

Severe fever with thrombocytopenia syndrome (SFTS) virus (SFTSV) is an emerging highly pathogenic phlebovirus. The syndrome is characterized by the substantial production of inflammatory cytokines and chemokines, described as a cytokine storm, which correlates with multiorgan failure and high mortality.
KEYWORDS: SFTSV NSs, NF-κB, cytokine storm, TBK1, viral pathogenesis, NF-κB
ABSTRACT
Severe fever with thrombocytopenia syndrome (SFTS) virus (SFTSV) is an emerging highly pathogenic phlebovirus. The syndrome is characterized by the substantial production of inflammatory cytokines and chemokines, described as a cytokine storm, which correlates with multiorgan failure and high mortality. SFSTV nonstructural (NSs) protein was suggested to mediate the pathogenesis by inhibiting antiviral interferon signaling in the host. However, whether SFTSV NSs protein mediates the induction of a fatal cytokine storm remains unaddressed. We demonstrated that SFTSV NSs promotes the hyperinduction of cytokine/chemokine genes in vitro, reminiscent of a cytokine storm. Using gene deletion and pharmacological intervention, we found that the induced cytokine storm is driven by the transcription factor NF-κB. Our investigation revealed that TANK-binding kinase 1 (TBK1) suppresses NF-κB signaling and cytokine/chemokine induction in a kinase activity-dependent manner and that NSs sequesters TBK1 to prevent it from suppressing NF-κB, thereby promoting the activation of NF-κB and its target cytokine/chemokine genes. Of note, NF-κB inhibition suppressed the induction of proinflammatory cytokines in SFTSV-infected type I interferon (IFN-I) receptor 1-deficient (Ifnar1−/−) mice. These findings establish the essential role of NSs in SFTS pathogenesis and suggest NF-κB as a possible therapeutic target.
INTRODUCTION
Severe fever with thrombocytopenia syndrome (SFTS) is a life-threatening hemorrhagic fever-like disease caused by tick-borne SFTS virus (SFTSV), a member of the Phlebovirus genus, Phenuiviridae family, Bunyavirales order (1, 2). SFTSV was first identified in China in 2009 and subsequently detected in South Korea and Japan (3–6). Moreover, another phlebovirus that is genetically closely related to SFTSV, Heartland virus, was isolated from patients in the United States a few years later (7).
Due to its high fatality rate (between 12 and 30%), the heavy disease burden, and the lack of specific and efficient medical countermeasures, SFTSV was listed by the World Health Organization as one of the top prioritized pathogens for research and development (8). Since its identification in 2009, significant progress has been achieved regarding our understanding of the molecular biology of the virus and the development of diagnostic assays and reagents. On the other hand, little progress has been made on its pathophysiology, immunological responses, and treatment regimens. Thus, there is a growing and urgent need for a detailed understanding of the pathology of SFTS, which is essential for the development of medical interventions.
The hallmarks of SFTSV infection are high fever and low platelet count (thrombocytopenia). Other commonly reported symptoms include vomiting, diarrhea, and elevated liver enzymes. The disease has three clinical phases: fever, multiorgan dysfunction, and convalescence. Pathological and clinical studies on SFTS revealed that the mechanisms behind the high fatality rate are the virus-induced cytokine storm, coagulopathy due to disseminated intravascular coagulation, and the progression of multiorgan dysfunction into multiorgan failure in severe cases (9, 10). The cytokine storm is an excessive inflammatory reaction characterized by the hyperproduction of proinflammatory cytokines and chemokines in response to virus detection. It is a sign of severe inflammation, which can cause serious pathological changes and result in multiorgan dysfunction commonly observed in Ebola, dengue, and Crimean-Congo hemorrhagic fever (11–13).
Several reports have described the cytokine storm in SFTS patients and the kinetics of cytokines and chemokine induction. Based on these reports, tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), interleukin-10 (IL-10), interleukin-6 (IL-6), and C-X-C motif ligand 10 (CXCL10), among others, were found to correlate with disease severity. Serum levels of TNF-α and monocyte chemoattractant protein 1 (MCP-1) were significantly higher in SFTS patients than in healthy persons, and in severe SFTS patients than in mild SFTS patients. Interleukin-8 (IL-8), among other chemokines, exhibited a unique pattern of increase in fatal cases but not in nonfatal cases (14–18). Moreover, increased levels of soluble interleukin-2 receptor alpha (IL-2Rα) correlated with a lower platelet count, decreased platelet-derived growth factor BB levels, and increased serum alanine aminotransferase, aspartate aminotransferase, and creatinine kinase levels in SFTS patients (17).
These data support the hypothesis that the cytokine storm is a prognostic marker in the acute phase of SFTSV infection.
Several transcription factors were reported to mediate the cytokine storm in response to virus detection, one of which is nuclear factor κB (NF-κB), which is a major regulator of the inflammatory response. NF-κB activation is a rapid event that does not require protein synthesis and functions in essential regulatory processes in the host cell. Indeed, many viruses have developed different strategies to alter the NF-κB pathway, including human immunodeficiency virus type 1, herpesviruses, human T-cell leukemia virus type 1 (HTLV-1), and influenza virus (19–25).
Of note, it has been reported that the nonstructural protein of SFTSV, NSs, forms cytoplasmic granules and adsorbs the TANK-binding kinase 1 (TBK1) therein to block its functions (26–28). NSs also targets retinoic acid-inducible gene І (RIG-I) and signal transducer and activator of transcription 1 and 2 (STAT1 and STAT2) (27, 29, 30). Thus, NSs is an important viral immune evasion component. However, its relationship with the cytokine storm is unclear.
Wild-type mice are resistant to SFTSV infection for lethal symptoms; however, type I interferon (IFN-I) receptor 1-deficient (Ifnar1−/−) mice are highly susceptible and used as a lethal SFTS model (31). SFTSV infection in Ifnar1−/− mice revealed the hyperproduction of inflammatory cytokines, resembling human SFTS. However, mice deficient for both mitochondrial antiviral-signaling protein (MAVS) and myeloid differentiation primary response protein (MyD88), which are responsible for both IFN-I and cytokine production, survived upon SFTSV infection (32). These results further emphasized the relationship between the fatality of SFTS and cytokine hyperproduction.
We aimed to delineate the mechanism of cytokine overexpression in response to SFTSV, particularly the role of the essential viral effector NSs. We demonstrated that NSs is involved as a hyperactivator of NF-κB, which mediated the cytokine storm in vitro. Of note, inhibition of NF-κB signaling rescued the cytokine storm by SFTSV infection both in vitro and in vivo. These findings present new insights into the pathogenesis of fatal SFTS and may promote the understanding of virus-host interactions and the development of antiviral therapeutics.
RESULTS
SFTSV-NSs altered the expression of cytokine and chemokine genes and their suppression by an NF-κB inhibitor.
As NSs of SFTSV has been linked to host immune alteration and evasion of innate antiviral immunity, we utilized a Flag-tagged SFTSV-NSs expression vector to transfect HeLa cells and investigated the effects of NSs on host immune gene expression related to cytokine/chemokine signaling. Overexpression of NSs marginally altered basal expression of the IL-8, IL-6, CXCL1, CXCL2, CCL2, and IL2RA genes (Fig. 1A). However, their expression induced by Sendai virus (SeV) infection was markedly increased by NSs. These results are consistent with the increased expression of MCP-1 (encoded by CCL2) and IL-2Rα in severe SFTS cases (Fig. 1B). Of note, an inhibitor of NF-κB, TPCA-1, strongly inhibited expression of these genes in HeLa cells, consistent with the reports that NF-κB plays an essential role in their regulation (33–38). To further demonstrate NF-κB involvement, we knocked down the p65 subunit of NF-κB by small interfering RNA (siRNA) (Fig. 1C). Efficient knockdown of p65 abolished the hyperactivation of the IL-8 gene by NSs. As an alternative approach, we generated mutant HeLa cells with an IKKA−/− IKKB−/− (IKKA,B double knockout [DKO]) or IKKG−/− (IKKG KO) genotype by gene editing. As expected, the strong IL-8 gene activation by NSs/SeV was undetectable in the mutant cells (Fig. 1D). These results prompted us to hypothesize that NSs targets NF-κB for hyperactivation of these genes.
FIG 1.

SFTSV NSs altered the expression of cytokine and chemokine genes and their suppression by an NF-κB inhibitor. (A) Expression of the IL-8, IL-6, CXCL1, CXCL2, CCL2, and IL2RA genes in HeLa cells expressing or not expressing NSs-Flag was quantified by reverse transcription-quantitative PCR (qRT-PCR) upon mock treatment or SeV infection for 9 h in the absence or presence of TPCA-1 (20 µM) (n = 3). DMSO, dimethyl sulfoxide. (B) Serum MCP-1 and IL-2Rα levels in SFTS patients. Comparison of fatal and nonfatal cases is shown. (C) (Left) HeLa cells were transfected with expression vector for control (siNC) or siRNA targeting p65 of NF-κB (sip65). Expression of p65 was examined by immunoblotting. (Right) Expression of the IL-8 gene in HeLa cells transfected with either sip65 or siNC in the presence or absence of NSs expression and SeV infection, as indicated (n = 3). (D) Wild-type HeLa cells and mutant HeLa cells deficient in both IKKα and -β (IKKA,B DKO) or IKKγ (IKKG KO) were stimulated by SeV infection in the presence or absence of NSs expression, as indicated. IL-8 gene expression was monitored by qRT-PCR (n = 3). Data are presented as means ± SEM. The Student t test was used for statistical analysis (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
NSs facilitated SeV- and TNF-α-induced activation of NF-κB.
Next, we examined the activation of NF-κB in the presence and absence of NSs. In unstimulated cells, NF-κB resides in the cytoplasm as an inert complex with its inhibitor IκB, and upon stimulation, NF-κB is liberated from inhibition and translocates to the nucleus. To monitor these processes, we performed subcellular fractionation and monitored nuclear NF-κB by immunoblotting (Fig. 2A). In unstimulated HeLa cells, the p65 subunit of NF-κB is absent in the nuclear fraction. Overexpression of NSs alone resulted in the detection of p65 in the nucleus, albeit at a lower level than that induced by SeV infection. Combined stimuli by NSs and SeV infection or NSs and TNF-α (Fig. 2B) further augmented the nuclear p65 translocation and the phosphorylation of NF-κB inhibitor IκB-α. It is noteworthy that such augmentation of nuclear NF-κB is absent in IKKA−/− IKKB−/− or IKKG−/− HeLa cells (Fig. 2B), indicating that NSs induces IKK complex-dependent nuclear translocation of NF-κB. As an alternative approach, we examined nuclear translocation of p65 by immunostaining and fluorescence quantification (Fig. 2C). Consistent with subcellular fractionation, p65 nuclear translocation induced by SeV was further increased by NSs. Moreover, TNF-α-induced p65 nuclear translocation was markedly augmented by NSs, suggesting that increased NF-κB nuclear translocation by NSs is stimulus independent. Next, the effects of NSs on transcriptional activity of NF-κB were examined using an NF-κB-dependent luciferase reporter gene (Fig. 2D). A small but significant increase was observed by overexpression of NSs alone (mock). Upon stimulation by different viral infections and TNF-α treatment, the reporter was activated and further enhanced by NSs. Taken together, the results show that NSs potently augments NF-κB activation induced by viral or TNF-α treatment.
FIG 2.

NSs accelerated the activation of NF-κB. (A) HeLa cells (lanes 1 and 3) or those transfected with the expression vector for NSs-Flag (lanes 2 and 4) were mock treated (lanes 1 and 2) or infected with SeV for 9 h (lanes 3 and 4). The cells were subjected to subcellular fractionation. Cytoplasmic and nuclear extracts were examined by immunoblotting using the indicated antibodies. (A) A representative results of three independent experiments is shown. (B) Wild-type, IKKA,B DKO, and IKKG KO HeLa cells (lanes 1 and 3) or those transfected with the expression vector for NSs-Flag (lanes 2 and 4) were mock treated (lanes 1 and 2) or treated with recombinant TNF-α (20 ng/ml) for 30 min (lanes 3 and 4). The cells were subjected to subcellular fractionation. Cytoplasmic and nuclear extracts were examined by immunoblotting using the indicated antibodies. Comparison of nuclear p65 and cytoplasmic p-IκB-α is shown, with a representative result of two independent experiments for each cell line. (C) HeLa cells were transfected with the empty or expression vector for Flag-NSs. Twenty-four hours later, cells were mock treated or infected with SeV for 9 h or treated with TNF-α (20 ng/ml) for 0.5 or 2 h. Cells were fixed and stained for p65 for microscopy (left). Nuclear intensities were quantified (right). A representative result of two independent experiments is shown. (D) HEK293T cells were cotransfected with the NF-κB-dependent reporter gene p55A2 and pRL-TK for normalization with or without NSs-Flag. Twenty-four hours later, cells were mock treated or infected with different viruses (SeV, 9 h; NDV, 9 h; and EMCV, 12 h) or treated with TNF-α (20 ng/ml) for 4 or 24 h (n = 3). Luciferase activity was assessed using pRL-TK activity as a reference. Data are presented as means ± SEM. The Student t test was used for statistical analysis.
TBK1 negatively regulates NF-κB.
We next investigated if the deletion of TBK1 causes hyperactivation of NF-κB. HeLa TBK1+/+ and HeLa TBK1−/− cells were mock treated or stimulated by SeV, and κB motif DNA binding activity was examined by electrophoretic mobility shift assay (EMSA) (Fig. 3A). Deletion of TBK1 resulted in hyperactivation of NF-κB DNA binding activity, which was confirmed by a blocking assay with specific antibodies. Moreover, immunoblotting revealed enhanced phosphorylation of IκB-α in stimulated and unstimulated TBK1−/− cells compared to their wild-type counterpart (Fig. 3B), suggesting that TBK1 attenuates phosphorylation of IκB-α. We also assessed the transcriptional activity of NF-κB using a κB motif-regulated reporter gene in HEK293T cells (Fig. 3C). Consistent with κB binding activity, deletion of TBK1 resulted in higher promoter activity stimulated by SeV. When TBK1 expression was complemented by transfection, the increased reporter activity was reversed. Of note, TBK1−/− cells exhibited basal κB binding (Fig. 3A) and reporter activities (Fig. 3C) compared with TBK1+/+ cells. We further compared TBK1+/+ and TBK1−/− cells for SeV-induced IL-8, CXCL1, and CXCL2 gene expression (Fig. 3D). Again, deletion of TBK1 resulted in hyperexpression of these genes and the NF-κB inhibitor TPCA-1 inhibited their expression. To further explore the involvement of the catalytic activity of TBK1 in increased gene expression, we used its pharmacological inhibitor, BX795 (Fig. 3E). The inhibition of TBK1 kinase activity was sufficient to reproduce hyper-gene expression caused by TBK1 deletion. As expected, inhibition of TBK1 abrogated SeV-induced IFNB1 gene induction. Collectively, these results suggest that TBK1 is an essential negative regulator of NF-κB in a catalytic-activity-dependent manner.
FIG 3.

TBK1 negatively regulates NF-κB. (A) Wild-type and mutant HeLa TBK1−/− cells were mock treated or infected with SeV for 9 h and subjected to subcellular fractionation. Nuclear extracts were prepared and subjected to EMSA using a γ-32P-labeled κB motif DNA probe. The complex of NF-κB and probe is indicated by an arrow. For blocking assay, the extracts were incubated with the indicated antibodies for 10 min prior to probe addition (left). EMSA bands intensities were quantified (right). A representative result of three independent experiments is shown. (B) Wild-type and mutant HeLa TBK1−/− cells were either mock treated, infected with SeV for 9 h, or treated with recombinant TNF-α (20 ng/ml) for 30 min before lysis. Whole-cell extracts were examined by immunoblotting using the indicated antibodies. (C) Wild-type and mutant HEK293T TBK1−/− cells were transfected with reporter genes p-55A2 and pRL-TK. Where indicated, cells were cotransfected with the expression vector for TBK1. Twenty-four hours later, cells were left unstimulated (mock) or stimulated with SeV infection as indicated. Luciferase activity was quantified using pRL-TK activity as an internal reference (n = 3). (D) Wild-type and mutant HeLa TBK1−/− cells were mock treated or infected with SeV for 9 h in the presence or absence of TPCA-1 (20 μM). The expression of the IL-8, CXCL1, and CXCL2 genes was measured by qRT-PCR (n = 3). (E) HeLa cells were mock treated or infected with SeV for 9 h in the presence or absence of BX795 (10 μM). The expression of the IL-8, CXCL1, CXCL2, and IFNB1 genes was measured by qRT-PCR (n = 3). Data are presented as means ± SEM. The Student t test was used for statistical analysis.
Physical interaction between TBK1 and IKKβ is interrupted by NSs.
As we found negative regulatory function of TBK1 against NF-κB activation, we sought to reveal the molecular mechanism. Physical interaction between TBK1 and IKKβ was examined by coimmunoprecipitation. First, we pulled down TBK1 from HEK293T IKKB−/− and IKKβ-reconstituted HEK293T IKKB−/− cells (Fig. 4A). This revealed an interaction between IKKβ and TBK1. Next, we used HEK293T TBK1−/− cells and their derivatives reconstituted with TBK1 (Fig. 4B). This revealed an interaction between IKKβ and TBK1, consistent with the previous result (39). To test whether NSs impacts this interaction, we used the latter cells and their derivatives transfected with NSs and pulled down with TBK1 antibody. Remarkably, the interaction between IKKβ and TBK1 was completely disrupted by NSs, a finding that may explain the potent activating effect of NSs on NF-κB.
FIG 4.

Physical interaction between TBK1 and IKKβ is disrupted by NSs. (A) Whole-cell lysates were prepared from HEK293T IKKB−/− cells and IKKβ-expressing HEK293T cells, prepared by stable complementation of the HEK293T IKKB−/− cells with an expression vector. The lysates were subjected to immunoprecipitation by anti-TBK1 antibody, followed by immunoblotting with anti-IKKβ or anti-TBK1 antibody. A representative result of two independent experiments is shown. (B) Whole-cell lysates were prepared from HEK293T TBK1−/− cells and TBK1-expressing cells, prepared by stable complementation of the HEK293T TBK1−/− cells with the expression vector for TBK1. The lysates were subjected to immunoprecipitation by anti-TBK1 antibody, followed by immunoblotting with anti-IKKβ or anti-TBK1 antibody. A representative result of two independent experiments is shown. (C) Whole-cell lysates (WCE) were prepared from HEK293T TBK1−/− cells and TBK1-expressing cells, prepared by stable complementation of the HEK293T TBK1−/− cells with the expression vector for TBK1. The cells were overexpressed with NSs-Flag for 24 h before lysis. The lysates were subjected to immunoprecipitation by anti-TBK1 antibody or IgG antibody as a control, followed by immunoblotting with the indicated antibodies. A representative result of two independent experiments is shown.
NF-κB inhibition ameliorates SFTSV-induced cytokine induction.
Finally, to address the physiological relevance of our observations, cytokine and chemokine gene expression was examined in an SFTSV infection model in vitro (Fig. 5A) and in vivo (Fig. 5B). Consistent with our observations, SFTSV strongly induced the expression of the IL-8, TNFA, and CCL2 genes in HEK293T cells, in addition to Il6, Cxcl1, Tnfa, and Ccl2 in type I interferon (IFN-I) receptor 1-deficient (Ifnar1−/−) mice, which was abrogated by TPCA-1. This was unlikely to be due to the inhibition of viral replication by TPCA-1 because TPCA-1 treatment did not affect viral nucleoprotein (NP) expression.
FIG 5.

NF-κB inhibitor inhibited the hyperinduction of cytokines by SFTSV infection. (A) HEK293T cells were mock treated or infected with SFTSV at a MOI of 1 × 103 or 1 × 104 TCID50 in the absence or presence of TPCA-1 (20 μM). The expression of the IL-8, TNFA, CCL2, and viral nucleoprotein NP genes was quantified by qRT-PCR at 24 and 48 h of infection. (B) Fourteen Ifnar−/− mice were intraperitoneally inoculated with 2 × 106 TCID50 of SFTSV on day 0. On day 2, half of the mice were given 1 mg of TPCA-1 intraperitoneally, and the rest were given PBS as a control. On day 3, the spleen was harvested from each mouse and RNA was extracted, purified, and reverse transcribed. Then, expression of Cxcl1, Il-6, Ccl2, Tnfa, and viral nucleoprotein NP genes was quantified by qRT-PCR. Data are presented as means ± SEM. The Student t test was used for statistical analysis.
DISCUSSION
Wild-type mice survive SFTSV infection after limited viral replication. Ifnar1−/− mice are used as a lethal infection model for SFTSV. Therefore, the failure of type I IFN (IFN-I) function was suspected as the basis of SFTS pathogenesis. However, it was previously reported that viral NSs strongly inhibits the production and activity of IFN-I (27, 40). These observations do not explain why Ifnar1+/+ mice resist fatal infection. On the other hand, SFTS patients and infected Ifnar1−/− mice exhibit hyperproduction of inflammatory cytokines and chemokines, termed the cytokine storm. In this study, we demonstrated that NSs promotes the SeV-induced cytokine storm in cell culture (Fig. 1), suggesting that NSs plays a central role in the hyperinflammation. Our analyses using a pharmacological inhibitor and gene knockdown and knockout revealed that NF-κB plays an essential role in the hyperactivation of cytokines and chemokines. Furthermore, we found that NSs strongly promotes SeV-induced activation of the nuclear translocation of NF-κB and NF-κB-dependent reporter gene activation (Fig. 2). Similarly, TNF-α-induced nuclear translocation of NF-κB and NF-κB-dependent reporter gene activation were promoted by NSs (Fig. 2).
In cells, NSs forms microscopic aggregates where TBK1 is sequestered, resulting in the blockade of signaling to activate IFN-I genes (26, 28). We investigated if TBK1 inhibition by NSs is also related to increased NF-κB activation. To address that, we generated TBK1−/− cells and observed the effect on NF-κB signaling. TBK1−/− cells exhibited enhanced phosphorylation of IκB-α, an event that inevitably precedes NF-κB nuclear localization (Fig. 3B), and increased NF-κB DNA binding activity (Fig. 3A). Furthermore, a pharmacological inhibitor of TBK1 resulted in hyperinduction of the cytokines and chemokines (Fig. 3E), suggesting that TBK1 negatively regulates NF-κB. Reconstitution of TBK1−/− cells with a TBK1 expression vector reversed the hyperactivation of NF-κB transcriptional activity (Fig. 3C). We demonstrated the physical interaction between TBK1 and IKKβ (Fig. 4). Alarmingly, NSs disrupted this interaction, as validated by pulldown and Western blot analysis (Fig. 4), suggesting that TBK1 inhibits the IKK complex through phosphorylation of a hypothetical substrate, which has yet to be identified, and that NSs releases the brakes on NF-κB by inhibiting TBK1 from interacting with IKK complex, thereby activating an NF-κB-dependent cytokine storm. Of note, phosphorylation of TBK1 by IKKβ is required for full TBK1 activation in TLR3/4-stimulated cells (39) and IKKβ inhibition inhibits IRF-3 activation. Thus, the cross talk between TBK1 and IKKβ is essential for homeostatic regulation. Our model is summarized in Fig. 6. Under physiological conditions, TBK1 attenuates NF-κB activity, thereby limiting the activation of cytokine and chemokine genes upon viral infection to avoid harmful inflammation. In SFTSV-infected cells, NSs strongly blocks TBK1, resulting in the hyperactivation of inflammation and blockade of IFN-I, causing the fatal cytokine storm. TNF-α, one of the inflammatory cytokines, may participate in positive feedback of NF-κB activation, as demonstrated in Fig. 2. This may explain why Tbk1−/− mice exhibit embryonic lethality (41).
FIG 6.
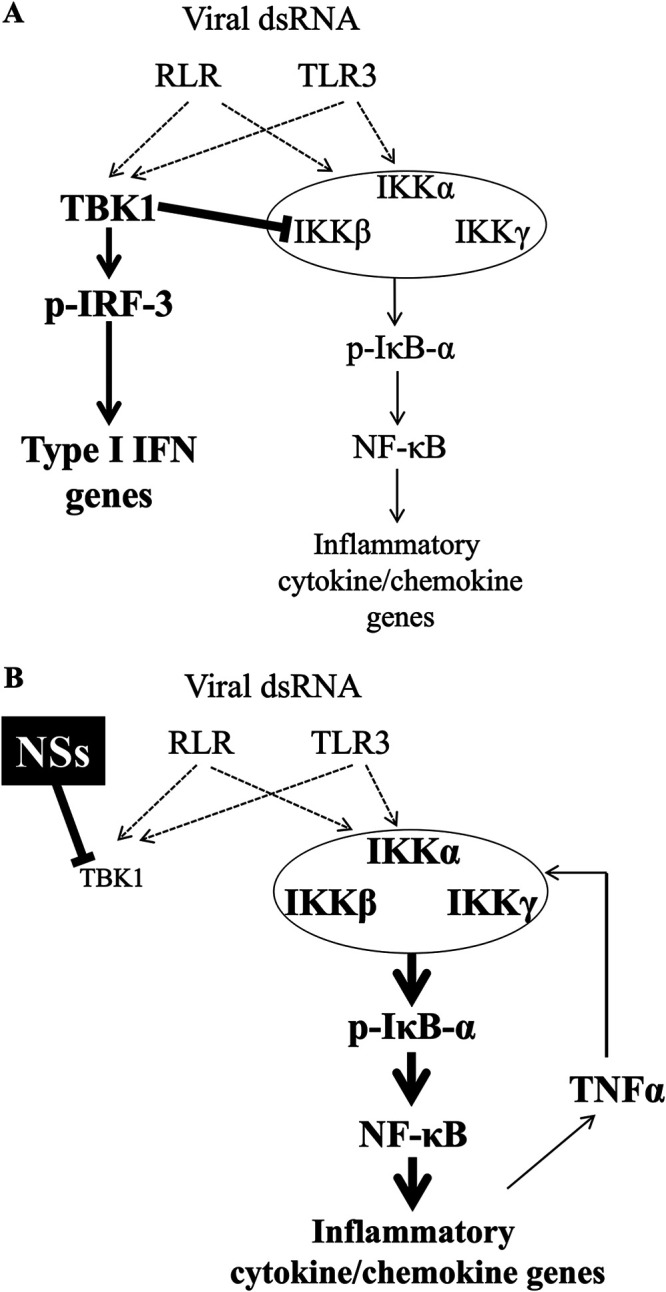
A model for the regulation of type I IFN genes and inflammatory cytokine and chemokine genes by TBK1. (A) Attenuation of NF-κB by TBK1. dsRNA, double-stranded RNA. (B) Derepression of NF-κB by NSs through TBK1 inhibition. See text.
Although several reports have addressed the activation of NF-κB by different viral regulatory and nonstructural proteins, the mechanism provided for SFTSV NSs is novel and unique. Other viruses activate NF-κB by yet-unknown mechanisms, such as human respiratory syncytial virus (HRSV) through its NS1 and NS2 proteins (42) and hepatitis C virus through its NS5A protein (43).On the other hand, the regulatory Tax-1 protein of human T-cell leukemia virus type 1 (HTLV-1) directly binds to the adapter protein NEMO, thus causing the constitutive activation of the IKK complex and NF-κB (44).
NF-κB-mediated inflammation was also reported in severe acute respiratory syndrome coronavirus (SARS-CoV) disease. NF-κB inhibitors reduced inflammation seen in infected lungs, with a decrease in the expression of proinflammatory cytokines and chemokines. Like NSs in SFTSV, the viral E protein induced this NF-κB-mediated inflammatory response, with a possible role of the SARS-CoV nucleoprotein N (45, 46). Another example is provided by the NS1 protein of dengue virus, which potentiates proinflammatory cytokine production by upregulating Toll-like receptor 2 (TLR-2) and TLR-6 (47). Collectively, these data show that the inflammatory cytokine storm can be elicited by different regulatory viral proteins through various mechanisms.
Our study highlights TBK1 as a critical regulator between antiviral and inflammatory responses upon viral infection. We propose the suppression of NF-κB as a promising therapy for SFTS, and compelling evidence comes from our experiment in Ifnar−/− mice (Fig. 5B). This idea is supported by the observation that SFTSV infection in Mavs−/− Myd88−/− mice suppresses the induction of inflammatory cytokines as well as IFN-I; however, no lethality was developed (32).
MATERIALS AND METHODS
Chemicals.
TPCA-1 and BX795 were purchased from TOCRIS.
RNA isolation and real-time qPCR.
Total RNA was harvested from cell lines using TRIzol reagent (Invitrogen) and subjected to DNase I treatment (Roche Diagnostics). cDNA was generated using a high-capacity cDNA reverse transcription kit (Applied Biosystems). Real-time quantitative PCR (qPCR) was performed on the Step One plus real-time PCR system (Applied Biosystems) using TaqMan fast universal PCR master mix (Applied Biosystems), Thunderbird probe qPCR mix (Toyobo), and Fast SYBR green master mix (Applied Biosystems) according to the manufacturers’ instructions. Sequences of the primers used in real-time PCR are listed in Table 1.
TABLE 1.
Sequences for SYBR green real-time PCR primers
| Target gene | SYBR green forward primer (5′→3′) | SYBR green reverse primer (5′→3′) |
|---|---|---|
| Human GAPDH | CTGCACCACCAACTGCTTAG | GTCTTCTGGGTGGCAGTGAT |
| Human IL-6 | CCAGTTCCTGCAGAAAAAGGC | AAGCTGCGCAGAATGAGATG |
| Human IL-8 | CTGCGCCAACACAGAAATTA | TGAATTCTCAGCCCTCTTCA |
| Human IFN-β | AGTCTCATTCCAGCCAGTGC | AGCTGCAGCAGTTCCAGAAG |
| Human CXCL1 | AACCGAAGTCATAGCCACAC | GTTGGATTTGTCACTGTTCAGC |
| Human CXCL2 | CCCATGGTTAAGAAAATCATCG | CTTCAGGAACAGCCACCAAT |
| Human CCL2 | AGCAAGTGTCCCAAAGAAGC | TCTTCGGAGTTTGGGTTTGC |
| Human IL-2Rα | TGGGAAAATGAAGCCACAGAG | TGAGGCTTCTCTTCACCTGGA |
| Human TNF-α | AGCCCATGTTGTAGCAAACC | TGAGGTACAGGCCCTCTGAT |
| Mouse GAPDH | CCCCAGCAAGGACACTGAGCAAG | GGGGTCTGGGATGGAAATTGTGAGG |
| Mouse CXCl1 | ACCCGCTCGCTTCTCTGT | AAGGGAGCTTCAGGGTCAAG |
| Mouse IL-6 | CATGTTCTCTGGGAAATCGTGG | GTACTCCAGGTAGCTATGGTAC |
| Mouse CCL2 | AAAAACCTGGATCGGAACCAA | CGGGTCAACTTCACATTCAAAG |
| Mouse TNF-α | GGCATGGATCTCAAAGACAACC | CAGGTATATGGGCTCATACCAG |
Immunostaining.
Cells in 8-well chamber plates (Matsunami) were fixed in 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and treated with 0.5% bovine serum albumin (BSA) for 30 min at room temperature before overnight incubation with the desired primary antibody at 4°C. A secondary antibody was applied and incubated with the samples for 1 h at room temperature. After the antibody treatment, cells were counterstained with DAPI (4′,6-diamidino-2-phenylindole) and later observed under a confocal microscope (TCS-SP8; Leica Microsystems). Fluorescence intensity was quantified by ImageJ software. For quantification figures, black background from mock cell images was considered the blank, and nuclear fluorescence in mock cells was quantified accordingly. Then, fluorescence from the nuclei of other cells was divided by fluorescence of mock nuclei to yield the folds of increase presented on the y axis in Fig. 2B.
Subcellular fractionation and electrophoretic mobility shift assay.
HeLa cells were subjected to the following subcellular fractionation protocol. Cell pellets obtained from centrifuging 106 cells from 10-cm dishes were suspended in 100 μl of 0.1% NP-40, mixed thoroughly, and immediately centrifuged. The supernatant was labeled as the cytoplasmic fraction. Pellets were washed at least twice with 500 μl of 0.1% NP-40 to prevent cytoplasmic contamination of the nuclear fraction. They were then lysed with 80 μl of radioimmunoprecipitation (RIPA) buffer (protease inhibitors added), mixed well, and incubated at 4°C for 30 min before the last centrifugation step. The supernatant was kept as the nuclear fraction of the sample.
Nuclear extracts (10 μg) were incubated for 30 min at room temperature with 0.1 pmol of γ−32P-labeled NF-κB oligonucleotide probe in a final reaction volume of 10 μl containing 10× binding buffer (containing 0.1 M Tris [pH 7.5], 0.5 M NaCl, 1 mM dithiothreitol [DTT], 10 mM EDTA [pH 8], and 50% glycerol) and 0.35 μg of herring sperm DNA as a nonspecific competitive inhibitor. For the blocking assay, 1 μl of phosphate-buffered saline (PBS)-diluted antibody (1:10) was incubated with the extract for 10 min before probe addition. Antibodies against the p50 and p65 subunits were produced by immunizing recombinant proteins produced by the baculovirus system into rabbits. These antibodies showed specific blocking of κB DNA binding of recombinant (p50)2 and (p65)2, respectively (48).
The 13% nondenaturing polyacrylamide gels were made and run using 10× Tris-borate-EDTA buffer. After electrophoresis, the gels were dried and radioactivity was visualized with an image analyzer (Fujifilm). Quantification of the bands was performed using ImageJ software.
Immunoblotting and immunoprecipitation.
Nuclear and cytoplasmic extracts were prepared as described above. Twenty-microgram volumes of lysates were resolved by SDS-PAGE and transferred to an Immobilon-P polyvinylidene difluoride (PVDF) membrane (Millipore) by semidry transfer at 100 V for 60 min. All membranes were blocked in 5% milk in Tris-buffered saline with 0.1% Tween 20 (TBST) and probed with the desired antibodies in 5% milk overnight. Primary antibodies included mouse monoclonal anti-Flag M2 (F1804) obtained from Sigma-Aldrich, goat polyclonal anti-lamin B (M-20) obtained from Santa Cruz Biotechnology, rabbit polyclonal anti-p65 (C-20) obtained from Santa Cruz Biotechnology, mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH; 6C5) obtained from Santa Cruz Biotechnology, rabbit monoclonal anti-IKK-beta (2C8) obtained from Cell Signaling, rabbit monoclonal anti-TBK1 (ab40676) obtained from Abcam, and rabbit monoclonal anti-pIκB-α obtained from Cell Signaling. Appropriate horseradish peroxidase-conjugated secondary antibodies were incubated on membranes in TBST at room temperature for 1 h, and bands were developed using the enhanced chemiluminescence reagent Chemi-lumi One Super (Nacalai Tesque) and imaged on an LAS-4000 imager (Fuji).
For immunoprecipitation, protein extracts from 6-well plates were incubated with 1 μg of the antibody per sample and rotated at 4°C overnight. Then, protein G Mag Sepharose (GE Healthcare) was added to each sample and rotated for 4 h at 4°C. After three washes with lysis buffer, immunocomplexes were denatured in 2× SDS sample buffer and analyzed as described for immunoblotting.
Luciferase reporter assay.
HEK293T cells seeded in 24-well plates were cotransfected with 100 ng of the NF-κB luciferase reporter plasmid, 10 ng of the TK-Renilla luciferase plasmid, and 50 ng of the plasmid encoding NSs protein. Twenty-four hours after transfection, cells were stimulated with virus infection or recombinant TNF-α (PeproTech; human TNF-α lot 0607B25). After stimulation, whole-cell lysates were prepared and subjected to the Dual-Glo luciferase assay according to the manufacturer’s instructions (Promega). Results are presented as firefly luciferase levels normalized by the Renilla luciferase levels.
Plasmids.
The coding sequence of NSs for the SFTSV SP010 strain was inserted into the multiple-cloning site of the backbone vector pRK5-FLAG as described previously (49, 50). Where indicated, an empty pRK5 vector was used as a control.
Generation of knockout cells by CRISPR/Cas9.
Knockout cell lines were generated by transient transfection of pSpCas9(BB)-2A-GFP (PX458) vector (Addgene) inserted with the double-strand oligonucleotides containing single-guide RNA (sgRNA) sequences (Table 2) according to the manufacturer’s protocol for 24 h. Cells were subjected to sorting by green fluorescent protein (GFP) expression levels using an SH800 cell sorter (Sony), followed by limiting dilution in 96-well culture plates to obtain single clones. Knockout of each gene was examined by immunoblotting.
TABLE 2.
Target sequences of sgRNAs for CRISPR-Cas9 gene knockout
| Primer name | Sequence (5′→3′) |
|---|---|
| IKKα-targeting sgRNA, forward | CACCGGTACCAAAAACAGAGAACGA |
| IKKα-targeting sgRNA, reverse | AAACTCGTTCTCTGTTTTTGGTACC |
| IKKβ-targeting sgRNA, forward | CACCGTCAGCCCCCGGAACCGAGAG |
| IKKβ-targeting sgRNA, reverse | AAACCTCTCGGTTCCGGGGGCTGAC |
| TBK1-targeting sgRNA, forward | CACCGAAATATCATGCGTGTTATAG |
| TBK1-targeting sgRNA, reverse | AAACCTATAACACGCATGATATTTC |
| NEMO-targeting sgRNA, forward | CACCGGCTGCACCTGCCTTCAGAACA |
| NEMO-targeting sgRNA, reverse | AAACTGTTCTGAAGGCAGGTGCAGCC |
Lentiviruses and complementation of knockout cells.
Lentiviruses encoding human IKKβ or mCherry-tagged human TBK1 were generated by cloning of the coding sequences into a pCSII-CMV-MCS-IRES2-Bsd vector, followed by the standard packaging protocol.
Complementation of the knockout cells was carried out by ectopic expression of TBK1 or IKKβ by lentiviral transduction. To generate cell lines stably expressing transgenes, lentiviral particles were produced by transfecting HEK293T cells with plasmids pCAG-HIVgp, pVSV-G-RSV-ReV, and pCSII-CMV-MCS-IRES2-Bsd containing genes of interest. Plasmids were transfected into HEK293T cells in 10-cm dishes at a confluency of 50% to 70% with Lipofectamine 2000 (Life Technologies). Medium was changed 8 h after transfection, and viral supernatants were collected 48 h after transfection. Viral supernatants were spun at 500 × g to remove cellular debris and then filtered (0.45 mm). Polybrene (Nacalai Tesque) was added to the filtered supernatants (10 µg/ml), and the viral supernatants were then used to infect the cells. The infected cell lines were then treated with blasticidin (10 µg/ml) 48 h after infection to select pCSII-CMV-MCS-IRES2-Bsd-containing cells.
Viruses.
SFTSV strain SPL010, an isolate from a Japanese SFTS patient (6), was used after propagation in Vero cells. Sendai virus (SeV; Cantell strain) and Newcastle disease virus (NDV) were propagated in 9-day embryonated chicken eggs for 2 days. The viral titer for SeV was measured by the hemagglutination test using chicken red blood cells. Virus was added to cells at 3.2 × 102 hemagglutination units (HAU). NDV was titrated by plaque assay using HEp-2 cells and added to cells at a multiplicity of infection (MOI) of 1. Encephalomyocarditis virus (EMCV) was propagated and titrated with L929 cells. Cells were infected with EMCV at 1 PFU/cell.
SFTSV infection in vivo.
We used a group of 14 Infar−/− mice from the C57BL/6 (B6) strain for the infection experiment. On day 0, all mice were intraperitoneally inoculated with 2 × 106 50% tissue culture infective doses (TCID50) of SFTSV, strain SPL010. On day2, half of the inoculated mice were given 1 mg of TPCA-1 intraperitoneally. The remaining were given PBS as a control. Twenty-four hours later, spleens were harvested and frozen with RNAlater. Later, spleens were moved to tubes containing 1 ml of TRIzol and two beads and shaken by a Qiagen TissueLyzer II for 1 min 6 times. RNA was purified as described above for cell lines. This experiment was performed in a biosafety level 3 (BSL3) facility at the National Institute of Infectious Diseases.
siRNA and transfection.
Lipofectamine 2000 agent (Invitrogen) was used as the transfection reagent in our experiments to effectively transfect HeLa or HEK293T cells. Small interfering RNA (siRNA) targeting human p65 (sequence, 5′ to 3′, [RNA] AAA CUC AUC AUA GUU GAU GGU GCU C; purchased from Life Technologies Japan) was transfected into HeLa cells using Lipofectamine 2000. After 48 h of incubation, cells were seeded into 12-well plates for transfection with pRK5-NSs-Flag vector the next day. Twenty-four hours after transfection with pRK5-Flag-NSs vector, the virus was introduced to the cells. An siRNA-negative control (silencer Select, catalog no. 4390843; Ambion) was included as a nontargeting 21-mer siRNA.
Serum cytokine assay.
Before the measurement of cytokines/chemokines, sera were mixed with Nonidet P-40 (1% final concentration) and exposed to UV light for 10 min (NTM-10 minitransilluminator; Funakoshi) to inactivate infectious SFTSV. A cytokine 25-plex human panel (Thermo Fisher Scientific) and Luminex 100 (Luminex) were used according to the manufacturer’s instructions.
Statistical analysis.
Statistical analyses were performed using Prism software (version 8). The two-tailed Student t test was used. A P value of <0.05 was considered significant.
ACKNOWLEDGMENTS
This study was supported by research grants from the Japan Agency for Medical Research and Development (Research Program on Emerging and Re-emerging Infectious Diseases [jp19fk0108081h1001 and jp20fk0108081h1202]) and from the Japan Society for the Promotion of Science, Fund for the Promotion of Joint International Research, Fostering Joint International Research [B] (18KK0232), and a Grant-in-Aid for Scientific Research B (18H02344).
J. Khalil performed the experiments, data analysis, and statistical analysis and wrote the initial manuscript. S. Yamada performed the SFTSV infection experiment in HEK293T cells. Y. Tsukamoto generated the NSs mutant plasmids. H. Abe generated the knockout cell lines. M. Shimojima performed the cytokine multiplex assay and infected the mouse model with SFTSV. H. Kato supervised the project and assisted in the study design. T. Fujita conceived the project, supervised it, analyzed the data, and wrote and revised the completed manuscript.
We declare no conflicts of interest.
REFERENCES
- 1.Yu XJ, Liang MF, Zhang SY, Liu Y, Li JD, Sun YL, Zhang L, Zhang QF, Popov VL, Li C, Qu J, Li Q, Zhang YP, Hai R, Wu W, Wang Q, Zhan FX, Wang XJ, Kan B, Wang SW, Wan KL, Jing HQ, Lu JX, Yin WW, Zhou H, Guan XH, Liu JF, Bi ZQ, Liu GH, Ren J, Wang H, Zhao Z, Song JD, He JR, Wan T, Zhang JS, Fu XP, Sun LN, Dong XP, Feng ZJ, Yang WZ, Hong T, Zhang Y, Walker DH, Wang Y, Li DX. 2011. Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med 364:1523–1532. doi: 10.1056/NEJMoa1010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu B, Liu L, Huang X, Ma H, Zhang Y, Du Y, Wang P, Tang X, Wang H, Kang K, Zhang S, Zhao G, Wu W, Yang Y, Chen H, Mu F, Chen W. 2011. Metagenomic analysis of fever, thrombocytopenia and leukopenia syndrome (FTLS) in Henan Province, China: discovery of a new bunyavirus. PLoS Pathog 7:e1002369. doi: 10.1371/journal.ppat.1002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silvas JA, Aguilar PV. 2017. The emergence of severe fever with thrombocytopenia syndrome virus. Am J Trop Med Hyg 97:992–996. doi: 10.4269/ajtmh.16-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Denic S, Janbeih J, Nair S, Conca W, Tariq WU, Al-Salam S. 2011. Acute thrombocytopenia, leucopenia, and multiorgan dysfunction: the first case of SFTS bunyavirus outside China? Case Rep Infect Dis 2011:204056. doi: 10.1155/2011/204056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim KH, Yi J, Kim G, Choi SJ, Jun KI, Kim NH, Choe PG, Kim NJ, Lee JK, Oh MD. 2013. Severe fever with thrombocytopenia syndrome, South Korea, 2012. Emerg Infect Dis 19:1892–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi T, Maeda K, Suzuki T, Ishido A, Shigeoka T, Tominaga T, Kamei T, Honda M, Ninomiya D, Sakai T, Senba T, Kaneyuki S, Sakaguchi S, Satoh A, Hosokawa T, Kawabe Y, Kurihara S, Izumikawa K, Kohno S, Azuma T, Suemori K, Yasukawa M, Mizutani T, Omatsu T, Katayama Y, Miyahara M, Ijuin M, Doi K, Okuda M, Umeki K, Saito T, Fukushima K, Nakajima K, Yoshikawa T, Tani H, Fukushi S, Fukuma A, Ogata M, Shimojima M, Nakajima N, Nagata N, Katano H, Fukumoto H, Sato Y, Hasegawa H, Yamagishi T, Oishi K, Kurane I, Morikawa S, Saijo M. 2014. The first identification and retrospective study of severe fever with thrombocytopenia syndrome in Japan. J Infect Dis 209:816–827. doi: 10.1093/infdis/jit603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stubbs AM, Steele MT. 2014. Heartland virus disease—United States, 2012–2013. Ann Emerg Med 64:314. doi: 10.1016/j.annemergmed.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 8.Word Health Organization. 2017. Annual review of diseases prioritized under the research and development blueprint. http://www.who.int/csr/research-and-development/en/.
- 9.Saijo M. 2018. Pathophysiology of severe fever with thrombocytopenia syndrome and development of specific antiviral therapy. J Infect Chemother 24:773–781. doi: 10.1016/j.jiac.2018.07.009. [DOI] [PubMed] [Google Scholar]
- 10.Reece LM, Beasley DW, Milligan GN, Sarathy VV, Barrett AD. 2018. Current status of severe fever with thrombocytopenia syndrome vaccine development. Curr Opin Virol 29:72–78. doi: 10.1016/j.coviro.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 11.Younan P, Iampietro M, Nishida A, Ramanathan P, Santos RI, Dutta M, Lubaki NM, Koup RA, Katze MG, Bukreyev A. 2017. Ebola virus binding to Tim-1 on T lymphocytes induces a cytokine storm. mBio 8:e00845-17. doi: 10.1128/mBio.00845-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang J, Liang W, Chen S, Zhu Y, Chen H, Mok CKP, Zhou Y. 2018. Serum cytokine profiles in patients with dengue fever at the acute infection phase. Dis Markers 2018:8403937. doi: 10.1155/2018/8403937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papa A, Tsergouli K, Çağlayık DY, Bino S, Como N, Uyar Y, Korukluoglu G. 2016. Cytokines as biomarkers of Crimean-Congo hemorrhagic fever. J Med Virol 88:21–27. doi: 10.1002/jmv.24312. [DOI] [PubMed] [Google Scholar]
- 14.Sun Y, Jin C, Zhan F, Wang X, Liang M, Zhang Q, Ding S, Guan X, Huo X, Li C, Qu J, Wang Q, Zhang S, Zhang Y, Wang S, Xu A, Bi Z, Li D. 2012. Host cytokine storm is associated with disease severity of severe fever with thrombocytopenia syndrome. J Infect Dis 206:1085–1094. doi: 10.1093/infdis/jis452. [DOI] [PubMed] [Google Scholar]
- 15.Deng B, Zhang S, Geng Y, Zhang Y, Wang Y, Yao W, Wen Y, Cui W, Zhou Y, Gu Q, Wang W, Wang Y, Shao Z, Wang Y, Li C, Wang D, Zhao Y, Liu P. 2012. Cytokine and chemokine levels in patients with severe fever with thrombocytopenia syndrome virus. PLoS One 7:e41365. doi: 10.1371/journal.pone.0041365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwon JS, Kim MC, Kim JY, Jeon NY, Ryu BH, Hong J, Kim MJ, Chong YP, Lee SO, Choi SH, Kim YS, Woo JH, Kim SH. 2018. Kinetics of viral load and cytokines in severe fever with thrombocytopenia syndrome. J Clin Virol 101:57–62. doi: 10.1016/j.jcv.2018.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding YP, Liang MF, Ye JB, Liu QH, Xiong CH, Long B, Lin WB, Cui N, Zou ZQ, Song YL, Zhang QF, Zhang S, Liu YZ, Song G, Ren YY, Li SH, Wang Y, Hou FQ, Yu H, Ding P, Ye F, Li DX, Wang GQ. 2014. Prognostic value of clinical and immunological markers in acute phase of SFTS virus infection. Clin Microbiol Infect 20:O870–O878. doi: 10.1111/1469-0691.12636. [DOI] [PubMed] [Google Scholar]
- 18.Liu MM, Lei XY, Yu H, Zhang JZ, Yu XJ. 2017. Correlation of cytokine level with the severity of severe fever with thrombocytopenia syndrome. Virol J 14:6. doi: 10.1186/s12985-016-0677-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmolke M, Viemann D, Roth J, Ludwig S. 2009. Essential impact of NF-kappaB signaling on the H5N1 influenza A virus-induced transcriptome. J Immunol 183:5180–5189. doi: 10.4049/jimmunol.0804198. [DOI] [PubMed] [Google Scholar]
- 20.Santoro MG, Rossi A, Amici C. 2003. NF-kappaB and virus infection: who controls whom. EMBO J 22:2552–2560. doi: 10.1093/emboj/cdg267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cahir-McFarland ED, Davidson DM, Schauer SL, Duong J, Kieff E. 2000. NF-kappa B inhibition causes spontaneous apoptosis in Epstein-Barr virus-transformed lymphoblastoid cells. Proc Natl Acad Sci U S A 97:6055–6060. doi: 10.1073/pnas.100119497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bour S, Perrin C, Akari H, Strebel K. 2001. The human immunodeficiency virus type 1 Vpu protein inhibits NF-kappa B activation by interfering with beta TrCP-mediated degradation of Ikappa B. J Biol Chem 276:15920–15928. doi: 10.1074/jbc.M010533200. [DOI] [PubMed] [Google Scholar]
- 23.Bargou RC, Emmerich F, Krappmann D, Bommert K, Mapara MY, Arnold W, Royer HD, Grinstein E, Greiner A, Scheidereit C, Dorken B. 1997. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J Clin Invest 100:2961–2969. doi: 10.1172/JCI119849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amici C, Belardo G, Rossi A, Santoro MG. 2001. Activation of I kappa b kinase by herpes simplex virus type 1. A novel target for anti-herpetic therapy. J Biol Chem 276:28759–28766. doi: 10.1074/jbc.M103408200. [DOI] [PubMed] [Google Scholar]
- 25.Akari H, Bour S, Kao S, Adachi A, Strebel K. 2001. The human immunodeficiency virus type 1 accessory protein Vpu induces apoptosis by suppressing the nuclear factor kappaB-dependent expression of antiapoptotic factors. J Exp Med 194:1299–1311. doi: 10.1084/jem.194.9.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qu B, Qi X, Wu X, Liang M, Li C, Cardona CJ, Xu W, Tang F, Li Z, Wu B, Powell K, Wegner M, Li D, Xing Z. 2012. Suppression of the interferon and NF-kappaB responses by severe fever with thrombocytopenia syndrome virus. J Virol 86:8388–8401. doi: 10.1128/JVI.00612-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ning YJ, Wang M, Deng M, Shen S, Liu W, Cao WC, Deng F, Wang YY, Hu Z, Wang H. 2014. Viral suppression of innate immunity via spatial isolation of TBK1/IKKepsilon from mitochondrial antiviral platform. J Mol Cell Biol 6:324–337. doi: 10.1093/jmcb/mju015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang S, Zheng B, Wang T, Li A, Wan J, Qu J, Li CH, Li D, Liang M. 2017. NSs protein of severe fever with thrombocytopenia syndrome virus suppresses interferon production through different mechanism than Rift Valley fever virus. Acta Virol 61:289–298. doi: 10.4149/av_2017_307. [DOI] [PubMed] [Google Scholar]
- 29.Santiago FW, Covaleda LM, Sanchez-Aparicio MT, Silvas JA, Diaz-Vizarreta AC, Patel JR, Popov V, Yu XJ, Garcia-Sastre A, Aguilar PV. 2014. Hijacking of RIG-I signaling proteins into virus-induced cytoplasmic structures correlates with the inhibition of type I interferon responses. J Virol 88:4572–4585. doi: 10.1128/JVI.03021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ning YJ, Feng K, Min YQ, Cao WC, Wang M, Deng F, Hu Z, Wang H. 2015. Disruption of type I interferon signaling by the nonstructural protein of severe fever with thrombocytopenia syndrome virus via the hijacking of STAT2 and STAT1 into inclusion bodies. J Virol 89:4227–4236. doi: 10.1128/JVI.00154-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Wu B, Paessler S, Walker DH, Tesh RB, Yu XJ. 2014. The pathogenesis of severe fever with thrombocytopenia syndrome virus infection in alpha/beta interferon knockout mice: insights into the pathologic mechanisms of a new viral hemorrhagic fever. J Virol 88:1781–1786. doi: 10.1128/JVI.02277-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamada S, Shimojima M, Narita R, Tsukamoto Y, Kato H, Saijo M, Fujita T. 2018. RIG-I-like receptor and Toll-like receptor signaling pathways cause aberrant production of inflammatory cytokines/chemokines in a severe fever with thrombocytopenia syndrome virus infection mouse model. J Virol 92:e02246-17. doi: 10.1128/JVI.02246-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Libermann TA, Baltimore D. 1990. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol 10:2327–2334. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu T, Zhang L, Joo D, Sun SC. 2017. NF-kappaB signaling in inflammation. Signal Transduct Target Ther 2:17023. doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burke SJ, Lu D, Sparer TE, Masi T, Goff MR, Karlstad MD, Collier JJ. 2014. NF-kappaB and STAT1 control CXCL1 and CXCL2 gene transcription. Am J Physiol Endocrinol Metab 306:E131–E149. doi: 10.1152/ajpendo.00347.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kunsch C, Rosen CA. 1993. NF-kappa B subunit-specific regulation of the interleukin-8 promoter. Mol Cell Biol 13:6137–6146. doi: 10.1128/mcb.13.10.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoyos B, Ballard DW, Bohnlein E, Siekevitz M, Greene WC. 1989. Kappa B-specific DNA binding proteins: role in the regulation of human interleukin-2 gene expression. Science 244:457–460. doi: 10.1126/science.2497518. [DOI] [PubMed] [Google Scholar]
- 38.Ishikado A, Nishio Y, Yamane K, Mukose A, Morino K, Murakami Y, Sekine O, Makino T, Maegawa H, Kashiwagi A. 2009. Soy phosphatidylcholine inhibited TLR4-mediated MCP-1 expression in vascular cells. Atherosclerosis 205:404–412. doi: 10.1016/j.atherosclerosis.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 39.Abe H, Satoh J, Shirasaka Y, Kogure A, Kato H, Ito S, Fujita T. 2020. Priming phosphorylation of TANK-binding kinase 1 by IkappaB kinase beta is essential in Toll-like receptor 3/4 signaling. Mol Cell Biol 40:e00509-19. doi: 10.1128/MCB.00509-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brennan B, Rezelj VV, Elliott RM. 2017. Mapping of transcription termination within the S segment of SFTS phlebovirus facilitated generation of NSs deletant viruses. J Virol 91:e00743-17. doi: 10.1128/JVI.00743-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonnard M, Mirtsos C, Suzuki S, Graham K, Huang J, Ng M, Itie A, Wakeham A, Shahinian A, Henzel WJ, Elia AJ, Shillinglaw W, Mak TW, Cao Z, Yeh WC. 2000. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J 19:4976–4985. doi: 10.1093/emboj/19.18.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spann KM, Tran KC, Collins PL. 2005. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-kappaB, and proinflammatory cytokines. J Virol 79:5353–5362. doi: 10.1128/JVI.79.9.5353-5362.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang YF, He B, Li NP, Ma J, Gong GZ, Zhang M. 2011. The oncogenic role of NS5A of hepatitis C virus is mediated by up-regulation of survivin gene expression in the hepatocellular cell through p53 and NF-κB pathways. Cell Biol Int 35:1225–1232. doi: 10.1042/CBI20110102. [DOI] [PubMed] [Google Scholar]
- 44.Jin DY, Giordano V, Kibler KV, Nakano H, Jeang KT. 1999. Role of adapter function in oncoprotein-mediated activation of NF-kappaB. Human T-cell leukemia virus type I Tax interacts directly with IkappaB kinase gamma. J Biol Chem 274:17402–17405. doi: 10.1074/jbc.274.25.17402. [DOI] [PubMed] [Google Scholar]
- 45.DeDiego ML, Nieto-Torres JL, Regla-Nava JA, Jimenez-Guardeño JM, Fernandez-Delgado R, Fett C, Castaño-Rodriguez C, Perlman S, Enjuanes L. 2014. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J Virol 88:913–924. doi: 10.1128/JVI.02576-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liao QJ, Ye LB, Timani KA, Zeng YC, She YL, Ye L, Wu ZH. 2005. Activation of NF-kappaB by the full-length nucleocapsid protein of the SARS coronavirus. Acta Biochim Biophys Sin (Shanghai) 37:607–612. doi: 10.1111/j.1745-7270.2005.00082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen J, Ng MM, Chu JJ. 2015. Activation of TLR2 and TLR6 by dengue NS1 protein and its implications in the immunopathogenesis of dengue virus infection. PLoS Pathog 11:e1005053. doi: 10.1371/journal.ppat.1005053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fujita T, Nolan GP, Ghosh S, Baltimore D. 1992. Independent modes of transcriptional activation by the p50 and p65 subunits of NF-kappa B. Genes Dev 6:775–787. doi: 10.1101/gad.6.5.775. [DOI] [PubMed] [Google Scholar]
- 49.Li L, Hailey DW, Soetandyo N, Li W, Lippincott-Schwartz J, Shu HB, Ye Y. 2008. Localization of A20 to a lysosome-associated compartment and its role in NFkappaB signaling. Biochim Biophys Acta 1783:1140–1149. doi: 10.1016/j.bbamcr.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y, Soetandyo N, Lee JG, Liu L, Xu Y, Clemons WM, Jr, Ye Y. 2014. USP13 antagonizes gp78 to maintain functionality of a chaperone in ER-associated degradation. Elife 3:e01369. doi: 10.7554/eLife.01369. [DOI] [PMC free article] [PubMed] [Google Scholar]