

Abstract
Post-translational modifications (PTMs) of histones are important epigenetic regulatory mechanisms that are often dysregulated in cancer. We employ middle-down proteomics to investigate the PTMs and proteoforms of histone H4 during cell cycle progression. We use pH gradient weak cation exchange-hydrophilic interaction liquid chromatography (WCX-HILIC) for on-line liquid chromatography-mass spectrometry analysis to separate and analyze the proteoforms of histone H4. This procedure provides enhanced separation of proteoforms, including positional isomers, and simplifies downstream data analysis. We use ultrahigh mass accuracy and resolution Fourier transform-ion cyclotron resonance (FT-ICR) mass spectrometer to unambiguously distinguish between acetylation and tri-methylation (Δm = 0.036 Da). In total, we identify and quantify 233 proteoforms of histone H4 in two breast cancer cell lines. We observe significant increases in S1 phosphorylation during mitosis, implicating an important role in mitotic chromatin condensation. A decrease of K20 unmodified proteoforms is observed as the cell cycle progresses, corresponding to an increase of K20 mono- and di-methylation. Acetylation at K5, K8, K12, and K16 declines as cells traverse from S phase to mitosis, suggesting cell cycle–dependence and an important role during chromatin replication and condensation. These new insights into the epigenetics of the cell cycle may provide new diagnostic and prognostic biomarkers.
Keywords: epigenetics, ETD, FT-ICR, mass spectrometry, PTM, WCX-HILIC
1. Introduction
Histone posttranslational modifications (PTMs) are crucial to the regulation and maintenance of eukaryotic genomes. The basic template of our heredity originates from genomic DNA. In the nuclei of all eukaryotic cells, genomic DNA is organized and compacted into chromatin that consists of repeating units of nucleosomes. Each nucleosome is comprised of an octamer (an H3-H4 tetramer and two H2A-H2B dimers) of core histone proteins wrapped around by ~146 base pairs of DNA.[1,2] Core histone proteins consist of a globular domain and a structurally dynamic N-terminal tail that is prone to a variety of PTMs. Commonly observed modifications include acetylation, methylation, and phosphorylation, and to a lesser extent ubiquitination, sumoylation, arginine deimination, proline isomerization, and ADP ribosylation.[3,4] These PTMs function in conjunction with histone variants, DNA methylation, and ATP-dependent chromatin remodeling.[5] These interrelated mechanisms generate a dynamic epigenetic machinery to modulate and regulate the chromatin structure and biological processes, such as transcription, DNA replication and repair, and chromatin condensation.[6]
Histone PTMs function in concert at the single molecule level. The histone code hypothesis proposes that histone PTMs may function in concert to orchestrate the regulation of gene expression.[6] This theory has been examined in numerous biological, biochemical, and biophysical studies. ChIP-chip and ChIP-seq techniques have been employed to demonstrate the genomic localization of histone modifications and suggest strong evidence of combinatorial effects of histone PTM marks.[3,7–9] However, these efforts often lack quantitative capacity and are not unbiased. The discovery of histone code “writers” and “erasers,” histone-modifying enzymes such as histone acetyltransferases (HATs), histone deacetylases (HDACs), and histone lysine methyltransferases (HKMTs), has revealed some basic elements of homeostasis of combinatorial patterns of histone modifications.[10] Previous studies reveal direct biophysical interactions that are mediated by multiple site-specific PTM recognition domains in proteins or protein complexes that function in tandem to recognize specific combinations of PTMs on single histone molecules.[11–15] These findings suggest that histone combinatorial PTMs serve as a signal platform to regulate the recruitment of downstream effectors or “readers,” often containing tandem PTM recognition domains, to determine the ultimate biological outcome.[6,16] Thus, development of a robust and effective analytical method to decipher the histone codes is highly desirable.
A proteoform is a protein product with all possible variations thereof (PTMs, sequence variants, etc.) unambiguously defined in combination at the single molecule level.[17] This terminology has recently been rapidly adopted in the scientific lexicon of the proteomics community because it concisely describes a complex concept that often requires continual repetition. For example, the histone H4 K20me2 PTM is distinct from the H4 K20me2 proteoform. The H4 K20me2 PTM simply indicates occupancy of a single residue with a PTM. The H4 K20me2 proteoform indicates that only that site is occupied and all other sites of variable modification are unambiguously unmodified. The concept of binary (or higher order) combinations expresses only how two (or more) PTMs exist or function in concert, without consideration of other PTMs. These concepts are exceptionally useful in describing the nuances of hyper-modified proteins such as histone H4. Note that we use proteoform in slight violation of this definition because we do not capture a few minor PTMs deep in the sequence; however, the utility of this concept remains essential to understanding the hyper-modified histone H4 N-terminal tail.
Liquid chromatography-mass spectrometry (LC-MS) methods, when properly designed, can enable the unbiased quantitation of single-molecule-level combinations of PTMs (proteoforms). Mass spectrometry (MS)-based methods are rapidly evolving and have become an important analysis platform for the characterization and analysis of histones and other proteins with PTMs due to its high mass accuracy, resolution, and selectivity.[18–21] Traditional bottom-up MS and antibody-based methods are incapable of maintaining the connectivity between modifications, making it impossible to observe single molecule combination of histone PTMs.[22] In addition, antibody-based techniques are susceptible to cross-reactivity and epitope occlusion.[3] Thus, the current most effective methods to connect two distal PTMs are via LC-MS approaches that achieve gas-phase sequencing of molecules containing both sites of variable modification.
Middle-down LC-MS provides an effective alternative to top-down LC-MS methods and maintains most of the single-molecule-level combinatorial information. Great efforts have been made with top-down MS studies of intact histone H4. However, they either suffer from an inability to couple on-line LC separation to high quality MS analysis or lack reliable quantification metrics to interpret the complicated spectra with re-producible quantitation.[23–28] Middle-down mass spectrometric analysis relies on digestion of the protein into large peptides (usually between 3 and 10 kDa) by use of proteases such as AspN and GluC, to provide a promising alternative approach to investigate histone proteins with high accuracy, sensitivity, and specificity.[29–32] Middle-down analysis maintains molecular connectivity over large stretches of sequence, and circumvents many of the challenges of top-down methods.
A quantitative understanding of histone H4 proteoforms in the cell cycle of breast cancer cells will contribute to an expanded understanding of breast cancer and fundamental biology. Breast cancer is the most commonly diagnosed cancer and the second leading cause of death for women throughout the world. Identification of new biomarkers of breast cancer, which can be evaluated as indicators of normal or pathologic processes, is challenging but exceptionally appealing. Crosstalk networks among histone modifications have been implicated in breast cancer.[33–36] Moreover, the current proteoform-level understanding of the fundamental biology of the cell cycle is very limited. Previously, our group has reported a significant increase in phosphorylation of two histone H1 variants in two breast cell lines as the cell cycle advances from S phase to mitosis,[36] suggesting that this modification is cell cycle-dependent and could serve as a marker for proliferation. Additionally, histone H4 PTMs, such as methylation, acetylation, and phosphorylation are known to change during cell cycle progression.[37–39] Further, increasing evidence indicates that these modifications function in concert rather than independently.[3,6–9] Studies have also shown that cell survival, cell death, and cell cycle progression are interconnected.[40] Thus, an investigation of the role of combinatorial histone PTMs in cell proliferation pathways during cell cycle progression will enhance our understanding of carcinogenesis. This may ultimately offer the potential to manipulate cancer cells into apoptosis or other cellular states prone to treatment and effectively control cancer progression.
Here, we present a robust, accurate, effective, and automated middle-down workflow to investigate the proteoforms of histone H4. This method uses chromatography with high selectivity to distinguish isomeric proteoforms, electron transfer dissociation (ETD) for extensive fragmentation of the highly basic H4 tail peptide, and automated quantitative data analysis. This approach enables us to make relative abundance comparisons with high quantitative accuracy. We apply this method to study the fluctuations of the combinatorial PTM patterns of histone H4 in two breast cancer cell lines during cell cycle progression. Our analysis reveals new insights on how histone H4 PTMs function in concert and how histone H4 proteoforms vary throughout the cell cycle in cultured breast cancer cells.
2. Experimental Section
Sample Preparation:
Breast cancer cells from MDA-MB-231 and MCF-10A cell lines were grown as previously described.[36] Two rounds of 2 mM thymidine (Sigma-Aldrich, St. Louis, MO, USA) were applied to the cell growth medium of each cell line to attain an S phase block and one round of 100 nM nocodazole (Sigma-Aldrich, St. Louis, MO, USA) to achieve a mitotic phase block. Cell nuclei from 50 million cells at each phase were isolated by standard NP-40 alternative-based methods. After nuclei isolation, the resulting pellets were acid-extracted with 0.4 N H2SO4, precipitated with 20% trichloroacetic acid (TCA), washed with acetone and air-dried overnight. Extracts from the previous two steps were purified and separated by use of reversed-phase high-performance liquid chromatography (RP-HPLC) with a C18 column (4.6 × 250 mm, 5 μm, 300 Å pore size, 218TP54, Grace, Columbia, Maryland) with a Dionex UltiMate 3000 × 2 Dual Analytical System (Thermo Scientific, Waltham, MA, USA). Solvent A consists of 5% CH3CN (LC grade) and 0.2% trifluoroacetic acid (TFA; LC-MS grade) and solvent B contains 95% CH3CN and 0.188% TFA. Fractions were eluted at a flow rate of 0.8 mL min−1 and collected with a linear gradient of solvent B as follows: 5% to 30% B in 5 min, 35% to 60% B in 105 min. Each fraction was collected with an autosampler and vacuum-dried. Histone H4 was subsequently digested with AspN endoprotease (Roche Applied Science) in 100 mM ammonium bicarbonate (pH = 8) buffer with a protein-to-enzyme ratio of 10:1 for 6 h at 37 °C. Digestion was quenched by freezing at −80 °C. The target 23-amino acid peptide of the N-terminal tail of histone H4 was further purified on a second C18 column (2.1 × 230 mm, 5 μm, 300 Å pore size, Grace, Columbia, Maryland) implemented on the second channel of the above HPLC system with a gradient of 1% B per min at a flow rate of 0.2 mL min−1.
WCX-HILIC-MS/MS:
The N-terminal tails of histone H4 were separated in the second dimension by pH gradient-driven weak cation exchange-hydrophilic interaction LC (WCX-HILIC) with a nanoLC system (Eksigent 2D-NanoLC Ultra), coupled on-line to a custom-built hybrid Velos Pro-14.5 or 21 tesla Fourier transform-ion cyclotron resonance (FT-ICR) mass spectrometer.[20,21] Even though the two instruments are highly similar in design except for magnet field strength, it could possibly account for part of the analytical differences we observed from two cell lines. The Velos Pro is equipped with front-end electron transfer dissociation (FETD), for which fluoranthene is used to generate reagent ions.[41] Approximately 1.0–2.0 μg of purified histone H4 1–23aa peptide sample was loaded onto a custom-packed HILIC column (~20 cm, 100 μm ID) with PolyCAT A resin (3 μm particles, 1500Å pore size, PolyLC, Columbia, MD, USA). Solvent A consists of 95% methanol (LC-MS grade), 5 mM propionic acid (>99.5%, Fluka Puriss, Sigma), and ammonium hydroxide (>99.99%) to bring the pH to 6.0. Solvent B is comprised of 4.5% methanol (LC-MS grade) with 0.5% formic acid (LC-MS grade) added to adjust the pH to 2.0. The gradient starts at 25% solvent B, followed by a linear gradient of 36–99.8% B in 100 min. A wash step of 99.8% B is maintained for 30 min before returning back to 25% B for 60 min for column reconditioning at a flow rate of 250 nL min−1. Data acquisition was performed by use of Xcalibur software (Thermo Fisher Scientific) with a mass range of m/z 210–2000 at a mass resolving power of 152 500 at m/z 406. Each full-range mass spectrum was followed by five data-dependent tandem ETD product ion spectra for the five most abundant precursor ions at a charge state of 6+ with 50 ms reaction period. The automatic gain control (AGC) target value was set at 1e6 for MS1 and 2e5 for MS2. For a large ion population, ten fills of the external quadrupole trap were employed to transfer more ions to the ICR cell for MS2 analysis. The isolation width was set at m/z 2.2 and maximum ion injection period of 500 ms. Precursor charge states 1+, 2+, and 3+ were and exclusion was throughout excluded dynamic disabled the analysis to maximize the number of MS2 spectra for the analytes of interest. This abundant and consistent sampling enables higher confidence and consistent partitioning of signal to proteoforms for quantitative analysis. All time-domain data were Hanning apodized, zero-filled once, fast Fourier transformed to magnitude mode, and then converted to mass-to-charge ratio spectra by a two-term calibration equation.[42]
Data Processing and Analysis:
Data processing is performed by a custom analysis suite adapted from DiMaggio, Jr. et al.[43] Briefly, theoretical MS2 fragment spectra are generated for all possible chosen PTM combinations. MS2 spectra are matched to theoretical spectra with 10 ppm error, and are within 6.8 Da window of the precursor mass (a di-methylated equivalent precursor will not be compared with a mono-methylated theoretical spectra). A mixed integer linear optimization solution is used to determine which combinations best account for the most intensity in a given spectrum. The matched MS2 peak magnitude specific to a given combination (only the magnitude of MS2 peaks which match a given assigned spectra) is multiplied by the MS1 precursor intensity and integrated with all identically matching MS2 spectra to determine a given proteoform’s abundance. Values are normalized to total percent of histone H4 and discrete and combinatorial values are obtained through marginalization of proteoform quantities. For example, the relative abundance of K16ac is simply the sum of the relative abundances of all proteoforms containing K16ac. The relative abundance of the single molecule binary combination of K16ac and K20me2 is simply the sum of the relative abundance of all proteoforms containing both K16ac and K20me2. Statistical analysis and figure generation is done by use of R and Python programming languages and packages (matplotlib). Statistical significance was defined as p < 0.05 derived from two-tail Student’s t-test and Pearson correlations were used to determine reproducibility.
3. Results and Discussion
3.1. Experimental Workflow
Off-line RP-HPLC is used to separate the crude histone acid extract into different families and the histone H4 fraction is subsequently digested and further purified. In Figure 1A, the core histones H2B, H2A, H4, and H3 and linker histone H1 are baseline-separated. After collection of each fraction, endoprotease AspN is used to digest the histone H4 fraction to produce the first 23-amino acid peptide of the N-terminal tail where the majority of PTMs are located. To reduce the sample complexity and facilitate data analysis, the digested peptides are subjected to a second offline RP-HPLC to further purify the target N-terminal peptide of 23 amino acids of histone H4 from the internal peptides. As demonstrated in Figure 1B, the N-terminal tail of histone H4 elutes starting from approximately 23 to 25 min. Proteoforms with different acetylations or other modifications are not well separated, thereby exposing the limitations of C18-based RP-HPLC for histone proteoform separation.
Figure 1.
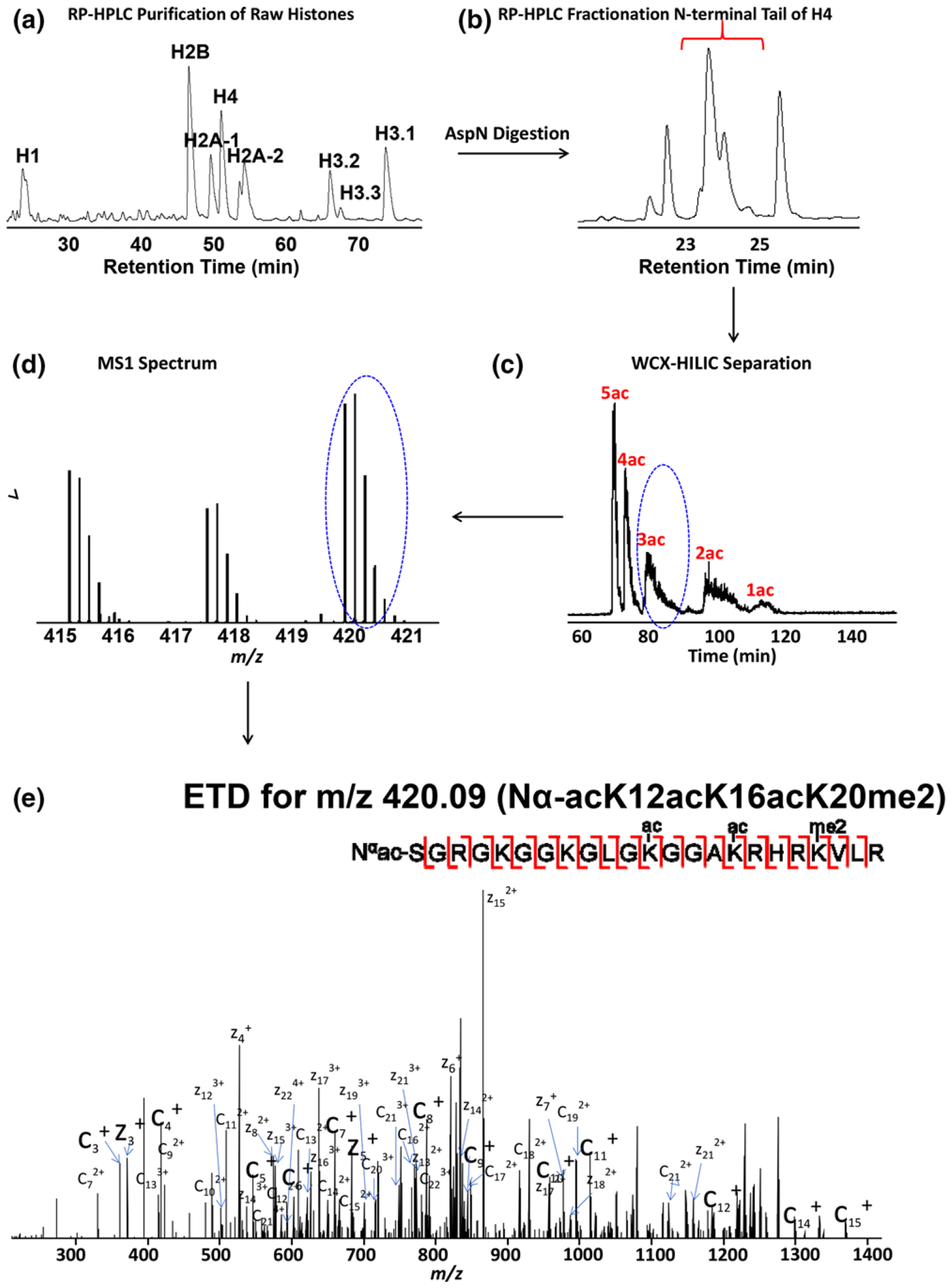
Overall experimental workflow for the H4 Nα-acK12acK16acK20me2 proteoform. a) Reversed-phase HPLC separates histones into different families (H2A, H2B, H3.1, H3.2, H3.3, H4, and H1). b) The target 23 amino acid peptide of the N-terminal tail is further purified by RP-HPLC. c) Isoforms are primarily separated by extent of acetylation, and then by methylation state and location by use of WCX-HILIC. d) Representative broadband mass spectrum of the desired 23 amino acid peptide of histone H4 at a retention time of 82.36 min. e) Representative ETD product ion spectrum for precursor ion m/z 420.09 with matched fragments marked.
To effectively resolve histone H4 proteoforms, on-line pH gradient weak cation exchange-hydrophilic interaction liquid chromatography (WCX-HILIC) is used to separate the proteoforms of histone H4 based on net charge state and hydrophilicity. Due to the charge neutralization of the acetyl groups, proteoforms are primarily separated by the extent of acetylation. They are further separated by methylation state and location of both acetylations and methylations (Figure 1C). The eluent is introduced by electrospray ionization (ESI) into a front-end electron transfer dissociation (ETD) equipped FT-ICR mass spectrometer[41]. The precursor masses are determined by high resolution MS1 (Figure 1D). After isolation, ETD is used for tandem mass spectrometry and localization of PTMs (Figure 1E). As an example, we present the annotated tandem mass spectrum of the N-terminal histone H4 tail proteoform Nα-acK12acK16acK20me2 PTM (Figure 1E). The fragmentation map suggests that our ETD induces sufficiently extensive fragmentation to confidently detect and quantify the PTMs and proteoforms, especially when further constrained by high resolution MS1 precursor mass.[30,43] One of the biggest challenges for histone investigations is to identify and characterize the positional isomers, which exhibit identical masses and very similar chromatography. Figure 1 shows separation based mainly on the degree of acetylation. However, both MS1 and MS2 results reveal that some of the isomerically modified peptides are eluted and isolated at different times. Thus, our separation technique does in fact increase the separation capability. For example, Figure 1E shows only the proteoform, Nα-acK12acK16acK20me2, from that ETD MS2 product ion mass spectrum; other positional isomers exist but are isolated and fragmented at different elution times and MS2 product ion mass spectra. The implemented 2D three-step separation technique greatly enhances our capability to effectively separate these positional isomers and reduces the complexity of data analysis. Notably, the ultrahigh mass accuracy and resolution FT-ICR MS permits us to distinguish unambiguously between acetylation and tri-methylation at the precursor mass level. These PTMs exhibit the same nominal mass and a mass difference (Δm = 0.036 Da) that is challenging to distinguish for most mass spectrometers for larger peptides and higher charge states observed here.
3.2. Bioinformatics and the Application to Explore the Dynamics of PTMs of Histone H4 during Breast Cancer Invasion
We employ this effective middle-down workflow to investigate changes in PTMs and proteoforms of histone H4 in two breast cancer cell lines during cell cycle progression. The cell lines include: MCF10A, a precancerous breast epithelial cell line that is often used as a surrogate of normal breast epithelial cells; and a highly invasive breast cancer cell line MDA-MB-231. For each cell line, thymidine and nocodazole are used to achieve an S phase and G2/M phase block. Asynchronous cells are also harvested, and represent a time-average of the cell cycle, but are substantially enriched for the G1 phase of the cell cycle. Two biological replicates of asynchronous cells and cells in S and G2/M phase for each cell line are subjected to 1–3 technical replicate runs depending on each sample amount. To examine the reproducibility, we plot a Pearson correlation matrix heat map for all sample runs. Color schemes are employed to show quantitative differences between data sets. As shown in Figure 2, cell lines and treatments mostly cluster together. Pearson correlations between technical replicates are generally about 0.9, indicating excellent technical reproducibility. One of the sample runs from nocodazole treatment in MDA-MB-231 cell line stands out as a mild outlier with a lower correlation to the other technical replicates, likely resulting from insufficient sample loading for the third technical replicate. We include it here as part of the complete data set. The ETD tandem mass spectra reveal that the acetylations occur on the N-terminus and at K5, K8, K12, and K16; phosphorylation at S1; monomethylation at R3; and un-, mono-, di-, and tri-methylation at K20, consistent with previous literature.
Figure 2.
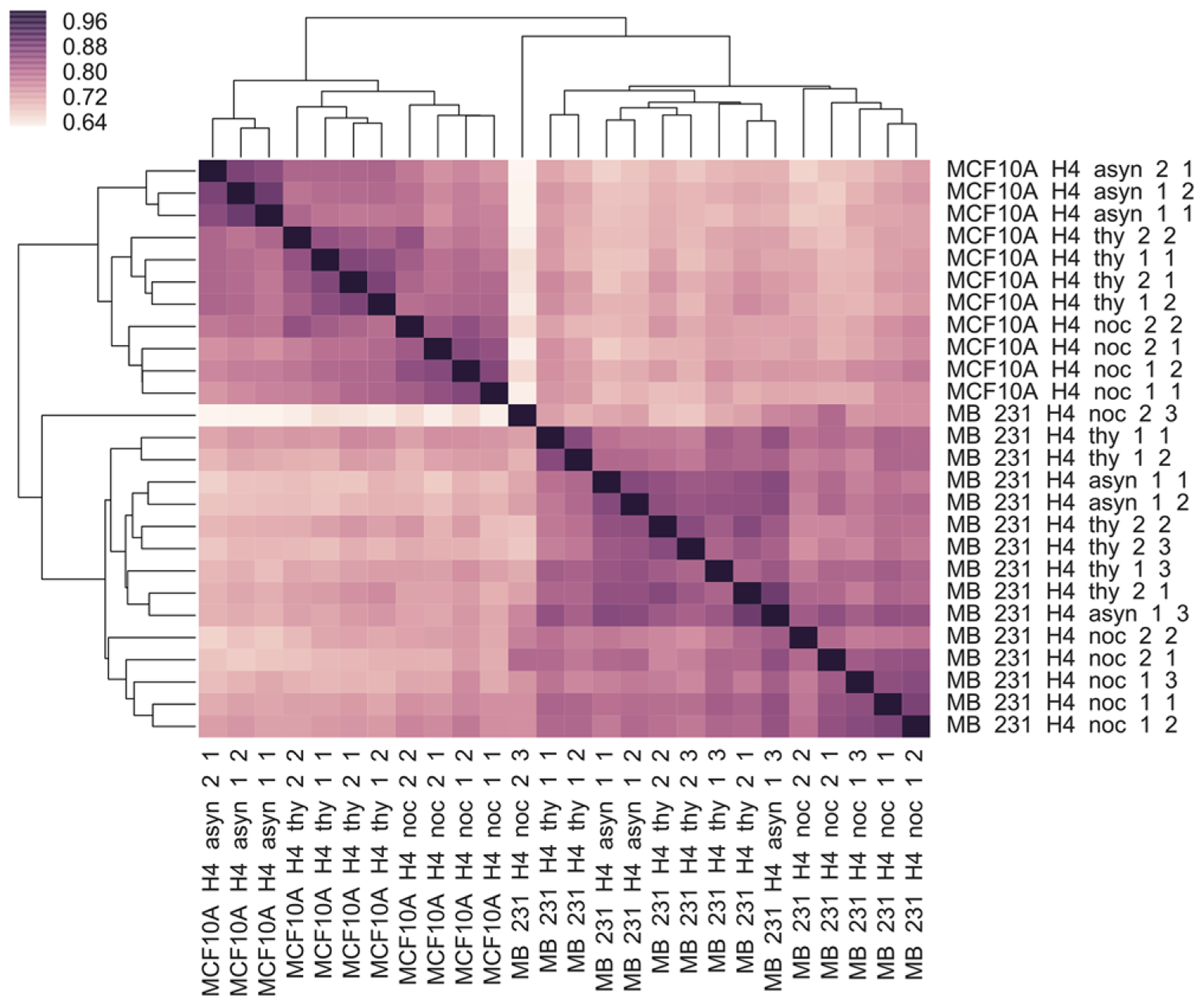
Pearson correlation matrix heat map for cell lines MDA-MB-231 and MCF-10A at different cell cycle stages.
We performed hierarchical clustering of all proteoforms of histone H4 commonly observed in both cell lines (Figure 3). The quantitative data is expressed in the form of log10 ratios. Our statistical analysis reveals distinct proteoform abundance differences between the MCF10A and MDA-MB-231 cell lines. Treatments with thymidine and nocodazole to achieve an S phase and G2/M phase blocks appear to not induce global proteoform disruption, but rather greatly influence a few proteoforms, such as proteoforms containing S1 phosphorylation. The differences between the two cell lines may result from differences in cellular and cancer phenotype; however, two cell lines are analyzed with two different FT-ICR mass spectrometers (14.5 T vs 21 T) on different days and may simply reflect analytical differences, even though the two instruments are highly similar in design except for magnet field strength.
Figure 3.
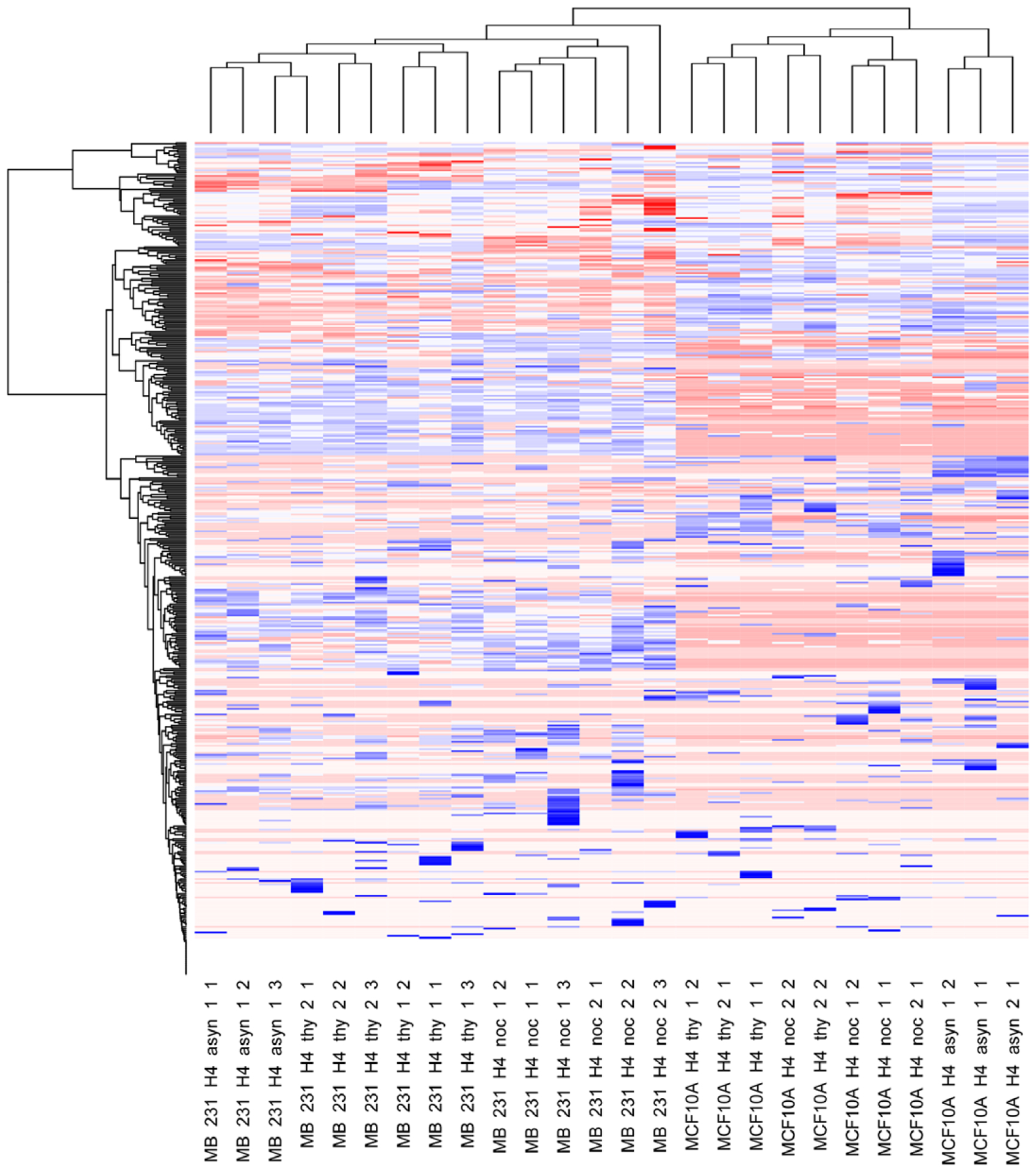
Cross-correlation analysis between two breast cancer cell lines (MDA-MB-231 and MCF-10A) and the histone H4 PTM log10(abundance). The y-axis represents individual proteoforms, which are too numerous to list explicitly in the figure and are included in Table S1, Supporting Information.
The proteoform data may be analyzed at many different levels, including the simplest which is discrete PTMs, without consideration of the presence of other PTMs (Table 1). Binary combinations of PTMs may be queried in a similar manner. Such reduced representations dispose of valuable information but are extremely helpful in identifying patterns and relationships that may be explored with more detailed questions regarding how these PTMs function in concert.
Table 1.
The percent abundance relative to the total histone H4 extracted from the cells for all discrete PTMs considered in our analysis with error expressed as standard deviation. Measurements are made on combinatorially modified peptides and these “discrete” PTM numbers are derived by summing those peptides containing each PTM while ignoring, or marginalizing, other PTMs. Note the large range of basal abundances of PTMs and that cell cycle dependence occurs for both abundant and scarce PTMs. Thus, fold change does not accurately represent the significance of all changes and absolute change is sometimes critical to understanding the function of PTMs.
| PTM | MCF10A Asyn/G1 | MCF10A Thy/S | MCF10A Noc/M | MB231 Asyn/G1 | MB231 Thy/S | MB231 Noc/M | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Average [%] | STDEV | Average [%] | STDEV | Average [%] STDEV | Average [%] | STDEV | Average [%] | STDEV | Average [%] | STDEV | ||
| N∝-ac | 99.57 | ±0.02 | 99.69 | ±0.08 | 99.48 | ±0.36 | 99.89 | ±0.03 | 95.87 | ±5.35 | 98.44 | ±1.05 |
| N∝-un | 0.43 | ±0.02 | 0.31 | ±0.08 | 0.52 | ±0.36 | 0.11 | ±0.03 | 4.14 | ±5.35 | 1.56 | ±1.05 |
| S1ph | 0.27 | ±0.02 | 0.34 | ±0.24 | 0.85 | ±0.36 | 0.09 | ±0.08 | 0.14 | ±0.09 | 0.50 | ±0.22 |
| S1un | 99.73 | ±0.02 | 99.66 | ±0.24 | 99.15 | ±0.36 | 99.91 | ±0.08 | 99.86 | ±0.09 | 99.50 | ±0.23 |
| R3me1 | 0.86 | ±0.10 | 0.98 | ±0.21 | 1.08 | ±0.16 | 0.57 | ±0.05 | 0.59 | ±0.14 | 0.85 | ±0.23 |
| R3un | 99.14 | ±0.10 | 99.02 | ±0.21 | 98.92 | ±0.16 | 99.44 | ±0.05 | 99.41 | ±0.14 | 99.15 | ±0.23 |
| K5ac | 4.38 | ±0.27 | 3.50 | ±0.44 | 2.74 | ±0.34 | 2.05 | ±0.12 | 2.25 | ±0.87 | 1.90 | ±0.53 |
| K5un | 95.62 | ±0.27 | 96.50 | ±0.44 | 97.26 | ±0.34 | 97.95 | ±0.12 | 97.75 | ±0.87 | 98.10 | ±0.53 |
| K8ac | 4.34 | ±0.35 | 4.20 | ±0.63 | 2.80 | ±0.19 | 3.38 | ±0.42 | 4.09 | ±1.10 | 2.20 | ±0.54 |
| K8un | 95.66 | ±0.35 | 95.80 | ±0.63 | 97.20 | ±0.19 | 96.62 | ±0.42 | 95.91 | ±1.10 | 97.80 | ±0.54 |
| K12ac | 9.67 | ±1.02 | 9.70 | ±1.02 | 9.55 | ±0.74 | 6.30 | ±0.32 | 6.63 | ±2.87 | 5.19 | ±1.94 |
| K12un | 90.33 | ±1.02 | 90.30 | ±1.02 | 90.45 | ±0.74 | 93.70 | ±0.32 | 93.37 | ±2.87 | 94.81 | ±1.94 |
| K16ac | 33.94 | ±2.30 | 29.17 | ±0.59 | 19.89 | ±1.90 | 41.03 | ±2.81 | 37.53 | ±5.16 | 33.77 | ±6.81 |
| K16un | 66.06 | ±2.30 | 70.83 | ±0.59 | 80.11 | ±1.90 | 58.97 | ±2.81 | 62.47 | ±5.16 | 66.23 | ±6.81 |
| K20me1 | 16.42 | ±0.48 | 4.53 | ±0.48 | 5.90 | ±0.27 | 13.87 | ±0.80 | 6.65 | ±0.57 | 11.48 | ±0.91 |
| K20me2 | 68.40 | ±0.89 | 84.17 | ±0.61 | 92.58 | ±0.53 | 72.33 | ±1.95 | 82.37 | ±1.50 | 86.67 | ±1.27 |
| K20me3 | 0.48 | ±0.08 | 1.64 | ±0.37 | 0.58 | ±0.17 | 0.06 | ±0.02 | 0.21 | ±0.09 | 0.17 | ±0.10 |
| K20un | 14.71 | ±1.23 | 9.66 | ±0.72 | 0.95 | ±0.12 | 13.74 | ±1.40 | 10.77 | ±1.72 | 1.67 | ±0.65 |
4. H4 S1 Phosphorylation
Phosphorylation at serine 1 of histone H4 has been found in a wide range of organisms, including worms, flies, and mammals, and is associated with chromatin condensation during mitosis.[44] As shown in Figure 4A, the abundance of S1 phosphorylation in both cell lines dramatically increases as the cell cycle traverses from S phase to G2/M phase, in agreement with previous studies,[44,45] and consistent with an important role for S1ph in mitotic chromatin condensation. In Figure 4B, all proteoform changes observed are represented as a volcano plot of −log10(p value) versus log10(fold change). Individual proteoforms that are above the statistical threshold of p < 0.05 and change by more than fivefold are considered meaningful. Each point on this graph corresponds to a row in Table S1, Supporting Information.
Figure 4.
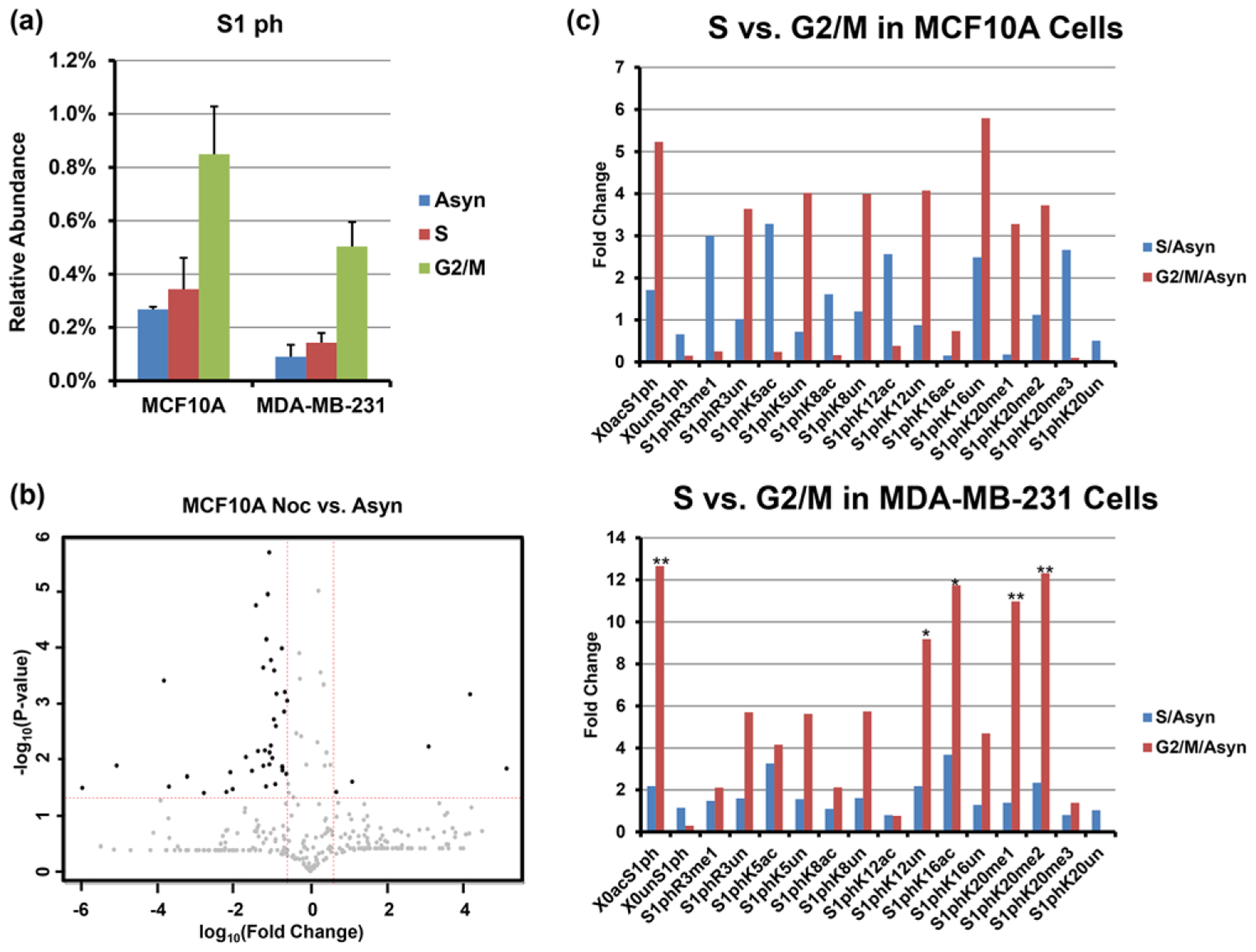
a) Relative abundances of total phosphorylation on serine 1 observed from cell lines MCF-10A and MDA-MB-231 at different cell cycle stages. Error bars represent standard error (S.E.) from two biological replicates in S and G2/M phase for each cell line, two biological replicates of asynchronous cells for cell line MDA-MB-231, and one biological replicate for cell line MCF-10A. Each biological replicate contains one to three technical replicates depending on sample amount. b) Volcano plot of proteoforms containing S1ph comparison between mitosis and asynchronous cells from cell line MCF-10A. c) Fold change of binary combinations of PTMs containing S1ph and another PTM relative to asynchronous cells. Top: MCF10A; Bottom: MB-231. Asterisks denote p values < 0.05 as determined by Student’s t-test.
4.1. H4S1ph ± ac
With our middle-down analysis, we are also able to explore the correlation between S1ph and other PTMs occurring on the N-terminus. From the fold change bar graph in binary combinations of PTMs shown in Figure 4C, we observe that the relative abundances of S1ph-containing binary combinations mostly dramatically increase during cell cycle progression in both cell lines. However, the specific deviations from this increase are the most interesting. The moderate increases in S1ph are mostly specific to lysine acetylation-containing proteoforms, with the exception of K16 in MDA-MB-231 cells. This behavior is most obvious in MCF10A cells, for which there is a dramatic shift from S1ph co-occurring with acetylation in S phase to co-occurring with unacetylation in G2/M. The increase in S1ph reaches an apex in G2/M and this increase is highly anticorrelated with Nε-lysine acetylation at all sites at the single molecule level, with the exception of K16ac only in MDA-MB-231 cells. MCF10A cells show strikingly opposite behavior for the binary combination of S1phK16ac. This single molecule combination is instead depleted relative to asynchronous cells (surrogate for G1) for both S and G2/M.
4.2. H4S1ph ± me
The relationship between S1ph and site of methylation is also interesting. Our results exhibit increases in S1phR3un, S1phK20me1, and S1phK20me2 and huge decrease for S1phK20un in both cell lines. Phosphorylation of S1 and the methylation of R3 site may be mutually impeded due to steric hindrance. The correlation between mono- and di-methylation of K20 and S1ph distinct from the unmodified state at K20 is interesting and may be due to localization of the kinase to regions enriched in these marks. K20me3 is characterized as heterochromatic and less accessible; however, the structural characteristics of K20un are not well understood.
5. H4 Acetylation
Acetylation is the most commonly observed modification in histone H4. We investigate the levels of universally conserved acetylation sites at each individual lysine residue of the N-terminal tail of histone H4 through the cell cycle. K16 is the most abundant Nε-lysine acetylation site, followed in order of decreasing abundance by K12, K8, and K5. We observe a pronounced decline for acetylation at K16 and slight decrease for acetylation at K5 and K8 whereas K12ac remains almost the same (Figure S1, Supporting Information), consequently resulting in a steady increase in the abundances of K5un, K8un, and K16un during the cell cycle progression. Previous reports have indicated a role for acetylation at K5 of histone H4 in histone deposition during chromatin replication and assembly in S phase.[46] The deacetylated form of K16 is pivotal to the formation of condensed chromatin during M phase.[47] Thus, our observed decrease of acetylation at these sites is consistent with previous findings.
5.1. H4 Nα-ac
We find that more than 99.5% of cells in MCF10A cell line, and 95.9% in MDA-MB-231 cell line are acetylated on the N-termi nus. This result contradicts the assumption in previous studies that histone H4 is constitutively N-terminally acetylated as a co-translational event[45] with 100% of histone H4 being N-terminally acetylated. Instead, we reproducibly observe a small but measurable amount of not N-terminally acetylated histone H4 and this effect varies across the cell cycle. This observation further demonstrates the precision and effectiveness of our implemented middle-down workflow.
5.2. H4ac ± ac
We also investigated the change in abundance of binary combinations of PTMs containing two acetylations. Figure S2, Supporting Information exhibits a universal decrease for all diacetylated marks for both cell lines. A similar trend is observed for proteoforms with three acetylations and four acetylations.
6. H4 Methylation
Both K20 methylation and R3 methylation are investigated here. Both of these events draw the methylation from the same physiological source, S-adenosylmethionine; however, the biological function of lysine acetylation and arginine methylation are not strongly similar.
6.1. K20 Methylation
K20 di-methylation is the predominant methylation state at K20, accounting for over 70% of all histone H4 in both cell lines, followed by K20 mono-methylated, unmodified, and K20 tri-methylated proteoforms. Histone H4 K20 methylation is involved in mitosis and other chromatin-regulated processes, including transcriptional activation, gene silencing, and DNA repair.[48–50] As shown in Figure 5, the relative abundances of both K20me2 and K20me1 in both cell lines increase between S phase to mitosis, resulting in a substantial decrease of K20 unmodified proteoforms and not an increase in K20me3. Also, we observe that Nα-acK20un, K5acK20un, K8acK20un, K12acK20un, and K16acK20un all decrease substantially, consistent with the overall decrease of K20 unmodified proteoforms. This observation suggests that K20un proteoforms play a role in histone replication and deposition processes in S phase and then they are extensively converted to K20me1 and K20me2 as the cell cycle progresses to G2/M phase. In contrast to the upregulation in K20me2 and K20me1, we notice a decrease for K20 tri-methylation as cells advance through the cell cycle, known to be mostly located in heterochromatic region and participating in modulating transcriptional silencing. And this decrease is more obvious in the MCF10A cell line and we find that the decline of K20me3 is mainly associated with other unmodified acetylation sites, such as K5unK20me3, K8unK20me3, K12unK20me3, and K16unK20me3 (Figure S3, Supporting Information). This result is consistent with the changes in the heterochromatic regions, because reduced acetylation is also associated with heterochromatin. Note that we do observe K20me3 in acetylated histone H4; however, such proteoforms do not decrease during the cell cycle.
Figure 5.
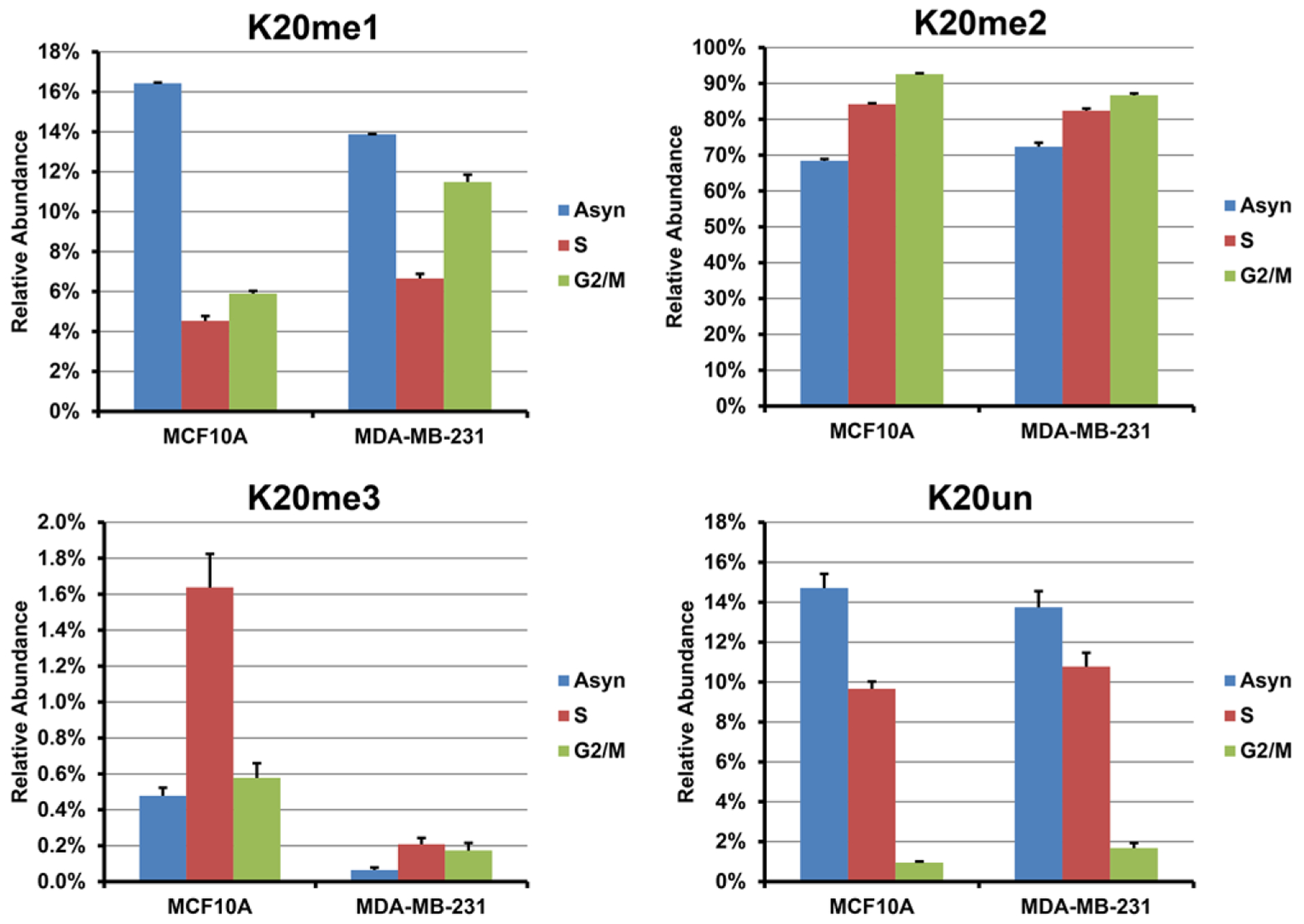
Relative abundances of mono-, di-, and tri-methylation and unmodified proteoforms on lysine 20 observed from cell lines MCF-10A and MDA-MB-231 at different cell cycle stages. Error bars represent S.E. from two biological replicates in S and G2/M phase for each cell line, two biological replicates of asynchronous cells for cell line MDA-MB-231, and one biological replicate for cell line MCF-10A. Each biological replicate contains one to three technical replicates depending on sample amount.
6.2. R3 Methylation
In addition to K20, methylation at R3 is also interrogated. The overall relative abundances of R3 mono-methylation and unmodified forms remain at a similar level during the cell cycle for both cell lines, suggesting that the regulation of methylation at R3 is cell cycle independent. We do not observe any di-methylation at this site. To explore the relationship between R3 and K20 methylation, we observe the presence of R3me1K20me1, R3me1K20me2, R3me1K20me3, and R3me1K20un, in contrast to a previous report that methylation of H4R3 is observed only in the presence of H4K20 di-methylation.[29] The discrepancy further suggests that our method is more advantageous and sensitive in detecting low-abundance proteoforms and combinations, given that the overall relative abundance of R3me1 is less than 1% and the relative abundances of R3me1K20me1, R3me1K20un, and R3me1K20me3 are orders of magnitude less and can be easily overlooked by other methods. Relative to the fluctuations in abundances of methylation at K20 through the cell cycle, we postulate that there is no direct interdependence between methylations at these two different sites.
7. Concluding Remarks
Here we present a robust and effective three-step 2D RP/WCX-HILIC-MS/MS workflow to investigate histone H4 at both the discrete PTMs and proteoform levels. We demonstrate the advantages of this method in separating isobaric and isomeric proteoforms, thereby greatly alleviating the complexity of spectra and the downstream data analysis to increase our quantitation confidence. Moreover, this optimized method requires approximately 1 μg sample, which, given the relative abundance of histone H4, makes many biological studies feasible. The study of two breast cancer cell lines through the cell cycle presented here reveals distinct epigenetic patterns in models of different progression states of breast cancer. At the PTM level, our results show dramatic increase for S1 phosphorylation, K20 mono- and di-methylation and downregulation for K20 unmodification and acetylation at K5, K8, K12, and K16 for both cell lines between S phase and G2/M phase, consistent with previous studies. N-acetylation and R3 mono-methylation remain almost the same. At the proteoform level, we analyze the fluctuation of combinatorial PTMs for both cell lines. In total, 233 proteoforms are reproducibly observed and quantitated for both cell lines. An additional 202 proteoforms were observed for a total of 435; however, these additional 202 proteoforms were not reproducibly detected and are considered below our limit of quantitation. Proteoforms that contain the “binary combinations” (note that other sites may or may not be modified): S1phK5un, S1phK8un, S1phK12un, and S1phK16un show clear synergistic effect during cell cycle progression, whereas R3 methylation and K20 methylation status appear uncoordinated. To further understand the mechanism of breast carcinogenesis, an extension of the research to combinatorial histone codes across different histone family members in each nucleosome or across different nucleosomes would be necessary. Additional follow-up with other molecular biology techniques, with these quantitative and single molecule specific results in mind, could provide complementary information to advance our understanding of the complexity of epigenetic regulations during the cell cycle of breast cancer cells.
Supplementary Material
Significance Statement.
In the present study, a middle-down approach is employed for the identification, characterization, and quantitation of the posttranslational modifications (PTMs) and proteoforms of histone H4 in two breast cancer cell lines. Our experimental strategy not only preserves the combinatorial attributes of most existing PTMs, but also enhances the separation ability and reduces the complexity of downstream data analysis. The fluctuations in the relative abundances of common modifications throughout the cell cycle, including phosphorylation, acetylation, and methylation, suggest that they play significant roles at different stages of the cell cycle in breast cancer and could participate in carcinogenesis. In summary, our analysis advances the understanding and elucidation of cancer epigenetics with preservation of single molecule connectivity between the major N-terminal PTMs and lays the foundation for the further study of histones or other proteins.
Acknowledgments
This work was supported by NSF Division of Materials Research through DMR 11-57490, and the State of Florida. This work was supported by NIH awards CA016058, OD018056 and support from the Ohio State University. Raw mass spectrometry data was deposited into ProteomeXchange via the PRIDE partner repository with the dataset identifier PXD008296.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Tingting Jiang, Department of Chemistry and Biochemistry, Florida State University, 95 Chieftain Way, Tallahassee, FL, 32306, USA.
Michael E. Hoover, Department of Cancer Biology and Genetics, Ohio State University, 460 West 12th Avenue, Columbus, OH, 43210, USA
Matthew V. Holt, Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Baylor College of Medicine, One Baylor Plaza, Houston, TX, 77030, USA
Michael A. Freitas, Department of Cancer Biology and Genetics, Ohio State University, 460 West 12th Avenue, Columbus, OH, 43210, USA
Alan G. Marshall, Department of Chemistry and Biochemistry, Florida State University, 95 Chieftain Way, Tallahassee, FL, 32306, USA Ion Cyclotron Resonance Program, National High Magnetic Field Laboratory, 1800 East Paul Dirac Drive, Tallahassee, FL, 32310, USA.
Nicolas L. Young, Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Baylor College of Medicine, One Baylor Plaza, Houston, TX, 77030, USA Department of Molecular and Cellular Biology, Baylor College of Medicine, One Baylor Plaza, Houston, TX, 77030, USA.
References
- [1].Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ, Nature 1997, 389, 251. [DOI] [PubMed] [Google Scholar]
- [2].Kornberg RD, Lorch YL, Cell 1999, 98, 285. [DOI] [PubMed] [Google Scholar]
- [3].Young NL, Dimaggio PA, Garcia BA, Cell. Mol. Life Sci 2010, 67, 3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bannister AJ, Kouzarides T, Cell Res. 2011, 21, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bonisch C, Nieratschker SM, Orfanos NK, Hake SB, Expert Rev. Proteomics 2008, 5, 105. [DOI] [PubMed] [Google Scholar]
- [6].Strahl BD, Allis CD, Nature 2000, 403, 41. [DOI] [PubMed] [Google Scholar]
- [7].Dion MF, Altschuler SJ, Wu LF, Rando OJ, Proc. Natl. Acad. Sci. U.S.A 2005, 102, 5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Liu CL, Kaplan T, Kim M, Buratowski S, Schreiber SL, Friedman N, Rando OJ, PLOS Biol. 2005, 3, 1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kurdistani SK, Tavazoie S, Grunstein M, Cell 2004, 117, 721. [DOI] [PubMed] [Google Scholar]
- [10].Chi P, Allis CD, Wang GG, Nat. Rev. Cancer 2010, 10, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li B, Gogol M, Carey M, Lee D, Seidel C, Workman JL, Science 2007, 316, 1050. [DOI] [PubMed] [Google Scholar]
- [12].Ruthenburg AJ, Li H, Patel DJ, Allis CD, Nat. Rev. Mol. Cell Biol 2007, 8, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Trojer P, Li G, Sims RJ, Vaquero A, Kalakonda N, Boccuni P, Lee D, Erdjument-Bromage H, Tempst P, Nimer SD, Wang YH, Reinberg D, Cell 2007, 129, 915. [DOI] [PubMed] [Google Scholar]
- [14].Moriniere J, Rousseaux S, Steuerwald U, Soler-Lopez M, Curtet S, Vitte AL, Govin J, Gaucher J, Sadoul K, Hart DJ, Krijgsveld J, Khochbin S, Muller CW, Petosa C, Nature 2009, 461, 664. [DOI] [PubMed] [Google Scholar]
- [15].Saksouk N, Avvakumov N, Champagne KS, Hung T, Doyon Y, Cayrou C, Paquet E, Ullah M, Landry AJ, Cote V, Yang XJ, Gozani O, Kutateladze TG, Cote J, Mol. Cell 2009, 33, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yun M, Wu J, Workman JL, Li B, Cell Res. 2011, 21, 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Smith LM, Kelleher NL, Proteomics CTD, Nat. Methods 2013, 10, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Marshall AG, Hendrickson CL, Jackson GS, Mass Spectrom. Rev 1998, 17, 1. [DOI] [PubMed] [Google Scholar]
- [19].Kaiser NK, Quinn JP, Blakney GT, Hendrickson CL, Marshall AG, J. Am. Soc. Mass Spectrom 2011, 22, 1343. [DOI] [PubMed] [Google Scholar]
- [20].Schaub TM, Hendrickson CL, Horning S, Quinn JP, Senko MW, Marshall AG, Anal. Chem 2008, 80, 3985. [DOI] [PubMed] [Google Scholar]
- [21].Hendrickson CL, Quinn JP, Kaiser NK, Smith DF, Blakney GT, Chen T, Marshall AG, Weisbrod CR, Beu SC, J. Am. Soc. Mass Spectrom 2015, 26, 1626. [DOI] [PubMed] [Google Scholar]
- [22].Fuchs SM, Krajewski K, Baker RW, Miller VL, Strahl BD, Curr. Biol 2011, 21, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kelleher NL, Anal. Chem 2004, 76, 196a. [PubMed] [Google Scholar]
- [24].Pesavento JJ, Mizzen CA, Kelleher NL, Anal. Chem 2006, 78, 4271. [DOI] [PubMed] [Google Scholar]
- [25].Pesavento JJ, Bullock CR, LeDuc RD, Mizzen CA, Kelleher NL, J. Biol. Chem 2008, 283, 14927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tian Z, Tolic N, Zhao R, Moore RJ, Hengel SM, Robinson EW, Stenoien DL, Wu S, Smith RD, Pasa-Tolic L, Genome Biol 2012, 13, R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dang X, Scotcher J, Wu S, Chu RK, Tolic N, Ntai I, Thomas PM, Fellers RT, Early BP, Zheng Y, Durbin KR, Leduc RD, Wolff JJ, Thompson CJ, Pan J, Han J, Shaw JB, Salisbury JP, Easterling M, Borchers CH, Brodbelt JS, Agar JN, Pasa-Tolic L, Kelleher NL, Young NL, Proteomics 2014, 14, 1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rea M, Jiang T, Eleazer R, Eckstein M, Marshall AG, Fondufe-Mittendorf YN, Mol. Cell. Proteomics 2016, 15, 2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Phanstiel D, Brumbaugh J, Berggren WT, Conard K, Feng X, Levenstein ME, McAlister GC, Thomson JA, Coon JJ, Proc. Natl. Acad. Sci. U.S.A 2008, 105, 4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Young NL, DiMaggio PA, Plazas-Mayorca MD, Baliban RC, Floudas CA, Garcia BA, Mol. Cell. Proteomics 2009, 8, 2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Moradian A, Kalli A, Sweredoski MJ, Hess S, Proteomics 2014, 14, 489. [DOI] [PubMed] [Google Scholar]
- [32].Sidoli S, Schwammle V, Ruminowicz C, Hansen TA, Wu X, Helin K, Jensen ON, Proteomics 2014, 14, 2200. [DOI] [PubMed] [Google Scholar]
- [33].Lo PK, Sukumar S, Pharmacogenomics 2008, 9, 1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sharma S, Kelly TK, Jones PA, Carcinogenesis 2010, 31, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Harshman SW, Hoover ME, Huang CS, Branson OE, Chaney SB, Cheney CM, Rosol TJ, Shapiro CL, Wysocki VH, Huebner K, Freitas MA, J. Proteome Res 2014, 13, 2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chen Y, Hoover ME, Dang X, Shomo AA, Guan X, Marshall AG, Freitas MA, Young NL, Mol. Cell. Proteomics 2016, 15, 818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Brustel J, Tardat M, Kirsh O, Grimaud C, Julien E, Trends Cell Biol. 2011, 21, 452. [DOI] [PubMed] [Google Scholar]
- [38].Kruhlak MJ, Hendzel MJ, Fischle W, Bertos NR, Hameed S, Yang XJ, Verdin E, Bazett-Jones DP, J. Biol. Chem 2001, 276, 38307. [DOI] [PubMed] [Google Scholar]
- [39].Banerjee T, Chakravarti D, Mol. Cell. Biol 2011, 31, 4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A, Eshraghi M, Manda KD, Wiechec E, Los M, Drug Resist. Updat 2007, 10, 13. [DOI] [PubMed] [Google Scholar]
- [41].Weisbrod CR, Kaiser NK, Syka JEP, Early L, Mullen C, Dunyach JJ, English AM, Anderson LC, Blakney GT, Shabanowitz J, Hendrickson CL, Marshall AG, Hunt DF, J. Am. Soc. Mass Spectrom 2017, 28, 1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ledford EB, Rempel DL, Gross ML, Anal. Chem 1984, 56, 2744. [DOI] [PubMed] [Google Scholar]
- [43].DiMaggio PA Jr., Young NL, Baliban RC, Garcia BA, Floudas CA, Mol. Cell. Proteomics 2009, 8, 2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Barber CM, Turner FB, Wang Y, Hagstrom K, Taverna SD, Mollah S, Ueberheide B, Meyer BJ, Hunt DF, Cheung P, Allis CD, Chromosoma 2004, 112, 360. [DOI] [PubMed] [Google Scholar]
- [45].Yamamoto K, Chikaoka Y, Hayashi G, Sakamoto R, Yamamoto R, Sugiyama A, Kodama T, Okamoto A, Kawamura T, Mass Spectrom. (Tokyo) 2015, 4, A0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Koprinarova MA, Russev GC, Cell Cycle 2008, 7, 414. [DOI] [PubMed] [Google Scholar]
- [47].Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW, Serrano L, Sternglanz R, Reinberg D, Genes Dev. 2006, 20, 1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Doerks T, Copley RR, Schultz J, Ponting CP, Bork P, Genome Res. 2002, 12, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pesavento JJ, Yang H, Kelleher NL, Mizzen CA, Mol. Cell. Biol 2008, 28, 468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jorgensen S, Schotta G, Sorensen CS, Nucleic Acids Res 2013, 41, 2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.