

Abstract
Leukocyte extravasation is an essential step during the immune response and requires the destabilization of endothelial junctions. We have shown previously that this process depends in vivo on the dephosphorylation of VE‐cadherin‐Y731. Here, we reveal the underlying mechanism. Leukocyte‐induced stimulation of PECAM‐1 triggers dissociation of the phosphatase SHP2 which then directly targets VE‐cadherin‐Y731. The binding site of PECAM‐1 for SHP2 is needed for VE‐cadherin dephosphorylation and subsequent endocytosis. Importantly, the contribution of PECAM‐1 to leukocyte diapedesis in vitro and in vivo was strictly dependent on the presence of Y731 of VE‐cadherin. In addition to SHP2, dephosphorylation of Y731 required Ca2+‐signaling, non‐muscle myosin II activation, and endothelial cell tension. Since we found that β‐catenin/plakoglobin mask VE‐cadherin‐Y731 and leukocyte docking to endothelial cells exert force on the VE‐cadherin–catenin complex, we propose that leukocytes destabilize junctions by PECAM‐1‐SHP2‐triggered dephosphorylation of VE‐cadherin‐Y731 which becomes accessible by actomyosin‐mediated mechanical force exerted on the VE‐cadherin–catenin complex.
Keywords: cell adhesion, endothelial junctions, leukocyte extravasation, mechanical tension, VE‐cadherin
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Immunology; Vascular Biology & Angiogenesis
Trans‐endothelial migration of leukocytes requires the release of phosphatase SHP‐2 from PECAM‐1 to trigger mechanical force‐dependent modification of VE‐cadherin and opening of junctions.
Introduction
The entry of leukocytes into tissue is essential for the defense against infections and a hallmark of inflammation. It is controlled by a sophisticated interplay of adhesion receptors and chemotactic signals that regulates the docking of leukocytes to the endothelial cell surface and mediates the diapedesis through the vessel wall of venules (Muller, 2011; Nourshargh & Alon, 2014; Vestweber, 2015). Leukocytes can pass through the endothelial barrier by migration through destabilized junctions between endothelial cells or directly by moving through endothelial cells on a transcellular route. The latter pathway, however, is only responsible for about 10% of the extravasation events in vivo (Schulte et al, 2011; Woodfin et al, 2011). Thus, a central question of the diapedesis process is how endothelial junctions are triggered to open.
Of the various adhesion molecules at endothelial junctions, VE‐cadherin is arguably the most important one for junction integrity (Dejana & Vestweber, 2013). Antibodies against VE‐cadherin are sufficient to destabilize endothelial junctions in vitro (Breviario et al, 1995) and in several vascular beds in vivo (Gotsch et al, 1997; Matsuyoshi et al, 1997; Corada et al, 1999). Several in vitro studies reported that leukocyte docking to endothelial cells modulates tyrosine phosphorylation of VE‐cadherin, and different tyrosine phosphorylation sites were reported as important for the diapedesis process in vitro (Allingham et al, 2007; Turowski et al, 2008). Based on an in vivo study with knock‐in mice expressing point mutated forms of VE‐cadherin with either tyrosine 685 (Y685) or Y731 replaced by phenylalanine, we showed previously that Y731, but not Y685, was required for leukocyte diapedesis (Wessel et al, 2014). In contrast, Y685 was needed for the induction of vascular permeability, but not for leukocyte extravasation. Furthermore, whereas the induction of Y685 phosphorylation by inflammatory mediators supported leak formation, Y731 was found to be constitutively phosphorylated and leukocytes triggered the dephosphorylation of Y731. This event required the presence of the phosphatase SHP2 and triggered endocytosis of VE‐cadherin, which supported neutrophil diapedesis (Wessel et al, 2014). How leukocytes trigger the dephosphorylation of VE‐cadherin‐Y731 is unknown.
Several adhesion receptors on endothelial cells are involved in leukocyte diapedesis. PECAM‐1 was found first to serve this function (Muller et al, 1993; Bogen et al, 1994), followed by CD99 (Schenkel et al, 2002; Bixel et al, 2004), CD99L2 (Bixel et al, 2010), the nectin‐like molecule poliovirus receptor (PVR) (Reymond et al, 2004; Sullivan et al, 2013), junctional adhesion molecule (JAM)‐A (Martin‐Padura et al, 1998; Aurrand‐Lions et al, 2002), and intercellular adhesion molecule (ICAM)‐2 (Huang et al, 2006; Woodfin et al, 2009). Whereas antibody blockage or deletion does interfere with the diapedesis process, the molecular mechanism by which they support diapedesis is thus far elusive. For PECAM‐1, it was shown that its stimulation was needed for the fusion of a tubulovesicular membrane compartment with the plasma membrane at cell contacts (Mamdouh et al, 2003; Mamdouh et al, 2008). This recycling mechanism was suggested to be involved in the leukocyte diapedesis process. Yet, for none of these players effects on VE‐cadherin or a role in leukocyte‐induced junction destabilization have been reported.
Here we investigated how leukocytes address the stability of the endothelial barrier during the diapedesis process. We found that leukocytes destabilize endothelial junctions by interacting with PECAM‐1, which releases SHP2 and targets VE‐cadherin in a force‐dependent manner.
Results
SHP2 supports leukocyte diapedesis by directly dephosphorylating VE‐cadherin‐Y731
We have shown previously that Y731 of VE‐cadherin is constitutively phosphorylated, required for efficient leukocyte diapedesis and becomes dephosphorylated upon leukocyte docking to endothelial cells in a SHP2‐dependent manner (Wessel et al, 2014). To investigate how SHP2 is involved in these processes, we performed in vitro transmigration assays with primary endothelial cells isolated from VE‐cadherin wild‐type (WT) or Y731F mice; each transfected with control or SHP2‐specific siRNAs. We found that silencing SHP2 reduced neutrophil transmigration in WT endothelial cells, but not in Y731F mutant cells (Fig 1A). Thus, the contribution of SHP2 to neutrophil transmigration depends on VE‐cadherin‐Y731. Specificity of RNA silencing (targeting the 3′UTR of SHP2 mRNA) was controlled by re‐expression of SHP2, which restored transmigration efficiency (Fig 1B). To test whether VE‐cadherin‐Y731 is a direct substrate of SHP2, we used a substrate trapping approach (Flint et al, 1997) with a trapping mutant of SHP2 that carries a D to A point mutation in its catalytic core, which stabilizes the enzyme‐substrate‐interaction by blocking the dephosphorylation activity while leaving substrate‐recognition intact. Our affinity precipitation studies showed that VE‐cadherin binds to GST‐SHP2‐DA, but not to GST‐SHP2‐WT or GST only (Fig 1C). Moreover, VE‐cadherin WT associated with GST‐SHP2‐DA to a much greater extent than VE‐cadherin‐Y731F, demonstrating that Y731 is a direct substrate of SHP2. Docking of T lymphocytes to endothelial cells enhanced the ability to co‐immunoprecipitate VE‐cadherin with SHP2 concomitantly to dephosphorylation of VE‐cadherin‐Y731 (Fig 1D). Collectively, our results demonstrate that leukocyte docking to endothelium recruits SHP2 to VE‐cadherin, where it directly dephosphorylates Y731.
Figure 1. SHP2 supports leukocyte diapedesis by directly dephosphorylating VE‐cadherin‐Y731.
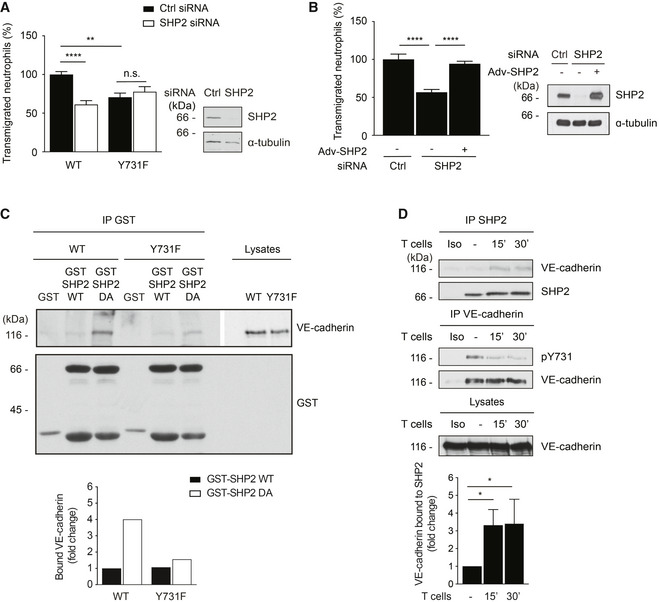
- Transmigration of mouse neutrophils toward the chemokine CXCL‐1 through TNF‐α‐stimulated VE‐cadherin WT (WT) or VE‐cadherin Y731F (Y731F) primary endothelial cells, each transfected with control or SHP2‐specific siRNA. The transmigration rate is presented relative to that of WT cells transfected with control siRNA, set as 100%. On the right, Western blot analysis of lysates from cells used in transmigration assays for the expression of SHP2 and α‐tubulin.
- Transmigration of human neutrophils toward the chemokine IL‐8 through TNF‐α‐stimulated human endothelial cells (HUVEC), transfected with control or SHP2‐specific siRNA, and were either not transduced (−) or transduced (+) with adenoviruses expressing human SHP2 (Adv‐SHP2). The transmigration rate is presented relative to that of cells transfected with control siRNA, set as 100%. On the right, Western blot analysis of lysates from cells used in transmigration assays for the expression of SHP2 and α‐tubulin.
- Lysates of WT or Y731F primary endothelial cells were submitted to pulldowns with GST, GST‐SHP2‐WT, or GST‐SHP2‐DA. Pulldowns and total cell lysates were analyzed by Western blotting for VE‐cadherin and GST. Graph below represents quantification of two independent experiments, presented as fold change of bound VE‐cadherin.
- TNF‐α‐activated bEnd.5 cells were incubated with mouse T cells for 0–30 min. SHP2 (above) or VE‐cadherin (below) were immunoprecipitated (IP) from cell lysates and precipitates as well as total cell lysates were analyzed by immunoblotting for indicated antigens. Molecular weight markers are indicated in kDa. Graph below represents quantification of five independent experiments, presented as fold change of bound VE‐cadherin.
Data information: Immunoblots are representative of two (C) or five (D) independent experiments and transmigration experiments represent data from four (A) and three (B) independent experiments with 3–6 values for each group per assay. Graphs represent mean ± SEM. Statistical significance was tested with one‐way ANOVA, (A, B) or Kruskal–Wallis test (D), *P < 0.05, **P < 0.01, ****P < 0.0001, n.s., not significant.
Source data are available online for this figure.
PECAM‐1 releases SHP2 and is required for leukocyte‐induced dephosphorylation of VE‐cadherin‐Y731
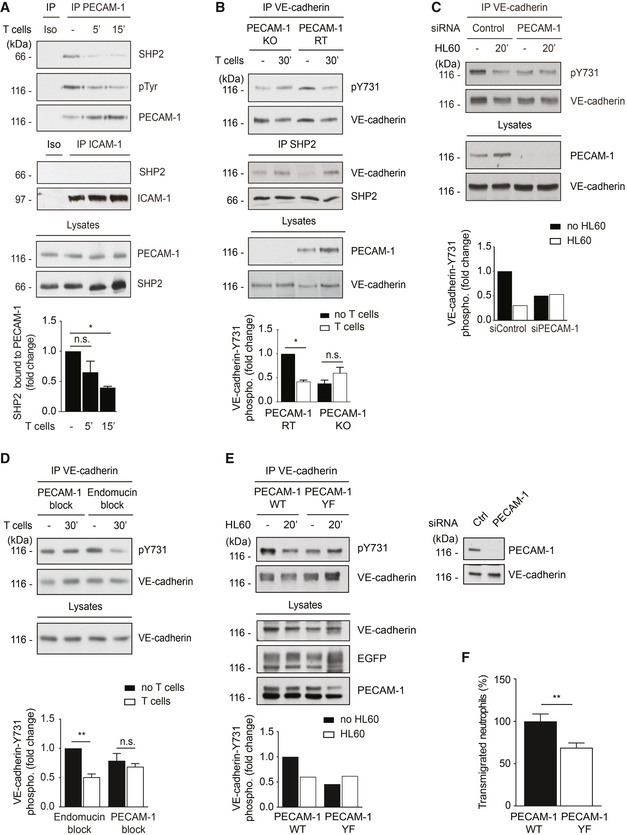
We next explored which adhesion receptor would trigger SHP2 mediated VE‐cadherin‐Y731 dephosphorylation. SHP2 was reported to associate with PECAM‐1 (Masuda et al, 1997) and ICAM‐1 (Pluskota et al, 2000). We found that we could co‐precipitate SHP2 from endothelioma cells with anti‐PECAM‐1, but not with anti‐ICAM‐1 antibodies (Fig 2A). Adhesion of T cells triggered rapid tyrosine dephosphorylation of PECAM‐1 concomitant with the dissociation of SHP2 (Fig 2A). This raised the possibility that PECAM‐1 may provide SHP2 for the dephosphorylation of VE‐cadherin‐Y731. Consistent with this hypothesis, endothelioma cells established from PECAM‐1 gene‐inactivated mice had lost the ability to dephosphorylate Y731 upon leukocyte interaction, whereas PECAM‐1 re‐expression rescued this phenotype (Fig 2B). Furthermore, silencing of PECAM‐1 by siRNA in HUVEC blocked the ability of HL60‐derived neutrophil‐like cells to induce VE‐cadherin‐Y731 dephosphorylation (Fig 2C). Finally, antibodies against Ig‐domain 2 of mouse PECAM‐1 blocked T cell‐induced VE‐cadherin‐Y731 dephosphorylation in endothelioma cells, whereas anti‐endomucin control antibodies had no effect (Fig 2D). Thus, PECAM‐1 is required for leukocyte‐induced VE‐cadherin‐Y731 dephosphorylation.
Figure 2. PECAM‐1 provides SHP2 and is required for leukocyte‐induced dephosphorylation of VE‐cadherin‐Y731.
-
A–EDifferent endothelial cells were stimulated with TNF‐α for 17 h prior to adding leukocytes for indicated time points followed by immunoprecipitations (antigens indicated above) and immunoblots (antigens indicated on the right) of precipitates and endothelial cell lysates (Iso = isotype control). (A) bEnd.5 cells were incubated with antigen‐stimulated mouse T cells for 0–15 min. (B) luEnd cells devoid of PECAM‐1 (KO) or re‐transfected with PECAM‐1 (RT) were incubated with T cells for 30 min. (C) HUVEC transfected with control or PECAM‐1‐specific siRNA were incubated with differentiated HL60 cells for 20 min. (D) bEnd.5 cells were pre‐treated with anti‐PECAM‐1 (Mec13.3) or anti‐endomucin antibodies for 30 min prior to incubation with T cells for 30 min. (E) HUVEC, siRNA depleted for endogenous PECAM‐1, were transduced with wild‐type PECAM‐1 (PECAM‐1 WT) or Y663,686F PECAM‐1 double mutant (PECAM‐1 YF) and incubated with HL60‐derived neutrophils for 20 min. On the right, immunoblot analysis of lysates from siRNA‐treated cells before PECAM‐1 re‐expression.
-
FTransmigration of human neutrophils toward IL‐8 through TNF‐α‐stimulated HUVEC expressing PECAM‐1 WT or PECAM‐1‐Y663,686F. The transmigration rate is presented relative to that of PECAM‐1 WT expressing cells, set as 100%.
Data information: Graphs below the immunoblots represent quantification of three (A, B), two (C, E), or four (D) independent experiments, presented as fold change in pY731‐VE‐cadherin signal intensities, adjusted to the amount of precipitated VE‐cadherin and normalized to control cell, set as 1. Immunoblots are representative of three (A, B) independent experiments and transmigration results from three (F) independent experiments with 6 values for each group per assay (mean ± SEM). Statistical significance was tested with one‐way ANOVA (A, B, D) or unpaired two‐tailed t‐test (F), *P < 0.05, **P < 0.01, n.s., not significant.
Source data are available online for this figure.
If PECAM‐1 provides SHP2 for this process, this should be impaired by mutating the binding sites of PECAM‐1 for SHP2. Tyrosine residues Y663 and Y686 of PECAM‐1 represent ITIM motifs for the binding of SHP2 (Jackson et al, 1997). Hence, we expressed either a PECAM‐1‐Y663,686F tyrosine mutant or WT PECAM‐1 by adenoviral transduction of HUVEC cells after depletion of endogenous PECAM‐1 by RNA interference. We found that HL60‐derived neutrophils induced VE‐cadherin‐Y731 dephosphorylation only in cells that re‐expressed WT PECAM‐1, but not in those that expressed the Y/F mutant form (Fig 2E). Importantly, transmigration of human neutrophils through HUVEC transduced with the mutant form of PECAM‐1 was reduced by 31% in comparison to HUVEC transduced with WT PECAM‐1 (Fig 2F). Collectively, our results suggest that PECAM‐1 releases SHP2, a phosphatase needed for leukocyte‐induced VE‐cadherin‐Y731 dephosphorylation and diapedesis.
PECAM‐1 is required for leukocyte‐induced endocytosis of VE‐cadherin
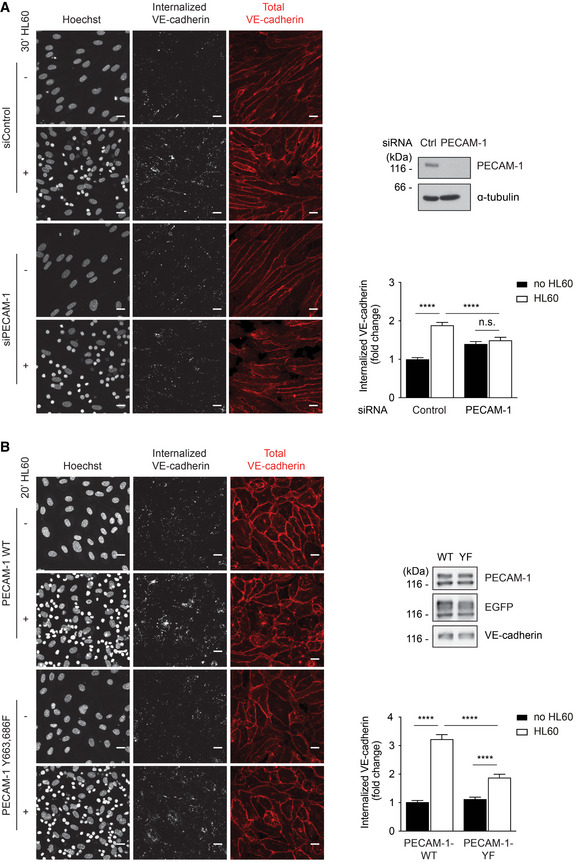
We have previously shown that leukocyte docking to endothelial cells triggers endocytosis of VE‐cadherin and that this is blocked upon mutating Y731 in VE‐cadherin (Wessel et al, 2014). Based on the essential role of PECAM‐1 for leukocyte‐induced VE‐cadherin‐Y731 dephosphorylation, we tested whether PECAM‐1 would be relevant for leukocyte‐induced internalization of VE‐cadherin. Endocytosis was monitored with the help of the mAb BV6 against the extracellular part of human VE‐cadherin. First, we compared HUVEC treated with PECAM‐1 siRNA and ctrl siRNA and found that HL60‐derived neutrophils enhanced VE‐cadherin internalization in ctrl HUVEC 1.9‐fold, but caused no significant increase in HUVEC silenced for the expression of PECAM‐1 (Fig 3A). Leukocyte‐induced VE‐cadherin internalization strongly recovered (3.2‐fold) upon re‐expression of WT PECAM‐1, but much less efficiently (1.7‐fold) upon expression of PECAM‐1‐Y663,686F (Fig 3B). Thus, leukocyte‐induced VE‐cadherin endocytosis depends to a significant extent on the PECAM‐1 binding site for SHP2.
Figure 3. PECAM‐1 is required for leukocyte‐induced internalization of VE‐cadherin.
-
A, BEndocytosis of VE‐cadherin in 17 h TNF‐α‐treated HUVEC was induced by adding HL60‐derived neutrophils for 20–30 min at 37°C and monitored by the internalization of pre‐incubated mAb BV6, which was visualized in fixed cells with a secondary fluorescent antibody (white). Total VE‐cadherin was stained with another mAb (red), nuclei stained with Hoechst (white). (A) HUVEC had been transfected prior to the experiment with control or PECAM‐1‐specific siRNA. Cell lysates were analyzed for expression levels of PECAM‐1 and tubulin (below). (B) Similar as A, except that PECAM‐1 siRNA‐treated HUVEC were transduced with WT PECAM‐1‐EGFP or PECAM‐1‐Y663,686F‐EGFP. Expression levels of transduced PECAM‐1 forms and of EGFP and VE‐cadherin are shown in immunoblots (on the right). Relative VE‐cadherin internalization efficiency is given on the right.
Data information: Graphs represent data from three independent experiments (mean ± SEM) (A, B). Statistical significance was tested with one‐way ANOVA, ****P < 0.0001, n.s., not significant. Scale bars 20 μm.
Source data are available online for this figure.
The contribution of PECAM‐1 to leukocyte diapedesis is dependent on Y731 of VE‐cadherin
To provide direct evidence that PECAM‐1 regulates diapedesis through the SHP2‐VE‐cadherin‐dependent mechanism described above, we tested next whether anti‐PECAM‐1 antibodies could still inhibit neutrophil diapedesis through endothelioma cells silenced for SHP2. We found that anti‐PECAM‐1 antibodies, when compared with control antibodies against endomucin, inhibited diapedesis in ctrl siRNA‐treated cells by 25%, whereas no inhibition was observed in SHP2 siRNA‐treated cells (Fig 4A). Likewise, anti‐PECAM‐1 antibodies inhibited diapedesis of neutrophils through primary endothelial cells from WT mice by 32%, but had no inhibitory effect on diapedesis through endothelial cells from Y731F knock‐in mice (Fig 4B). A similar result was found when we analyzed the effects of anti‐PECAM‐1 antibodies on IL‐1β‐induced neutrophil extravasation in the cremaster of WT and Y731F mice. As shown in Fig 4C, anti‐PECAM‐1 inhibited extravasation by 31% in WT mice, but had no effect in Y731F mice. As expected, no inhibitory effects were observed for neutrophil adhesion or rolling flux fraction (Fig 4D and E). Collectively, these results reveal that the contribution of PECAM‐1 to neutrophil diapedesis in vitro and in vivo relies on SHP2‐mediated dephosphorylation of VE‐cadherin‐Y731.
Figure 4. PECAM‐1 mediated leukocyte diapedesis is in vivo and in vitro dependent on VE‐cadherin‐Y731.
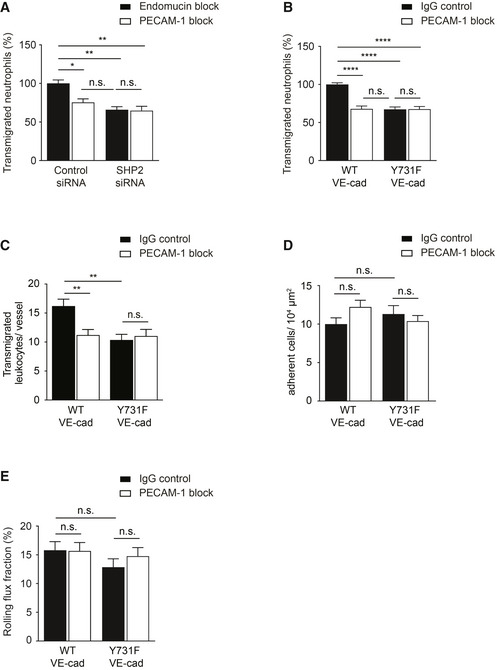
-
ATransmigration of mouse neutrophils toward CXCL‐1 through TNF‐α‐stimulated bEnd.5 cells transfected with control or SHP2‐specific siRNA and pre‐treated with anti‐endomucin (7C7.1) or anti‐PECAM‐1 (Mec13.3) antibodies for 30 min at 37°C before addition of neutrophils. The transmigration rate is presented relative to that of control cells, set as 100%.
-
BTransmigration of mouse neutrophils toward CXCL‐1 through TNF‐α‐stimulated WT or Y731F primary mouse endothelial cells pre‐treated with isotype control or anti‐PECAM‐1 (2H8) antibodies for 30 min at 37°C prior to addition of neutrophils. The transmigration rate is presented relative to that of control cells, set as 100%.
-
C–EExtravasated leukocytes (C), adherent leukocytes (D), and rolling flux fraction of leukocytes (E) in cremaster tissue from WT or Y731F VE‐cadherin mice stimulated intrascrotally with IL‐1β and treated i.v. with isotype control or anti‐PECAM‐1 (2H8) antibodies for 4 h before intravital microscopy.
Data information: Graphs represent data from three (A, B) independent experiments in triplicate for each group (mean ± SEM). Data in (C–E) are of 38 vessels from four mice, 39 vessels from four mice, 31 vessels from three mice and 43 vessels from four mice analyzed in WT + IgG, WT + 2H8, Y731F + IgG, and Y731F + 2H8 treatment groups, respectively (mean ± SEM). Statistical significance was tested with one‐way ANOVA, *P < 0.05, **P < 0.01, ****P < 0.0001, n.s., not significant.
VE‐cadherin‐Y731 becomes phosphorylated before it reaches the cell surface and is masked by catenins
In the course of investigating the role of SHP2 in VE‐cadherin phosphorylation, we found to our surprise that overexpression of SHP2 in HUVEC by adenoviral transduction did not lead to the dephosphorylation of VE‐cadherin‐Y731 (Fig 5A). This raised the idea that in resting endothelial cells, Y731 of mature VE‐cadherin may not be freely accessible. Hence, we tested whether Y731 may become phosphorylated during its post‐translational processing before it reaches the cell surface. Cadherins are known to undergo cleavage of its prodomain in the Golgi (Posthaus et al, 1998). To determine, whether phosphorylation of Y731 already occurs at the proform stage, we generated antibodies against the prodomain of VE‐cadherin. As shown in Fig 5B, these antibodies specifically recognized a form of VE‐cadherin with slightly higher mobility in PAGE. Reduction of the apparent molecular weight of this proform in comparison with mature VE‐cadherin was due to its not yet fully processed glycosylation, as was determined by blocking N‐glycosylation with tunicamycin (Fig EV1). Importantly, the proform of VE‐cadherin was already associated with the catenins and phosphorylated at Y731 (Fig 5B).
Figure 5. VE‐cadherin‐Y731 becomes phosphorylated before it reaches the cell surface and is masked by catenins.
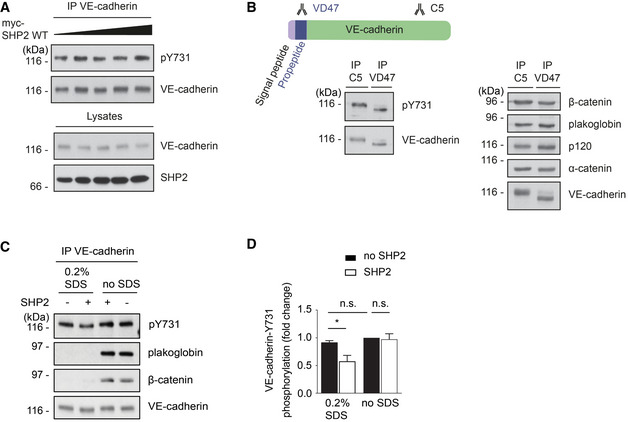
- HUVEC were transduced with increasing titers of adenovirus expressing human SHP2. VE‐cadherin was immunoprecipitated from cell lysates and precipitates as well as total cell lysates were analyzed by immunoblots for indicated antigens.
- bEnd.3 cells were lysed, and VE‐cadherin or immature VE‐cadherin was immunoprecipitated, respectively, with C5 or VD47 antibodies. Precipitates were blotted against pY731‐VE‐cadherin, β‐catenin, plakoglobin, p120, α‐catenin, and VE‐cadherin.
- VE‐cadherin–catenin complex was immunoprecipitated from HUVEC lysates and was either left untreated (no SDS) or treated with 0.2% SDS for 30 min at room temperature, followed by removal of SDS and addition of 2 μg recombinant human SHP2 for 30 min at 30°C. Precipitates were analyzed in immunoblots for pY731‐VE‐cadherin, plakoglobin, β‐catenin, and VE‐cadherin.
- Quantification of pY731 blot signals of three independent experiments (from C), with the signal intensity for untreated VE‐cadherin set as 1. Molecular weight markers are indicated in kDa.
Data information: Immunoblots are representative of at least three (A, B‐right panel, C) or five (B‐left panel) independent experiments (mean ± SEM). Statistical significance was tested with unpaired two‐tailed t‐test, *P < 0.05, n.s., not significant.
Source data are available online for this figure.
Figure EV1. Reduced apparent molecular weight of VE‐cadherin proform is due to immature glycosylation.
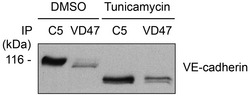
bEnd.5 cells were treated with tunicamycin or DMSO for 48 h prior to cell lysis. VE‐cadherin was precipitated from cell lysates with the antibodies C5 (mature VE‐cadherin) or VD47 (proform of VE‐cadherin). Precipitates were analyzed by Western blotting with antibodies against VE‐cadherin. Molecular weight marker is indicated in kDa.
To test directly, whether β‐catenin or plakoglobin impair access of SHP2 to Y731 of VE‐cadherin, we performed in vitro phosphatase assays with recombinant, purified SHP2, and immunoprecipitated VE‐cadherin–catenin complexes. By subsequent immunoblotting, we found that Y731 phosphorylation could indeed not be reduced by this treatment (Fig 5C). Only when the immunoprecipitated VE‐cadherin–catenin complexes were treated with 0.2% SDS to dissociate catenins from VE‐cadherin prior to incubation with SHP2, we observed a reduction in Y731 phosphorylation (Fig 5C and D). These results suggest that the catenins mask the phosphorylation site of VE‐cadherin at Y731, which requires additional mechanisms in cells to make this site available for SHP2 during leukocyte diapedesis.
Dephosphorylation of VE‐cadherin‐Y731 during leukocyte diapedesis requires Ca2+ mobilization and actomyosin contraction in endothelial cells
Since leukocyte docking to endothelial cells was not accompanied by dissociation of β‐catenin or plakoglobin from VE‐cadherin (Fig EV2), we searched for more subtle ways of demasking pY731. It is well documented that leukocyte transmigration requires Ca2+‐signaling and the activation of non‐muscle myosin II (NM‐II) (Hixenbaugh et al, 1997; Garcia et al, 1998; Saito et al, 1998; Saito et al, 2002) which enhance contractility in endothelial cells. We speculated that the catenin‐mediated link of VE‐cadherin to actin may allow mechanical forces to modulate the catenin‐VE–cadherin interaction thereby rendering Y731 accessible for SHP2. Thus, we tested whether blocking of Ca2+‐signaling by the chelator MAPTAM or of myosin by the inhibitor blebbistatin would not only inhibit leukocyte transmigration, but also leukocyte‐induced dephosphorylation of VE‐cadherin‐Y731. We found that MAPTAM inhibited transmigration of neutrophils through primary mouse endothelial cells of WT mice, but not if endothelial cells originated from Y731F mice (Fig 6A). In line with this, we found that T cell‐induced VE‐cadherin‐Y731 dephosphorylation was blocked by MAPTAM (Fig 6B). In both assays, MAPTAM had been washed away before adding leukocytes. Similar results as with MAPTAM were found with the myosin inhibitor blebbistatin (Fig 6C and D). Again, blebbistatin was washed away before adding leukocytes. Next, we tested whether stimulation of stress fiber‐mediated force would enhance accessibility of VE‐cadherin‐Y731. To this end, we tested whether thrombin would assist overexpressed SHP2 in HUVEC to dephosphorylate Y731. As described above, overexpression of SHP2 alone was not sufficient to dephosphorylate Y731 (Fig 5A), but in combination with thrombin, phosphorylation of Y731 was strongly reduced (Fig 6E). No significant effect was observed when lacZ overexpressing cells were treated with thrombin. Collectively, these results reveal that Ca2+‐signaling and activation of NM‐II is required for leukocyte‐induced dephosphorylation of VE‐cadherin‐Y731 and for diapedesis, which is in agreement with the concept that leukocyte docking exerts force on VE‐cadherin, which may render the catenin‐masked Y731 accessible for SHP2.
Figure EV2. Leukocytes do not trigger the dissociation of β‐catenin or plakoglobin from VE‐cadherin.
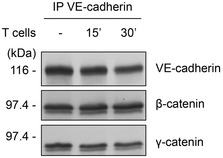
bEnd.5 cells were stimulated with TNF‐α for 18 h prior to incubation with antigen‐stimulated mouse T cells for 0–30 min. VE‐cadherin was immunoprecipitated from cell lysates, and precipitates were analyzed by immunoblots for indicated antigens.
Figure 6. Dephosphorylation of VE‐cadherin‐Y731 requires Ca2+ mobilization and actomyosin contraction during leukocyte diapedesis.
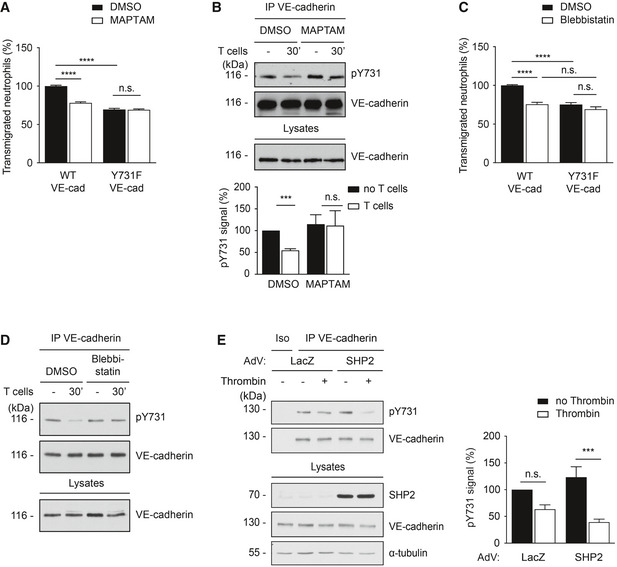
- Transmigration of mouse neutrophils toward the chemokine CXCL‐1 through TNF‐α‐stimulated WT or Y731F primary endothelial cells, either pre‐treated with vehicle control (DMSO) or Ca2+ chelator (MAPTAM) for 30 min at 37°C prior to addition of neutrophils. The transmigration rate is presented relative to DMSO‐treated endothelial cells expressing WT VE‐cadherin.
- TNF‐α‐activated bEnd.5 cells were treated with DMSO or MAPTAM, prior to incubation with antigen‐stimulated mouse T cells for 30 min. VE‐cadherin was immunoprecipitated from cell lysates and precipitates as well as total cell lysates were analyzed by immunoblots for indicated antigens. Quantification of pY731 blot signals of five independent experiments is shown below, with the signal intensity for the control sample (no T cells, vehicle control) set as 100%.
- Transmigration assays as in (A), except for replacing MAPTAM treatment with Blebbistatin.
- Similar as for (B), except for replacing MAPTAM treatment with Blebbistatin.
- HUVEC transduced with lacZ or with SHP2 were either untreated (−) or treated (+) with thrombin for 30 min followed by immunoblotting either VE‐cadherin immunoprecipitates or cell lysates for the indicated antigens on the right. A quantification of four independent experiments is shown on the right. Molecular weight markers are indicated in kDa.
Data information: The immunoblots are representative of five (B) or four (D, E) independent experiments, and the quantifications of transmigration experiments represent data from three (A, C) independent experiments in triplicate for each group per assay. Bars and error bars indicate mean ± SEM. Statistical significance was tested with one‐way ANOVA (B, E) or 2‐way ANOVA (A, C), ***P < 0.001, ****P < 0.0001, n.s., not significant.
Source data are available online for this figure.
Leukocyte migration through endothelium generates tension on VE‐cadherin required for dephosphorylation of VE‐cadherin‐Y731
To test whether leukocyte diapedesis enhances tension across VE‐cadherin, we generated a novel VE‐cadherin tension sensor by inserting a ferredoxin‐like (FL) linker‐based Förster resonance energy transfer (FRET) module (Austen et al, 2015; Cost et al, 2015; Ringer et al, 2017) between the p120 and the β‐catenin‐binding site of VE‐cadherin. With this sensor, tension above the threshold of 4 pN extends the elastic FL linker resulting in separation of two fluorophores YPet and mCherry and thereby reducing FRET. A VE‐cadherin tailless sensor (FL‐TS) lacking the catenin‐binding region of the cytoplasmic tail of VE‐cadherin behind the fused FRET module was used as negative control (Fig 7A). The constructs were expressed by adenoviral transduction in HUVEC where endogenous VE‐cadherin had been silenced by siRNA, targeting the untranslated region of VE‐cadherin mRNA. Comparable expression levels of each construct to endogenous VE‐cadherin levels in ctrl siRNA‐treated cells were confirmed by immunoblotting (Fig 7B). Immunofluorescence staining of VE‐cadherin confirmed the downregulation of endogenous VE‐cadherin in HUVEC as well as the correct localization of both VE‐cadherin tension sensors (FL and FL‐TS) at cell junctions (Fig 7C). The interaction of the VE‐cadherin tension sensor with catenins and thereby its ability to be connected to actin was demonstrated by co‐immunoprecipitating α‐catenin (Fig 7D). As expected, no such interaction was found for the negative control construct VE‐cad FL‐TS.
Figure 7. VE‐cadherin tension sensor is suitable to measure induction of force across VE‐cadherin during thrombin stimulation.
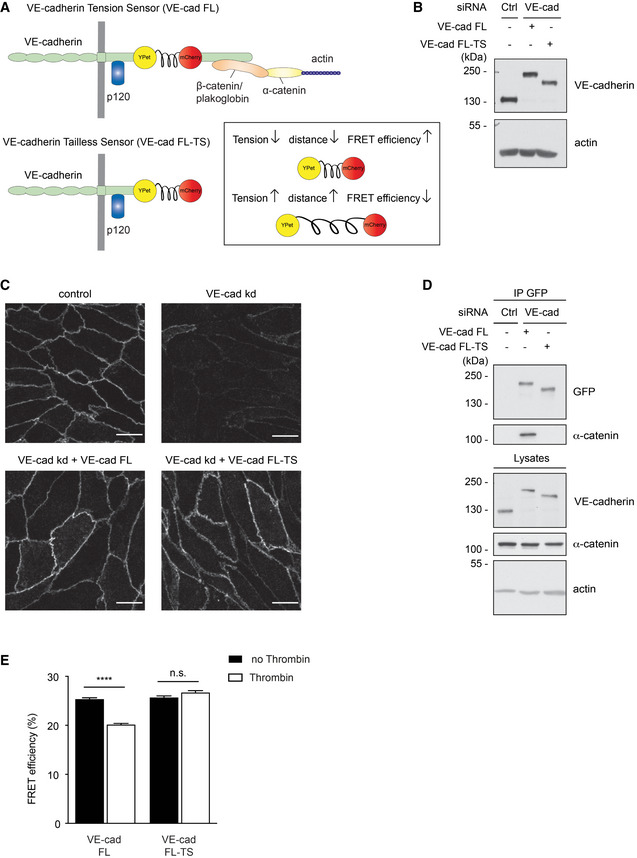
- Schematic representation of VE‐cadherin tension sensor (VE‐cad FL) and VE‐cadherin tailless sensor (VE‐cad FL‐TS). VE‐cadherin tension sensor has a ferredoxin‐like (FL) linker‐based FRET module inserted within VE‐cadherin between the p120 and the β‐catenin‐binding site, whereas VE‐cadherin tailless sensor lacks the catenin‐binding region of VE‐cadherin. The FRET module (depicted in box) consists of two fluorophores, YPet and mCherry separated by an elastic domain. When the module is under tension, FRET from YPet to mCherry decreases.
- HUVEC were transfected with control or VE‐cadherin‐specific siRNA and transduced with VE‐cad FL or VE‐cad FL‐TS. Cell lysates were immunoblotted for VE‐cadherin and actin.
- Immunofluorescence staining for VE‐cadherin of HUVEC transfected with control or VE‐cadherin‐specific siRNA (VE‐cad kd) and transduced with VE‐cad FL or VE‐cad FL‐TS.
- HUVEC were either transfected with control or VE‐cadherin targeting siRNA and transduced with VE‐cad FL or VE‐cad FL‐TS. The tension sensor was immunoprecipitated from cell lysates using an anti‐GFP antibody. Precipitates as well as total cell lysates were analyzed by immunoblotting for indicated antigens. Molecular weight markers are indicated in kDa.
- Quantification of FRET efficiency (percentage) in junctions of HUVEC expressing VE‐cad FL or VE‐cad FL‐TS, after thrombin stimulation (1 U/ml) or under control conditions.
Data information: (C, D) Representative pictures from three independent experiments. (E) Graph represents measurements (n = 60, 61, 52, 53) pooled from three independent experiments. Statistical significance was tested with unpaired t‐test, ****P < 0.0001, n.s., not significant. Scale bars 20 µm.
Source data are available online for this figure.
To characterize functionality of our VE‐cadherin tension sensor, we first analyzed the effect of thrombin. By determining donor fluorescence lifetime using fluorescent lifetime imaging microscopy (FLIM) of living, thrombin‐stimulated HUVEC, we showed clear reduction of YPet lifetime resulting in a drop of FRET efficiency by 20% for VE‐cad FL, whereas the FRET efficiency of VE‐cad FL‐TS did not change upon thrombin treatment (Fig 7E). Thus, our FRET sensor is suitable to detect changes in tension across VE‐cadherin.
Next, we used FLIM‐FRET measurements under live conditions to analyze whether transmigrating PMNs would affect tension across VE‐cadherin. Comparing the junctions of the same endothelial cells before starting flow and under flow conditions (1 dyn/cm2) resulted in no significant difference in FRET efficiency (Fig 8A). In contrast, in experiments where PMNs were added under flow conditions, a significant reduction in FRET efficiency was detected in endothelial cells expressing the VE‐cadherin FL tension sensor (Fig 8B), whereas no such effect was seen in cells expressing the VE‐cadherin FL‐TS control construct, which is unable to bind to catenins (Fig 8C). Since we imaged live cells, analyzing the vicinity of single, rapidly transmigrating leukocytes did not yield sufficient photon counts for reliable measurements. Therefore, areas of endothelial cells with multiple PMNs were analyzed, causing the problem that the signals of large junctional areas were also included, which were devoid of transmigrating neutrophils. This “diluted” our measured effects of PMNs on FRET efficiency.
Figure 8. Leukocyte migration through endothelium generates tension on VE‐cadherin required for dephosphorylation of VE‐cadherin‐Y731.
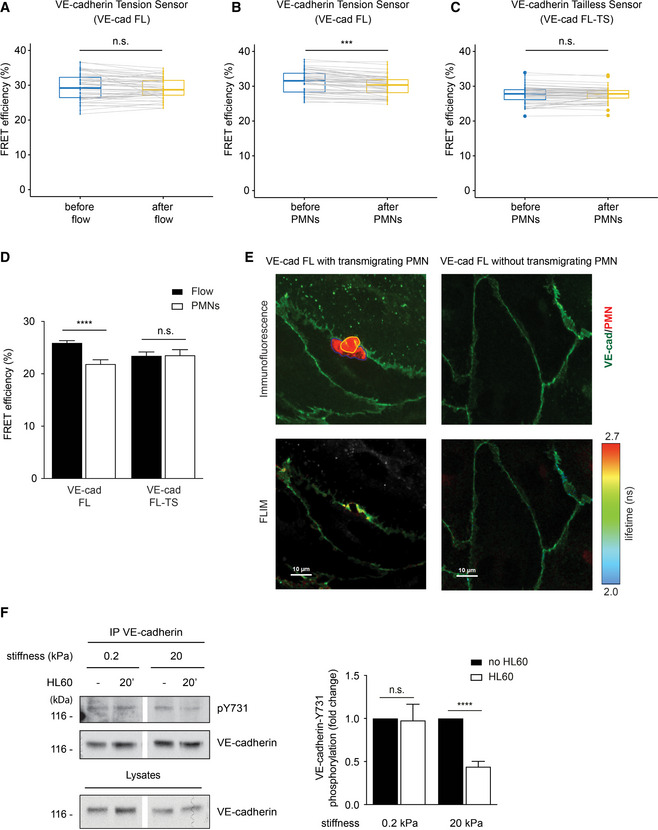
-
A–CQuantification of FRET efficiency (percentage) in junctions of HUVEC expressing VE‐cad FL (A, B) or VE‐cad FL‐TS (C). HUVEC were stimulated 4 h with TNF‐α prior to adding IL‐8 for 4 min, followed by flow with buffer alone (A) or together with PMNs (B, C). The connected data points represent FRET efficiency of same junctions compared before and after addition of flow/PMNs.
-
DQuantification of FRET efficiency (percentage) in junctions of HUVEC expressing VE‐cad FL or VE‐cad FL‐TS. Cells were either exposed to flow and PMNs or to flow alone, fixed using 4% PFA, and washed with PBS. FLIM measurements were performed at sites of transmigration (PMNs) or at junctions without PMNs (Flow).
-
ERepresentative images of HUVEC expressing VE‐cad FL at a site of PMN transmigration (left micrographs) or without PMN (right micrographs). Maximum intensity projection of a Z‐stack of YPet fluorescence (VE‐cadherin in green) and CellTracker DeepRed (PMN in red) is shown for the upper micrographs. The part of the PMN above the HUVEC monolayer is encircled in yellow and the part underneath in blue. The amplitude averaged lifetime of YPet per pixel (FLIM measurement) of the same cells is shown in the bottom micrographs.
-
FHUVEC were grown on collagen‐coated polyacrylamide gels of varying physiologic stiffness of 0.2 and 20 kPa and stimulated with TNF‐α for 17 h prior to adding HL60‐derived neutrophils for 20 min. VE‐cadherin was immunoprecipitated and precipitates as well as total cell lysates were analyzed by Western blotting for indicated antigens. Relative quantifications of pY731 blot signals adjusted to the amount of precipitated VE‐cadherin are shown on the right (from seven independent experiments).
Data information: Graphs represent data from n = 50 (A, B) and 51 (C) measurements pooled from 10 independent experiments. Graph in (D) represents measurements (n = 32, 30, 14, 14) of three independent experiments. Graph in (F) represents data of seven independent experiments. Bars and error bars indicate mean ± SEM. Statistical significance was tested with paired t‐test (A, B, C), and unpaired t‐test (D, F), ****P < 0.0001, ***P < 0.001, n.s., not significant.
Source data are available online for this figure.
To circumvent this problem, we analyzed HUVEC monolayers fixed with paraformaldehyde during the process of PMN transmigration. Direct comparison of junctions at sites of PMN transmigration with junctions of HUVEC without PMNs revealed a 15.4% drop in FRET efficiency, whereas no such effect was seen when HUVEC expressed the VE‐cadherin FL‐TS control construct (Fig 8D). Fluorescence imaging based on pixel‐wise FLIM analysis showed a local increase in tension around the site of PMN diapedesis (Fig 8E). Thus, neutrophils stimulate mechanisms that cause pulling forces on VE‐cadherin, which require its catenin‐mediated link to actin.
Similar FLIM‐FRET experiments with HUVEC silenced for SHP2 and expressing the tension sensor showed no impairment of the reduction in FRET efficiency at sites of PMN diapedesis, demonstrating that SHP2 acts downstream of leukocyte‐induced tension exerted on VE‐cadherin (Fig EV3).
Figure EV3. Leukocyte‐induced tension across VE‐cadherin is independent of SHP2, and tension is not required for SHP2 dissociation from PECAM‐1.
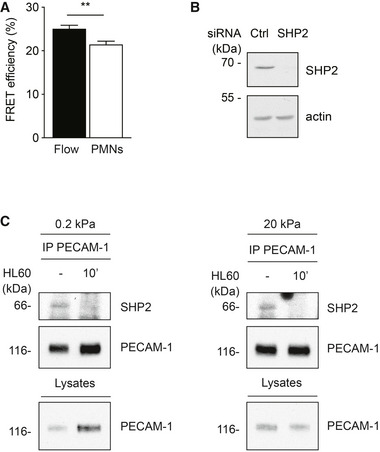
- Quantification of FRET efficiency (percentage) at junctions of HUVEC treated with siRNAs for SHP2 and VE‐cadherin prior to transducing VE‐cadherin‐FL. Cells were exposed either to flow alone or to flow in the presence of PMNs and then fixed with 4% PFA followed by FLIM measurements at sites of PMN transmigration (PMN) or at junctions without PMNs (Flow).
- Representative immunoblot for SHP2 and actin expression of HUVEC used for the experiments in (A).
- HUVEC were grown on collagen‐coated polyacrylamide gels of varying physiologic stiffness of 0.2 kPa (left) and 20 kPa (right) and stimulated with TNF‐α for 17 h prior to adding HL60‐derived neutrophils for 20 min. PECAM‐1 immunoprecipitates and cell lysates were immunoblotted for the indicated antigens. Note that leukocytes triggered SHP2 dissociation from PECAM‐1 independent of substrate stiffness.
Data information: Graph in (A) represents n = 21 and 20 measurements in a total of four independent experiments. Bars and error bars indicate mean ± SEM. Statistical significance was tested with unpaired t‐test, **P < 0.01. Blots are representative for 3 experiments (C).
To test whether mechanical tension would indeed support leukocyte‐induced dephosphorylation of VE‐cadherin‐Y731, we cultured HUVEC on substrates of 0.2 and 20 kPa stiffness. When HL60‐derived neutrophils adhered to these cells, we found that dephosphorylation of VE‐cadherin‐Y731 was only observed at high substrate stiffness (Fig 8F). Thus, tension across endothelial cells is needed to stimulate Y731 dephosphorylation, which supports the concept that pulling forces on the VE‐cadherin–catenin complex are needed. Importantly, tension was not needed for leukocyte‐induced dissociation of SHP2 from PECAM‐1, as was tested with HL60‐derived neutrophils and HUVEC grown on 0.2 and 20 kPa substrates (Fig EV3).
Discussion
Here we show that PECAM‐1 mediates leukocyte diapedesis in vivo and in vitro by interfering with VE‐cadherin. This is based on the following findings: Blocking PECAM‐1 expression or function abolished the ability of leukocytes to induce VE‐cadherin‐Y731 dephosphorylation and VE‐cadherin internalization. Likewise, mutating the binding site of PECAM‐1 for SHP2 had similar effects. Furthermore, leukocytes triggered the dissociation of SHP2 from PECAM‐1 and its association with VE‐cadherin, where Y731 is a direct substrate of SHP2. Finally, blocking of PECAM‐1 inhibited neutrophil diapedesis in vitro and in vivo only if the endothelium expressed WT VE‐cadherin, but not if Y731 was mutated. We conclude that PECAM‐1 supports leukocyte diapedesis by triggering Y731 dephosphorylation and internalization of VE‐cadherin (Fig 9).
Figure 9. Proposed model for the molecular mechanisms promoting opening of junctions during leukocyte diapedesis.
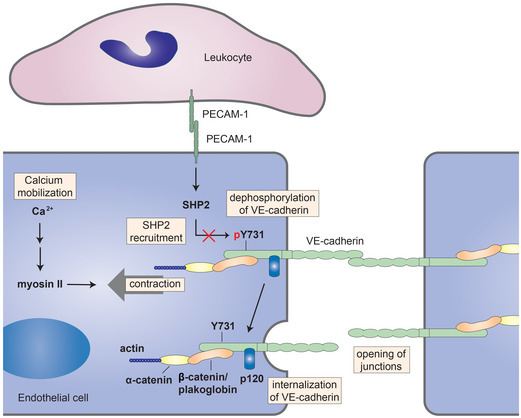
Leukocytes destabilize endothelial junctions by stimulating PECAM‐1. Leukocyte‐induced stimulation of PECAM‐1 triggers the release of SHP2 that directly interacts with and dephosphorylates VE‐cadherin‐Y731, which is required for the internalization of VE‐cadherin. In addition to mobilizing SHP2, leukocytes stimulate Ca2+‐signals in endothelial cells, and activate actomyosin‐based tension across the VE‐cadherin–catenin complex which is needed for Y731 dephosphorylation to occur. Since β‐catenin/plakoglobin mask the accessibility of Y731 for SHP2, we propose that leukocytes destabilize junctions by PECAM‐1‐SHP2‐triggered dephosphorylation of VE‐cadherin‐Y731 which becomes accessible by actomyosin‐mediated mechanical force exerted on the VE‐cadherin–catenin complex.
A second central requirement for the dephosphorylation of Y731, in addition to SHP2, is a mechanism which renders Y731 accessible. We found that the catenins mask Y731 and that its phosphorylation occurs in cells already before the prodomain of VE‐cadherin is cleaved off, suggesting that phosphorylation of Y731 may take place before catenins are associated. Searching for a potential demasking mechanism we found: First, inhibition of Ca2+‐signaling and non‐muscle myosin activity in endothelial cells blocked leukocyte‐induced Y731 dephosphorylation and inhibited leukocyte diapedesis in a Y731‐dependent manner. Second, induction of Y731 dephosphorylation needed cellular tension. Third, leukocyte stimulation of endothelial cells exerted force on the VE‐cadherin–catenin complex, as demonstrated by FLIM. Collectively, these results suggest that leukocyte–endothelial interactions exert force on the VE‐cadherin–catenin complex, which could assist in making Y731 accessible (Fig 9).
According to our results, PECAM‐1 contributes to the diapedesis process by destabilizing junctions via triggering the endocytosis of VE‐cadherin. Although PECAM‐1 is known since decades to support the diapedesis process (Muller et al, 1993; Bogen et al, 1994), until today it has not yet been shown whether or how it triggers destabilization of endothelial junctions. PECAM‐1 was found to stimulate in vitro leukocyte‐induced recycling of a tubulovesicular structure, called lateral border recycling compartment (LBRC), to the plasma membrane at cell contacts (Mamdouh et al, 2003; Mamdouh et al, 2008). Analysis of the protein composition of these vesicles did not allow to identify unique markers, but showed the lack of VE‐cadherin (Sullivan et al, 2014). Recycling of these vesicles to cell contacts was suggested to enlarge the cell surface area at junctions which would help to accommodate the body of the transmigrating leukocyte (Mamdouh et al, 2003). A role of the LBRC in leukocyte diapedesis is based on the correlation that several proteins which are involved in LBRC recycling, such as PECAM‐1, CD99, and the adaptor IQGAP1 are also involved in the diapedesis process (Muller, 2011; Sullivan et al, 2019). It is presently difficult to think of a selective and specific way of inhibiting LBRC recycling without affecting other cellular functions. Once specific means of interference with the LBRC are known, it will be possible to test the relation of LBRC recycling to the function of PECAM‐1‐triggered opening of junctions, which we have demonstrated here. We assume that the mobilization of LBRC vesicles to junctions occurs after PECAM‐1 triggers internalization of VE‐cadherin and thereby destabilization of junctions.
In addition to transmigration through the endothelial barrier, PECAM‐1 acts at a later step in extravasation. Blocking PECAM‐1 with antibodies against more membrane proximal Ig domains as well as gene inactivation causes the accumulation of leukocytes between endothelial cells and the basement membrane in mice and rats (Liao et al, 1995; Wakelin et al, 1996; Thompson et al, 2001; Woodfin et al, 2007). The molecular mechanism by which PECAM‐1 supports the exit of leukocytes from the endothelial cell basement membrane interphase is elusive.
It is well documented that neutrophil and lymphocyte diapedesis depend on Ca2+‐signaling in endothelial cells (Huang et al, 1993; Pfau et al, 1995; Etienne‐Manneville et al, 2000; Su et al, 2000; Kielbassa‐Schnepp et al, 2001), and different Ca2+‐channels such as TRPC6 (Weber et al, 2015) and TRPC7 (Mittal et al, 2017) have been suggested to be involved. In addition, neutrophil diapedesis requires the activation of endothelial myosin light chain kinase and the phosphorylation of non‐muscle myosin regulatory light chain (Hixenbaugh et al, 1997; Garcia et al, 1998), which is triggered by PMN‐stimulated Ca2+‐signaling and calmodulin activation (Saito et al, 1998). These mechanisms were accompanied by enhanced formation of radial stress fibers and it was therefore suggested that leukocytes induce signaling mechanisms within endothelial cells which trigger pulling forces on endothelial junctions that support gap formation. In line with this, a stiff cellular substrate, without which tension cannot form (Urbano et al, 2017), is needed for leukocyte diapedesis (Stroka & Aranda‐Espinoza, 2011) and mechanical forces in endothelial cells are induced by transmigrating leukocytes (Rabodzey et al, 2008; Liu et al, 2010). According to a recently proposed alternative mechanism, leukocytes rather push their way through endothelial junctions, whereas endothelial myosin activity was considered obsolete (Barzilai et al, 2017). In light of this alternative concept, it is important that our study shows for the first time, that neutrophils indeed trigger signals within endothelial cells which enhance the tension on the VE‐cadherin–catenin complex. We propose that this force renders VE‐cadherin‐Y731 accessible for SHP2, which leads to VE‐cadherin internalization and junction destabilization as outlined above. The comparably small increase which we observed in average VE‐cadherin tension in live cells indicates that it is a subset of VE‐cadherin molecules that are involved in this process. This makes sense, since the effects are expected to be local at sites of transmigration whereas junctions at other sites should be unaffected. This is strongly supported by our results with fixed endothelial cells where we could show that tension across VE‐cadherin was locally enhanced within the vicinity of transmigrating PMNs. In addition to effects in Y731 accessibility, leukocyte‐induced tension on VE‐cadherin is of course also likely to provide direct mechanical support for the formation of gaps at endothelial junctions. Intriguingly, the here described modulation of VE‐cadherin mechanics appears to be distinct from a previously described process, in which application of shear flow leads to a decrease in VE‐cadherin tension (Conway et al, 2013; Conway et al, 2017). Potential reasons why we did not detect flow induced effects on VE‐cadherin tension could be based on the fact that we performed our experiments at much lower shear force (1 instead of 15 dyne/cm2). In addition, the previously reported sensor reacts gradually to forces from below 1–6 pN, whereas the sensor we used here responds to forces above 4 pN (Ringer et al, 2017).
SHP2 is an important regulator of numerous signaling processes in many cell types in physiology and pathophysiology (Mohi & Neel, 2007; Tajan et al, 2015). Its binding to the phosphorylated Y663 and 686 residues of PECAM‐1 is well documented (Pumphrey et al, 1999). In resting endothelial cells, SHP2 was reported to preserve junction integrity by inactivating the tyrosine kinase Met and the GTPase Arf1 (Zhang et al, 2019). Thrombin was reported to trigger the dissociation of SHP2 from VE‐cadherin and thereby enhance VE‐cadherin phosphorylation and destabilize junctions (Ukropec et al, 2000). On the other hand, thrombin was found to enhance the interaction of SHP2 with β‐catenin which supported the recovery of junctions during reassembly (Timmerman et al, 2012). Inhibition of SHP2 was suggested to enhance leukocyte–endothelial adhesion, but impair the diapedesis process (Yan et al, 2017). In line with the former result, SHP2 was found to become inactivated in sepsis which supported IL‐1β induced NFkB‐driven pro‐inflammatory transcriptional activity and enhanced expression of ICAM‐1 and VCAM‐1 (Heun et al, 2019). Thus, SHP2 regulates multiple signaling mechanisms in endothelial cells which affect inflammatory processes in different ways. With respect to its role in leukocyte diapedesis, we show here that it mediates the PECAM‐1‐triggered dephosphorylation of VE‐cadherin‐Y731, which leads to the internalization of VE‐cadherin.
Taken together, we have shown that the contribution of PECAM‐1 to leukocyte transmigration through endothelium is in vivo based on targeting VE‐cadherin and destabilizing the endothelial barrier. Our results support a model in which mechanical signals cooperate with signaling‐induced enzymatic activities in a synergistic fashion which modulates post‐translational modifications of VE‐cadherin and thereby endothelial junction permeance for leukocytes.
Materials and Methods
Cell cultures
Primary endothelial cells from wild‐type VE‐cadherin or VE‐cadherin‐Y731F knock‐in mice were isolated and cultured as described (Frye et al, 2015). Human umbilical vein endothelial cells (HUVEC) were maintained in EBM‐2 medium supplemented with SingleQuots (Lonza) and cultured on Corning® CellBIND® dishes (3296) at 37°C and 5% CO2. bEnd.3, bEnd.5 (ECACC 96091929 & 96091930), and PECAM‐1−/− lung endothelioma (luEnd) cells were cultured as described previously (Nottebaum et al, 2008). OT‐II T cells isolated from OT‐II mice (The Jackson Laboratory) were maintained in RPMI medium containing 20% FCS, 4 mM l‐glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 mM sodium‐pyruvate, 1% NEAA, 50 μM β‐mercaptoethanol, and 1.5% IL‐2. Mouse polymorphonuclear neutrophils (PMNs) were isolated from bone marrow of C57BL/6 mice (Janvier) mice as described (Bixel et al, 2007). Human PMNs were isolated from blood derived from healthy donors (with formal consent) via density gradient centrifugation (Histopaque®‐1077, Histopaque®‐1119). HL60 cells were cultured in Iscove's modified Dulbecco's medium (IMDM, Gibco) containing 20% FCS, 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C and 5% CO2. Neutrophil‐like cells were derived from HL60 cells through differentiation with 1.3% DMSO for 7 days.
Mice
VE‐cadherin‐WT and VE‐cadherin‐Y731F knock‐in mice (Wessel et al, 2014) on C57BL/6 background were raised in a barrier facility under special pathogen‐free conditions. All experiments were carried out under the German legislation for the protection of animals and were approved by the Landesamt fuer Natur, Umwelt, und Verbraucherschutz Nordrhein‐Westfalen.
Antibodies, chelators, and inhibitors
VD‐47 directed against the propeptide of VE‐cadherin (C‐GPNFPQIDTPNMLPAHH) was obtained by the immunization of rabbits using a described method (Ebnet et al, 2000). Rabbit polyclonal antibodies C5 and VE42 against mouse VE‐cadherin and monoclonal antibody against VE‐cadherin‐pY731 were described (Gotsch et al, 1997; Broermann et al, 2011; Wessel et al, 2014). All commercial antibodies are listed in Table EV1. To deplete cells from Ca2+, primary endothelial cells were treated with 1 µM MAPTAM (Sigma, 16609) and endothelioma cells with 100 µM MAPTAM for 30 min at 37°C. HUVEC cells were stimulated with 1 U/ml thrombin (Calbiochem, 605195) for the indicated time periods at 37°C and 5% CO2. To inhibit myosin II, endothelial cells were treated with 50 μM blebbistatin (B0560) for 30 min at 37°C. To block the N‐linked glycosylation of proteins, endothelioma cells were treated with 1 μg/ml tunicamycin (Sigma, T7765) for 48 h at 37°C. DMSO was used as vehicle control.
Plasmids, DNA constructs, and adenoviral expression
The plasmid pCMV_PECAM‐1‐GFPSpark was purchased from Sino Biological Inc., and point mutations at Y663 and Y686 were introduced into the PECAM‐1 cDNA with mutagenesis primers Y663F (5′‐TCAGACGTGCAGTTCACGGAAGTTCAA‐3′) and Y686F (5′‐ACAGAGACAGTGTTCAGTGAAGTCCGG‐3′) using QuikChange Lightning Site‐directed Mutagenesis Kit (Agilent Technologies). Sequences were cloned into the gateway vector pENTR2B (Invitrogen) and transferred via site–specific recombination into the destination vector pAd/CMV‐DEST (Invitrogen). Viral lysates were obtained by transfecting the producer cell line HEK293A with the adenoviral destination vector using lipofectamine 2000 (Invitrogen). To generate VE‐cadherin tension sensor constructs (VE‐cad FL, VE‐cad FL‐TS, and VE‐cad YPet), human VE‐cadherin cDNA was cloned into pENTR2B. Restriction sites for AgeI and MluI were added after the codon for glycine 669 by mutagenesis PCR using the mutagenesis primers: VEC‐FL‐F (5′‐CAACTCGGTGCGCCGCGGCACCGGTGTC ACGCGTGGGGCCAAGCCCCC GCGGC‐3′) and VEC‐FL‐R (5′‐GCCGCGGGGGC TTGGCCCCACGCGTGACACCGGTGCCGCGGCGCACCGAGTTG‐3′). VE‐cadherin tension sensor constructs (VE‐cad FL, VE‐cad FL‐TS, and VE‐cad YPet) were amplified using tension sensor cDNA (YPet‐FL‐mCherry) (Ringer et al, 2017) as a template. VE‐cad FL was amplified using primers: YPet‐F (5′‐GGACCGGTATGGTGAGCAAAGGCGAA GAGC‐3′) and mCherry‐R (5′‐TTACGCGTCTTGTACAGCTCGTCCATGCC‐3′). The primers used for VE‐cad FL‐TS amplification were YPet‐F and mCherry‐STOP‐R (5′‐TTACGCGTTTACTTGTACAGCT CGTCCATGCC‐3′). The primers used for VE‐cad YPet amplification were YPet‐F and YPet‐R (5′‐TTACGCGTGGCGGCGGTCAGGAACTCC‐3′). All three constructs were inserted into modified human VE‐cadherin‐pENTR2B using digestion with AgeI and MluI and subsequent ligation. All three constructs were transferred into the destination vector pAd/CMV‐DEST, and viral lysates were produced as described above. SHP2‐WT and SHP2‐DA cDNA (mutation at Asp425 to Ala425, substrate trapping mutant) were gifts from Jaap van Buul, cloned in the laboratory of Anton Bennett (Kolli et al, 2004). Adenoviruses expressing the SHP2 variants were prepared in the same way as described above.
RNA‐mediated interference
For interference with mouse SHP2 expression, the following siRNAs were used in combination: Mm_Ptpn11_7 (5′‐GCGUGUUAGGAACGUCAAATT‐3′; Qiagen) and Mm_Ptpn11_8 (5′‐GAGAUGUUAUCGAGCUCAATT‐3′; Qiagen). For interference with human SHP2 expression, Hs_PTPN11_11 (5′‐GGGAAACUUAGACUAUAGATT‐3′, Qiagen) was used. For RNA‐mediated interference with human PECAM‐1, Hs_PECAM1_7 (5′‐CAGCAAUUCCUCAGGCUAATT‐3′) by Qiagen was used. For interference with human VE‐cadherin expression, CDH5 siRNA by Invitrogen was used (5′‐GGGUUUUUGCAUAAU AAGCTT‐3′). AllStars negative control siRNA (Qiagen, sequence not provided) that does not target any known mammalian gene was used as control. Routinely, 1 × 106 mouse primary endothelial cells or HUVEC were transfected respectively with 20 nM (mouse SHP2) or 60–80 nM (hSHP2, hVE‐cadherin, or hPECAM‐1) siRNAs for 48–72 h using INTERFERin® (Polyplus) according to manufacturer’s instructions. The transfection of bEnd.5 cells with siRNA was achieved by electroporation using Amaxa™ Nucleofector™ kit (Lonza) as per manufacturer’s guidelines.
Immunoprecipitation and immunoblotting
Cells were lysed in lysis buffer containing 10 mM Na3PO4, 150 mM NaCl, 1% Nonidet P‐40 substitute, 2 mM EDTA, 1 mM Na3VO4, and 12.5× cOmplete™ proteinase inhibitor (Roche). Lysates were clarified by centrifugation at 20,817 g and 4°C for 20 min. If necessary, cleared cell lysates were first incubated with Protein A/G Sepharose coupled to isotype control antibodies to reduce unspecific background binding. Aliquots of cell lysates were set aside for direct blot analysis, and aliquots for immunoprecipitation were incubated for 2 h at 4°C with Protein A/G Sepharose loaded with the respective antibodies. GST‐tagged proteins were pulled down using Glutathione Sepharose 4B beads (GSH beads, GE Healthcare). Immunocomplexes were washed five times with lysis buffer. For denaturing conditions, precipitated proteins and total cell lysates were mixed with SDS sample buffer (200 mM Tris–HCl pH 6.8, 6% SDS, 30% glycerol, 0.02% bromophenol blue, and 150 mM dithiothreitol) and boiled for 5 min at 95°C. Proteins were separated by SDS–PAGE and transferred on nitrocellulose membranes (Schleicher & Schuell) by wet blotting. Membranes were rinsed with TBS‐Tween (0.1%), and unspecific binding was minimized by blocking with 2% BSA in TBS‐T (including 200 µM Na3VO4 for phosphotyrosine detection) for 1 h at room temperature. Blots were analyzed as described (Ebnet et al, 2000).
Stimulation of endothelial cells with leukocytes
The different endothelial cell types were seeded in a number that allowed confluency on the day of assay. Cells were activated with 5 nM TNF‐α (Peprotech, 300‐01A) 16–18 h prior to the assay. Cells were washed twice with culture medium to remove residual TNF‐α prior to leukocytes addition. Differentiated HL60 cells were added to HUVEC in a ratio of 4:1, and T cells were added to bEnd.5 monolayers in a ratio of 5:1 (T cells: bEnd.5 cells), for 20 and 30 min, respectively, at 37°C. Unbound leukocytes were removed from endothelium by washing the cells twice with PBS prior to lysis, and lysates were subjected to immunoprecipitation.
Leukocyte transmigration assay
Endothelial cells were plated on fibronectin (100 µg/ml) coated transwell filters (5 µm pore size, Corning) and grown for 48 h to confluent monolayers. Cells were stimulated with 5 nM TNF‐α 16–18 h prior to the assay. If required, cells were pre‐treated with MAPTAM, blebbistatin (as described above) or blocking antibodies prior to neutrophils addition. 1 × 105 mouse neutrophils were added to mouse primary endothelial cells or bEnd.5 cells, whereas 2 × 105 freshly isolated human neutrophils were added to HUVEC monolayers. For assays with mouse neutrophils, 40 ng/ml CXCL1 in transmigration medium [DMEM, 5% FCS, 2% L‐glutamine, and 25 mM HEPES (pH 7.3)] was used as chemoattractant, and for human neutrophils, 5 ng/ml IL‐8 (Peprotech, 200‐08M) in HUVEC culture medium was used as chemoattractant in lower chambers. Mouse or human neutrophils were allowed to transmigrate for 1 h or 40 min, respectively at 37°C. The number of transmigrated neutrophils was counted with a CASY cell counter.
For transmigration in combination with SHP2 depletion, endothelial cells were transfected with SHP2‐specific siRNA (as described above) prior to cell seeding on transwell filters. For blocking PECAM‐1, 6 µg Mec13.3 or 10 µg 2H8 antibodies was added per transwell filter and the same amount of endomucin or isotype control antibodies was used as control, respectively, for 30 min at 37°C.
SHP2 substrate trapping assay
Primary endothelial cells isolated from VE‐cadherin WT or Y731F knock‐in mice were lysed in lysis buffer containing 20 mM Tris–HCl pH 7.5, 100 mM NaCl, 2 mM CaCl2, 1% Triton X‐100, 10% glycerol, 5 mM iodoacetamide (IAA), and 1× Complete protease inhibitor EDTA‐free (Roche) for 30 min at 4°C. IAA was blocked with 10 mM dithiothreitol (DTT) for 15 min at 4°C. Lysates were pre‐cleared by incubation with 5 μg GST coupled glutathione sepharose beads for 1 h at 4°C and subsequently incubated with 10 μg GST, GST‐SHP2‐WT or GST‐SHP2‐DA coupled glutathione sepharose for 3 h at 4°C. Affinity complexes were washed three times with washing buffer (20 mM Tris–HCl pH 7.5, 100 mM NaCl, 2 mM CaCl2, 1% Triton X‐100, 10% glycerin, and 1 mM DTT) and were analysed by SDS–PAGE and immunoblotting.
Internalization assay
Endocytosis of VE‐cadherin was assayed essentially as described (Nanes et al, 2012). HUVEC depleted of endogenous PECAM‐1 via siRNA transfection were seeded on fibronectin (100 µg/ml) coated 8‐well chamber slides, if required followed by adenoviral expression of PECAM‐1‐WT‐EGFP or PECAM‐1‐Y663F, Y686F‐EGFP. Cells were stimulated with 5 nM TNF‐α for 18 h prior to labeling of VE‐cadherin with BV6 antibody for 1 h at 4°C. To allow internalization to occur, cells were incubated with differentiated HL60 cells (3 × 105 cells/well) for 20 or 30 min at 37°C. Afterward, cells were rinsed to remove unbound leukocytes and the remaining cell surface‐bound antibodies were removed by acid wash (100 mM glycine‐HCl, pH 2.5) for 15 min at 4°C. Cells were then washed with PBS, fixed with 4% paraformaldehyde at room temperature (RT) for 5 min, rinsed with PBS containing 0.01% Tween (PBST), and permeabilized with 0.5% Triton X‐100 for 5 min at RT. To minimize unspecific binding of the antibodies, cells were incubated in PBST containing 3% BSA for 1 h. Total VE‐cadherin was detected using VE‐cadherin D87F2 antibody (1:400) for 1 h at RT. Primary antibodies were labeled with 0.6 µg per well Alexa Fluor 488–donkey anti‐mouse (internalized VE‐cadherin) and Alexa Fluor 568–donkey anti‐rabbit (membrane‐associated VE‐cadherin) for 90 min at RT. Cells were washed with PBST and DNA was stained with Hoechst 33342 (0.2 µg/ml, Invitrogen). Images were obtained using a Zeiss LSM780 confocal microscope. Endocytosed VE‐cadherin was quantified as the number of internalized VE‐cadherin vesicles per area using Fiji‐ImageJ software.
Intravital microscopy
VE‐cadherin WT or VE‐cadherin Y731F knock‐in mice (14–16 weeks old) were injected intrascrotally with 50 ng IL‐1β in 0.3 ml saline. In parallel, 100 µg 2H8 or isotype control antibodies were injected intravenously. Four hours later, mice were anaesthetized with an intraperitoneal injection of ketamine hydrochloride (125 mg/kg), xylazine (12.5 mg/kg), and atropine sulfate (0.025 mg/kg). Surgical preparation of cremaster muscles and intravital microscopy were carried out as described (Zarbock et al, 2007; Wessel et al, 2014). Adhesion, transmigration, and rolling flux fraction of leukocytes were analyzed in 4 mice per group. Vessels with diameter of 20–30 µm were investigated. Analysis was performed in a “blinding” to group allocation approach.
In vitro phosphatase treatment of VE‐cadherin–catenin complex
HUVEC were lysed and VE‐cadherin was immunoprecipitated from lysates for 2 h at 4°C, followed by washing five times with lysis buffer. For dissociation of catenins from VE‐cadherin in vitro, immunoprecipitated VE‐cadherin complex was incubated in 100 µl lysis buffer containing 0.2% SDS (or no SDS as control) for 30 min at RT. The samples were diluted 1:10 with lysis buffer and then incubated for 2 h at 4°C to allow rebinding of VE‐cadherin to the antibodies. Precipitated VE‐cadherin was washed three times with lysis buffer to remove dissociated catenins from the sample, followed by a final washing step with phosphatase buffer (25 mM HEPES pH 7.2, 50 mM NaCl, 5 mM EDTA, 10 mM DTT). VE‐cadherin was either incubated with phosphatase buffer only or with phosphatase buffer containing 2 µg recombinant human SHP2 (Sigma, SRP0217) for 30 min at 300 rpm and 30°C in a thermo‐shaker. Reactions were terminated by addition of 1.5× SDS sample buffer and analyzed by SDS–PAGE followed by immunoblotting.
Testing substrate stiffness effects
Polyacrylamide hydrogels attached to 35 mm glass coverslips were prepared according to a modified literature protocol (Tse & Engler, 2010). In brief, solutions with varying concentrations of acrylamide/bis‐acrylamide (3%/0.03% and 8%/0.264%) were mixed to yield hydrogels of different stiffness (Young’s moduli of 0.2 and 20 kPa, respectively). To allow cell attachment, collagen I was conjugated to the hydrogel surface using the heterobifunctional linker sulfo‐SANPAH. A 1 mg/ml solution of sulfo‐SANPAH (Sigma) in milli‐Q water was added to the hydrogel surface and gels were irradiated with 365 nm UV light (intensity of 20 mW/cm2) for 2 min. Substrates were washed with PBS and incubated with 50 μg/ml rat‐tail collagen I (BD Biosciences) solution in PBS for 2 h at 37°C. Substrates were rinsed with PBS before seeding HUVEC. Cells were activated with 5 nM TNF‐α 16–18 h prior to the assay. HL60‐derived neutrophils (6 x 106 cells/coverslip) were added to the cells for 20 min at 37°C. Coverslips were placed on ice, and cells were washed twice with cold PBS to remove unbound leukocytes. Cells were lysed in place with 1 ml lysis buffer/coverslip for 10 min, collected, and incubated additional 10 min in the overhead shaker at 4°C. VE‐cadherin was immunoprecipitated using 6 µg F‐8 antibody for 2 h at 4°C. Immunocomplexes were washed three times with lysis buffer and analyzed by SDS–PAGE and immunoblotting.
Imaging of the expression of VE‐cadherin tension sensor by immunofluorescence
HUVEC transfected with either control siRNA or VE‐cadherin targeting siRNA were seeded on fibronectin (100 µg/ml) coated ibidi 8‐well µ‐chamber slides with 3 × 104 cells per chamber, if required followed by re‐expression of VE‐cad FL or VE‐cad FL‐TS for 24 h. Cells were washed with PBS, fixed with 4% paraformaldehyde at room temperature for 15 min, rinsed with PBS, and permeabilized with 0.5% Triton X‐100 for 5 min at RT. VE‐cadherin was detected using anti‐VE‐cadherin antibody (75/cadherin‐5, 1.25 µg/ml) for 1 h at RT. Primary antibody was labeled with Alexa Fluor 647‐donkey anti‐mouse (5 µg/ml) for 45 min at RT. Cells were washed with PBS, and images were obtained using a ZEISS LSM880 confocal microscope. Maximum intensity projection was performed using Fiji‐ImageJ software. For better visualization, brightness and contrast were adjusted identically for all measurements.
Time‐correlated single‐photon‐counting fluorescence lifetime microscopy (TCSPC‐FLIM)
All TCSPC‐FLIM experiments were performed on a confocal laser scanning microscope (Zeiss LSM880) equipped with a 40× water immersion objective (C‐Apochromat 40×/1.20 W Korr M27) with 485 nm pulse excitation (LDH‐D‐C‐485) at 20 MHz for FLIM measurements under live conditions and 40 MHz for FLIM measurements under fixed conditions with 67% laser intensity. The emitted fluorescence was filtered with a 520/35 band pass filter. Photon arrival was detected with a TCSPC module with 80 ps time resolution (MultiHarp 150 4N). Live measurements were performed at constant 37°C and 5% CO2. Measurements of fixed samples were performed at constant 30°C.
FLIM‐FRET analysis via VE‐cadherin tension sensor during thrombin stimulation
HUVEC depleted of endogenous VE‐cadherin were seeded on fibronectin coated ibidi 8‐well µ‐chamber slides with 3 × 104 cells per chamber, followed by re‐expression of VE‐cad FL, VE‐cad FL‐TS or VE‐cad YPet for 24 h. FLIM measurements were performed before and 5–15 min after stimulation with thrombin. Images were acquired using ZEN 2.3 (Zeiss) with SymPhoTime 64 2.4 (Picoquant) at 1× zoom covering 212.55 × 212.55 μm in 512 × 512 pixel using a scanning speed of 16.38 μs/pixel or 65.5 µs/pixel and a pinhole of 6.5 AU. For FLIM analysis, photons of cell–cell junctions were included manually for subsequent data fitting. One‐exponential tailfit (VE‐cad YPet) and two‐exponential tailfit (VE‐cad FL and VE‐cad FL‐TS) were performed using SymPhoTime (Picoquant).
FLIM‐FRET analysis via VE‐cadherin tension sensor during leukocyte transmigration under live conditions
HUVEC depleted of endogenous VE‐cadherin were seeded on fibronectin coated ibidi VI (0.4) flow slides with 3 × 104 cells per lane, followed by re‐expression of VE‐cad FL, VE‐cad FL‐TS or VE‐cad YPet for 24 h. Cells were stimulated with 5 nM TNF‐α 4 h prior to measurements. Freshly isolated human PMNs (0.25 × 106 cells/ml) were resuspended in flow buffer HBSS +/+ (5 mg/ml BSA, 25 mM HEPES). Measurements were started 5–11 frames prior to IL‐8 addition to assess the baseline FRET efficiency. Cells were first exposed to IL‐8 (5 ng/ml) containing flow buffer at 0.75 dyn/cm2 for 4 min, followed by freshly isolated human PMNs at 1 dyn/cm2 or only flow buffer at 1 dyn/cm2 for 12 min. FRET measurements were recorded after 4 min of flow with or without PMNs for 44 frames. Images were acquired using ZEN 2.3 (Zeiss) with SymPhoTime 64 2.4 (Picoquant) at 1× zoom covering 212.55 × 212.55 μm in 512 × 512 pixel using a scanning speed of 16.38 μs/pixel and a pinhole of 6.5 AU. One‐exponential reconvolution (VE‐cad YPet) and two‐exponential reconvolution (VE‐cad FL and VE‐cad FL‐TS) with calculated instrument response function (IRF) were performed using SymPhoTime (Picoquant). The same junctions were analyzed before and after application of PMNs under flow. To exclude altered fitting results upon differences in the number of photons included in the analysis before and after PMN addition, the 44 frames after PMN addition were splitted into 4 times 11 frames with FRET efficiency calculation for each of the four splitted datasets. Finally, the average lifetime was assessed for statistical analysis.
FLIM‐FRET analysis via VE‐cadherin tension sensor during leukocyte transmigration under fixed conditions
HUVEC depleted of endogenous VE‐cadherin and, if required, SHP2, were seeded on fibronectin coated ibidi VI (0.4) flow slides with 3 × 104 cells per lane, followed by re‐expression of VE‐cad FL, VE‐cad FL‐TS or VE‐cad YPet for 24 h. Cells were stimulated with 5 nM TNF‐α 4 h prior to measurements. Freshly isolated human PMNs were stained with CellTracker Deep Red Dye (Thermo, C34565) 2.5 µM in HBSS for 20 min at 37°C. After washing, freshly stained PMNs were resuspended in flow buffer HBSS +/+ (5 mg/ml BSA, 25 mM HEPES) to a final concentration of 0.25 × 106 cells/ml. HUVEC were flushed with flow buffer at 1 dyn/cm2 for 1 min followed by 4.5 min of flow buffer with or without stained PMNs at 1 dyn/cm2. After washing of the cells for 1.5 min at 1 dyn/cm2 with PBS, cells were fixed for 8 min using 4% paraformaldehyde in PBS at 37°C. Fixation was stopped by washing the cells with PBS at 1 dyn/cm2 for 3 min. All steps were performed at 37°C and 5% CO2. Subsequent measurement of fixed cells was performed at 30°C using ZEN 2.3 (Zeiss) with SymPhoTime 64 2.4 (Picoquant) at 3× zoom covering 70.71 × 70.71 μm in 512 × 512 pixel using a scanning speed of 131 μs/pixel and a pinhole of 6.5 AU. Prior each measurement, a z‐stack of the fluorescence signal of YPet and CellTracker Deep Red was performed using 488 and 641 nm constant laser with 4.1 µs/pixel and a pinhole of 2 AU within the same field of view as the FLIM acquisition. For the analysis of FLIM‐FRET data, photons of cell–cell junctions in close proximity to a transmigrating PMN were included manually for subsequent data fitting. One‐exponential tailfit (VE‐cad YPet) and two‐exponential tailfit (VE‐cad FL and VE‐cad FL‐TS) were performed using SymPhoTime (Picoquant). For pixel‐wise data fitting, a 2 × 2 pixel binning was performed with subsequent bi‐exponential tailfitting using preset values for the two lifetimes t 1 = 3.1 ns and t 2 = 1.4 ns.
FLIM‐FRET data analysis
The average lifetime of VE‐cad YPet was used for each experimental day to determine τD. FRET efficiency was calculated for each measurement, using the amplitude weighted lifetime of VE‐cad FL and VE‐cad FL‐TS to determine τDA, with the following calculation:
Statistical analysis and software
Statistical significance was analyzed using two‐tailed Student's t‐test, one‐way ANOVA, or two‐way ANOVA for independent samples. Tukey's multiple comparison test was applied to correct for multiple comparisons. For FLIM‐FRET data, outliers were identified using Robust regression and outlier removal in combination (ROUT) and significance was analyzed with a paired or unpaired t‐test including Bonferroni correction for multiple testing using R. GraphPad Prism7 or R (version 3.6.0) software was used for statistical analysis. P‐values less than 0.05 were considered significant. P‐values are indicated by asterisks: *P < 0.05; **P < 0.01; ***P < 0.001 and ****P < 0.0001. Results are shown as mean ± SEM.
For obtaining microscope data via confocal Zeiss microscopes, Zen 2.3 SPI Black 64‐bit software was used. Quantification of Western blot intensities and VE‐cadherin internalization was performed using Fiji‐ImageJ. Measurement and analysis of FLIM‐FRET were performed using SymPhoTime 64. For analysis and plotting of FLIM‐FRET data, R‐studio was used in combination with the packages ggplot2, ggpubr, and Cairo.
Author contributions
NA drafted the manuscript and carried out and analyzed experiments. MZ and AN carried out experiments and conceptualized large parts of the study ‡. DT and KS carried out experiments and analyzed results. RB designed the FRET constructs and designed, performed, and analyzed FRET experiments. Y‐TL advised on techniques. BT and CG analyzed results, revised the manuscript, and provided central reagents. DV supervised and conceptualized the study, analyzed results, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
We thank Anna‐Lena Schweizer for valuable discussions, Tanja Möller for excellent technical assistance, and Jaap van Buul (Amsterdam) for the SHP2 cDNA constructs. This work was supported by funds from the Max Planck Society (D.V.) by funds from the CIM‐IMPRS graduate school and grants from the Deutsche Forschungsgemeinschaft (SFB1348, B01; SFB1009, A01) to D.V. Open Access funding enabled and organized by ProjektDEAL.
The EMBO Journal (2021) 40: e106113.
Footnotes
Correction added on 3 May 2021, after first online publication: AN was moved from ‘carried out experiments and analyzed results’ to ‘carried out experiments and conceptualized large parts of the study’.
Data availability
This study includes no data deposited in external repositories.
References
- Allingham MJ, van Buul JD, Burridge K (2007) ICAM‐1‐mediated, Src‐ and Pyk2‐dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol 179: 4053–4064 [DOI] [PubMed] [Google Scholar]
- Aurrand‐Lions M, Johnson‐Leger C, Lamagna C, Ozaki H, Kita T, Imhof BA (2002) Junctional adhesion molecules and interendothelial junctions. Cells Tissues Organs 172: 152–160 [DOI] [PubMed] [Google Scholar]
- Austen K, Ringer P, Mehlich A, Chrostek‐Grashoff A, Kluger C, Klingner C, Sabass B, Zent R, Rief M, Grashoff C (2015) Extracellular rigidity sensing by talin isoform‐specific mechanical linkages. Nat Cell Biol 17: 1597–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai S, Yadav SK, Morrell S, Roncato F, Klein E, Stoler‐Barak L, Golani O, Feigelson SW, Zemel A, Nourshargh S et al (2017) Leukocytes breach endothelial barriers by insertion of nuclear lobes and disassembly of endothelial actin filaments. Cell Rep 18: 685–699 [DOI] [PubMed] [Google Scholar]
- Bixel G, Kloep S, Butz S, Petri B, Engelhardt B, Vestweber D (2004) Mouse CD99 participates in T cell recruitment into inflamed skin. Blood 104: 3205–3213 [DOI] [PubMed] [Google Scholar]
- Bixel MG, Petri B, Khandoga AG, Khandoga A, Wolburg‐Buchholz K, Wolburg H, Marz S, Krombach F, Vestweber D (2007) A CD99‐related antigen on endothelial cells mediates neutrophil but not lymphocyte extravasation in vivo . Blood 109: 5327–5336 [DOI] [PubMed] [Google Scholar]
- Bixel MG, Li H, Petri B, Khandoga AG, Khandoga A, Zarbock A, Wolburg‐Buchholz K, Wolburg H, Sorokin L, Zeuschner D et al (2010) CD99 and CD99L2 act at the same site as, but independently of, PECAM‐1 during leukocyte diapedesis. Blood 116: 1172–1184 [DOI] [PubMed] [Google Scholar]
- Bogen S, Pak J, Garifallou M, Deng X, Muller WA (1994) Monoclonal antibody to murine PECAM‐1 (CD31) blocks acute inflammation in vivo . J Exp Med 179: 1059–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breviario F, Caveda L, Corada M, Martin Padura I, Navarro P, Golay J, Introna M, Gulino D, Lampugnani MG, Dejana E (1995) Functional properties of human vascular endothelial cadherin (7B4/cadherin‐5), an endothelium‐specific cadherin. Arterioscler Thromb Vasc Biol 15: 1229–1239 [DOI] [PubMed] [Google Scholar]
- Broermann A, Winderlich M, Block H, Frye M, Rossaint J, Zarbock A, Cagna G, Linnepe R, Schulte D, Nottebaum AF et al (2011) Dissociation of VE‐PTP from VE‐cadherin is required for leukocyte extravasation and for VEGF‐induced vascular permeability in vivo . J Exp Med 208: 2393–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway DE, Breckenridge MT, Hinde E, Gratton E, Chen CS, Schwartz MA (2013) Fluid shear stress on endothelial cells modulates mechanical tension across VE‐cadherin and PECAM‐1. Curr Biol 23: 1024–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway DE, Coon BG, Budatha M, Arsenovic PT, Orsenigo F, Wessel F, Zhang J, Zhuang Z, Dejana E, Vestweber D et al (2017) VE‐cadherin phosphorylation regulates endothelial fluid shear stress responses through the polarity protein LGN. Curr Biol 27: 2219–2225 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corada M, Mariotti M, Thurston G, Smith K, Kunkel R, Brockhaus M, Lampugnani MG, Martin‐Padura I, Stoppacciaro A, Ruco L et al (1999) Vascular endothelial‐cadherin is an important determinant of microvascular integrity in vivo . Proc Natl Acad Sci USA 96: 9815–9820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cost AL, Ringer P, Chrostek‐Grashoff A, Grashoff C (2015) How to measure molecular forces in cells: a guide to evaluating genetically‐encoded FRET‐based tension sensors. Cell Mol Bioeng 8: 96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E, Vestweber D (2013) The role of VE‐cadherin in vascular morphogenesis and permeability control, In The molecular biology of cadherins , van Roy F (ed.), pp 119–144., Amsterdam: Elsevier; [DOI] [PubMed] [Google Scholar]
- Ebnet K, Schulz CU, Meyer‐zu‐Brickwedde M‐K, Pendl GG, Vestweber D (2000) Junctional Adhesion Molecule (JAM) interacts with the PDZ domain containing proteins AF‐6 and ZO‐1. J Biol Chem 275: 27979–27988 [DOI] [PubMed] [Google Scholar]
- Etienne‐Manneville S, Manneville JB, Adamson P, Wilbourn B, Greenwood J, Couraud PO (2000) ICAM‐1‐coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J Immunol 165: 3375–3383 [DOI] [PubMed] [Google Scholar]
- Flint AJ, Tiganis T, Barford D, Tonks NK (1997) Development of “substrate‐trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci USA 94: 1680–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye M, Dierkes M, Küppers V, Vockel M, Tomm J, Zeuschner D, Rossaint J, Zarbock A, Koh GY, Peters K et al (2015) Interfering with VE‐PTP stabilizes endothelial junctions in vivo via Tie‐2 in the absence of VE‐cadherin. J Exp Med 212: 2267–2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JG, Verin AD, Herenyiova M, English D (1998) Adherent neutrophils activate endothelial myosin light chain kinase: role in transendothelial migration. J Appl Physiol 84: 1817–1821 [DOI] [PubMed] [Google Scholar]
- Gotsch U, Borges E, Bosse R, Böggemeyer E, Simon M, Mossmann H, Vestweber D (1997) VE‐cadherin antibody accelerates neutrophil recruiment in vivo . J Cell Sci 110: 583–588 [DOI] [PubMed] [Google Scholar]
- Heun Y, Pircher J, Czermak T, Bluem P, Hupel G, Bohmer M, Kraemer BF, Pogoda K, Pfeifer A, Woernle M et al (2019) Inactivation of the tyrosine phosphatase SHP‐2 drives vascular dysfunction in Sepsis. EBioMedicine 42: 120–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hixenbaugh EA, Goeckeler ZM, Papaiya NN, Wysolmerski RB, Silverstein SC, Huang AJ (1997) Stimulated neutrophils induce myosin light chain phosphorylation and isometric tension in endothelial cells. Am J Physiol 273: H981–H988 [DOI] [PubMed] [Google Scholar]
- Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR, Silverstein SC (1993) Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells. J Cell Biol 120: 1371–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MT, Larbi KY, Scheiermann C, Woodfin A, Gerwin N, Haskard DO, Nourshargh S (2006) ICAM‐2 mediates neutrophil transmigration in vivo: evidence for stimulus‐specificity and a role in PECAM‐1‐independent transmigration. Blood 107: 4721–4727 [DOI] [PubMed] [Google Scholar]
- Jackson DE, Ward CM, Wang R, Newman PJ (1997) The protein‐tyrosine phosphatase SHP‐2 binds platelet/endothelial cell adhesion molecule‐1 (PECAM‐1) and forms a distinct signaling complex during platelet aggregation. Evidence for a mechanistic link between PECAM‐1‐ and integrin‐mediated cellular signaling. J Biol Chem 272: 6986–6993 [DOI] [PubMed] [Google Scholar]
- Kielbassa‐Schnepp K, Strey A, Janning A, Missiaen L, Nilius B, Gerke V (2001) Endothelial intracellular Ca2+ release following monocyte adhesion is required for the transendothelial migration of monocytes. Cell Calcium 30: 29–40 [DOI] [PubMed] [Google Scholar]
- Kolli S, Zito CI, Mossink MH, Wiemer EA, Bennett AM (2004) The major vault protein is a novel substrate for the tyrosine phosphatase SHP‐2 and scaffold protein in epidermal growth factor signaling. J Biol Chem 279: 29374–29385 [DOI] [PubMed] [Google Scholar]
- Liao F, Huynh HK, Eiroa A, Greene T, Polizzi E, Muller WA (1995) Migration of monocytes across endothelium and passage through extracellular matrix involve separate molecular domains of PECAM‐1. J Exp Med 182: 1337–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Sniadecki NJ, Chen CS (2010) Mechanical forces in endothelial cell during firm adhesion and early transmigartion of human monocytes. Cell Mol Bioeng 3: 50–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamdouh Z, Chen X, Pierini LM, Maxfield FR, Muller WA (2003) Targeted recycling of PECAM from endothelial surface‐connected compartments during diapedesis. Nature 421: 748–753 [DOI] [PubMed] [Google Scholar]
- Mamdouh Z, Kreitzer GE, Muller WA (2008) Leukocyte transmigration requires kinesin‐mediatet microtubule‐dependent membrane trafficking from the lateral border recycling compartment. J Exp Med 205: 951–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A et al (1998) Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol 142: 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda M, Osawa M, Shigematsu H, Harada N, Fujiwara K (1997) Platelet endothelial cell adhesion molecule‐1 is a major SH‐PTP2 binding protein in vascular endothelial cells. FEBS Lett 408: 331–336 [DOI] [PubMed] [Google Scholar]
- Matsuyoshi N, Toda K, Horiguchi Y, Tanaka T, Nakagawa S, Takeichi M, Imamura S (1997) In vivo evidence of the critical role of cadherin‐5 in murine vascular integrity. Proc Assoc Am Physicians 109: 362–371 [PubMed] [Google Scholar]
- Mittal M, Nepal S, Tsukasaki Y, Hecquet CM, Soni D, Rehman J, Tiruppathi C, Malik AB (2017) Neutrophil Activation of Endothelial Cell‐Expressed TRPM2 Mediates Transendothelial Neutrophil Migration and Vascular Injury. Circ Res 121: 1081–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohi MG, Neel BG (2007) The role of Shp2 (PTPN11) in cancer. Curr Opin Genet Dev 17: 23–30 [DOI] [PubMed] [Google Scholar]
- Muller WA, Weigl SA, Deng X, Phillips DM (1993) PECAM‐1 is required for transendothelial migration of leukocytes. J Exp Med 178: 449–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA (2011) Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol 6: 323–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanes BA, Chiasson‐MacKenzie C, Lowery AM, Ishiyama N, Faundez V, Ikura M, Vincent PA, Kowalczyk AP (2012) p120‐catenin binding masks an endocytic signal conserved in classical cadherins. J Cell Biol 199: 365–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottebaum AF, Cagna G, Winderlich M, Gamp AC, Linnepe R, Polaschegg C, Filippova K, Lyck R, Engelhardt B, Kamenyeva O et al (2008) VE‐PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE‐cadherin by leukocytes and by VEGF. J Exp Med 205: 2929–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourshargh S, Alon R (2014) Leukocyte migration into inflamed tissues. Immunity 41: 694–707 [DOI] [PubMed] [Google Scholar]
- Pfau S, Leitenberg D, Rinder H, Smith BR, Pardi R, Bender JR (1995) Lymphocyte adhesion‐dependent calcium signaling in human endothelial cells. J Cell Biol 128: 969–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluskota E, Chen Y, D'Souza SE (2000) Src homology domain 2‐containing tyrosine phosphatase 2 associates with intercellular adhesion molecule 1 to regulate cell survival. J Biol Chem 275: 30029–30036 [DOI] [PubMed] [Google Scholar]
- Posthaus H, Dubois CM, Laprise MH, Grondin F, Suter MM, Muller E (1998) Proprotein cleavage of E‐cadherin by furin in baculovirus over‐expres‐sion system: potential role of other convertases in mammalian cells. FEBS Lett 438: 306–310 [DOI] [PubMed] [Google Scholar]
- Pumphrey NJ, Taylor V, Freeman S, Douglas MR, Bradfield PF, Young SP, Lord JM, Wakelam MJ, Bird IN, Salmon M et al (1999) Differential association of cytoplasmic signalling molecules SHP‐1, SHP‐2, SHIP and phospholipase C‐gamma1 with PECAM‐1/CD31. FEBS Lett 450: 77–83 [DOI] [PubMed] [Google Scholar]
- Rabodzey A, Alcaide P, Luscinskas FW, Ladoux B (2008) Mechanical forces induced by the transendothelial migration of human neutrophils. Biophys J 95: 1428–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond N, Imbert AM, Devilard E, Fabre S, Chabannon C, Xerri L, Farnarier C, Cantoni C, Bottino C, Moretta A et al (2004) DNAM‐1 and PVR regulate monocyte migration through endothelial junctions. J Exp Med 199: 1331–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringer P, Weißl A, Cost AL, Freikamp A, Sabass B, Mehlich A, Tramier M, Rief M, Grashoff C (2017) Multiplexing molecular tension sensors reveals piconewton force gradient across talin‐1. Nat Methods 14: 1090–1096 [DOI] [PubMed] [Google Scholar]
- Saito H, Minamiya Y, Kitamura M, Saito S, Enomoto K, Terada K, Ogawa J (1998) Endothelial myosin light chain kinase regulates neutrophil migration across human umbilical vein endothelial cell monolayer. J Immunol 161: 1533–1540 [PubMed] [Google Scholar]
- Saito H, Minamiya Y, Saito S, Ogawa J (2002) Endothelial Rho and Rho kinase regulate neutrophil migration via endothelial myosin light chain phosphorylation. J Leuk Biol 72: 829–836 [PubMed] [Google Scholar]
- Schenkel AR, Mamdouh Z, Chen X, Liebman RM, Muller WA (2002) CD99 plays a major role in the migration of monocytes through endothelial junctions. NatImmunol 3: 143–150 [DOI] [PubMed] [Google Scholar]
- Schulte D, Küppers V, Dartsch N, Broermann A, Li H, Zarbock A, Kamenyeva O, Kiefer F, Khandoga A, Massberg S et al (2011) Stabilizing the VE‐cadherin‐catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J 30: 4157–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroka KM, Aranda‐Espinoza H (2011) Endothelial cell substrate stiffness influences neutrophil transmigration via myosin light chain kinase‐dependent cell contraction. Blood 118: 1632–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su WH, Chen HI, Huang JP, Jen CJ (2000) Endothelial [Ca(2+)](i) signaling during transmigration of polymorphonuclear leukocytes. Blood 96: 3816–3822 [PubMed] [Google Scholar]
- Sullivan DP, Seidman MA, Muller WA (2013) Poliovirus receptor (CD155) regulates a step in transendothelial migration between PECAM and CD99. Am J Pathol 182: 1031–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan DP, Rüffer C, Muller WA (2014) Isolation of the lateral border recycling compartment using a diaminobenzidine‐induced density shift. Traffic 15: 1016–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan DP, Dalal PJ, Jaulin F, Sacks DB, Kreitzer G, Muller WA (2019) Endothelial IQGAP1 regulates leukocyte transmigration by directing the LBRC to the site of diapedesis. J Exp Med 216: 2582–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajan M, de Rocca SA, Valet P, Edouard T, Yart A (2015) SHP2 sails from physiology to pathology. Eur J Med Genet 58: 509–525 [DOI] [PubMed] [Google Scholar]
- Thompson RD, Noble KE, Larbi KY, Dewar A, Duncan GS, Mak TW, Nourshargh S (2001) Platelet‐endothelial cell adhesion molecule‐1 (PECAM‐1)‐deficient mice demonstrate a transient and cytokine‐specific role for PECAM‐1 in leukocyte migration through the perivascular basement membrane. Blood 97: 1854–1860 [DOI] [PubMed] [Google Scholar]
- Timmerman I, Hoogenboezem M, Bennett AM, Geerts D, Hordijk PL, van Buul JD (2012) The tyrosine phosphatase SHP2 regulates recovery of endothelial adherens junctions through control of β‐catenin phosphorylation. Mol Biol Cell 23: 4212–4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse JR, Engler AJ (2010) Preparation of hydrogel substrates with tunable mechanical properties. Curr Protoc Cell Biol Chapter 10: Unit 10 16 [DOI] [PubMed] [Google Scholar]
- Turowski P, Martinelli R, Crawford R, Wateridge D, Papagiorgiou A‐P, Lampugnani MG, Gamp AC, Vestweber D, Adamson P, Dejana E et al (2008) Phosphorylation of vascular endothelial cadherin controls lymphocyte emigration. J Cell Sci 121: 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukropec JA, Hollinger MK, Salva SM, Woolkalis MJ (2000) SHP2 association with VE‐cadherin complexes in human endothelial cells is regulated by thrombin. J Biol Chem 275: 5983–5986 [DOI] [PubMed] [Google Scholar]
- Urbano RL, Furia C, Baseshore S, Clyne AM (2017) Stiff substrates increase inflammation‐induced endothelial monolayer tension and permeability. Biophys J 113: 645–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestweber D (2015) How leukocytes cross the vascular endothelium. Nat Rev Immunol 15: 692–704 [DOI] [PubMed] [Google Scholar]
- Wakelin MW, Sanz MJ, Dewar A, Albelda SM, Larkin SW, Boughton Smith N, Williams TJ, Nourshargh S (1996) An anti‐platelet‐endothelial cell adhesion molecule‐1 antibody inhibits leukocyte extravasation from mesenteric microvessels in vivo by blocking the passage through the basement membrane. J Exp Med 184: 229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber EW, Han F, Tauseef M, Birnbaumer L, Mehta D, Muller WA (2015) TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. J Exp Med 212: 1883–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel F, Winderlich M, Holm M, Frye M, Rivera‐Galdos R, Vockel M, Linnepe R, Ipe U, Stadtmann A, Zarbock A et al (2014) Leukocyte extravasation and vascular permeability are each controlled in vivo by a different tyrosine residue of VE‐cadherin. Nat Immunol 15: 223–230 [DOI] [PubMed] [Google Scholar]
- Woodfin A, Reichel CA, Khandoga A, Corada M, Voisin MB, Scheiermann C, Haskard DO, Dejana E, Krombach F, Nourshargh S (2007) JAM‐A mediates neutrophil transmigration in a stimulus‐specific manner in vivo: evidence for sequential roles for JAM‐A and PECAM‐1 in neutrophil transmigration. Blood 110: 1848–1856 [DOI] [PubMed] [Google Scholar]
- Woodfin A, Voisin MB, Imhof BA, Dejana E, Engelhardt B, Nourshargh S (2009) Endothelial cell activation leads to neutrophil transmigration as supported by the sequential roles of ICAM‐2, JAM‐A and PECAM‐1. Blood 113: 6246–6257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE et al (2011) The junctional adhesion molecule JAM‐C regulates polarized transendothelial migration of neutrophils in vivo . Nat Immunol 12: 761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Zhang X, Chen A, Gu W, Liu J, Ren X, Zhang J, Wu X, Place AT, Minshall RD et al (2017) Endothelial cell SHP‐2 negatively regulates neutrophil adhesion and promotes transmigration by enhancing ICAM‐1‐VE‐cadherin interaction. FASEB J 31: 4759–4769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarbock A, Lowell CA, Ley K (2007) Spleen tyrosine kinase Syk is necessary for E‐selectin‐induced alpha(L)beta(2) integrin‐mediated rolling on intercellular adhesion molecule‐1. Immunity 26: 773–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Huang J, Qi T, Huang Y, Lu Y, Zhan T, Gong H, Zhu Z, Shi Y, Zhou J et al (2019) SHP2 protects endothelial cell barrier through suppressing VE‐cadherin internalization regulated by MET‐ARF1. FASEB J 33: 1124–1137 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Data Availability Statement
This study includes no data deposited in external repositories.