

Streptococcus pneumoniae remains a leading cause of bacterial pneumonia despite the widespread use of vaccines. While vaccines are effective at reducing the incidence of most serotypes included in vaccines, a rise in infection due to nonvaccine serotypes and moderate efficacy against some vaccine serotypes have contributed to high disease incidence.
KEYWORDS: Streptococcus pneumoniae, monoclonal antibodies, pneumococcal infection
ABSTRACT
Streptococcus pneumoniae remains a leading cause of bacterial pneumonia despite the widespread use of vaccines. While vaccines are effective at reducing the incidence of most serotypes included in vaccines, a rise in infection due to nonvaccine serotypes and moderate efficacy against some vaccine serotypes have contributed to high disease incidence. Additionally, numerous isolates of S. pneumoniae are antibiotic or multidrug resistant. Several conserved pneumococcal proteins prevalent in the majority of serotypes have been examined for their potential as vaccines in preclinical and clinical trials. An additional, yet-unexplored tool for disease prevention and treatment is the use of human monoclonal antibodies (MAbs) targeting conserved pneumococcal proteins. Here, we isolated the first human MAbs (PhtD3, PhtD6, PhtD7, PhtD8, and PspA16) against the pneumococcal histidine triad protein (PhtD) and the pneumococcal surface protein A (PspA), two conserved and protective antigens. MAbs to PhtD target diverse epitopes on PhtD, and MAb PspA16 targets the N-terminal segment of PspA. The PhtD-specific MAbs bind to multiple serotypes, while MAb PspA16 serotype breadth is limited. MAbs PhtD3 and PhtD8 prolong the survival of mice infected with pneumococcal serotype 3. Furthermore, MAb PhtD3 prolongs the survival of mice in intranasal and intravenous infection models with pneumococcal serotype 4 and in mice infected with pneumococcal serotype 3 when administered 24 h after pneumococcal infection. All PhtD and PspA MAbs demonstrate opsonophagocytic activity, suggesting a potential mechanism of protection. Our results identify new human MAbs for pneumococcal disease prevention and treatment and identify epitopes on PhtD and PspA recognized by human B cells.
INTRODUCTION
Streptococcus pneumoniae remains a leading cause of infectious morbidity and mortality despite the widespread use of two vaccines for disease prevention (1). The World Health Organization estimates that over 1 million deaths occur worldwide each year due to pneumococcal infection (2). Similar to other respiratory pathogens, individuals below the age of 2 and above the age of 65 are more susceptible to invasive pneumococcal disease (3). In addition, there is also an increased frequency and risk of severe infection in individuals with preexisting conditions, including those with diabetes, chronic obstructive pulmonary disease, cardiovascular diseases, and human immunodeficiency virus (4). Although vaccination is widespread in the developed world, pneumococcal infection is responsible for 30% of adult pneumonia and has a mortality rate of 11 to 40% (5). Furthermore, in regions of the world with high childhood mortality rates, pneumococcal pneumonia is the cause of 20 to 50% of deaths in children (6).
S. pneumoniae is a common resident of the upper respiratory tract (7), and pneumococcal carriage precedes active infection (8). In young children, carriage rates of S. pneumoniae can be as high as 60% (9). Colonization is typically asymptomatic; however, S. pneumoniae can rapidly disseminate, often following a primary infection such as influenza (10) or coronavirus disease 2019 (COVID-19) (11), to cause pneumonia and invasive disease. Repeated colonization with S. pneumoniae typically results in immunization, and several studies have determined that colonization induces serum antibody responses to the capsular polysaccharide (12) and both serum antibody (13–17) and cellular immune responses to protein antigens (18, 19). These antibody levels in serum increase during the first few years of life (16) but tend to decrease in the elderly (20), which may contribute to the higher risk of disease in children and the elderly.
The majority of S. pneumoniae isolates are encapsulated, and 100 capsular serotypes have been identified (21), which are based on differences in the chemical structures of the capsular polysaccharide in each serotype (22). Current vaccines are based on eliciting opsonophagocytic antibody responses to the capsular polysaccharide and utilize either a 13-valent diphtheria toxoid conjugate vaccine to elicit T-dependent, high-affinity, and class-switched antibody responses (PCV13) or a 23-valent capsular polysaccharide mixture (PPSV23) to elicit T-independent antibody responses or as a booster to PCV13. Antiglycan antibodies produced in response to the vaccine are serotype specific due to the distinct chemical structures of the capsular polysaccharides (23). Although vaccines have been highly effective at reducing the incidence of pneumococcal disease, a rise in the incidence of nonvaccine serotypes has occurred, termed serotype replacement (24). In addition, the incidence of invasive disease due to serotypes 3 and 19A have persisted in some reports despite widespread vaccination (25). In terms of treatment, antibiotic resistance among nonvaccine serotypes has risen, and this presents challenges in treating pneumococcal infection (26). Based on the limitations of current vaccines and treatments, additional options are currently being explored. For many years, such research has focused on developing vaccines that are broadly reactive, primarily based on the idea that conserved protein antigens present in the majority of pneumococcal serotypes would be effective at preventing disease independent of serotype (27). Multiple antigens have been tested in preclinical infection models, with several entering clinical trials, including the toxin pneumolysin, pneumococcal surface protein A (PspA), pneumococcal surface antigen A (PsaA), pneumococcal choline binding protein A (PcpA), PcsB, serine threonine kinase protein (StkP), and pneumococcal histidine triad protein (PhtD) (28).
PhtD is a member of a group of conserved surface proteins on S. pneumoniae that also includes PhtA, PhtB, and PhtC, all of which share histidine triad motifs (29). The proteins have high sequence homology to each other, and PhtB and PhtD share 87% sequence homology (30). PhtD is highly conserved, with amino acid sequence identity varying from 91 to 98% among strains isolated from invasive disease cases in children (31). One study of 107 pneumococcal strains showed that PhtD was expressed in 100% of tested serotypes (30), while other studies have found that PhtD is widely prevalent but is absent in a subset of isolated strains (30, 32, 33). The function of the Pht family of proteins has not been fully elucidated, although data have implicated the proteins in attachment of S. pneumoniae to respiratory epithelial cells (34, 35). In addition, the first histidine triad motif of PhtD has been shown to be important for zinc acquisition and bacterial homeostasis (36). Although the full structure of PhtD has not been determined, a crystal structure of the third histidine triad motif bound to Zn2+, and a solution nuclear magnetic resonance (NMR) structure of the N-terminal fragment of PhtD has been determined (37, 38).
All Pht proteins are immunogenic and induce protective humoral immunity, and vaccination with these proteins was shown to reduce colonization, sepsis, and pneumonia (29, 39, 40). PhtD has been shown to protect against systemic pneumococcal disease in a mouse model (29), and immunization of rhesus macaques with PhtD along with detoxified pneumolysin protected the animals against pneumococcal infection (41). Fragments of PhtD have also been assessed for protective efficacy, and somewhat conflicting reports have demonstrated that both the N and C terminus are immunogenic and protective (42, 43). PhtD was recently used as an antigen in a phase IIb clinical trial, demonstrating that PhtD remains an antigen of interest in pneumococcal vaccinology, although PhtD was administered along with PCV13, so a direct comparison of PhtD versus PCV13 was not accomplished (44). Mouse monoclonal antibodies to PhtD were shown to protect mice using a macrophage- and complement-dependent mechanism (45), and human polyclonal antibodies to PhtD were shown to reduce adherence of the pneumococcus to lung epithelial cells and reduce murine nasopharyngeal colonization (46). Human polyclonal antibodies generated in response to alum-adjuvanted PhtD vaccination were also shown to protect mice from pneumococcal disease (47).
Another vaccine antigen, PspA, is an important virulence factor of S. pneumoniae and one of the most abundant surface proteins (48). As with PhtD, PspA is found in the majority of examined clinical isolates (33, 49). PspA mutant strains are cleared faster from the blood of mice compared to intact strains (50), and vaccination with PspA protects mice from pneumococcal infection (51–57). PspA is less conserved than PhtD and is grouped into three families with >55% identity and six clades with >75% identity (58). PspA has four distinct structural domains, including the alpha-helical region, the proline-rich region, the choline-binding repeat domain, and the cytoplasmic tail, of which the proline-rich region is highly conserved across clades, while the N-terminal alpha-helical region is more variable (59). PspA has been shown to inhibit complement deposition (60–62) and has shown specificity for binding of human lactoferrin, although the importance of this binding is unclear (63). An X-ray crystal structure of the lactoferrin-binding domain of PspA in complex with the N-terminal region of human lactoferrin has been determined (64). Mouse monoclonal antibodies (MAbs) to PspA have been shown to prolong survival of mice and improve efficacy of antibiotic treatment (63). Additionally, antibodies isolated from humans following immunization with recombinant PspA are broadly cross-reactive and protect mice from pneumococcal infection with heterologous PspA (65, 66). A clinical trial of a recombinant attenuated Salmonella enterica serovar Typhi vaccine vector producing PspA has been completed (NCT01033409), and a protein-based phase Ia clinical trial incorporating PspA is under way (NCT04087460).
It is well defined that antibodies can prevent pneumococcal infection based on the success of antibody-based pneumococcal vaccines. Since both PspA and PhtD are protective antigens and elicit protective antibodies, it is reasonable to assume that human MAbs to these antigens would be protective. As these proteins are highly conserved across pneumococcal serotypes, MAbs to PhtD and PspA could prevent and possibly treat disease caused by a broad spectrum of pneumococcal serotypes. Human MAbs are promising as therapeutics for bacterial pathogens, as bezlotoxumab was FDA approved for prevention of recurrent Clostridioides (formerly Clostridium) difficile infection (67). However, no human MAbs to any pneumococcal protein antigens have been isolated. Serum antibodies to PhtD and PspA are elicited in response to pneumococcal carriage (16, 68, 69), and in this study, we generated human monoclonal antibodies to PhtD and PspA from healthy human subjects. We determined the serotype breadth and epitope specificity of the MAbs and demonstrated the protective efficacy of PhtD-specific human MAbs in multiple mouse models of pneumococcal infection.
RESULTS
Isolation of pneumococcal protein-specific human MAbs.
To identify PhtD and PspA-specific human MAbs, we recombinantly expressed His-tagged PhtD and PspA from strain TCH8431 (serotype 19A) in Escherichia coli (Fig. 1A) and utilized these proteins to screen stimulated B cells from human donor peripheral blood mononuclear cells (PBMCs) as previously described (70). PBMCs from healthy human subjects were plated onto a feeder layer expressing human CD40L, human interleukin 21 (IL-21), and human B-cell-activating factor (BAFF) for 6 days to stimulate B cell growth and antibody secretion. Cell supernatants from the stimulated B cells were screened against recombinant PhtD and PspA by enzyme-linked immunosorbent assay (ELISA). Responses to the recombinant proteins varied between subjects, as shown in an example in Fig. 1B. From five subjects’ PBMCs, we fused several reactive wells for generation of human hybridomas and subsequent human MAb isolation. Four hybridoma lines, each from a unique donor, were successfully generated and biologically cloned by single-cell sorting for PhtD, and one MAb was generated for PspA from an independent subject. The MAbs to PhtD had similar 50% effective concentrations (EC50s) for binding, as determined by ELISA (Fig. 1C), and MAbs to PhtD and PspA bound with high avidity, with EC50s ranging from 26 to 45 ng/ml (Fig. 1C and D). To determine the V, D, and J genes utilized by each MAb, the hybridomas were sequenced by reverse transcription-PCR (RT-PCR) followed by TA cloning; the results are shown in Table 1, and the sequences are provided in the supplemental material. MAbs PhtD3 and PhtD6 utilize kappa light chains, while MAbs PhtD7 and PhtD8 use lambda light chains. All MAbs were of the IgG1 isotype based on isotyping data determined by ELISA. All MAbs utilize unique heavy-chain and light-chain V genes, with the exception of MAbs PhtD7 and PhtD8, as these share predicted VL and JL gene usage, although complementarity-determining region 3 of the light chain (LCDR3) sequences share little sequence identity. MAbs PhtD7 and PhtD8 share VH and JH gene usage, although they vary in the use of the DH gene, which leads to stark differences in CDR3 lengths, with MAbs PhtD7 and PhtD8 having 20-amino-acid and 8-amino-acid lengths of CDR3 of the heavy chain (HCDR3), respectively. MAb PspA16 shares V gene usage with MAbs PhtD7 and PhtD8.
FIG 1.
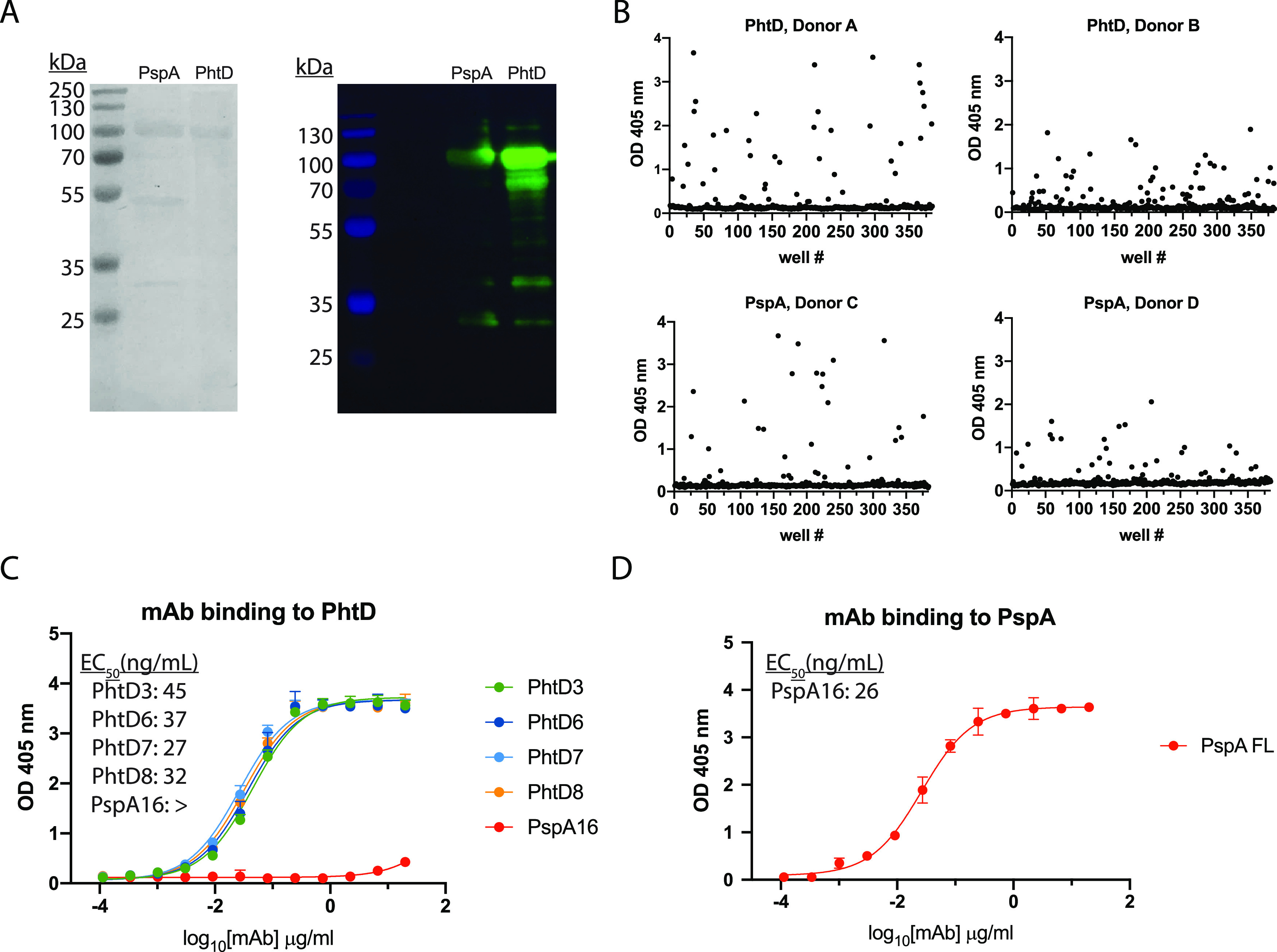
Antibody responses and MAb binding properties to recombinant PhtD and PspA proteins. (A) SDS-PAGE (left) and Western blot (right) of purified recombinantly expressed PspA and PhtD. Both proteins were pure, with the appearance of degradation products. (B) ELISA binding responses from the supernatant of stimulated B cells to recombinant PhtD and PspA proteins. (C) ELISA binding curves of anti-PhtD MAbs against recombinant PhtD protein. PspA16 was utilized as a negative control. The symbol “>” indicates that no binding was observed at an OD405 greater than 1 at the highest concentration. (D) Binding of PspA16 to recombinant PspA. For panels C and D, computed EC50s are reported from a nonlinear regression curve fit (agonist). Data points are averages of four replicates from one of at least two independent experiments. Error bars indicate 95% confidence intervals.
TABLE 1.
Summary of genetic characteristics of pneumococcus-specific MAbsa
| MAb | Isotype | VH gene (% mutation) | DH | JH | HCDR3 sequence | VL (% mutation) | JL | LCDR3 sequence |
|---|---|---|---|---|---|---|---|---|
| PhtD3 | IgG1, κ | V1-69*18 (85) | D3-16*01 | J3*01 | ARDGHIMRTTLSDAALDV | V3-20*01 (91) | J4*01 | QQYQNSPFT |
| PhtD6 | IgG1, κ | V1-8*01 (93) | D2-15*01 | J5*02 | ARGPYWVENWFDT | V1-39*01 (92) | J1*01 | QQSYSNQKT |
| PhtD7 | IgG1, λ | V1-2*02 (91) | D3-16*01 | J4*02 | ARVLRGSYDFRGNYPHDFDY | V4-69*01 (88) | J3*02 | QTWDTGLQGV |
| PhtD8 | IgG1, λ | V1-2*02 (93) | D2-15*01 | J4*02 | ARGGTLDH | V4-69*01 (94) | J3*02 | HTWVTNIHLV |
| PspA16 | IgG1, κ | V1-2*02 (93) | D3-10*01 | J1*01 | ARAWAPGAEYLHH | V3-20*01 (94) | J3*01 | QQHDHSPFT |
Analysis was performed using IMGT/V-Quest.
Epitope mapping of the human MAbs.
To identify the specific regions of PhtD targeted by the human MAbs, we generated truncated fragments of PhtD based on a secondary structure predictor. The fragments were fused to the maltose-binding protein (MBP) to ensure solubility and were expressed in E. coli and purified using amylose resin. The majority of the fragments were >90% pure with the exception of free MBP protein for the MBP fusion proteins (Fig. 2A). To identify the specific regions of PhtD targeted by the isolated MAbs, we measured ELISA binding of MAbs to fragments of PhtD. Since there are no previously generated MAbs to these proteins with defined epitopes, the generated fragments provide rough estimates of MAb epitopes. Each of the four MAbs binds to a unique region on the PhtD protein (Fig. 2B). MAbs PhtD3 and PhtD6 bind the N-terminal portion of the protein, while MAb PhtD8 binds the C-terminal portion. MAb PhtD7 appears to target a unique conformational epitope that is dependent on amino acids 341 to 838, but this MAb does not bind the fragments from amino acids 341 to 647 or 645 to 838. We next assessed the epitopes of the MAbs by competitive biolayer interferometry to compare the binding epitopes between MAbs. Anti-penta-His biosensors were loaded with His-tagged PhtD protein, and MAbs were competed for binding sequentially (Fig. 2C and D). The MAbs bind distinct regions with limited competition similar to results from the fragment ELISA data. MAbs PhtD3 and PhtD6 show intermediate competition, and the epitopes for these MAbs also overlap in our fragment ELISA data. To map the binding region of MAb PspA16, we fragmented PspA into several truncations based on previously determined domains (Fig. 3A) (59). MAb PspA16 had high avidity to recombinant PspA and bound to the N-terminal fragment from amino acids 1 to 247, based on positive binding to fragments from amino acids 1 to 438 and 1 to 512 and negative binding to amino acids 247 to 512, 436 to 725, and 247 to 725 (Fig. 3B).
FIG 2.

Epitope mapping of anti-PhtD MAbs. (A) SDS-PAGE of the purified maltose-binding protein (MBP)–PhtD fragment fusion proteins. Each fusion protein was pure after purification with the exception of free MBP. (B) ELISA binding curves of the PhtD MAbs to each MBP-PhtD fragment. A summary of the binding curves is displayed below the binding curves, where cells in blue indicate binding and those in white indicate no binding. Data points are averages of four replicates from one of at least two independent experiments. Error bars indicate 95% confidence intervals. (C) Example of an epitope mapping experiment for the anti-PhtD MAbs. The top graph displays the signal from biolayer interferometry of the first MAb loaded onto immobilized PhtD protein. The signal for each MAb is colored according to the legend. The bottom graph displays the signal from loading of the second MAb in the presence of the first MAb, MAb PhtD3 in this example. A decrease in signal compared to the top graph is observed for MAbs PhtD3 and PhtD8, as MAb PhtD3 competes with itself and MAbs PhtD3 and PhtD6 have partially overlapping epitopes. (D) Epitope mapping of the PhtD-specific MAbs. Data are percent binding of the competing antibody in the presence of the primary antibody, compared with the competing antibody alone. Cells in black indicate full competition, in which ≤33% of the uncompeted signal was observed; gray indicates intermediate competition, in which the signal was between 33% and 66%; and white indicates noncompetition, where the signal was ≥66%.
FIG 3.
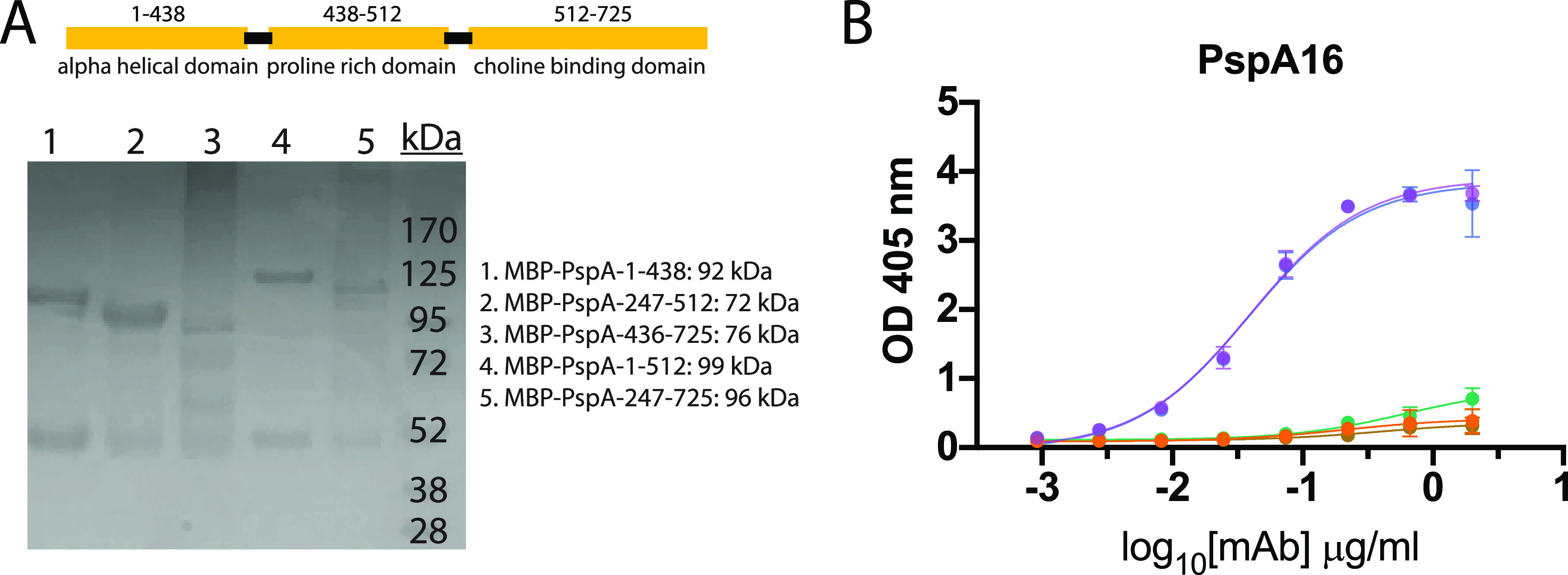
Epitope mapping of PspA16. (A) SDS-PAGE of recombinant MBP PspA fragment fusion proteins. Fragments 1, 2, 4, and 5 purified well, with only visible MBP protein as a contaminant. PspA fragment 3 has multiple copurified bands and/or degradation products. (B) ELISA binding curves for PspA16 to each fragment. PspA16 bound to fragment 1 and fragment 4, but not others, suggesting the epitope lies within amino acids 1 to 247. Data points are averages of four replicates from one of at least two independent experiments. Error bars indicate 95% confidence intervals.
Serotype breadth of the isolated PhtD-specific MAbs.
Pneumococcal surface proteins PhtD and PspA are conserved across serotypes and are widely prevalent in the majority of serotypes. As such, human MAbs to these antigens could have the potential to treat pneumococcal infection from multiple serotypes. In order to determine the serotype breadth of the isolated MAbs, we initially assessed MAb binding to strains of two diverse pneumococcal serotypes, strain TCH8431 (serotype 19A), from which the genes for recombinant PhtD and PspA proteins were cloned and expressed, and the commonly used laboratory strain TIGR4 (serotype 4). PspA shares 88% amino acid sequence identity between TCH8431 and TIGR4, although significant variability is present in the N-terminal domain, with 70% identity in amino acids 1 to 247. In contrast, PhtD shares 98% amino acid sequence identity between these two strains. We conducted Western blotting by probing bacterial lysates from TIGR4 and TCH8431 with MAbs PhtD3 and PspA16. MAb PspA16 labels only PspA protein from strain TCH8431 (Fig. 4A), while MAb PhtD3 is able to label PhtD protein from both pneumococcal strains. However, as the bacterial lysis likely results in protein denaturation, it is possible that the epitope for MAb PspA16 is altered during denaturation. We next determined if MAbs isolated against each of the recombinant proteins bind whole bacteria. We conducted ELISAs by coating plates with fixed bacteria and measuring MAb binding by ELISA. MAbs PhtD3, PhtD6, PhtD7, and PhtD8 were broadly reactive across multiple unrelated pneumococcal serotypes, and MAbs PhtD3, PhtD6, and PhtD7 had higher avidity for fixed bacteria than PhtD8 (Fig. 4B). In contrast, PspA16 bound only to strain TCH8431, similar to results from the Western blot experiments. Since PspA16 binds to the most variable region of PspA, the reduced binding to divergent serotypes was expected. In a third experiment, we assessed binding of the MAbs to a panel of pneumococcal serotypes by flow cytometry. As shown in Fig. 4C and Fig. S1, we utilized serum from a donor vaccinated 21 days previously with Prevnar-13 as a positive control. The PhtD MAbs bound to the majority of tested serotypes, with MAbs PhtD3 and PhtD8 showing the broadest binding. In contrast, PspA16 bound only to TCH8431 and the serotype 3 strain WU2.
FIG 4.
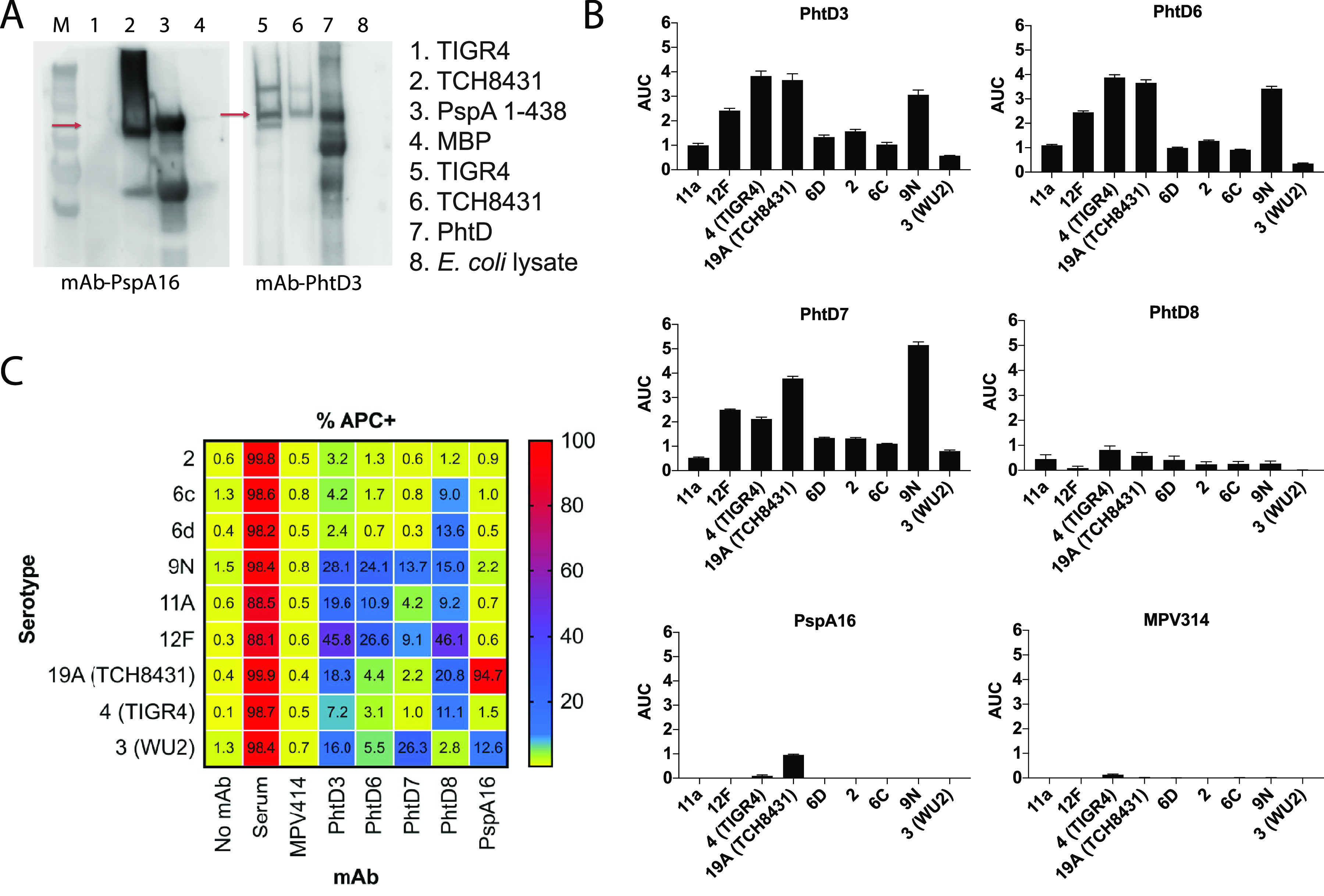
Serotype breadth of the isolated MAbs. (A) Western blot of TIGR4 and TCH8431 strains with PspA16 and PhtD3 as the primary antibodies. In the Western blot for PspA16, the PspA fragment from aa 1 to 438 fused to the MBP was used as the positive control, and MBP was used as the negative control. In the PhtD3 Western blot, recombinant PhtD was used as the positive control, and E. coli lysates were used as the negative control. (B) Area-under-the-curve (AUC) values calculated from ELISA binding curves of serially diluted MAbs against plates coated with fixed bacteria. The ELISA binding curves were the averages of four data points from one of at least two independent experiments. The baseline for the AUC calculation was set as the average of the signal for the highest concentration (20 μg/ml) of the negative control MAb MPV314. Error bars show standard errors of the AUC calculation. (C) Example gating strategy for antibody binding to bacteria. Bacteria were labeled with CFSE, and antibodies were labeled with APC. (D) Heat map and percentages for antibody binding to each pneumococcal serotype. Data are averages from 3 or 4 experiments and are the percentage of bacteria that are APC positive. MPV314 and MPV414 are human antibodies specific to the human metapneumovirus fusion protein, and these were used as negative controls.
PhtD3 protects mice from fatal pneumococcal infection.
As MAbs PhtD3 and PhtD8 exhibited the highest overall breadth in the serotype binding analysis by flow cytometry, these MAbs were further analyzed for protective efficacy in the mouse model. In addition, these MAbs were chosen in order to determine whether the epitope specificity of MAbs to PhtD affects protective efficacy, as they target nonoverlapping epitopes. Mouse MAbs to PhtD (45) and polyclonal human antibodies from both healthy human subjects (46) and PhtD-vaccinated humans (47) have been shown to protect against colonization or disease in mouse models of pneumococcal infection. However, no human MAbs have been examined for protective efficacy. To determine if the PhtD-specific MAbs protect against infection, we examined the efficacy of MAbs PhtD3 and PhtD8 in a mouse model of pneumococcal pneumonia with a serotype 3 strain (WU2), as serotype 3 is a leading cause of invasive pneumococcal disease (71). Since the MAbs were isolated from human hybridomas, and thus have authentic human Fc regions, we isotype-switched the Fc region to the closest mouse homolog (human IgG1 became mouse IgG2a). MAbs PhtD3 and PhtD8 chimeras with mouse IgG2a Fc regions (PhtD3-IgG2a and PhtD8-IgG2a) were recombinantly expressed in HEK293F cells for testing in the mouse model. As a control for the study, we purchased a mouse IgG2a isotype control antibody.
We first examined the binding of the MAbs to ensure that binding was still observed for the recombinant PhtD3-IgG2a and PhtD8-IgG2a MAbs and that no binding was observed for the isotype control MAb. As expected, PhtD3-IgG2a and PhtD8-IgG2a had binding avidity for recombinant PhtD similar to that of hybridoma-derived PhtD, while the isotype control showed no binding (Fig. 5A). We first tested the prophylactic efficacy of PhtD3-IgG2a and PhtD8-IgG2a in a pneumonia model with pneumococcal serotype 3. Both MAbs prolonged the survival of mice compared to the isotype control, although mice treated with MAb PhtD3 demonstrated higher survival (80% versus 30%) (Fig. 5B and C). As MAb PhtD3-IgG2a protected a larger percentage of mice, we chose this MAb for further analysis.
FIG 5.
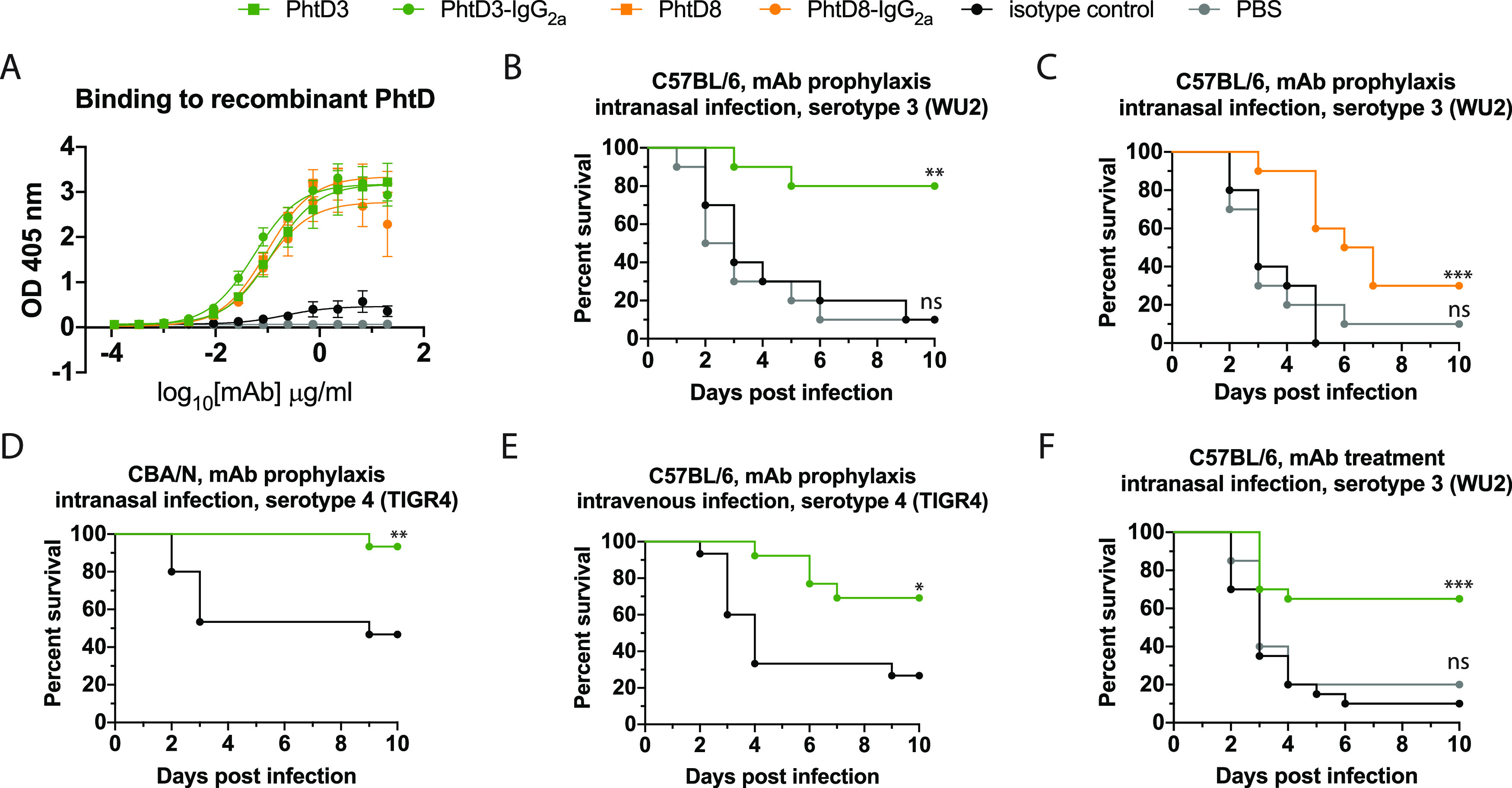
Protective efficacy of anti-PhtD MAbs. (A) ELISA binding curve of MAb PhtD3, the isotype-switched MAb PhtD-IgG2a, and an IgG2a isotype control. Data points are averages of four replicates from one of at least two independent experiments. Error bars indicate 95% confidence intervals. (B) Prophylactic efficacy of MAb PhtD3 in an intranasal infection model of pneumococcal serotype 3 (strain WU2) in C57BL/6 mice. **, P = 0.0012; ns, not significant via log-rank (Mantel-Cox) test. n = 10 mice/group. (C) Prophylactic efficacy of MAb PhtD8 in an intranasal infection model of pneumococcal serotype 3 (strain WU2) in C57BL/6 mice. ***, P = 0.0009; ns, not significant via log-rank (Mantel-Cox) test. n = 10 mice/group. (C) Prophylactic efficacy of MAb PhtD3 in an intranasal infection model of pneumococcal serotype 4 (strain TIGR4) in CBA/N mice. **, P = 0.0045 via log-rank (Mantel-Cox) test. n = 15 mice/group. (D) Prophylactic efficacy of MAb PhtD3 in an intravenous infection model of pneumococcal serotype 4 (strain TIGR4) in C57BL/6 mice. **, P = 0.0101 via log-rank (Mantel-Cox) test. n = 13 to 15 mice/group. (E) Treatment efficacy of MAb PhtD3 in an intranasal infection model of pneumococcal serotype 3 (strain WU2) in C57BL/6 mice. ***, P = 0.0002; ns, not significant via log-rank (Mantel-Cox) test. n = 20 mice/group.
MAb PhtD3-IgG2a was tested for protective efficacy against pneumococcal serotype 4 (TIGR4) to determine whether the broad binding correlates to broad protection. In experiments with TIGR4, we used only an isotype control MAb group, since no significant difference was observed between the phosphate-buffered saline (PBS) and isotype control MAb groups in the serotype 3 experiments. For this serotype, we used CBA/N mice for the intranasal infection model, as TIGR4 was not sufficiently lethal by intranasal infection in C57BL/6 mice. CBA/N mice have previously been shown to be susceptible to serotype 4 (72). PhtD3-IgG2a prolonged survival of mice, providing 93% protection, compared to 47% for the isotype control (Fig. 5C). As we were not able to test PhtD3-IgG2a in an intranasal infection model with TIGR4 in C57BL/6 mice, we conducted an experiment in C57BL/6 mice in which mice were intravenously infected with TIGR4 to model septic pneumococcal infection. In this study, PhtD3-IgG2a prolonged survival of mice with 69% efficacy, compared to 27% survival with the isotype control (Fig. 5D). The most clinically relevant scenario for MAb treatment of pneumococcal infection would be administration after pneumococcal infection. To model such a scenario, we infected mice with pneumococcal serotype 3 and administered MAb PhtD3-IgG2a 24 h after infection. In this model, 65% of PhtD3-IgG2a-treated mice survived the infection, compared to 10% for the isotype control group (Fig. 5E).
PhtD-specific human MAbs have opsonophagocytic activity.
The correlate of protection for current pneumococcal vaccines is based on the elicitation of anti-capsule antibodies that opsonize bacteria, leading to their phagocytosis by host immune cells and subsequent bacterial killing (73, 74). Mouse MAbs isolated by vaccination with PhtD were previously shown to induce bacterial opsonophagocytosis, which was dependent on complement and macrophages (45). To determine a potential mechanism of protection by PhtD3 and additional PhtD MAbs, we utilized established opsonophagocytosis killing assays (OPKAs) using the HL-60 cell line. We tested the MAbs against serotype 4 (strain TIGR4), serotype 3 (strain WU2), and serotype 19A (strain TCH8431), from which our PhtD and PspA constructs were cloned. These MAbs were also compared to purified IgG obtained from a human subject previously vaccinated with Prevnar-13 21 days before blood collection, as the OPKA is the standard to measure vaccine uptake (75). All PhtD MAbs resulted in a decrease in the number of CFU of all three serotypes compared to no antibody and an irrelevant MAb to human metapneumovirus (Fig. 6A). PspA16 also decreased CFU against all three serotypes, although the efficacy against serotype 4 was lower, as expected based on the serotype binding data.
FIG 6.
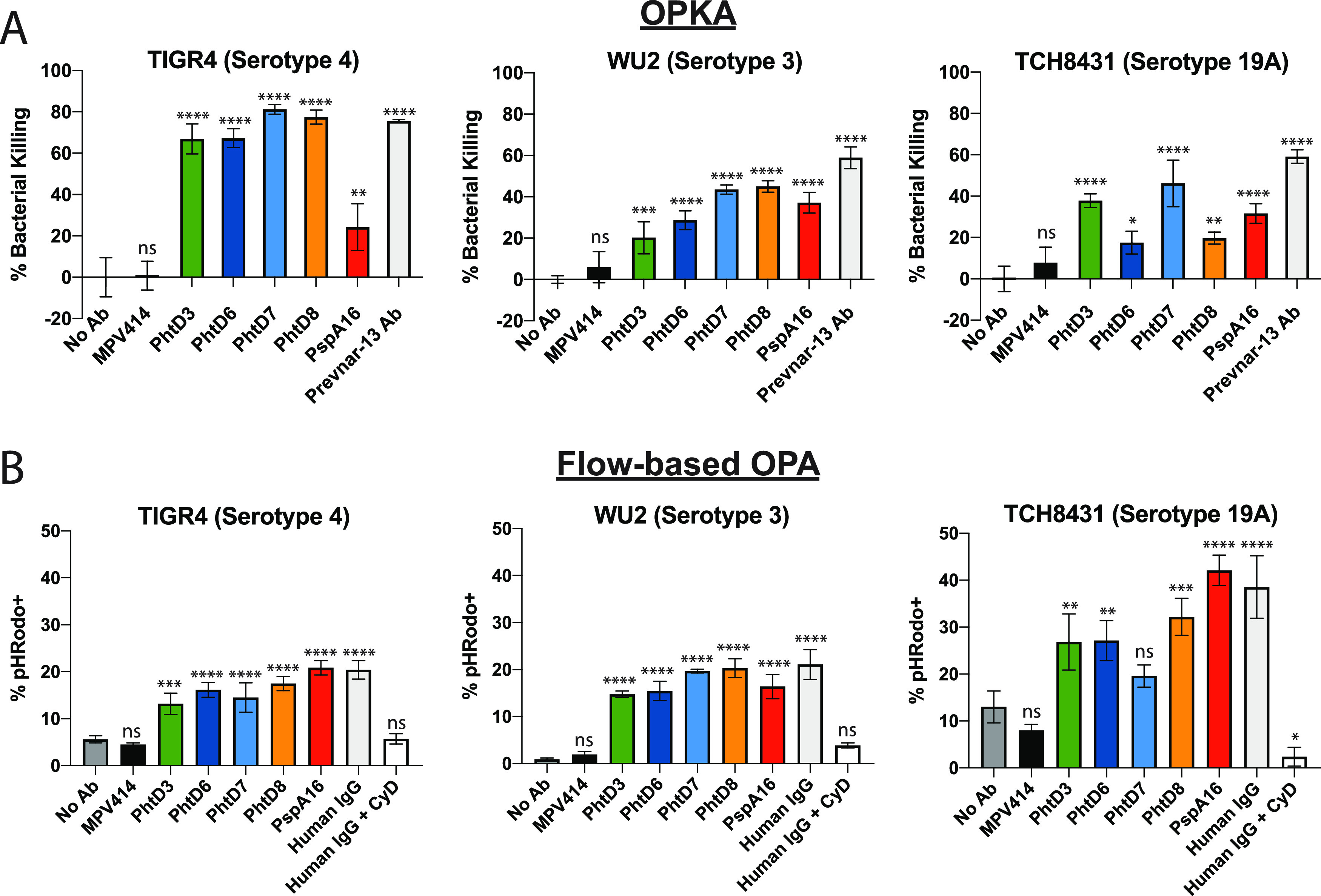
Opsonophagocytic activity of PhtD-specific human MAbs. (A) MAbs and serum were tested in a standard OPA assay using differentiated HL-60 cells. Bacteria were opsonized with antibodies, and subsequently HL-60 cells were added before plating on blood agar plates. Plates were incubated overnight and CFU counted. Data are averages of three replicates from one experiment. Error bars show ranges. Percent bacterial killing was calculated as the counted CFU value of each triplicate normalized against the average of the no-Ab control. One-way analysis of variance (ANOVA) with Dunnett’s multiple-comparison test was used to determine significance. ns, not significant; ****, P < 0.0001. (B) MAbs and serum were tested in a flow-based opsonophagocytosis assay. pHRodo-labeled bacteria were opsonized with antibodies and incubated with HL-60 cells before being subjected to analysis by flow cytometry. Data are percentages of CD38+ CD11b+ HL-60 cells that are pHRodo+. Each bar graph shows averages of three experimental replicates, and error bars show standard deviations. ns, not significant; ***, P = 0.0001 to 0.0006; ****, P < 0.0001 via one-way ANOVA with Dunnett’s multiple-comparison test. MPV414 is a human MAb specific to the human metapneumovirus fusion protein.
To confirm these findings, we adopted a flow-based assay previously shown to work for group B streptococcus (76). HL-60 cells were incubated with opsonized bacteria that were labeled with pHRodo, which leads to fluorescent HL-60 cells upon phagocytosis of labeled bacteria. Similar to our results from the OPKA, all PhtD MAbs induced an increase in pHRodo+ HL-60 cells compared to no-antibody and isotype control antibody analyses (Fig. 6B). Purified IgG from an unvaccinated donor showed the highest number of pHRodo+ cells, as human IgG contains antibodies to multiple pneumococcal surface proteins. Interestingly, PspA16 also induced increased uptake to all three serotypes in this assay, although the highest activity was observed for serotype 19A, the serotype from which we cloned our PspA gene.
DISCUSSION
In this study, we isolated and determined the binding affinity, epitope specificity, serotype breadth, and protective properties of the first human MAbs to any pneumococcal surface protein. Both PhtD and PspA have been examined in depth as vaccine candidates for prevention of pneumococcal infection, although the current outlook for progress of these antigens in the era of conjugate vaccines remains uncertain. However, human MAbs to these conserved antigens offer the ability for pan-serotype recognition and potentially disease prevention and treatment. In contrast, human MAbs isolated following vaccination with pneumococcal polysaccharide vaccines are highly serotype specific (23) and would therefore offer limited use in the clinic.
Based on the B cell stimulation and screening results, it is clear that healthy individuals have circulating B cells specific to pneumococcal antigens PhtD and PspA. One drawback of our study is the lack of knowledge on the infection history of the human subjects used in the study. It was shown previously that colonization by S. pneumoniae results in immunization, and it is unknown whether all of these donors were previously infected with S. pneumoniae. Therefore, the MAbs isolated here are likely the result of pneumococcal colonization, which resulted in class-switched B cells with 85 to 94% somatic mutation, which is similar to previous work in our lab studying healthy individuals who were presumed to be previously infected with human metapneumovirus (70, 77). Each of the PhtD-specific MAbs was isolated from unique human subjects, and MAbs PhtD3, PhtD6, and PhtD7/8 utilized different heavy- and light-chain V genes. Interestingly, MAbs PhtD7 and PhtD8 utilize the same V gene in both heavy and light chains yet differ in the predicted heavy-chain J gene, which leads to very different CDR3 lengths. Although these two MAbs share heavy- and light-chain V genes, the epitopes for these MAbs do not compete and have only partial overlap based on the binding experiments with truncated protein fragments.
It is a striking observation that the N-terminal specificity of MAbs PhtD3 and PhtD6 correlates with higher binding to whole-cell bacteria than does that of PhtD8, as the N-terminal region of the protein is predicted to be attached to the bacterial surface, leaving the C-terminal half more surface exposed (34). Further mapping experiments through X-ray crystallographic analysis will help clarify this observation. Previous work identified specific linear peptide epitopes that are immunodominant in pediatric patients with invasive pneumococcal disease, and these included amino acids (aa) 88 to 107, aa 172 to 191, and aa 200 to 219 (78). These peptides overlap the epitopes for MAbs PhtD3 and PhtD6, although we have not yet determined if these MAbs bind these peptide epitopes.
Overall, these data suggest that the human antibody response to PhtD targets multiple epitopes. For PspA16, the MAb targets the N-terminal region of PspA, which has a high number of negatively charged residues, and has been shown to be protective in several studies (79). Mouse MAbs isolated against PspA were determined to target the N-terminal fragment as well, suggesting that this domain is immunogenic in both mice and humans (79, 80). Although MAb PspA16 has limited serotype breadth, it is unclear if other human MAbs to PspA, even those targeting the N-terminal fragment, will be more broadly reactive. It is well established that the N-terminal region of PspA is more variable than the proline-rich region, and further studies will determine whether other PspA MAbs are more broadly reactive than PspA16, as MAb PspA16 binds outside the highly conserved proline-rich region (58). Limitations of the present study include the limited number of MAbs isolated for PhtD and PspA. Isolation of additional MAbs will help to determine whether the epitopes and gene usage of the MAbs described here are common in multiple donors.
Antipneumococcal MAbs have potential for use in the clinic, as current vaccines cover only a subset of current serotypes (although they are the most prevalent in invasive disease), and a rise in nonvaccine serotypes has occurred following vaccine introduction (24, 81). The prophylactic efficacy of MAbs PhtD3 and PhtD8 was demonstrated against pneumococcal serotype 3, a leading cause of invasive pneumococcal disease (24). We have also assessed the prophylactic efficacy of MAb PhtD3 against serotype 4 in both intranasal and intravenous infection studies, to model pneumococcal pneumonia and sepsis. Furthermore, we have demonstrated that MAb PhtD3 prolongs survival of mice treated with the MAb 24 h after infection with pneumococcal serotype 3. As MAbs PhtD3 and PhtD8 target unique epitopes on the N-terminal and C-terminal regions of PhtD, respectively, the higher survival of MAb PhtD3-treated mice suggests a potential role of epitope specificity in protective efficacy, although other factors, such as functional activity, decreased binding to serotype 3 bacteria in the ELISA and flow binding assays compared to MAb PhtD3, CDR length and percent somatic hypermutation, and corresponding binding modes, may be important for the observed differences. Further studies will need to be completed to determine the efficacy of other PhtD MAbs, to examine whether the specific epitope on PhtD indeed influences the protective efficacy of these MAbs and to determine whether the MAbs protect against infection with additional serotypes. In addition, the delivery timing of the MAbs for optimal protection, the potential use of MAbs in combinations for improved protective efficacy, and the use of MAbs in concert with antibiotic treatment will need to be examined. Furthermore, as secondary pneumococcal infection is prominent following influenza (10) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (11), another potentially useful scenario for use of antipneumococcal MAbs would be administration following primary viral infection, to prevent secondary pneumococcal infection. Further studies will need to be completed to determine if MAb PhtD3 or other MAbs will protect against secondary infection.
A potential mechanism of protection for MAbs PhtD3 and PhtD8 and the functional activity of the other PhtD- and PspA-specific MAbs was assessed in opsonophagocytic assays. While showing activity in these in vitro assays, the mechanism of protection in vivo was not determined and will need to be further explored. MAbs to the pneumococcal capsular polysaccharide have been shown to be protective through multiple mechanisms, with even nonopsonic MAbs demonstrating protective efficacy and the ability to reduce pneumococcal colonization (82, 83). PhtD has been shown to be important for pneumococcal adherence (35, 84), and MAbs to PhtD have also been shown to limit adherence of bacteria (46). Therefore, additional protective mechanisms for anti-PhtD MAbs exist and may work in concert with opsonophagocytosis.
Overall, our study adds support for using human MAbs to highly conserved surface antigens for prevention and treatment of pneumococcal infection. In addition, the application of human MAbs for other bacterial infections, particularly those that entail concerns about antibiotic resistance, is an important path forward for the development of new therapies. Further defining the epitope specificity of protective human MAbs to conserved pneumococcal surface proteins would also facilitate the development of an epitope-based and potentially multiantigen and broadly protective pneumococcal vaccine.
MATERIALS AND METHODS
Ethics statement.
This study was approved by the University of Georgia Institutional Review Board as STUDY00005127 and STUDY00005368. Healthy human donors were recruited to the University of Georgia Clinical and Translational Research Unit, and written informed consent was obtained. For the samples from Prevnar-13-vaccinated individuals, healthy subjects were recruited for vaccination with Prevnar-13, and a single blood sample was collected 21 to 28 days following immunization. All animal studies performed were in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Georgia.
Blood draws and isolation of PBMCs.
After informed consent had been obtained, 90 ml of blood was drawn by venipuncture into 9 heparin-coated tubes, and 10 ml of blood was collected into a serum separator tube. Peripheral blood mononuclear cells (PBMCs) were isolated from human donor blood samples using Ficoll-Histopaque density gradient centrifugation, and PBMCs were frozen in the liquid nitrogen vapor phase until further use.
Pneumococcal protein cloning and expression.
PspA and PhtD full-length proteins and fragments were cloned from the genome of S. pneumoniae strain TCH8431 (serotype 19A) with the primers listed in Table 2. The full-length PspA and PhtD were ligated into the pET28a vector, while the fragments were ligated into the pMAL-c5x vector. The sequences of all constructed plasmids were confirmed by sequencing and then transformed into E. coli BL21(DE3) for protein expression. Single colonies of transformed E. coli were picked and cultured in 5 ml of LB medium supplemented with antibiotic (50 μg/ml kanamycin for pET28a and 100 μg/ml ampicillin for pMAL-c5x) overnight in a shaking incubator at 37°C. The overnight culture was then expanded at a 1:100 ratio in 2× yeast tryptone (YT) medium with antibiotic and cultured at 37°C. After the optical density at 600 nm (OD600) reached 0.5 to 0.7, the culture was induced with 50 μM isopropyl-d-thiogalactopyranoside for 12 to 16 h at room temperature. Bacteria pellets were collected by centrifugation at 6,000 × g for 10 min and frozen at −80°C. Thawed E. coli pellets were resuspended in 10 ml of buffer containing 20 mM Tris (pH 7.5) and 500 mM NaCl and then lysed by sonication. Cell lysates were centrifuged at 12,000 × g for 30 min, and the supernatant was subsequently used for protein purification through a HisTrap column (His-tagged full-length proteins; GE Healthcare) or amylose resin (MBP-tagged fragments; New England Biolabs) following the manufacturer’s protocols.
TABLE 2.
Summary of primers used for cloning of PspA and PhtD genes
| Primer | Sequence (5′→3′)a |
|---|---|
| PspA1-F | CGCCATATGATGGCTAATAAGAAAAAAATGATTTT |
| PspA247-F | CGCCATATGGAGCTAAACGCTAAACAA |
| PspA436-F | CGCCATATGGATGAAGAAGAAACTCCAGCG |
| PspA438-R | TAGCGGCCGTTAATGGTGATGGTGATGGTGTTCTTCATCTCCATCAGGGC |
| PspA512-R | TAGCGGCCGTTAATGGTGATGGTGATGGTGTTTTGGAGTGGCTGGTTTTTC |
| PspA725-R | TAGCGGCCGTTAATGGTGATGGTGATGGTGAACCCATTCACCATTGGCAT |
| PhtD1-F | CATGCCATGGCCATGAAAATAAATAAAAAATATCTAGCAGG |
| PhtD168-F | CATGCCATGGCCGCAGTAAATGCTGTTGCTG |
| PhtD341-F | CATGCCATGGCCTATCGTTCAAACCATTGGGT |
| PhtD645-F | CATGCCATGGCCGACCATTAACCATAACATCAAATTTG |
| PhtD170-R | CCCAAGCTTTTAATGGTGATGGTGATGGTGATTATCTGCTCTTGAGTTATGATTATG |
| PhtD343-R | CCCAAGCTTTTAATGGTGATGGTGATGGTGTGAACGATAACGAAGGGGAAT |
| PhtD647-R | CCCAAGCTTTTAATGGTGATGGTGATGGTGGTAATGGTCATAATGAGGTATGATTAAA |
| PhtD838-R | CCCAAGCTTTTAATGGTGATGGTGATGGTGCTGTATAGGAGCCGGTTGA |
Restriction enzyme sites are in bold; His tags are underlined.
Enzyme-linked immunosorbent assay for binding to pneumococcal proteins.
For recombinant-protein capture ELISAs, 384-well plates were treated with 2 μg/ml of antigen in PBS for 1 h at 37°C or overnight at 4°C. Following this, plates were washed once with distilled water before blocking for 1 h with 2% nonfat milk–2% goat serum in 0.05% PBS-Tween (PBS-T) (blocking buffer). Plates were washed with water three times before serially diluted primary MAbs in PBS were applied for 1 h. Following this, plates were washed with water three times before application of 25 μl of secondary antibody (goat anti-human IgG Fc; Meridian Life Science) at a dilution of 1:4,000 in blocking solution. After incubation for 1 h, the plates were washed five times with PBS-T, and 25 μl of a PNPP (p-nitrophenyl phosphate) solution (1 mg/ml PNPP in 1 M Tris base) was added to each well. The plates were incubated at room temperature for 1 h before reading the optical density at 405 nm on a BioTek plate reader. Binding assay data were analyzed in GraphPad Prism using a nonlinear regression curve fit and the log(agonist)-versus-response function to calculate the binding EC50s.
Generation of pneumococcus-specific hybridomas.
For hybridoma generation, 10 million peripheral blood mononuclear cells purified from the blood of human donors were mixed with 8 million previously frozen and gamma-irradiated NIH 3T3 cells modified to express human CD40L, human interleukin 21 (IL-21), and human BAFF (70) in 80 ml StemCell medium A (StemCell Technologies) containing 6.3 μg/ml of CpG (phosphorothioate-modified oligodeoxynucleotide ZOEZOEZZZZZOEEZOEZZZT; Invitrogen) and 1 μg/ml of cyclosporine (Millipore-Sigma). The mixture of cells was plated in four 96-well plates at 200 μl per well in StemCell medium A. After 6 days, culture supernatants were screened by ELISA for binding to recombinant pneumococcal protein, and cells from positive wells were electrofused to generate hybridomas and biologically cloned as previously described (70).
Human MAb expression and purification.
For hybridoma-derived MAbs, hybridoma cell lines were expanded in StemCell medium A until they were 80% confluent in 75-cm2 flasks. Cells from one 75-cm2 cell culture flask were collected with a cell scraper and expanded to 225-cm2 cell culture flasks in serum-free medium (Hybridoma-SFM; Thermo Fisher Scientific). Recombinant cultures from transfection were stopped after 5 to 7 days, and hybridoma cultures were stopped after 30 days. For recombinant PhtD3-IgG2a, plasmids encoding cDNAs for the heavy and light chain sequences of PhtD3-IgG2a were synthesized (GenScript) and cloned into pCDNA3.1+. MAbs were obtained by transfection of plasmids into Expi293F cells by transfection. For each milliliter of transfection, 1 μg of plasmid DNA was mixed with 4 μg of 25,000-molecular-weight polyethylenimine (PEI; PolySciences Inc.) in 66.67 μl Opti-MEM cell culture medium (Gibco). After 30 min, the DNA-PEI mixture was added to the Expi293F cells, and cells were cultured for 5 to 6 days for protein expression. Culture supernatants from both approaches were filtered using 0.45-μm filters to remove cell debris. MAbs were purified directly from culture supernatants using HiTrap protein G columns (GE Healthcare Life Sciences) according to the manufacturer’s protocol.
Isotype determination for human MAbs.
For determination of MAb isotypes, 96-well Immulon 4HBX plates (Thermo Fisher Scientific) were coated with 2 μg/ml of each MAb in PBS (duplicate wells for each sample). The plates were incubated at 4°C overnight and then washed once with water. Plates were blocked with blocking buffer and then incubated for 1 h at room temperature. After incubation, the plates were washed three times with water. Isotype-specific antibodies obtained from Southern Biotech (goat anti-human kappa–alkaline phosphatase [AP] [catalog number 100244-340], goat anti-human lambda–AP [catalog number 100244-376], mouse anti-human IgG1 [Fc]–AP [catalog number 100245714], mouse anti-human IgG2 [Fc]–AP [catalog number 100245-734], mouse anti-human IgG3 [hinge]–AP [catalog number 100245-824], and mouse anti-human IgG4 [Fc]–AP [catalog number 100245-812]) were diluted 1:1,000 in blocking buffer, and 50 μl of each solution was added to the respective wells. Plates were incubated for 1 h at room temperature and then washed five times with PBS-T. The PNPP substrate was prepared at 1 mg/ml in substrate buffer (1 M Tris base, 0.5 mM MgCl2 [pH 9.8]), and 100 μl of this solution was added to each well. Plates were incubated for 1 h at room temperature and read at 405 nm on a BioTek plate reader.
RT-PCR for hybridoma MAb variable gamma chain and variable light chain.
RNA was isolated from expanded hybridoma cells using the EZNA total RNA kit (Omega BioTek) according to the manufacturer’s protocol. cDNA was obtained using the Superscript IV reverse transcriptase kit. Following this, PCR was conducted in two steps using established primers for the heavy chain and kappa and lambda light chains (85). Samples were analyzed by agarose gel electrophoresis and purified PCR products (EZNA Cycle Pure kit; Omega Bio-Tek) were cloned into the pCR2.1 vector using the original TA Cloning kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Plasmids were purified from positive DH5α colonies with an EZNA plasmid DNA minikit (Omega Bio-Tek) and submitted to Genewiz for sequencing. Sequences were analyzed using IMGT/V-Quest (86).
Experimental setup for biolayer interferometry.
For all biosensors, an initial baseline in running buffer (PBS, 0.5% bovine serum albumin [BSA], 0.05% Tween 20, 0.04% thimerosal) was obtained. For epitope mapping, 100 μg/ml of His-tagged PhtD protein was immobilized on anti-penta-HIS biosensor tips (FortéBio) for 120 s. For binding competition, the baseline signal was measured again for 60 s before biosensor tips were immersed in wells containing 100 μg/ml of primary antibody for 300 s. Following this, biosensors were immersed in wells containing 100 μg/ml of a second MAb for 300 s. Percent binding of the second MAb in the presence of the first MAb was determined by comparing the maximal signal of the second MAb after the first MAb was added to the maximum signal of the second MAb alone. MAbs were considered noncompeting if maximum binding of the second MAb was ≥66% of its uncompeted binding. A level between 33% and 66% of its uncompeted binding was considered intermediate competition, and ≤33% was considered competition.
Bacterial strains and growth conditions.
Pneumococcal strains were grown at 37°C in 5% CO2 in Todd-Hewitt broth (BD, Franklin Lakes NJ) supplemented with 0.5% yeast extract for 12 h. Ten percent glycerol was added to the medium, and 500-μl aliquots were made. Cultures were kept at −80°C until used, and cultures were washed twice with PBS before being used in experiments. Colonies were grown on BD Trypticase soy agar II with 5% sheep blood (BD, Franklin Lakes NJ). The numbers of CFU per milliliter of these stocks were determined, after the aliquots had been frozen, by plating a single quick-thawed diluted aliquot on sheep blood agar plates. The calculated number of CFU was subsequently used to make dilutions for experiments from aliquots thawed at later times. In each experiment, the actual number of CFU administered was determined by plating on blood agar at the time of the assay. Strains used in this study are listed in Table 3.
TABLE 3.
Summary of pneumococcal strains used in this study
| Pneumococcal strain | Serotype | Source |
|---|---|---|
| SPEC 1 | 1 | BEI NR-13388 |
| STREP2 | 2 | BEI NR-31700 |
| WU2 | 3 | Gift from Moon Nahm, University of Alabama Birmingham |
| TIGR4 | 4 | Gift from Larry McDaniel, University of Mississippi Medical Center |
| SPEC6C | 6C | BEI NR-20805 |
| SPEC6D | 6D | BEI NR-20806 |
| STREP8 | 8 | BEI NR-31701 |
| SPEC9N | 9N | BEI NR-31702 |
| OREP10A | 10A | BEI NR-31703 |
| TREP11A | 11A | BEI NR-31705 |
| TREP12F | 12F | BEI NR-31704 |
| TREP15B | 15B | BEI NR-33666 |
| OREP17F | 17F | BEI NR-31706 |
| TCH8431 | 19A | BEI HM-145 |
| SPEC20B | 20B | BEI NR-33664 |
| TREP22F | 22F | BEI NR-31707 |
| STREP33F | 33F | BEI NR-33665 |
Western blotting.
Pneumococcal strains were mixed with nonreducing loading buffer (Laemmli SDS sample buffer, nonreducing 6×) and loaded on a 4 to 12% bis-Tris gel (Invitrogen). Samples were then transferred to polyvinylidene difluoride (PVDF) membranes via an iBlot system (Invitrogen) and then blocked with 5% blocking buffer (5% nonfat milk in PBS-T) for 1 h at room temperature or at 4°C overnight. The membrane was washed three times in 5-min intervals on an orbital shaker with 0.05% PBS-T. Then, primary antibodies were added at dilutions of 1 μg/ml in PBS for 1 h at room temperature. The membranes were then washed three time at 5-min intervals with PBS-T on an orbital shaker and soaked in the secondary antibody at a 1:8,000 dilution in blocking buffer for 1 h. Next, the membranes were washed five times at 5-min intervals on the orbital shaker with PBS-T, substrate (Pierce ECL Western blotting substrate; Thermo Scientific) was added, and an image was taken immediately with the ChemiDoc imaging system (Bio-Rad).
Enzyme-linked immunosorbent assay of fixed pneumococcus.
For ELISAs, 384-well plates were treated with 15 μl (∼107 CFU) of whole-cell pneumococcus in PBS in each well. Cell density was checked by microscope to ensure that a confluent layer of pneumococcus was present. The bacteria were then fixed with 15 μl of 4% paraformaldehyde in each well and placed on a plate shaker for 10 min to mix. The 384-well plates were incubated at 4°C for 24 to 48 h to allow the bacteria to fix to the bottom of the plates. Following this, the plates were washed once with 75 μl of PBS-T in each well. The plates were then blocked with 2% blocking buffer for 1 h at room temperature and washed three times with PBS-T. Next, 25 μl of serially diluted primary antibodies was applied to the wells for 1 h at room temperature; then, plates were washed with PBS-T three times. Following this, 25 μl of secondary antibody (goat anti-human IgG Fc; Meridian Life Science) at a 1:4,000 dilution in blocking buffer was applied to each well for 1 h at room temperature. After the plates were washed with PBS-T five times, 25 μl of PNPP (p-nitrophenyl phosphate) solution (1 mg/ml PNPP in 1 M Tris base) was added to each well for 1 h at room temperature. After 1 h the optical density was read at 405 nm on a BioTek plate reader. Binding assay data were analyzed in GraphPad Prism.
Binding of antibodies to bacteria by flow cytometry.
The ability of MAbs to bind antigen exposed on the surface of S. pneumoniae was determined by flow cytometry. Bacteria were stained with 10 μM CFSE (carboxyfluorescein succinimidyl ester; Millipore Sigma) for 1 h at 37°C. Bacteria were then washed with Hanks’ balanced salt solution (HBSS) containing 1% BSA to remove excess stain. Following this, 1 × 106 bacteria were incubated with 10 μg/ml of antibody for 30 min at 37°C. Bacteria were then washed twice with HBSS plus 1% BSA. Antibody binding was detected using allophycocyanin (APC)-conjugated anti-human IgG Fc (BioLegend) at a 1:100 dilution incubated for 1 h with the bacteria. Cells were washed with HBSS–1% BSA and fixed in 2% paraformaldehyde (PFA) in PBS prior to analysis on a NovoCyte Quanteon flow cytometer.
Determination of MAb efficacy.
For the intranasal challenge study with TIGR4, 5- to 7-week-old CBA/CaHN-Btkxid/J (CBA/N) mice (The Jackson Laboratory, Bar Harbor, ME) were used. Mice were intraperitoneally inoculated with antibody treatments 2 h prior to pneumococcal infection. For infection, mice were anesthetized by inhalation of 5% isoflurane and intranasally challenged with 40 μl of PBS containing 105 CFU of TIGR4. Mice were weighed and assessed daily and were considered moribund when >20% of body weight was lost or they were nonresponsive to manual stimulation or exhibited respiratory distress. Mice were euthanized by CO2 asphyxiation followed by cervical dislocation. For the intravenous challenge with TIGR4, C57BL/6 mice 5 to 7 weeks old (Charles River) were used. Mice were intraperitoneally inoculated with antibody treatments 2 h prior to pneumococcal infection and infected intravenously with 106 CFU of TIGR4 via the tail vein. Mice were monitored and euthanized as described above. For intranasal challenge studies with WU2, C57BL/6 mice 5 to 7 weeks old (Charles River) were used. For intranasal infection, mice were anesthetized by inhalation of 5% isoflurane and intranasally challenged with 40 μl of PBS containing 106 CFU of WU2. Mice were treated either 2 h before infection or 24 h postinfection by intraperitoneal inoculation with antibody. In prophylactic studies, mice were euthanized based on the humane endpoints above. For treatment studies, mice were euthanized when >30% of preinfection body weight was lost or they were nonresponsive to manual stimulation or exhibited respiratory distress. Actual doses delivered to mice in all studies were determined by titrating the bacteria after delivery.
Opsonophagocytic killing assay.
An opsonophagocytic killing assay was performed as described previously (74, 87), as adapted from an earlier protocol with modifications (88). TIGR4 stocks were incubated in triplicate wells in a 96-well round-bottom plate for 1 h at 37°C with the indicated antibodies (10 μg of antibody per well in a final volume of 100 μl per well) in opsonization buffer B (OBB; sterile 1× PBS with Ca2+/Mg2+, 0.1% gelatin, and 5% heat-inactivated FetalClone [HyClone]), with heat-inactivated FetalClone-only-treated TGR4 cells serving as a control. Cells of the human promyelocytic leukemia cell line HL-60 (ATCC) were cultured in RPMI with 10% heat-inactivated FetalClone, and 1% l-glutamine. HL-60 cells were differentiated using 0.6% N,N-dimethylformamide (DMF [Fisher]) for 3 days before the opsonophagocytosis assay (OPA), harvested, and resuspended in OBB. Baby rabbit complement (Pel-Freez) was added to HL-60 cells at a 1:5 final volume. The HL-60–complement mixture was added to the bacteria at 5 × 105 cells/well. The final reaction mixtures were incubated at 37°C for 1 h with shaking. The reactions were stopped by incubating the samples on ice for approximately 20 min. Then, 10 μl of each reaction mixture (in triplicate) was diluted to a final volume of 50 μl and plated on blood agar plates. Plates were incubated overnight at 30°C, and CFU were counted the next day. The percentage of bacterial killing was calculated as the value for each sample replicate normalized to the mean value obtained for the control samples, subtracted from 100 (with no-Ab control samples representing 0% survival).
Flow-based opsonophagocytosis assay.
Pneumococcal cells were stained with pHRodo succinimidyl ester (Invitrogen) following the manufacturer’s protocol. Approximately, ∼108 CFU of bacteria were fixed with 1% paraformaldehyde in PBS for 30 min at room temperature. Fixed bacteria were washed twice with PBS and resuspended with 0.5 ml freshly prepared 100 mM NaHCO3 (pH 8.5). Immediately before use, the contents of a 0.1-mg vial of pHRodo iFL amine-reactive dye were dissolved in 10 μl of dimethyl sulfoxide (DMSO) to prepare a 10 mM stock solution. pHRodo was diluted in the bacterial suspension at a final concentration of 0.1 mM, and bacteria were stained for 1 h at room temperature. Stained bacteria were washed twice with Hanks’ balanced salt solution with Ca2+ and Mg2+ (HBSS; Gibco), resuspended with 0.5 ml HBSS, and stored in the dark at 4°C.
The opsonophagocytosis assay was performed in 96-well U-bottom plates in a total volume of 120 μl per well. First, 20 μl of pHRodo labeled bacteria (∼107 CFU/well) was mixed with 40 μl of sterile-filtered MAbs (50 μg/well) and incubated on a shaker at 37°C for 30 min. Bacteria were mixed with HBSS as a negative control, and purified human serum IgG was used as a positive control. Differentiated HL-60 cells were washed twice with HBSS and mixed with baby rabbit complement (Pel-Freez Biologicals) at a final concentration of 10% in each well. Following this, 60 μl (1 × 106 viable cells) of differentiated HL60 cells and complement were added to the mixture of bacteria and antibodies and incubated on a shaker at 37°C for 60 min. The plate was then centrifuged at 1,300 rpm for 5 min at 4°C to remove the supernatant, and the pellet was washed twice with 200 μl of HBSS. After the second wash, the pellet was resuspended in a 50-μl mixture of phycoerythrin (PE)-conjugated anti-human CD11b (Southern Biotech; 10 μl/million cells), Alexa Fluor 647–anti-human CD35 (BD Biosciences; 5 μl/million cells), and DAPI (4′,6-diamidino-2-phenylindole; Invitrogen, 50 ng/million cells) in PBS containing 1% BSA. After a 30-min incubation at 4°C in the dark, the plate was washed twice with 200 μl of PBS, and cells were resuspended in 100 μl of PBS. Cells were analyzed with a NovoCyte Quanteon flow cytometer. Single fluorophore-stained differentiated HL60 cells and pHRodo-stained bacteria were used to calculate the compensation matrix. A total of 10,000 ungated events were collected from each sample well, and data were analyzed by FlowJo.
Supplementary Material
ACKNOWLEDGMENTS
These studies were supported by startup funding from the University of Georgia Office of the Vice President for Research, a Junior Faculty Seed Grant from the University of Georgia Office of Research, and National Institutes of Health grants 1K01OD026569 (J.J.M.) and R01AI123383 (F.Y.A.). Human subject studies were partially funded by the National Center for Advancing Translational Sciences award number UL1TR002378. F.R. was supported by National Institutes of Health NIGMS grant GM109435, Post-Baccalaureate Training in Infectious Diseases Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We thank the University of Georgia Clinical and Translational Research Unit for assistance with donor identification and blood draws and the University of Georgia Center for Tropical and Emerging Global Diseases and College of Veterinary Medicine flow cytometry cores for assistance with cell sorting and analysis.
J.H., A.D.G., F.R., F.Y.A., and J.J.M. are inventors on a provisional patent application filed describing the sequences of the monoclonal antibodies.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Levine OS, O'Brien KL, Knoll M, Adegbola RA, Black S, Cherian T, Dagan R, Goldblatt D, Grange A, Greenwood B, Hennessy T, Klugman KP, Madhi SA, Mulholland K, Nohynek H, Santosham M, Saha SK, Scott JA, Sow S, Whitney CG, Cutts F. 2006. Pneumococcal vaccination in developing countries. Lancet 367:1880–1882. 10.1016/S0140-6736(06)68703-5. [DOI] [PubMed] [Google Scholar]
- 2.WHO. 2003. Pneumococcal vaccines. Wkly Epidemiol Rec 78:110–119. [Google Scholar]
- 3.Advisory Committee on Immunization Practices. 2000. Preventing pneumococcal disease among infants and young children. MMWR Recomm Rep 49:1–35. [PubMed] [Google Scholar]
- 4.van der Poll T, Opal SM. 2009. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 374:1543–1556. 10.1016/S0140-6736(09)61114-4. [DOI] [PubMed] [Google Scholar]
- 5.Bridy-Pappas AE, Margolis MB, Center KJ, Isaacman DJ. 2005. Streptococcus pneumoniae: description of the pathogen, disease epidemiology, treatment, and prevention. Pharmacotherapy 25:1193–1212. 10.1592/phco.2005.25.9.1193. [DOI] [PubMed] [Google Scholar]
- 6.Williams BG, Gouws E, Boschi-Pinto C, Bryce J, Dye C. 2002. Estimates of world-wide distribution of child deaths from acute respiratory infections. Lancet Infect Dis 2:25–32. 10.1016/s1473-3099(01)00170-0. [DOI] [PubMed] [Google Scholar]
- 7.Sulikowska A, Grzesiowski P, Sadowy E, Fiett J, Hryniewicz W. 2004. Characteristics of Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis isolated from the nasopharynges of asymptomatic children and molecular analysis of S. pneumoniae and H. influenzae strain replacement in the nasopharynx. J Clin Microbiol 42:3942–3949. 10.1128/JCM.42.9.3942-3949.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simell B, Auranen K, Käyhty H, Goldblatt D, Dagan R, O'Brien KL, Pneumococcal Carriage Group. 2012. The fundamental link between pneumococcal carriage and disease. Expert Rev Vaccines 11:841–855. 10.1586/erv.12.53. [DOI] [PubMed] [Google Scholar]
- 9.Cardozo DM, Nascimento-Carvalho CM, Andrade AL, Silvany-Neto AM, Daltro CH, Brandao MA, Brandao AP, Brandileone MC. 2008. Prevalence and risk factors for nasopharyngeal carriage of Streptococcus pneumoniae among adolescents. J Med Microbiol 57:185–189. 10.1099/jmm.0.47470-0. [DOI] [PubMed] [Google Scholar]
- 10.Shrestha S, Foxman B, Weinberger DM, Steiner C, Viboud C, Rohani P. 2013. Identifying the interaction between influenza and pneumococcal pneumonia using incidence data. Sci Transl Med 5:191ra84. 10.1126/scitranslmed.3005982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu X, Ge Y, Wu T, Zhao K, Chen Y, Wu B, Zhu F, Zhu B, Cui L. 2020. Co-infection with respiratory pathogens among COVID-2019 cases. Virus Res 285:198005. 10.1016/j.virusres.2020.198005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinberger DM, Dagan R, Givon‐Lavi N, Regev‐Yochay G, Malley R, Lipsitch M. 2008. Epidemiologic evidence for serotype‐specific acquired immunity to pneumococcal carriage. J Infect Dis 197:1511–1518. 10.1086/587941. [DOI] [PubMed] [Google Scholar]
- 13.Goldblatt D, Hussain M, Andrews N, Ashton L, Virta C, Melegaro A, Pebody R, George R, Soininen A, Edmunds J, Gay N, Kayhty H, Miller E. 2005. Antibody responses to nasopharyngeal carriage of Streptococcus pneumoniae in adults: a longitudinal household study. J Infect Dis 192:387–393. 10.1086/431524. [DOI] [PubMed] [Google Scholar]
- 14.McCool TL, Cate TR, Moy G, Weiser JN. 2002. The immune response to pneumococcal proteins during experimental human carriage. J Exp Med 195:359–365. 10.1084/jem.20011576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prevaes SMPJ, van Wamel WJB, de Vogel CP, Veenhoven RH, van Gils EJM, van Belkum A, Sanders EAM, Bogaert D. 2012. Nasopharyngeal colonization elicits antibody responses to staphylococcal and pneumococcal proteins that are not associated with a reduced risk of subsequent carriage. Infect Immun 80:2186–2193. 10.1128/IAI.00037-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turner P, Turner C, Green N, Ashton L, Lwe E, Jankhot A, Day NP, White NJ, Nosten F, Goldblatt D. 2013. Serum antibody responses to pneumococcal colonization in the first 2 years of life: results from an SE Asian longitudinal cohort study. Clin Microbiol Infect 19:E551–E558. 10.1111/1469-0691.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Q, Bernatoniene J, Bagrade L, Pollard AJ, Mitchell TJ, Paton JC, Finn A. 2006. Serum and mucosal antibody responses to pneumococcal protein antigens in children: relationships with carriage status. Eur J Immunol 36:46–57. 10.1002/eji.200535101. [DOI] [PubMed] [Google Scholar]
- 18.Mureithi MW, Finn A, Ota MO, Zhang Q, Davenport V, Mitchell TJ, Williams NA, Adegbola RA, Heyderman RS. 2009. T cell memory response to pneumococcal protein antigens in an area of high pneumococcal carriage and disease. J Infect Dis 200:783–793. 10.1086/605023. [DOI] [PubMed] [Google Scholar]
- 19.Wright AKA, Bangert M, Gritzfeld JF, Ferreira DM, Jambo KC, Wright AD, Collins AM, Gordon SB. 2013. Experimental human pneumococcal carriage augments IL-17A-dependent T-cell defence of the lung. PLoS Pathog 9:e1003274. 10.1371/journal.ppat.1003274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simell B, Lahdenkari M, Reunanen A, Käyhty H, Väkeväinen M. 2008. Effects of ageing and gender on naturally acquired antibodies to pneumococcal capsular polysaccharides and virulence-associated proteins. Clin Vaccine Immunol 15:1391–1397. 10.1128/CVI.00110-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganaie F, Saad JS, McGee L, van Tonder AJ, Bentley SD, Lo SW, Gladstone RA, Turner P, Keenan JD, Breiman RF, Nahm MH. 2020. A new pneumococcal capsule type, 10D, is the 100th serotype and has a large cps fragment from an oral streptococcus. mBio 11:e00937-20. 10.1128/mBio.00937-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geno KA, Gilbert GL, Song JY, Skovsted IC, Klugman KP, Jones C, Konradsen HB, Nahm MH. 2015. Pneumococcal capsules and their types: past, present, and future. Clin Microbiol Rev 28:871–899. 10.1128/CMR.00024-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Z, Cox KS, Tang A, Roman J, Fink M, Kaufhold RM, Guan L, Xie A, Boddicker MA, McGuinness D, Xiao X, Li H, Skinner JM, Verch T, Retzlaff M, Vora KA. 2018. Human monoclonal antibodies isolated from a primary pneumococcal conjugate vaccinee demonstrates the expansion of an antigen-driven hypermutated memory B cell response. BMC Infect Dis 18:613. 10.1186/s12879-018-3517-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wantuch PL, Avci FY. 2018. Current status and future directions of invasive pneumococcal diseases and prophylactic approaches to control them. Hum Vaccin Immunother 14:2303–2309. 10.1080/21645515.2018.1470726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linley E, Bell A, Gritzfeld JF, Borrow R. 2019. Should pneumococcal serotype 3 be included in serotype-specific immunoassays? Vaccines 7:4. 10.3390/vaccines7010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lo SW, Gladstone RA, van Tonder AJ, Lees JA, Du Plessis M, Benisty R, Givon-Lavi N, Hawkins PA, Cornick JE, Kwambana-Adams B, Law PY, Ho PL, Antonio M, Everett DB, Dagan R, von Gottberg A, Klugman KP, McGee L, Breiman RF, Bentley SD, Brooks AW, Corso A, Davydov A, Maguire A, Pollard A, Kiran A, Skoczynska A, Moiane B, Beall B, Sigauque B, Aanensen D, Lehmann D, Faccone D, Foster-Nyarko E, Bojang E, Egorova E, Voropaeva E, Sampane-Donkor E, Sadowy E, Bigogo G, Mucavele H, Belabbès H, Diawara I, Moïsi J, Verani J, Keenan J, Nair Thulasee Bhai JN, Ndlangisa KM, Zerouali K, Ravikumar KL, et al. 2019. Pneumococcal lineages associated with serotype replacement and antibiotic resistance in childhood invasive pneumococcal disease in the post-PCV13 era: an international whole-genome sequencing study. Lancet Infect Dis 19:759–769. 10.1016/S1473-3099(19)30297-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daniels CC, Rogers PD, Shelton CM. 2016. A review of pneumococcal vaccines: current polysaccharide vaccine recommendations and future protein antigens. J Pediatr Pharmacol Ther 21:27–35. 10.5863/1551-6776-21.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lagousi T, Basdeki P, Routsias J, Spoulou V. 2019. Novel protein-based pneumococcal vaccines: ssessing the use of distinct protein fragments instead of full-length proteins as vaccine antigens. Vaccines 7:9. 10.3390/vaccines7010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adamou JE, Heinrichs JH, Erwin AL, Walsh W, Gayle T, Dormitzer M, Dagan R, Brewah YA, Barren P, Lathigra R, Langermann S, Koenig S, Johnson S. 2001. Identification and characterization of a novel family of pneumococcal proteins that are protective against sepsis. Infect Immun 69:949–958. 10.1128/IAI.69.2.949-958.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rioux S, Neyt C, Di Paolo E, Turpin L, Charland N, Labbé S, Mortier MC, Mitchell TJ, Feron C, Martin D, Poolman JT. 2011. Transcriptional regulation, occurrence and putative role of the Pht family of Streptococcus pneumoniae. Microbiology (Reading) 157:336–348. 10.1099/mic.0.042184-0. [DOI] [PubMed] [Google Scholar]
- 31.Yun KW, Lee H, Choi EH, Lee HJ. 2015. Diversity of pneumolysin and pneumococcal histidine triad protein D of Streptococcus pneumoniae isolated from invasive diseases in Korean children. PLoS One 10:e0134055. 10.1371/journal.pone.0134055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawaguchiya M, Urushibara N, Aung MS, Shinagawa M, Takahashi S, Kobayashi N. 2019. Prevalence of various vaccine candidate proteins in clinical isolates of Streptococcus pneumoniae: characterization of the novel Pht fusion proteins PhtA/B and PhtA/D. Pathogens (Basel) 8:162. 10.3390/pathogens8040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blumental S, Granger-Farbos A, Moïsi JC, Soullié B, Leroy P, Njanpop-Lafourcade B-M, Yaro S, Nacro B, Hallin M, Koeck J-L. 2015. Virulence factors of Streptococcus pneumoniae. comparison between African and French invasive isolates and implication for future vaccines. PLoS One 10:e0133885. 10.1371/journal.pone.0133885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plumptre CD, Ogunniyi AD, Paton JC. 2013. Surface association of Pht proteins of Streptococcus pneumoniae. Infect Immun 81:3644–3651. 10.1128/IAI.00562-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kallio A, Sepponen K, Hermand P, Denoël P, Godfroid F, Melin M. 2014. Role of Pht proteins in attachment of Streptococcus pneumoniae to respiratory epithelial cells. Infect Immun 82:1683–1691. 10.1128/IAI.00699-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eijkelkamp BA, Pederick VG, Plumptre CD, Harvey RM, Hughes CE, Paton JC, McDevitt CA. 2016. The first histidine triad motif of PhtD is critical for zinc homeostasis in Streptococcus pneumoniae. Infect Immun 84:407–415. 10.1128/IAI.01082-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo Z, Pederick VG, Paton JC, McDevitt CA, Kobe B. 2018. Structural characterisation of the HT3 motif of the polyhistidine triad protein D from Streptococcus pneumoniae. FEBS Lett 592:2341–2350. 10.1002/1873-3468.13122. [DOI] [PubMed] [Google Scholar]
- 38.Bersch B, Bougault C, Roux L, Favier A, Vernet T, Durmort C. 2013. New insights into histidine triad proteins: solution structure of a Streptococcus pneumoniae PhtD domain and zinc transfer to AdcAII. PLoS One 8:e81168. 10.1371/journal.pone.0081168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Godfroid F, Hermand P, Verlant V, Denoël P, Poolman JT. 2011. Preclinical evaluation of the Pht proteins as potential cross-protective pneumococcal vaccine antigens. Infect Immun 79:238–245. 10.1128/IAI.00378-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wizemann TM, Heinrichs JH, Adamou JE, Erwin AL, Kunsch C, Choi GH, Barash SC, Rosen CA, Masure HR, Tuomanen E, Gayle A, Brewah YA, Walsh W, Barren P, Lathigra R, Hanson M, Langermann S, Johnson S, Koenig S. 2001. Use of a whole genome approach to identify vaccine molecules affording protection against Streptococcus pneumoniae infection. Infect Immun 69:1593–1598. 10.1128/IAI.69.3.1593-1598.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Denoël P, Philipp MT, Doyle L, Martin D, Carletti G, Poolman JT. 2011. A protein-based pneumococcal vaccine protects rhesus macaques from pneumonia after experimental infection with Streptococcus pneumoniae. Vaccine 29:5495–5501. 10.1016/j.vaccine.2011.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plumptre CD, Ogunniyi AD, Paton JC. 2013. Vaccination against Streptococcus pneumoniae using truncated derivatives of polyhistidine triad protein D. PLoS One 8:e78916. 10.1371/journal.pone.0078916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.André GO, Assoni L, Rodriguez D, Leite LCC, dos Santos TEP, Ferraz LFC, Converso TR, Darrieux M. 2020. Immunization with PhtD truncated fragments reduces nasopharyngeal colonization by Streptococcus pneumoniae. Vaccine 38:4146–4153. 10.1016/j.vaccine.2020.04.050. [DOI] [PubMed] [Google Scholar]
- 44.Hammitt LL, Campbell JC, Borys D, Weatherholtz RC, Reid R, Goklish N, Moulton LH, Traskine M, Song Y, Swinnen K, Santosham M, Brien KLO. 2019. Efficacy, safety and immunogenicity of a pneumococcal protein-based vaccine co-administered with 13-valent pneumococcal conjugate vaccine against acute otitis media in young children: a phase IIb randomized study. Vaccine 37:7482–7492. 10.1016/j.vaccine.2019.09.076. [DOI] [PubMed] [Google Scholar]
- 45.Visan L, Rouleau N, Proust E, Peyrot L, Donadieu A, Ochs M. 2018. Antibodies to PcpA and PhtD protect mice against Streptococcus pneumoniae by a macrophage- and complement-dependent mechanism. Hum Vaccin Immunother 14:489–494. 10.1080/21645515.2017.1403698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaur R, Surendran N, Ochs M, Pichichero ME. 2014. Human antibodies to PhtD, PcpA, and ply reduce adherence to human lung epithelial cells and murine nasopharyngeal colonization by Streptococcus pneumoniae. Infect Immun 82:5069–5075. 10.1128/IAI.02124-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brookes RH, Ming M, Williams K, Hopfer R, Gurunathan S, Gallichan S, Tang M, Ochs MM. 2015. Passive protection of mice against Streptococcus pneumoniae challenge by naturally occurring and vaccine-induced human anti-PhtD antibodies. Hum Vaccin Immunother 11:1836–1839. 10.1080/21645515.2015.1039210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosenow C, Ryan P, Weiser JN, Johnson S, Fontan P, Ortqvist A, Masure HR. 1997. Contribution of novel choline-binding proteins to adherence, colonization and immunogenicity of Streptococcus pneumoniae. Mol Microbiol 25:819–829. 10.1111/j.1365-2958.1997.mmi494.x. [DOI] [PubMed] [Google Scholar]
- 49.Hollingshead SK, Baril L, Ferro S, King J, Coan P, Briles DE, Group TPPES. 2006. Pneumococcal surface protein A (PspA) family distribution among clinical isolates from adults over 50 years of age collected in seven countries. J Med Microbiol 55:215–221. 10.1099/jmm.0.46268-0. [DOI] [PubMed] [Google Scholar]
- 50.Briles DE, Yother J, McDaniel LS. 1988. Role of pneumococcal surface protein A in the virulence of Streptococcus pneumoniae. Rev Infect Dis 10(Suppl 2):S372–S374. 10.1093/cid/10.supplement_2.s372. [DOI] [PubMed] [Google Scholar]
- 51.Bosarge JR, Watt JM, McDaniel DO, Swiatlo E, McDaniel LS. 2001. Genetic immunization with the region encoding the alpha-helical domain of PspA elicits protective immunity against Streptococcus pneumoniae. Infect Immun 69:5456–5463. 10.1128/iai.69.9.5456-5463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McDaniel LS, Sheffield JS, Delucchi P, Briles DE. 1991. PspA, a surface protein of Streptococcus pneumoniae, is capable of eliciting protection against pneumococci of more than one capsular type. Infect Immun 59:222–228. 10.1128/IAI.59.1.222-228.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McDaniel LS, McDaniel DO, Hollingshead SK, Briles DE. 1998. Comparison of the PspA sequence from Streptococcus pneumoniae EF5668 to the previously identified PspA sequence from strain Rx1 and ability of PspA from EF5668 to elicit protection against pneumococci of different capsular types. Infect Immun 66:4748–4754. 10.1128/IAI.66.10.4748-4754.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kong IG, Sato A, Yuki Y, Nochi T, Takahashi H, Sawada S, Mejima M, Kurokawa S, Okada K, Sato S, Briles DE, Kunisawa J, Inoue Y, Yamamoto M, Akiyoshi K, Kiyono H. 2013. Nanogel-based PspA intranasal vaccine prevents invasive disease and nasal colonization by Streptococcus pneumoniae. Infect Immun 81:1625–1634. 10.1128/IAI.00240-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Piao Z, Akeda Y, Takeuchi D, Ishii KJ, Ubukata K, Briles DE, Tomono K, Oishi K. 2014. Protective properties of a fusion pneumococcal surface protein A (PspA) vaccine against pneumococcal challenge by five different PspA clades in mice. Vaccine 32:5607–5613. 10.1016/j.vaccine.2014.07.108. [DOI] [PubMed] [Google Scholar]
- 56.Fukuyama Y, Yuki Y, Katakai Y, Harada N, Takahashi H, Takeda S, Mejima M, Joo S, Kurokawa S, Sawada S, Shibata H, Park EJ, Fujihashi K, Briles DE, Yasutomi Y, Tsukada H, Akiyoshi K, Kiyono H. 2015. Nanogel-based pneumococcal surface protein A nasal vaccine induces microRNA-associated Th17 cell responses with neutralizing antibodies against Streptococcus pneumoniae in macaques. Mucosal Immunol 8:1144–1153. 10.1038/mi.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arulanandam BP, Lynch JM, Briles DE, Hollingshead S, Metzger DW. 2001. Intranasal vaccination with pneumococcal surface protein A and interleukin-12 augments antibody-mediated opsonization and protective immunity against Streptococcus pneumoniae infection. Infect Immun 69:6718–6724. 10.1128/IAI.69.11.6718-6724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hollingshead SK, Becker R, Briles DE. 2000. Diversity of PspA: mosaic genes and evidence for past recombination in Streptococcus pneumoniae. Infect Immun 68:5889–5900. 10.1128/iai.68.10.5889-5900.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Khan N, Jan AT. 2017. Towards identifying protective B-cell epitopes: the PspA story. Front Microbiol 8:742. 10.3389/fmicb.2017.00742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ren B, Szalai AJ, Hollingshead SK, Briles DE. 2004. Effects of PspA and antibodies to PspA on activation and deposition of complement on the pneumococcal surface. Infect Immun 72:114–122. 10.1128/iai.72.1.114-122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mukerji R, Mirza S, Roche AM, Widener RW, Croney CM, Rhee D-K, Weiser JN, Szalai AJ, Briles DE. 2012. Pneumococcal surface protein A inhibits complement deposition on the pneumococcal surface by competing with the binding of C-reactive protein to cell-surface phosphocholine. J Immunol 189:5327–5335. 10.4049/jimmunol.1201967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tu A-HT, Fulgham RL, McCrory MA, Briles DE, Szalai AJ. 1999. Pneumococcal surface protein A inhibits complement activation by Streptococcus pneumoniae. Infect Immun 67:4720–4724. 10.1128/IAI.67.9.4720-4724.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Håkansson A, Roche H, Mirza S, McDaniel LS, Brooks-Walter A, Briles DE. 2001. Characterization of binding of human lactoferrin to pneumococcal surface protein A. Infect Immun 69:3372–3381. 10.1128/IAI.69.5.3372-3381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Senkovich O, Cook WJ, Mirza S, Hollingshead SK, Protasevich II, Briles DE, Chattopadhyay D. 2007. Structure of a complex of human lactoferrin N-lobe with pneumococcal surface protein A provides insight into microbial defense mechanism. J Mol Biol 370:701–713. 10.1016/j.jmb.2007.04.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nabors GS, Braun PA, Herrmann DJ, Heise ML, Pyle DJ, Gravenstein S, Schilling M, Ferguson LM, Hollingshead SK, Briles DE, Becker RS. 2000. Immunization of healthy adults with a single recombinant pneumococcal surface protein A (PspA) variant stimulates broadly cross-reactive antibodies to heterologous PspA molecules. Vaccine 18:1743–1754. 10.1016/s0264-410x(99)00530-7. [DOI] [PubMed] [Google Scholar]
- 66.Briles DE, Hollingshead SK, King J, Swift A, Braun PA, Park MK, Ferguson LM, Nahm MH, Nabors GS. 2000. Immunization of humans with recombinant pneumococcal surface protein A (rPspA) elicits antibodies that passively protect mice from fatal infection with Streptococcus pneumoniae bearing heterologous PspA. J Infect Dis 182:1694–1701. 10.1086/317602. [DOI] [PubMed] [Google Scholar]
- 67.Wilcox MH, Gerding DN, Poxton IR, Kelly C, Nathan R, Birch T, Cornely OA, Rahav G, Bouza E, Lee C, Jenkin G, Jensen W, Kim YS, Yoshida J, Gabryelski L, Pedley A, Eves K, Tipping R, Guris D, Kartsonis N, Dorr MB. 2017. Bezlotoxumab for prevention of recurrent Clostridium difficile infection. N Engl J Med 376:305–317. 10.1056/NEJMoa1602615. [DOI] [PubMed] [Google Scholar]
- 68.Wilson R, Cohen JM, Reglinski M, Jose RJ, Chan WY, Marshall H, de Vogel C, Gordon S, Goldblatt D, Petersen FC, Baxendale H, Brown JS. 2017. Naturally acquired human immunity to pneumococcus is dependent on antibody to protein antigens. PLoS Pathog 13:e1006137. 10.1371/journal.ppat.1006137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Simell B, Ahokas P, Lahdenkari M, Poolman J, Henckaerts I, Kilpi TM, Käyhty H. 2009. Pneumococcal carriage and acute otitis media induce serum antibodies to pneumococcal surface proteins CbpA and PhtD in children. Vaccine 27:4615–4621. 10.1016/j.vaccine.2009.05.071. [DOI] [PubMed] [Google Scholar]
- 70.Bar-Peled Y, Diaz D, Pena-Briseno A, Murray J, Huang J, Tripp RA, Mousa JJ. 2019. A potent neutralizing site III-specific human antibody neutralizes human metapneumovirus in vivo. J Virol 93:e00342-19. 10.1128/JVI.00342-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wantuch PL, Avci FY. 2019. Invasive pneumococcal disease in relation to vaccine type serotypes. Hum Vaccin Immunother 15:874–875. 10.1080/21645515.2018.1564444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sandgren A, Albiger B, Orihuela CJ, Tuomanen E, Normark S, Henriques-Normark B. 2005. Virulence in mice of pneumococcal clonal types with known invasive disease potential in humans. J Infect Dis 192:791–800. 10.1086/432513. [DOI] [PubMed] [Google Scholar]
- 73.Romero-Steiner S, Frasch CE, Carlone G, Fleck RA, Goldblatt D, Nahm MH. 2006. Use of opsonophagocytosis for serological evaluation of pneumococcal vaccines. Clin Vaccine Immunol 13:165–169. 10.1128/CVI.13.2.165-169.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Paschall AV, Middleton DR, Avci FY. 2019. Opsonophagocytic killing assay to assess immunological responses against bacterial pathogens. J Vis Exp 2019(146):e59400. 10.3791/59400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pilishvili T, Bennett NM. 2015. Pneumococcal disease prevention among adults: strategies for the use of pneumococcal vaccines. Vaccine 33:D60–D65. 10.1016/j.vaccine.2015.05.102. [DOI] [PubMed] [Google Scholar]
- 76.Fabbrini M, Sammicheli C, Margarit I, Maione D, Grandi G, Giuliani MM, Mori E, Nuti S. 2012. A new flow-cytometry-based opsonophagocytosis assay for the rapid measurement of functional antibody levels against group B Streptococcus. J Immunol Methods 378:11–19. 10.1016/j.jim.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 77.Huang J, Diaz D, Mousa JJ. 2020. Antibody recognition of the Pneumovirus fusion protein trimer interface. PLoS Pathog 16:e1008942. 10.1371/journal.ppat.1008942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lagousi T, Routsias J, Piperi C, Tsakris A, Chrousos G, Theodoridou M, Spoulou V. 2015. Discovery of immunodominant B cell epitopes within surface pneumococcal virulence proteins in pediatric patients with invasive pneumococcal disease. J Biol Chem 290:27500–27510. 10.1074/jbc.M115.666818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McDaniel LS, Ralph BA, McDaniel DO, Briles DE. 1994. Localization of protection-eliciting epitopes on PspA of Streptococcus pneumoniae between amino acid residues 192 and 260. Microb Pathog 17:323–337. 10.1006/mpat.1994.1078. [DOI] [PubMed] [Google Scholar]
- 80.Kolberg J, Aase A, Rødal G, Littlejohn JE, Jedrzejas MJ. 2003. Epitope mapping of pneumococcal surface protein A of strain Rx1 using monoclonal antibodies and molecular structure modelling. FEMS Immunol Med Microbiol 39:265–273. 10.1016/S0928-8244(03)00255-4. [DOI] [PubMed] [Google Scholar]
- 81.Keller LE, Robinson DA, McDaniel LS. 2016. Nonencapsulated Streptococcus pneumoniae: emergence and pathogenesis. mBio 7:e01792. 10.1128/mBio.01792-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Doyle CR, Pirofski L. 2016. Reduction of Streptococcus pneumoniae colonization and dissemination by a nonopsonic capsular polysaccharide antibody. mBio 7:e02260-15. 10.1128/mBio.02260-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tian H, Weber S, Thorkildson P, Kozel TR, Pirofski LA. 2009. Efficacy of opsonic and nonopsonic serotype 3 pneumococcal capsular polysaccharide-specific monoclonal antibodies against intranasal challenge with streptococcus pneumoniae in mice. Infect Immun 77:1502–1513. 10.1128/IAI.01075-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Khan MN, Pichichero ME. 2012. Vaccine candidates PhtD and PhtE of Streptococcus pneumoniae are adhesins that elicit functional antibodies in humans. Vaccine 30:2900–2907. 10.1016/j.vaccine.2012.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guthmiller JJ, Dugan HL, Neu KE, Lan LY-L, Wilson PC. 2019. An efficient method to generate monoclonal antibodies from human B cells. Methods Mol Biol 1904:109–145. 10.1007/978-1-4939-8958-4_5. [DOI] [PubMed] [Google Scholar]
- 86.Brochet X, Lefranc MP, Giudicelli V. 2008. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res 36:503–508. 10.1093/nar/gkn316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Middleton DR, Paschall AV, Duke JA, Avci FY. 2018. Enzymatic hydrolysis of pneumococcal capsular polysaccharide renders the bacterium vulnerable to host defense. Infect Immun 86:e00316-18. 10.1128/IAI.00316-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Burton RL, Nahm MH. 2012. Development of a fourfold multiplexed opsonophagocytosis assay for pneumococcal antibodies against additional serotypes and discovery of serological subtypes in Streptococcus pneumoniae serotype 20. Clin Vaccine Immunol 19:835–841. 10.1128/CVI.00086-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.