

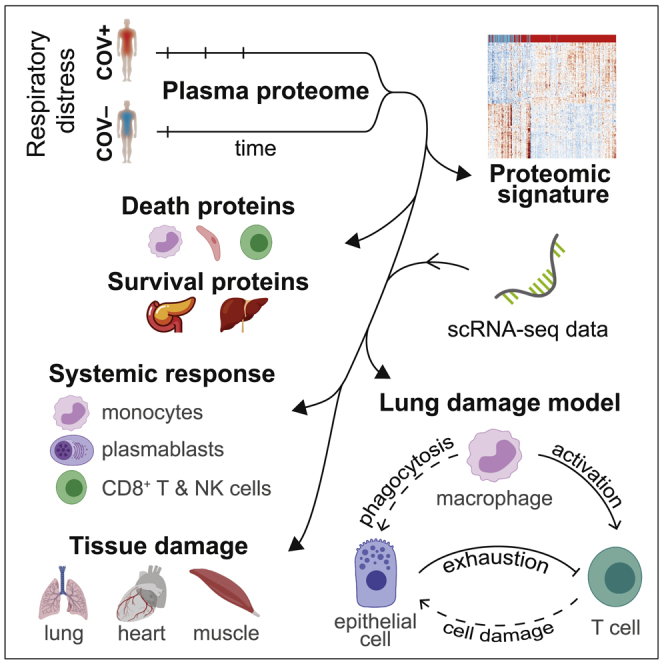
Summary
Mechanisms underlying severe coronavirus disease 2019 (COVID-19) disease remain poorly understood. We analyze several thousand plasma proteins longitudinally in 306 COVID-19 patients and 78 symptomatic controls, uncovering immune and non-immune proteins linked to COVID-19. Deconvolution of our plasma proteome data using published scRNA-seq datasets reveals contributions from circulating immune and tissue cells. Sixteen percent of patients display reduced inflammation yet comparably poor outcomes. Comparison of patients who died to severely ill survivors identifies dynamic immune-cell-derived and tissue-associated proteins associated with survival, including exocrine pancreatic proteases. Using derived tissue-specific and cell-type-specific intracellular death signatures, cellular angiotensin-converting enzyme 2 (ACE2) expression, and our data, we infer whether organ damage resulted from direct or indirect effects of infection. We propose a model in which interactions among myeloid, epithelial, and T cells drive tissue damage. These datasets provide important insights and a rich resource for analysis of mechanisms of severe COVID-19 disease.
Keywords: COVID-19 severity, death versus survival, plasma proteomics, lung epithelial cells, T cell activation, lung monocyte/macrophages, pancreatic exocrine proteases, longitudinal, acute respiratory distress syndrome, ARDS, intracellular death signatures
Graphical abstract
Highlights
16% of COVID-19 patients display an atypical low-inflammatory plasma proteome
Severe COVID-19 is associated with heterogeneous plasma proteomic responses
Death of virus-infected lung epithelial cells is a key feature of severe disease
Lung monocyte/macrophages drive T cell activation, together promoting epithelial damage
Filbin et al. use plasma proteomics in 306 coronavirus disease 2019 (COVID-19) patients and 78 symptomatic controls over time to better understand the role of circulating immune cells and tissue cells in inflammation, disease severity, and survival. They propose a model in which interactions among myeloid, epithelial, and T cells drive tissue damage.
Introduction
Coronavirus disease 2019 (COVID-19) has caused >1 million deaths globally. Disease varies considerably,1, 2, 3, 4 ranging from an asymptomatic carrier state to severe illness, organ dysfunction, and death.5 Implicated in the pathophysiology of severe disease is immune dysfunction, involving both hyper-immune responses (activated inflammatory cascades, cytokine storm, tissue infiltrates, damage) and hypo-immune responses (relative lymphopenia, impaired T cell function, impaired interferon [IFN] antiviral responses, reduced viral clearance).5, 6, 7, 8 To date, many studies addressing the immune response to Severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) are limited by small sample sizes or analyze narrow sets of immune mediators,2,4,9, 10, 11, 12, 13 although multi-omic approaches are beginning to overcome these limitations.14 By analyzing responses to SARS-CoV-2 using two unbiased plasma proteomic methodologies in a large cohort of acutely ill patients presenting to a large urban emergency department (ED), we uncover protein signatures associated with COVID-19 infection, severity, and death. To gain insights into underlying disease mechanisms, we map these to specific cell types in the context of relevant clinical phenotypes.
Results
Viral response and IFN pathway proteins
We enrolled 384 unique subjects who presented with acute respiratory distress suspected or known to be due to COVID-19 infection. A total of 306 patients were subsequently confirmed to be COVID-19 infected. We classified patients by acuity levels A1–A5 on days 0, 3, 7, and 28 (based on the World Health Organization [WHO] ordinal outcomes scale15: A1, died; A2, intubated, survived; A3, hospitalized on oxygen; A4, hospitalized without oxygen; A5, discharged), with the primary outcome of maximal acuity (Acuitymax) within 28 days of enrollment (Figure 1A; Table S1). COVID-19+ patients were younger than COVID-19− patients (median age 58 versus 67 years, respectively), with a wide age distribution (Figure S1B), and were predominantly Hispanic (54% versus 15%, respectively). Clinically measured non-specific inflammatory markers, including C-reactive protein (CRP) and ferritin, were significantly higher in COVID-19+ than COVID-19− patients; 28-day outcomes were similar (Figure S1B). Given that enrollment occurred early in the pandemic, few patients received targeted therapies that may be expected to alter the disease course; 6 received remdesivir versus placebo and 22 received anti-interleukin-6 (IL-6) receptor monoclonal antibody versus placebo (both as study protocols), and dexamethasone was not administered as usual care for COVID-19.
Figure 1.

SARS-CoV-2 infection induces viral response and IFN-pathway proteins detected in patient plasma
(A) Schematic of study cohort: 306 COVID-19-infected patients and 78 symptomatic COVID-19− controls. Inclusion criteria are indicated. Shown are maximal acuity level within 28 days (Acuitymax) for COVID-19-infected patients (A1, most severe; A5, least severe), N, proportion of patients, and severe versus non-severe group.
(B) Schematic of study methodology.
(C–E) Differentially expressed proteins by COVID-19 status. Linear model fitting each Olink protein, with COVID-19 status as a main effect and putative confounders as covariates (see STAR Methods). p values calculated to account for false discovery rate (FDR) < 0.05, Benjamini-Hochberg method.
(C) Heatmap of top 200 differentially expressed proteins between COVID-19+ and COVID-19− patients. Each row represents the expression of an individual protein over the entire cohort; each cell represents the Z score of protein expression for all measurements across a row. COVID-19 signature scores calculated by taking the mean Z score of the top 25 differentially expressed proteins in COVID-19+ patients minus the top 25 differentially expressed proteins in COVID-19− patients.
(D) Volcano plot of differentially expressed proteins based on mean normalized protein expression (NPX) values between COVID-19+ and COVID-19− patients. Blue circles, significantly differentially expressed proteins. All of the proteins are shown.
(E) Boxplots of select differentially expressed viral response and interferon (IFN) pathway proteins (from D), including IFN-γ, DDX58 (or RIG-I), IFN-λ1, and chemokines CXCL10, CXCL11, CCL7, CCL16, and CCL24.
(F) Inference of cell of origin by mapping gene expression of differentially expressed plasma proteins elevated in COVID-19+ versus COVID-19− patients in a scRNA-seq peripheral blood cell COVID-19 dataset.16 Heatmaps of mean expression of COVID-19-related proteins (y axis) in immune cell subtypes (x axis). gd T cells, γδ T cells; pDCs, plasmacytoid dendritic cells.
See also Figures S1–S3 and S5 and Tables S1 and S2.
We analyzed 1,472 unique plasma proteins measured by proximity extension assay (PEA) using the Olink platform (Olink Explore 1536) for all patients on day 0 (D0, N = 383, one assay outlier excluded) and for COVID-19+ patients still hospitalized on D3 (N = 217) and D7 (N = 143) (Tables S2 and S3). Time since symptom onset at presentation ranged from 0 to 31 days (median 7 days). Unsupervised clustering of D0 protein levels shows clustering by COVID-19 status, age, acuity, ethnicity, and kidney disease (Figure S1A).
To identify proteins differential between COVID-19+ and COVID-19− patients, linear models were fit to each protein at D0, with COVID-19 status as a main effect and adjusted for age, demographics, and key comorbidities (Figures 1B and S2; Table S3). Hierarchical clustering of patients using these differentially expressed proteins demonstrated a clear separation of the majority of COVID-19+ from COVID-19− patients (Figures 1C and S1C–S1E). COVID-19+ patients displayed a higher expression of viral response and IFN pathway proteins, including DDX58 (RIG-I), type II (IFN-γ), and type III (IFN-λ1) IFNs, and the proinflammatory cytokines CCL7, CXCL10, and CXCL11 (Figures 1D and 1E), with the enrichment of proteins in pathways associated with vaccine response, innate immune activation, and T cell function (Figures S1F). Fifty (16%) COVID-19+ patients clustered with COVID-19− patients, displaying lower levels of the typical COVID-19+ inflammatory signature (Figure 1C), yet with mortality similar to the main cluster of COVID-19+ patients (Table S1). Although significantly older than the main COVID-19+ patients (median age 69 versus 57 years), with more cardiac and kidney comorbidities, this subset is comparably ill, with a distinct low-inflammatory proteomic signature.
To derive potential immune cell subtype origins of key proteins, we mapped the differential protein expression (Olink assay) in COVID-19-infected patients to published single-cell RNA sequencing (scRNA-seq) profiles from peripheral blood mononuclear cells (PBMCs) and bronchoalveolar lavage (BAL) samples from COVID-19-infected patients (Figures 1F,S3A, and S3B).8,16,17 The majority of proteins were selectively expressed in circulating plasmablasts (e.g., RRM2, WARS, PRDX1) and myeloid cells (e.g., CD14, SIGLEC1, SIGLEC10, IL-1RN, CCL8, CXCL10), particularly monocytes and neutrophils, which is consistent with the reported remodeling of these cell types in infected patients.8,16,17 A smaller group of proteins, expressed strongly in peripheral CD8+ T cells and natural killer (NK) cells, reflected cytotoxic responses, including IFN-γ, granzymes B and H (GZMB, GZMH), which trigger cell death upon delivery into target cells, and the receptor LAG3 (Figure 1F). Whereas membrane-embedded LAG3 inhibits T cell activation, soluble LAG3, such as we observed in plasma, functions as an immune adjuvant.18,19 A set of proteins was found to be overlapping within BAL cells and circulating myeloid and T cells; BAL lung epithelial cells additionally expressed proteins not detected in the plasma (Figure S3B). As these datasets were generated from distinct cohorts of patients, the conclusions drawn will require validation in individuals in whom plasma proteomics is performed in parallel with scRNA-seq of PBMCs and BAL samples.
Heterogeneous phenotypes associated with severity
Similar to previous reports,20, 21, 22 Acuitymax of COVID-19 patients was significantly correlated with age, D0 acute kidney dysfunction, lactate dehydrogenase (LDH), lymphopenia, acute inflammatory markers (erythrocyte sedimentation rate [ESR], C-reactive protein [CRP], d-dimer, ferritin), and the preexisting comorbidities kidney disease, diabetes, smoking, and heart disease (Figures 2A and S3C–S3E). Distinct from some reports,20, 21, 22 Acuitymax was not significantly correlated with race, ethnicity, or body mass index (BMI). Virus neutralization activity by plasma was highly correlated with inflammatory markers, absolute neutrophil count (ANC), and COVID-19+ status, but not with Acuitymax (Figures 2A, 3A, S3C, and S3D).
Figure 2.

Plasma proteomic biomarkers and predictors of disease severity
(A) Pairwise correlation heatmap of clinically annotated variables for COVID-19+ patients showing correlations having p < 0.05.
(B) Unsupervised clustering by uniform manifold approximation and projection (UMAP) for COVID-19+ patients, color-coded (left to right) by day of sample collection (D0, D3, D7), Acuitymax by D28, severity, age decile, gender, and ethnicity. E, event-driven samples (see STAR Methods).
(C) Linear mixed model fitting each Olink protein, with severity, time point, and the interaction of the 2 terms as main effects and putative confounders as covariates (see STAR Methods). Heatmap of significant differentially expressed proteins between severe and non-severe patients at D0. Significance of the 3 model terms determined with an F test, Satterthwaite degrees of freedom, and type III sum of squares. p values for the 3 model terms of interest calculated to account for FDR < 0.05 using the Benjamini-Hochberg method for multiple hypothesis correction. Group differences calculated for each significant protein; p values adjusted using Tukey method.
(D) Linear mixed model fitting each Olink protein, with severity, time point, and the interaction of the 2 terms as main effects and putative confounders as covariates (see STAR Methods). Volcano plots of differentially expressed proteins between severe and non-severe COVID-19+ patients by time point, with number (N) indicated. Blue circles, proteins that are significantly differentially expressed. All of the proteins are shown.
(E) Distribution of patient samples by acuity level on day of collection and as a function of time. N, number of individual patient samples.
(F) Point range plots over time of selected set of proteins significant for interaction term in the model described in (D), color-coded by disease severity.
(G) Receiver operating characteristic (ROC) curve of predictive performance of an elastic net logistic regression classifier of disease severity, for Olink proteins of each patient at D0. Performance was evaluated using 100 repeats of 5-fold cross-validation. Mean area under the curve (AUC) with 95% confidence intervals (CIs). Neutralization, virus neutralization activity by plasma.
See also Figures S2–S5 and Table S2.
Figure 3.

Predictors of neutralization and its association with disease severity and age
(A) Boxplot of SARS-CoV-2 Spike pseudovirus neutralization levels for COVID-19− and COVID-19+ patients at D0. Box edges, interquartile range (IQR); middle line, median.
(B) Point-range plots of neutralization levels in non-severe and severe COVID-19+ patients over time. Color-coding by neutralization level at D3, grouped into 0%–25%, 25%–50%, 50%–75%, and 75%–100%.
(C) Proportion of patients with neutralization levels as in (B), over time and by severity level.
(D) Boxplots of neutralization levels in non-severe and severe patients over time. Box edges, IQR; middle line, median.
(E) Scatterplot of the correlation of age with rate of change in neutralization level over time in A2 (left) and A1 (right) patients. Rate of change is the negative of the regression line slope through log2(fold change) in GFP levels at each time point compared to controls.
(F) Proportion of patients aged £65 years (left) or >65 years (right) achieving neutralization titers of ³50% (blue) or 75% (orange) at D3. Error bars, 95% CI of proportion.
(G) Lasso regression model for prediction of D3 neutralization level (above or below 75%) using Olink plasma proteins at D0 across all COVID-19+ patients. Prediction performed with 5-fold cross-validation over 100 iterations; AUC 0.83 (95% CI 0.80–0.85).
(H) Heatmap of Olink plasma protein expression of each of the top selected features from the predictor in (G) that did not overlap with the top severity-associated proteins from the linear mixed model (see STAR Methods).
(I) Volcano plot of differentially expressed proteins based on mean NPX values between high and low viral neutralization titers (>0.75 versus <0.75) across COVID-19+ patients. Blue circles, significantly differentially expressed proteins. All of the proteins are shown.
Unsupervised clustering of COVID-19+ patient samples demonstrated the separation of patient samples by Acuitymax, severity (severe: Acuitymax A1–A2; non-severe: Acuitymax A3–A5), age, and time point (Figures 2B and S3F). To identify proteins associated with Acuitymax levels and severity, we fit linear mixed models (LMMs), which correct for non-independence of time course data, to protein values with time and either Acuitymax or severity as main effects, and with covariates age, demographics, and key comorbidities (Figures 2C and S4A–S4E; Table S3B). At D0, 251 Olink plasma proteins were differentially expressed between severe and non-severe patients, 694 at D3, and 767 at D7 (Figures 2D–2F; Table S3B). Because many patients with mild disease were discharged from the hospital within 3 days of admission, and D3 and D7 samples were collected from the subset of patients who remained hospitalized at these time points, D3 and D7 samples represent a generally sicker population than do D0 samples (Figure 2E).
The increased numbers of severity-associated proteins at the D3 and D7 time points indicate that even though the population is generally sicker, the differences between those with severe disease and those with non-severe disease become more pronounced with time; these dynamic changes likely reflect clinically relevant phenotypes and underlying disease processes. These severity proteins showed signals for enrichment in pathways implicated in the COVID-19 inflammatory response, including IFN-γ, IL-6, and tumor necrosis factor (TNF) signaling, and in tissue remodeling, including KRAS signaling and epithelial-to-mesenchymal transitions (Table S4). Hierarchical clustering of patients by D0 severity-associated proteins revealed multiple distinct clusters of severe patients (Figure 2C), indicating that severe disease is phenotypically heterogeneous and underscoring the presence of multiple phenotypes of patients with severe disease, beyond the single subgroup described above that displayed a low-inflammatory proteomic signature (Figure 1C). Similar to proteins associated with COVID-19+ status (Figure 1), the majority of circulating proteins associated with severity were most highly transcriptionally expressed in myeloid and plasmablast subsets (Figure S5).
Plasma proteomic prediction of severity
To test whether D0 plasma proteins predict subsequent disease severity, we built a classifier of severe disease (Acuitymax A1 or A2, Olink data) using elastic-net logistic regression with cross-validation; the classifier yielded good predictive performance (area under the curve [AUC] 0.85, 95% confidence interval (CI) 0.81–0.86) (Figures 2G, S4F, and S4G), although an independent validation dataset is needed. Among the strongest weighted proteins in the predictor were IL-6, IL-1RL1, PTX3, and the IL-1 receptor inhibitor IL-1RN, consistent with our LMM results, the epithelial damage marker keratin-19 (KRT19, a predominantly intracellular cytoskeletal protein23), and the apoptosis inhibitor TRIAP1 (Figure S4F).24 The strength and weighting of this predictor highlight that disease severity can be accurately predicted at the time of presentation to the hospital, that proinflammatory signatures are associated with severity, and that severity-associated proteins identified both here (PTX3, IL-1RN) and previously (IL-62, 3, 4,9,10,25,26) contribute to a robust predictor.
SARS-CoV-2 pseudovirus neutralization activity and age
Virus neutralization activity was detected in plasma from nearly all COVID-19+ patients (Figure 3A; Table S5), consistent with previous reports.27,28 Consistent with the observed lack of correlation with Acuitymax (Figure 2A), neutralization activity increased over time among the majority of both severe and non-severe patients (Figures 3B–3D), indicating that, as previously described,27 neutralization activity per se does not predict milder disease. However, neutralization activity was inversely correlated with age and age-related comorbidities (Figure 2A), as previously observed,27 displaying age-associated decreases in both the rate of increase over time and the level of neutralization activity achieved (Figures 3E and 3F). The negative impact of age on the rate of increase in neutralization over time was observed only among patients who died (A1) (Figure 3E), suggesting that disease processes present in severe illness contribute to impaired adaptive immune responses.
D0 plasma protein levels (Olink assay) predicted neutralization levels at D3 (AUC 0.83, CI 0.80–0.85), with many proteins contributing to the prediction being independent of those associated with severity (Figures 3G and 3H), although this needs to be validated in an independent dataset. Among the proteins most often selected in predicting neutralization were those involved in the induction of apoptosis (TNF superfamily members TNFSF10, TNFSF8, and galectin-7 [LGALS7B]), phagocytosis (BRK1), T cell proliferation (IL-2), and tissue regeneration and proliferation (EGFR, PTEN, PLA2G10, DKK3, RRM2). To identify plasma proteins differentially expressed between patients with high and low neutralization titers, we also used a LMM with neutralization level and time as main effects (Figure 3I); this identified several proteins expressed in plasma cells, (e.g., MZB1, SDC1), and others known to be important for priming (e.g., CD40LG). Among the proteins most significantly highly expressed in patients with low neutralization titers were CXCL10, which has recently been implicated to be negatively correlated with CD4+ T cell features associated with antibody titers,27 and GPA33, a marker of thymic regulatory T cells (Figure 3I).29 These findings indicate that elderly patients who do poorly display distinctive neutralization activity-associated protein profiles that may be useful in clinical prediction algorithms, vaccine response prediction, and identifying subsets of patients most appropriate for trials of antibody-based therapy.
Decreased anti-inflammatory proteomic profile in ARDS
ARDS is the leading cause of death in COVID-19. To gain insight into processes that may underlie the development of ARDS, we compared patients who died (A1, median time to death 9 days [interquartile range {IQR} 4–17]) to those receiving mechanical ventilation yet surviving (A2) (Figure 4; Table S3C); by clinical criteria, essentially all of the patients in both groups had ARDS, although not all who died were mechanically ventilated. At D7, 24 plasma proteins were significantly differentially expressed between the 2 groups (Figure 4A); among those elevated in patients who died were previously reported proinflammatory proteins (proinflammatory cytokines IL-6,2, 3, 4,9,10,25,26 IL-8,2,3,9,10,26 and CXCL102,9,10,26), chemokines that attract monocytes/T cells (CCL2, CCL7, CCL8, CCL20), a receptor for IL-33 that activates T cells and mast cells (IL-1RL1), regulators of innate immunity (PTX3), the endothelial and monocyte receptor for the growth factors vascular endothelial growth factor (VEGF) and placental growth factor (PGF) (FLT1), and a multi-functional cytokine (IL-24). Most of these proinflammatory proteins showed similar upward trajectories in survivor and non-survivor groups through D3, but diverged at D7, with a decline in survivors and a sustained elevation in those who died (Figure 4C); D0 plasma levels were associated with survival (Figure 4B). Whereas an upward trajectory at D3 could result from the sicker composition of the D3 patient population compared with the D0 population, the subsequent divergence in trajectories observed at D7 between survivors and those who died instead likely represents relevant biological processes associated with death. Several exocrine pancreas proteases and protease inhibitors (CTRC, CELA3A, CPA2, CTRB1, AMY2A, AMY2B) were reduced in the plasma of those who died relative to survivors (A2) (Figures 4A and 4D); whereas their relevance in COVID-19 remains uncertain, many display anti-inflammatory effects in mouse models.30, 31, 32, 33 Few patients received the anti-inflammatory dexamethasone because it was not yet the standard of care at the time of patient recruitment. These findings suggest that survival from COVID-19 ARDS is associated with decreased proinflammatory and increased anti-inflammatory responses over time.
Figure 4.

Patients with ARDS who survive display reduced inflammatory markers and increased anti-inflammatory pancreatic proteases
(A) Differentially expressed proteins at day 7 between patients who had Acuitymax of A1 (death) versus A2 (ARDS but survived). Linear mixed model fitting each Olink protein, with Acuitymax, time point, and the interaction between the 2 terms as main effects. Covariates and statistical analysis as in Figure 2C.
(B) Kaplan-Meier curves for overall survival of patients stratified by higher or lower than median expression of indicated proteins from (A).
(C and D) Point-range plots for select proteins from (A) with positive (C) or negative (D) NPX differences.
See also Table S3.
Tissue-specific signatures mark toxicity
To elucidate patterns of tissue damage, we calculated gene expression signatures associated with specific tissues using the Genotype-Tissue Expression (GTEx)34 dataset and confirmed using published scRNA-seq datasets that these signatures were expressed primarily in non-immune cells that compose the structure of tissues (Figures S6A–S6F). Because of the breadth of the database, we chose for analysis of these signatures the SomaScan platform,35 which detects >4,400 proteins. We confirmed a high degree of overlap in differentially expressed proteins between severe and non-severe patients using this platform as compared to the Olink platform (Table S6; STAR Methods). We identified plasma proteins that overlap with these tissue signatures and filtered for intracellular proteins (Table S7A), based on the principle that intracellular proteins found in the circulation represent the release of cellular cytosolic contents in the setting of tissue damage. When possible, we validated our tissue-specific signatures against clinically measured laboratory values, finding significant correlations with tissue-specific clinical markers of damage (Figures 5B and S6I–S6L).
Figure 5.

Severe COVID-19+ patients display elevated plasma markers of cell death from heart, lung, and skeletal muscle
(A) Expression of tissue-specific plasma protein signatures in non-severe versus severe patients at each time point.
(B) Scatterplot of the correlation of the D0 plasma heart signature as derived in (A) with D0 clinical troponin measurements.
(C) Kaplan-Meier curve of overall survival of patients with high or low expression (above or below median expression level) of the derived plasma heart signature in (A).
(D) Heatmap of mean gene expression per cell type of severity-associated intracellular plasma proteins at D0 derived from SomaScan data that map to scRNA-seq of BAL fluid,36 with TMPRSS2 and ACE2 expression indicated.
(E and F) Scatterplots of the difference between severe and non-severe patients of lung (E) and heart (F) cell-specific intracellular death scores, derived from expression of differentially expressed proteins at each time point versus cell-type-specific ACE2 and TMPRSS2 expression levels from scRNA-seq of BAL fluid36 (E) or heart single-nucleus RNA-seq data37 (F). AT2, alveolar type 2 epithelial cells.
See also Figures S5–S7, and Tables S6 and S7.
In patients with severe COVID-19 (A1, A2), among the organ-specific signatures, heart, lung, and skeletal muscle intracellular plasma protein signatures were elevated as early as D0 and remained elevated to D7 (Figures 5A and S6G). Elevated D0 heart and skeletal muscle protein signatures portended poor overall survival (Figure 5C; Table S7C). Our lung signature contained only one protein, the intracellular cytoskeletal protein keratin-7 (KRT7); therefore, this particular signature should be interpreted with caution. Our tissue damage signatures suggest that COVID-19 illness drives organ damage that can be detected in the circulation upon hospital presentation.
Lung damage due to epithelial death
We mapped intracellular severity-associated plasma proteins to organ-specific cell types using published scRNA-seq datasets. Datasets from healthy lung, kidney, pancreas, and liver revealed that D0 severity-associated intracellular proteins found in plasma are expressed predominantly in macrophage subsets and epithelial cells, with higher expression in kidney proximal tubule cells; pancreatic stellate, ductal, and acinar cells; and hepatocytes (data not shown). Parallel analysis in single cells of BAL fluid and upper airways from COVID-19-infected patients,36,38 more disease-specific contexts, showed distinct clusters of proteins expressed within lung epithelial cells and T cells, with lower expression in tissue-associated myeloid or B cells (Figures 5D and S7C).
Within lung epithelial cells from COVID-19 BAL cells,36 the expression of severity-associated intracellular proteins correlated with the expression of the SARS-Co-V-2 receptor angiotensin-converting enzyme 2 (ACE2) (R = 0.49, p = 0.04), but not of the SARS-CoV-2 priming protease TMPRSS2 (Figure 5E; Table S7B), which suggests that the increased levels of these proteins in plasma may result from SARS-CoV-2 infection-induced cell death and is consistent with proteases other than TMPRSS2 being involved in spike protein processing during viral entry. Consistent with this hypothesis of lung epithelial death, plasma levels of alveolar cell markers advanced glycosylation end product-specific receptor (RAGE) and pulmonary surfactant-associated proteins A1, A2, and D are significantly elevated at D0 in severe versus mild patients (data not shown).
In contrast, heart cell-type plasma signatures did not correlate with ACE2 expression (Figure 5F), suggesting that heart damage may be largely an indirect effect of the disease process (assuming that ACE2 expression is similar in healthy individuals and COVID-19-infected patients); the implications of the observed correlation with TMPRSS2 expression on cells (R = 0.62, p < 0.001; Figure 5F) are unclear. Unlike that in circulating immune cells (Figure S5A–S5D), the expression of a larger subset of severity-associated intracellular plasma proteins was found in effector and cytotoxic CD8+ T cells and NK cells located within the lung (Figure 5D). Intracellular plasma signatures from cell subsets that do not express ACE2 or TMPRSS2 (Figure 5D), including these cytotoxic CD8+ T cells and NK cells, epithelial progenitors, and alveolar macrophages, may result from bystander cell death. These findings suggest that immune-mediated death of virus-infected lung epithelial cells is a key feature of severe disease, that damage to several other cell types is indirect, and that cell death is detectable in the circulating proteome.
Lung epithelial-immune communication
To gain insights into immune activation in severe disease, we looked for enrichment of inflammatory pathways among plasma proteins that are normally secreted or membrane bound. Within the D0 Olink severity-associated proteome (and consistent with SomaScan results), we analyzed enriched pathways against the entire measured protein set and found enrichment in signaling by cytokines IL-6 and IL-10, activation of myeloid and T cells by the cytokine IL-17, airway pathology in chronic obstructive pulmonary disease (COPD), cardiac hypertrophy signaling, signaling by the proinflammatory danger-associated molecular pattern (DAMP) molecule HMGB1, and signaling via the glucocorticoid receptor (Table S4A). Analysis of upstream regulators revealed TNF to be the most significant putative regulator of these pathways (Table S4B). To identify cellular mechanisms regulated by severity-associated proteins, we analyzed ligand-receptor interactions39 using the BAL fluid cell dataset from COVID-19-infected patients (Figure 6).36 From D0 to D3, the number of predicted ligand-receptor interactions increased dramatically (Figure 6A), predominantly represented by ligand-receptor interactions occurring in lung epithelial cells, T cells, and mast cells (Figure 6B).
Figure 6.

Interactions among lung epithelial cells, monocytes, and T cells drive disease severity and tissue damage
(A) Heatmap of the total number of ligand-receptor interactions at D0 and D3 inferred from BAL fluid scRNA-seq data36 using only ligands differentially expressed in the plasma of severe versus non-severe COVID-19+ patients.
(B) Heatmap of fold change from D0 to D3 in the number of ligand-receptor interactions between each cell type identified from BAL fluid scRNA-seq data.36
(C) Ligand-receptor contact map between D0 severity-associated ligands expressed by lung epithelial cells per BAL fluid scRNA-seq data36 (left) and the respective receptors for these ligands with their cell-specific expression from the same BAL dataset (right).
(D) Ligand-receptor contact map between receptors expressed on lung epithelial cells in BAL fluid36 (right) and their respective severity-associated plasma ligands from our data (left). Ligand-receptor pairs are those for which the ligand was significantly associated with severity at D0.
(E) Ligand-receptor contact map between ligands expressed on monocytes/macrophages in BAL fluid scRNA-seq data36 (left) and the respective receptors for these ligands with their cell-specific expression from the same BAL dataset (right).
(F) As in (D), but ligand-receptor pairs selected for receptors expressed on T cells in BAL fluid.
In (C)–(F), each cell in the heatmaps represents expression of the listed ligand or protein relative to its expression across all cell types. Ligands and receptors are color-coded (vertical color bar) by the cell type that demonstrates their highest expression. Ligand-receptor pairs and their connecting lines are color-coded by time point (D3 only, or both D0 and D3) at which the interaction was present. Key to cell type color-coding applies to (C)–(F). Trm, resident memory CD8+ T cells; DC, dendritic cell; Mon-derived mac, monocyte-derived macrophages; mac, macrophages; Tfh, T follicular helper cell; Tregs, regulatory T cells.
See also Figures S5–S7, and Tables S2 and S4.
Most of the dramatic changes in terms of fold change were in mast cells, although the total number of interactions was lower than other cell types. This was driven by their interactions with other mast cells, CD4+ and CD8+ T cell subsets, and epithelial progenitors (Figure 6B). Consistent with this, the mast cell function marker tryptase was differentially expressed between severe and non-severe patients over time (Figure S6H). Mast cell activity in lung tissue may be related to signaling by the proinflammatory cytokine IL-18,40 with release of proinflammatory cytokines IL-4 and IL-13,41 and may play a role in local tissue damage. Mast cells have been implicated in the vascular leak and coagulopathy observed in infections due to dengue and certain other viruses,42,43 which, together with the increased mast cell activity we observe in lung tissue, suggests that further investigation into their role in COVID-19 infection is warranted.
To better understand the specific pathways mediating disease severity, we constructed mappings of key ligand-receptor relationships of cells in BAL fluid and the airways with D0 and D3 plasma severity-associated ligands (Figures 6C–6F, S7A, and S7B). We observed within the lung predominantly epithelial and myeloid cell ligands interacting with epithelial, T cell, and NK cell receptors. Pairings of ligands from lung epithelial cells with receptors on other lung epithelial cells identified pathways involved in alveolar maintenance and protection, growth factor signaling, and tissue regeneration (including HGF-MET, TGFA-EGFR, DKK1-LRP6, KITLG-KIT, and semaphorin-PLXNA receptors; Figure 6C). Several T cell-activating and -exhaustion signals were upregulated and may originate from lung epithelial cells, including, as early as D0, poliovirus receptor (PVR) triggering of the receptors TIGIT and CD96, which induces an immunosuppressive and non-cytotoxic response, and at D3, IL-18, and IL-7 (Figure 6C), which dampen T cell exhaustion44 and maintain non-exhausted T cells,45 respectively. IL-18 is a predominant effector released upon inflammasome activation and pyroptotic cell death; the observed increase in IL-18 here thus suggests increased inflammasome activation in severe COVID-19.
We examined lung epithelial cell receptor interactions with severity-associated ligands (Figure 6D); a correlation matrix of plasma ligand abundance identified co-regulated groups of proteins that act on lung epithelial cells, including protein modules for regeneration and growth factor signaling (module 1: growth factors EGF, transforming growth factor-β1 [TGF-β1], and VEGFA, and anti-apoptotic factor Dickkopf WNT signaling pathway inhibitor 1 [DKK1]; module 2: growth factors bone morphogenic protein 6 [BMP6] and hepatocyte growth factor [HGF], and Wnt signaling pathway activators RSPO3 and RSPO1) and for IL-6 pathway signaling (IL-6 and the IL-6 family cytokines oncostatin M [OSM] and leukemia inhibitory factor [LIF]). The direct effects of IL-6 signaling on lung epithelial cells in COVID-19 are unknown.
Many severity-associated ligands were expressed in lung-resident monocytes/macrophages and function in T cell recruitment, activation, and exhaustion, with some proteins found as early as D0 (e.g., ligand-receptor interactions CXCL9-CXCR3, CXCL10-CXCR3, IL-15-IL-2R, CD74-LAG3, CD274-PDCD1, IL-18-IL-8R, IL-15-IL-2R, PVR-TIGIT, and CD96; CXCL16-CXCR6, CD74-LAG3, and CD27-CD70; Figures 6E) and often co-regulated in association with patient death (CCL2, CCL7, CCL8, and CXCL10; Figures 4A, 4B, and 6F). Activated T cells and NK cells express granzyme proteins and may cause direct and indirect killing of cells in the lung. As in lung epithelial cells (Figure 6C), IL-18 interactions suggest severity-associated inflammasome activation in lung monocyte/macrophages (Figure 6E). Additional ligand-receptor interactions between monocyte/macrophages and lung epithelial cells and other myeloid cells, most apparent at D3, may drive later-stage damage, immune suppression, and regulation of phagocytosis (e.g., ligand-receptor interactions TGFβ1-ITGβ6 and -ITGβ8, secreted phosphoprotein 1 [SPP1]-integrin αv [ITGAV], signal regulatory protein alpha [SIRPA]-CD47; Figure 6E). The interaction of TGF-β1 proprotein with ITGβ6/8 on lung epithelial cells likely releases active TGF-β1,46,47 which inhibits cytotoxic T cells and naive T cell and B cell proliferation and enhances Treg differentiation.48 The interaction of SPP1 with its receptor integrin ITGAV is associated with lung fibrosis and is proposed to inhibit apoptosis.49 The interaction of CD47, which is ubiquitously expressed on cell surfaces, with SIRPA on macrophages inhibits phagocytosis.
Based on these data, we propose a model of COVID-19-induced immune and cellular responses and cell death within the lower airways. We posit that early monocyte activation drives T cell recruitment, activation, and exhaustion. This is followed by a temporally delayed activation of additional proinflammatory monocyte pathways and repair and regeneration within lung epithelial cells (Figure 7). In patients who die, there is increased expression over time of severity-associated, monocyte-secreted ligands that interact with T cells (e.g., IL-18, IL-7, IL-15), suggesting an inability to contain proinflammatory immune responses.
Figure 7.

Model of contributions to the plasma proteome from circulating immune cells (primarily monocytes, plasmablasts, CD8+ T, NK cells) and damaged tissues
Temporally ordered interaction network between monocyte/macrophages, T cells, and lung epithelial cells that drives disease severity.
Discussion
This plasma proteomic analysis provides a comprehensive longitudinal summary of the systemic host response to SARS-CoV-2. COVID-19 patients have dramatically different plasma proteomic profiles than acutely ill COVID-19− controls (Figure 1). The large size of our cohort enabled the identification of a substantial subset (16%) of COVID-19-infected patients with inflammatory signatures similar to COVID-19− controls but outcomes similar to those of other COVID-19+ patients. In these patients, the muted levels of circulating inflammatory proteins suggest that much of the underlying pathology is due to viral infection itself and preexisting comorbidities in the setting of advanced age rather than immune-mediated processes. In this case, clinical response to immune-targeted therapies, including dexamethasone, could be suboptimal, and antiviral and other interventions may have more of an impact.
Over 250 proteins were independently associated with COVID-19 severity, with multiple inflammatory mediators associated with death in ARDS patients, including previously identified markers (IL-6,2, 3, 4,9,10,25,26 IL-8,2,3,9,10,26 and CXCL102,9,10,26) and several other markers (CCL2, CCL7, CCL8, CCL20, AREG, IL-1RL1, FLT1, IL-24) (Figures 2 and 4), with some recently reported in a smaller study of hemodialysis-dependent COVID-19 patients from a distinct geographic region,50 independently validating our findings. Of note, several exocrine pancreas proteases and other proteases were significantly associated with the survival of patients with ARDS. Determining whether these proteases are markers of underlying processes that contribute to survival or are directly contributing to a beneficial anti-inflammatory response will require further investigation.
Prior proteomics studies have included fewer COVID+ patients (N = 46, N = 22, N = 48) compared with ours (N = 306) and have not obtained a sample at point of hospital arrival, yet these studies have the advantage of using unbiased methods (e.g., liquid chromatography/mass spectrometry) for protein discovery.11,13,51 Among the few overlapping proteins from these prior datasets, our findings are consistent, yet compared to these other works, our data show overall stronger associations of pro-inflammatory cytokines and chemokines with severity and death, and less strong associations with complement activation and coagulation signals. These differences may in part reflect an enrichment in our panel of proteins of immune-mediated markers. This enrichment enables us to better infer immune cell function and cellular communication at play in severe COVID-19. Our classifier of severity did not perform as well as in the above-mentioned studies.13,51 Decreased classifier performance may reflect increased heterogeneity of our population with respect to comorbidities and treatments received, resulting in less distinct proteomic signatures in severe versus mild COVID-19, or it may be a limitation of the finite number of proteins assayed on our platform.
We observed a strong association between advanced age and attenuated neutralizing antibody production and identified discrete plasma protein signatures associated with the neutralization response (Figure 3), which may predict vaccination response and have implications for vaccination strategies. The strong predictive value of D0 plasma proteins highlights the presence of severity-associated pathways that may be amenable to early therapeutic intervention. The incorporation of derived biomarkers into diagnostics could stratify high-risk patients for tailored therapies.
Proteins end up in the plasma via a variety of routes. Many, including cytokines, interferons, and growth factors, are secreted from effector cells. Some, including IL-18, are released from the cytosol during programmed cell death of immune cells, whereas others that are also normally cytosolic, including KRT7, are released from the cytosol of dying cells. Less clear are the mechanisms of observed increases in plasma membrane proteins in the plasma. Some, including LAG3, exist as both soluble and membrane-embedded forms; our primary data do not enable the determination of the contribution of each. However, the assignment of proteins to signatures we derived for specific tissues and cells provides context for many relevant plasma proteins, enabling inference of their origins, with implications for underlying processes in the pathogenesis of COVID-19 infection.
By leveraging scRNA-seq datasets from PBMCs of COVID-19 patients and from healthy tissues, we deconvoluted the relative contribution of different compartments to the plasma proteome, finding major subsets of severity-associated plasma proteins expressed in circulating monocytes and plasmablasts and a smaller subset in circulating T cells and NK cells. In contrast, plasma severity-associated proteins were enriched in T cell and NK cell expression in BAL samples, implicating a role of these cell types in tissue inflammation in the lung. By deriving tissue-specific intracellular death signatures, we show that severe patients have early signals of heart and skeletal muscle tissue damage (Figure 5). Examination of the expression profiles of cells from BAL fluid reveals that the severity-associated proteome is significantly associated with cell-type-specific ACE2 gene expression, implying that direct infection of lung epithelial cells may be driving cell death that is measurable in plasma (Figure 5). Concomitant elevation of epithelial cell markers in our severe COVID-19 patients supports lung epithelial cell damage, although the role of ACE2 in catalyzing this process through direct viral infection remains speculative, particularly given the low proportion of lung epithelial cells that express ACE2.52 Our derived protein signatures correlate with clinical metrics of tissue-specific cellular damage and, by using scRNA-seq data, primarily show gene expression in epithelial cells within the respective tissues, supporting their validity. These plasma tissue-specific damage signatures will have broader utility as liquid biopsies for organ damage and will enhance interpretation of the plasma proteome in settings of tissue-specific cell death and inflammation.
Analysis of the interactions of circulating ligands with receptors within cells in BAL fluid identified a temporal order of cellular communication in the lung associated with disease severity (Figure 6), acknowledging that circulating factors are also produced by tissues besides the lung. In severe patients, we propose that early activation of monocytes/macrophages leads to (1) recruitment of neutrophils, monocytes, dendritic cells (DCs), and T cells; (2) activation and expression of exhaustion markers on T cells; (3) the death of lung epithelial cells; and (4) regeneration and growth factor signaling in lung cells (Figure 7). This model is consistent with the spatial colocalization of macrophages and T cells in autopsy tissue53,54 and ligand/receptor expression patterns in COVID-19 patients with severe versus mild disease derived from single-cell profiles of immune and lung epithelial cells.36,38,55 Many severity-associated proteins were also associated with the nuclear factor κB (NF-κB) pathway, showing substantial overlap with published bronchial and nasopharyngeal cells collected from patients,38,55 genes induced by TNF-α in monocytes in vitro,56 and a TNF-α pathway signature observed by scRNA-seq in COVID-19 severity-associated monocytes.17,57 Few severity-associated proteins were part of the type I IFN response, in agreement with published data4,7 and with the association of COVID-19 severity with genetic variants that weaken IFN-related viral sensing.58 Our proteomic analysis of a large cohort of COVID-19 patients reveals COVID-19 severity- and mortality-associated pathways that may serve as potential therapeutic targets and provide the basis for diagnostics to stratify high-risk patients for tailored therapies and earlier interventions. The proteomic datasets we generated, which are freely available for investigators from Mendeley Data: https://data.mendeley.com/datasets/nf853r8xsj/1, will serve as a valuable resource in COVID-19 discovery.
Limitations of study
First, because it was not feasible to collect a second cohort for the validation of our findings, trends seen here will need to be corroborated in future studies, especially at other institutions. Second, blood collections at later time points were biased toward sicker patients, as they were more likely to remain hospitalized, thus skewing the balance of severity groups over time, affecting the comparison of differentially expressed proteins, and limiting the ability to interpret effect estimate trends. Third, relative contributions to the plasma proteome from circulating immune cells or lung-resident cells were inferred from mapping to published scRNA-seq data from PBMC and BAL datasets, respectively. Whereas consistent patterns of co-expression were observed between our data and published scRNA-seq datasets, because circulating plasma proteins may have multiple sources, confirmation of cell or tissue origin will require validation in individuals in whom plasma proteomics is performed in parallel with scRNA-seq of PBMC and BAL samples. Fourth, the mapping of peripheral plasma proteins onto tissue expression was done using scRNA-seq data from normal, healthy tissues that may not reflect expression profiles in SARS-CoV-2 infection. Fifth, in LMMs, we used significance in the interaction term of severity × time to define our subset of severity-associated proteins; thus, significant association of a protein with severity required a dynamic effect over time, and proteins stably differentially expressed between severity groups over time or at particular time points, may not have been identified as significant. All of the terms of our LMMs are included in Table S1. Patient characteristics by 28-day outcome category in this cohort, related to Figures 1 and S1 (A) Clinical data summary. (B) Subject-level metadata. (C) Annotations, Table S2. List of proteins assayed using the Olink proteomics platform, related to Figures 1, 2, 3, 6, S1–S5, and S7 (A) All Olink proteins assayed. (B) Alphabetical list of proteins included in Olink platform. (C) Protein expression matrix for Olink analysis given as sample ID versus protein levels in normalized protein expression values (NPX), Table S3. Olink models, related to Figures 1, 2, 4, S1, and S4 (A) Linear model with COVID status as a main effect and age, gender, ethnicity, heart disease, diabetes, hypertension, hyperlipidemia, pulmonary disease, kidney disease, immuno-compromised status as covariates. (B) Linear mixed model with severity and time as a main effects and age, gender, ethnicity, heart disease, diabetes, hypertension, hyperlipidemia, pulmonary disease, kidney disease, immunocompromised status as covariates. (C) Linear mixed model with Acuity_max and time as a main effects and age, gender, ethnicity, heart disease, diabetes, hypertension, hyperlipidemia, pulmonary disease, kidney disease, immuno-compromised status as covariates. (D) Protein expression matrix of residual values from a linear model fit to all comorbidities for Olink data given as sample name versus protein (common protein names for each OlinkID is supplied in Table S2). See Method details for derivation, Table S4. Ingenuity Pathway Analysis of severity-associated proteins in Olink assay, related to Figures 2, 6, and S5 (A) Ingenuity Pathway Analysis (QIAGEN) of all Olink severity-associated proteins. (B) Ingenuity Pathway Analysis (QIAGEN) of all Olink severity-associated proteins with upstream analysis, Table S5. Virus neutralization assay data, related to Figure 3, Table S6. SomaScan models of severity, related to Figure 5 Linear mixed model for SomaScan data with severity and time as a main effects and age, gender, ethnicity, heart disease, diabetes, hypertension, hyperlipidemia, pulmonary disease, kidney disease, immunocompromised status as covariates, Table S7. Derived organ-specific intracellular plasma protein signatures, related to Figure 5 (A) GTEx organ-specific proteins that overlap with SomaScan proteins (all) filtered for those that are intracellular. (B) Top differentially expressed genes per lung cell type obtained from the subset of severity-associated intracellular plasma proteins at D0. ACE2 and TMPRSS2 expression indicated by orange and blue circles, respectively. (C) p values for Kaplan-Meier survival analysis using median cut-offs for expression of each organ signature. Lastly, gene set enrichment and pathway analyses may be biased by the preselected set of proteins available on the proteomic platforms used for this study, which have been selected for association with particular diseases and pathways of interest.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| ACK buffer 10x500ml | Quality Biological INC | 118-156-101 |
| CryoStor Cs10 | HemaCare | 210102 |
| Neutrophil isolation kit (StemCell) | STEMCELL | 19666 |
| SepMate 15ml tubes (100/pk) | STEMCELL | 85415 |
| SepMate 50ml tubes (500/cs) | STEMCELL | 85460 |
| TCL buffer 125ml | QIAGEN | 1031576 |
| 96 round bottom plates | WestNet | 3788 |
| Deposited data | ||
| scRNA-sequencing data PBMCs | Wilk et al. (2020)16 COVID-19 atlas | Database: https://www.covid19cellatlas.org/#wilk20 |
| scRNA-sequencing data PBMCs | Lee et al. (2020)8 | GEO accession - GSE149689 |
| scRNA-sequencing data PBMCs | Arunachalam et al. (2020)4 | GEO accession - GSE155673 |
| scRNA-sequencing data PBMCs | Schulte-Schrepping et al. (2020)17 | EGA accession - EGAS00001004571 |
| scRNA-sequencing data BAL | Bost et al. (2020)36 | GEO accession - GSE145926 and GSE149443 |
| scRNA-sequencing data BAL | Chua et al. (2020)38 | Database: https://figshare.com/articles/COVID-19_severity_correlates_with_airway_epithelium-immune_cell_interactions_identified_by_single-cell_analysis/12436517 |
| scRNA-sequencing data heart | Tucker et al. (2020)37 | Broad Institute’s Single Cell Portal study - ID SCP498 |
| scRNA-sequencing data kidney | Menon et al. (2020)59 | GEO accession - GSE140989 |
| scRNA-sequencing data liver | MacParland et al. (2018)60 | GEO accession number - GSE115469 |
| scRNA-sequencing data pancreas | Baron et al. (2016)61 | expression matrix obtained from the Itai Yanai lab |
| Olink proteomic dataset | This study | Mendeley Data: https://data.mendeley.com/datasets/nf853r8xsj/1 |
| SomaScan proteomic dataset | This study | Mendeley Data: https://data.mendeley.com/datasets/nf853r8xsj/1 |
| Experimental models: Cell lines | ||
| 293T ACE2 TMPRSS2 | This paper | N/A |
| Recombinant DNA | ||
| pCMV-SARS2ΔC-gp41 | This paper | N/A |
| psPAX2 | Addgene | RRID: Addgene_12260 |
| pCMV-VSV-G | Addgene | RRID: Addgene_8454 |
| pTRIP-SFFV-GFP-NLS | Addgene | RRID: Addgene_86677 |
| pTRIP-SFFV-Hygro-2A-TMPRSS2 | This paper | N/A |
| Software and algorithms | ||
| FlowJo v10.7.1 | BD | N/A |
| RStudio | Database: https://www.rstudio.com/ | v1.4 |
| R | Database: https://cran.r-project.org/ | v4.0.4 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marcia B. Goldberg (marcia.goldberg@mgh.harvard.edu).
Materials availability
Plasmids generated in this study have been deposited to Addgene: pTRIP-SFFV-Hygro-2A-TMRPSS2, ID 170390, and pCMV-SARS2SΔC-H2gp41, ID 170389. 293T ACE2/TMPRSS2 are available upon request from the Lead Contact without restriction.
Data and code availability
Original proteomic data have been deposited to Mendeley Data: http://dx.doi.org/10.17632/nf853r8xsj. Single-cell RNAseq datasets were obtained as directed in the references for each dataset. All code used for analysis will be available without restriction from the Lead Contact; examples needed to replicate analysis of proteomic data have been deposited to github at https://github.com/arnav-mehta/covid19-proteomics. Original Supplemental Tables data have been deposited to Mendeley Data: http://dx.doi.org/10.17632/nf853r8xsj.
Experimental model and subject details
Patient cohort and clinical data collection
Patients were enrolled in the Emergency Department (ED) of a large, urban, academic hospital in Boston from 3/24/2020 to 4/30/2020 during the peak of a COVID-19 surge. All study procedures involving human subjects were approved by the Mass General Brigham (formerly Partners) Human Research Committee, the governing institutional review board at Massachusetts General Hospital. A waiver of informed consent was approved in compliance with the Code of Federal Regulations (45CFR 46, 2018 Common Rule). Included were patients 18 years or older with a clinical concern upon ED arrival for COVID-19 and with acute respiratory distress, with at least one of the following: 1) tachypnea (22 breaths per minute), 2) oxygen saturation ≤92% on room air, 3) a requirement for supplemental oxygen, or 4) positive-pressure ventilation. The day 0 blood sample (N = 384) was obtained concurrent with the initial clinical blood draw in the ED, and day 3 (N = 217) and day 7 (N = 143) samples were obtained for COVID-19-positive patients, if still hospitalized at those times, yielding 744 samples. In addition, blood was collected from some patients at the time of substantial clinical deterioration (44 samples); these event-driven samples were excluded from linear models. Clinical course was followed to 28 days post-enrollment or until hospital discharge, if that occurred after 28 days.
Patients were classified by acuity levels A1-A5 on days 0, 3, 7, and 28 (WHO Ordinal Outcomes Scale15) where the acuity levels are described as follows: A1, death within 28 days (N = 42, 14%); A2, intubation, mechanical ventilation, and survival to ≥28 days (N = 67, 22%); A3, hospitalized and requiring supplemental oxygen (N = 133, 43%); A4, hospitalized without requiring supplemental oxygen (N = 41, 13%); and A5, discharged directly from the ED without subsequently returning and requiring admission within 28 days (N = 23, 8%). A1 and A2 were classified as severe (N = 109) and A3-A5 as non-severe (N = 197).
Of all 384 enrolled, 78 (20%) tested negative for SARS-CoV-2; among these, for 50 (64%), suspicion for COVID-19 was very low based on careful retrospective chart review by MRF and RPB, an emergency physician and infectious diseases physician, respectively. Among the remaining 28 patients, COVID-19 was a diagnostic possibility, yet most had multiple negative PCR tests during their hospital course. These 78 subjects were categorized as controls. We dichotomized COVID-19 subjects by illness severity and outcome into severe (A1-A2) and less severe (A3-A5) groups. Of the 42 COVID-19 patients who died, 24 (57%) received mechanical ventilation and 18 (43%) did not. The latter group was significantly older, many with advanced directives to withhold aggressive care. Demographic, past medical history and clinical data were collected and summarized for each outcome group, using medians with interquartile ranges and proportions with 95% confidence intervals, where appropriate. Detailed clinical data, including age, gender, ethnicity, and race, are summarized for all outcome cohorts in Table S1. Patient-level clinical data are available from Mendeley Data: https://data.mendeley.com/datasets/nf853r8xsj/1. To protect the identity of individual subjects, public posting of patient-level demographic information is limited as required by the Mass General Brigham Human Research Committee.
Human cell lines
293T ACE2/TMPRSS2 were derived from 293T, a kidney cell line. Culture methods and transductions are detailed in the paragraph “Measurement of neutralization levels.” Cells were regularly tested for mycoplasma contamination.
Method details
Plasma collection and processing
Blood samples were collected in EDTA tubes and processed no more than 3 hours post blood draw in a Biosafety Level 2+ laboratory on site. Whole blood was diluted with room temperature RPMI medium in a 1:2 ratio to facilitate cell separation for other analyses using the SepMate PBMC isolation tubes (STEMCELL) containing 16 mL Ficoll (GE Healthcare). Diluted whole blood was centrifuged at 1200 g for 20 minutes at 20°C. After centrifugation, plasma (5 mL) was pipetted into 15 mL conical tubes and placed on ice during PBMC separation procedures, centrifuged at 1000 g for 5 min at 4°C, aliquoted into cryovials, and stored at −80°C. Study samples (45 μL) were randomly allocated onto 96-well plates based on disease outcome grouping and were treated with 1% Triton X-100 for virus inactivation at room temperature for 2 hr.
Olink plasma proteomic assays
The Olink Proximity Extension Assay (PEA) is a technology developed for high-multiplex analysis of proteins using 1 μL of sample. In PEA, oligonucleotide-labeled monoclonal or polyclonal antibodies (PEA probes) are used to bind target proteins in a pairwise manner thereby preventing all cross-reactive events. Upon binding, the oligonucleotides come in close proximity and hybridize followed by extension generating a unique sequence used for digital identification of the specific protein assay. With recent developments, PEA enables an increased number of 384 multiplex assays and higher throughput using next-generation sequencing (NGS) as a readout method. PEA probe design is based on addition of Illumina adaptor sequences, unique barcodes for protein identification and indexes to distinguish samples in multiplex sequencing. The protocol has also been miniaturized and automated using liquid handlers to further improve robustness and maximize output.
The full library (Olink® Explore 1536) consists of 1472 proteins and 48 controls assays divided into four 384-plex panels focused on inflammation, oncology, cardiometabolic and neurology proteins. In each of the four 384-plex panels, overlapping assays of IL6, IL8 (CXCL8), and TNF are included for quality control (QC) purposes. Library content is based on target selection of low-abundant inflammation proteins, actively secreted proteins, organ-specific proteins leaked into circulation, drug targets (established and from ongoing clinical trials), and proteins detected in blood by mass spectrometry. Selection, classification, and categorization of proteins were based on using various databases (e.g., Gene Ontology), the Blood Atlas – the human secretome (Database: www.proteinatlas.org), a collaboration with the Institute of Systems Biology, Seattle WA, for tissue-specific proteins, Database: https://www.clinicaltrials.gov for mapping of drug targets, detection of proteins in blood measured by mass spectrometry and finally, various text-mining approaches identifying protein biomarkers described in the literature. The analytical performance of PEA is carefully validated for each protein assay; performance data are available at Database: https://www.olink.com. Technical criteria include assessing sensitivity, dynamic range, specificity, precision, scalability, endogenous interference, and detectability in healthy and pathological plasma and serum samples.
In the immune reaction, 2.8 μL of sample is mixed with PEA probes and incubated overnight at 4°C. Then, a combined extension and pre-amplification mix is added to the incubated samples at room temperature for PCR. The PCR products are pooled before a second PCR step following addition of individual sample index sequences. All samples are thereafter pooled, followed by bead purification and QC of the generated libraries on a Bioanalyzer. Finally, sequencing is performed on a NovaSeq 6000 system using two S1 flow cells with 2 × 50 base read lengths. Counts of known sequences are thereafter translated into normalized protein expression (NPX) units through a QC and normalization process developed and provided by Olink.
Quality control, Olink plasma proteomics
The Olink PEA QC process consists of specifically engineered controls to monitor the performance of the main steps of the assays (immunoreaction, extension and amplification/detection) as well as the individual samples. Internal controls are spiked into each sample and represent a control using a non-human assay, an extension control composed of an antibody coupled to a unique DNA-pair always in proximity and, finally, a detection control based on a double stranded DNA amplicon. In addition, each plate run with Olink includes a control strip with sample controls used to estimate precision (intra- and inter-coefficient of variation). A negative control (buffer) run in triplicate is utilized to set background levels and calculate limit of detection (LOD), a plate control (plasma pool) is run in triplicate to adjust levels between plates, and a sample control (reference plasma) is included in duplicate to estimate CV between runs.
NPX is Olink’s relative protein quantification unit on a log2 scale and values are calculated from the number of matched counts on the NovaSeq run. Data generation of NPX consists of normalization to the extension control (known standard), log2-transformation, and level adjustment using the plate control (plasma sample).
SomaScan plasma proteomic assays
The SomaScan Platform for proteomic profiling uses 4979 SOMAmer reagents, single-stranded DNA aptamers, to 4776 unique human protein targets. The modified aptamer binding reagents,35 SomaScan assay,35,62 its performance characteristics,63,64 and specificity65,66 to human targets have been previously described. The assay used standard controls, including 12 hybridization normalization control sequences to control for variability in the Agilent readout process and 5 human calibrator control pooled replicates and 3 quality control pooled replicates to mitigate batch effects and verify the quality of the assay run using standard acceptance criteria.
Quality control, SomaScan plasma proteomics
The SomaScan Assay is run using 96-well plates; 11 wells are allocated for control samples used to control for batch effects and to estimate the accuracy, precision, and buffer background of the assay over time. Five pooled Calibrator replicates, three pooled QC replicates, and three buffer replicates are run on every plate. The readout is performed using Agilent hybridization, scan, and feature extraction technology. Twelve Hybridization Control SOMAmers are added alongside SOMAmers to be measured from the biological samples and controls of each well during the SOMAmer elution step to control for readout variability. The control samples are run repeatedly during assay qualification and robust point estimates are generated and stored as references for each SOMAmer result for the Calibrator and QC samples. The results are used as references throughout the life of the SOMAscan V4 Assay. Plate Calibration is performed by calculating the ratio of the Calibrator Reference RFU value to the plate-specific Calibrator replicate median RFU value for each SOMAmer. The resulting ratio distribution is decomposed into a Plate Scale factor defined by the median of the distribution and a vector of SOMAmer-specific Calibration Scale Factors. Normalization of QC replicates and samples is performed using adaptive normalization by maximum likelihood (ANML) with point and variance estimates from a normal U.S. population. Post calibration accuracy is estimated using the ratio of the QC reference RFU value to the plate-specific QC replicate median RFU value for each SOMAmer. The resulting QC ratio distribution provides a robust estimate of accuracy for each SOMAmer on every plate. Plate-specific Acceptance Criteria: Plate Scale Factor between 0.4-2.5 and 85% of QC ratios between 0.8 and 1.2 must be met prior to release.
Measurement of neutralization levels
Constructs
SARS-CoV-2 S was amplified by PCR (Q5 High-Fidelity 2X Master Mix, New England Biolabs) from pUC57-nCoV-S (gift of Jonathan Abraham), in which the C-terminal 27 amino acids of SARS-CoV-2 S are replaced by the NRVRQGYS sequence of HIV-1, a strategy previously described for retroviruses pseudotyped with SARS-CoV S.67 The truncated SARS-CoV-2 S fused to gp41 was cloned into pCMV by Gibson assembly to obtain pCMV-SARS2ΔC-gp41. psPAX2 and pCMV-VSV-G were previously described.68 pTRIP-SFFV-EGFP-NLS was previously described69 (Addgene plasmid #86677). cDNA for human TMPRSS2 and the hygromycin resistance gene were generated by synthesis (Integrated DNA Technologies). pTRIP-SFFV-Hygro-2A-TMPRSS2 was generated by Gibson assembly.
Cell culture
293T cells were cultured in DMEM, 10% FBS (ThermoFisher Scientific), and PenStrep (ThermoFisher Scientific). 293T ACE2 cells (gift of Michael Farzan) were transduced with pTRIP-SFFV-Hygro-TMPRSS2 using TransIT®-293 Transfection Reagent (Mirus Bio, MIR 2700) to obtain 293T ACE2/TMPRSS2 cells, which were selected with 320 μg/ml of hygromycin (Invivogen) and used as a target in pseudotyped SARS-CoV-2 S lentivirus neutralization assays.
Pseudotyped SARS-CoV-2 lentivirus production
The protocol for lentiviral production was previously described.68 Briefly, 293T cells were seeded at 0.8 × 106 cells per well in a 6-well plate and were transfected the same day with a mix of DNA containing 1 μg psPAX, 1.6 μg pTRIP-SFFV-EGFP-NLS, and 0.4 μg pCMV-SARS2ΔC-gp41 using TransIT®-293 Transfection Reagent. After overnight incubation, the medium was changed. SARS-CoV-2 S pseudotyped lentiviral particles were collected 30-34 hr post medium exchange and filtered using a 0.45 μm syringe filter. To transduce 293T ACE2 cells, the same protocol was followed, with a mix containing 1 μg psPAX, 1.6 μg pTRIP-SFFV-Hygro-2A-TMPRSS2, and 0.4 μg pCMV-VSV-G.
SARS-CoV-2 S antibody neutralization assay
The day before the experiment, 293T ACE2/TMPRSS2 cells were seeded at 5 × 103 cells in 100 μl per well in 96-well plates. On the day of lentiviral harvest, 100 μl SARS-CoV-2 S pseudotyped lentivirus was incubated with 50 μl of plasma diluted in medium to a final concentration of 1:100. Medium was then removed from 293T ACE2/TMPRSS2 cells and replaced with 150 μl of the mix of plasma and pseudotyped lentivirus. Wells in the outermost rows of the 96-well plate were excluded from the assay. After overnight incubation, medium was changed to 100 μl of fresh medium. Cells were harvested 40-44 hr post infection with TrypLE (Thermo Fisher), washed in medium, and fixed in FACS buffer containing 1% PFA (Electron Microscopy Sciences). Percentage GFP was quantified on a Cytoflex LX (Beckman Coulter), and data was analyzed with FlowJo.
Quantification and statistical analysis
Data analysis and visualization
All statistical analyses for the clinical and proteomics data in this cohort was performed using R version 4.0.2. All plots were generated using the ggplot2 package in R with the exception that the correlation plots were generated using the corrplot() function in R. Pairwise Pearson correlations were calculated for all proteins, and rows and columns of correlation plots were ordered based on hierarchical clustering. All heatmaps were generated using the heatmap370 package and NPX values for each protein centered to have a mean of 0 and scaled to have a standard deviation of 1 within each protein. Scaled data greater than either 4 or 5 standard deviations from the mean were truncated at ± 4 or 5. Rows and columns were ordered based on hierarchical clustering.
Unsupervised clustering
Principal components analysis (PCA) was performed using all proteins and all samples using the prcomp() function in R. Unsupervised clustering by UMAP was performed using all proteins, and either all samples or just day 0 samples, using the umap() function in R, and UMAP coordinates were plotted using the ggplot2 package. Unsupervised clustering by tSNE was by first performing dimensionality reduction by PCA and then taking the top principal components for a tSNE embedding using the Rtsne package and the argument pca = TRUE. k-nearest neighbor (KNN) graphs and Louvain community detection was performed using custom code and the FNN package provided in R.
Linear models
Linear regression models were fit independently to each protein using the lm package in R with protein values (NPX for Olink data) as the dependent variable. The models included a term for COVID-19 status and covariates for age, gender, ethnicity, heart disease, diabetes, hypertension, hyperlipidemia, pulmonary disease, kidney disease, immuno-compromised status to control for any potential confounding. P values were adjusted to control the false discovery rate (FDR) at 5% using the Benjamini-Hochberg method implemented in the emmeans package in R.
Linear mixed models
Linear mixed effects models (LMMs) were fit independently to each protein using the lme469 package in R with protein values (NPX for Olink data) as the dependent variable. The model for severity included a main effect of time, a main effect of severity, the interaction between these two terms, and a random effect of patient ID to account for the correlation between samples coming from the same patient. Covariates for age, gender, ethnicity, heart disease, diabetes, hypertension, hyperlipidemia, pulmonary disease, kidney disease, and immuno-compromised status were included in the model to control for any potential confounding. Significance of the three model terms was determined with an F-test using Satterthwaite degrees of freedom and type III sum of squares implemented with the lmerTest71 package in R. P values for the three model terms of interest were adjusted to control the FDR at 5% using the Benjamini-Hochberg method. Group differences were calculated for each protein passing the FDR threshold with p values adjusted using the Tukey method implemented by the emmeans package in R. Group differences with Tukey adjusted p values less than 0.05 were considered statistically significant. Note, all other models were run similarly with time in addition to either Acuitymax, age, or both age and severity as main effects instead of severity.
For SomaScan data, LMMs for severity and time as main effects were run as was done for Olink. Overall, significant proteins were found to be partially overlapping with those found for Olink (hypergeometric test p = 0.002) (Tables S3B and S6); for example, at D0, of the 1085 overlapping assays between the two platforms, 779 proteins were significant for severity or interaction term in Olink data, and 669 in the SomaScan data, with 460 proteins overlapping between the two sets. In other words, 69% of the SomaScan severity-associated proteins overlapped with those identified by Olink data. The non-overlapping assays in part due to a narrower dynamic range for some of the SomaScan assays.
Residuals
Model residual values were extracted from LMMs (as described above) independently fit to every protein using NPX as the dependent variable, age, gender, ethnicity, heart disease, diabetes, hypertension, hyperlipidemia, pulmonary disease, kidney disease, and immuno-compromised status as covariates and a random effect of patient ID to account for the correlation between samples taken from the same patient. These residuals represent the remaining unexplained variance in the protein expression after accounting for the effects of the included covariates.
Permutation controls
For the Olink assay, the likelihood of observing 1131 statistically significant proteins for the Acuitymax model term and 963 statistically significant proteins for the time and Acuitymax interaction term from the linear mixed models was evaluated using permutation testing. Acuitymax group was randomly permuted 100 times among patients and for each permutation the full LMM procedure was followed. None of the permutations produced as many statistically significant results as were observed when using the true Acuitymax groupings.
Gene set enrichment and pathway analysis
For analysis of functional pathways, two different strategies were employed: (i) gene set enrichment analysis72 using the ClusterProfiler package in R using the C7 immunologic signature gene set from the molecular signatures database v7.2 (Database: https://www.gsea-msigdb.org/gsea/msigdb); and (ii) Ingenuity Pathway Analysis (QIAGEN) on our gene lists using default parameters from the vendor. Pathways were visualized in dot plots and bar plots using the ggplot2 package in R.
Prediction of severity
Predictive performance of severity within 28 days was performed using all proteins and model covariates and was estimated using elastic net logistic regression implemented by the glmnet73 package in R and 100 repeats of 5-fold cross validation. Model tuning was performed using the caret package in R. Variable scaling, model tuning, and feature selection was performed independently for each held-out fold such that the predictive model was never exposed to the held-out data. Measures of predictive performance are reported as medians and 95% confidence intervals calculated from the 100 repeats of the cross validation. Features were ranked by how frequently they were chosen to be included in the model.
Prediction of neutralization level
Generalized linear models with lasso regularization were trained (using the R caret package) on COVID-19-positive patient proteome samples (consisting of 1472 Olink protein features) from each selected day (0, 3, and 7) to neutralization levels (≤ or > 75%). For percent neutralization predictions, protein levels at day 0 were used to predict binned neutralization categories at day 3. Repeated 5-fold cross validation (with a hyperparameter scan from 0.0001 to 1 to select the lambda constant yielding the greatest prediction accuracy) was replicated 100 times to obtain a confidence interval for the area under the ROC curve (where ROC curves were generated using each patient’s estimated probability while serving as the held-out fold). The average feature weights of the final models from each of the 100 rounds of 5-fold cross validation were used to identify proteins of importance. Orthogonally, 10-fold cross validation was used to train and validate a random forest model (with default ntree = 500 and mtry = 38) to predict neutralization quartiles (0%–25%, 25%–50%, 50%–75%, 75%–100%) and important proteins were identified based on mean decrease in Gini. To identify protein features that were independent from or overlapping with severity markers, the union of the top 50 important features from the lasso and random forest models were intersected with significantly variable proteins between severity groups on day 0 (from the LMM described above).
scRNA-sequencing data processing and analysis
We analyzed 4 publicly available scRNA-seq PBMC datasets from COVID-19 patients, which were obtained from: 1) Wilk et al., 2020,16 COVID-19 atlas, Database: https://www.covid19cellatlas.org/#wilk20; 2) Lee et al., 2020,8 GEO: GSE149689; 3) Arunachalam et al., 2020,4 GEO: GSE155673; and 4) Schulte-Schrepping et al., 2020,17 EGA accession EGAS00001004571. Gene expression matrices after filtering low quality cells were used as provided by the respective investigators, and annotations were used as described in each of the studies. scRNA-seq data from BAL fluid and lower airways of COVID-19 patients were obtained from 1) Bost et al., 2020,36 GEO: GSE145926 and GEO: GSE149443 and 2) Chua et al., 2020,38 FigShare Database: https://figshare.com/articles/COVID-19_severity_correlates_with_airway_epithelium-immune_cell_interactions_identified_by_single-cell_analysis/12436517. Cell-type specific expression in lung tissue was derived as described below. scRNA-seq data from other tissues were obtained from the following sources: 1) heart from Tucker et al., 2020,37 Broad Institute’s Single Cell Portal study ID SCP498; 2) kidney from Menon et al., 2020,59 GEO: GSE140989; 3) liver from MacParland et al., 2018,60 GEO: GSE115469; and 4) pancreas from Baron et al., 2016,61 expression matrix obtained from the Itai Yanai lab.
Expression data generation, lung cell subsets
To generate lung cell-type specific signatures, we collected and aggregated scRNA-seq studies, normalized each dataset, harmonized the published cell type annotations, and trained a multiclass logistic regression model.
Dataset selection: Only studies with scRNA-seq data from primary tissue (including healthy, fibrotic, and COVID-19 donors), sequenced using the 10X Genomics platform, and published annotations were included. Two additional studies (chosen to maximize the number of cell types in the test set) were held out for cross validation to test cell type predictions and tune hyperparameters. The training datasets were Adams et al., 2020,74 Chua et al., 2020,38 Habermann et al., 2020,75 Travaglini et al., 2020,76 and two unpublished datasets. The test datasets used were Vieira et al., 201977 and Laio et al., 2020.78
Normalization: Single cell/nuclei RNA-seq datasets from individual studies were aggregated and normalized using Scanpy.79 Each study was subjected to identical pre-processing steps. First, UMI count values were winsorized, those above the 99th percentile of non-zero counts were reduced to the value of the 99th percentile (13 counts). Winsorized count data were normalized, so that UMI counts per cell/nucleus summed to 10,000, and then were logged, resulting in log(1+10,000∗UMIs / total UMIs) for each cell/nuclei (“logtp10k”). Then the aggregated expression data were scaled using the scanpy `scale` function with zero_center = False. To prepare cell type labels, we mapped each annotation to a common reference list before training. Cells labeled with cell types with ambiguous mappings (e.g., “T cell” or “myeloid”) were excluded from training.
Signature extraction: Cell type signatures were learned using an L2 penalized logistic regression model trained to predict the cell type from a single cell gene expression profile. The model was trained using SciKitLearn’s LogisticRegression function with the default parameters with the exception of C = 0.1, max_iter = 30, and multi_class = ‘ovr’. During fitting, individual cells were weighted to balance with respect to both cell type and study. Model coefficients learned were used as cell type signatures.
Analysis of scRNA-seq data
All scRNA-seq gene expression data was analyzed in R version 4.0.2 using custom code to look at average expression of genes of interest in each cell type. Genes of interest were selected from the proteomic analysis, and the tissue distribution of these genes (or groups of genes) were evaluated in the different scRNA-seq datasets. For visualization, gene expression was normalized across cell types (rows) with Z-scores and visualized in heatmaps using the heatmap3 function in R with hierarchical clustering of both cell types and genes. Where cell types were annotated on heatmaps, this was done by identifying cell types with the highest relative expression by Z-scores. The cell-type-specific intracellular gene list was defined as the top 20 genes with the highest relative expression for that cell type.
Derivation tissue-specific protein signatures
Organ specific protein signatures were defined using RNA sequencing data from the Genotype-Tissue Expression (GTEx) Portal (Database: https://www.gtexportal.org/home/). The median transcripts per million (TPM) of 56,200 genes across 54 non-diseased tissue sites were obtained. For each tissue site, the intersection of the top 500 highest TPM genes and the top 500 most variable genes (based on coefficient of variation across tissue types) was identified. Proteins that were also measured by SomaScan were extracted, validated for high tissue specific expression, and consolidated across related tissues for each organ of interest. Organ signatures were split based on localization (intracellular versus membrane/secreted) using UniProt and literature annotations. The values for each protein across all COVID-19-positive patients were scaled to Z scores, and the mean Z score of all proteins in an organ set was used as an overall signature score for a given patient.
Ligand-receptor analysis
Single-cell RNA-seq expression profiles (10X genomics) of immune cells isolated from BAL fluid of healthy and COVID-19-infected patients of varying severity from Bost et al., 202036 was obtained from GEO: GSE145926 and GEO: GSE149443. Python 3.8 was used to run the python package Cellphonedb v2.1.4 with the following parameters: database v2.0.0, statistical method analysis, 1000 iterations, 6000 cell subsampling. The metadata cluster identities were previously assigned based on the published annotations. Analysis of specific ligands and receptors was performed from a curated list of known ligand-receptor pairs, and cell types were assigned to particular ligands and receptors by identifying cell types with the highest relative expression by Z-scores.
Acknowledgments
We owe deep gratitude to the study participants, Translational and Clinical Research Center (TCRC), and nursing staff, in particular Grace Holland, RN, Katherine Broderick, RN, and Siobhan Boyce, RN, and Kathryn Hall, NP, for sample collection. We thank the Massachusetts General Hospital for institutional support to enable enrollment when access to clinical spaces was limited. We thank the Departments of Emergency Medicine and Medicine for maintaining needed staffing levels during enrollment, when many research funding sources were suspended. We thank Caroline Beakes and Nicole Russell for assistance with data entry, and Jayaraj Rajagopal, Itai Yanai, Patrick Ellinor, and Mark Chaffin for access to processed Single-cell RNA-sequencing datasets. We thank Arthur, Sandra, and Sarah Irving for a gift that enabled this study and funded the David P. Ryan, MD Endowed Chair in Cancer Research (to N.H.). We are grateful for the generous contributions of Olink Proteomics and Novartis (in collaboration with SomaLogic) for providing in-kind proteomics assays. We acknowledge the following funding sources: B.L., Cystic Fibrosis Foundation Postdoctoral Fellowship, LIN19F0; N.H., NIH/NIAID U19 AI082630, Chair and gift from Sandra, Sarah, and Arthur Irving; M.B.G., M.R.F., and N.H., American Lung Association (COVID-19 Action Initiative); M.B.G. and M.R.F., Executive Committee on Research at MGH; A.-C.V., Chan Zuckerberg Initiative; and Harvard Catalyst/Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, National Institutes of Health Awards UL1 TR 001102 and UL1 TR 002541-01).
Author contributions
Conceptualization, M.R.F., A.M., N.H., and M.B.G.; resources, M.R.F., N.H., M.B.G., I. Grundberg, and L.L.J.; methodology, A.M., N.H., M.R.F., M.B.G., M.S.-F., A.-C.V., B.A.P., R.P.B., L.L.J., I. Grundberg, and R.E.G.; investigation, all of the authors; formal analysis, A.M., A.M.S., J.R.G., M.G., B.G.F., and B.L.; writing – original draft, A.M., M.R.F., N.H., and M.B.G.; writing – review & editing, M.R.F., A.M., N.H., M.B.G., A.M.S., R.E.G., B.G.F., I. Gushterova, L.L.J., A.-C.V., M.S.-F., A.S.Z., T.E.W., B.C.R., R.P.B., and B.A.P.; supervision, M.R.F., A.M., N.H., and M.B.G.
Declaration of interests
A.M. is a consultant for Third Rock Ventures. J.R.G. and I. Gushterova are employees of Olink Proteomics. G.S.H. is an employee of Genentech (as of November 2020). L.L.J. is an employee and stockholder of Novartis. N.H. holds equity in BioNTech and is a consultant for Related Sciences.
Inclusion and diversity
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Published: April 30, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100287.
Contributor Information
Michael R. Filbin, Email: mfilbin@mgh.harvard.edu.
Arnav Mehta, Email: amehta@broadinstitute.org.
Nir Hacohen, Email: nhacohen@mgh.harvard.edu.
Marcia B. Goldberg, Email: marcia.goldberg@mgh.harvard.edu.
Supplemental information
References
- 1.Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D’Andrea K., UPenn COVID Processing Unit Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369:eabc8511. doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lucas C., Wong P., Klein J., Castro T.B.R., Silva J., Sundaram M., Ellingson M.K., Mao T., Oh J.E., Israelow B., Yale IMPACT Team Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Del Valle D.M., Kim-Schulze S., Huang H.H., Beckmann N.D., Nirenberg S., Wang B., Lavin Y., Swartz T.H., Madduri D., Stock A. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020;26:1636–1643. doi: 10.1038/s41591-020-1051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arunachalam P.S., Wimmers F., Mok C.K.P., Perera R.A.P.M., Scott M., Hagan T., Sigal N., Feng Y., Bristow L., Tak-Yin Tsang O. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science. 2020;369:1210–1220. doi: 10.1126/science.abc6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williamson E.J., Walker A.J., Bhaskaran K., Bacon S., Bates C., Morton C.E., Curtis H.J., Mehrkar A., Evans D., Inglesby P. Factors associated with COVID-19-related death using OpenSAFELY. Nature. 2020;584:430–436. doi: 10.1038/s41586-020-2521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuri-Cervantes L., Pampena M.B., Meng W., Rosenfeld A.M., Ittner C.A.G., Weisman A.R., Agyekum R.S., Mathew D., Baxter A.E., Vella L.A. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020;5:eabd7114. doi: 10.1126/sciimmunol.abd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee J.S., Park S., Jeong H.W., Ahn J.Y., Choi S.J., Lee H., Choi B., Nam S.K., Sa M., Kwon J.S. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. 2020;5:eabd1554. doi: 10.1126/sciimmunol.abd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodriguez L., Pekkarinen P.T., Lakshmikanth T., Tan Z., Consiglio C.R., Pou C., Chen Y., Mugabo C.H., Nguyen N.A., Nowlan K. Systems-Level Immunomonitoring from Acute to Recovery Phase of Severe COVID-19. Cell Rep. Med. 2020;1:100078. doi: 10.1016/j.xcrm.2020.100078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long Q.X., Tang X.J., Shi Q.L., Li Q., Deng H.J., Yuan J., Hu J.L., Xu W., Zhang Y., Lv F.J. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat. Med. 2020;26:1200–1204. doi: 10.1038/s41591-020-0965-6. [DOI] [PubMed] [Google Scholar]
- 11.Messner C.B., Demichev V., Wendisch D., Michalick L., White M., Freiwald A., Textoris-Taube K., Vernardis S.I., Egger A.S., Kreidl M. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst. 2020;11:11–24.e4. doi: 10.1016/j.cels.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson J.G., Simpson L.J., Ferreira A.M., Rustagi A., Roque J., Asuni A., Ranganath T., Grant P.M., Subramanian A., Rosenberg-Hasson Y. Cytokine profile in plasma of severe COVID-19 does not differ from ARDS and sepsis. JCI Insight. 2020;5:e140289. doi: 10.1172/jci.insight.140289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen B., Yi X., Sun Y., Bi X., Du J., Zhang C., Quan S., Zhang F., Sun R., Qian L. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell. 2020;182:59–72.e15. doi: 10.1016/j.cell.2020.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su Y., Chen D., Yuan D., Lausted C., Choi J., Dai C.L., Voillet V., Duvvuri V.R., Scherler K., Troisch P., ISB-Swedish COVID19 Biobanking Unit Multi-Omics Resolves a Sharp Disease-State Shift between Mild and Moderate COVID-19. Cell. 2020;183:1479–1495.e20. doi: 10.1016/j.cell.2020.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.World Health Organization . 2020. WHO R&D Blueprint. Novel Coronavirus. COVID-19 Therapeutic Trial Synopsis.https://cdn.who.int/media/docs/default-source/blue-print/covid-19-therapeutic-trial-synopsis.pdf?sfvrsn=44b83344_1&download=true [Google Scholar]
- 16.Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020;26:1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., Deutsche COVID-19 OMICS Initiative (DeCOI) Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prigent P., El Mir S., Dréano M., Triebel F. Lymphocyte activation gene-3 induces tumor regression and antitumor immune responses. Eur. J. Immunol. 1999;29:3867–3876. doi: 10.1002/(SICI)1521-4141(199912)29:12<3867::AID-IMMU3867>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 19.Casati C., Camisaschi C., Rini F., Arienti F., Rivoltini L., Triebel F., Parmiani G., Castelli C. Soluble human LAG-3 molecule amplifies the in vitro generation of type 1 tumor-specific immunity. Cancer Res. 2006;66:4450–4460. doi: 10.1158/0008-5472.CAN-05-2728. [DOI] [PubMed] [Google Scholar]
- 20.Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu C., Chen X., Cai Y., Xia J., Zhou X., Xu S., Huang H., Zhang L., Zhou X., Du C. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020;180:934–943. doi: 10.1001/jamainternmed.2020.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu F., Liu M., Wang A., Lu L., Wang Q., Gu C., Chen J., Wu Y., Xia S., Ling Y. Evaluating the Association of Clinical Characteristics With Neutralizing Antibody Levels in Patients Who Have Recovered From Mild COVID-19 in Shanghai, China. JAMA Intern. Med. 2020;180:1356–1362. doi: 10.1001/jamainternmed.2020.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stern J.B., Paugam C., Validire P., Adle-Biassette H., Jaffré S., Dehoux M., Crestani B. Cytokeratin 19 fragments in patients with acute lung injury: a preliminary observation. Intensive Care Med. 2006;32:910–914. doi: 10.1007/s00134-006-0124-7. [DOI] [PubMed] [Google Scholar]
- 24.Potting C., Tatsuta T., König T., Haag M., Wai T., Aaltonen M.J., Langer T. TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab. 2013;18:287–295. doi: 10.1016/j.cmet.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 25.Fraser D.D., Cepinskas G., Patterson E.K., Slessarev M., Martin C., Daley M., Patel M.A., Miller M.R., O’Gorman D.B., Gill S.E. Novel Outcome Biomarkers Identified With Targeted Proteomic Analyses of Plasma From Critically Ill Coronavirus Disease 2019 Patients. Crit. Care Explor. 2020;2:e0189. doi: 10.1097/CCE.0000000000000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laing A.G., Lorenc A., Del Molino Del Barrio I., Das A., Fish M., Monin L., Muñoz-Ruiz M., McKenzie D.R., Hayday T.S., Francos-Quijorna I. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020;26:1623–1635. doi: 10.1038/s41591-020-1038-6. [DOI] [PubMed] [Google Scholar]
- 27.Rydyznski Moderbacher C., Ramirez S.I., Dan J.M., Grifoni A., Hastie K.M., Weiskopf D., Belanger S., Abbott R.K., Kim C., Choi J. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell. 2020;183:996–1012.e19. doi: 10.1016/j.cell.2020.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suthar M.S., Zimmerman M.G., Kauffman R.C., Mantus G., Linderman S.L., Hudson W.H., Vanderheiden A., Nyhoff L., Davis C.W., Adekunle O. Rapid Generation of Neutralizing Antibody Responses in COVID-19 Patients. Cell Rep. Med. 2020;1:100040. doi: 10.1016/j.xcrm.2020.100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cuadrado E., van den Biggelaar M., de Kivit S., Chen Y.Y., Slot M., Doubal I., Meijer A., van Lier R.A.W., Borst J., Amsen D. Proteomic Analyses of Human Regulatory T Cells Reveal Adaptations in Signaling Pathways that Protect Cellular Identity. Immunity. 2018;48:1046–1059.e6. doi: 10.1016/j.immuni.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 30.Jancsó Z., Hegyi E., Sahin-Tóth M. Chymotrypsin Reduces the Severity of Secretagogue-Induced Pancreatitis in Mice. Gastroenterology. 2018;155:1017–1021. doi: 10.1053/j.gastro.2018.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bär F., Föh B., Pagel R., Schröder T., Schlichting H., Hirose M., Lemcke S., Klinger A., König P., Karsten C.M. Carboxypeptidase E modulates intestinal immune homeostasis and protects against experimental colitis in mice. PLoS ONE. 2014;9:e102347. doi: 10.1371/journal.pone.0102347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leung L.L., Myles T., Nishimura T., Song J.J., Robinson W.H. Regulation of tissue inflammation by thrombin-activatable carboxypeptidase B (or TAFI) Mol. Immunol. 2008;45:4080–4083. doi: 10.1016/j.molimm.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geisz A., Jancsó Z., Németh B.C., Hegyi E., Sahin-Tóth M. Natural single-nucleotide deletion in chymotrypsinogen C gene increases severity of secretagogue-induced pancreatitis in C57BL/6 mice. JCI Insight. 2019;4:e129717. doi: 10.1172/jci.insight.129717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferraro N.M., Strober B.J., Einson J., Abell N.S., Aguet F., Barbeira A.N., Brandt M., Bucan M., Castel S.E., Davis J.R., TOPMed Lipids Working Group. GTEx Consortium Transcriptomic signatures across human tissues identify functional rare genetic variation. Science. 2020;369:eaaz5900. doi: 10.1126/science.aaz5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rohloff J.C., Gelinas A.D., Jarvis T.C., Ochsner U.A., Schneider D.J., Gold L., Janjic N. Nucleic Acid Ligands With Protein-like Side Chains: Modified Aptamers and Their Use as Diagnostic and Therapeutic Agents. Mol. Ther. Nucleic Acids. 2014;3:e201. doi: 10.1038/mtna.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bost P., Giladi A., Liu Y., Bendjelal Y., Xu G., David E., Blecher-Gonen R., Cohen M., Medaglia C., Li H. Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients. Cell. 2020;181:1475–1488.e12. doi: 10.1016/j.cell.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tucker N.R., Chaffin M., Fleming S.J., Hall A.W., Parsons V.A., Bedi K.C., Jr., Akkad A.D., Herndon C.N., Arduini A., Papangeli I. Transcriptional and Cellular Diversity of the Human Heart. Circulation. 2020;142:466–482. doi: 10.1161/CIRCULATIONAHA.119.045401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chua R.L., Lukassen S., Trump S., Hennig B.P., Wendisch D., Pott F., Debnath O., Thürmann L., Kurth F., Völker M.T. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020;38:970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- 39.Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 40.Sandersa N.L., Venkateshaiah S.U., Manohar M., Verma A.K., Kandikattu H.K., Mishra A. Interleukin-18 has an Important Role in Differentiation and Maturation of Mucosal Mast Cells. J. Mucosal Immunol. Res. 2018;2:109. [PMC free article] [PubMed] [Google Scholar]
- 41.McLeod J.J., Baker B., Ryan J.J. Mast cell production and response to IL-4 and IL-13. Cytokine. 2015;75:57–61. doi: 10.1016/j.cyto.2015.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rathore A.P., St John A.L. Protective and pathogenic roles for mast cells during viral infections. Curr. Opin. Immunol. 2020;66:74–81. doi: 10.1016/j.coi.2020.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boesiger J., Tsai M., Maurer M., Yamaguchi M., Brown L.F., Claffey K.P., Dvorak H.F., Galli S.J. Mast cells can secrete vascular permeability factor/ vascular endothelial cell growth factor and exhibit enhanced release after immunoglobulin E-dependent upregulation of fc epsilon receptor I expression. J. Exp. Med. 1998;188:1135–1145. doi: 10.1084/jem.188.6.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou T., Damsky W., Weizman O.E., McGeary M.K., Hartmann K.P., Rosen C.E., Fischer S., Jackson R., Flavell R.A., Wang J. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature. 2020;583:609–614. doi: 10.1038/s41586-020-2422-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lang K.S., Recher M., Navarini A.A., Harris N.L., Löhning M., Junt T., Probst H.C., Hengartner H., Zinkernagel R.M. Inverse correlation between IL-7 receptor expression and CD8 T cell exhaustion during persistent antigen stimulation. Eur. J. Immunol. 2005;35:738–745. doi: 10.1002/eji.200425828. [DOI] [PubMed] [Google Scholar]
- 46.Dong X., Zhao B., Iacob R.E., Zhu J., Koksal A.C., Lu C., Engen J.R., Springer T.A. Force interacts with macromolecular structure in activation of TGF-β. Nature. 2017;542:55–59. doi: 10.1038/nature21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang R., Zhu J., Dong X., Shi M., Lu C., Springer T.A. GARP regulates the bioavailability and activation of TGFβ. Mol. Biol. Cell. 2012;23:1129–1139. doi: 10.1091/mbc.E11-12-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanjabi S., Oh S.A., Li M.O. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017;9:a022236. doi: 10.1101/cshperspect.a022236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang C., Wu M., Zhang L., Shang L.R., Fang J.H., Zhuang S.M. Fibrotic microenvironment promotes the metastatic seeding of tumor cells via activating the fibronectin 1/secreted phosphoprotein 1-integrin signaling. Oncotarget. 2016;7:45702–45714. doi: 10.18632/oncotarget.10157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gisby J., Clarke C.L., Medjeral-Thomas N., Malik T.H., Papadaki A., Mortimer P.M., Buang N.B., Lewis S., Pereira M., Toulza F. Longitudinal proteomic profiling of dialysis patients with COVID-19 reveals markers of severity and predictors of death. eLife. 2021;10:e64827. doi: 10.7554/eLife.64827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shu T., Ning W., Wu D., Xu J., Han Q., Huang M., Zou X., Yang Q., Yuan Y., Bie Y. Plasma Proteomics Identify Biomarkers and Pathogenesis of COVID-19. Immunity. 2020;53:1108–1122.e5. doi: 10.1016/j.immuni.2020.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao Y., Zhao Z., Wang Y., Zhou Y., Ma Y., Zuo W. Single-Cell RNA Expression Profiling of ACE2, the Receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020;202:756–759. doi: 10.1164/rccm.202001-0179LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Desai N., Neyaz A., Szabolcs A., Shih A.R., Chen J.H., Thapar V., Nieman L.T., Solovyov A., Mehta A., Lieb D.J. Temporal and spatial heterogeneity of host response to SARS-CoV-2 pulmonary infection. Nat. Commun. 2020;11:6319. doi: 10.1038/s41467-020-20139-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dorward D.A., Russell C.D., Um I.H., Elshani M., Armstrong S.D., Penrice-Randal R., Millar T., Lerpiniere C.E.B., Tagliavini G., Hartley C.S. Tissue-Specific Immunopathology in Fatal COVID-19. Am. J. Respir. Crit. Care Med. 2021;203:192–201. doi: 10.1164/rccm.202008-3265OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang F., Mears J.R., Shakib L., Beynor J.I., Shanaj S., Korsunsky I., Nathan A., Donlin L.T., Raychaudhuri S. 2020. IFN-γ and TNF-α drive a CXCL10 + CCL2 + macrophage phenotype expanded in severe COVID-19 and other diseases with tissue inflammation. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reyes M., Filbin M.R., Bhattacharyya R.P., Sonny A., Mehta A., Billman K., Kays K.R., Pinilla-Vera M., Benson M.E., Cosimi L.A. 2020. Induction of a regulatory myeloid program in bacterial sepsis and severe COVID-19. bioRxiv. [DOI] [Google Scholar]
- 58.Zhang Q., Bastard P., Liu Z., Le Pen J., Moncada-Velez M., Chen J., Ogishi M., Sabli I.K.D., Hodeib S., Korol C., COVID-STORM Clinicians. COVID Clinicians. Imagine COVID Group. French COVID Cohort Study Group. CoV-Contact Cohort. Amsterdam UMC Covid-19 Biobank. COVID Human Genetic Effort. NIAID-USUHS/TAGC COVID Immunity Group Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370:eabd4570. doi: 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Menon R., Otto E.A., Hoover P., Eddy S., Mariani L., Godfrey B., Berthier C.C., Eichinger F., Subramanian L., Harder J., Nephrotic Syndrome Study Network (NEPTUNE) Single cell transcriptomics identifies focal segmental glomerulosclerosis remission endothelial biomarker. JCI Insight. 2020;5:e133267. doi: 10.1172/jci.insight.133267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.MacParland S.A., Liu J.C., Ma X.Z., Innes B.T., Bartczak A.M., Gage B.K., Manuel J., Khuu N., Echeverri J., Linares I. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018;9:4383. doi: 10.1038/s41467-018-06318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baron M., Veres A., Wolock S.L., Faust A.L., Gaujoux R., Vetere A., Ryu J.H., Wagner B.K., Shen-Orr S.S., Klein A.M. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst. 2016;3:346–360.e4. doi: 10.1016/j.cels.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gold L., Ayers D., Bertino J., Bock C., Bock A., Brody E.N., Carter J., Dalby A.B., Eaton B.E., Fitzwater T. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE. 2010;5:e15004. doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim C.H., Tworoger S.S., Stampfer M.J., Dillon S.T., Gu X., Sawyer S.J., Chan A.T., Libermann T.A., Eliassen A.H. Stability and reproducibility of proteomic profiles measured with an aptamer-based platform. Sci. Rep. 2018;8:8382. doi: 10.1038/s41598-018-26640-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Williams S.A., Kivimaki M., Langenberg C., Hingorani A.D., Casas J.P., Bouchard C., Jonasson C., Sarzynski M.A., Shipley M.J., Alexander L. Plasma protein patterns as comprehensive indicators of health. Nat. Med. 2019;25:1851–1857. doi: 10.1038/s41591-019-0665-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun B.B., Maranville J.C., Peters J.E., Stacey D., Staley J.R., Blackshaw J., Burgess S., Jiang T., Paige E., Surendran P. Genomic atlas of the human plasma proteome. Nature. 2018;558:73–79. doi: 10.1038/s41586-018-0175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Emilsson V., Ilkov M., Lamb J.R., Finkel N., Gudmundsson E.F., Pitts R., Hoover H., Gudmundsdottir V., Horman S.R., Aspelund T. Co-regulatory networks of human serum proteins link genetics to disease. Science. 2018;361:769–773. doi: 10.1126/science.aaq1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moore M.J., Dorfman T., Li W., Wong S.K., Li Y., Kuhn J.H., Coderre J., Vasilieva N., Han Z., Greenough T.C. Retroviruses pseudotyped with the severe acute respiratory syndrome coronavirus spike protein efficiently infect cells expressing angiotensin-converting enzyme 2. J. Virol. 2004;78:10628–10635. doi: 10.1128/JVI.78.19.10628-10635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gentili M., Kowal J., Tkach M., Satoh T., Lahaye X., Conrad C., Boyron M., Lombard B., Durand S., Kroemer G. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 2015;349:1232–1236. doi: 10.1126/science.aab3628. [DOI] [PubMed] [Google Scholar]
- 69.Raab M., Gentili M., de Belly H., Thiam H.R., Vargas P., Jimenez A.J., Lautenschlaeger F., Voituriez R., Lennon-Duménil A.M., Manel N., Piel M. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science. 2016;352:359–362. doi: 10.1126/science.aad7611. [DOI] [PubMed] [Google Scholar]
- 70.Zhao S., Guo Y., Sheng Q., Shyr Y. Heatmap3: an improved heatmap package with more powerful and convenient features. BMC Bioinformatics. 2014;15:16. [Google Scholar]
- 71.Kuznetsova A., Brockhoff P.B., Christensen R.H.B. lmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw. 2017;82 doi: 10.18637/jss.v082.i13. [DOI] [Google Scholar]
- 72.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Friedman J., Hastie T., Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 74.Adams T.S., Schupp J.C., Poli S., Ayaub E.A., Neumark N., Ahangari F., Chu S.G., Raby B.A., DeIuliis G., Januszyk M. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020;6:eaba1983. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Habermann A.C., Gutierrez A.J., Bui L.T., Yahn S.L., Winters N.I., Calvi C.L., Peter L., Chung M.I., Taylor C.J., Jetter C. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020;6:eaba1972. doi: 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Travaglini K.J., Nabhan A.N., Penland L., Sinha R., Gillich A., Sit R.V., Chang S., Conley S.D., Mori Y., Seita J. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature. 2020;587:619–625. doi: 10.1038/s41586-020-2922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vieira Braga F.A., Kar G., Berg M., Carpaij O.A., Polanski K., Simon L.M., Brouwer S., Gomes T., Hesse L., Jiang J. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 2019;25:1153–1163. doi: 10.1038/s41591-019-0468-5. [DOI] [PubMed] [Google Scholar]
- 78.Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 79.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original proteomic data have been deposited to Mendeley Data: http://dx.doi.org/10.17632/nf853r8xsj. Single-cell RNAseq datasets were obtained as directed in the references for each dataset. All code used for analysis will be available without restriction from the Lead Contact; examples needed to replicate analysis of proteomic data have been deposited to github at https://github.com/arnav-mehta/covid19-proteomics. Original Supplemental Tables data have been deposited to Mendeley Data: http://dx.doi.org/10.17632/nf853r8xsj.