

Abstract
Chronic metabolic diseases, including diabetes and obesity, have become a major global health threat of the twenty-first century. Maintaining glucose homeostasis is essential for survival in mammals. Complex and highly coordinated interactions between glucose-sensing mechanisms and multiple effector systems are essential for controlling glucose levels in the blood. The central nervous system (CNS) plays a crucial role in regulating glucose homeostasis. Growing evidence indicates that disruption of glucose sensing in selective CNS areas, such as the hypothalamus, is closely interlinked with the pathogenesis of obesity and type 2 diabetes mellitus. However, the underlying intracellular mechanisms of glucose sensing in the hypothalamus remain elusive. Here, we review the current literature on hypothalamic glucose-sensing mechanisms and discuss the impact of alterations of these mechanisms on the pathogenesis of diabetes.
Keywords: Astrocytes, Brain, Counterregulatory responses, Diabetes, Glucose-sensing, Hypothalamus, Neurons, Obesity, Review, Tanycytes
Introduction
The escalating global epidemic of obesity and metabolic disorders, such as type 2 diabetes mellitus, represents one of the most pressing and costly biomedical challenges facing modern society. Obesity-associated type 2 diabetes is characterised by impaired glucose homeostasis [1]. However, much about the pathogenesis of these metabolic disorders remains unclear. The maintenance of glucose homeostasis is essential for sustaining life in mammals. Although essential, glucose is minimally stored in the body and its circulating levels are maintained within a tight range, accomplished by the complex and highly coordinated interplay between glucose-sensing mechanisms and multiple effector systems in the body [2]. The central nervous system (CNS) plays a critical role in continuously monitoring circulating glucose levels and appropriately communicating to peripheral organs in order to maintain glucose levels within a tight range.
Notably, CNS glucose levels are significantly lower than those in the circulation. It has been estimated that brain glucose levels range between 1 mmol/l and 2.5 mmol/l, and that they may go down to 0.5 mmol/l in hypoglycaemia, while, in hyperglycaemia, they may reach a concentration of 5 mmol/l [3]. In the CNS, the hypothalamus is the main area involved in glucose regulation. It is comprised of several nuclei that contain distinct neuronal populations, which express a variety of neuropeptides and neurotransmitters that are involved in the regulation of energy homeostasis. The ability of the hypothalamus to control glucose and energy metabolism is due to the ability of its neuronal populations to sense, integrate and respond to a variety of metabolic changes [4], including changes in glucose levels. In this review, we summarise the current evidence on hypothalamic sensing mechanisms and their role in the development of type 2 diabetes and hypoglycaemia unawareness in type 1 diabetic patients.
Hypothalamic neuronal glucose-sensing mechanisms
The hypothalamus plays a critical role in regulating blood glucose levels due to its ability to sense, integrate and respond to changes in circulating signals. This complex and highly coordinated function of the hypothalamus with the periphery is accomplished through its control of the autonomic nervous system (ANS) and the endocrine system. Two types of glucose-sensing neurons are present in the hypothalamus: glucose-excited and glucose-inhibited neurons. Glucose-excited neurons are activated by increased glucose levels, whereas hypoglycaemic conditions cause activation of glucose-inhibited neurons [5]. Several hypothalamic nuclei have been identified and studied for their roles in glucose metabolism, including the arcuate (ARC) and the ventromedial hypothalamic (VMH) nuclei [6]. How neurons in these nuclei sense changes in glucose levels has been the object of studies in rodents and humans.
Although we are focusing on neuronal glucose-sensing mechanisms in these two hypothalamic nuclei, it is important to consider that glucose-excited and glucose-inhibited neurons are also present in other hypothalamic areas, such as the lateral hypothalamus, and in extrahypothalamic areas, such as the brain stem, including the nucleus of the solitary tract (nucleus tractus solitarius [NTS]) and the dorsal motor nucleus of the vagus (DMX).
ARC neurons
The ARC plays a fundamental role in sensing whole-body energy status. The ARC contains two main neuronal populations, each characterised by the expression of specific neuropeptides: neuropeptide Y (NPY)/Agouti-related protein (AgRP) neurons and pro-opiomelanocortin (POMC) neurons. NPY/AgRP neurons and POMC neurons regulate energy and glucose homeostasis in an opposite manner [7]. During positive energy balance and when circulating glucose levels are elevated, POMC neurons are activated, leading to the secretion of α-melanocyte stimulating hormone (α-MSH). α-MSH is a POMC-derived peptide, which, by binding to and activating melanocortin receptors, including those located in the paraventricular nucleus (PVN), induces an anorexigenic effect and regulation of peripheral glucose metabolism [8]. On the contrary, during negative energy balance, such as food deprivation and reduced circulating glucose levels, NPY/AgRP neurons are activated, AgRP is released and, by acting as a melanocortin receptor inverse agonist, inhibits α-MSH functions [9]. The ability of these two ARC neuronal populations to alter metabolism is due to their sensitivity to several circulating signals, including hormones and nutrients, such as glucose (Fig. 1). Indeed, electrophysiological studies have shown that glucose alters excitability of NPY/AgRP and POMC neurons. A decrease in glucose levels in ARC glucose-inhibited NPY/AgRP neurons leads to Ca2+ oscillations [10] and neuronal depolarisation, possibly mediated by plasma membrane Cl− channels [11]. In this context, the cellular energy sensor 5-AMP-activated protein kinase (AMPK) seems to play a key role in mediating glucose sensing. This depolarisation, in turn, promotes food intake and restoration of blood glucose levels [12].
Fig. 1.
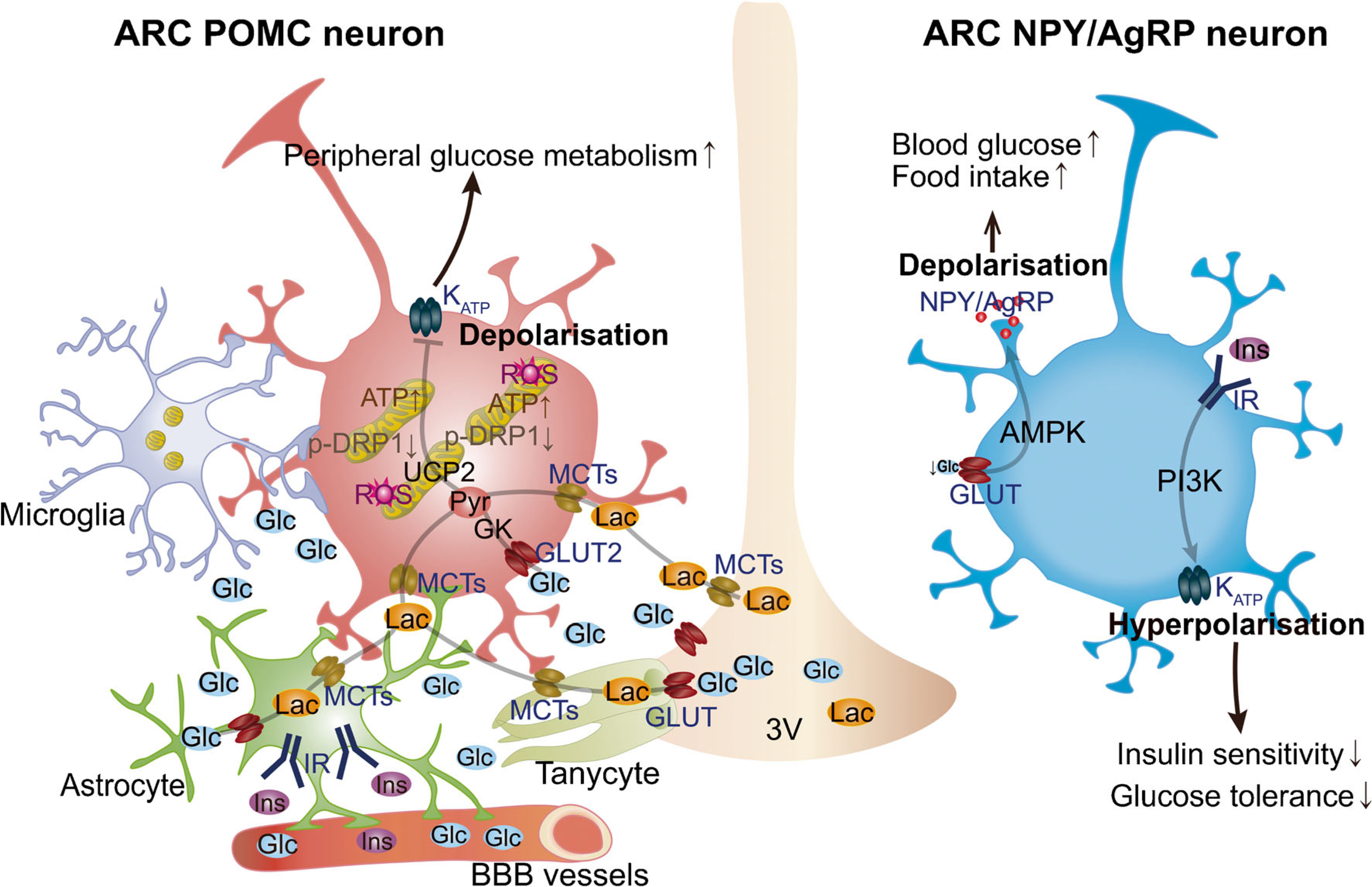
Glucose-sensing mechanisms in the hypothalamic ARC. The ability of POMC and NPY/AgRP neuronal populations in ARC to alter energy metabolism is due to their sensitivity to several circulating signals, including hormones and nutrients (e.g. glucose and lactate). Hypothalamic astrocytes provide neurons with structural support and nutrients. Astrocytic insulin signalling regulates glucose sensing via control of glucose uptake across the blood–brain barrier (BBB). Glucose is delivered to the extracellular space and transported to glial cells, such as astrocytes and tanycytes, through GLUTs and then metabolised to lactate. When released, lactate is taken up by neurons via monocarboxylate transporters (MCTs) to serve as a glycolytic substrate and metabolised into pyruvate. Hypothalamic microglia have been shown to affect POMC neuronal activation and synaptic input organisation in diet-induced obesity. Changes in glucose levels have been shown to affect POMC activity via KATP channels. Mitochondrial dynamics in hypothalamic POMC neurons has been also shown to play a pivotal role in regulating neuronal activity in response to glucose fluctuations. The activity of the ARC glucose-inhibited NPY/AgRP neurons is regulated by AMPK activity in response to decreased glucose levels, while insulin has been shown to induce hyperpolarisation of these neurons. Glc, glucose; Ins, insulin; IR, insulin receptor; Lac, lactate; PI3K, phosphoinositide 3-kinase; Pyr, pyruvate; ROS, reactive oxygen species; 3V, third ventricle.
In addition to the impact of glucose on AgRP neurons in regulating peripheral glucose metabolism, insulin signalling in these neurons has also been shown to play a role in the control of circulating glucose levels by regulating peripheral insulin sensitivity [13]. Insulin has been reported to cause hyperpolarisation and alteration of the frequency of action potentials of AgRP neurons. Exposure to the ATP-sensitive potassium (KATP) channel blocker tolbutamide has been shown to reverse these effects [14]. Furthermore, in mice with selective deletion of insulin receptors in AgRP neurons, insulin failed to alter membrane potential and frequency of action potentials, as well as failing to suppress hepatic glucose production [14], thus indicating a critical role of insulin signalling in AgRP neurons in the regulation of peripheral glucose metabolism. In addition to this, acute activation of AgRP neurons has been shown to cause peripheral insulin resistance through impairment of insulin-stimulated glucose uptake into brown adipose tissue (BAT) via a different neuronal circuit to that responsible for food intake [15], thus suggesting that AgRP neurons are able to rapidly coordinate hunger with glucose homeostasis through different circuits (Fig. 1).
ARC glucose-excited POMC neurons have been shown to share similar glucose-sensing mechanisms to those observed in pancreatic beta cells (Fig. 1). GLUT2, glucokinase (GK; a critical regulator of glycolytic ATP production) and KATP channels have been shown to be expressed in a large subpopulation of POMC neurons [16]. Once transported inside POMC neurons, glucose can be converted into pyruvate via glycolysis and, by increasing ATP levels, it can induce the closure of KATP channels and depolarise glucose-excited POMC neurons [16]. In support of this, inhibition or activation of KATP channels alters POMC neuronal responses to changes in glucose levels [17] and selective deletion of the KATP channel Kir6.2 subunit in POMC neurons has a significant impact on glucose sensing [18]. In addition to this mechanism, it has been recently proposed that there is a role for transient receptor potential canonical type 3 (TRPC3) channels in mediating the effect of increased glucose levels on glucose-excited neurons of the mediobasal hypothalamus (MBH) via a mechanism that depends on free radical levels [19]. However, the identity of the neurons within the MBH that express TRPC3 remains to be determined [19].
A mitochondrial mechanism involving the mitochondrial uncoupling protein 2 (UCP2) was suggested in diet-induced POMC neuron glucose insensitivity [18]. Several studies have since demonstrated the role of mitochondria in POMC neuron glucose sensing, albeit in a UCP2-independent manner. Mitochondria are highly dynamic cellular organelles that undergo morphological changes through fission and fusion processes, defined as mitochondrial dynamics, in response to various metabolic stimuli [20, 21]. Recent studies have demonstrated that mitochondrial dynamics in hypothalamic neurons are critical events in mediating neural activity in response to changes in metabolic states. This suggests that mitochondrial dysfunction in the hypothalamus might be strongly associated with the development of metabolic disorders, such as type 2 diabetes and obesity [22]. Increased glucose levels are associated with mitochondrial fusion in glucose-excited POMC neurons, while decreased glucose levels induce mitochondrial fission [19]. In support of a pivotal role of mitochondrial dynamics in glucose sensing, when the gene (Dnm1l) encoding the mitochondrial fission protein dynamin-related protein 1 (DRP1) was selectively deleted in adult POMC neurons, animals showed increased POMC neuron glucose sensing and improved glucose metabolism. Mechanistically, it was found that altered reactive oxygen species (ROS) levels, mostly produced during mitochondrial substrate oxidation, affect peroxisome proliferator-activated receptor γ (PPARγ) target genes, such as the KATP channel Kcnj11 gene, which, in turn, affects POMC neuronal responsiveness to changes in glucose levels [23]. On the other hand, when the mitochondrial fusion protein mitofusin 1 was selectively deleted from POMC neurons, glucose sensing was negatively affected and defective pancreatic insulin release was observed [21]. Similar to POMC neurons, mitochondrial dynamics in pancreatic beta cells have very recently been shown to play a role in glucose sensing and insulin secretion [24]. However, unlike POMC neurons, selective deletion of Dnm1l in beta cells impaired glucose metabolism by affecting glucose-stimulated amplification of insulin secretion [24].
Ventromedial nucleus neurons
The VMH has been identified as a crucial brain region in monitoring and regulating glucose homeostasis [5] (Fig. 2). Stanley and colleagues reported that neuronal modulation of GK-expressing hypothalamic VMH neurons alters blood glucose levels and feeding behaviour by regulating pancreatic hormone levels, such as insulin and glucagon [25]. These findings indicate that VMH glucose-sensing neurons play a crucial role in communicating to peripheral tissues to maintain blood glucose levels within a tight range. Several studies have pointed to the VMH glucose-inhibited neurons as being critical players in counterregulatory responses (CRRs) to hypoglycaemia, which involve detection of declining blood glucose levels and release of several hormones, including glucagon, glucocorticoids and catecholamines, to restore blood glucose levels. More recent studies have demonstrated that VMH glucose-excited neurons play an important role in regulating insulin secretion in response to hyperglycaemia [13].
Fig. 2.
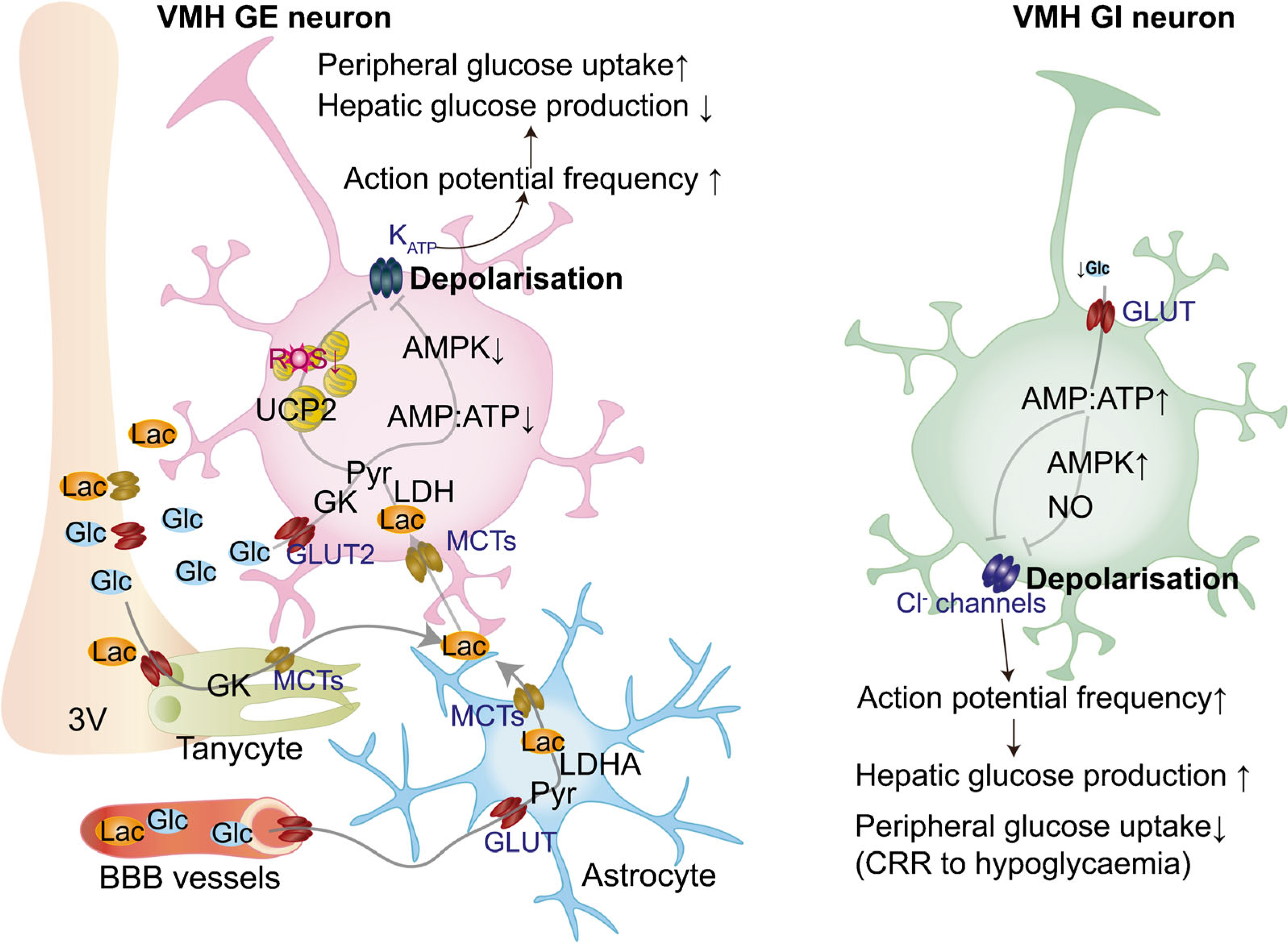
Glucose-sensing mechanisms in the hypothalamic VMH. In vivo, glucose is released in the extracellular space and transported to glial cells, such as astrocytes and tanycytes, through GLUTs and then metabolised to lactate. Once released, lactate enters neurons via monocarboxylate transporters (MCTs) to serve as a glycolytic substrate and is metabolised into pyruvate via the enzyme LDH. Mitochondrial pyruvate uptake leads to increased ATP levels that, in turn, affect ATP-dependent K+ (KATP) channels, resulting in plasma membrane depolarisation. Glucose-excited (GE) neurons can also uptake glucose through GLUT2, leading to the phosphorylation of glucose by GK, a decrease in the AMP:ATP ratio, decreased AMPK activity and closing of KATP channel. Closure of KATP channels leads to membrane depolarisation and increased action potential frequency. The activation of VMH GE neurons leads to decreased hepatic glucose production and increased peripheral glucose uptake. VMH glucose-inhibited (GI) neurons are activated in response to hypoglycaemia. Decreased glucose entry into neurons leads to the rise in the cellular AMP:ATP ratio and activation of AMPK, which induces formation of NO. Increased AMP:ATP also inhibits Cl− channels, leading to membrane depolarisation and increased action potential frequency, which leads to activation of CRRs to hypoglycaemia to increase hepatic glucose production and decrease peripheral glucose uptake. BBB, blood–brain barrier; Glc, glucose; Lac, lactate; LDHA, LDH isoenzyme A; Pyr, pyruvate; ROS, reactive oxygen species; 3V, third ventricle.
The molecular signatures that distinguish the ability of these VMH neuronal populations to respond to changes in glucose levels are still unknown. Although VMH neurons may be identified due to their expression of several known markers, including receptors for several hormones (e.g., leptin, insulin and oestrogen) and growth/transcription factors, such as brain derived neurotrophic factor (BDNF) and steroidogenic factor 1 (SF1), the identity of glucose-excited and glucose-inhibited neurons and the intracellular signals that enable them to sense and respond to changes in glucose levels are yet to be fully defined. Similar to ARC glucose-inhibited NPY/AgRP neurons, decreased glucose levels in VMH glucose-inhibited neurons lead to the activation of AMPK. In turn, AMPK causes neuronal depolarisation, increased frequency of action potential and neurotransmitter release, including release of glutamate, which leads to the activation of CRRs to hypoglycaemia [26]. Stimulation of AMPK in the VMH has been shown to amplify hormonal CRRs and increase endogenous glucose production [27]. Activated AMPK in glucose-inhibited neurons may also lead to the phosphorylation of neuronal nitric oxide synthase (nNOS) and, thus, increased production of NO, which induces closure of the Cl− channel cystic fibrosis transmembrane regulator (CFTR; Fig. 2) [28].
In VMH glucose-excited neurons, similar to ARC POMC neurons, increased glucose levels have been shown to lead to a decrease in AMP:ATP ratio, thus inducing closure of KATP channels, depolarisation, increased frequency of action potential, and neurotransmitter release (e.g., release of γ-aminobutyric acid [GABA]), resulting in lower hepatic glucose production and increase peripheral glucose uptake [29]. Furthermore, as with glucose-excited POMC neurons, recent evidence has shown a role for mitochondrial dynamics in VMH glucose-excited neuron glucose sensing [30]. However, unlike POMC neurons, it was found that systemic glucose administration in mice promotes DRP1-mediated mitochondrial fission in VMH glucose-excited neurons, which plays a pivotal role in glucose sensing and, thus, in the regulation of glucose homeostasis [30]. Furthermore, in contrast to glucose-excited POMC neurons, it was found that this mitochondrial fission-associated glucose-sensing mechanism was dependent on mitochondrial UCP2 [30]. Further studies are warranted to understand the opposing roles of mitochondria in glucose-excited neurons of the ARC vs the VMH.
Glucose-sensing mechanisms in non-neuronal cells
Whether glucose-sensing neurons directly sense extracellular glucose that is converted intracellularly into pyruvate or sense and utilise lactate derived from the glycolytic processes of glucose within astrocytes remains controversial [31]. Astrocytes are the most abundant glial cells in the brain and have many functions; for example, they provide nutrients and protection for neurons. Indeed, hypothalamic astrocytes play a pivotal role in the formation of cell-to-cell communication in networks through gap junctions, which allow intercellular exchange of metabolic signals, such as glucose and lactate [32, 33]. Growing experimental evidence has shown that astroglial gap junctions, composed of connexin (CX)30 and CX43, are altered by metabolic status, leading to modulation of the glucose response in neurons [34].
It has also been demonstrated that insulin signalling in hypothalamic astrocytes plays a role in the regulation of CNS and systemic glucose metabolism via glucose and insulin entry into the brain by changes in blood–brain barrier (BBB) permeability (Fig. 1) [35].
Moreover, it has been shown that hypothalamic astrocytic glucose transporters, GLUT1 and GLUT2, are crucial for glucose sensing in the regulation of glucose homeostasis. Once astrocytes uptake glucose, they can store it as glycogen and/or convert it to lactate [36]. The transfer of glucose-derived lactate from astrocytes to neurons is referred to as the astrocyte–neuron lactate shuttle hypothesis (Fig. 1 and Fig. 2) [31, 37]. Extracellular lactate can be utilised by neurons for pyruvate production via lactate dehydrogenase (LDH). Interestingly, glucose-excited neurons are also activated by lactate [38], suggesting that hypothalamic astrocytes play a crucial role in glucose metabolism by sensing circulating glucose levels and via lactate production, conveying the metabolic status of the organism to neurons involved in the regulation of glucose homeostasis. In vivo studies have indeed demonstrated that intracerebroventricular (ICV) or i.v. injection of lactate decrease blood glucose levels, while this effect is abolished when lactate is co-injected with oxamate, an LDH inhibitor, indicating that lactate conversion into pyruvate may play a role in peripheral glucose metabolism [39]. In support of this, a recent study has shown that ICV leptin administration enhances LDH isoenzyme A (LDHA [converts pyruvate to lactate])-dependent hypothalamic glucose sensing, inducing a decrease in blood glucose levels in high-fat-diet (HFD)-induced obese rats (Fig. 2) [40].
In addition to astrocytes, studies have shown that tanycytes, specialised ependymal cells that have cell bodies located on the wall of the third ventricle, may play a role in nutrient sensing by projecting their long processes into several hypothalamic nuclei that control metabolism [41]. Moreover, tanycytes express several important genes including those encoding GLUT2, GK and monocarboxylate transporters (MCTs; lactate transporter), suggesting their involvement in glucose-sensing mechanisms (Fig. 1 and Fig. 2) [42]. Indeed, a previous study showed that injection of the third ventricle with alloxan, a GK inhibitor and toxin that induces tanycyte ablation, impaired CRRs to hypoglycaemia [43]. However, a more recent study has shown that ablation of tanycytes in a genetic mouse model had no effect on glucose levels [44]. Thus, further studies are needed to understand the role of tanycytes in glucose sensing and in the regulation of glucose metabolism.
Altogether, these studies support the emerging new concept that non-neuronal cells may play a crucial role in regulating glucose sensing in response to fluctuations of blood glucose levels via metabolic crosstalk with glucose-sensing neurons in order to control glucose homeostasis.
The link between impaired hypothalamic glucose-sensing mechanisms and metabolic disorders
HFD-induced obesity is linked to defective hypothalamic glucose sensing by decreased levels of GLUT2 and inappropriate activation of AMPK in the hypothalamus, leading to impaired energy and glucose homeostasis [45]. Depolarisation of glucose-excited neurons in response to increases in extracellular glucose is driven by ATP-mediated closure of KATP channels [46]. Although glucose-sensing mechanisms in neurons are being extensively investigated, the relevance and contribution of glucose sensing by neurons to type 2 diabetes and obesity remain elusive. Previous studies show that disruption of glucose sensing in POMC neurons can be caused by HFD-induced obesity, indicating that defects in glucose-sensing mechanisms contribute to the development of type 2 diabetes [18]. Obesity-induced disruption of glucose sensing in POMC neurons has been suggested to be mediated by UCP2, which is highly expressed in the hypothalamus, and increased expression of UCP2 has been observed in obese animal models [18]. However, to date, no studies have reported on the specific role of UCP2 in POMC neurons in the regulation of metabolism.
Diabetes mellitus is considered a chronic metabolic disease. BMI is strongly associated with the development of insulin resistance and diabetes mellitus [47]. Many studies have revealed that factors that are involved in the development of insulin resistance [48], such as increased NEFA, elevated serum triacylglycerol and proinflammatory cytokines, are observed in individuals who are overweight and obese [49]. Similar outcomes are observed in animal models of obesity. It has been established that HFD triggers neuroinflammation, including reactive gliosis and cytokine secretion, which precedes hypothalamic neuronal dysfunction and the development of the obese and diabetic phenotype [50]. However, it remains unclear how HFD feeding triggers neuroinflammation and how this, in turn, induces neuronal dysfunction. We have recently demonstrated that HFD feeding affects microglial mitochondrial function due to changes in fuel availability [51]. Once activated, microglial cells affect the function of hypothalamic glucose-excited POMC neurons by rearranging the synaptic input organisation on their cell bodies. These results indicate that HFD-induced hypothalamic gliosis plays a critical role in the regulation of glucose and energy homeostasis, causing alterations in susceptibility to metabolic disorders, such as diabetes and obesity [51].
Modulation of hypothalamic glucose sensing
Hypothalamic glucose-sensing mechanisms can be acutely altered by circulating signals, such as hormones. For example, insulin has been shown to acutely activate KATP channels in hypothalamic glucose-sensing neurons and alteration of CNS insulin signalling has been observed in individuals with type 2 diabetes [13]. In addition, Diggs-Andrews et al. [52] showed that neuronal deletion of the insulin receptor impairs glucose sensing in VMH glucose-inhibited neurons and that PVN neurons are activated in response to hypoglycaemia; these findings suggest that insulin signalling in the VMH plays a role in CRRs to hypoglycaemia.
Recurrent hypoglycaemic events have been shown to impair VMH glucose-sensing neurons, resulting in failure to suppress insulin levels and enhance glucagon secretion during hypoglycaemia in type 1 diabetes and, to some extent, in type 2 diabetes. This may also lead to impaired sympatho-adrenal responses, thus initiating a vicious cycle that may lead to hypoglycaemia unawareness.
Alterations in glucose transport and/or metabolism have been hypothesised to be responsible for failure of CRRs due to repeated hypoglycaemic events. Studies in rats have shown increased expression of glucose transporters after acute or recurrent hypoglycaemia. In addition, GK expression [53] and hypothalamic glucose phosphorylation [54] have also been observed to be upregulated after recurrent hypoglycaemic events. Together, these observations suggest that recurrent hypoglycaemia may increase the capacity for glucose metabolism during subsequent hypoglycaemic events, thus inducing hypoglycaemia unawareness at the cellular level in glucose-sensing neurons and resulting in a diminished sympatho-adrenal response.
Although altered glucose transport/metabolism as a cause for hypoglycaemia unawareness has been suggested in animal models, these data have not been substantiated in humans. It has been shown that, during hypoglycaemia, glucose metabolism in the brain is similar in individuals with type 1 diabetes as compared with healthy control participants [55]. Furthermore, individuals with hypoglycaemia unawareness showed normal global brain glucose metabolism, although studies have identified brain regions with reduced glucose uptake in these individuals, such as the hypothalamus, prefrontal cortex and amygdala [56, 57]. Future clinical studies utilising enhanced imaging technologies to examine brain glucose uptake/metabolism in individuals with type 1 diabetes with and without hypoglycaemia unawareness are needed to define brain areas involved in this pathological condition. Findings from these studies may offer new targets for the development of therapeutic strategies to prevent hypoglycaemia unawareness in individuals with type 1 diabetes.
Conclusions and future directions
Studies focusing on identifying central glucose-sensing mechanisms involved in the regulation of glucose metabolism have highlighted the complexity and variety of these mechanisms, both at the cellular and circuit levels, which include multiple brain sites. In addition to the mechanisms discussed, it is important to consider other nutrient- and hormone-sensing mechanisms that may interact with glucose-sensing pathways in the modulation of glucose metabolism. Finally, as the majority of the findings highlighting these mechanisms have been performed in mouse and rat models (Table 1), investigations into their relevance in humans are warranted. Thus, until we fully understand the different components of these sensing mechanisms, how they interact with each other and their relevance to humans, it is unlikely that major therapeutic approaches aiming to centrally prevent and treat diabetes will be developed.
Table 1.
Models used in studies of hypothalamic glucose-sensing mechanisms
| Characteristics/model description | Species | References |
|---|---|---|
| T2DM | ||
| Obesity | Mouse and human | [1, 43, 44, 45] |
| HFD | Mouse | [12, 16, 20, 23, 40, 41, 46, 47] |
| HFD | Rat | [35, 48] |
| High sucrose diet | Mouse | [23] |
| Other (transgenic animal models) | ||
| Insulin receptor knockout in AgRP | Mouse | [12] |
| DREADD hM3DG inserted in AgRP-Cre | Mouse | [13] |
| POMC-GFP | Mouse | [15] |
| Mutant KATP channel subunit Kir6.2 in POMC | Mouse | [16] |
| TRPC3 knockout | Mouse | [17] |
| Mfn1 knockout in POMC | Mouse | [19] |
| Opa1 knockout in POMC | Mouse | [19] |
| Dnm1l knockout in POMC | Mouse | [21] |
| vGlut2 knockout in SF1 | Mouse | [23] |
| Ucp2 knockin in SF1; Ucp2 knockout | Mouse | [26] |
| Insulin receptor knockout in astrocytes | Mouse | [31] |
| Eno2 knockout in tanycytes | Mouse | [40] |
| Ucp2 knockout in microglia | Mouse | [47] |
| Insulin receptor knockout in neurons | Mouse | [48] |
DREADD, designer receptors exclusively activated by designer drugs; SF1, steroidogenic factor 1; T2DM, type 2 diabetes
Funding
Work in the authors’ laboratory is supported by NIH DK097566, DK107293, DK120321 grants.
Abbreviations
- AgRP
Agouti-related protein
- AMPK
AMP-activated protein kinase
- ARC
Arcuate nucleus
- CNS
Central nervous system
- CRR
Counterregulatory response
- CX
Connexin
- DRP1
Dynamin-related protein 1
- GK
Glucokinase
- HFD
High-fat diet
- ICV
Intracerebroventricular
- KATP
ATP-sensitive potassium (channel)
- LDH
Lactate dehydrogenase
- MBH
Mediobasal hypothalamus
- α-MSH
α-Melanocyte stimulating hormone
- NPY
Neuropeptide Y
- POMC
Pro-opiomelanocortin
- PVN
Paraventricular nucleus
- TRPC3
Transient receptor potential canonical type 3
- UCP2
Uncoupling protein 2
- VMH
Ventromedial hypothalamic nucleus
Footnotes
Authors’ relationships and activities The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Supplementary Information The online version contains a slideset of the figures for download available at https://doi.org/10.1007/s00125-021-05395-6.
References
- 1.Czech MP (2017) Insulin action and resistance in obesity and type2 diabetes. Nat Med 23(7):804–814. 10.1038/nm.4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roder PV, Wu B, Liu Y, Han W (2016) Pancreatic regulation of glucose homeostasis. Exp Mol Med 48:e219. 10.1038/emm.2016.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ritter S (2017) Monitoring and maintenance of brain glucose supply importance of hindbrain catecholamine neurons in this multifaceted task. In: Harris RBS (ed) Appetite and food intake: Central control, 2nd edn. CRC Press/Taylor& Francis, Boca Raton, pp 177–204 [PubMed] [Google Scholar]
- 4.Timper K, Bruning JC (2017) Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis Model Mech 10(6):679–689. 10.1242/dmm.026609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Routh VH (2010) Glucose sensing neurons in the ventromedial hypothalamus. Sensors 10(10):9002–9025. 10.3390/s101009002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao Q, Horvath TL (2008) Neuronal control of energy homeostasis. FEBS Lett 582(1):132–141. 10.1016/j.febslet.2007.11.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varela L, Horvath TL (2012) Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep 13(12):1079–1086. 10.1038/embor.2012.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG (2000) Central nervous system control of food intake. Nature 404(6778):661–671. 10.1038/35007534 [DOI] [PubMed] [Google Scholar]
- 9.Nijenhuis WA, Oosterom J, Adan RA (2001) AgRP(83–132) acts as an inverse agonist on the human-melanocortin-4 receptor. Mol Endocrinol 15(1):164–171. 10.1210/mend.15.1.0578 [DOI] [PubMed] [Google Scholar]
- 10.Mountjoy PD, Bailey SJ, Rutter GA (2007) Inhibition by glucose or leptin of hypothalamic neurons expressing neuropeptide Y requires changes in AMP-activated protein kinase activity. Diabetologia 50(1):168–177. 10.1007/s00125-006-0473-3 [DOI] [PubMed] [Google Scholar]
- 11.Fioramonti X, Contie S, Song Z, Routh VH, Lorsignol A, Penicaud L (2007) Characterization of glucosensing neuron subpopulations in the arcuate nucleus: Integration in neuropeptide Y and pro-opio melanocortin networks? Diabetes 56(5):1219–1227. 10.2337/db06-0567 [DOI] [PubMed] [Google Scholar]
- 12.Murphy BA, Fioramonti X, Jochnowitz N et al. (2009) Fasting enhances the response of arcuate neuropeptide Y-glucose-inhibited neurons to decreased extracellular glucose. Am J Physiol Cell Physiol 296(4):C746–C756. 10.1152/ajpcell.00641.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruud J, Steculorum SM, Bruning JC (2017) Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat Commun 8:15259. 10.1038/ncomms15259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konner AC, Janoschek R, Plum L et al. (2007) Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab 5(6):438–449. 10.1016/j.cmet.2007.05.004 [DOI] [PubMed] [Google Scholar]
- 15.Steculorum SM, Ruud J, Karakasilioti I et al. (2016) AgRP neurons control systemic insulin sensitivity via myostatin expression in brown adipose tissue. Cell 165(1):125–138. 10.1016/j.cell.2016.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pozo M, Claret M (2018) Hypothalamic control of systemic glucose homeostasis: The pancreas connection. Trends Endocrinol Metab 29(8):581–594. 10.1016/j.tem.2018.05.001 [DOI] [PubMed] [Google Scholar]
- 17.Ibrahim N, Bosch MA, Smart JL et al. (2003) Hypothalamic proopiomelanocortin neurons are glucose responsive and express KATP channels. Endocrinology 144(4):1331–1340. 10.1210/en.2002-221033 [DOI] [PubMed] [Google Scholar]
- 18.Parton LE, Ye CP, Coppari R et al. (2007) Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 449(7159):228–232. 10.1038/nature06098 [DOI] [PubMed] [Google Scholar]
- 19.Chretien C, Fenech C, Lienard F et al. (2017) Transient receptor potential canonical 3 (TRPC3) channels are required for hypothalamic glucose detection and energy homeostasis. Diabetes 66(2): 314–324. 10.2337/db16-1114 [DOI] [PubMed] [Google Scholar]
- 20.Wai T, Langer T (2016) Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab 27(2):105–117. 10.1016/j.tem.2015.12.001 [DOI] [PubMed] [Google Scholar]
- 21.Ramirez S, Gomez-Valades AG, Schneeberger M et al. (2017) Mitochondrial dynamics mediated by Mitofusin 1 is required for POMC neuron glucose-sensing and insulin release control. Cell Metab 25(6):1390–1399 e6. 10.1016/j.cmet.2017.05.010 [DOI] [PubMed] [Google Scholar]
- 22.Jin S, Diano S (2018) Mitochondrial dynamics and hypothalamic regulation of metabolism. Endocrinology 159(10):3596–3604. 10.1210/en.2018-00667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santoro A, Campolo M, Liu C et al. (2017) DRP1 suppresses leptin and glucose sensing of POMC neurons. Cell Metab 25(3):647–660. 10.1016/j.cmet.2017.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hennings TG, Chopra DG, DeLeon ER et al. (2018) In vivo deletion of beta-cell Drp1 impairs insulin secretion without affecting islet oxygen consumption. Endocrinology. 159(9):3245–3256. 10.1210/en.2018-00445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stanley SA, Kelly L, Latcha KN et al. (2016) Bidirectional electromagnetic control of the hypothalamus regulates feeding and metabolism. Nature 531(7596):647–650. 10.1038/nature17183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tong Q, Ye C, McCrimmon RJ et al. (2007) Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab 5(5):383–393. 10.1016/j.cmet.2007.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCrimmon RJ, Fan X, Cheng H et al. (2006) Activation of AMP-activated protein kinase within the ventromedial hypothalamus amplifies counterregulatory hormone responses in rats with defective counterregulation. Diabetes 55(6):1755–1760. 10.2337/db05-1359 [DOI] [PubMed] [Google Scholar]
- 28.Murphy BA, Fakira KA, Song Z, Beuve A, Routh VH (2009) AMP-activated protein kinase and nitric oxide regulate the glucose sensitivity of ventromedial hypothalamic glucose-inhibited neurons. Am J Physiol Cell Physiol 297(3):C750–C758. 10.1152/ajpcell.00127.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan O, Cheng H, Herzog R et al. (2008) Increased GABAergic tone in the ventromedial hypothalamus contributes to suppression of counterregulatory responses after antecedent hypoglycemia. Diabetes 57(5):1363–1370. 10.2337/db07-1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toda C, Kim JD, Impellizzeri D, Cuzzocrea S, Liu ZW, Diano S (2016) UCP2 regulates mitochondrial fission and ventromedial nucleus control of glucose responsiveness. Cell. 164(5):872–883. 10.1016/j.cell.2016.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pellerin L, Magistretti PJ (2004) Neuroscience. Let there be (NADH) light. Science 305(5680):50–52. 10.1126/science.1100428 [DOI] [PubMed] [Google Scholar]
- 32.Harris AL (2007) Connexin channel permeability to cytoplasmic molecules. Prog Biophys Mol Biol 94(1–2):120–143. 10.1016/j.pbiomolbio.2007.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sohl G, Willecke K (2004) Gap junctions and the connexin protein family. Cardiovasc Res 62(2):228–232. 10.1016/j.cardiores.2003.11.013 [DOI] [PubMed] [Google Scholar]
- 34.Allard C, Carneiro L, Grall S et al. (2014) Hypothalamic astroglial connexins are required for brain glucose sensing-induced insulin secretion. J Cereb Blood Flow Metab 34(2):339–346. 10.1038/jcbfm.2013.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia-Caceres C, Quarta C, Varela L et al. (2016) Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 166(4):867–880. 10.1016/j.cell.2016.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belanger M, Allaman I, Magistretti PJ (2011) Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab 14(6):724–738. 10.1016/j.cmet.2011.08.016 [DOI] [PubMed] [Google Scholar]
- 37.Ainscow EK, Mirshamsi S, Tang T, Ashford ML, Rutter GA (2002) Dynamic imaging of free cytosolic ATP concentration during fuel sensing by rat hypothalamic neurones: Evidence for ATP-independent control of ATP-sensitive K+ channels. J Physiol 544(2):429–445. 10.1113/jphysiol.2002.022434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song Z, Routh VH (2005) Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 54(1):15–22. 10.2337/diabetes.54.1.15 [DOI] [PubMed] [Google Scholar]
- 39.Lam TK, Gutierrez-Juarez R, Pocai A et al. (2007) Brain glucose metabolism controls the hepatic secretion of triglyceride-rich lipoproteins. Nat Med 13(2):171–180. 10.1038/nm1540 [DOI] [PubMed] [Google Scholar]
- 40.Abraham MA, Rasti M, Bauer PV, Lam TKT (2018) Leptin enhances hypothalamic lactate dehydrogenase A (LDHA)-dependent glucose sensing to lower glucose production in high-fat-fed rats. J Biol Chem 293(11):4159–4166. 10.1074/jbc.RA117.000838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elizondo-Vega R, Cortes-Campos C, Barahona MJ, Oyarce KA, Carril CA, Garcia-Robles MA (2015) The role of tanycytes in hypothalamic glucosensing. J Cell Mol Med 19(7):1471–1482. 10.1111/jcmm.12590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elizondo-Vega RJ, Recabal A, Oyarce K (2019) Nutrient sensing by hypothalamic tanycytes. Front Endocrinol 10:244. 10.3389/fendo.2019.00244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanders NM, Dunn-Meynell AA, Levin BE (2004) Third ventricular alloxan reversibly impairs glucose counterregulatory responses. Diabetes 53(5):1230–1236. 10.2337/diabetes.53.5.1230 [DOI] [PubMed] [Google Scholar]
- 44.Yoo S, Cha D, Kim S et al. (2020) Tanycyte ablation in the arcuate nucleus and median eminence increases obesity susceptibility by increasing body fat content in male mice. Glia 68(10):1987–2000. 10.1002/glia.23817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Andrade IS, Zemdegs JC, de Souza AP et al. (2015) Diet-induced obesity impairs hypothalamic glucose sensing but not glucose hypothalamic extracellular levels, as measured by microdialysis. Nutr Diabetes 5:e162. 10.1038/nutd.2015.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ashford ML, Boden PR, Treherne JM (1990) Glucose-induced excitation of hypothalamic neurones is mediated by ATP-sensitive K+ channels. Pflugers Arch 415(4):479–483. 10.1007/BF00373626 [DOI] [PubMed] [Google Scholar]
- 47.Taylor R (2012) Insulin resistance and type 2 diabetes. Diabetes 61(4):778–779. 10.2337/db12-0073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Magnan C, Levin BE, Luquet S (2015) Brain lipid sensing and the neural control of energy balance. Mol Cell Endocrinol 418(Pt 1):3–8. 10.1016/j.mce.2015.09.019 [DOI] [PubMed] [Google Scholar]
- 49.Wilcox G (2005) Insulin and insulin resistance. Clin Biochem Rev 26(2):19–39 [PMC free article] [PubMed] [Google Scholar]
- 50.Thaler JP, Yi CX, Schur EA et al. (2012) Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest 122(1): 153–162. 10.1172/JCI59660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim JD, Yoon NA, Jin S, Diano S (2019) Microglial UCP2 mediates inflammation and obesity induced by high-fat feeding. Cell Metab 30(5):952–962 e5. 10.1016/j.cmet.2019.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diggs-Andrews KA, Zhang X, Song Z, Daphna-Iken D, Routh VH, Fisher SJ (2010) Brain insulin action regulates hypothalamic glucose sensing and the counterregulatory response to hypoglycemia. Diabetes 59(9):2271–2280. 10.2337/db10-0401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dunn-Meynell AA, Routh VH, Kang L, Gaspers L, Levin BE (2002) Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes 51(7):2056–2065. 10.2337/diabetes.51.7.2056 [DOI] [PubMed] [Google Scholar]
- 54.Osundiji MA, Evans ML (2011) Hypothalamic glucose sensing and glycaemic disease. Curr Diabetes Rev 7(2):84–98. 10.2174/157339911794940701 [DOI] [PubMed] [Google Scholar]
- 55.van de Ven KC, van der Graaf M, Tack CJ, Heerschap A, de Galan BE (2012) Steady-state brain glucose concentrations during hypoglycemia in healthy humans and patients with type 1 diabetes. Diabetes 61(8):1974–1977. 10.2337/db11-1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cranston I, Reed LJ, Marsden PK, Amiel SA (2001) Changes in regional brain 18F-fluorodeoxyglucose uptake at hypoglycemia in type 1 diabetic men associated with hypoglycemia unawareness and counter-regulatory failure. Diabetes 50(10):2329–2336. 10.2337/diabetes.50.10.2329 [DOI] [PubMed] [Google Scholar]
- 57.Bingham EM, Dunn JT, Smith D et al. (2005) Differential changes in brain glucose metabolism during hypoglycaemia accompany loss of hypoglycaemia awareness in men with type 1 diabetes mellitus. An [11C]-3-O-methyl-D-glucose PET study. Diabetologia 48(10): 2080–2089. 10.1007/s00125-005-1900-6 [DOI] [PubMed] [Google Scholar]