

How lipids travel between membranes of diderm bacteria is a challenging mechanistic question because lipids, which are hydrophobic molecules, must traverse a hydrophilic periplasm. This question is even more complex for mycobacteria, which have a unique cell envelope that is highly impermeable to molecules.
KEYWORDS: Mce, Mycobacterium, cell wall, cholesterol, transporters
ABSTRACT
Mycobacteria possess Mce transporters that import lipids and are thought to function analogously to ATP-binding cassette (ABC) transporters. However, whereas ABC transporters import substrates using a single solute-binding protein (SBP) to deliver a substrate to permease proteins in the membrane, mycobacterial Mce transporters have a potential for six SBPs (MceA to MceF) working with a pair of permeases (YrbEA and YrbEB), a cytoplasmic ATPase (MceG), and multiple Mce-associated membrane (Mam) and orphaned Mam (Omam) proteins to transport lipids. In this study, we used the model mycobacterium Mycobacterium smegmatis to study the requirement for individual Mce, Mam, and Omam proteins in Mce4 transport of cholesterol. All of the Mce4 and Mam4 proteins we investigated were required for cholesterol uptake. However, not all Omam proteins, which are encoded by genes outside mce loci, proved to contribute to cholesterol import. OmamA and OmamB were required for cholesterol import, while OmamC, OmamD, OmamE, and OmamF were not. In the absence of any single Mce4, Mam4, or Omam protein that we tested, the abundance of Mce4A and Mce4E declined. This relationship between the levels of Mce4A and Mce4E and these additional proteins suggests a network of interactions that assemble and/or stabilize a multiprotein Mce4 transporter complex. Further support for Mce transporters being multiprotein complexes was obtained by immunoprecipitation-mass spectrometry, in which we identified every single Mce, YrbE, MceG, Mam, and Omam protein with a role in cholesterol transport as associating with Mce4A. This study represents the first time any of these Mce4 transporter proteins has been shown to associate.
IMPORTANCE How lipids travel between membranes of diderm bacteria is a challenging mechanistic question because lipids, which are hydrophobic molecules, must traverse a hydrophilic periplasm. This question is even more complex for mycobacteria, which have a unique cell envelope that is highly impermeable to molecules. A growing body of knowledge identifies Mce transporters as lipid importers for mycobacteria. Here, using protein stability experiments and immunoprecipitation-mass spectrometry, we provide evidence for mycobacterial Mce transporters existing as multiprotein complexes.
INTRODUCTION
Lipids play important roles in bacterial physiology, where they serve as nutrient sources or as components of the protective cell envelope (1, 2). However, lipid transport across the bacterial cell envelope remains a poorly understood process (3–5). In diderm (two-membrane-containing) bacteria, such as Gram-negative Escherichia coli, or actinobacteria, such as mycobacteria, there is a cytoplasmic inner membrane (IM) and an outer membrane (OM). Although the OMs of Gram-negative bacteria and actinobacteria have different compositions, in both cases they are hydrophobic lipid barriers that are separated from the IM by a hydrophilic periplasm (5–7). Thus, lipids moving across the cell envelope, in either direction (in or out), traverse an environment in which they are not soluble, necessitating specialized transport systems. In E. coli, macromolecular systems for transporting lipids across the cell envelope to the OM have been identified, including the Lpt transporter, which delivers lipopolysaccharide (8, 9), and the Lol system, which delivers lipoproteins (10). Both Lpt and Lol systems involve a dedicated ATP-binding cassette (ABC) transporter in the IM. Most recently, transporters that traffic phospholipids to or from the inner leaflet of the E. coli OM were identified. A notable feature of E. coli phospholipid transporters is that they involve a Mce domain-containing protein (11, 12). So far, the best characterized of these Mce transporters in E. coli is the Mla system, which is itself an ABC transporter-dependent system (11, 13–15).
In the Mla system, the Mce domain-containing protein is the IM-anchored periplasmic MlaD, which acts as a solute-binding protein (SBP) that binds the phospholipid (13, 14, 16). SBPs are periplasmic proteins that deliver solutes to ABC transporters (17). There is evidence for MlaD and other E. coli Mce domain proteins (YebT/LetB and PqiB), which function in separate phospholipid transport pathways, forming a homohexameric tunnel (13, 14, 18, 19) for translocating and delivering the phospholipid to the core ABC transporter components (the IM permease MlaE and the cytoplasmic ATPase MlaF) (16). The Mla system also involves the cytoplasmic MlaB protein, which stabilizes the ATP-binding domain of MlaF, and a periplasmic MlaC protein, which acts as a chaperone (15, 20, 21), ferrying phospholipids between MlaD and an OM lipoprotein, MlaA (20, 22).
The very first Mce protein discovered was the Mce1A protein of Mycobacterium tuberculosis, which was initially identified as a protein that promotes “mammalian cell entry” when expressed in heterologous bacteria (23). Only later were actinobacterial Mce proteins recognized to function in lipid transport (24–27). The best characterized actinobacterial Mce system is the Mce4 transporter of Rhodococcus jostii and mycobacteria, which imports cholesterol from the environment into the bacterial cytoplasm (2, 24, 25). M. tuberculosis contains four Mce transporters, Mce1 to Mce4, and the model mycobacterial organism, Mycobacterium smegmatis, contains six Mce transporters (28, 29). It is likely that each of the mycobacterial Mce transporters imports different lipids. For instance, the Mce1 transporter imports oleic and palmitic fatty acids, whereas the Mce4 transporter imports cholesterol (25–27). Fatty acids and cholesterol are metabolized by M. tuberculosis during infection, and both Mce1 and Mce4 contribute to the pathogenesis of M. tuberculosis (2, 25, 30–36).
Mycobacterial Mce transporters appear to be more complex than E. coli Mce domain-containing transporters (Fig. 1). The added complexity may reflect unique barriers to lipid transport across the mycobacterial cell envelope, which includes a covalent network of peptidoglycan and arabinogalactan and a unique mycobacterial OM (MOM) (6). Each mycobacterial mce locus has an operon made up of genes encoding predicted permeases YrbEA and YrbEB and six Mce proteins (MceA to MceF) (Fig. 1) proposed to function as SBPs (not just one, as in typical ABC transporters) (28). Moreover, compared to MlaD, which is composed of a transmembrane domain followed by a Mce domain, Mce proteins of mycobacterial systems are larger, possessing beyond the Mce domain a second conserved domain, Mce4_CUP1, followed by a variable region (see Fig. S1 in the supplemental material). Immediately adjacent to nearly all mce operons are genes encoding Mce-associated membrane (Mam) proteins, which have a transmembrane domain at their N terminus, followed by a periplasmic domain. In addition, there are orphaned Mam (Omam) proteins, with predicted structural similarity to Mam proteins, that are encoded by genes that are located distal to mce operons (37). We previously established that one of these Omam proteins, OmamA, contributes to the function of multiple Mce transporters (37). Finally, MceG is an ATPase that is predicted to work with all of the Mce transporters of mycobacteria (32, 38). Apart from MceG, which is cytoplasmic, all of these proteins have been demonstrated or are predicted to reside in the cell envelope (28, 33, 39, 40). How these components interact and work together to import lipids through the mycobacterial cell envelope is largely unknown.
FIG 1.

Mce domain-containing operon organization. (A) Depiction of the mycobacterial mce4 operon and other genes associated with Mce4 transporter function. The red C indicates the lipobox cysteine conserved within MceE proteins. Solid-color genes are shared between Mycobacterium tuberculosis and Mycobacterium smegmatis. Hatched-color genes are found only in Mycobacterium smegmatis. Genes located within boxes are outside the mce4 locus. (B) Depiction of the E. coli mla operon and other genes associated with Mla transporter function. Genes located within boxes are outside the mla operon.
Here, we investigated a set of M. smegmatis mce4, mam4, and omam mutants and demonstrated nonredundant roles for individual components in cholesterol transport using growth in cholesterol and radioactive cholesterol uptake assays. However, not all of the predicted Omam proteins in mycobacteria proved to be required for cholesterol import. While OmamA and OmamB contributed to Mce4 cholesterol import, OmamC, OmamD, OmamE, and OmamF were not required. We then demonstrated that Mce4A and Mce4E protein levels were reduced in every one of the individual mce4, mam4, and omam mutants that had cholesterol uptake defects. Because the stability of proteins in multiprotein complexes often relies on protein interactions, this dependence of Mce4A and Mce4E levels on the presence of individual Mce4, Mam4, and Omam proteins suggests an extensive network of interactions that serve to assemble and/or to stabilize a multiprotein Mce4 transporter complex. Further, using immunoprecipitation-mass spectrometry (IP-MS) to identify Mce4A-associated proteins, we identified Mce4B to Mce4F, YrbE4A, YrbE4B, Mam4A, Mam4B, OmamA, OmamB, and MceG. Together, our results provide the first evidence for interactions between Mce4 transporter proteins and for Mce transporters being large multiprotein complexes in the mycobacterial cell envelope.
RESULTS
Individual Mce4 and Mam4 proteins have required functions in Mce4 transport of cholesterol.
As is the case for all Mce transporter systems, the mce4 operon encodes six Mce proteins (Mce4A to Mce4F) (Fig. 1), all with approximately 20% identity to one another (see Table S1 in the supplemental material). To address the functions of individual Mce proteins, we generated an in-frame, unmarked deletion in mce4A (codons for amino acids 7 to 392 were deleted; Mce4A contains 400 amino acids) in the M. smegmatis genome. We also constructed an in-frame, unmarked deletion of mce4E (codons for amino acids 27 to 363 were deleted; Mce4E contains 382 amino acids). Along with Mce4A, we chose to study Mce4E because, in nearly every mycobacterial Mce system, the MceE protein is a predicted lipoprotein, while all of the other Mce proteins, including MceA, are not predicted lipoproteins (28, 41–44). To evaluate the contributions of Mce4A and Mce4E to cholesterol import, the Δmce4A and Δmce4E mutants were each tested for growth in medium containing cholesterol as the sole carbon source (Fig. 2A and B). The ability of metabolically active mycobacteria to reduce resazurin dye, as measured by fluorescence, was used to monitor growth in culture (45). While the parental wild-type (WT) M. smegmatis strain grew in M9 medium with cholesterol as the sole carbon source, the Δmce4A mutant and the Δmce4E mutant exhibited little to no growth. In parallel, we tested Δmce4A and Δmce4E strains carrying multicopy plasmids expressing the WT copy of the deleted mce gene from the constitutive hsp60 promoter. The growth defect in cholesterol was reversed in these complemented strains, demonstrating that the phenotype is due to the mutation of the single gene (Fig. 2A and B). All strains grew equally well on medium with glucose and glycerol as carbon sources, eliminating the possibility that the mutants have a more general growth defect (Fig. S2A and B). We further tested our Δmce4A and Δmce4E mutants and complemented strains for their ability to import 14C-radiolabeled cholesterol (Fig. 2F). WT, mutant, and complemented strains were incubated in M9 medium with 14C-radiolabeled cholesterol for 3 h, after which the mycobacterial strains were washed and the radioactivity taken up by the cells was measured. The level of radiolabeled cholesterol taken up by the Δmce4A and Δmce4E mutants was significantly reduced, compared to the WT and complemented strains (Fig. 2F). Together, these experiments demonstrated that Mce4A and Mce4E are each individually required for cholesterol uptake.
FIG 2.

Individual Mce4 proteins, Mam proteins, and OmamA and OmamB proteins are required for M. smegmatis to grow on and take up cholesterol. (A to E) Growth of 104 CFU of M. smegmatis strains with cholesterol as the sole carbon source was measured over time, using resazurin reduction as a fluorescent readout of metabolic activity. Cholesterol growth curves measured by resazurin reduction by Δmce4A and complemented strains (A), Δmce4E, Δmce4Epolar, and complemented strains (B), Δmam4Apolar and complemented strains (C), ΔomamA, ΔomamB, and complemented strains (D), and ΔomamCD and ΔomamEF mutant M. smegmatis strains (E) are depicted. (F and G) Cholesterol uptake was measured by incubating M. smegmatis strains with [4-14C]cholesterol for 3 h and measuring the cell-associated radioactivity using a scintillation counter. Measurements of cholesterol uptake were normalized as a percentage of the value of WT M. smegmatis, which was set to 100%. All strains were complemented with multicopy (mc) or single-copy (sc) plasmids as indicated. Mutant strains contain empty vectors (EV) as indicated. Error bars represent standard deviations. *, P < 0.001, compared to WT results. Results are representative of at least three independent experiments.
We also constructed a larger Δmce4E deletion mutant that extended to the second to last codon (codons for amino acids 27 to 381 were deleted). This larger deletion Δmce4E mutant was also unable to grow on or take up cholesterol (Fig. 2B and F). However, with this mutant the same mce4E complementation plasmid as used above did not restore growth on cholesterol or uptake of radiolabeled cholesterol, although a plasmid expressing both mce4E and mce4F was able to reverse the cholesterol defects (Fig. 2B and F). These results indicated that the larger deletion has a polar effect on the downstream mce4F and that Mce4F is additionally required for cholesterol uptake. Because the stop codon in mce4E and the start codon in mce4F are immediately adjacent to one another, we suspect that the polar effect resulted from the larger in-frame Δmce4E deletion, hereafter called Δmce4Epolar, disrupting translation initiation of the downstream mce4F. In this report, this larger Δmce4Epolar mutant strain carrying the mce4E complementation plasmid used above (i.e., complemented for the mce4E deficiency only) is referred to as a mce4F* mutant. All of the mce4Epolar mutant strains grew equally well on glucose and glycerol (Fig. S2B).
We also investigated the roles of the Mam4A and Mam4B proteins in cholesterol uptake. Mam proteins are encoded, often in pairs, at the end of mce loci. We constructed an in-frame deletion of mam4A (codons for amino acids 10 to 209 were deleted; Mam4A contains 210 amino acids). This Δmam4A mutant was unable to grow on cholesterol or take up radiolabeled cholesterol, compared to the WT strain (Fig. 2C and F). However, similar to the Δmce4Epolar mutation, the Δmam4A in-frame deletion had a polar effect on mam4B. Introduction of a multicopy plasmid expressing mam4A alone was unable to complement the mutant phenotypes, while a multicopy plasmid expressing mam4AB reversed the phenotypes. Further, expression of mam4B alone in the Δmam4Apolar mutant was unable to rescue cholesterol growth and import defects but, when the Δmam4Apolar mutant carried pairs of multicopy and single-copy (i.e., integrating) plasmids that in combination expressed both mam4A and mam4B, cholesterol defects were fully restored. These experiments demonstrated that both Mam4A and Mam4B are required for cholesterol import. The strains with different combinations of plasmids also served to demonstrate that the plasmids carrying mam4A or mam4B expressed functional protein (Fig. 2C and F). In this report, the Δmam4Apolar mutant carrying the multicopy mam4A plasmid is referred to as a mam4B* mutant and the Δmam4Apolar mutant with the multicopy mam4B plasmid is referred to as a mam4A* mutant. All of the mam4Apolar mutant strains grew equally well on glucose and glycerol (Fig. S2C).
Together, these results demonstrated that Mce4A, MceE, MceF, Mam4A, and MamB each have essential functions in cholesterol uptake. The results further indicated that, at least under the conditions tested, orthologous genes from other mce operons (e.g., mce1A) are unable to compensate for mce4 locus genes.
OmamA and OmamB proteins contribute to Mce4 transport of cholesterol but OmamC to OmamF do not.
In addition to the Mam proteins that are encoded by genes at the ends of mce loci, we previously identified Omam proteins in mycobacteria that are encoded by genes distal to mce operons (28, 37). Mam and Omam proteins have predicted structural and membrane topology similarity, and studies of omamA mutants of M. smegmatis and M. tuberculosis demonstrated a role for OmamA in Mce4 and Mce1 transporters (37). There are 10 omam genes in the M. smegmatis genome (omamA to omamJ), of which five are conserved in M. tuberculosis (omamA to omamE) (37). Genes encoding Omam proteins often appear in pairs, as is the case for most mam genes. To determine whether the Omam proteins beyond OmamA are similarly involved in Mce transport, we evaluated whether the additional M. smegmatis Omam proteins that are conserved in M. tuberculosis (M. smegmatis OmamB to OmamE) are required for cholesterol uptake. We constructed an in-frame, unmarked deletion of omamB in M. smegmatis, as well as deletion mutants in which both omam genes of a pair were deleted, i.e., ΔomamCD and ΔomamEF double mutants. As seen with the ΔomamA mutant, ΔomamB mutants had a defect in growth on and uptake of radiolabeled cholesterol, compared to the WT strain, and these phenotypes were reversed when an omamB complementation plasmid was present (Fig. 2D and G). The ΔomamA and ΔomamB mutants exhibited normal growth on glucose and glycerol, comparable to that of the WT strain (Fig. S2D). Unlike ΔomamA and ΔomamB mutants, the other omam mutants (ΔomamCD and ΔomamEF) grew on and imported cholesterol at WT levels, indicating that not all Omam proteins are required for cholesterol uptake (Fig. 2E).
Effects of Mce, Mam, and Omam proteins on the abundance of Mce4A and Mce4E.
Given the many Mce, Mam, and Omam proteins required for cholesterol import, all of which are predicted to localize to the cell envelope (28, 33, 39, 40), we hypothesized that some or all of these proteins interact in a Mce4 transporter complex. Using antibodies raised to unique peptides of Mce4A and Mce4E, we confirmed a cell envelope location of these specific Mce proteins by immunoblotting subcellular fractions of M. smegmatis (Fig. S3C). Mce4A and Mce4E were primarily localized to the cell wall fraction, where the MspA porin resides, with lesser amounts associated with the membrane fraction, where the integral IM protein SecY is localized (Fig. S3C to E). These antibodies are specific for their respective Mce4 proteins and do not cross-react with Mce proteins of other transporters, as shown by immunoblot analysis of mutant strains (Fig. S3A and B).
A common property of multiprotein complexes is that the stability of individual proteins in the complex depends on interactions among the components (46–48). Therefore, as an approach to predict proteins in a Mce4 transporter complex, we determined whether the steady-state levels of Mce4A and Mce4E depended on individual Mce, Mam, or Omam proteins by performing immunoblot analysis of whole-cell lysates (WCLs) of our mce4, mam4, and omam mutants with cholesterol uptake defects. We started by testing the effect of Mce4E on Mce4A protein levels (Fig. 3A). Immunoblotting with anti-Mce4A antibodies of WT strain, Δmce4E mutant, and complemented strain WCLs revealed reduced levels of Mce4A in the Δmce4E mutant, compared to the WT and complemented strains. The reciprocal experiment reinforced the relationship between Mce4A and Mce4E protein levels (Fig. 3B). The WCL of the Δmce4A mutant had reduced levels of Mce4E, compared to the WT and complemented strains. We also assessed Mce4A levels in the mce4F* mutant. Here too, the level of Mce4A in the WCL was reduced in the mce4F* mutant (Fig. 3A). Given the nature of our mce4F* mutant (i.e., the Δmce4Epolar mutant carrying a mce4E complementation plasmid), it was not possible to determine the effect of Mce4F on Mce4E levels. Next, we tested whether Mam4A and Mam4B had an effect on Mce4A and Mce4E levels. WCLs of mam4A* and mam4B* mutants also exhibited reduced Mce4A and Mce4E protein levels, compared to the WT and complemented strains (Fig. 3). Finally, Mce4A and Mce4E protein levels were also reduced in ΔomamA and ΔomamB mutants, compared to the WT and complemented strains (Fig. 3). GroEL levels were used as a protein loading control in these experiments. These results indicated that the levels of Mce4A and Mce4E are reduced when any Mce4A, MceE, MceF, Mam4A, MamB, OmamA, or OmamB protein is absent. These results are consistent with a network of interactions among these proteins that leads to either assembly or stability of Mce4A- and Mce4E-containing complexes.
FIG 3.

Mce4A and Mce4E protein levels are reduced in the absence of individual Mce4, Mam4, OmamA, and OmamB proteins. Protein levels of Mce4A (A) and Mce4E (B) were assessed in WT, mutant, and complemented M. smegmatis strains via immunoblotting. The arrow indicates the Mce4A band, to distinguish it from the higher species cross-reacting band. Mutant strains contain empty vectors (EV) as indicated. Protein levels of GroEL were assessed to ensure equal protein loading across strains. Results are representative of at least three independent replicates.
Conserved lipobox cysteine of Mce4E is not necessary for Mce4 transporter function.
The results described above suggested that individual Mce4 proteins associate and that each is required to play a specific role in cholesterol import. The fifth Mce protein (MceE) encoded by nearly every mce locus is distinct from other Mce proteins in being a predicted lipoprotein (28) due to the presence of a lipoprotein signal peptide with a lipobox motif, including the invariant cysteine that is the site of lipid modification (Fig. 4A). In contrast to MceE, none of the other five Mce proteins possesses a lipobox. We hypothesized that the role of MceE in the Mce4 transporter would depend on the lipobox cysteine of Mce4E, potentially because the lipid modification on Mce4E would tether the protein to the IM or MOM as a prerequisite for assembly, stabilization, or proper localization of the Mce4 transporter complex. Using site-directed mutagenesis, we constructed a plasmid expressing a mutant mce4EC16A that encodes a Mce4E with the lipobox cysteine substituted with an alanine (Fig. 4B). This substitution eliminates the lipobox motif and converts the export signal peptide of Mce4E to a standard Sec signal peptide (41, 42). To our surprise, Mce4EC16A was functional. Introduction of a plasmid carrying the mce4EC16A allele into the Δmce4E mutant fully restored growth on cholesterol and radioactive cholesterol uptake (Fig. 4C and D). Additionally, Mce4A protein levels were comparable to WT levels in the presence of Mce4EC16A (Fig. 4E). In contrast to our predictions, these results indicated that Mce4E does not require lipidation for its function in the Mce4 transporter, despite conservation of a lipobox cysteine across MceE proteins of mycobacterial species.
FIG 4.

The lipobox cysteine of Mce4E is not necessary for Mce4 transporter function. (A) PRALINE multiple sequence alignment of Mycobacterium smegmatis MceE proteins, with lipobox cysteines highlighted in green (63). (B) Immunoblot confirming expression by mce4E complementation plasmids. (C) Growth of 104 CFU of M. smegmatis strains on cholesterol was measured using resazurin reduction as a fluorescent readout of metabolic activity. (D) Radioactive cholesterol uptake was measured using a scintillation counter. Measurements of cholesterol uptake were normalized as a percentage of the value of WT M. smegmatis, which was set to 100%. Error bars represent standard deviations. *, P < 0.001, compared to WT results. Results are representative of at least three independent experiments. (E) Mce4A protein levels were assessed in WT, mutant, and complemented M. smegmatis strains via immunoblotting. The arrow indicates the Mce4A band, to distinguish it from the higher species cross-reacting band. GroEL levels were used as a loading control across strains. Mutant strains contain empty vectors (EV) as indicated.
Mce4A and Mce4E interact.
The finding that Mce4A and Mce4E are mutually required to stabilize one another suggested an interaction between the proteins. To test for an interaction between Mce4A and Mce4E in mycobacteria, we performed a coimmunoprecipitation experiment with the WCL of a Δmce4A mutant strain engineered to produce Mce4A-hemagglutinin (HA) fusion protein. Following immunoprecipitation with anti-HA-agarose beads, the immunoprecipitated sample was immunoblotted for the presence of native Mce4E protein using anti-Mce4E antibody. Mce4E was observed to coimmunoprecipitate with Mce4A-HA (Fig. 5A). As a control, Mce4E was not recovered when the HA immunoprecipitation was performed with the WCL of a strain producing untagged Mce4A (Fig. 5A). Further evidence for an interaction between Mce4A and Mce4E was obtained with the reciprocal coimmunoprecipitation experiment. Native Mce4A coimmunoprecipitated with Mce4E-HA (Fig. 5B). As a control, we also immunoblotted the immunoprecipitate for MspA, a cell wall porin (49). MspA did not coimmunoprecipitate with Mce4E-HA (Fig. 5B), confirming the specificity of the Mce4A-Mce4E interaction. The interaction between Mce4A and Mce4E was observed regardless of whether multicopy or single-copy (i.e., chromosomally integrated) expression plasmids were used to produce the HA-tagged proteins (Fig. 5C and D). The HA-tagged Mce4A and Mce4E proteins were functional, as shown by their ability to promote cholesterol utilization when expressed in the Δmce4A and Δmce4E strain backgrounds (Fig. S4).
FIG 5.

Mce4A and Mce4E interact. A clarified WCL generated by passage through a French pressure cell was used for coimmunoprecipitation with anti-HA-conjugated beads, followed by immunoblot analysis. The amount of clarified WCL (Lys) loaded represents one-fifth of the amount of protein used for the immunoprecipitation (IP) sample. (A) The strains used for immunoprecipitation were M. smegmatis Δmce4A expressing either Mce4A-HAmc or Mce4Amc without a tag as a control. An HA antibody was used to detect Mce4A-HA and an Mce4E antibody was used to detect Mce4E in the coimmunoprecipitate. (B) The strains used for immunoprecipitation were M. smegmatis Δmce4E expressing either Mce4E-HAmc or Mce4Emc without a tag as a control. An Mce4E antibody was used to detect Mce4E-HA and an Mce4A antibody was used to detect Mce4A in the coimmunoprecipitate. The arrow indicates the Mce4A band, to distinguish it from the higher species cross-reacting band. An MspA antibody was used to detect MspA as a negative control. (C) The strains used for immunoprecipitation were M. smegmatis Δmce4A expressing Mce4A-HAsc or the WT strain expressing an empty vector (EV) with an HA tag. An HA antibody was used to detect Mce4A-HA and an Mce4E antibody was used to detect Mce4E in the coimmunoprecipitate. (D) The strains used for immunoprecipitation were M. smegmatis Δmce4E expressing Mce4E-HAsc or the WT strain expressing an empty vector (EV) with an HA tag. An HA antibody was used to detect Mce4E-HA and an Mce4A antibody was used to detect Mce4A in the coimmunoprecipitate. The arrow indicates the Mce4A band, to distinguish it from the higher species cross-reacting band. Mutant strains contain empty vectors (EV) as indicated.
IP-MS identifies additional components of a Mce4A-containing complex.
To identify additional proteins that exist in a complex with Mce4A, we repeated the Mce4A-HA immunoprecipitation and analyzed the samples using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Immunoprecipitated samples prepared from strains expressing Mce4A-HA or HA only as a control (Fig. 5C) were subjected to trypsin digestion followed by LC-MS/MS. Label-free quantitation (LFQ), using the area under the curve, was performed on proteins identified by two or more unique peptides, and significant differences in LFQ intensity of proteins in the Mce4A-HA immunoprecipitation versus the control were determined using Student's t test. Proteins with P values of <0.05 and a minimum of 2-fold enrichment (log2 = 1.0) in the Mce4A-HA immunoprecipitate, compared to control, were considered interactors (Fig. 6; also see Data Set S1). Among the seven most highly enriched proteins (28- to 481-fold enrichment) in the Mce4A-HA immunoprecipitation were all six of the Mce4 proteins (Mce4A to Mce4F). Six additional proteins with roles in Mce4 transport of cholesterol, all of which are demonstrated or predicted to reside in the cell envelope, met our criteria for interacting with Mce4A-HA, namely, YrbE4A, YrbE4B, Mam4A, Mam4B, OmamA, and OmamB. In addition, the cytoplasmic ATPase MceG was identified as interacting with Mce4A-HA (Fig. 6 and Data Set S1). All of these additional proteins met our cutoff values for an interaction; however, the level of enrichment was less than that observed for the Mce4A to Mce4F proteins, which may reflect less stable interactions. Combined with the dependence of Mce4A and Mce4E stability on the presence of individual Mce4, Mam4, OmamA, and OmamB proteins, the identification of these proteins as associating with Mce4A by IP-MS indicates that this collection of proteins work together to assemble and/or to function as multiprotein Mce4 transporter complexes. In addition, five Mce1 transporter proteins (Mce1C, Mce1D, Mce1E, Mce1F, and Mam1A) met our significance cutoff values and were identified as associating with Mce4A (Fig. 6 and Data Set S1).
FIG 6.
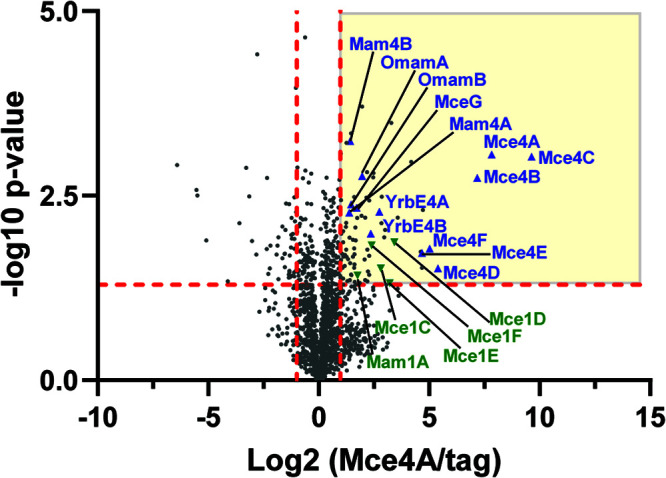
LFQ of proteins enriched in the Mce4A-HA immunoprecipitation. Proteins identified by two or more unique peptides in an Mce4A-HA immunoprecipitation, compared to an HA-only control, are shown plotted by log2(Mce4A-HA/control) and −log10(P value). Proteins with P values of <0.05 and a minimum of 2-fold enrichment (log2 = 1.0) in the Mce4A-HA immunoprecipitate, compared to the control, were considered interactors (yellow quadrant). Mce4 transporter component proteins are marked on the plot with blue triangles, and Mce1 transporter components are marked with green inverted triangles.
DISCUSSION
How Mce transporters work to import lipids in mycobacteria is unknown. Here, we constructed a set of mutants and used them to demonstrate the requirement of individual Mce4, Mam4, OmamA, and OmamB proteins for the cholesterol uptake function of the Mce4 transporter (Fig. 2). We further demonstrated that not all Omam proteins are required for cholesterol uptake by M. smegmatis, at least under the conditions tested (Fig. 2). Moreover, we demonstrated that the individual Mce4, Mam4, OmamA, and OmamB proteins are required for stabilization of Mce4A and Mce4E proteins (Fig. 3). Because this was the case for every protein associated with the Mce4 transporter system that we tested, it suggests a network of interactions between these proteins that serves to assemble and/or to stabilize multiprotein Mce4 transporter complexes. Additionally, through IP-MS experiments, we identified the Mce4A protein as interacting with every single Mce, Mam, and Omam protein required for the Mce4 transporter to import cholesterol (Fig. 6). Together, these results argue for the Mce4 transporter functioning as a large multiprotein complex.
Mce4 transporter components required for cholesterol growth and uptake.
By inactivating mce4A, mce4E, mce4F, mam4A, and mam4B, we showed that Mce4A, Mce4E, Mce4F, Mam4A, and Mam4B proteins are necessary for M. smegmatis to grow on cholesterol and, more specifically, to take up cholesterol (Fig. 2). Previously, García-Fernández et al. (50) created in-frame mutants of all of the genes in the mce4 locus and demonstrated that they were defective for growth on cholesterol. In that same study, a Δmce4A mutant was demonstrated to be unable to take up radiolabeled cholesterol. However, the other mutants in that study were not similarly evaluated for cholesterol uptake (50), which is an important detail since growth on cholesterol reflects more events than just cholesterol uptake. Furthermore, the process of Mce4-dependent import of cholesterol was previously proposed to involve dissociable events (i.e., entry into the cell envelope and translocation across the IM) (27). In fact, a M. tuberculosis Δmam4B::hyg mutant was reported to be competent in cholesterol uptake but defective for growth on cholesterol (27). However, in contrast to this result in M. tuberculosis, our M. smegmatis Δmam4B* mutant exhibited both a cholesterol growth defect and an uptake defect (Fig. 2). While these different outcomes remain to be resolved, the cholesterol uptake defect we observed with our Δmam4B* mutant is consistent with our finding that the levels of Mce4A and Mce4E proteins, both of which are themselves required for cholesterol uptake, were severely reduced in the Δmam4B* mutant (Fig. 3). Taking advantage of the cholesterol utilization and uptake phenotypes of the Δmce4E mutant, we also investigated the significance of the conserved lipobox cysteine in Mce4E. We had hypothesized that lipidation of Mce4E would be required for its proper location and function in the transporter. To our surprise, the lipobox cysteine was not required for Mce4E function, at least under the conditions tested (Fig. 4). The subcellular localization of the Mce4EC16A variant, in comparison to WT Mce4E, was also unchanged (data not shown), which is consistent with our finding that the mutation did not eliminate Mce4E function.
We also evaluated the role of Omam proteins, which are not linked to mce operons, in the Mce4 transporter function of cholesterol uptake. Despite relatively low sequence similarity, Omam proteins have predicted structural and membrane topology similarity to Mam proteins, and we previously showed that OmamA is required for Mce4 uptake of cholesterol (37). OmamA also has a role with the Mce1 transporter (37, 51). In the absence of OmamA, Mce1 proteins as well as Mce4 proteins, with the latter being shown in this report for the first time (Fig. 3), are unstable (37). These results suggest that Omam proteins have roles in multiple Mce transporters. There are 10 omam genes (omamA to omamJ) in M. smegmatis, five of which (omamA to omamE) are conserved in M. tuberculosis. Besides OmamB of M. tuberculosis being shown to be required for Mce1 transporter uptake of fatty acids (51), other Omam proteins have not been studied. Here, we assessed the role of the five Omam proteins that are conserved in M. tuberculosis (OmamA to OmamE), as well as OmamF in M. smegmatis. Only ΔomamA and ΔomamB mutants were impaired in growth on cholesterol and in cholesterol uptake (Fig. 2D, E, and G).
The structural prediction program Phyre 2 (52) identified similarity between Omam and Mam proteins and VirB8 proteins from Brucella suis and Agrobacteria tumefaciens (37). VirB8 is important for the stability and function of the large multiprotein type IV secretion system (T4SS) complexes (46–48, 53). We hypothesize that Omam and Mam proteins function as VirB8 equivalents in mycobacteria, serving to stabilize large multiprotein complexes. More specifically, we hypothesize that Mam proteins stabilize the Mce transporter with which their genes are linked in the genome, whereas OmamA and OmamB work with and stabilize multiple or all Mce transporters. This hypothesis is supported by the observation that OmamA and OmamB affect Mce1 and Mce4 transport but Mam4B affects only Mce4 transport (27, 37, 51). Due to the lack of knowledge about the other Mce transporters, it is not yet possible to test whether the functions of OmamA and OmamB extend to them as well. Because omamCD and omamEF mutants were fully competent in cholesterol uptake (Fig. 2E), we speculate that these and the other Omam proteins likely stabilize multiprotein complexes other than Mce transporters in the mycobacterial cell envelope.
Interactions among Mce4 transporter proteins.
With the exception of the cytoplasmic MceG ATPase, a cell envelope location is either predicted or proven for all of the proteins so far identified as playing roles in Mce transporters (Fig. S3) (28, 33, 39, 40). Similar to lipid transporters of Gram-negative bacteria, we hypothesized that Mce4 transporters would exist as multiprotein complexes in the cell envelope (8–10, 16, 18–20, 22). For proteins that interact in a complex, it is common for them to stabilize one another (46–48). As an approach to predict proteins that associate in a Mce4 transporter complex, we used anti-Mce4A and anti-Mce4E antibodies to examine the levels of Mce4A and Mce4E in our collection of mce4, mam4, omamA, and omamB mutants. In every case, the levels of Mce4A and Mce4E were reduced and these phenotypes could be complemented (Fig. 3). These results suggest the existence of an extensive network of interactions between Mce4A, Mce4E, Mce4F, Mam4A, Mam4B, OmamA, and OmamB proteins that may serve to assemble and/or associate in multiprotein Mce4 transporter complexes. We were unable to assess the stability of additional Mce4-associated proteins for our mutants due to the lack of available antibodies. However, given our results so far, we consider it likely that all of the proteins involved in Mce4 transport of cholesterol are dependent on one another for stability.
To further explore interactions between Mce4, Mam4, OmamA, and OmamB proteins, we performed coimmunoprecipitation experiments. First, by immunoprecipitating Mce4A or Mce4E and immunoblotting for the presence of the other Mce4 protein, we were able to demonstrate that Mce4A and Mce4E interact (Fig. 5). We subsequently extended this analysis and performed IP-MS on Mce4A-immunoprecipitated samples. In the IP-MS data, we identified all mce4 locus-encoded proteins (YrbE4A, YrbE4B, Mce4A to Mce4F, Mam4A, and Mam4B) as being significantly enriched and meeting the significance cutoff values for an interaction in the Mce4A immunoprecipitation versus the control. In addition, we identified OmamA and OmamB as interacting with Mce4A. The Mce4A to Mce4F proteins were six of the seven most enriched proteins in the immunoprecipitated samples (Fig. 6; also see Data Set S1 in the supplemental material). Based on structural studies of E. coli Mce domain-containing proteins, which form homohexameric rings (13, 14, 18, 19), we speculate that the Mce4A to Mce4F proteins may form highly stable heterohexameric rings or that each Mce protein may form a homohexameric ring and the rings stack on top of one another to form a highly stable complex. The cytoplasmic ATPase MceG was also identified as associating with the cell envelope-localized Mce4A (Fig. 6), indicating the existence of a stable complex that spans the IM. While all of the proteins were previously assumed to work together to import cholesterol, this is the first time any of these proteins were demonstrated to associate with one another. Given the many proteins identified as associating with Mce4A, we consider it unlikely that they all participate in direct protein-protein interactions with Mce4A. Instead, we suspect they are members of large complexes in which Mce4A is a component. In addition, it is currently not possible to know whether the IP-MS data reflect a single large complex or reflect subcomplexes in which Mce4A participates (such as an assembly complex and a separate structural transporter complex). Future studies will be needed to define the nature of the associations with Mce4A.
The IP-MS analysis also identified five Mce1-associated proteins (Mce1C to Mce1F and Mam1A) as interacting with Mce4A (Fig. 6). This result indicates the potential for interactions between Mce systems. However, the biological significance of such interactions remains to be determined since, at least under the conditions employed in our mutant analysis, there was no indication that Mce1 components could compensate for loss of the specific Mce4 or Mam4 proteins we investigated (Fig. 2). Finally, there were additional proteins, besides Mce-related proteins, that met our criteria for associating with Mce4A (Fig. 6 and Data Set S1). Although future studies are required to validate these interactors, it is possible that these proteins also play roles in Mce transporters.
LucA was the one protein that was previously reported to function in cholesterol uptake by the Mce4 transporter that we did not identify in IP-MS. In M. tuberculosis, LucA is required for lipid uptake by Mce4 and Mce1 transporters and, using a bacterial two-hybrid system, a domain of LucA was found to interact with Oman and Mam proteins (27). It is possible that LucA is not a stable member of the transporter complex and instead has a more transient interaction, perhaps in regulation or assembly of the transporter, or it has a role specific to Omam and Mam proteins.
In summary, in this report we demonstrated individual contributions of Mce4, Mam4, and Omam proteins in cholesterol uptake and for the first time identified associations between these proteins. Our data indicate that the Mce4 transporter is a stable, multiprotein complex involving a minimum of 13 proteins that function together to import cholesterol. The added complexity of the mycobacterial Mce transporter versus the Mce domain-containing lipid transport systems of Gram-negative bacteria may reflect the need for a system that can accommodate the unique challenges posed by the mycobacterial cell wall. While further studies are needed to address the issue of direct protein-protein interactions, and whether all of the proteins found to associate with Mce4A reflect Mce4A subcomplexes or a single large complex remains to be resolved, our data provide important evidence for Mce transporters being another example of a multiprotein complex built to transport lipids across the bacterial cell envelope.
MATERIALS AND METHODS
Bacterial strains and plasmids.
In this study, we used the bacterial strains listed in Table S2 and the plasmids listed in Table S3 in the supplemental material. Primers used for plasmid construction are listed in Table S4.
Bacterial growth conditions.
M. smegmatis liquid cultures were grown at 37°C in either Middlebrook 7H9 broth (BD Difco) supplemented with 0.2% glucose, 0.5% glycerol, and 0.05% Tween 80 (Thermo Fisher Scientific), Mueller-Hinton medium (BD Difco) supplemented with 0.05% Tween 80, M9 medium supplemented with 0.2% glucose, 0.5% glycerol, and 0.05% tyloxapol (Sigma), or M9 medium supplemented with 0.5 mM cholesterol stock solution and 0.05% tyloxapol. M9 medium was prepared as follows: 1 liter distilled H2O, 12.8 g Na2HPO4, 3 g KH2PO4, 0.5 g NaCl, 1 g NH4Cl, 25 μl 1 M CaCl2, 500 μl 1 M MgSO4, and 2.5 ml 10% tyloxapol. To make the cholesterol stock solution, cholesterol was solubilized in cyclodextrin, as described previously (37). Briefly, 1 g methyl-β-cyclodextrin (Sigma) was dissolved in 11 ml phosphate-buffered saline (PBS) (0.09 g/ml) and heated to 80°C with continuous stirring. Thirty milligrams of cholesterol (Sigma) was dissolved in 400 μl isopropanol-chloroform (2:1). The cholesterol solution was added to the cyclodextrin in 50-μl aliquots, with continuous stirring. The solution was cooled slowly and filter sterilized. M. smegmatis was grown at 37°C on Middlebrook 7H10 (BD Difco) agar plates supplemented with 0.2% glucose, 0.5% glycerol, and 0.05% Tween 80. Plasmids were maintained in M. smegmatis with appropriate antibiotics for selection either in liquid culture or on plates (50 μg/ml hygromycin [Roche] or 20 μg/ml kanamycin [Sigma]).
E. coli DH5α was grown at 37°C in Luria-Bertani (LB) broth (Thermo Fisher Scientific) or on LB agar plates. Plasmids were maintained in E. coli with appropriate antibiotics for selection either in liquid culture or on plates (150 μg/ml hygromycin [Roche] or 50 μg/ml kanamycin [Sigma]).
M. smegmatis mutant construction.
The M. smegmatis unmarked Δmce4A mutant was created by two-step allelic exchange, as described previously (54). Briefly, the mce4A knockout suicide plasmid contained a hygromycin resistance marker, a sacB counter-selectable marker, and flanking regions for mce4A. The plasmid was transformed into M. smegmatis strain mc2155, and the first recombination event was selected for by resistance to hygromycin. Bacteria in which a second recombination event occurred were selected by plating on Middlebrook 7H10 agar supplemented with 0.2% glucose and 4.5% sucrose.
The M. smegmatis unmarked Δmce4E, Δmce4Epolar, Δmam4Apolar, ΔomamB, ΔomamCD, and ΔomamEF strains were created using recombineering, as described previously (55). Briefly, recombineering plasmids were digested with restriction enzymes to create a linear fragment containing the upstream flank, hygromycin resistance cassette, and downstream flank. This linear DNA fragment was transformed via electroporation into the mc2155 strain containing the recombinase plasmid pJV53. Double crossover allelic exchange recombinants were selected for by resistance to hygromycin. To create unmarked mutants, strains were cured of pJV53 by passaging three or four times in the absence of kanamycin. Plasmid-cured strains were then transformed with the resolvase-expressing pMP854 plasmid to remove the hygromycin marker, which is flanked by res sites in the deletion cassette. All mutants were confirmed using Sanger sequencing (Genewiz).
Cholesterol growth assay.
Cholesterol growth assays were performed as described previously (37). M. smegmatis strains were grown to an optical density at 600 nm (OD600) of 1 in M9 medium supplemented with 0.2% glucose, 0.5% glycerol, and 0.05% tyloxapol. Strains were washed three times by pelleting cells by centrifugation at 3,000 × g for 10 min at 4°C and resuspending the pellets in 10 ml in M9 medium with tyloxapol (no carbon). After washing, cells were diluted in M9 medium with tyloxapol (no carbon) to 105 CFU/ml, and 100 μl was plated into 96-well plates with M9 medium containing 0.2% glucose and 0.5% glycerol or 0.5 mM cholesterol stock solution. Plates were incubated at 37°C overnight with shaking, after which resazurin (Sigma) was added to a final concentration of 0.0125 mg/ml. Florescence was monitored every 1 h using a SpectraMax M2 microplate reader, with excitation at 544 nm and emission at 590 nm (45).
Cholesterol uptake assay.
Cholesterol uptake assays were performed as described previously (26, 37). Briefly, M. smegmatis strains were grown to an OD600 of 1 in M9 medium supplemented with 0.2% glucose, 0.5% glycerol, and 0.05% tyloxapol. Strains were washed three times by pelleting cells by centrifugation at 3,000 × g for 10 min at 4°C and resuspending the pellets in 10 ml of M9 medium with tyloxapol (no carbon). After the final wash, strains were resuspended in M9 medium with 0.05% tyloxapol (no carbon) to an OD600 of 0.5, to which 0.04 μCi [4-14C]cholesterol (NEC018050UC; PerkinElmer) was added, and cells were incubated for 3 h at 37°C. After incubation, cells were pelleted and washed three times with M9 medium with tyloxapol, and cell-associated radioactivity was measured with a scintillation counter.
Mce4A and Mce4E antiserum production.
Antipeptide antibodies for Mce4A and Mce4E in M. smegmatis were prepared by using peptides specific for each of the proteins. The peptides were synthesized, conjugated to keyhole limpet hemocyanin, and used to immunize specific-pathogen-free New Zealand White rabbits, using TiterMax Gold adjuvant, at Pierce Custom Antibody Services (Rockton, IL). For anti-Mce4A antibodies, the peptide C-DAPATLQFLFNGAFAERDDF was used. For anti-Mce4E antibodies, the peptide C-SGQAADPFKIPPGTA was used. Antibodies were purified by affinity chromatography.
Immunoblotting.
Equal protein amounts, as determined by the bicinchoninic acid assay (Pierce, Thermo Fisher Scientific), for all fractions and strains were separated by SDS‐PAGE and transferred to nitrocellulose membranes. Proteins were detected using the following antibodies: Mce4 proteins were detected using anti-Mce4A (1:500), anti-Mce4E (1:500), and anti‐HA (1:25,000) (Covance). GroEL2 was detected using an anti‐His antibody (1:10,000) (Abgent), as described previously (56), that recognizes a string of histidines in M. smegmatis GroEL2. MspA was detected using an anti-MspA antibody (1:2,000) provided by Michael Niederweis. Anti‐mouse IgG and anti‐rabbit IgG conjugated to horseradish peroxidase (HRP) (1:25,000) (Bio-Rad) were used as secondary antibodies, as appropriate. HRP signal was detected using Western Lightning chemiluminescent detection reagent (PerkinElmer). For Fig. S3, SecA1 was detected using an anti-SecA1 antibody (1:10,000), as described previously (58). SecY was detected using an anti-SecY antibody (1:150), as described previously (59). MspA was detected using an anti-MspA antibody (1:2,000) provided by Michael Niederweis. GroEL2 was detected using an anti‐His antibody (1:10,000) (Abgent), as described previously (56).
Subcellular fractionation.
Subcellular fractions were generated for Fig. S3 as described previously (57). Briefly, M. smegmatis strains were grown in 50 ml Mueller-Hinton broth to an OD600 of 1.0. Cells were centrifuged for 10 min at 3,000 × g and resuspended in 2.5 ml 1× PBS buffer containing protease inhibitor cocktail used at 1:500 dilution. Cells were lysed by three passages through a French pressure cell. Unlysed cells were removed by centrifugation at 3,000 × g for 30 min to generate clarified WCLs. The WCLs were centrifuged at 27,000 × g for 30 min to pellet the cell wall fraction only. The supernatant following cell wall isolation was centrifuged at 100,000 × g for 2 h to separate the membrane fraction and to collect the soluble cytoplasm-containing fraction. Fractions were separated by SDS-PAGE, and the total amounts of cell wall, membrane, and soluble fractions shown are equivalent to the amount of WCL loaded.
Site-directed mutagenesis of mce4E.
The cysteine at position 16 of Mce4E was changed to an alanine using site-directed mutagenesis to generate Mce4EC16A. The mce4E expression plasmid pLR106 was used as a template. The primer sequences used are listed in Table S4. DpnI (New England Biolabs) was added to degrade the methylated template prior to transformation into E. coli. The mutation was confirmed by Sanger sequencing (Genewiz).
Coimmunoprecipitation assay.
M. smegmatis cells were transformed with plasmids expressing either HA fusion proteins or control plasmids. Strains were grown in 50 ml Mueller-Hinton broth to an OD600 of 0.5. Cells were pelleted for 10 min at 3,000 × g and resuspended in 2.5 ml 1× PBS buffer containing protease inhibitor cocktail used at 1:500 dilution. Cells were lysed by two passages through a French pressure cell. Unlysed cells were removed by centrifugation at 3,000 × g for 30 min to generate clarified WCLs. Equal amounts of lysates (normalized by OD) or totals of 1 mg of protein for samples used for LC-MS/MS were diluted in 1 ml of 1× PBS with protease inhibitors, to which 30 μl of anti-HA-agarose (Sigma-Aldrich) was added, and subsequently mixed end to end at 4°C for 4 h. The immunoprecipitates were pelleted and washed four times with 1 ml 1× PBS with protease inhibitors. After the final wash, the agarose beads were resuspended in 25 μl of 1× SDS-PAGE buffer, boiled, run on 15% SDS-PAGE gels, transferred onto nitrocellulose membranes, and immunoblotted or were resuspended in 30 μl of 1× PBS for LC-MS/MS analysis.
Immunoprecipitation and on-bead LC-MS/MS.
(i) Sample preparation. Immunoprecipitated samples (prepared as described above; n = 2) were subjected to on-bead trypsin digestion, as described previously (60). After the last wash step, 50 μl of 50 mM ammonium bicarbonate (pH 8) containing 1 μg trypsin (Promega) was added to the beads overnight at 37°C with shaking. The next day, 500 ng of trypsin was added for an additional 3 h at 37°C with shaking. Supernatants from pelleted beads were transferred, and then the beads were washed twice with 50 μl LC/MS-grade water. These rinses were combined with the original supernatant and then acidified with 2% formic acid. Peptides were desalted with peptide-desalting spin columns (Thermo Fisher Scientific) and dried via vacuum centrifugation.
(ii) LC-MS/MS analysis. The peptide samples were analyzed by LC-MS/MS using an Easy-nLC 1200 system coupled to a Q Exactive HF mass spectrometer (Thermo Fisher Scientific). Samples were injected onto an EASY-Spray PepMap C18 column (75 μm i.d. by 25 cm; 2-μm particle size) (Thermo Fisher Scientific) and separated over 120 min. The gradient for separation consisted of 5 to 40% mobile phase B at a flow rate of 250 nl/min; mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in 80% acetonitrile. The Q Exactive HF mass spectrometer was operated in data-dependent mode in which the 15 most intense precursors were selected for subsequent fragmentation. Resolution for the precursor scan (m/z 350 to 1,600) was set to 120,000 with a target value of 3 × 106 ions. The MS/MS scan resolution was set to 15,000 with a target value of 5 × 104 ions and a 60-ms maximum injection time. The normalized collision energy was set to 27% for higher-energy collisional dissociation (HCD). Dynamic exclusion was set to 30 s, peptide match was set to preferred, and precursors with unknown charge or a charge state of 1 and ≥ 7 were excluded.
(iii) Data analysis. Raw data were processed using the MaxQuant software suite (version 1.6.3.4) for identification and LFQ (61). Data were searched against a UniProt Mycobacterium smegmatis (strain ATCC 700084/mc2155) database (containing 6,602 entries) using the integrated Andromeda search engine. A maximum of two missed tryptic cleavages were allowed. The variable modification specified was oxidation of methionine and N-terminal acetylation. LFQ was enabled. Results were filtered to a false discovery rate of 1% at the unique peptide level and grouped into proteins within MaxQuant (61). Perseus software (version 1.6.14.0) was used for further processing. Only proteins with >1 unique+razor peptide were used for LFQ analysis. Proteins with >50% missing values were removed, and missing values were replaced from normal distribution within Perseus. Log2 fold change ratios of the Mce4A-HA samples versus the HA control samples were calculated using the averaged log2 LFQ intensities, and proteins with log2 fold changes of ≥1 were considered significant. Student's t test was performed, and P values were calculated; proteins with P values of <0.05 (i.e., −log10 P values of >1.33) were considered significant. All results can be found in Data Set S1 in the supplemental material.
Data availability.
The MS proteomics data have been deposited at the ProteomeXchange Consortium via the PRIDE partner repository (62), with the data set identifier PXD023082.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from NIAID grant R21 AI135899 (to M.B.). This research is based in part on work conducted using the UNC Proteomics Core Facility, which is supported in part by a Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center (grant P30 CA016086).
We thank Alan Schmalstig and Christian Xander of the Braunstein laboratory for experimental advice and critical reading of this manuscript, Michael Niederweis for the MspA antibody, and Fabiana Bigi for the Δ6×mce M. smegmatis strain.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Sohlenkamp C, Geiger O. 2016. Bacterial membrane lipids: diversity in structures and pathways. FEMS Microbiol Rev 40:133–159. 10.1093/femsre/fuv008. [DOI] [PubMed] [Google Scholar]
- 2.Wilburn KM, Fieweger RA, VanderVen BC. 2018. Cholesterol and fatty acids grease the wheels of Mycobacterium tuberculosis pathogenesis. Pathog Dis 76:fty021. 10.1093/femspd/fty021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shrivastava R, Chng S-S. 2019. Lipid trafficking across the Gram-negative cell envelope. J Biol Chem 294:14175–14184. 10.1074/jbc.AW119.008139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.May KL, Silhavy TJ. 2017. Making a membrane on the other side of the wall. Biochim Biophys Acta Mol Cell Biol Lipids 1862:1386–1393. 10.1016/j.bbalip.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Touchette MH, Seeliger JC. 2017. Transport of outer membrane lipids in mycobacteria. Biochim Biophys Acta Mol Cell Biol Lipids 1862:1340–1354. 10.1016/j.bbalip.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Daffé M, Marrakchi H. 2019. Unraveling the structure of the mycobacterial envelope. Microbiol Spectr 7:GPP3-0027-2018. 10.1128/microbiolspec.GPP3-0027-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb Perspect Biol 2:a000414. 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Putker F, Bos MP, Tommassen J. 2015. Transport of lipopolysaccharide to the Gram-negative bacterial cell surface. FEMS Microbiol Rev 39:985–1002. 10.1093/femsre/fuv026. [DOI] [PubMed] [Google Scholar]
- 9.Simpson BW, May JM, Sherman DJ, Kahne D, Ruiz N. 2015. Lipopolysaccharide transport to the cell surface: biosynthesis and extraction from the inner membrane. Philos Trans R Soc B 370:20150029. 10.1098/rstb.2015.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okuda S, Tokuda H. 2011. Lipoprotein sorting in bacteria. Annu Rev Microbiol 65:239–259. 10.1146/annurev-micro-090110-102859. [DOI] [PubMed] [Google Scholar]
- 11.Malinverni JC, Silhavy TJ. 2009. An ABC transport system that maintains lipid asymmetry in the Gram-negative outer membrane. Proc Natl Acad Sci U S A 106:8009–8014. 10.1073/pnas.0903229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakayama T, Zhang-Akiyama Q-M. 2017. pqiABC and yebST, putative mce operons of Escherichia coli, encode transport pathways and contribute to membrane integrity. J Bacteriol 199:e00606-16. 10.1128/JB.00606-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ekiert DC, Bhabha G, Isom GL, Greenan G, Ovchinnikov S, Henderson IR, Cox JS, Vale RD. 2017. Architectures of lipid transport systems for the bacterial outer membrane. Cell 169:273–285.e17. 10.1016/j.cell.2017.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thong S, Ercan B, Torta F, Fong ZY, Wong HYA, Wenk MR, Chng S-S. 2016. Defining key roles for auxiliary proteins in an ABC transporter that maintains bacterial outer membrane lipid asymmetry. Elife 5:e19042. 10.7554/eLife.19042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hughes GW, Hall SCL, Laxton CS, Sridhar P, Mahadi AH, Hatton C, Piggot TJ, Wotherspoon PJ, Leney AC, Ward DG, Jamshad M, Spana V, Cadby IT, Harding C, Isom GL, Bryant JA, Parr RJ, Yakub Y, Jeeves M, Huber D, Henderson IR, Clifton LA, Lovering AL, Knowles TJ. 2019. Evidence for phospholipid export from the bacterial inner membrane by the Mla ABC transport system. Nat Microbiol 4:1692–1705. 10.1038/s41564-019-0481-y. [DOI] [PubMed] [Google Scholar]
- 16.Coudray N, Isom GL, MacRae MR, Saiduddin MN, Bhabha G, Ekiert DC. 2020. Structure of bacterial phospholipid transporter MlaFEDB with substrate bound. Elife 9:e62518. 10.7554/eLife.62518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davidson AL, Chen J. 2004. ATP-binding cassette transporters in bacteria. Annu Rev Biochem 73:241–268. 10.1146/annurev.biochem.73.011303.073626. [DOI] [PubMed] [Google Scholar]
- 18.Liu C, Ma J, Wang J, Wang H, Zhang L. 2020. Cryo-EM structure of a bacterial lipid transporter YebT. J Mol Biol 432:1008–1019. 10.1016/j.jmb.2019.12.008. [DOI] [PubMed] [Google Scholar]
- 19.Isom GL, Coudray N, MacRae MR, McManus CT, Ekiert DC, Bhabha G. 2020. LetB structure reveals a tunnel for lipid transport across the bacterial envelope. Cell 181:653–664.e19. 10.1016/j.cell.2020.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ercan B, Low W-Y, Liu X, Chng S-S. 2019. Characterization of interactions and phospholipid transfer between substrate binding proteins of the OmpC-Mla system. Biochemistry 58:114–119. 10.1021/acs.biochem.8b00897. [DOI] [PubMed] [Google Scholar]
- 21.Kolich LR, Chang Y-T, Coudray N, Giacometti SI, MacRae MR, Isom GL, Teran EM, Bhabha G, Ekiert DC. 2020. Structure of MlaFB uncovers novel mechanisms of ABC transporter regulation. Elife 9:e60030. 10.7554/eLife.60030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chong Z-S, Woo W-F, Chng S-S. 2015. Osmoporin OmpC forms a complex with MlaA to maintain outer membrane lipid asymmetry in Escherichia coli. Mol Microbiol 98:1133–1146. 10.1111/mmi.13202. [DOI] [PubMed] [Google Scholar]
- 23.Arruda S, Bomfim G, Knights R, Huima-Byron T, Riley LW. 1993. Cloning of an M. tuberculosis DNA fragment associated with entry and survival inside cells. Science 261:1454–1457. 10.1126/science.8367727. [DOI] [PubMed] [Google Scholar]
- 24.Mohn WW, van der Geize R, Stewart GR, Okamoto S, Liu J, Dijkhuizen L, Eltis LD. 2008. The actinobacterial mce4 locus encodes a steroid transporter. J Biol Chem 283:35368–35374. 10.1074/jbc.M805496200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pandey AK, Sassetti CM. 2008. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A 105:4376–4380. 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klepp LI, Forrellad MA, Osella AV, Blanco FC, Stella EJ, Bianco MV, Santangelo M, de la P, Sassetti C, Jackson M, Cataldi AA, Bigi F, Morbidoni HR. 2012. Impact of the deletion of the six mce operons in Mycobacterium smegmatis. Microbes Infect 14:590–599. 10.1016/j.micinf.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nazarova EV, Montague CR, La T, Wilburn KM, Sukumar N, Lee W, Caldwell S, Russell DG, VanderVen BC. 2017. Rv3723/LucA coordinates fatty acid and cholesterol uptake in Mycobacterium tuberculosis. Elife 6:e26969. 10.7554/eLife.26969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Casali N, Riley LW. 2007. A phylogenomic analysis of the Actinomycetales mce operons. BMC Genomics 8:60. 10.1186/1471-2164-8-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar A, Chandolia A, Chaudhry U, Brahmachari V, Bose M. 2005. Comparison of mammalian cell entry operons of mycobacteria: in silico analysis and expression profiling. FEMS Immunol Med Microbiol 43:185–195. 10.1016/j.femsim.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 30.Sassetti CM, Rubin EJ. 2003. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A 100:12989–12994. 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gioffré A, Infante E, Aguilar D, Santangelo MP, Klepp L, Amadio A, Meikle V, Etchechoury I, Romano MI, Cataldi A, Hernández RP, Bigi F. 2005. Mutation in mce operons attenuates Mycobacterium tuberculosis virulence. Microbes Infect 7:325–334. 10.1016/j.micinf.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 32.Joshi SM, Pandey AK, Capite N, Fortune SM, Rubin EJ, Sassetti CM. 2006. Characterization of mycobacterial virulence genes through genetic interaction mapping. Proc Natl Acad Sci U S A 103:11760–11765. 10.1073/pnas.0603179103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forrellad MA, Klepp LI, Gioffré A, Sabio y García J, Morbidoni HR, Santangelo M, de la P, Cataldi AA, Bigi F. 2013. Virulence factors of the Mycobacterium tuberculosis complex. Virulence 4:3–66. 10.4161/viru.22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimono N, Morici L, Casali N, Cantrell S, Sidders B, Ehrt S, Riley LW. 2003. Hypervirulent mutant of Mycobacterium tuberculosis resulting from disruption of the mce1 operon. Proc Natl Acad Sci U S A 100:15918–15923. 10.1073/pnas.2433882100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lima P, Sidders B, Morici L, Reader R, Senaratne R, Casali N, Riley LW. 2007. Enhanced mortality despite control of lung infection in mice aerogenically infected with a Mycobacterium tuberculosis mce1 operon mutant. Microbes Infect 9:1285–1290. 10.1016/j.micinf.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Senaratne RH, Sidders B, Sequeira P, Saunders G, Dunphy K, Marjanovic O, Reader JR, Lima P, Chan S, Kendall S, McFadden J, Riley LW. 2008. Mycobacterium tuberculosis strains disrupted in mce3 and mce4 operons are attenuated in mice. J Med Microbiol 57:164–170. 10.1099/jmm.0.47454-0. [DOI] [PubMed] [Google Scholar]
- 37.Perkowski EF, Miller BK, McCann JR, Sullivan JT, Malik S, Allen IC, Godfrey V, Hayden JD, Braunstein M. 2016. An orphaned Mce-associated membrane protein of Mycobacterium tuberculosis is a virulence factor that stabilizes Mce transporters. Mol Microbiol 100:90–107. 10.1111/mmi.13303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.García-Fernández J, Papavinasasundaram K, Galán B, Sassetti CM, García JL. 2017. Unravelling the pleiotropic role of the MceG ATPase in Mycobacterium smegmatis. Environ Microbiol 19:2564–2576. 10.1111/1462-2920.13771. [DOI] [PubMed] [Google Scholar]
- 39.Perkowski EF, Zulauf KE, Weerakoon D, Hayden JD, Ioerger TR, Oreper D, Gomez SM, Sacchettini JC, Braunstein M. 2017. The EXIT strategy: an approach for identifying bacterial proteins exported during host infection. mBio 8:e00333-17. 10.1128/mBio.00333-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feltcher ME, Gunawardena HP, Zulauf KE, Malik S, Griffin JE, Sassetti CM, Chen X, Braunstein M. 2015. Label-free quantitative proteomics reveals a role for the Mycobacterium tuberculosis SecA2 pathway in exporting solute binding proteins and Mce transporters to the cell wall. Mol Cell Proteomics 14:1501–1516. 10.1074/mcp.M114.044685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Braun V, Wu HC. 1994. Lipoproteins, structure, function, biosynthesis and model for protein export, p 319–341. In Ghuysen J-M, Hakenbeck R (ed), New comprehensive biochemistry, vol 27. Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 42.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 43.Juncker AS, Willenbrock H, von Heijne G, Brunak S, Nielsen H, Krogh A. 2003. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci 12:1652–1662. 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bagos PG, Tsirigos KD, Liakopoulos TD, Hamodrakas SJ. 2008. Prediction of lipoprotein signal peptides in Gram-positive bacteria with a hidden Markov model. J Proteome Res 7:5082–5093. 10.1021/pr800162c. [DOI] [PubMed] [Google Scholar]
- 45.Hayden JD, Brown LR, Gunawardena HP, Perkowski EF, Chen X, Braunstein M. 2013. Reversible acetylation regulates acetate and propionate metabolism in Mycobacterium smegmatis. Microbiology (Reading) 159:1986–1999. 10.1099/mic.0.068585-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vincent CD, Friedman JR, Jeong KC, Buford EC, Miller JL, Vogel JP. 2006. Identification of the core transmembrane complex of the Legionella Dot/Icm type IV secretion system. Mol Microbiol 62:1278–1291. 10.1111/j.1365-2958.2006.05446.x. [DOI] [PubMed] [Google Scholar]
- 47.den Hartigh AB, Rolán HG, de Jong MF, Tsolis RM. 2008. VirB3 to VirB6 and VirB8 to VirB11, but not VirB7, are essential for mediating persistence of Brucella in the reticuloendothelial system. J Bacteriol 190:4427–4436. 10.1128/JB.00406-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sivanesan D, Baron C. 2011. The dimer interface of Agrobacterium tumefaciens VirB8 is important for type IV secretion system function, stability, and association of VirB2 with the core complex. J Bacteriol 193:2097–2106. 10.1128/JB.00907-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stahl C, Kubetzko S, Kaps I, Seeber S, Engelhardt H, Niederweis M. 2001. MspA provides the main hydrophilic pathway through the cell wall of Mycobacterium smegmatis. Mol Microbiol 40:451–464. 10.1046/j.1365-2958.2001.02394.x. [DOI] [PubMed] [Google Scholar]
- 50.García‐Fernández J, Papavinasasundaram K, Galán B, Sassetti CM, García JL. 2017. Molecular and functional analysis of the mce4 operon in Mycobacterium smegmatis. Environ Microbiol 19:3689–3699. 10.1111/1462-2920.13869. [DOI] [PubMed] [Google Scholar]
- 51.Nazarova EV, Montague CR, Huang L, La T, Russell D, VanderVen BC. 2019. The genetic requirements of fatty acid import by Mycobacterium tuberculosis within macrophages. Elife 8:e43621. 10.7554/eLife.43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baron C. 2006. VirB8: a conserved type IV secretion system assembly factor and drug target. Biochem Cell Biol 84:890–899. 10.1139/o06-148. [DOI] [PubMed] [Google Scholar]
- 54.Braunstein M, Brown AM, Kurtz S, Jacobs WR. 2001. Two nonredundant SecA homologues function in mycobacteria. J Bacteriol 183:6979–6990. 10.1128/JB.183.24.6979-6990.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Kessel JC, Marinelli LJ, Hatfull GF. 2008. Recombineering mycobacteria and their phages. Nat Rev Microbiol 6:851–857. 10.1038/nrmicro2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rengarajan J, Murphy E, Park A, Krone CL, Hett EC, Bloom BR, Glimcher LH, Rubin EJ. 2008. Mycobacterium tuberculosis Rv2224c modulates innate immune responses. Proc Natl Acad Sci U S A 105:264–269. 10.1073/pnas.0710601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feltcher ME, Gibbons HS, Ligon LS, Braunstein M. 2013. Protein export by the mycobacterial SecA2 system is determined by the preprotein mature domain. J Bacteriol 195:672–681. 10.1128/JB.02032-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo XV, Monteleone M, Klotzsche M, Kamionka A, Hillen W, Braunstein M, Ehrt S, Schnappinger D. 2007. Silencing essential protein secretion in Mycobacterium smegmatis by using tetracycline repressors. J Bacteriol 189:4614–4623. 10.1128/JB.00216-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ligon LS, Rigel NW, Romanchuk A, Jones CD, Braunstein M. 2013. Suppressor analysis reveals a role for SecY in the SecA2-dependent protein export pathway of mycobacteria. J Bacteriol 195:4456–4465. 10.1128/JB.00630-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Youn J-Y, Dunham WH, Hong SJ, Knight JDR, Bashkurov M, Chen GI, Bagci H, Rathod B, MacLeod G, Eng SWM, Angers S, Morris Q, Fabian M, Côté J-F, Gingras A-C. 2018. High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol Cell 69:517–532.e11. 10.1016/j.molcel.2017.12.020. [DOI] [PubMed] [Google Scholar]
- 61.Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 62.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, Pérez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yılmaz Ş, Tiwary S, Cox J, Audain E, Walzer M, Jarnuczak AF, Ternent T, Brazma A, Vizcaíno JA. 2019. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47:D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Simossis VA, Heringa J. 2005. PRALINE: a multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res 33:W289–W294. 10.1093/nar/gki390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data have been deposited at the ProteomeXchange Consortium via the PRIDE partner repository (62), with the data set identifier PXD023082.