

Abstract
The genomes of eukaryotes are full of parasitic sequences known as transposable elements (TEs). Here, we report the discovery of a putative giant tyrosine-recombinase-mobilized DNA transposon, Enterprise, from the model fungus Podospora anserina. Previously, we described a large genomic feature called the Spok block which is notable due to the presence of meiotic drive genes of the Spok gene family. The Spok block ranges from 110 kb to 247 kb and can be present in at least four different genomic locations within P. anserina, despite what is an otherwise highly conserved genome structure. We propose that the reason for its varying positions is that the Spok block is not only capable of meiotic drive but is also capable of transposition. More precisely, the Spok block represents a unique case where the Enterprise has captured the Spoks, thereby parasitizing a resident genomic parasite to become a genomic hyperparasite. Furthermore, we demonstrate that Enterprise (without the Spoks) is found in other fungal lineages, where it can be as large as 70 kb. Lastly, we provide experimental evidence that the Spok block is deleterious, with detrimental effects on spore production in strains which carry it. This union of meiotic drivers and a transposon has created a selfish element of impressive size in Podospora, challenging our perception of how TEs influence genome evolution and broadening the horizons in terms of what the upper limit of transposition may be.
Transposable elements (TEs) are major agents of change in eukaryotic genomes. Their ability to selfishly parasitize their host replication machinery has large impacts on both genome size and on gene regulation (Chénais et al. 2012). In extreme cases, TEs can contribute up to 85% of genomic content (Schnable et al. 2009), and expansion and reduction of TEs can result in rapid changes in both genome size and architecture (Haas et al. 2009; Möller and Stukenbrock 2017; Talla et al. 2017). Generally, TEs have small sizes (∼50–12,000 bp) and accomplish these large-scale changes through their sheer number. For example, there are ∼1.1 million Alu elements in the human genome, which have had a large impact on genome evolution (Jurka 2004; Bennett et al. 2008). The largest known cases among Class I retrotransposons are long terminal repeat (LTR) elements that can be as large as 30 kb, but among Class II DNA transposons, Mavericks/Polintons are known to grow as large as 40 kb through the capture of additional open reading frames (ORFs) (Arkhipova and Yushenova 2019). Recently, a behemoth TE named Teratorn was described in teleost fish; it can be up to 182 kb in length, dwarfing all other known TEs. Teratorn has achieved this impressive size by fusing a piggyBac DNA transposon with a herpesvirus, thereby blurring the line between TEs and viruses (Inoue et al. 2017, 2018). Truly massive transposons may be lurking in the depths of many eukaryotic genomes, but the limitations of short-read genome sequencing technologies and the lack of population-level high-quality assemblies may make them difficult to identify.
The Spok block is a large genomic feature that was first identified thanks to the presence of the spore killing (Spok) genes in species from the genus Podospora (Grognet et al. 2014; Vogan et al. 2019). The Spoks are selfish genetic elements that bias their transmission to the next generation in a process known as meiotic drive. Here, drive occurs by inducing the death of spores that do not inherit them, through a single protein that operates as both a toxin and an antidote (Grognet et al. 2014; Vogan et al. 2019). The first Spok gene described, Spok1, was discovered in Podospora comata (Grognet et al. 2014). In P. anserina, the homologous gene Spok2 is found at high population frequencies, whereas two other genes of the Spok family, Spok3 and Spok4, are at low to intermediate frequencies (Vogan et al. 2019). Unlike Spok1 and Spok2, however, Spok3 and Spok4 are always associated with a large genomic region (the Spok block). The Spok block can be located at different chromosomal locations in different individuals but is never found more than once in natural strains. The number of Spok genes and the location of the Spok block (which carries Spok3, Spok4, or both) define the overall meiotic driver behavior of a given genome, which can be classified into the so-called Podospora spore killers or Psks (van der Gaag et al. 2000; Vogan et al. 2019). The Spok block stands out not only because of its size, typically around 150 kb, but also because there is otherwise high genome collinearity among strains of P. anserina and with the related species P. comata and P. pauciseta (Vogan et al. 2019).
The fact that the Spok block is found at unique genomic positions between otherwise highly similar strains is of prime interest as each novel Spok block position creates a unique meiotic drive type (Psk) due to the intricacies of meiosis in Podospora (Vogan et al. 2019). We therefore set out to determine the mechanism through which the Spok block relocates throughout the genome. Additionally, we annotated the gene content of the various Spok blocks to describe their composition and understand what represents the minimal component of the Spok block. Lastly, we conducted fitness assays to investigate whether the presence of the Spok block imparts any detrimental effects upon the host.
Results
Newly isolated Podospora strains reveal a novel spore killer type
We isolated and sequenced with Illumina HiSeq technology a strain of P. anserina (named Wa137) and two strains of P. comata (Wa131 and Wa139) collected in Wageningen, the Netherlands. The strains Wa137 and Wa139 were also sequenced using MinION Oxford Nanopore technology, achieving similar quality to published chromosome-level assemblies (Supplemental Table S1). Additionally, we included in our analyses all published long-read genomes of P. anserina (10 strains), P. pauciseta (one), and P. comata (one) (Espagne et al. 2008; Silar et al. 2019; Vogan et al. 2019), which are mostly assembled at chromosome level, with respective genome sizes around 36 Mb (Supplemental Table S1). Previous work demonstrated that the Spok block can be found at three unique chromosomal positions among characterized P. anserina strains, defining the killer types Psk-1/5 (with the Spok block located at Chromosome 3), Psk-2 (right arm of Chromosome 5), and Psk-7/8 (left arm of Chromosome 5) (Table 1; Vogan et al. 2019). The newly isolated strain Wa137 was found to have a Spok block with the largest size yet reported (247 kb) and in a novel position (Chromosome 1), conferring it a new spore killing phenotype that we named Psk-9 (killing percentage of 70% against Psk-S) (see Vogan et al. 2019 for the principle of nomenclature). The published P. pauciseta genome has a Spok block in Chromosome 4 (Psk-P1), but it seems to represent a fragmented version of the P. anserina blocks (Vogan et al. 2019). Unlike P. anserina and P. pauciseta, the newly isolated strains of P. comata were found to have a single full-copy Spok gene (Spok1) which is not associated with any Spok block-like features, in agreement with the previously published reference genome of P. comata (Table 1; Grognet et al. 2014; Silar et al. 2019).
Table 1.
Strains used in this study

The Spok block is inferred to move via transposition
A number of mechanisms exist by which a sequence can move within a genome, including reciprocal translocation, ectopic recombination, and transposition (Mieczkowski et al. 2006). We can rule out reciprocal translocations in the case of the Spok block, as overall chromosome collinearity is preserved (Vogan et al. 2019). Ectopic recombination is often mediated by TEs, and transposition is, by definition, the process of TE mobility. In order to determine a candidate mode by which the Spok block moves, we examined the four unique Spok block insertion sites. This analysis revealed that no specific TE was present at the insertion site but showed that six base pairs (RGGTAG) are always present and are repeated at the end of the Spok block (Fig. 1A,B). This repeated insertion-flanking motif occurs at typical genomic background frequencies (Supplemental Fig. S1) and may represent a target site duplication (TSD), which is a hallmark of transposition mechanisms (Wicker et al. 2007). Additionally, the Psk-1 and Psk-9 Spok block insertion sites constitute a partially palindromic sequence ATACYT||AGGTAG (Fig. 1B), a characteristic of some TE target sites (Linheiro and Bergman 2012). Together, these findings support an explanation whereby the Spok block is mobilized like a TE via transposition.
Figure 1.

Genomic context of the Spok block. (A) An alignment of the region where the Spok block is located in the strain Wa137 (Psk-9) from all P. anserina genomes analyzed herein. The putative target site duplication (TSD) is highlighted. Only the ends of the Spok block are shown for clarity. (B) An alignment of the ends of four versions of the Spok block displaying the TSD (pink trapezoid) and an imperfect palindromic motif (magenta). The flanks of the Spok block that relate to the unclassified repeats bufo and schoutedenella are underlined in red and blue, respectively. Note that for Psk-7, the Spok block is inserted immediately next to a truncated bufo element.
The terminal sequences of TEs are often composed of structural features that are intrinsic to the transposition mechanism; LTRs for LTR retrotransposons and terminal inverted repeats (TIRs) for transposase-mobilized DNA transposons, for example (Wicker et al. 2007). These features can thus be used to determine the type of transposition or transposon underlying a given translocation. With this expectation, we examined the ends of the Spok block (which are very similar across all instances) (Vogan et al. 2019) but found no such structural features flanked by the TSDs. The ends of the Spok block have been previously identified as two unclassified repetitive elements called bufo and schoutedenella (Fig. 1B; Espagne et al. 2008). Neither of these elements appears to have features indicative of TEs, such as TSDs, terminal inverted repeats, or transposases. We suspect that these unrelated elements actually represent the two ends of the Spok block (bufo representing the first 463 bp and schoutedenella representing the last 231 bp) and are found on their own in the genome as remains of previous, partially deleted, Spok block copies. To test this assumption, we mapped the location of all bufo and schoutedenella elements present in all the Podospora genomes. We found that both elements can be identified throughout the chromosomes, with a tendency to locate in TE-rich areas in all species (Fig. 2; Supplemental Fig. S2). In agreement with the postulate that the genomic copies of bufo and schoutedenella represent past Spok block insertions, we found that aligning the flanking sequences of these two elements often reveals extended homology with the Spok block (Supplemental Fig. S3). For example, at the Psk-7 insertion site, there is ∼1200 bp of sequence that is homologous to the beginning of the Spok block. This is nearly 700 bp longer than what bufo alone represents and further implies that the Psk-7 Spok block was inserted at the same site as a previous, now largely deleted copy. Moreover, the RGGTAG motif is only ever found at the 5′ end of bufo and at the 3′ end of schoutedenella, as defined in our repeat library. Together, these findings further support the idea that the Spok block inserts at the specific target site sequence RGGTAG which is duplicated during transposition (Fig. 1B).
Figure 2.

A Circos plot of the genome of Wa137 (light gray track, size in Mb) aligned against its own Spok block (dark gray, enlarged by 50×). The tracks on the Spok block, from outside inward, represent: the conservation of regions among the different iterations of the Spok block where the height of the conservation track is equivalent to the number of blocks that have a given position (light blue); gene models from manual annotation of the Spok block, indicating genes with homologs within the reference genome of P. anserina (orange) and those without homologs (purple); and annotated repeated elements (red). The tracks on the chromosomal scaffolds show the coverage of genic regions (orange) and repeats (red) calculated in sliding windows of 50 kb with steps of 10 kb. Green lines connect homologous segments based on MUMmer alignments. The unclassified repetitive elements bufo and schoutedenella are connected in dark red and blue segments, respectively, based on BLASTN searches, highlighting potential past Spok block or closely related Enterprise element insertion points.
The Spok block accumulates sequence from the genome
As there were no structural features to guide us in our attempt to determine the mechanism through which the Spok block translocates, we examined the composition of the Spok block to identify candidate genes capable of transposing it. We annotated all genome assemblies using a modified version of the annotation pipeline in Vogan et al. (2019), relying on additional RNA-seq data, as well as an improved, manually curated repeat library (see Supplemental Methods). The content of the different copies of the Spok block is largely overlapping (Vogan et al. 2019), although the Wa137 Spok block has a large region of unique sequence, resulting in a total size of 247 kb (Fig. 2; Supplemental Fig. S4). Very few genes were predicted within the Spok block by this methodology (as evidenced by the dip in the orange track on the Chr 1.2 scaffold in Fig. 2), so we manually annotated the Spok block of representative strains (Wa53, Wa28, Wa58, and Wa137), which identified numerous hypothetical protein-coding genes (e.g., 67 in the Wa137 Spok block), many of which appear to be pseudogenes or gene fragments. During this process of manual annotation, it became apparent that, although we had previously reported that a pseudogenized Spok gene (SpokΨ1) on Chromosome 5 of some strains was not located in a known Spok block (Vogan et al. 2019), this region does in fact appear to correspond to a Spok block in an advanced state of degradation (Supplemental Fig. S5).
The blocks do not generally exhibit an unusually high TE load. For example, the Spok block in Wa137 is only composed of 10.5% annotated TEs (compared to genome-wide estimates of 3%–6%) (Supplemental Table S1), nor do they appear to have a strong signature of repeat-induced point mutation (RIP). RIP is a process that operates in numerous fungi, including Podospora (Graïa et al. 2001), by specifically inducing C-to-T mutations of any repeated sequence within the genome (Selker et al. 1987; Cambareri et al. 1989) and results in a drop of GC proportion in repetitive elements. Such a GC-drop is clear in many regions of the Podospora genome but is conspicuously absent within the Spok block (Vogan et al. 2019). Whereas some of the predicted genes had identifiable homologs within the P. anserina reference genome (orange features in Fig. 2), many are absent from the reference but have sequence similarity to genes from other fungi (purple features in Fig. 2). These could either represent genes which were copied into the block from the main genome and then subsequently lost from all strains, or they could be the result of horizontal gene transfer from other fungi to P. anserina. Of note, many of the predicted genes have potential roles in various metabolic pathways, like metal tolerance or antimicrobial resistance (Supplemental Table S2). It thus seems likely that the Spok block has grown to its large size, at least in part, by accumulating nonrepetitive sequence from elsewhere within the genome. The accumulation does not appear to have been a single event but was more likely many individual events, as the genomic homologs are found on all chromosomes (green links in Fig. 2).
The Enterprise
The gene annotation provided us with very few clues as to the agent(s) responsible for the translocation of the Spok block, so we turned to another approach. We hypothesize that the Spok block was a much smaller element in the past and that such an element could be still transposing in the genomes of Podospora species. The closely related species P. comata possesses both bufo and schoutedenella repeats and appears to have large areas of homology to the Spok block (Supplemental Fig. S2B). We discovered that the P. comata strain Wa139 contains a 39-kb subtelomeric region on Chromosome 5 (henceforth referred to as the Enterprise) that is nearly completely composed of TEs and sequence homologous to the Spok block (Supplemental Fig. S4). The ends of the Enterprise are consistent with the Spok block (both bufo and schoutedenella are present), and it possesses the RGGTAG target site duplication (Fig. 3A; Supplemental Fig. S6A). Enterprise is absent at the orthologous location in P. anserina and P. pauciseta, is much smaller in the other P. comata strain TD (5.5 kb), and unassembled in Wa131. The other P. comata strains possess bufo and schoutedenella at this location, suggesting that this specific Enterprise insertion is now polymorphic for its state of degradation. However, it is difficult to fully recapitulate the history of the region due to the limited number of strains and the fact that Wa139 also has another schoutedenella copy at the insertion site that is absent in the other strains (Supplemental Fig. S6A). Manual annotation revealed five putative genes within the Wa139 Enterprise, most of which are homologous to the genes within the Spok block and thus represent good candidates for involvement in the transposition of Enterprise and therefore the Spok block (Fig. 3A).
Figure 3.

The Enterprise and the crew. (A) Cartoon models representing the structure and gene content of four Spok blocks and of Enterprise from Wa139. Relevant genes and features are annotated. Not to scale. (B) Cartoon model of the hierarchical nomenclature of Enterprise-like elements, Enterprise, and the Spok block. (TSD) Target site duplication.
None of these genes have a known function and none are Spok homologs. We examined their predicted protein domains, searched the P. anserina genomes for homologs, and scanned genomic databases for similar sequences in an attempt to discern whether they may be integral to the movement of the Enterprise (Supplemental Table S2). Only one of the genes is a likely candidate for enacting the transposition of the Enterprise. We have named this gene Kirc, for “spore killing related crypton” (see below). Kirc is always present as the first ORF in the Spok block but is degraded and interrupted by multiple TEs in Wa139, suggesting that this copy would no longer be active. The ORF possesses multiple domains which may be characteristic of specific transposons, as determined by bioinformatic analysis (see Supplemental Methods): a domain of unknown function called DUF3435, a zinc finger domain, and a predicted CHROMO domain (Fig. 4A). Zinc finger domains are important for DNA binding, and CHROMO domains are implicated in histone binding and may allow TEs to be targeted to specific regions of the genome (Kordiš 2005). There is strong overlap between the DUF3435 domain and tyrosine recombinases (YRs), a group of enzymes important for DNA integration of other selfish genetic elements known as Cryptons, parasitic plasmids, and bacteria (Kojima and Jurka 2011). YRs are extremely divergent, and there is little conserved sequence identity between this gene and known YRs, but importantly, YRs are known to have a catalytic pentad consisting of an R-K-H-R-Y motif (Esposito and Scocca 1997; Ma et al. 2007), which is overlapping with the DUF3435 domain (Fig. 4A,B).
Figure 4.

Molecular characterization of Kirc. (A) Domain annotation of the Kirc protein obtained using HHPred. Four domains were identified: an N-terminal tyrosine recombinase domain with the associated core-binding domain, a central Zn-finger domain, and a C-terminal chromodomain. An alignment of 279 Kirc homologs generated by Gremlin with HHBlits was used to create sequence logo of relevant regions of the Kirc sequences, including the active site regions of the tyrosine recombinase domain (i and ii), the Zn-finger region (iii), and the chromodomain region (iv) using WebLogo. Residues corresponding to the catalytic pentad of the tyrosine recombinase are highlighted in yellow. (B) Alignment of the Kirc sequence with an active site region of known tyrosine recombinases together with the sequence of a Kirc homolog from Melanconium sp. strain NRRL 54901. Alignment is based on the HHPred output. Active site residues are marked with an asterisk. (Kirc) Kirc gene in the Spok block of strain Wa58 (sites 309–344), (Kirc_Msp) CE55503_10527 (110–145), (lambda) lambda integrase PDB: 5J0N (308–342), (Cre) recombinase enterobacteriaphage P1, PDB: 1XO0 (270–305), (XerD) XerD site-specific recombinase E. coli, PDB: 1A0P (242–277).
Although we suspect that Kirc plays the primary role in the transposition of the Enterprise and the Spok block, the other genes found in Wa139 and conserved in the Spok block may have accessory roles in the transposition mechanism. We have named these genes Scoty, Chekof, and Uhera. A fifth, unnamed ORF seems to be a fragment of the gene Pa_6_8905 present in the reference genome (see Espagne et al. 2008 for Podospora gene notation) but otherwise absent from the Spok blocks. The new gene Scoty is distantly related to Pa_6_8905 (29% amino acid similarity), and neither it nor its homologs in other species have known protein domains or obvious features indicative of a putative TE. Chekof shows the highest similarity to Pa_6_2440, but there are another three additional divergent paralogs in P. anserina. It possesses a kinase domain but otherwise has no known function or putative TE features. The final gene in the block is named Uhera and is unusual in a number of ways. It has very few homologs among closely related fungi, including none within the reference genome for P. anserina, yet is present within the Spok block multiple times (Fig. 3). In the strain TG, up to four Uhera copies can be found within its Spok block due to a segmental duplication (Vogan et al. 2019). In fact, we previously showed that the Spok genes themselves are associated with segmental duplications within the block (Vogan et al. 2019), and Uhera is found within these duplications as well. Uhera possesses a putative DEAD-like helicase, which could implicate it in either DNA repair or as a Helitron transposon, but there are no other transposon features. Additionally, it is difficult to discern a proper gene model for the ORF as there is an apparent stop codon near the beginning of the gene conserved in other homologs even outside Podospora. RNA-seq data indicate RNA editing of the stop codon and other bases, and hence transcripts of this gene might still be intact (Supplemental Fig. S7).
From the evidence presented here, we propose that the Spok block and the homologous region of Wa139 represent copies of a previously unknown group of DNA transposons, which we name Enterprise. Cryptons are a type of DNA transposon defined only by the presence of a YR domain (Wicker et al. 2007) and may possess a TSD or not (Kojima and Jurka 2011); thus, Enterprise can be classified as a novel group of Crypton. We define Enterprise as being composed of a YR-encoding gene homologous to Kirc and possessing a TSD (Fig. 3B). The Spok block therefore represents a specific version of Enterprise that contains Spok genes.
To support our hypothesis that Enterprise is capable of transposition and selfish replication, we mined fungal genomes available on JGI MycoCosm for homologous proteins of Kirc that are present in multiple copies within a single genome. We identified such a case in Melanconium sp. NRRL 54901. In this genome, a homolog of Kirc (∼60% amino acid similarity) (Fig. 5) is found at the beginning of a ∼70-kb region that is present in four copies in the genome (see Supplemental Fig. S8). Only one of the four copies is well annotated for genes; the other three appear to be degraded and have only ∼30% GC content, suggesting that they have been the target of RIP. This region is flanked by the same TSD as the Spok block, and the target site is in its full palindromic context CTACCT||AGGTAG (Supplemental Fig. S6B). The only gene homologous with the Enterprise in Podospora is the Kirc homolog (Supplemental Fig. S8B), thereby confirming that the minimal feature for transposition is the putative YR-encoding gene, Kirc, and classifying this region of Melanconium sp. as an Enterprise.
Figure 5.

Unrooted maximum likelihood phylogenetic tree showing the relationship between Kirc, the homolog from Melanconium sp., and other homologs from fungi. Bootstrap support values are shown above branches. Branch lengths are proportional to the scale bar (amino acid substitution per site). Taxonomic rank is indicated with colored highlights. The sequences present in Podospora anserina and in the basidiomycete Fomitopsis rosea are marked with cartoons of the corresponding fruiting bodies. Nomenclature is formatted as species_strain_protein code.
Kirc is widespread in filamentous fungi
Given the fact that Enterprise is present in at least one species outside of Podospora, we queried the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) with the sequence of Kirc using BLASTP to determine how widespread Enterprise is. We recovered a total of 481 protein hits, with given strains possessing between one and 21 copies (Supplemental Table S3). The hits were mostly from genomes of Pezizomycotina, although putative homologs were identified from 10 basidiomycete genomes as well. A phylogeny of a representative set of sequences shows that the relationships among the homologs do not follow the expected species phylogeny (Fig. 5). Numerous species have multiple Kirc homologs in the same genome, some of which are not closely related. For example, within P. anserina, one homolog was recovered, Pa_5_10116, which appears to be distantly related to Kirc, yet highly similar to homologs from Fusarium. Furthermore, Pa_5_10116 is pseudogenized and absent in the close relatives of P. anserina, P. comata and P. pauciseta, which is more consistent with it being a transposable element, as opposed to performing cellular functions. The Melanconium sp. also has an additional homolog that is not associated with its Enterprise element (Melsp1_19023). Attempts to describe TSDs in other fungi were largely unsuccessful. The CTACCT||AGGTAG motif can be found in some copies at the beginning of the element, but we have not been able to find ones with the RGGTAG motif preserved at the other terminus, suggesting that either these are 3′ truncated copies or potentially that the terminal RGGTAG motif in Podospora and Melanconium is part of the elements instead of a TSD. In general, Cryptons appear to vary in whether or not they produce TSDs upon insertion, so this is not necessarily unexpected (Kojima and Jurka 2011). Thus, in the absence of additional evidence from these fungal species, we consider these as Enterprise-like elements. Regardless, these results suggest that Enterprise is a YR-mobilized group of DNA transposons, related to Cryptons, that is spread throughout fungal genomes.
The Spok block can be deleterious
In P. anserina, wild strains with more than one full copy of the Spok block have never been found, although it is easy to generate them in the laboratory via crosses (Vogan et al. 2019). As observed in other meiotic drive systems (Lindholm et al. 2016), it is possible that the Spok block imposes a fitness cost to strains which possess it. To evaluate this hypothesis, we made use of backcrosses of two of the Psk strains (Vogan et al. 2019). The backcrossed strains Psk1xS14 and Psk7xS14 are isogenic with the reference strain S, except that S has no Spok block. Psk1xS14 has the Spok block on Chromosome 3 and induces killing in 90% of meioses, and Psk7xS14 has the Spok block on the left arm of Chromosome 5 and induces spore killing in 50% of meioses. We crossed strains either to themselves (no spore killing) or to strain S (spore killing) (Fig. 6A) and evaluated three traits assumed to correlate to fitness (Pringle and Taylor 2002). Radial growth and percentage of germinated spores produced by the matings showed no variation between crosses (Supplemental Table S4); however, significant differences were observed among the amount of spores produced by a cross (Fig. 6B). Specifically, crosses from selfings of Psk1xS14 produced significantly fewer spores than from selfings of either S or Psk7xS14, despite the fact that no spore killing occurs, indicating that the Psk-1 Spok block inhibits spore production. This effect was even greater in crosses between S and Psk1xS14, and was most prominent when Psk1xS14 was used as the female, suggesting a maternal effect. With both Psk1xS14 and Psk7xS14, more spores were produced in the killing crosses than expected given the proportion of killing per ascus (Vogan et al. 2019). As there were no significant differences in the amount of spores produced by Psk7xS14 than by S, this result suggests that the general presence of the Spok block itself is not deleterious but rather that the negative effect is due to the specific content and/or to the genomic location of a given Spok block.
Figure 6.
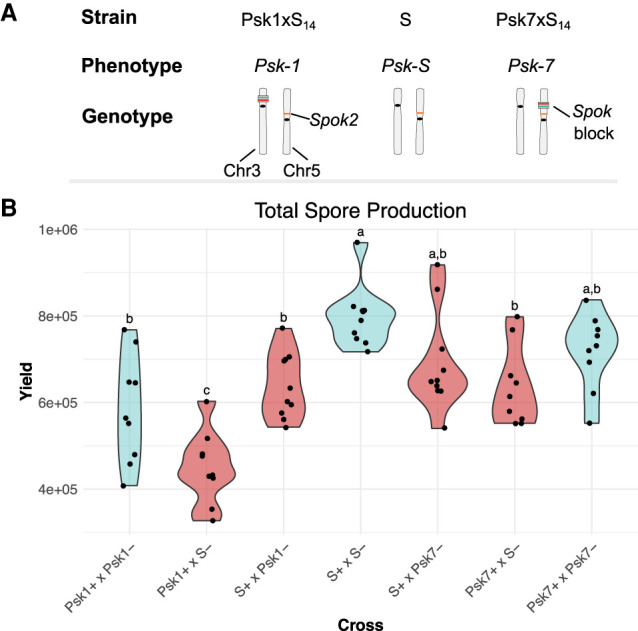
Fitness effects of the Spok block. (A) Diagram of the genotypes and phenotypes of strains used for the fitness assay. Genotypes are illustrated with location of the Spok genes and Spok blocks on their respective chromosomes. Within the Spok block, Spok3 is marked in green and Spok4 is marked with red. Spok2 (orange) is not located within a Spok block but is present in all strains, so it does not cause spore killing. (B) Violin plots showing the total amount of ascospores collected from crosses of the isogenic strains possessing various iterations of the Spok block, where Psk1 and Psk7 refer to strains Psk1xS14 and Psk7xS14, respectively. The mating type is indicated by a + or − after the strain name. The mat+ parent was always used as the maternal strain in crosses. Yields obtained in crosses where spore killing occurs are shown in red; crosses with no spore killing are in blue. Letters mark the results of an ANOVA as analyzed with a Tukey's HSD test. Dots indicate replicate values (jittered).
Discussion
Here, we provide evidence that the Spok block is a variant of a newly described TE, Enterprise, that likely moves throughout the genome of P. anserina by means of YR-mediated transposition. In addition to this ability to move, the Spok block is also capable of meiotic drive due to the presence of the Spok genes. Given the union of these two selfish properties, it can be asked who is parasitizing whom. One possibility is that Enterprise has hijacked the Spoks in order to increase its rate of transmission, thereby parasitizing a resident genomic parasite and becoming a genomic hyperparasite. Support for this commandeering can be found in the distribution of the Spok genes. In both P. anserina and P. comata, the Spok genes that are not found in association with the Spok block are at high frequency (Spok2 is found in 98% of strains isolated in Wageningen [Vogan et al. 2019], and so far Spok1 has been found in all analyzed strains of P. comata). The Spok block is comparatively rare (∼18% of strains from Wageningen) (Vogan et al. 2019), indicating that it may prevent the Spoks from reaching high frequencies. Nevertheless, it is possible that this low percentage still represents a higher frequency than Enterprise could have achieved without the Spok genes. Alternatively, the Spok genes may generally benefit from moving throughout the genome. Spok1 and Spok2 are found at different locations in the genome and are surrounded by TEs, suggesting that they may have moved through other mechanisms like TE-mediated ectopic recombination (Vogan et al. 2019). It may thus be advantageous for the Spoks to mobilize within TEs, like Enterprise, in order to change their genomic position on a regular basis, due to the fact that this relocation will result in a novel spore killing phenotype. Additionally, as the distance of the Spok genes to the centromere directly modifies the percentage of spores killed (spore killing approaches 100% as Spoks are located closer to centromeres) (Vogan et al. 2019), a given Spok gene may be able to increase its transmission distortion by relocating with the help of Enterprise. We have no direct evidence that this occurs, but Spok2 has a relatively low killing percentage (40%), whereas all Spok blocks are located at positions which result in killing frequencies of 50%–90%. Presumably, the population dynamics of meiotic drive ultimately decide the fate of the Spoks (Nauta and Hoekstra 1993), but the confederation of the Spoks and Enterprise as the Spok block may fundamentally change how effective selection is at controlling either element.
Given that the results of the fitness experiments suggest that the Spok block can be deleterious, there may be strong selection to purge any copies of Enterprise, but this may ultimately be dependent on genomic context as insinuated by the differences in phenotypic effect of carrying the Psk-1 and Psk-7 Spok blocks. The Psk-7 Spok block is larger by nearly 50 kb, some of which includes known retrotransposons. Yet, only Psk-1 showed a significant decrease in the amount of spores produced. It thus seems probable that the location of the block has a stronger deleterious effect rather than its content or size. The Psk-1 Spok block is located close to the centromere on the left arm of Chromosome 3. It is possible that the increased amount of killing in Psk-1 results in its poorer performance regarding spore production, although this cannot explain the observed maternal effect. The rDNA cluster resides on the same chromosome arm as the Psk-1 block (Espagne et al. 2008). Given that this arm is only ∼700 kb and the block itself is 113 kb, the Spok block insertion might interfere with recombination, either through changes of 3D chromosome structure or epigenetic marks, which is necessary for proper segregation of rDNA (Tomson et al. 2006) and could inhibit the ability of the strain to produce viable spores. Moreover, the observation that there is not a large decrease in spore production for Psk-1 or Pks-7 strains when involved in spore killing suggests that strains are able to compensate for the lost spores in some way. This latter result has significant implications to our understanding of spore killing as meiotic drive, as it shifts the system away from providing the killer genotype a relative fitness advantage to an absolute fitness advantage (Lyttle 1991; Nauta and Hoekstra 1993; Martinossi-Allibert et al. 2021). Explicitly, in the naive expectation, a spore killer that is 100% efficient would reduce the total amount of spores produced in a killing cross to half. Thus, it does not produce more total offspring with its genotype than a non-spore killing gene (relative advantage). However, if the strain carrying the spore killer is able to compensate for the loss of spore production and produce the same or similar number of spores as in a nonkilling cross (as observed here), the spore killer observes an absolute advantage, as is the case for other types of meiotic drive, like female meiotic drive. Therefore, spore killers may be more successful at invading and driving through populations than previously thought.
The Enterprise clearly has the ability to move large amounts of genetic material around the genome, and TEs are known to be agents of horizontal gene transfer (HGT) (Schaack et al. 2010; Gilbert and Feschotte 2018). As such, it is plausible that Enterprise and related YR-mobilized DNA transposons may jump between species and thereby transport any additional genes they may carry. The phylogeny of the Kirc homologs is indicative of HGT as it shows closely related Kirc homologs distributed among unrelated fungi. In at least two cases, fungal TEs have been implicated as the vehicles for gene mobility of adaptive genes. In the first case, a hAT element is associated with the HGT of toxin genes among cereal pathogens (McDonald et al. 2019). The second case comes from a recent publication which described a TE named HEPHAESTUS in the fungus Paecilomyces (Urquhart et al. 2020). It carries multiple genes that provide resistance to at least five different heavy metals and shows evidence of transfer with a distantly related species of Penicillium. In the latter example, the TE responsible for enacting the transposition is unidentified, but a gene with a DUF3435 domain is present at the start of HEPHAESTUS (annotated as hhpA). We find little similarity between Kirc and hhpA at the protein level, but our analyses here strongly suggest that hhpA is also a YR and may be responsible for the reported transposition. Whether the Enterprise itself can also play a role in adaptive HGT is unknown as of yet, but the potential certainly exists. We note that most of the fungal genomes in which HGT has been reported (Wisecaver and Rokas 2015) possess genes containing DUF3435 domains which, together with the data from HEPHAESTUS, suggests a convergent role in this type of TE as a vector for HGT of adaptive genes in fungi.
The constant “tug-of-war” between TEs linking themselves to host genes and the actions of genome defense and selection to purge them is of key importance to the evolution of genome architecture. It is likely that we are witnessing this fight play out to the extreme in P. anserina, with the high effectiveness of both RIP and the Spok block, making Podospora an ideal system in which to continue to study genomic conflict. This study not only changes the perception of how TEs influence genome evolution but also broadens the horizon in terms of what may be possible through genetic manipulations in the laboratory. Understanding the molecular mechanism of YR-mediated transposition and conducting laboratory assays to confirm that Kirc is indeed capable of transposing with such a large amount of sequence should thus be of highest priority for future research.
Methods
An extensive description of the methodology is available in the Supplemental Methods.
DNA extraction, genome sequencing, and assembly
Briefly, the DNA of the newly isolated strains Wa131, Wa137, and Wa139 was extracted and sequenced with paired-end Illumina HiSeq X technology (all), RNA-seq (Wa131), and MinION Oxford Nanopore (Wa137 and Wa139). The MinION reads were filtered and assembled with minimap2 v. 2.11 and Miniasm v. 0.2 (Li, 2016, 2018). The resulting assemblies were polished with Racon v.1.3.1 (Vaser et al. 2017) and Pilon v. 1.22 (Walker et al. 2014). We used SPAdes v. 3.12.0 (Bankevich et al. 2012) to assemble cleaned reads of Wa131. These new genome assemblies were used in conjunction with previously published assemblies of other strains (Espagne et al. 2008; Silar et al. 2019; Vogan et al. 2019).
Comparative genomics and phylogenetics
For genome annotation, we produced a manually curated repeat library and ran an updated version of the pipeline in Vogan et al. (2019) that relies on MAKER v. 3.01.2 and on the RNA-seq data. We further performed manual curation of the annotation of the Spok blocks and related elements. To assess the abundance of the TSD motif typical of the Spok block, we used Jellyfish v. 2.2.10 (Marçais and Kingsford 2011). Genome comparisons were made using the MUMmer package v. 4.0.0beta2 (Kurtz et al. 2004), the BLAST suite 2.9.0 (Camacho et al. 2009), and Circos v. 0.69.6 (Krzywinski et al. 2009). Maximum likelihood analyses were performed using IQ-TREE v. 1.6.8 (Nguyen et al. 2015; Kalyaanamoorthy et al. 2017).
Fitness assays
To measure the spore yield as a proxy for fitness, we grew strains S and backcrosses Psk1xS14 and Psk7xS14 (Vogan et al. 2019) in 35-mm Petri dishes with HPM medium (Vogan et al. 2019). After 7 d, these colonies acted as female recipients for fertilization with a microconidial suspension (King 2013) of different combinations of the same strains, simulating selfing or outcrossing (with subsequent spore killing). The fertilized mycelia were incubated under standard conditions (27°C, 12/12 light/dark cycle) (Vogan et al. 2019) and monitored for the shooting of spores. At 12 d postfertilization, spores were harvested from the lids of each crossed culture and used for estimating total spore yield. Ten replicate crosses were conducted for each cross.
Data access
The short- and long-read sequencing data generated in this study, as well as the genome assemblies, have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA523441 (Biosamples SAMN15214446, SAMN15214447, and SAMN15214448). Genome assemblies, annotation files, the TE library, and the spore yield data generated in this study have been submitted to the Dryad Digital Repository (https://datadryad.org/stash/dataset/doi:10.5061/dryad.4tmpg4f8m). Reference sequences of Kirc, Scoty, Chekof, and Uhera were submitted to the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) (accession numbers MW262968–MW262970 and MW591707). Main bioinformatic pipelines were written in Snakemake v. 5.4.4 (Köster and Rahmann 2012) and are available in a GitHub repository (https://github.com/johannessonlab/SpokBlockPaper) and as Supplemental Code.
Supplementary Material
Acknowledgments
This work was supported by the European Research Council (ERC) grant ERC-2014-CoG (project 648143, SpoKiGen) and The Swedish Research Council (project 2015-04649) to H.J., and by the Lars Hierta Memorial Foundation and The Nilsson-Ehle Endowments of the Royal Physiographic Society of Lund to S.L.A.-V. We thank the National Genomics Infrastructure (NGI)/Uppsala Genome Center for support on massive parallel DNA sequencing. The computations were performed on resources provided by SNIC through the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under the projects SNIC 2017/1-567 and SNIC 2019/8-371. The sequence data of Melanconium sp. NRRL 54901 was produced by the U.S. Department of Energy Joint Genome Institute (http://www.jgi.doe.gov/) in collaboration with the user community. We also thank Sergio Tusso and the TE Jamboree of the Suh Lab for useful advice, and three anonymous reviewers for comments that improved this manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.267609.120.
Freely available online through the Genome Research Open Access option.
Competing interest statement
The authors declare no competing interests.
References
- Arkhipova IR, Yushenova IA. 2019. Giant transposons in eukaryotes: is bigger better? Genome Biol Evol 11: 906–918. 10.1093/gbe/evz041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19: 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett EA, Keller H, Mills RE, Schmidt S, Moran JV, Weichenrieder O, Devine SE. 2008. Active Alu retrotransposons in the human genome. Genome Res 18: 1875–1883. 10.1101/gr.081737.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10: 421. 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambareri E, Jensen B, Schabtach E, Selker E. 1989. Repeat-induced G-C to A-T mutations in Neurospora. Science 244: 1571–1575. 10.1126/science.2544994 [DOI] [PubMed] [Google Scholar]
- Chénais B, Caruso A, Hiard S, Casse N. 2012. The impact of transposable elements on eukaryotic genomes: from genome size increase to genetic adaptation to stressful environments. Gene 509: 7–15. 10.1016/j.gene.2012.07.042 [DOI] [PubMed] [Google Scholar]
- Espagne E, Lespinet O, Malagnac F, Da Silva C, Jaillon O, Porcel BM, Couloux A, Aury J-M, Ségurens B, Poulain J, et al. 2008. The genome sequence of the model ascomycete fungus Podospora anserina. Genome Biol 9: R77. 10.1186/gb-2008-9-5-r77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito D, Scocca JJ. 1997. The integrase family of tyrosine recombinases: evolution of a conserved active site domain. Nucleic Acids Res 25: 3605–3614. 10.1093/nar/25.18.3605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C, Feschotte C. 2018. Horizontal acquisition of transposable elements and viral sequences: patterns and consequences. Curr Opin Genet Dev 49: 15–24. 10.1016/j.gde.2018.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graïa F, Lespinet O, Rimbault B, Dequard-Chablat M, Coppin E, Picard M. 2001. Genome quality control: RIP (repeat-induced point mutation) comes to Podospora. Mol Microbiol 40: 586–595. 10.1046/j.1365-2958.2001.02367.x [DOI] [PubMed] [Google Scholar]
- Grognet P, Lalucque H, Malagnac F, Silar P. 2014. Genes that bias Mendelian segregation. PLoS Genet 10: e1004387. 10.1371/journal.pgen.1004387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Kamoun S, Zody MC, Jiang RHY, Handsaker RE, Cano LM, Grabherr M, Kodira CD, Raffaele S, Torto-Alalibo T, et al. 2009. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 461: 393–398. 10.1038/nature08358 [DOI] [PubMed] [Google Scholar]
- Inoue Y, Saga T, Aikawa T, Kumagai M, Shimada A, Kawaguchi Y, Naruse K, Morishita S, Koga A, Takeda H. 2017. Complete fusion of a transposon and herpesvirus created the Teratorn mobile element in medaka fish. Nat Commun 8: 551. 10.1038/s41467-017-00527-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Kumagai M, Zhang X, Saga T, Wang D, Koga A, Takeda H. 2018. Fusion of piggyBac-like transposons and herpesviruses occurs frequently in teleosts Zoological Lett 4: 6. 10.1186/s40851-018-0089-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J. 2004. Evolutionary impact of human Alu repetitive elements. Curr Opin Genet Dev 14: 603–608. 10.1016/j.gde.2004.08.008 [DOI] [PubMed] [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14: 587–589. 10.1038/nmeth.4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RC. 2013. Handbook of genetics: volume 1 bacteria, bacteriophages, and fungi. Springer, New York. [Google Scholar]
- Kojima KK, Jurka J. 2011. Crypton transposons: identification of new diverse families and ancient domestication events. Mob DNA 2: 12. 10.1186/1759-8753-2-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordiš D. 2005. A genomic perspective on the chromodomain-containing retrotransposons: chromoviruses. Gene 347: 161–173. 10.1016/j.gene.2004.12.017 [DOI] [PubMed] [Google Scholar]
- Köster J, Rahmann S. 2012. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 28: 2520–2522. 10.1093/bioinformatics/bts480 [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. 10.1101/gr.092759.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol 5: R12. 10.1186/gb-2004-5-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2016. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32: 2103–2110. 10.1093/bioinformatics/btw152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm AK, Dyer KA, Firman RC, Fishman L, Forstmeier W, Holman L, Johannesson H, Knief U, Kokko H, Larracuente AM, et al. 2016. The ecology and evolutionary dynamics of meiotic drive. Trends Ecol Evol 31: 315–326. 10.1016/j.tree.2016.02.001 [DOI] [PubMed] [Google Scholar]
- Linheiro RS, Bergman CM. 2012. Whole genome resequencing reveals natural target site preferences of transposable elements in Drosophila melanogaster. PLoS One 7: e30008. 10.1371/journal.pone.0030008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyttle TW. 1991. Segregation distorters. Annu Rev Genet 25: 511–581. 10.1146/annurev.ge.25.120191.002455 [DOI] [PubMed] [Google Scholar]
- Ma C-H, Kwiatek A, Bolusani S, Voziyanov Y, Jayaram M. 2007. Unveiling hidden catalytic contributions of the conserved His/Trp-III in tyrosine recombinases: assembly of a novel active site in Flp recombinase harboring alanine at this position. J Mol Biol 368: 183–196. 10.1016/j.jmb.2007.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais G, Kingsford C. 2011. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27: 764–770. 10.1093/bioinformatics/btr011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinossi-Allibert I, Veller C, Ament-Velásquez SL, Vogan AA, Rueffler C, Johannesson H. 2021. Invasion and maintenance of meiotic drivers in populations of ascomycete fungi. Evolution 10.1111/evo.14214 [DOI] [PubMed]
- McDonald MC, Taranto AP, Hill E, Schwessinger B, Liu Z, Simpfendorfer S, Milgate A, Solomon PS. 2019. Transposon-mediated horizontal transfer of the host-specific virulence protein ToxA between three fungal wheat pathogens. mBio 10: e01515-19. 10.1128/mBio.01515-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieczkowski PA, Lemoine FJ, Petes TD. 2006. Recombination between retrotransposons as a source of chromosome rearrangements in the yeast Saccharomyces cerevisiae. DNA Repair (Amst) 5: 1010–1020. 10.1016/j.dnarep.2006.05.027 [DOI] [PubMed] [Google Scholar]
- Möller M, Stukenbrock EH. 2017. Evolution and genome architecture in fungal plant pathogens. Nat Rev Microbiol 15: 756–771. 10.1038/nrmicro.2017.76 [DOI] [PubMed] [Google Scholar]
- Nauta MJ, Hoekstra RF. 1993. Evolutionary dynamics of spore killers. Genetics 135: 923–930. 10.1093/genetics/135.3.923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol 32: 268–274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle A, Taylor JW. 2002. The fitness of filamentous fungi. Trends Microbiol 10: 474–481. 10.1016/S0966-842X(02)02447-2 [DOI] [PubMed] [Google Scholar]
- Schaack S, Gilbert C, Feschotte C. 2010. Promiscuous DNA: horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol Evol 25: 537–546. 10.1016/j.tree.2010.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. 2009. The B73 maize genome: complexity, diversity, and dynamics. Science 326: 1112–1115. 10.1126/science.1178534 [DOI] [PubMed] [Google Scholar]
- Selker EU, Cambareri EB, Jensen BC, Haack KR. 1987. Rearrangement of duplicated DNA in specialized cells of Neurospora. Cell 51: 741–752. 10.1016/0092-8674(87)90097-3 [DOI] [PubMed] [Google Scholar]
- Silar P, Dauget J-M, Gautier V, Grognet P, Chablat M, Hermann-Le Denmat S, Couloux A, Wincker P, Debuchy R. 2019. A gene graveyard in the genome of the fungus Podospora comata. Mol Genet Genomics 294: 177–190. 10.1007/s00438-018-1497-3 [DOI] [PubMed] [Google Scholar]
- Talla V, Suh A, Kalsoom F, Dincă V, Vila R, Friberg M, Wiklund C, Backström N. 2017. Rapid increase in genome size as a consequence of transposable element hyperactivity in wood-white (Leptidea) butterflies. Genome Biol Evol 9: 2491–2505. 10.1093/gbe/evx163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomson BN, D'Amours D, Adamson BS, Aragon L, Amon A. 2006. Ribosomal DNA transcription-dependent processes interfere with chromosome segregation. Mol Cell Biol 26: 6239–6247. 10.1128/MCB.00693-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urquhart AS, Chong NF, Yang Y, Idnurm A. 2020. Eukaryotic transposable elements as “cargo carriers”: the forging of metal resistance in the fungus Paecilomyces variotii. bioRxiv 10.1101/2020.03.06.981548 [DOI] [PubMed]
- van der Gaag M, Debets AJ, Oosterhof J, Slakhorst M, Thijssen JA, Hoekstra RF. 2000. Spore-killing meiotic drive factors in a natural population of the fungus Podospora anserina. Genetics 156: 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaser R, Sović I, Nagarajan N, Šikić M. 2017. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27: 737–746. 10.1101/gr.214270.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogan AA, Ament-Velásquez SL, Granger-Farbos A, Svedberg J, Bastiaans E, Debets AJM, Coustou V, Yvanne H, Clavé C, Saupe SJ, et al. 2019. Combinations of Spok genes create multiple meiotic drivers in Podospora. eLife 8: e46454. 10.7554/elife.46454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9: e112963. 10.1371/journal.pone.0112963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, et al. 2007. A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8: 973–982. 10.1038/nrg2165 [DOI] [PubMed] [Google Scholar]
- Wisecaver JH, Rokas A. 2015. Fungal metabolic gene clusters—caravans traveling across genomes and environments. Front Microbiol 6: 161. 10.3389/fmicb.2015.00161 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.