

Summary
Antibiotic resistance is a growing problem, but bacteria can evade antibiotic treatment via tolerance and persistence. Antibiotic persisters are a small subpopulation of bacteria that tolerate antibiotics due to a physiologically dormant state. Hence, persistence is considered a major contributor to the evolution of antibiotic-resistant and relapsing infections. Here, we used the synthetically developed minimal cell Mycoplasma mycoides JCVI-Syn3B to examine essential mechanisms of antibiotic survival. The minimal cell contains only 473 genes, and most genes are essential. Its reduced complexity helps to reveal hidden phenomenon and fundamental biological principles can be explored because of less redundancy and feedback between systems compared to natural cells. We found that Syn3B evolves antibiotic resistance to different types of antibiotics expeditiously. The minimal cell also tolerates and persists against multiple antibiotics. It contains a few already identified persister-related genes, although lacking many systems previously linked to persistence (e.g. toxin-antitoxin systems, ribosome hibernation genes).
Subject areas: Microbiology, Microbiofilms
Graphical abstract
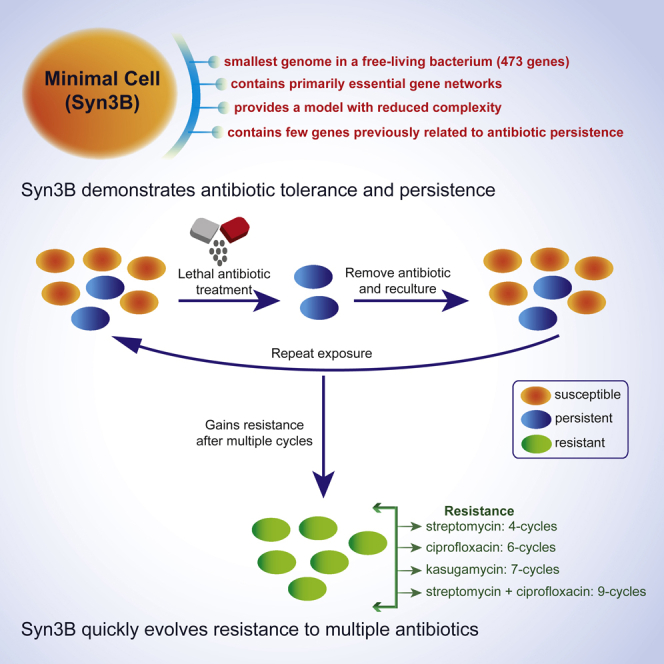
Highlights
-
•
Syn3B can form antibiotic tolerant and persister subpopulations with only 473 genes
-
•
Syn3B evolves expeditiously to gain resistance against multiple antibiotics
-
•
Syn3B contains a few systems previously linked to persistence (e.g., protease)
-
•
Syn3B lacks systems previously linked to persistence (e.g., ribosome dimerization)
Microbiology ; Microbiofilms
Introduction
The evolution of antibiotic resistance is a pressing public health concern in the 21st century; antibiotic resistance results from one or more genetic mutations that counteract an antibiotic (van Den Bergh et al., 2016). Resistance is regarded as the primary culprit of antibiotic treatment failure, but bacteria also employ less publicized strategies to evade antibiotic treatment, namely antibiotic tolerance and persistence (Harms et al., 2016; Fisher et al., 2017; Lewis, 2010). Persisters are a subpopulation of tolerant cells, which can sustain longer against antibiotic treatment in comparison to slow-growing dying cells by entering a metabolically repressed state (non-multiplying cells) (Cabral et al., 2018; Fisher et al., 2017). Most antibiotics are only effective against growing cells, and by not growing, the persister population can survive longer even without being genetically resistant. Here, we define persisters based on a kill curve and the original paper where persistence was proposed (Bigger, 1944) (our definition of persistence is explained in detail in the discussion). Two types of persisters may exist, triggered persisters (formed by environmental stimuli) and spontaneous persisters (generated stochastically) (Balaban et al., 2004, 2019; Sulaiman and Lam, 2020; Uruén et al., 2021), although spontaneous persister formation is controversial and evidence is sparse (Orman and Brynildsen, 2013; Keren et al., 2004; Kim and Wood, 2016). What makes persisters medically relevant is that they can revive and give rise to a new progeny after antibiotic treatment; the new progeny can be genetically identical to the original susceptible kin, and this process plays a pivotal role in recurring infections (Wilmaerts et al., 2019b) (Figure 1). Furthermore, evolutionary studies have determined that repeated exposure to antibiotics over many generations rapidly increases tolerance leading to antibiotic resistance (van Den Bergh et al., 2016, Fridman et al., 2014; Sulaiman and Lam, 2020; Cohen et al., 2013; Balaban and Liu, 2019; Liu et al., 2020; Schumacher et al., 2015).
Figure 1.

Tolerance permits antibiotic resistance
Persisters survive antibiotic treatment, reestablish the population when antibiotics are removed, and increase the odds of gaining resistance. Presumably, viable but nonculturable cells (VBNCs) can do this too (although we did not study VBNCs in this work).
The precise molecular mechanisms underlying persistence are still debated (multiple mechanisms are likely to exist), albeit several genes have been implicated (Wilmaerts et al., 2019a; Wu et al., 2015). Toxin-antitoxin (TA) systems have been implicated in persistence and were previously thought to be the key players for persistence (Wang and Wood, 2011; Lewis, 2010, 2012). Particularly important is the HipAB TA system because a toxin mutant, hipA7, increases persistence by impeding cellular growth in the absence of its cognate antitoxin hipB (Moyed and Bertrand, 1983). Subsequent studies also report that overexpression of other TA systems' toxins increased persistence (Korch and Hill, 2006; Correia et al., 2006; Kim and Wood, 2010). In contrast, new research found that the deletion of 10 TA systems (~22% of TA system in the genome) in E. coli does not have an apparent effect on persistence both at the population and single-cell levels (Goormaghtigh et al., 2018), but E. coli can have more than 45 TA systems (Horesh et al., 2018; Karp et al., 2014; Xie et al., 2018). Although this work supports that TA systems are not controlling persistence, the remaining 35 TA systems (~78%) could be controlling this phenomenon. Mauricio et al. studied persistence on Δ12TA Salmonella enterica and demonstrated that TA systems are dispensable (Pontes and Groisman, 2019), although this strain has 18 predicted TA systems based on the TAfinder tool (Xie et al., 2018). Another knockout study on Staphylococcus aureus deleted three copies of type II TA system from the Newman strain (Conlon et al., 2016). But further studies identified several type-I (sprG1/sprF1, sprG2/sprF2, sprG4/sprF4) (Riffaud et al., 2019) and type-II (SavRS) (Wen et al., 2018) TA systems in HG003 and NCTC-8325 strains, respectively. These are the parental strains of Newman, and these TA genes are also found in the Newman strain (Sassi et al., 2015). Moreover, a recent study showed that antitoxin sprF1 mediates translation inhibition to promote persister formation in S. aureus (Pinel-Marie et al., 2021). These findings raise many questions about TA systems' implication in bacterial persistence (Goormaghtigh et al., 2018; Tsilibaris et al., 2007; Pontes and Groisman, 2019; Kim and Wood, 2016). There are major limitations of studying TA systems in native bacteria due to their high abundance in most bacterial genomes; they often have redundant and overlapping functions and can have interdependencies within network clusters (Ronneau and Helaine, 2019; Harms et al., 2018; Wang et al., 2013). Additionally, TA systems respond to a variety of stresses (e.g. bacteriophage infection, oxidative stress, etc.), which creates a hurdle to probe their connection with any phenomenon related to stress response, namely antibiotic tolerance and persistence (Ronneau and Helaine, 2019; Harms et al., 2018; Kang et al., 2018). TA systems are also involved in other mechanisms in the cell that respond to stress, such as the stringent and SOS responses (Ronneau and Helaine, 2019), virulence (de la Cruz et al., 2013), and the regulation of pathogen intracellular lifestyle in varied host cell types (Lobato-Marquez et al., 2015). Furthermore, new types of TA systems may be yet unidentified. These challenges could be resolved using a strain that lacks canonical TA systems. But TA systems are naturally abundant, and large-scale knockouts are both error-prone and labor-intensive. We took advantage of the recently developed minimal cell that does not encode any sequences displaying homology to known TA systems and showed that it can still form persisters.
Several other mechanisms have also been considered in persister research including SOS response, oxidative stress response, etc (Wilmaerts et al., 2019a, 2019b; Trastoy et al., 2018). Two extensively studied phenomenon related to persistence are cellular ATP levels and (p)ppGpp levels. The accumulation of (p)ppGpp (stress sensing alarmone) mediates the stringent response, which controls a stress-related persistence mechanism (Harms et al., 2016). (p)ppGpp regulates many networks (such as ribosome dimerization) that can cause cells to go into dormancy (Song and Wood, 2020; Gaca et al., 2015; Wood and Song, 2020). Another well-studied model, ATP depletion increases persistence, has drawn much attention in persister research (Conlon et al., 2016). This finding is consistent with recently published result by Pu and colleagues (Pu et al., 2019), which demonstrated that lower ATP levels lead to protein aggregation and increased tolerance. This result is also coherent with our recently published data, which established that interfering with protein degradation by forming a proteolytic queue at ClpXP will increase tolerance levels dramatically (Deter et al., 2019). Transcriptomic analysis of queuing-tolerant population showed upregulation of genes related to metabolism and energy (Deter et al., 2021). However, other studies reported that ppGpp and ATP reduction are not essential for persistence (Pontes and Groisman, 2019; Chowdhury et al., 2016; Bhaskar et al., 2018). These contradictory studies are common in persister research, and we hypothesize these inconsistencies are due to the interconnection of gene networks surrounding persistence.
One goal of our study is to clarify mechanisms using a minimal, simpler system. Research over the last several years has resulted in a lot of discussion concerning genes essential for persistence (Wilmaerts et al., 2019a, 2019b; Ronneau and Helaine, 2019; Pontes and Groisman, 2019). Since persistence and tolerance are present in phylogenetically diversified bacterial species (Wilmaerts et al., 2019b; Meylan et al., 2018), it is feasible that an underlying genetic mechanism is conserved in evolutionarily related microorganisms, and the most likely candidates are essential genes or the disruption of crucial networks. In our recent work, we demonstrate that antibiotic tolerance in Escherichia coli may result from a whole-cell response and can occur through multiple pathways or networks, which may work simultaneously and cooperatively to survive against antibiotics (Deter et al., 2021).
Our strategy to study the underlying mechanisms of persistence was to use a bacterial species that contains mainly essential genes and networks with reduced complexity and fewer networks. The minimal system can reveal hidden phenomena (Glass et al., 2017) of antibiotic survival. For example, genes and networks that have previously been identified can be eliminated as causal if the genome lacks them. In contrast, genes present in Syn3B that were previously identified in other organisms can become the focus of the work. The reduced complexity has its limits as there are likely several methods microbes use to survive antibiotics, and not all methods will be in a minimal system. With these limitations well-understood, we explored antibiotic tolerance, persistence, and resistance in the minimal cell Mycoplasma mycoides JCVI-Syn3B (called Syn3B throughout), a synthetic genetic background that contains the least number of genes and smallest genome of any known free-living organism (Syn3B contains 473 genes and its genome is ~531 Kbp long, while E. coli contains >4,000 genes and its genome is ~4,600 Kbp long). The Syn3B genome was minimized from Syn1.0 (contains a chemically synthesized genome of M. mycoides subspecies capri with some watermarks and vector sequences) by removing non-essential genes (Hutchison et al., 2016; Gibson et al., 2010). Syn3B consists predominantly of essential and a few non-essential (added for ease of genome manipulation) and quasi-essential genes (required for robust growth) (Hutchison et al., 2016). This microbe was designed to have a minimal number of genes and networks with the expectation that many of the first principles of cellular life could be explored in the simplest biological systems (Glass et al., 2017). We show that Syn3B populations contain both persister and tolerant cells, and its genome contains a few previously identified persister-related genes. This work establishes that many systems previously shown to be related to bacterial persistence, such as TA systems and ribosome dimerization, are not essential for persistence in the minimal cell, and it demonstrates the effectiveness of using the minimal cell to study antibiotic survival.
Result
Whole-genome analysis of the evolved minimal cell
Although the minimal genome (Syn3B) was designed for ease of genetic manipulation, Syn3B grows slowly and to a low cell density. We adjusted the original SP4 media (Tully et al., 1979) to address these limitations. The new media, SP16, allows for faster cell growth and higher yields in liquid cultures. We noticed that Syn3B growth was slightly better after every subculture. Thus, we did a cyclic subculture of Syn3B in our optimized media by passing cells during logarithmic growth. After 26 passages, we observed better growth and isolated a single colony named Syn3B P026 (Pass 26). This strain has a shorter lag phase and an increased growth rate; the average doubling time of P026 is approximately 2.5 hr compared to the ancestral Syn3B doubling time of ~6 hr under the same conditions (Figure S1A and B). We performed a whole-genome analysis of Syn3B P026 to examine the genetic basis of these changes. Most of the mutations (9 of 11) in P026 are intergenic except one synonymous (fakA) and one non-synonymous (dnaA) (Table 1). fakA is a fatty acid kinase, and there is less evidence to suggest a direct connection to substantial alterations in bacterial growth. dnaA is a positive regulator of DNA replication initiation. Considering that bacterial growth rate is dependent on the frequency of DNA replication and that dnaA mutants have been known to result in over-replication (Skarstad and Boye, 1994), we hypothesize that the mutation of dnaA in P026 could be responsible for the higher growth rate.
Table 1.
Mutation of Mycoplasma mycoides JCVI-Syn3B, and Whole Genome Sequence (WGS) analysis identified the mutations (bold and underlined)
| Syn3B Strains |
Intergenic mutationa | Mutation positionsb | Genotype change | Amino acid substitution | Gene | Gene annotation |
|---|---|---|---|---|---|---|
| Parent∗ | ||||||
| P026 | 9 | 547 | GCA → ACA | Ala → Thr | dnaA+ | Chromosomal replication initiator protein DnaA |
| 274928 | TAC → TAT | Tyr → Tyr | fakAc+ | Dihydroxyacetone kinase | ||
| KsgR | ||||||
| PK07-L1 | 8 | 479174 | CCT → ACT | Pro → Thr | rpoC+ | DNA-directed RNA polymerase subunit beta |
| 3322 | GGA → GAA | Gly → Glu | ksgA+ | 16S rRNA (adenine(1518)-N(6)/adenine(1519)-N(6))- dimethyltransferase | ||
| PK07-L2 | 8 | 479174 | CCT → ACT | Pro → Thr | rpoC+ | |
| 3322 | GGA → GAA | Gly → Glu | ksgA+ | |||
| StrepR | ||||||
| PS04-L1 | 4 | 101452 | AAA → AGA | Lys → Arg | rpsL+ | 30S ribosomal protein S12 |
| 33,652 | GGA → AGA | Gly → Arg | CDS_6+ | Unknown | ||
| PS04-L2 | 3 | 101452 | AAA → AGA | Lys → Arg | rpsL+ | |
| 147803 | AGC → ATC | Ser → Ile; | rpsD+ | 30S ribosomal protein S4 | ||
| 33,652 | GGA → AGA | Gly → Arg | CDS 6+ | |||
| CipR | ||||||
| PC06-L1 | 4 | 7735 | GAA → AAA | Glu→ Lys | gyrA+ | DNA gyrase subunit A |
| 305613 | GAT → AAT | Asp→ Asn | parC+ | DNA topoisomerase 4 subunit A | ||
| PC06-L2 | 4 | 7735 | GAA → AAA | Glu→ Lys | gyrA+ | |
| 305613 | GAT → AAT | Asp→ Asn | parC+ | |||
| 304707 | GAT → GCT | Asp→ Ala | parE+ | DNA topoisomerase 4 subunit B | ||
| 33,652 | GGA → AGA | Gly → Arg | CDS_6+ | |||
| StrepR-CipR | ||||||
| PSC09-L1 | 4 | 7725 | AGT → AGG | Ser→ Arg | gyrA+ | |
| 101452 | AAA → AGA | Lys → Arg | rpsL+ | |||
| 33,652 | GGA → AGA | Gly → Arg | CDS_6+ | |||
| 305602 | AGT → ATT | Ser→ Ile | parC+ | |||
| PSC09-L2 | 4 | 7725 | AGT → AGG | Ser→ Arg | gyrA+ | |
| 101452 | AAA → AGA | Lys → Arg | rpsL+ | |||
| 33,652 | GGA → AGA | Gly → Arg | CDS_6+ | |||
| 305602 | AGT → ATT | Ser→ Ile | parC+ | |||
All antibiotic-resistant mutant strains were named based on the number of passes (P) in a specific antibiotic (K: Ksg, S: Strep, C: Cip, SC: Strep-Cip). All antibiotic-resistant mutants were selected from two separate evolutionary lineages, and named L1 for lineage 1 or L2 for linage 2.
∗Parent: High growth rate mutant parent to all of the strains in the table.
+Mutated genes are likely to be functional, although the mutated genes' functionality has not been tested in this study.
Mutation in the non-coding DNA sequences located between genes.
Genomic position numberings correspond to Mycoplasma mycoides JCVI-Syn3B and P026 (CP053944).
Synonymous mutation: mutation does not change in the encoded amino acid sequence.
Syn3B and evolved minimal cell P026 display antibiotic tolerance and persistence
We assessed tolerance and persistence (Figure 2) of Syn3B (parent strain) and Syn3B P026 cultures using two bactericidal antibiotics, ciprofloxacin (a fluoroquinolone) and streptomycin (an aminoglycoside). Ciprofloxacin inhibits DNA replication by targeting DNA gyrase and topoisomerase IV activity (Sanders, 1988). Streptomycin blocks protein synthesis by irreversibly binding to the 16s rRNA of the 30S ribosomal subunit (Luzzatto et al., 1968). Syn3B and P026 were grown to stationary phase and diluted into fresh media containing the antibiotics to observe population decay (see methods). Both parent strain and P026 cultures showed a typical biphasic death curve from stationary phase; the death rate became slower at ~20-24 h treatment with both antibiotics compared to the earlier stage of population decay, which indicates Syn3B displays persistence (Figures 2B and C). Survival was slightly higher in the Syn3B parent strain than P026 for both antibiotic treatments. We posit that it could be due to the slower growth rate of ancestor strain, which is consistent with the literature, as several research groups already establish that growth rate has a strong correlation with antibiotic susceptibility (Pontes and Groisman, 2019; Abshire and Neidhardt, 1993; Tuomanen et al., 1986; Lee et al., 2018). As we observed tolerance and persistence in both the ancestor strain and Syn3B P026, we decided to do further analysis with Syn3B P026, because it was much easier to work.
Figure 2.

Population decay during antibiotic treatment shows Syn3B persistence
(A) A simplified model of population decay having two phases: short-term tolerance phase and long-term tolerance phase. Both phases contain heterogeneous populations. The short-term tolerant phase contains susceptible cells, slow-growing cells, transient tolerant cells, persister and viable but nonculturable cells (VBNCs). Transient tolerant cells survive longer than fast-growing susceptible cells, while transient tolerant cells die quicker than the persisters and VBNCs (VBNCs and persisters were not distinguished in this study). The long-term tolerant phase contains both persisters and VBNCs.
(B) Population decay of Syn3B (parent strain). Overnight cultures of Syn3B (parent strain) were grown to stationary phase, diluted to 1:10, and then treated with streptomycin (100 μg/mL) or ciprofloxacin (1 μg/mL) and sampled over time. Error bars represent SEM (n ≥ 3).
(C and D) Population decay of Syn3B P026. Syn3B P026 cells were treated with streptomycin (100 μg/mL) or ciprofloxacin (1 μg/mL) and sampled over time. (C) Stationary phase cells were diluted 1:10 into fresh media containing antibiotics. Error bars represent SEM (n ≥ 6). (D) Exponential phase cells were treated with streptomycin (100 μg/mL) or ciprofloxacin (1 μg/mL) and sampled over time. Error bars represent SEM (n ≥ 3). There is 100% survival at time zero because percent survival is determined by the surviving CFU/ml compared to the CFU/ml at time zero.
Persister cells have been identified in native bacterial species in both stationary and exponential phase cultures, and we tested if this was also true for the minimal cell. P026 was grown to exponential phase and treated with antibiotics to determine whether the minimal genome showed a similar biphasic death curve in exponential phase. As expected, we observed a similar biphasic killing curve in exponential phase with slightly lower survival in exponential phase compared to stationary phase for both antibiotics (Figure 2D). Moreover, we performed resistance assays through the course of this work to rule out the possibility of resistance instead of tolerance or persistence (see methods), and no resistant colonies were identified. We then tested if the surviving population had increased persister levels after antibiotic treatment. After 48 h of antibiotic treatment, the culture was passed into fresh media and then grown to stationary phase. The culture was then re-exposed to the same antibiotic under the same condition, and as expected, no significant difference between the two death curves was observed (Figure S2).
At this point, we have demonstrated that tolerance and persistence can be detected in Syn3B cultures during both stationary and exponential phase, and this is consistent with native bacterial species. Another well-known phenotype of antibiotic survival is that the tolerant population increases with co-treatment of bacteriostatic and bactericidal antibiotics (Lewin and Smith, 1988) (pre-treatment is another method (Kwan et al., 2013)). To use the minimal cell as a model of antibiotic survival, this phenomenon should also be observed. Bacteriostatic antibiotics can counteract the bactericidal antibiotic's killing activity by arresting the growth of rapidly growing cells, which increases tolerance to the antibiotics (Kwan et al., 2013). Chloramphenicol is a bacteriostatic antibiotic that inhibits translation by binding to bacterial ribosomes and inhibiting protein synthesis, thereby inhibiting bacterial growth (Volkov et al., 2019). We co-treated with the bacteriostatic antibiotic chloramphenicol and either streptomycin or ciprofloxacin. As expected, the overall percent survival with chloramphenicol co-treatment increased compared to streptomycin or ciprofloxacin alone after 24 h and 48 h (Figure 3). These results support that inhibition of translation by co-treating the cell with chloramphenicol increases tolerance in P026.
Figure 3.

Co-treatment of bactericidal and bacteriostatic antibiotic increases survival percentage
The co-treatment of bactericidal antibiotic (streptomycin (Strep) or ciprofloxacin (Cip)) with a bacteriostatic antibiotic (chloramphenicol (Cm)) shows increased survival of cells for 24 h (left) and 48 h (right) in Syn3B P026. Error bars represent SEM (n ≥ 3). ∗p < 0.05. ∗∗p < 0.01.
The minimal genome contains previously identified genes related to tolerance and persistence
The mechanisms of persister formation are complex, but recent studies identify several genes involved in this process (Cameron et al., 2018; Wilmaerts et al., 2019a). A few of these genes are present in the minimal genome. We identified known and predicted genes related to tolerance and persistence in the Syn3B genome (Table 2). During nutrient limitation, bacteria generally switch their gene expression profile from rapid growth to a survival state by (p)ppGpp levels (a hallmark of the stringent response), which is regulated by the RelA protein in E. coli (Boutte and Crosson, 2013). Another important gene in the stringent response is phoU, a global negative regulator, which is deactivated upon inorganic phosphate starvation. Mutation or deletion of this gene in other microorganisms leads to a metabolically hyperactive state and decreased persistence (Li and Zhang, 2007; Zhang and Li, 2010). The SOS response is another signaling pathway that upregulates DNA repair functions, which appears to be linked to bacterial persistence. Genes related to the SOS response have been found upregulated in E. coli persister (e.g. uvrA, uvrB) (Keren et al., 2004), and other mutants (such as xseB) have lost their ability to induce detectable levels of persistence when pretreated with rifampicin, a bacteriostatic antibiotic (Cui et al., 2018).
Table 2.
Syn3B contains genes previously shown to be related to E.coli persistence and tolerance
| Gene | Protein/RNA | Major function | REF |
|---|---|---|---|
| dnaK, dnaJ, clpB | Chaperon | Global regulator | (Wu et al., 2015; Hansen et al., 2008) |
| phoU | Phosphate-specific transport system accessory protein | Global regulator | (Li and Zhang, 2007) |
| xseB | Exodeoxyribonuclease | DNA mismatch repair and recombination | (Cui et al., 2018) |
| uvrA, uvrB | Endonuclease | SOS response | (Wu et al., 2015; Cui et al., 2018; Keren et al., 2004) |
| relA | GTP pyrophosphokinase | Stringent response | (Wu et al., 2015) |
| lon | Protease | Protease | (Wu et al., 2015) |
| glyA | Serine hydroxymethyltransferase | Metabolism | (Cui et al., 2018) |
| atpA, atpC, atpF | ATP synthase | ATP synthesis | (Girgis et al., 2012; Lobritz et al., 2015; Kiss, 2000) |
| galU | UTP–glucose-1-phosphate uridylyltransferase | Lipopolysaccharide precursor synthesis | (Girgis et al., 2012) |
| trmE; efp; ybeY | tRNA modification GTPase; Elongation factor P; Endoribonuclease | Translation | (Cui et al., 2018) |
| ssrA; smpB | Transfer messenger RNA (tmRNA); Small Protein B | Trans-translation | (Wu et al., 2015; Yamasaki et al., 2020) |
Other literature advocates the connection of persistence with energy metabolism, although the outcomes are often inconsistent between models (Wilmaerts et al., 2019a). For example, the deletion of atpA (encoding for ATP synthase subunit alpha) decreases the persister fraction (Lobritz et al., 2015), but deletion of genes encoding other parts of the ATP synthase (atpC and atpF) increases the persister fraction (Girgis et al., 2012; Kiss, 2000). Several heat shock proteins, mainly proteases (e.g. lon (Wu et al., 2015; Pu et al., 2016)) and chaperons (e.g. dnaK (Wu et al., 2015), dnaJ (Hansen et al., 2008), clpB (Wu et al., 2015)), are related to persistence considering that deletion of those genes decreases persistence.
Another survival mechanism connected to persistence is trans-translation, which allows bacteria to recycle stalled ribosomes and tag unfinished polypeptides for degradation, which in E. coli requires tmRNA (encoded by ssrA) and a small protein (smpB). The deletion of these genes causes persister-reduction in E.coli (Liu et al., 2017; Wu et al., 2015; Yamasaki et al., 2020). Additional genes glyA (Cui et al., 2018), galU (Girgis et al., 2012), trmE (Cui et al., 2018), efp (Cui et al., 2018), ybeY (Cui et al., 2018), etc.; see Table 2) are indirectly related to stress response or metabolism and have been reported to affect persistence. Overall, this shortlist (Table 2 compare to Table S1) of genes demonstrates that there are far fewer genes to explore in the minimal cell than any other known free-living microorganism on the planet. However, further exploration is needed to test if there is a relationship between genes in Table 2 to antibiotic survival in the minimal cell.
Evolution of the minimal cell against different types of antibiotic and whole-genome analysis of the resistant strain
Up until this point, we have focused on antibiotic tolerance and persistence. For the minimal system to be also useful as a model of antibiotic resistance, it must be able to evolve resistance without horizontal gene transfer (i.e. isolated from other organisms). We hypothesize that due to lack of complexity and less control of mutation, simpler cells likely evolve faster than more complex organisms; a simpler, slimmed-down genome allows for rapid evolution to selective pressures. We selected different classes of bactericidal antibiotics including streptomycin (Strep), ciprofloxacin (Cip), a combination of streptomycin and ciprofloxacin (Strep-Cip), and one bacteriostatic antibiotic-kasugamycin (Ksg), to study the evolution of antibiotic resistance in the minimal genome. We applied cyclic antibiotic treatments where we repeatedly regrew and retreated the culture with the lethal dose of the same antibiotic for a few cycles with two biological replicates (Figure S4B.) to make resistant mutants. In seven passes or less (nine passes with two antibiotics), the minimal genome rapidly evolved resistance to all antibiotics tested. We isolated single colonies (two separate evolutionary lineages named L1 for lineage 1 or L2 for linage 2) from each resistant mutants named Syn3B PS04 (4 Passes in Streptomycin), PC06 (6 Passes in Ciprofloxacin), PSC09 (9 Passes in Streptomycin and Ciprofloxacin), and PK07 (7 Passes in Kasugamycin). We then analyzed the whole genome for mutations. Most mutations were in intergenic regions of the resistant mutants, similar to Syn3B P026 (Table 1).
We observed Syn3B P026 quickly evolved against streptomycin, as it only took 4 passes to become resistant. Only two non-synonymous mutations were found in both lineages. The first one is in the rpsL gene (encoding the S12 ribosomal protein), which is frequently identified in streptomycin-resistant strains (Villellas et al., 2013), and the other one is the CDS_6 gene, the function of which is not yet known. In PS04_L2, we also observed another mutation in the rpsD gene (encoding the S4 ribosomal protein). We found that Syn3B P026 took 6 and 9 cycles to gain resistance against ciprofloxacin and a combination of both streptomycin and ciprofloxacin, respectively. In both replicates of PC06, we observed mutations in gyrA (encoding the DNA gyrase subunit A) and parC (encoding DNA topoisomerase 4 subunit A). In PC06_L2, we observed another two mutations, one in parE (encoding DNA topoisomerase 4 subunit B) and another in the CDS_6 gene. For the combined streptomycin-ciprofloxacin resistant strains, both lineages showed similar mutations in rpsL, gyrA, parC, and CDS_6 genes. In the kasugamycin-resistant strain PK07, only two nonsynonymous mutations were found. The first one is in the ksgA gene, which encodes a methylase that modifies 16S rRNA, and inhibition of this protein by the antibiotic kasugamycin causes susceptibility. Therefore, when ksgA is inactivated, cells became kasugamycin-resistant (van Gemen et al., 1987; Mariscal et al., 2018). We reasoned that the nonsynonymous mutation in ksgA in PK07 makes the protein inactive and able to confer kasugamycin resistance. The other detected non-synonymous mutation is in rpoC (RNA polymerase subunit).
Discussion
Overall, we have demonstrated that the minimal cell contains both antibiotic tolerant and persistent subpopulations, and it can quickly evolve to gain resistance. We have successfully generated an evolved strain of the minimal cell Syn3B P026, which has a better growth rate than the ancestral strain and overcomes one of the major difficulties of working with the minimal cell. We show that Syn3B can be used as a model for studying antibiotic survival, especially when we consider that the minimal genome's core proteins are present in many other microorganisms including human pathogens. Syn3B has over 50% similarity to human pathogens that have been identified as a concern with respect to the growing antibiotic-resistance problem (Table S2) (Centers for Disease Control and Preventions, 2019). For example, out of 455 protein-encoding genes of Syn3B, 338 of them (74%) have homologs to S. aureus NCTC 8325 proteins. S. aureus causes meningitis and pneumonia, and antibiotic resistance is a major problem of this organism (Foster, 2017).
The definition of persistence is often debated, but we argue that the original definition proposed by Biggers in his seminal 1944 paper (Bigger, 1944), where persisters were first classified and named, is sufficient. Syn3B meets 9 out of 10 characteristics defined by Bigger. The only characteristic not found in Syn3B (point 4 in Table 3) was an untested hypothesis that Bigger put forward about persisters, which states that persisters can be induced without antibiotic stress (now known as spontaneous persisters). This characteristic has yet to be resolved and was not addressed in our work. Bigger made it clear that persister identification should occur over several days and suggested 72 h (Bigger, 1944). We tested Syn3B for 72 h (Figures 2B–D), and it formed persisters. Though we agree with Bigger's original assertion that prolonged antibiotic treatment is required to demonstrate persistence, the originally 72 h cutoff does not consider different dividing times of bacteria, which is likely to affect killing rates.
Table 3.
In Bigger's 1944 paper, he identified cells that survived antibiotics longer, named them persisters, and listed 10 characteristics of the persister subpopulation
| Bigger (1944) persister definition | Syn3B persisters | |
|---|---|---|
| 1. | An antibiotic failed to kill a small population of bacteria. | Multiple antibiotics failed to kill a small population of Syn3B. |
| 2. | Persisters are a small population initially present in the population. | Syn3B contains a small persister subpopulation among the susceptible population. |
| 3. | Persister can be induced or changed based on the environment. | The persistence (surviving population) level is different when the culture originated from a stationary phase or exponential phase. The persistence level is different when cultures were co-treated with bacteriostatic and bactericidal antibiotics compared to the bactericidal antibiotic only; chloramphenicol and streptomycin, and chloramphenicol and ciprofloxacin, compared to the antibiotics alone. |
| 4.a | Untested hypothesis: No evidence showed that persisters were produced by the presence of an antibiotic. | This was not tested for Syn3B persisters. |
| 5. | Bacteriocidal concentrations of an antibiotic killed the susceptible population, but not persisters. | The Syn3B susceptible population was killed by bacteriocidal concentrations of more than one antibiotic type, but persisters were not. |
| 6. | Persisters are likely insensitive to an antibiotic because they are dormant and non-dividing. The antibiotic used, penicillin, kills bacteria only when they divide. | Syn3B persisters also appear to be dormant and likely non-dividing, because the antibiotics used are more effective against dividing cells. |
| 7. | Descendants of persisters are no more resistant to an antibiotic than the original population. | Syn3B descendants of persisters are no more resistant to an antibiotic than the original population. |
| 8. | When the antibiotic is destroyed, the majority of persisters will emerge and grow normally. | Syn3B persisters grew once the antibiotic was removed or diluted below the MIC. |
| 9. | The antibiotic prolongs the dormant phase but not indefinitely. | Syn3B persisters continue to die, but slowly, the longer they are exposed to antibiotics. There is no plateau but a slow decline in cell death. |
| 10.a | Untested hypothesis: Persisters may be coming out of the dormant state, attempting to divide and being killed by the antibiotic. | This was not tested for Syn3B persisters. Syn3B persisters continue to die, but slowly, the longer they are exposed to antibiotics. There is no plateau but a slow decline in cell death. |
Syn3B meets 9 of 10 characteristics defined by Bigger. Left column: summary of Bigger's definition. Right column: similar characteristics of Syn3B persisters. We provided no evidence related to point 4 but observed a similar phenotype as Bigger to support point 10.
Points 4 and 10 were untested hypotheses of Bigger and were not tested in our work.
A more recent definition of tolerance and persistence was put forth by a consensus statement released after a discussion panel with 121 researchers (Balaban et al., 2019). They defined persistence similarly to the original Bigger's paper with some slight changes. They defined persistence as a subpopulation of tolerance, and this is generally agreed upon in the literature. They then stated that the “tolerance state” could be distinguished from the “persistence state” based on a killing curve. We agree that persisters can be distinguished from other populations using a kill curve (Figure 2A), and we showed that Syn3B also has a distinguished death curve (Figures 2B–D). However, we do not call the first death phase “tolerance,” as proposed by Balaban (Balaban et al., 2019), because naming this subpopulation tolerance is easily confused when the entire population is tolerant. We, instead, label the first phase (faster death rate) of the death curve as short-term tolerance phase contains heterogeneous population includes susceptible cells, transient tolerant cells (caused by slow-growth or heterogeneity in gene expression (Lee et al., 2019; El Meouche and Dunlop, 2018)), persisters and VBNCs. Susceptible cells are included because it will take time to kill off these cells regardless of the antibiotic used. Transient tolerant cells can tolerate antibiotics longer than susceptible cells, but not as long as VBNCs or persisters (VBNCs were not investigated in this study). We termed the second phase (slower death rate) as a long-term tolerance phase containing persister and VBNCs. We described in our definition that a death curve can distinguish short-term tolerance and persistence.
Few researchers have put forth that the definition for persisters should include a rapid decline in the short-term tolerance stage and then a persister plateau (Mulcahy et al., 2010; Bartell et al., 2020; Sahukhal et al., 2017, Song and Wood, 2021). Depiction of persisters as a plateau may underrepresent the heterogeneity of the population (Kaldalu et al., 2016), and a true plateau have a slope of zero for an extended period of time, which we do not see in our Syn3B and E.coli data. We first tested if a plateau is present in the model organism E. coli. Since other researchers (Song et al., 2019; Mohiuddin et al., 2020; Aedo et al., 2019) and ourselves (Deter et al., 2019) have shown that E. coli persisters can be detected after three hours of ampicillin treatment at bactericidal concentrations (100 μg/mL treatment), we reanalyzed our previously published data (Deter et al., 2019) and included 24 ampicillin exposure data to check for a plateau using a simple t test point comparison (Figure S3 and Table S3A). The hypothesis is there is a slow decline in persistence after 3 hr of antibiotic treatment (thus, there is no plateau and the slope of the line is not zero). Mathematically, a plateau has a slope at or near zero. The null hypothesis is the population reaches a steady-state with no significant decay after 3 h (thus, there is a plateau and the slope of the line is zero). Comparing the p values of 3 h ampicillin exposure to later exposure times could test if the population is a plateau (no significant difference) or a slow decline (significant difference). As we suspected, the long-term E. coli death curve slope is not zero, the cells are dying slowly. This is evident by the fact that 3 h ampicillin treatment is significantly different (p < 0.05) than the longer exposures of ampicillin treatment: the p values for 6 h, 8 h, and 24 h, were 6.5 × 10−5, 4.6 × 10−4, and 3.3 × 10−6, respectively, compared to the 3 h (n ≥ 3) (Table S3A).
There is no significant change in persister levels (p > 0.5) between some closed time points, e.g. 6 h–8 h. This is not surprising because the decay rate is slow. However, overall, there is a slow decline of persisters, e.g. 3 h–24 h (p < 0.5; Table S3; Figure S3). This work underscores the importance of doing long-term kill curves as stated in recent consensus statement (Balaban et al., 2019). Next, we tested Syn3B P026 persisters and as expected, there was no plateau, the slope is not zero for different antibiotics (Figure 2C), and there is a slow decline in persistence over time. This is evident by the fact that 24 h ciprofloxacin treatment was significantly different (p < 0.05) than longer exposures of ciprofloxacin treatment: the p values for 48 h and 72 h were 5.2 × 10−5 and 3.6 × 10−2 compared to the 24 h (n ≥ 3) (Table S3B). Similar results were obtained for streptomycin (100 μg/mL); the p value for 48 h and 72 h treatment compared to 24 h is 4.2 × 10−3 and 3.3 × 10−2 (n ≥ 3) (Table S3C). Similar to E. coli, there is no significant change in persister levels (p > 0.5) between some closed time points, but overall, there is a slow decline of persisters. This again underscores the importance of doing long-term kill curves. Our results support a rapid decline in the short-term tolerance stage and then a slow decline in the persister stage, not a plateau, which aligns with the historical data (Bigger, 1944) and modern definition (Balaban et al., 2019).
After establishing that Syn3B forms persisters, we looked into genes previously shown to be related to tolerance in other organisms. We also find that the Syn3B genome lacks homologs of several genes and pathways reported in earlier research to be related to tolerance and persistence (Table S1). For instance, TA systems are often implicated for persistence and are still referenced as causing persistence (Moreno-del Álamo et al., 2020; Riffaud et al., 2020; Shenkutie et al., 2020), although recent research provided evidence of a lack of causation between persister formation and TA system (Pontes and Groisman, 2019; Conlon et al., 2016). In our study, we identify no known TA systems or homologous genes based on the results from TADB 2.0 (a database designed to search for TA systems based on homology) (Xie et al., 2018) in Syn3B. Our study clearly shows that TA systems are not required for tolerance or persistence in the minimal cell, and it seems likely that most bacteria do not require them either. It is not surprising that the minimal genome does not contain TA systems because the genome was designed by researchers and not subject to the natural environment. Natural TA systems are often described as “addictive” systems that are hard to lose once acquired; they often have overlapping toxin and antitoxin genes, making mutations less likely to be selected. Curiously, we observed some overlapping genes that are not at all similar to TA systems (they are also much longer in sequence than traditional TA systems, and are not homologous to any known TA system), and whose functions are not yet defined (Table S4).
Recently, the Wood group (Song and Wood, 2020; Wood and Song, 2020) proposed the ppGpp ribosome dimerization persister (PRDP) model for entering and exiting the persister state where ppGpp induces hpf, rmf, and raiA, which converts active ribosomes into their inactive state (such as 100s ribosome) to reduce translation, and consequently cells enter into persistence. Upon removal of antibiotic stress, cAMP level is reduced and HflX production is stimulated, which dissociates inactive 100S ribosomes into active 70S ribosomes and growth resumes. However, we did not detect homologs to the required genes, raiA, rmf, hpf, and hflX, for ribosome dimerization in the minimal cell genome, which means ribosome dimerization is not an essential mechanism for this organism (or another unknown mechanism for ribosome dimerization exists in Syn3B). (p)ppGpp may still play a role in Syn3B persistence, the genome contains relA (converts ATP and GTP to (p)ppGpp; Table 2). A potentially fruitful study would be to study the effects on survival by altering (p)ppGpp of Syn3B cultures.
This strain shows that TA systems and ribosome hibernation genes are not required for bacterial tolerance or persistence. Moreover, there are far fewer genes (less than 20 genes) present in Syn3B, which have been shown related to persistence. Thus, if specific genes or regulons are responsible for persistence, then Syn3B should be very useful in identifying them in future studies because it has a minimal number of genes. It is possible that persister formation and maintenance results from slowed or disrupted cellular networks (this is the hypothesis we most prescribe to), rather than the activity of specific genes or regulons. If tolerance and persistence results from slowed or disrupted cellular networks, then Syn3B is an ideal model organism to use because it has a minimal number of networks. While different genes and networks are likely responsible for persistence depending on the antibiotic, strain, or bacterial species, identifying genes in this minimal system should be applicable to a number of other microorganisms including the pathogen Mycoplasma mycoides from which Syn3B was derived. Regardless of how cells enter the dormant state, Syn3B provides a new model to study the genes and networks that allow cells to survive antibiotic treatment and could pave the way for finding new drugs that target the persister and tolerant subpopulations.
Limitations of the study
We observed that despite controlling methodology to the greatest extent possible, persister levels varied considerably (far more than observed in our previous work with E. coli) (Deter et al., 2019) between experiments for both streptomycin and ciprofloxacin treatments (Table S3). We hypothesize that this variability might be due to the stochastic fluctuations (noise) in gene expression levels, which results in protein level variations even among genetically identical cells in a similar environment (Soltani et al., 2016). We expect that gene expression and protein production to be more erratic in Syn3B compared to natural microorganisms because this cell has a designed genome lacking many control mechanisms and did not evolve to achieve some level of internal cellular “equilibrium” like native cells have.
We did not test for other subpopulations that have been identified during antibiotic treatment. We did not test for Syn3B transient tolerant cells, VBNCs, or spontaneous persisters.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to Nicholas C. Butzin (nicholas.butzin@sdstate.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data that supports the findings of this study are available from the corresponding author upon request. Whole genomes data of P026, PK07_L1, PK07_L2, PS04_L1, PS04_L2, PC06_L1, PC06_L2, PSC09_L1 and PSC09_L2 strains has been deposited on the NCBI Genome Bank in BioProject PRJNA635211 with the accession number CP053944, CP069339, CP069340, CP069341, CP069342, CP069343, CP069344, CP069345, CP069346, respectively. The code used for colony counting is available on GitHub at https://github.com/hdeter/CountColonies.
Methods
All methods can be found in the accompanying transparent methods supplemental file.
Acknowledgments
Mycoplasma mycoides JCVI-Syn3B was a generous gift from Dr. John I. Glass from J. Craig Venter Institute (JCVI), La Jolla, CA, USA. This work is supported by the National Science Foundation award Numbers 1922542 and 1849206, and by a USDA National Institute of Food and Agriculture Hatch project grant number SD00H653-18/project accession no. 1015687.
Author contributions
T.H. wrote the manuscript and performed most experiments. H.S.D. repeated streptomycin persister assay and developed custom code for colony counting, growth rate, and statistical analyses. E.P. performed cyclic antibiotic treatment experiments. N.C.B. planned and directed the project. All authors contributed to discussing and editing the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102391.
Supplemental information
References
- Abshire K.Z., Neidhardt F.C. Growth rate paradox of Salmonella typhimurium within host macrophages. J. Bacteriol. 1993;175:3744–3748. doi: 10.1128/jb.175.12.3744-3748.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aedo S.J., Orman M.A., Brynildsen M.P. Stationary phase persister formation in Escherichia coli can be suppressed by piperacillin and PBP3 inhibition. BMC Microbiol. 2019;19:1–12. doi: 10.1186/s12866-019-1506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban N.Q., Helaine S., Lewis K., Ackermann M., Aldridge B., Andersson D.I., Brynildsen M.P., Bumann D., Camilli A., Collins J.J. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 2019;17:441–448. doi: 10.1038/s41579-019-0196-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban N.Q., Liu J. Persister Cells Infectious Disease, Springer, Cham. 2019:1–17. [Google Scholar]
- Balaban N.Q., Merrin J., Chait R., Kowalik L., Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- Bartell J.A., Cameron D.R., Mojsoska B., Haagensen J.A.J., Pressler T., Sommer L.M., Lewis K., Molin S., Johansen H.K. Bacterial persisters in long-term infection: emergence and fitness in a complex host environment. PLoS Pathog. 2020;16:e1009112. doi: 10.1371/journal.ppat.1009112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar A., de Piano C., Gelman E., Mckinney J.D., Dhar N. Elucidating the role of (p) ppGpp in mycobacterial persistence against antibiotics. IUBMB life. 2018;70:836–844. doi: 10.1002/iub.1888. [DOI] [PubMed] [Google Scholar]
- Bigger J. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet. 1944;244:497–500. [Google Scholar]
- Boutte C.C., Crosson S. Bacterial lifestyle shapes stringent response activation. Trends Microbiol. 2013;21:174–180. doi: 10.1016/j.tim.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral D.J., Wurster J.I., Belenky P. Antibiotic persistence as a metabolic adaptation: stress, metabolism, the host, and new directions. Pharmaceuticals. 2018;11:14. doi: 10.3390/ph11010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron D.R., Shan Y., Zalis E.A., Isabella V., Lewis K. A genetic determinant of persister cell formation in bacterial pathogens. J. Bacteriol. 2018;200:e00303–e00318. doi: 10.1128/JB.00303-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Preventions Biggest threats and data [Online] 2019. https://www.cdc.gov/drugresistance/biggest-threats.html
- Chowdhury N., Kwan B.W., Wood T.K. Persistence increases in the absence of the alarmone guanosine tetraphosphate by reducing cell growth. Sci. Rep. 2016;6:20519. doi: 10.1038/srep20519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen N.R., Lobritz M.A., Collins J.J. Microbial persistence and the road to drug resistance. Cell Host Microbe. 2013;13:632–642. doi: 10.1016/j.chom.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon B.P., Rowe S.E., Gandt A.B., Nuxoll A.S., Donegan N.P., Zalis E.A., Clair G., Adkins J.N., Cheung A.L., Lewis K. Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol. 2016;1:1–7. doi: 10.1038/nmicrobiol.2016.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia F.F., D'onofrio A., Rejtar T., LI L., Karger B.L., Makarova K., Koonin E.V., Lewis K. Kinase activity of overexpressed HipA is required for growth arrest and multidrug tolerance in Escherichia coli. J. Bacteriol. 2006;188:8360–8367. doi: 10.1128/JB.01237-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui P., Niu H., Shi W., Zhang S., Zhang W., Zhang Y. Identification of genes involved in bacteriostatic antibiotic-induced persister formation. Front. Microbiol. 2018;9:413. doi: 10.3389/fmicb.2018.00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz M.A., Zhao W., Farenc C., Gimenez G., Raoult D., Cambillau C., Gorvel J.P., Meresse S. A toxin-antitoxin module of Salmonella promotes virulence in mice. PLoS Pathog. 2013;9:e1003827. doi: 10.1371/journal.ppat.1003827. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Deter H.S., Abualrahi A.H., Jadhav P., Schweer E.K., Ogle C.T., Butzin N.C. Proteolytic queues at ClpXP increase antibiotic tolerance. ACS Synth. Biol. 2019;9:95–103. doi: 10.1021/acssynbio.9b00358. [DOI] [PubMed] [Google Scholar]
- Deter H.S., Hossain T., Butzin N.C. Antibiotic tolerance is associated with a broad and complex transcriptional response in E. coli. Sci. Rep. 2021;11:1–15. doi: 10.1038/s41598-021-85509-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Meouche I., Dunlop M.J. Heterogeneity in efflux pump expression predisposes antibiotic-resistant cells to mutation. Science. 2018;362:686–690. doi: 10.1126/science.aar7981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R.A., Gollan B., Helaine S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017;15:453. doi: 10.1038/nrmicro.2017.42. [DOI] [PubMed] [Google Scholar]
- Foster T.J. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 2017;41:430–449. doi: 10.1093/femsre/fux007. [DOI] [PubMed] [Google Scholar]
- Fridman O., Goldberg A., Ronin I., Shoresh N., Balaban N.Q. Optimization of lag time underlies antibiotic tolerance in evolved bacterial populations. Nature. 2014;513:418–421. doi: 10.1038/nature13469. [DOI] [PubMed] [Google Scholar]
- Gaca A.O., Colomer-Winter C., Lemos J.A. Many means to a common end: the intricacies of (p) ppGpp metabolism and its control of bacterial homeostasis. J. Bacteriol. 2015;197:1146–1156. doi: 10.1128/JB.02577-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D.G., Glass J.I., Lartigue C., Noskov V.N., Chuang R.-Y., Algire M.A., Benders G.A., Montague M.G., Ma L., Moodie M.M. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 2010;329:52–56. doi: 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- Girgis H.S., Harris K., Tavazoie S. Large mutational target size for rapid emergence of bacterial persistence. Proc. Natl. Acad. Sci. U S A. 2012;109:12740–12745. doi: 10.1073/pnas.1205124109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass J.I., Merryman C., Wise K.S., Hutchison C.A., 3rd, Smith H.O. Minimal cells-real and imagined. Cold Spring Harb. Perspect. Biol. 2017;9:a023861. doi: 10.1101/cshperspect.a023861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goormaghtigh F., Fraikin N., Putrins M., Hallaert T., Hauryliuk V., Garcia-Pino A., Sjodin A., Kasvandik S., Udekwu K., Tenson T. Reassessing the role of type II toxin-antitoxin systems in formation of Escherichia coli type II persister cells. mBio. 2018;9 doi: 10.1128/mBio.00640-18. e00640–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen S., Lewis K., Vulić M. Role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrob. Agents Chemother. 2008;52:2718–2726. doi: 10.1128/AAC.00144-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms A., Brodersen D.E., Mitarai N., Gerdes K. Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol. Cell. 2018;70:768–784. doi: 10.1016/j.molcel.2018.01.003. [DOI] [PubMed] [Google Scholar]
- Harms A., Maisonneuve E., Gerdes K. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science. 2016;354:aaf4268. doi: 10.1126/science.aaf4268. [DOI] [PubMed] [Google Scholar]
- Horesh G., Harms A., Fino C., Parts L., Gerdes K., Heinz E., Thomson N.R. SLING: a tool to search for linked genes in bacterial datasets. Nucleic Acids Res. 2018;46:e128. doi: 10.1093/nar/gky738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison C.A., 3rd, Chuang R.Y., Noskov V.N., Assad-Garcia N., Deerinck T.J., Ellisman M.H., Gill J., Kannan K., Karas B.J., Ma L. Design and synthesis of a minimal bacterial genome. Science. 2016;351:aad6253. doi: 10.1126/science.aad6253. [DOI] [PubMed] [Google Scholar]
- Kaldalu N., Hauryliuk V., Tenson T. Persisters—as elusive as ever. Appl. Microbiol. Biotechnol. 2016;100:6545–6553. doi: 10.1007/s00253-016-7648-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S.M., Kim D.H., Jin C., Lee B.J. A systematic overview of type II and III toxin-antitoxin systems with a focus on druggability. Toxins. 2018;10:515. doi: 10.3390/toxins10120515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp P.D., Weaver D., Paley S., Fulcher C., Kubo A., Kothari A., Krummenacker M., Subhraveti P., Weerasinghe D., Gama-Castro S. The EcoCyc database. EcoSal Plus. 2014;6:1–13. doi: 10.1128/ecosalplus.ESP-0009-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren I., Shah D., Spoering A., Kaldalu N., Lewis K. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 2004;186:8172–8180. doi: 10.1128/JB.186.24.8172-8180.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-S., Wood T.K. Persistent persister misperceptions. Front. Microbiol. 2016;7:2134. doi: 10.3389/fmicb.2016.02134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Wood T.K. Toxins Hha and CspD and small RNA regulator Hfq are involved in persister cell formation through MqsR in Escherichia coli. Biochem. Biophysical Res. Commun. 2010;391:209–213. doi: 10.1016/j.bbrc.2009.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss P. Results concerning products and sums of the terms of linear recurrences. Acta Acad. Agriensis Sectio Math. 2000;27:1–7. [Google Scholar]
- Korch S.B., Hill T.M. Ectopic overexpression of wild-type and mutant hipA genes in Escherichia coli: effects on macromolecular synthesis and persister formation. J. Bacteriol. 2006;188:3826–3836. doi: 10.1128/JB.01740-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan B.W., Valenta J.A., Benedik M.J., Wood T.K. Arrested protein synthesis increases persister-like cell formation. Antimicrob. Agents Chemother. 2013;57:1468–1473. doi: 10.1128/AAC.02135-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.J., Wang S., Meredith H.R., Zhuang B., Dai Z., You L. Robust, linear correlations between growth rates and β-lactam–mediated lysis rates. Proc. Natl. Acad. Sci. U S A. 2018;115:4069–4074. doi: 10.1073/pnas.1719504115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.J., Lee S.-K., Song N., Nathan T.O., Swarts B.M., Eum S.-Y., Ehrt S., Cho S.-N., Eoh H. Transient drug-tolerance and permanent drug-resistance rely on the trehalose-catalytic shift in Mycobacterium tuberculosis. Nat. Commun. 2019;10:1–12. doi: 10.1038/s41467-019-10975-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin C.S., Smith J.T. Bactericidal mechanisms of ofloxacin. J. Antimicrob. Chemother. 1988;22(Suppl C):1–8. doi: 10.1093/jac/22.supplement_c.1. [DOI] [PubMed] [Google Scholar]
- Lewis K. Persister cells. Annu. Rev. Microbiol. 2010;64:357–372. doi: 10.1146/annurev.micro.112408.134306. [DOI] [PubMed] [Google Scholar]
- Lewis K. Persister cells: molecular mechanisms related to antibiotic tolerance. Handb Exp. Pharmacol. 2012:121–133. doi: 10.1007/978-3-642-28951-4_8. [DOI] [PubMed] [Google Scholar]
- Li Y., Zhang Y. PhoU is a persistence switch involved in persister formation and tolerance to multiple antibiotics and stresses in Escherichia coli. Antimicrob. Agents Chemother. 2007;51:2092–2099. doi: 10.1128/AAC.00052-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Gefen O., Ronin I., Bar-Meir M., Balaban N.Q. Effect of tolerance on the evolution of antibiotic resistance under drug combinations. Science. 2020;367:200–204. doi: 10.1126/science.aay3041. [DOI] [PubMed] [Google Scholar]
- Liu S., Wu N., Zhang S., Yuan Y., Zhang W., Zhang Y. Variable persister gene interactions with (p) ppGpp for persister formation in Escherichia coli. Front. Microbiol. 2017;8:1795. doi: 10.3389/fmicb.2017.01795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobato-Marquez D., Moreno-Cordoba I., Figueroa V., Diaz-Orejas R., Garcia-Del Portillo F. Distinct type I and type II toxin-antitoxin modules control Salmonella lifestyle inside eukaryotic cells. Sci. Rep. 2015;5:9374. doi: 10.1038/srep09374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobritz M.A., Belenky P., Porter C.B., Gutierrez A., Yang J.H., Schwarz E.G., Dwyer D.J., Khalil A.S., Collins J.J. Antibiotic efficacy is linked to bacterial cellular respiration. Proc. Natl. Acad. Sci. U S A. 2015;112:8173–8180. doi: 10.1073/pnas.1509743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzatto L., Apirion D., Schlessinger D. Mechanism of action of streptomycin in E. coli: interruption of the ribosome cycle at the initiation of protein synthesis. Proc. Natl. Acad. Sci. U S A. 1968;60:873. doi: 10.1073/pnas.60.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariscal A.M., Kakizawa S., Hsu J.Y., Tanaka K., Gonzalez-Gonzalez L., Broto A., Querol E., Lluch-Senar M., Pinero-Lambea C., Sun L. Tuning gene activity by inducible and targeted regulation of gene expression in minimal bacterial cells. ACS Synth. Biol. 2018;7:1538–1552. doi: 10.1021/acssynbio.8b00028. [DOI] [PubMed] [Google Scholar]
- Meylan S., Andrews I.W., Collins J.J. Targeting antibiotic tolerance, pathogen by pathogen. Cell. 2018;172:1228–1238. doi: 10.1016/j.cell.2018.01.037. [DOI] [PubMed] [Google Scholar]
- Mohiuddin S.G., Kavousi P., Orman M.A. Flow-cytometry analysis reveals persister resuscitation characteristics. BMC Microbiol. 2020;20:1–13. doi: 10.1186/s12866-020-01888-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-del Álamo M., Marchisone C., Alonso J.C. Antitoxin ε reverses toxin ζ-facilitated ampicillin dormants. Toxins. 2020;12:801. doi: 10.3390/toxins12120801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyed H.S., Bertrand K.P. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 1983;155:768–775. doi: 10.1128/jb.155.2.768-775.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy L.R., Burns J.L., Lory S., Lewis K. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol. 2010;192:6191–6199. doi: 10.1128/JB.01651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orman M.A., Brynildsen M.P. Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob. Agents Chemother. 2013;57:3230–3239. doi: 10.1128/AAC.00243-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinel-Marie M.-L., Brielle R., Riffaud C., Germain-Amiot N., Polacek N., Felden B. RNA antitoxin SprF1 binds ribosomes to attenuate translation and promote persister cell formation in Staphylococcus aureus. Nat. Microbiol. 2021:1–12. doi: 10.1038/s41564-020-00819-2. [DOI] [PubMed] [Google Scholar]
- Pontes M.H., Groisman E.A. Slow growth determines nonheritable antibiotic resistance in Salmonella enterica. Sci. Signal. 2019;12:eaax3938. doi: 10.1126/scisignal.aax3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu Y., Li Y., Jin X., Tian T., Ma Q., Zhao Z., Lin S.Y., Chen Z., Li B., Yao G. ATP-dependent dynamic protein aggregation regulates bacterial dormancy depth critical for antibiotic tolerance. Mol. Cell. 2019;73:143–156 e4. doi: 10.1016/j.molcel.2018.10.022. [DOI] [PubMed] [Google Scholar]
- Pu Y., Zhao Z., Li Y., Zou J., Ma Q., Zhao Y., Ke Y., Zhu Y., Chen H., Baker M.A. Enhanced efflux activity facilitates drug tolerance in dormant bacterial cells. Mol. Cell. 2016;62:284–294. doi: 10.1016/j.molcel.2016.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riffaud C., Pinel-Marie M.-L., Felden B. Cross-regulations between bacterial toxin–antitoxin systems: evidence of an interconnected regulatory network? Trends Microbiol. 2020;28:851–866. doi: 10.1016/j.tim.2020.05.016. [DOI] [PubMed] [Google Scholar]
- Riffaud C., Pinel-Marie M.-L., Pascreau G., Felden B. Functionality and cross-regulation of the four SprG/SprF type I toxin–antitoxin systems in Staphylococcus aureus. Nucleic Acids Res. 2019;47:1740–1758. doi: 10.1093/nar/gky1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronneau S., Helaine S. Clarifying the link between toxin-antitoxin modules and bacterial persistence. J. Mol. Biol. 2019;431:3462–3471. doi: 10.1016/j.jmb.2019.03.019. [DOI] [PubMed] [Google Scholar]
- Sahukhal G.S., Pandey S., Elasri M.O. msaABCR operon is involved in persister cell formation in Staphylococcus aureus. BMC Microbiol. 2017;17:218. doi: 10.1186/s12866-017-1129-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders C.C. Ciprofloxacin: in vitro activity, mechanism of action, and resistance. Rev. Infect. Dis. 1988;10:516–527. doi: 10.1093/clinids/10.3.516. [DOI] [PubMed] [Google Scholar]
- Sassi M., Augagneur Y., Mauro T., Ivain L., Chabelskaya S., Hallier M., Sallou O., Felden B. SRD: a Staphylococcus regulatory RNA database. Rna. 2015;21:1005–1017. doi: 10.1261/rna.049346.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher M.A., Balani P., Min J., Chinnam N.B., Hansen S., Vulić M., Lewis K., Brennan R.G. HipBA–promoter structures reveal the basis of heritable multidrug tolerance. Nature. 2015;524:59–64. doi: 10.1038/nature14662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenkutie A.M., Yao M.Z., Siu G.K.-H., Wong B.K.C., Leung P.H.-M. Biofilm-induced antibiotic resistance in clinical acinetobacter baumannii isolates. Antibiotics. 2020;9:817. doi: 10.3390/antibiotics9110817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarstad K., Boye E. The initiator protein DnaA: evolution, properties and function. Biochim. Biophys. Acta. 1994;1217:111–130. doi: 10.1016/0167-4781(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Soltani M., Vargas-Garcia C.A., Antunes D., Singh A. Intercellular variability in protein levels from stochastic expression and noisy cell cycle processes. PLoS Comput. Biol. 2016;12:e1004972. doi: 10.1371/journal.pcbi.1004972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S., Gong T., Yamasaki R., Kim J.S., Wood T.K. Identification of a potent indigoid persister antimicrobial by screening dormant cells. Biotechnol. Bioeng. 2019;116:2263–2274. doi: 10.1002/bit.27078. [DOI] [PubMed] [Google Scholar]
- Song S., Wood T.K. ppGpp ribosome dimerization model for bacterial persister formation and resuscitation. Biochem. Biophysical Res. Commun. 2020;523:281–286. doi: 10.1016/j.bbrc.2020.01.102. [DOI] [PubMed] [Google Scholar]
- Song S., Wood T.K. Are we really studying persister cells? Environ. Microbiol. Rep. 2021;13:3–7. doi: 10.1111/1758-2229.12849. [DOI] [PubMed] [Google Scholar]
- Sulaiman J.E., Lam H. Proteomic investigation of tolerant Escherichia coli populations from cyclic antibiotic treatment. J. Proteome Res. 2020;19:900–913. doi: 10.1021/acs.jproteome.9b00687. [DOI] [PubMed] [Google Scholar]
- Trastoy R., Manso T., Fernandez-Garcia L., blasco L., Ambroa A., del Molino M.P., Bou G., Garcia-Contreras R., Wood T., Tomas M. Mechanisms of bacterial tolerance and persistence in the gastrointestinal and respiratory environments. Clin. Microbiol. Rev. 2018;31 doi: 10.1128/CMR.00023-18. e00023–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsilibaris V., Maenhaut-Michel G., Mine N., van Melderen L. What is the benefit to <em>Escherichia coli</em> of having multiple toxin-antitoxin systems in its genome? J. Bacteriol. 2007;189:6101–6108. doi: 10.1128/JB.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully J.G., Rose D.L., Whitcomb R., Wenzel R. Enhanced isolation of Mycoplasma pneumoniae from throat washings with a newly modified culture medium. J. Infect. Dis. 1979;139:478–482. doi: 10.1093/infdis/139.4.478. [DOI] [PubMed] [Google Scholar]
- Tuomanen E., Cozens R., Tosch W., Zak O., Tomasz A. The rate of killing of Escherichia coli byβ-lactam antibiotics is strictly proportional to the rate of bacterial growth. Microbiology. 1986;132:1297–1304. doi: 10.1099/00221287-132-5-1297. [DOI] [PubMed] [Google Scholar]
- Uruén C., Chopo-Escuin G., Tommassen J., Mainar-Jaime R.C., Arenas J. Biofilms as promoters of bacterial antibiotic resistance and tolerance. Antibiotics. 2021;10:3. doi: 10.3390/antibiotics10010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Den Bergh B., Michiels J.E., Wenseleers T., Windels E.M., Boer P.V., Kestemont D., de Meester L., Verstrepen K.J., Verstraeten N., Fauvart M., Michiels J. Frequency of antibiotic application drives rapid evolutionary adaptation of Escherichia coli persistence. Nat. Microbiol. 2016;1:16020. doi: 10.1038/nmicrobiol.2016.20. [DOI] [PubMed] [Google Scholar]
- van Gemen B., Koets H.J., Plooy C.A., Bodlaender J., van Knippenberg P.H. Characterization of the ksgA gene of Escherichia coli determining kasugamycin sensitivity. Biochimie. 1987;69:841–848. doi: 10.1016/0300-9084(87)90210-0. [DOI] [PubMed] [Google Scholar]
- Villellas C., Aristimuño L., Vitoria M.-A., Prat C., Blanco S., DE Viedma D.G., Domínguez J., Samper S., Aínsa J.A. Analysis of mutations in streptomycin-resistant strains reveals a simple and reliable genetic marker for identification of the Mycobacterium tuberculosis Beijing genotype. J. Clin. Microbiol. 2013;51:2124–2130. doi: 10.1128/JCM.01944-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov I.L., Seefeldt A.C., Johansson M. Tracking of single tRNAs for translation kinetics measurements in chloramphenicol treated bacteria. Methods. 2019;162:23–30. doi: 10.1016/j.ymeth.2019.02.004. [DOI] [PubMed] [Google Scholar]
- Wang X., Lord D.M., Hong S.H., Peti W., Benedik M.J., Page R., Wood T.K. Type II toxin/antitoxin MqsR/MqsA controls type V toxin/antitoxin GhoT/GhoS. Environ. Microbiol. 2013;15:1734–1744. doi: 10.1111/1462-2920.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Wood T.K. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 2011;77:5577–5583. doi: 10.1128/AEM.05068-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W., Liu B., Xue L., Zhu Z., Niu L., Sun B. Autoregulation and virulence control by the toxin-antitoxin system SavRS in Staphylococcus aureus. Infect. Immun. 2018;86 doi: 10.1128/IAI.00032-18. e00032–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmaerts D., Herpels P., Michiels J., Verstraeten N. Genetic determinants of persistence in Escherichia coli. Persister Cells Infect. Dis. 2019:133–180. [Google Scholar]
- Wilmaerts D., Windels E.M., Verstraeten N., Michiels J. General mechanisms leading to persister formation and awakening. Trends Genet. 2019;35:401–411. doi: 10.1016/j.tig.2019.03.007. [DOI] [PubMed] [Google Scholar]
- Wood T.K., Song S. Forming and waking dormant cells: the ppGpp ribosome dimerization persister model. Biofilm. 2020;2:100018. doi: 10.1016/j.bioflm.2019.100018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N., He L., Cui P., Wang W., Yuan Y., Liu S., Xu T., Zhang S., Wu J., Zhang W. Ranking of persister genes in the same Escherichia coli genetic background demonstrates varying importance of individual persister genes in tolerance to different antibiotics. Front. Microbiol. 2015;6:1003. doi: 10.3389/fmicb.2015.01003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y., Wei Y., Shen Y., Li X., Zhou H., Tai C., Deng Z., Ou H.Y. Tadb 2.0: an updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res. 2018;46:D749–D753. doi: 10.1093/nar/gkx1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki R., Song S., Benedik M.J., Wood T.K. Persister cells resuscitate using membrane sensors that activate chemotaxis, lower cAMP levels, and revive ribosomes. Iscience. 2020;23:100792. doi: 10.1016/j.isci.2019.100792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. & Li, Y. 2010. PhoU (PerF), A persistence switch involved in persister formation and tolerance to multiple antibiotics and stresses as a drug target for persister bacteria. Google Patents. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data that supports the findings of this study are available from the corresponding author upon request. Whole genomes data of P026, PK07_L1, PK07_L2, PS04_L1, PS04_L2, PC06_L1, PC06_L2, PSC09_L1 and PSC09_L2 strains has been deposited on the NCBI Genome Bank in BioProject PRJNA635211 with the accession number CP053944, CP069339, CP069340, CP069341, CP069342, CP069343, CP069344, CP069345, CP069346, respectively. The code used for colony counting is available on GitHub at https://github.com/hdeter/CountColonies.