiple family members have been solved, a full-length structure has remained elusive. A partial crystal structure of PKCβII has been reported, however there was insufficient electron density to resolve all the domains (12). Previous SAXS for two conventional (PKCα and βII) and of two novel PKC (δ and η) revealed similar scattering curves, with a radius of gyration of 33-34 Å calculated for both classes of isozymes (13). Ab initio calculations of molecular envelopes showed similar globular shapes, suggesting a conserved tertiary structure of both types of PKC. This raised the intriguing question of how the same 3D architecture could be obtained given the different topologies of the C2 domains and their different order in the primary sequence of conventional vs. novel PKC isozymes. Reasoning that contact residues at the regulatory domain:kinase interface would be conserved amongst isozymes, Leonard and colleagues recently proposed potential interaction surfaces of each domain with the kinase (13). However, a model of the architecture for full-length PKC was not proposed.
Here we considered the extensive mechanistic information on PKC, the existing structural data, and the properties of individual domains to guide molecular docking of each of the domains on kinase domain. We paid particular attention to the ‘plug in’ property of each regulatory module: the N and C termini are on the same face of the domain, in close proximity (Figure 1B). Therefore, they can be ‘plugged’ into specific sockets of the structure (14, 15). The proximity of the N and C termini is a well-studied phenomenon and is an important feature of protein domain evolution (16, 17). This property immediately suggested that both types of C2 domain could occupy the same position on the kinase domain but their ‘plugs’ would be on opposite sides. This would provide an explanation for why they connect to different modules in the primary structure (hinge leading to kinase domain for cPKC and pseudosubstrate for nPKC; Figure 1A).
The schematic in Figure 1C illustrates the rationale for a general docking of the domains for conventional PKCs. The pseudosubstrate peptide binds into the active site of the kinase with its C-terminus directed towards the C-helix. The linker between the P+1 residue and the C1A domain is only 9 residues long, therefore, we reasoned that it may be positioned at the C-helix surface of the kinase. Positioning of the C1B domain was the major challenge as the proposed position of C1A is incompatible with the previously published structure of PKCβII (12). As the linker between C1A and C1B is only 15 residues long, it is impossible to circumvent the N-lobe of the kinase and connect it to the N-terminus of C1B that was positioned behind the C-terminal tail. The latter contains an important regulatory NFD-helix that was shown to be in a direct contact with the C1B domain of PKCβII (12). If we consider that the N and C termini of the C1A domain are in close proximity, it would be logical to suggest that the C1A-C1B linker must be antiparallel to the pseudosubstrate peptide. Following this logic, we positioned the C1B domain near the N-terminus of the pseudosubstrate, next to the NFD-helix in the C-terminal tail, albeit at its “front” side, opposite to the position proposed earlier (12) (Figure 2, compare A and D). To position the C2 domain we followed our biochemically validated model that suggests binding of the C2 over the pseudosubstrate region covering the active site of the kinase (18). As the N-terminus of the C2 must be close to the C-terminus of C1B (6 residue linker), we concluded that the C2 “plug” must be facing the N-lobe of the kinase, as shown in Figure 1C. This position would satisfy not only the constraints for the connection between the C1B and C2 domains, but would easily accommodate a long linker between the C2 and the N-terminus of the kinase domain.
Figure 2.
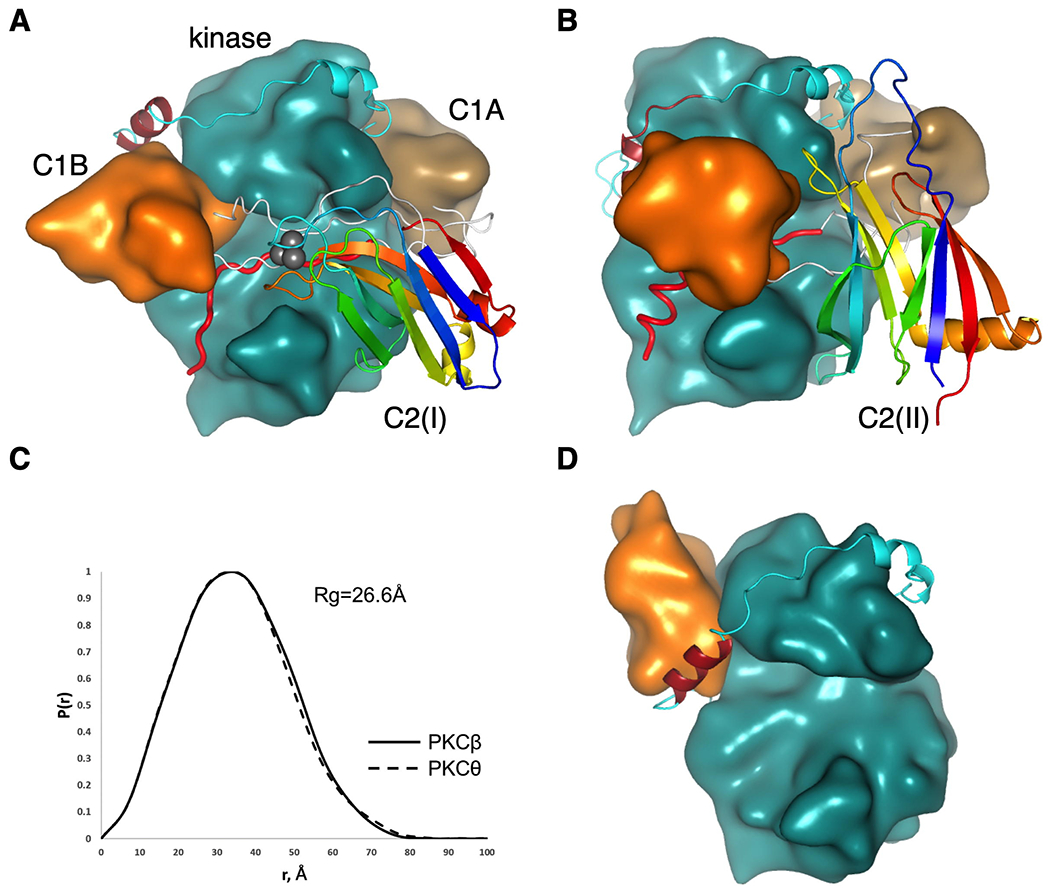
Conserved architecture of conventional and novel PKC isozymes. (A) Proposed structure of PKCβII highlighting positions of the C1B (orange), NFD helix (burgundy) in the kinase domain (cyan), C1A domain (sand) and C2 domain (ribbon diagram colored from blue (N terminus) to red (C terminus) to indicate topology; Ca2+ ions in grey). Linkers connecting domains are shown in white with the pseudosubstrate (red) positioned in the active site. (B) Proposed structure of PKCθ, colored as in (A) showing inverted C2-domain topology yet same overall structure. (C) Pair distance distribution function P(r) computed from the proposed models showing similar shapes and radii of gyration of conventional PKCβ and novel PKCθ. (D) Previously reported structure of PKCβII (12) (pdb:3PFQ) showing C1B domain (orange) positioned on top of NFD helix (burgundy).
With this domain positioning in mind, we discovered that the same logic applied to the novel PKCs results in a structure with the same overall geometry (Figure 1D). As the C2 in this case precedes the pseudosubstrate and the “plug” of the C2 has to be reversed with respect to the conventional C2 domains, its C-terminus should face the C-lobe of the kinase and, thus, can be easily connected the N-terminus of the pseudosubstrate. Positioning of the latter and the two C1 domains can be the same as in the previous case with the only difference that the C-terminus of the C1B will lead towards the N-terminus of the kinase domain instead of the C2 domain.
We then proceeded to dock each domain on the structure of PKC. We used the ClusPro protein docking server (19) with spatial restrictions to achieve positions close to the proposed models (the restrained residues had to be within 15Å proximity). For the docking of the C1A of PKCβ, the restriction was applied to Phe43, which was suggested to be at a protein-protein binding interface (13). For the C1B docking in PKCβ, Asp116 was restricted to a close proximity to the NFD helix as it was also considered to be a conserved C1B-specific residue (13). The C2 domain docking was based on our previous model with the correction to the “plug” orientation (18). Flexible linkers between the pseudosubstrate, the C1 domains and the C2 domain were generated using UCSF Chimera (20) and Modeller (21). The model for PKCθ was created using the PKCβ model as a template using PKCθ known domain structures.
Discussion
Here we present a unifying model for both conventional and novel PKCs that accounts for the inverted topology and order of the Type I vs Type II C2 domains. This model is consistent with existing biochemical data (Table 1) (11–13, 18, 22) and defines new interfaces that can now be verified experimentally. This conserved architecture not only underscores the role of each domain in tethering the pseudosubstrate in the kinase domain, but also draws attention to linker regions as playing key roles in maintaining the autoinhibited conformation.
Table 1.
Comparison of existing biochemical data with proposed model predictions. Disruption of autoinhibition upon mutation of the indicated residues in conventional or novel PKC isozymes was determined experimentally either by assessing increased membrane translocation (11-13) or increased basal activity (18) in cells, or increased cofactor-independent activity of pure protein in vitro (22). Mutated residues were examined in the new structural model and predictions were made about inter- and intradomain interactions that may affect autoinhibition. Residues are numbered according to PKCβII.
| Domain | Residue | Isozyme(s) | Disrupted Autoinhibition? | Prediction with New Model |
|---|---|---|---|---|
| C1A | R42 | α | YES11 | Interaction with E655 in C-Tail |
| F43 | α, βII,δ, η, θ | YES11,13 | Interaction with F656, F659, or F661 in C-Tail | |
| T54 | βII | NO13 | No predicted interactions | |
| D55 | α, βII | YES11,13 | Interaction with R76, domain unfolding | |
| F72 | α | YES11 | interaction with F84, domain unfolding | |
| C1B | T108 | βII | NO13 | No predicted interactions |
| L125 | βII | NO12* | Disrupt phorbol-binding pocket | |
| C2 | K236 | βII | NO22 | No predicted interactions |
| R238 | βII | NO22 | No predicted interactions | |
| Kinase | L358 | α, βII, δ, η, θ | YES12,13 | Could interact with Y422 or M145 |
| L367 | βII | YES12 | L367D could unfold N-lobe due to DE(D) negative patch | |
| D382 | βII | YES18 | Interaction with K91 (between C1A and C1B) | |
| Y422 | βII | YES12 | Could interact with L358 / in nucleotide binding region | |
| Y430 | βII | YES12 | Could pi-stack with H140 in C1B | |
| C-Tail | F629 | α, βII, δ, η, θ | YES12,13/NO18 | No predicted interactions |
| F633 | α, βII, δ, η, θ | YES12,13 | Could pi-stack with F114 in C1B | |
| E655 | βII | YES18 | Interaction with R42 in C1A |
Phorbol-induced membrane translocation impaired
The estimated radius of gyration for our models of both conventional and novel PKC isozymes estimated by CRYSOL and GNOM programs from ATSAS 2.8 package (23) is the same (Figure 2C), consistent with experimental observations (13). At 27 □ it is slightly smaller than the reported 33-34 □ (13). The maximum dimension of our models (around 80 □) is also smaller than the experimentally measured 110-115 □ (13); this difference can be explained by the absence of the large linker between the C2 and the kinase domain in PKCβ and between C1B and the kinase domain in PKCθ in our models.
The proposed model for the tertiary structure of conventional and novel PKC isozymes differs in several important aspects from the partial crystal structure of PKCβII (12) (Figure 2D) and a re-interpretation of this structure (18). First, the C1B in our model binds to the NFD-helix at the side proximal to the active site of the kinase, whereas Hurley and colleagues placed it on the back-side positioned to clamp on to Phe629 of the NFD helix. In our model, Phe629 is not predicted to interact with the C1B. Consistent with this lack of interaction, mutation of this residue in PKCβII was shown to have no detectable effect on the basal activity or activation in cells (18). Instead, our model positions the C1B to interact with Phe633 in the C-Tail. Indeed mutation of Phe633 has been shown to increase the rate of membrane translocation of PKCβII, consistent with reduced autoinhibition (13). Furthermore, Biondi and colleagues found a similar position for the C1A domain of atypical PKCs based on 1H/2H exchange, which showed that the C1A domain also clamps over the αC helix and hydrophobic motif segment of the C-tail (24). In a re-interpretation of the crystal packing (18), the C2 domain was proposed to clamp over the pseudosubstrate to stabilize the autoinhibited conformation. In our new model, the C2 domain occupies the same overall position to clamp the pseudosubstrate in place but is rotated. Residues in the C2 domain previously proposed to be at the kinase interface, Lys205 and Lys209 (18), are also predicted to be at the C2:kinase interface in our new model. These basic residues, which are part of a basic PIP2-sensing face (25, 26), were proposed to interact with Asp382 in the kinase domain and Glu655 in the C-Tail: mutation of either of these acidic residues to Lys resulted in an accelerated rate of membrane translocation, consistent with unmasking of the regulatory domains and reduced autoinhibition. While co-mutation of the ‘partner’ basic residues in the C2 domain to acidic residues (to reverse the predicted ion pairs) reduced the rate of membrane translocation back to that observed for wild-type enzyme, the C2 mutations disabled the plasma membrane ‘sensor’ confounding interpretation. In the current model, Asp382 is predicted to bind a conserved Lys in the linker between the C1A and C1B, and Glu655 is predicted to interact with a conserved Arg in the C1A (Table 1). Our model is also consistent with the conserved residues in conventional and novel PKC isozymes proposed by Lucic et al. (13) as mediating interdomain interactions. For example, Phe43 in the C1A is predicted to interact with the C-Tail and Leu358 in the kinase domain could interact with Met145 or Tyr422.
An important feature of this model is the role of the linker regions in coordinating domain interactions. The C1B-C2 linker has previously been shown to contribute to autoinhibition (9); whereas a GST-fusion of the regulatory moiety of PKCα effectively inhibited the activity of full-length PKCα, mutation of residues in this linker to a stretch of Ala decreased the ability of a the regulatory domain to inhibit PKCα. In our model of PKCβII, the C1A-C1B linker crisscrosses over the pseudosubstrate to reinforce its interaction in the substrate binding cavity. Interactions between the conserved Lys91 in the C1A-C1B linker with Asp382 in the kinase domain may provide an additional layer of autoinhibition. Thus, the pseudosubstrate is secured in the substrate-binding cavity by the C2 domain and by linkers that weave over the bound pseudosubstrate.
In a physiological context, aberrant PKC that is not properly autoinhibited is unstable and shunted to degradation. This property of PKC provides a major mechanism by which cancer-associated mutations in PKC isozymes are loss-of-function (27): perturbation of interdomain surfaces that activate the enzyme are paradoxically loss-of-function because the aberrant PKC is degraded by quality control mechanisms (28). Indeed, the pseudosubstrate of PKCβ is a functional hotspot for mutations (29), either because mutation of a residue strengthens the affinity for the pseudosubstrate for the kinase (resulting in decreased activity) or because the mutation decreases autoinhibition to promote the degradation of the enzyme and lower steady-state levels (28). Our model accounts for why cancer-associated mutations occur throughout the domain structure of the enzyme and exemplify the exquisite conformational control driven by a coordinated network of domains and linkers.
Acknowledgements:
This work was supported by NIH R03 TR002914 (A.C.N.) and R35 GM122523 (A.C.N). A.C.J. was supported in part by the University of California, San Diego, Graduate Training Program in Cellular and Molecular Pharmacology (T32 GM007752) and the National Science Foundation Graduate Research Fellowship (#DGE-1650112). This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. (DGE-1650112). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. The authors declare no conflicts of interest.
Abbreviations:
- DG
diacylglycerol
- PIP2
phosphatidylinositol-4,5-bisphosphate
- PKC
protein kinase C
- cPKC
conventional protein kinase C
- nPKC
novel protein kinase C
- SAXS
small angle X-ray scattering
References
- 1.Nishizuka Y (1995) Protein kinase C and lipid signaling for sustained cellular responses. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 9(7):484–496. [PubMed] [Google Scholar]
- 2.Newton AC (2018) Protein kinase C: perfectly balanced. Critical reviews in biochemistry and molecular biology 53(2):208–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antal CE, Violin JD, Kunkel MT, Skovso S, & Newton AC (2014) Intramolecular Conformational Changes Optimize Protein Kinase C Signaling. Chemistry & biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nalefski EA & Falke JJ (1996) The C2 domain calcium-binding motif: structural and functional diversity. Prot. Sci. 5:2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kannan N, Haste N, Taylor SS, & Neuwald AF (2007) The hallmark of AGC kinase functional divergence is its C-terminal tail, a cis-acting regulatory module. Proc Natl Acad Sci U S A 104(4):1272–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.House C & Kemp BE (1987) Protein Kinase C Contains a Pseudosubstrate Prototope in Its Regulatory Domain. Science 238:1726–1728. [DOI] [PubMed] [Google Scholar]
- 7.Parissenti AM, Kirwan AF, Kim SA, Colantonio CM, & Schimmer BP (1998) Inhibitory properties of the regulatory domains of human protein kinase Calpha and mouse protein kinase Cepsilon. The Journal of biological chemistry 273(15):8940–8945. [DOI] [PubMed] [Google Scholar]
- 8.Lopez-Garcia LA, et al. (2011) Allosteric regulation of protein kinase PKCzeta by the N-terminal C1 domain and small compounds to the PIF-pocket. Chemistry & biology 18(11):1463–1473. [DOI] [PubMed] [Google Scholar]
- 9.Kirwan AF, et al. (2003) Inhibition of protein kinase C catalytic activity by additional regions within the human protein kinase Calpha-regulatory domain lying outside of the pseudosubstrate sequence. The Biochemical journal 373(Pt 2):571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slater SJ, et al. (2002) Regulation of PKC alpha activity by C1-C2 domain interactions. The Journal of biological chemistry 277(18):15277–15285. [DOI] [PubMed] [Google Scholar]
- 11.Sommese RF, Ritt M, Swanson CJ, & Sivaramakrishnan S (2017) The Role of Regulatory Domains in Maintaining Autoinhibition in the Multidomain Kinase PKCalpha. J Biol Chem 292(7):2873–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leonard TA, Rozycki B, Saidi LF, Hummer G, & Hurley JH (2011) Crystal structure and allosteric activation of protein kinase C betaII. Cell 144(1):55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lucic I, Truebestein L, & Leonard TA (2016) Novel Features of DAG-Activated PKC Isozymes Reveal a Conserved 3-D Architecture. J Mol Biol 428(1):121–141. [DOI] [PubMed] [Google Scholar]
- 14.Thornton JM & Sibanda BL (1983) Amino and carboxy-terminal regions in globular proteins. J Mol Biol 167(2):443–460. [DOI] [PubMed] [Google Scholar]
- 15.Russell RB (1994) Domain insertion. Protein Eng 7(12):1407–1410. [DOI] [PubMed] [Google Scholar]
- 16.Bjorklund AK, Ekman D, Light S, Frey-Skott J, & Elofsson A (2005) Domain rearrangements in protein evolution. J Mol Biol 353(4):911–923. [DOI] [PubMed] [Google Scholar]
- 17.Yang S & Bourne PE (2009) The evolutionary history of protein domains viewed by species phylogeny. PLoS One 4(12):e8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antal CE, Callender JA, Kornev AP, Taylor SS, & Newton AC (2015) Intramolecular C2 Domain-Mediated Autoinhibition of Protein Kinase C betaII. Cell Rep 12(8):1252–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozakov D, et al. (2017) The ClusPro web server for protein-protein docking. Nat Protoc 12(2):255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pettersen EF, et al. (2004) UCSF chimera - A visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612. [DOI] [PubMed] [Google Scholar]
- 21.Fiser A, Do RKG, & Sali A (2000) Modeling of loops in protein structures. Protein Sci. 9(9):1753–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson JE, Edwards AS, & Newton AC (1997) A putative phosphatidylserine binding motif is not involved in the lipid regulation of protein kinase C. The Journal of biological chemistry 272(49):30787–30792. [DOI] [PubMed] [Google Scholar]
- 23.Franke D, et al. (2017) ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. Journal of Applied Crystallography 50:1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H, et al. (2014) Molecular mechanism of regulation of the atypical protein kinase C by N-terminal domains and an allosteric small compound. Chemistry & biology 21(6):754–765. [DOI] [PubMed] [Google Scholar]
- 25.Corbalan-Garcia S, Garcia-Garcia J, Rodriguez-Alfaro JA, & Gomez-Fernandez JC (2003) A new phosphatidylinositol 4,5-bisphosphate-binding site located in the C2 domain of protein kinase Calpha. The Journal of biological chemistry 278(7):4972–4980. [DOI] [PubMed] [Google Scholar]
- 26.Evans JH, Murray D, Leslie CC, & Falke JJ (2006) Specific translocation of protein kinase Calpha to the plasma membrane requires both Ca2+ and PIP2 recognition by its C2 domain. Molecular biology of the cell 17(1):56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antal CE, et al. (2015) Cancer-Associated Protein Kinase C Mutations Reveal Kinase’s Role as Tumor Suppressor. Cell 160(3):489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baffi TR, Van AN, Zhao W, Mills GB, & Newton AC (2019) Protein Kinase C Quality Control by Phosphatase PHLPP1 Unveils Loss-of-Function Mechanism in Cancer. Molecular cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao J, et al. (2017) 3D clusters of somatic mutations in cancer reveal numerous rare mutations as functional targets. Genome medicine 9(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]