

Abstract
Cancer is a complex disease characterized by loss of cellular homeostasis through genetic and epigenetic alterations. Emerging evidence highlights a role for histone variants and their dedicated chaperones in cancer initiation and progression. Histone variants are involved in processes as diverse as maintenance of genome integrity, nuclear architecture and cell identity. On a molecular level, histone variants add a layer of complexity to the dynamic regulation of transcription, DNA replication and repair, and mitotic chromosome segregation. Because these functions are critical to ensure normal proliferation and maintenance of cellular fate, cancer cells are defined by their capacity to subvert them. Hijacking histone variants and their chaperones is emerging as a common means to disrupt homeostasis across a wide range of cancers, particularly solid tumours. Here we discuss histone variants and histone chaperones as tumour-promoting or tumour-suppressive players in the pathogenesis of cancer.
Histone variants and their dedicated chaperones, which escort histones throughout the cell and deposit them into chromatin (FIG. 1), are altered in both paediatric and adult solid tumours (TABLE 1). The discovery of ‘oncohistones’, histone mutations found at high frequency in specific tumour types, has dramatically shifted the paradigm of histone variant function from cellular housekeeping and developmental processes to cancer initiation and progression. Ongoing efforts to understand the biological and clinical consequences of epigenome rewiring by abnormal histone incorporation may reveal novel tumour vulnerabilities that can be leveraged with anticancer therapeutics.
Fig. 1 |. Localization and deposition pathways of histone variants across the genome.
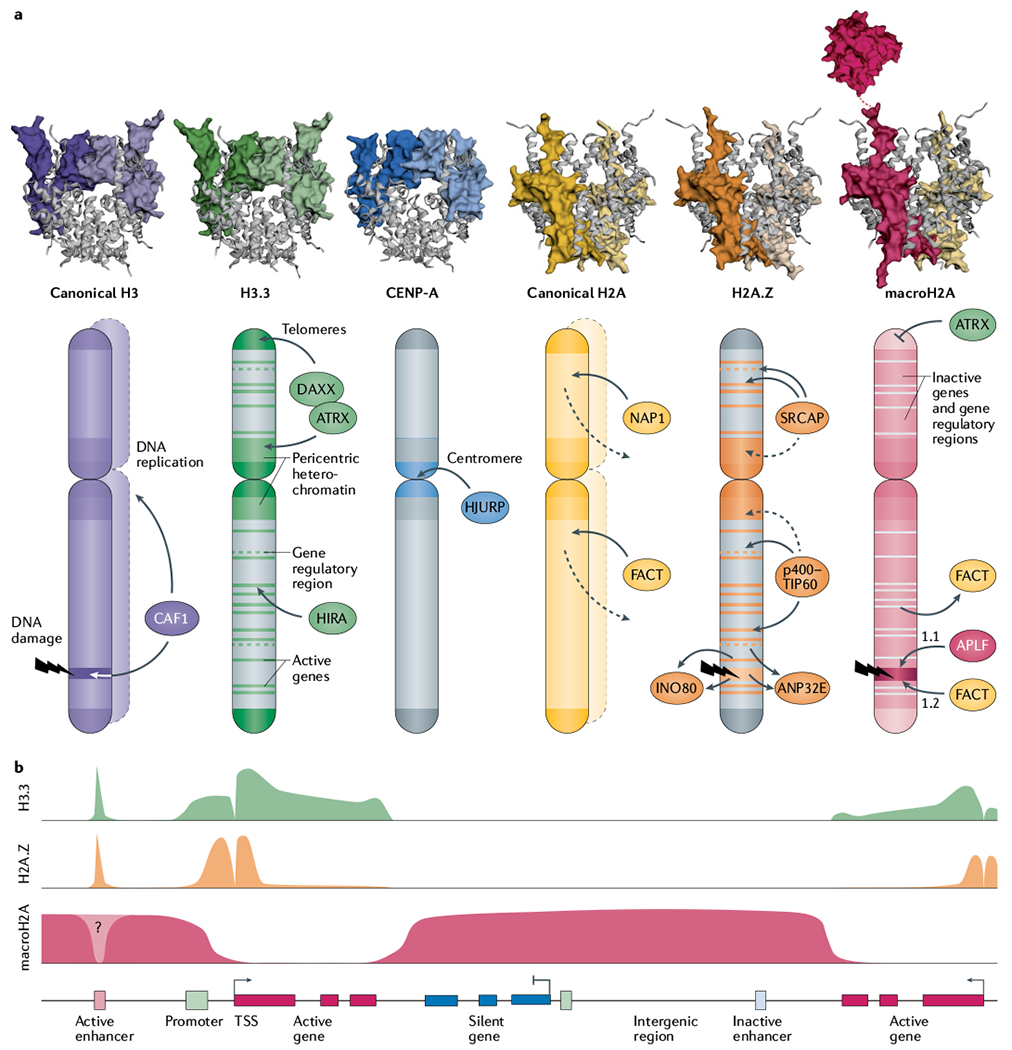
a | Histone variants are structurally similar to their canonical counterparts, with the exception of macroH2A’s non-histone macrodomain, which protrudes from the nucleosome particle via its linker region. Histone variants are differentially enriched across genomic landmarks as a function of histone chaperones. The CAF1 (chromatin assembly factor 1) complex deposits canonical H3 variants H3.1 and H3.2 during DNA replication or repair. H3.3 is deposited at active genes and at gene regulatory and nucleosome-depleted regions by the HIRA (histone cell cycle regulation-defective homologue A) complex, and at pericentric heterochromatin and subtelomeric regions by death domain-associated protein 6 (DAXX)–ATRX (α-thalassaemia/mental retardation syndrome X-linked). Centromeric protein A (CENP-A) is deposited at the active centromere by Holliday junction recognition protein (HJURP). NAP1 and the facilitates chromatin transcription (FACT) complex catalyse incorporation of canonical H2A during replication, and throughout the cell cycle, counterbalancing its continuous turnover (dashed lines). H2A.Z is deposited at active genes and regulatory regions by the SNF2-related CBP activator protein (SRCAP) and p400–TIP60 complexes, but is also enriched at pericentric heterochromatin through an unclear mechanism. The INO80 remodeling complex and ANP32E chaperone exchange H2A.Z for H2A at active genes, gene regulatory elements and DNA damage sites. APLF and FACT promote the accumulation of macroH2A1.1 and macroH2A1.2, respectively, at DNA damage sites. MacroH2A enrichment is negatively regulated by ATRX at telomeres and by FACT-mediated active eviction at transcribed genes. Crystal structures of canonical histones and H3.3 (REF.283), CENP-A284, H2A.Z12, macroH2A–H2A heterotypic nucleosome285 and macroH2A macrodomain76 were rendered with use of EzMol286. b | At gene-level resolution, H3.3 and H2A.Z are enriched at active regulatory elements (enhancers) and around the promoter of active genes, with the exception of the nucleosome-free region at the transcription start site (TSS). H3.3 is additionally present across transcribed gene bodies and transcription stop sites. MacroH2A forms large domains limited by actively transcribed regions.
Table 1 |.
Tumour-specific alterations in histone variants and chaperones and their mechanisms of action
| Histone variant or chaperone | Alteration in cancer | Clinical variables associated with alteration | Mechanism of action |
|---|---|---|---|
| Pan-tumour studies | |||
| MacroH2A1.1 (REF.88) | Downregulation | Increased Ki67 positivity, higher grade | MacroH2A1.1 loss promotes proliferation |
| H2A.Z258 | Upregulation | High-grade tumours | Not known |
| CENP-A and HJURP221,227 | Upregulation | Tumour vs normal cells, late vs early lesions, increased genomic instability, shorter patient survival | Not known |
| TP53 gene deficiency | p53 represses the CENPA and HJURP genes, which are required to sustain centromere propagation | ||
| Breast cancer | |||
| H2A.Z41,42,259,260 | Upregulation | High grade | Oestrogen induces H2A.Z expression synergistically with MYC; H2A.Z is deposited at ERα target genes and poised enhancers |
| MacroH2A1/macroH2A1.1 (REFS93,94) | Ubiquitylation/downregulation | Increased cancer cell proliferation, EMT | Degradation by SKP2 activates CDK8-dependent proliferation and migration pathways; reduced expression during EMT induction |
| MacroH2A1.2 (REF.91) | Upregulation | Increased cancer cell invasion and migration | ERα cofactors DDX17 and DDX5 favour splicing of macroH2A1.2, increasing breast cancer cell invasiveness |
| CENP-A224,261,262 | Upregulation | Higher grade, invasion, relapse, ERα-negative status | Not known |
| HJURP222,262 | Upregulation | Higher grade, proliferation, shorter patient survival, radiosensitivity, ERα-and PR-negative status, increased Ki67 positivity | Not known |
| Melanoma | |||
| H2A.Z.2 (REF.13) | Upregulation | Higher proliferative potential | Promotes expression of E2F target genes |
| MacroH2A97,263 | Downregulation | Increased proliferation and metastasis | Increased CDK8 Levels promote cell proliferation and metastatic burden |
| Hepatocellular carcinoma | |||
| H2A.Z36 | Upregulation | Increased cell proliferation | Promotes cell cycle and EMT gene expression |
| MacroH2A1 (REFS103,104) | Downregulation | Maintenance of stem cell features | Resistance to proliferation inhibition by altering lipid and glucose metabolism |
| Upregulation | Ageing, senescence-associated β-galactosidase marker | Escape from senescence through p38-IL-8 pathway | |
| CENP-A223 | Upregulation | Higher histological grade, Ki67 status, p53 positive | Necessary for cancer cell proliferation and survival |
| HJURP264 | Upregulation | Shorter patient survival, less differentiated, increased size and stage | Promotes cancer cell proliferation |
| Prostate cancer | |||
| H2A.Z46,49,56 | Acetylation/upregulation | Increased proliferation | Present at enhancers of genes associated with tumorigenesis, promotes AR-dependent transcription |
| SRCAP40 | Upregulation | Increased proliferation | Associated with AR to regulate PSA promoter |
| MacroH2A1.1 (REF.90) | Downregulation | Higher tumour Gleason score | Not known |
| Lung cancer (NSCLC unless otherwise specified) | |||
| GAS41 (SRCAP or p400 complexes)53 | Amplification and upregulation | Level elevated in NSCLC vs normal tissue | Promotes histone H2A.Z deposition to allow expression of cell cycle genes |
| MacroH2A1.1/macroH2A2 (REF.87) | Downregulation | Higher tumour proliferation rate | MacroH2A leveis correlate with senescence |
| CENP-A265 | Upregulation | Higher grade, proliferation, invasion of lung adenocarcinoma samples | Not known |
| HJURP266 | Upregulation | Shorter patient survival | Not known |
| Colorectal cancer | |||
| TIP60-p400 (REFS54,55) | Decreased TIP60/p400 ratio | Decreased apoptosis, high proliferation | TIP60 prevents β-catenin signalling, while p400 promotes expression of WNT target genes |
| MacroH2A1 (REF.267) | Downregulation | Increased proliferation | Increased expression of ZEB1 and DKK1, escape senescence |
| CENP-A268 | Upregulation | Higher expression in tumour vs. normal tissue | Not known |
| Glioma | |||
| MacroH2A269 | Ubiquitylation/degradation | Increased Ki67 levels, presence of CD44 (cancer stem cell marker) | MacroH2A1 ubiquitylation by TRIM59 enhances STAT3 signalling activation and tumorigenesis |
| H3.3 (REFS127–131,133,135,136,139,142–145,161–163) | Missense mutation | K27M: prevalent in paediatric midline gliomas; fatal prognosis, less differentiated, more invasive tumours | Inhibition of PRC2 and loss or redistribution of H3K27me3, altering expression of neurogenesis, differentiation or cell proliferation genes |
| G34R/V: biased towards occurrence in the cerebral hemispheres in teenagers and young adults | Inhibition of SETD2 methyltransferase and decrease of H3K36me3 | ||
| CENP-A270 | Upregulation | Shorter patient survival in the mesenchymal subtype | Not known |
| ATRX127,131 | Mutations | High-grade tumours in adolescent patients, associated with TP53 and IDH1 mutations, positive correlation with ALT | Not known |
| HJURP271,272 | Upregulation | Grade, shorter patient survival | Necessary for cancer cell proliferation |
| Pancreatic neuroendocrine tumours | |||
| DAXX-ATRX196,198,199 | Mutation | High-grade tumours, poor patient prognosis | ATRX and DAXX mutations associated with ALT |
| Neuroblastoma | |||
| ATRX182,200,201 | Mutation/in-frame fusions | Indolent disease, shorter patient survival | Suppression of neuronal gene expression programmes, sensitivity to EZH2 inhibitors |
| Ovarian cancer | |||
| CENP-A and HJURP273,274 | Upregulation | Higher tumour grade, higher tumour stage, shorter patient survival, metastasis | Not known |
| Endometrial cancer | |||
| HJURP275 | Upregulation | Shorter patient survival | Not known |
| Sarcomas | |||
| CENP-A225 | Osteosarcoma: upregulation | Increased tumour size and proliferation, chemoresistance, metastasis, shorter patient survival | Not known |
| ATRX276–282 | Osteosarcoma: point mutations, deletions | ALT positivity, co-occurs with TP53 mutations | DAXX-KIFC3 gene fusion impairs DAXX function, associated with ALT |
| Leiomyosarcoma: nonsense and frameshift mutations | ALT positivity, poor differentiation, shorter patient survival | Not known | |
| Pleomorphic and myxofibrosarcomas: loss of expression | Highly correlated with ALT | Not known | |
| Giant cell-rich skeletal tumours | |||
| H3.3 (REFS143,155,157,159,169) | Missense mutation | K36M: chondroblastoma | Inhibition of K36 dimethylation and trimethylation by NSD2 and SETD2, redistribution of K27me3, impaired gene expression and DNA repair |
| G34W/L: GCTB, increased proliferation and invasive capacity | Inhibition of K36 methylation by SETD2, impaired K27 methylation and HIRA binding | ||
ALT, alternative lengthening of telomeres; AR, androgen receptor; ATRX, α-thalassaemia/mental retardation syndrome X-linked; CDK, cyclin-dependent kinase; CENP-A, centromeric protein A; EMT, epithelial–mesenchymal transition; ERα, oestrogen receptor-α; GCTB, giant cell tumour of the bone; HJURP, Holliday junction recognition protein; NSCLC, non-small-cell lung cancer; PR, progesterone receptor; PRC2, Polycomb repressive complex 2; SRCAP, SNF2-related CBP activator protein.
Histones play a fundamental role in the process of compacting DNA into the nucleosomal fibre, and actively proliferating cells ensure this function by loading large amounts of canonical histones onto DNA during replication (BOX 1). Histone variants, which share protein sequence homology with canonical histones, are encoded by single genes dispersed throughout the genome, synthesized and incorporated independently of DNA replication1. Canonical and variant histones can harbour a similar repertoire of post-translational modifications (PTMs) at key residues, which engage chromatin ‘readers’ to regulate chromatin accessibility, gene expression or other processes. What sets histone variants apart from their canonical counterparts is their unique biophysical properties, genomic localization patterns (FIG. 1) and binding partners, conferred by differences in primary amino acid sequence and structure2–4. Histone variants can replace existing canonical histones and their associated PTMs, thus remodelling the epigenome throughout the cell cycle and during development5.
Box 1 |. Replicative histones and their deposition pathway.
Histones are distinguished by their expression pattern throughout the cell cycle and by a dedicated network of histone chaperones ensuring their transit throughout the cell and assembly into chromatin. Canonical histones H2A, H2B, H3 and H4, also known as replicative histones, form the nucleosomal core287. They peak in expression during S phase and are incorporated into chromatin in a DNA synthesis-coupled manner by specific chaperones. To meet the requirement for extensive incorporation throughout the genome during S phase2,4, replicative histones have unique genomic organization and transcriptional control. They are present in multiple copies organized in clusters; they lack intronic regions and are not polyadenylated, but instead have a conserved RNA processing mechanism involving the stem-loop structure and purine-rich sequence at their 3’ end, providing a unique means to control their expression1. Chromatin assembly factor 1 (CAF1) is an H3–H4 chaperone complex that associates specifically with H3.1 and H3.2 replicative histones288,289 and incorporates them into chromatin in tandem with replication fork passage, by directly associating with the DNA polymerase processivity factor known as proliferating cell nuclear antigen (PCNA)290,291. Equally important is ASF1, an H3-H4 chaperone capable of binding heterodimers containing all H3 variants, functioning as an intermediate to hand them over to CAF1 or HIRA (histone cell cycle regulation-defective homologue A) complexes292–294. By directly interacting with the MCM helicase, ASF1 regulates the reincorporation of pre-existing histones displaced by the replication fork and the integration of newly synthesized histones294,295.
In recent years, a growing number of studies have connected histone variant mutation, deregulation or misincorporation to various aspects of tumour biology. Here we review the normal and cancer-associated functions of histone variants and their dedicated chaperones, delineate outstanding questions in the field and discuss potential therapeutic avenues. We focus our discussion on members of the diverse H2A and H3 histone variant families, including H2A.Z, macroH2A, H3.3 and centromeric protein A (CENP-A), where a strong body of evidence for their involvement in solid tumours is available. Other aspects of histone biology in cancer are reviewed in detail elsewhere6–8.
The H2A histone family
H2A.Z
Functions and partners.
H2A.Z is a highly conserved histone variant with 60% similarity to H2 A. In mammals, two paralogues of H2A.Z exist: H2A.Z.1 and H2A.Z.2, encoded by the genes H2AFZ (also known as H2AZ1) and H2AFV (also known as H2AZ2), respectively9,10. These are expressed in most tissues, with H2A.Z.1 being predominant. While H2A.Z isoforms differ by only three amino acids distributed throughout the protein, they have different effects on chromatin properties and display tissue-specific functions11–16. Overall, H2A.Z has been associated with transcriptional activation (FIG. 1), possibly due to the presence of an extended acidic patch that decreases the electrostatic interaction between DNA and histone17. Additionally, H2A.Z can co-occur with H3.3 in the same nucleosome, generating a highly salt-labile particle enriched at regions of high histone turnover, which likely facilitates the access of transcription factors to the underlying DNA18,19. Similarly to other histones, H2A.Z is subjected to PTMs such as acetylation, methylation and ubiquitylation that differentially modulate gene expression; for example, hyperacetylated H2A.Z is found near transcription start sites (TSSs) and at enhancers of active genes, correlating with transcriptional activation20,21.
Deposition of H2A.Z into specific genomic regions is achieved by either of the ATP-dependent chaperone complexes SNF2-related CBP activator protein (SRCAP) or p400–TIP60 (FIG. 1a). Both complexes contain several subunits essential for proper H2A.Z deposition and distribution throughout the genome, many of which are shared between the two complexes (reviewed elsewhere22). Besides histone deposition, these complexes can also modify histones. For example, TIP60 is a lysine acetyltransferase that is required for H2A.Z acetylation in mammals23,24, as well as for canonical H2A acetylation as a prerequisite for H2A.Z loading25. The p400–TIP60 and SRCAP complexes also contain ‘reader’ activities that bind to specific histone PTMs. For instance, GAS41, common to both complexes, contains a YEATS domain that binds diacetylated H3 residues directing H2A.Z deposition26,27. On the other hand, eviction of H2A.Z is orchestrated by the INO80 and ANP32E chaperone complexes (FiG. 1a). ANP32E has been shown to negatively regulate H2A.Z enrichment around the TSS, at enhancers and at insulators28, while both the ANP32E and INO80 complexes remove H2A.Z from sites of DNA damage to allow efficient homologous recombination29,30.
H2A.Z has been studied mostly in the context of transcriptional regulation; however it is also implicated in chromosome segregation, pericentric heterochromatin formation during the cell cycle, and cellular fate26,31–33. Regarding the last of these, H2A.Z is incorporated in bivalent chromatin domains in embryonic stem cells (ES cells) via GAS41, and allows the recruitment of Polycomb repressive complex 2 (PRC2) to lineage-specific genes26,34,35. Taken together, these data show that H2A.Z is an important player in arbitrating cell fate transitions and lineage commitment, which are directly relevant to cancer.
H2A.Z: transcriptional regulator of cancer-associated genes.
While H2A.Z.1 and H2AZ.2 are not frequently mutated, their expression is commonly upregulated in several tumour types (FiG. 2; TABLE 1). Our group demonstrated a specific role for H2A.Z.2 in sustaining the proliferation of melanoma cells in vitro, whereby the transcription factor E2F and the bromodomain and extra-terminal (BET) protein BRD2 are recruited to cell cycle genes in an H2A.Z.2-dependent fashion to promote their expression13. Similar effects on cell proliferation were observed in hepatocellular carcinoma (HCC) cell lines. Here, H2A.Z.1 selectively modulates cell division through suppression of cell cycle inhibitors, while also being required for expression of cyclin-dependent kinases (CDKs)36. In colon cancer cell lines, H2A.Z.1 expression is positively regulated by the WNT–β-catenin pathway, and its depletion impairs proliferation and induces expression of enterocyte differentiation markers37.
Fig. 2 |. Histone variant mutational spectrum and altered expression across solid tumours.
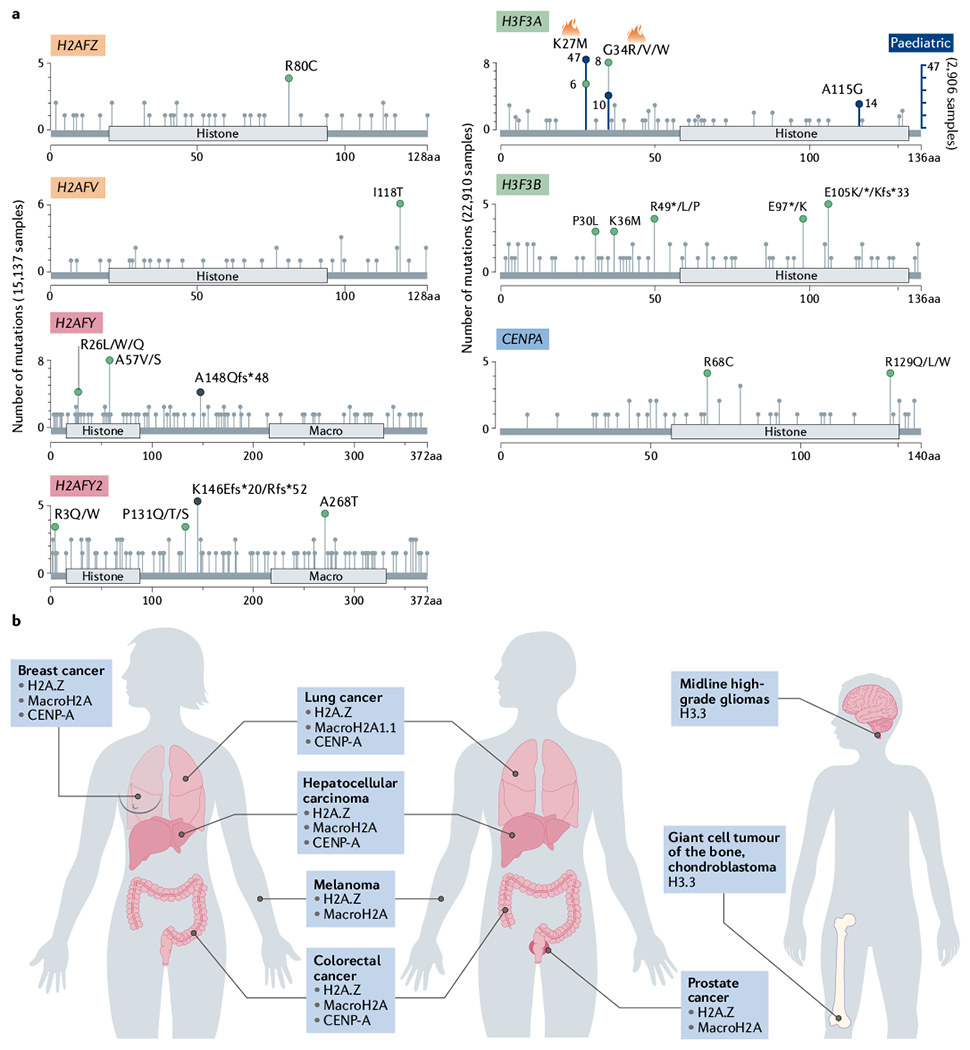
a | From pan-cancer studies (The Cancer Genome Atlas cohorts used are listed in Supplementary Box 1), the mutational spectra of histone variant genes H2AFZ (encoding H2A.Z.1), H2AFV (encoding H2A.Z.2), H2AFY (encoding macroH2A1; both isoforms, macroH2A1.1 and macroH2A1.2, are included), H2AFY2 (encoding macroH2A2), H3F3A (encoding H3.3), H3F3B (encoding H3.3) and CENPA are shown. Most histone variants show a low frequency of mutation. H3F3A has hotspot mutations (denoted by flame symbol) that represent K27M/R and G34R/V mutations. While in adult cancers these mutations are diluted, cohorts of paediatric tumours (blue axis; right) have higher mutation frequency in these hotspots. Histones are not drawn to scale. Green dots represent missense somatic mutations, while black dots indicate truncations. b | Histone variant alterations across the human body by location; H2A.Z, centromeric protein A (CENP-A) and macroH2A display altered regulation in diverse adult cancers, while H3.3 alterations are restricted to paediatric tumours. aa, amino acids.
A large body of evidence highlights the importance of H2A.Z.1 in hormone-regulated cancers, such as breast and prostate cancer (FIG. 2b; TABLE 1). Here, H2A.Z.1 incorporation can be guided to specific genomic regions by a variety of transcription factors, such as MYC, oestrogen receptor-α (ERα) or androgen receptor (AR), allowing specific and context-dependent gene expression regulation by H2A.Z.1 (REFS38–40). In the context of ERα+ breast cancer, modelled in MCF-7 cells, the presence of canonical oestrogen response elements in the H2AFZ promoter enhances expression synergistically with MYC binding41,42. This signalling is further amplified by p400–TIP60-mediated H2A.Z.1 incorporation at enhancers of ERα-regulated genes and recruitment of the transcription factor FOXA1, promoting increased expression of these genes in a positive-feedback loop43. Also in ERα+ breast cancer, the lysine methyltransferase SMYD3 methylates H2A.Z at lysine 101, stabilizing the variant at the TSS of the gene encoding cyclin A1 (CCNA1) driving cell cycle progression44. Due to its direct regulation by ERα, it is assumed that H2A.Z.1 plays a recurrent role in hormone-positive breast cancers versus other breast cancer subtypes, while the role of H2A.Z.2 remains unknown.
A similar mechanism of action occurs in prostate cancer. Although the H2AFZ gene itself is not controlled by androgen response elements (AREs), H2A.Z.1 is present at several AREs driving cancer progression45,46. Androgen signalling increases MYC binding at the H2AFZ promoter, leading to H2A.Z.1 overexpression in a positive-feedback loop47. H2A.Z.1 accumulates at androgen-activated enhancers and promotes gene expression in prostate cancer cells, contributing to the stabilization of a poised enhancer state that persists after withdrawal of androgen stimulation46. Acetylated H2A.Z can drive abnormal gene expression, and TIP60 has been implicated in acetylating H2A.Z in ARE regions near enhancers48. In addition, H2A.Z acetylation is a key modification present at the TSS of active genes in prostate cancer cell lines, while unacetylated H2A.Z in promoters of tumour suppressor genes negatively regulates their expression49.
Besides acetylation, H2A.Z can also undergo monoubiquitylation by the PRC1 ubiquitin ligase RING1B, an activity associated with gene repression50. Deubiquitylation by USP10 promotes the transition of the gene encoding prostate-specific antigen (PSA, also known as KLK3) and other genes from a poised state to being actively transcribed driving tumorigenesis51. Thus, in both breast cancer and prostate cancer, H2A.Z.1 drives transcription of key oncogenic factors and its function is modulated by its PTM status.
H2A.Z chaperones in cancer.
Deregulation of histone chaperone complexes can contribute to tumour development, and in the case of H2A.Z, both its chaperone complexes, SRCAP and p400–TIP60, are affected. Components of the SRCAP complex, including the SRCAP helicase, YL1, GAS41, RUVBL1 and RUVBL2, are upregulated in several tumours, such as ovarian tumours, breast tumours, thyroid tumours, prostate tumours, melanoma, bladder tumours and glioma (cBioPortal for Cancer Genomics52). Although pan-cancer studies report mutations in these components (cBioPortal for Cancer Genomics52), functional effects of the mutations have not been described to date. In contrast, downstream consequences of altered expression of SRCAP complex components have been described. GAS41, for example, is essential to modulate deposition of H2A.Z at cell cycle and DNA replication genes in non-small-cell lung cancer cells, where it promotes expression of these genes, and in turn, proliferation53.
The level of the TIP60 subunit of the p400–TIP60 complex is also altered in cancer. In contrast with subunits of the SRCAP complex however, TIP60 exhibits a tumour-suppressive role. In human colorectal cancer cell lines, downregulation of TIP60 brings about a low ratio of TIP60 to p400, which reduces the DNA damage response, resulting in decreased apoptosis and increased proliferation54. In a mouse model of colon cancer, TIP60 loss promoted β-catenin activity, whereas concomitant p400 loss rescued this phenotype, suggesting opposing roles for these two complex subunits and highlighting the importance of the TIP60/p400 ratio for tumour outcome55.
While there are many reports of increased levels of H2A.Z in tumour cells, it remains to be established whether deregulation of the deposition machinery, including chaperones, is responsible for the gain or redistribution of H2A.Z isoforms across the genome. The interplay between H2A.Z, its PTMs and its chaperones will need to be further unravelled if we are to harness these therapeutically.
Therapeutic opportunities for H2A.Z-driven tumours.
Direct therapeutic targeting of H2A.Z and its chaperones remains elusive; however, strategies to counteract H2A.Z function are emerging. Since H2A.Z acetylation may promote tumorigenesis and is found in hyperacetylated nucleosomes13,56, BET inhibitors (BETi) are a potential therapeutic strategy to counter this effect. BETi block the ability of BET family proteins (BRD2, BRD3, BRD4 and BRDT) to bind acetylated H4, among other acetylated histone residues, through their bromodomains57. Our laboratory showed that H2A.Z.2 depletion from cell cycle genes in combination with JQ1 (a BETi) selectively drove melanoma cell apoptosis due to the inability of BRD2 to bind acetylated histones at promoters of these genes, hindering expression and thus reducing proliferation13. In intrahepatic cholangiocarcinoma cell lines, knockdown of H2A.Z.1 promoted tumour cell sensitivity to cisplatin by amplifying drug-induced cell cycle arrest and apoptosis58, possibly by means of H2A.Z’s recently discovered role in regulating DNA replication origin firing59. Taken together, these data show that H2A.Z loss sensitizes tumour cells to both conventional and epigenetic therapies, suggesting that H2A.Z levels might be useful to stratify patients for therapy. As we continue to learn more about the structure and function of H2A.Z chaperones, therapeutic possibilities emerge, such as the development of small molecules to inhibit epigenetic readers and writers of chaperone complexes, or inhibitors of protein-protein interactions. To this end, the crystal structure of the H2A.Z/H2B dimer and YL1 (H2A.Z/H2B-YL1) was recently solved60,61; as YL1 is a subunit of both the SRCAP complex and the p400–TIP60 complexes, targeting its interaction with H2A.Z isoforms might be a potential therapeutic opportunity for H2A.Z-driven cancers.
MacroH2A
Structure and function.
MacroH2A is a divergent member of the H2A family characterized by a tripartite structure, containing a H2A-like domain, a lysine-rich linker region and a distinct macrodomain that protrudes from the nucleosome62,63 (FIG. 1). MacroH2A1.1 and macroH2A1.2 are isoforms resulting from alternative splicing of the H2AFY gene, and macroH2A2 is a third isoform, encoded by H2AFY2 (the genes were recently renamed MACROH2A1 and MACROH2A2, respectively)64–66. MacroH2A variants are enriched on the inactive X chromosome, in senescence-associated heterochromatin foci, on repetitive DNA sequences and at inactive genes, and often colocalize with H3K27me3 or H3K9me3 genomic occupancy, collectively suggesting a role in transcriptional repression and heterochromatin structure65,67–74 (FIG. 1; BOX 2).
Box 2 |. Histone variants in senescence.
Senescence is a potent tumour-suppressive mechanism limiting proliferation of aged or damaged cells, elicited by a variety of cellular stressors, including oncogenic stimuli. It involves an irreversible exit from the cell cycle as well as metabolic and microenvironment reprogramming, including a senescence-associated secretory phenotype (SASP). SASP reinforces cell cycle arrest and contributes to senescent cell clearance by the immune system, but it can also support protumorigenic inflammation296. In addition, senescent cells undergo dramatic chromatin reorganization through formation of senescence-associated heterochromatin foci (SAHF), enriched in repressive histone methylation (H3K9me3 and H3K27me3), heterochromatin binding protein 1 (HP1), and macroH2A67,297. While macroH2A is not critical for initiation of SAHF, it plays a role in their maintenance67, and macroH2A1.1 specifically contributes to senescence by activating a subset of SASP genes257. Furthermore, H2A.Z regulates senescence induction, as its eviction from the CDKN1A promoter allows expression of its product p21, leading to growth arrest43,298.
Our group demonstrated that the H3.3 amino-terminal tail undergoes proteolytic clipping by the protease cathepsin L1 (REF.299), thereby inducing an irreversible loss of post-translational modifications involved in transcriptional activation, including K4me3. Concurrent with loss of H3K4me3 by JARID1A- or JARID1B-mediated demethylation300, this leads to downregulation of cell cycle genes and exit from the cell cycle299. H3 chaperones have a twofold impact on senescence. On the one hand, HIRA (histone cell cycle regulation-defective homologue A) and ASF1 contribute to recruitment of heterochromatin factors such as HP1 to SAHF67. However, it is unlikely they actively deposit H3.3 at these domains, as the histone variant is excluded from SAHF299 and new H3.3 appears to be incorporated into promyelocytic leukaemia bodies mainly by death domain-associated protein 6 (DAXX) in oncogene-induced senescent cells301. On the other hand, HIRA is required for H3.3 deposition at active genes in senescent cells, which in turn prevents loss of H4 K16 acetylation, a mark required to inhibit oncogene-induced transformation302.
Of the three isoforms, only the macroH2A1.1 macrodomain is capable of binding ADP-ribose metabolites and ADP-ribosylated proteins, including poly(ADP-ribose) polymerase 1 (PARP1)75–77. When bound to macroH2A1.1 in its ADP-ribosylated form, PARP1’s enzymatic activity is inhibited, affecting its downstream processes such as transcriptional repression, DNA damage repair and stress response78–80. MacroH2A1.1 has also been shown to limit nuclear NAD+ consumption by inhibiting PARP1, which in turn hinders mitochondrial function81. In addition, macroH2A1.1 interacts with O-acetyl-ADP-ribose, a by-product of the NAD+-dependent deacetylases (sirtuins)82, as well as autoribosylated sirtuins77. MacroH2A1.2 and macroH2A2 also inhibit PARP1 activity in vitro83, but it remains to be determined whether these can also bind metabolic cofactors, or what the consequences of this binding may be in vivo and in the context of cancer.
In the developing mouse embryo, macroH2A is highly enriched in the trophectoderm and in differentiated cell types84. MacroH2A prevents the expression of key pluripotency genes in somatic cells when reprogrammed towards pluripotency, and accordingly, depletion of macroH2A isoforms enhances reprogramming efficiency72,85,86. The finding that macroH2A ‘locks’ the cell in a differentiated state suggested that it might prevent cellular plasticity, thus acting as a tumour suppressor. While this holds true for many solid tumours, in specific contexts, contradictory findings have been reported, as discussed next.
MacroH2A: a putative tumour suppressor.
Like H2A.Z, macroH2A expression is altered in cancer, but the genes encoding macroH2A variants are not frequently mutated (FIG. 2; TABLE 1). MacroH2A levels are decreased in tumours relative to normal tissues across a spectrum of cancers87,88; however, whether the levels of the macroH2A isoforms impact tumorigenesis differentially is unresolved. In general, macroH2A1.1 is considered tumour suppressive, as downregulation of this variant is observed in several tumour types (TABLE 1). Splicing factors control the switch between macroH2A1 splice variants in tumour cells. QKI, for example, regulates splicing of H2AFY in favor of the macroH2A1.1 isoform over macroH2A1.2, and is lost in cancer, leading to decreased levels of macroH2A1.1 (REFS88–90). The RNA helicases DDX17 and DDX5 are transcriptional co-regulators of ERα that splice H2AFY producing the macroH2A1.2 variant (thus decreasing relative macroH2A1.1 levels), and promote breast cancer cell invasiveness in vitro91. In splicing factor-mutant myelodysplastic syndromes, macroH2A1 alternative splicing leads to a reduction in macroH2A1.1 levels, impacting differentiation along the haematopoietic lineage92. Beyond splicing regulation, macroH2A1.1 is ubiquitylated by the E3 ligase SKP2 in breast cancer cells, leading to its degradation and consequent tumour progression by increased CDK8 expression93. Additional support for its tumour-suppressive function comes from evidence that ectopic expression of macroH2A1.1 suppresses epithelial-mesenchymal transition in human mammary epithelial cells94.
Although there is general consensus on macroH2A1.1, the role of macroH2A1.2 in cancer is largely context dependent. While alternative splicing in several tumour types revealed macroH2A1.2 to have an oncogenic role, suppression of metastasis by macroH2A1.2 has also been described. Both in breast cancer cells and in prostate cancer cells, it inhibits the expression of osteoclastogenic soluble factors which are required for remodelling of the metastatic niche95,96. In melanoma, ectopic expression of macroH2A1.2 in the highly metastatic B16 cell line dramatically reduced the lung metastatic burden in mice97. Hence, more studies are required to delineate the mechanism of action of each macroH2A1 splice variant, which may have cell type-specific or even cancer subtype-specific consequences.
MacroH2A1 has also been studied extensively in liver cancer and precursor lesions such as hepatic steatosis, with somewhat conflicting findings. MacroH2A1-deficient mice present a non-penetrant phenotype in which a subset of mice develop steatosis98; however, this observation was not reported in two other liver-focused studies99,100, raising the possibility of mouse strain-specific effects. Furthermore, while macroH2A1 and macroH2A2 double knockout mice have altered expression of hepatic genes related to lipid metabolism101, any overt lipid-related pathology which could explain the steatosis phenotype was lacking. In contrast, macroH2A levels were increased in steatosis/HCC models. MacroH2A1.1 and macroH2A1.2 were upregulated in mice fed a high-fat diet with PTEN-driven HCC compared with control mice, and in human steatotic liver, cirrhotic liver and HCC samples102,103. In line with this, macroH2A1 was shown to prevent senescence induced by a DNA-demethylating agent in HCC cell lines103 (BOX 2). However, the same group found macroH2A1 knockdown in HCC cell lines promotes insensitivity to confluency-dependent and serum starvation-dependent inhibition of proliferation104, a common feature of cancer stem cells, raising questions as to how these conflicting observations hold true in the same tumour type.
Regarding a role for macroH2A2 in cancer, our team demonstrated that this variant is downregulated in metastatic melanoma, and plays an active role in suppressing proliferation and metastasis97. While less well studied than macroH2A1 variants, low levels of macroH2A2 are also associated with lung cancer recurrence, breast cancer proliferation and anal neoplasm progression in patient samples87,105.
No dedicated histone chaperones have been identified for macroH2A variants, and current evidence largely suggests that the genome-wide distribution pattern of macroH2A is a consequence of its active exclusion74 (FIG. 1). Co-transcriptional exclusion of macroH2A is enabled by the facilitates chromatin transcription (FACT) complex74, a general H2A chaperone. Increased expression of FACT is associated with poorly differentiated and more aggressive tumours, such as triple-negative breast cancer and HER2+ breast cancer106, yet it remains to be determined whether FACT upregulation is linked to macroH2A exclusion in this context.
Most studies on the tumour-suppressive function of macroH2A were performed in cell lines, patient samples or xenograft models. However, unlike the other histone variants, simultaneous ablation of all macroH2A isoforms in mice is not lethal and mice do not develop spontaneous tumours101, suggesting macroH2A loss on its own does not initiate tumorigenesis. In a constitutive knockout model, loss of all macroH2A variants led to increased proliferative and clonogenic potential of intestinal organoids. Although their repopulating capacity after experimental injury was reduced, when crossed to an Apc-mutant intestinal adenoma mouse model, the increased number of reserve stem cells did not translate into increased adenoma initiation107. Nevertheless, since macroH2A deregulation occurs in other tumours such as breast cancer, HCC and melanoma, in vivo studies modelling these tumour types are essential to unravel the contribution of this histone variant to tumour initiation and progression.
Therapeutic targeting of macroH2A.
Chromatin incorporation and removal of macroH2A involves essential multitasking protein complexes such as FACT and ATRX, which are not specific for macroH2A71,74; therefore, targeting these factors can be challenging. However, macroH2A1.1 and macroH2A2 levels can be used as valuable biomarkers of metastatic or undifferentiated cancers that could guide the use of targeted therapies87,88,97. For example, FDA-approved PARP inhibitors are currently being used against cancers with deficient homologous recombination repair108, and might be a targeted therapeutic strategy against macroH2A1.1-deficient tumours to restore PARP inhibition, and in turn reverse the consequences of macroH2A loss for DNA repair and metabolism.
The H3 histone family
H3.3
Functions and partners.
Encoded by two paralogues, H3F3A and H3F3B (recently renamed H3-3A and H3-3B, respectively), H3.3 differs from canonical H3.1 and H3.2 by only four and five amino acids, respectively. H3.3 is expressed throughout the cell cycle and across all cell types, and forms most of the H3 pool in post-mitotic cells109–111. H3.3 has two distinct chaperone systems that place it into euchromatin and heterochromatin, respectively (FIG. 1 a). The HIRA (histone cell cycle regulation-defective homologue A) complex deposits H3.3 in regions of high nucleosome turnover, such as active genes and gene regulatory elements, through its ability to bind nucleosome-depleted DNA, RNA polymerase II or single-stranded DNA-binding proteins112–114. HIRA-mediated H3.3 deposition plays both structural and gene regulatory roles, preventing persistence of nucleosome gaps in chromatin113, promoting transcriptional recovery after DNA damage115 and maintaining a bivalent state at developmental gene promoters in ES cells116. Numerous gene expression programmes induced during development involve H3.3 deposition at promoters and enhancers (reviewed elsewhere117), which may causally contribute to transcriptional activation. Together with H2A.Z, H3.3 decreases nucleosomal stability and primes chromatin decompaction19,118, while phosphorylation of S31 (an H3.3-specific residue) at stimulation-induced genes recruits chromatin remodelling activities to enhance accessibility of the transcription machinery119.
Death domain-associated protein 6 (DAXX) in complex with ATRX (α-thalassaemia/mental retardation syndrome X-linked) deposits H3.3 at heterochromatin regions, including pericentric and telomeric repeats, endogenous retroviruses and imprinted genes112,120–122. In contrast to HIRA, H3.3 deposited by DAXX–ATRX at repetitive elements forms a substrate for K9 trimethylation to ensure silencing of telomeric repeat RNA and retrotransposition112,121–124. The HIRA and DAXX–ATRX chaperone complexes as well as H3.3 itself are involved in cellular senescence (BOX 2).
Within the last decade, next-generation sequencing studies of tumours have identified multiple mutations within the histone genes (FIG. 2a). H3.3 stands out due to its remarkably high frequency of recurrent point mutations in a set of rare cancers in children and adolescents. These somatic heterozygous but clonally homogenous mutations disrupt PTMs of key histone tail residues, with pervasive effects on the chromatin landscape and gene expression (TABLE 1). Such characteristics demonstrated for the first time a ‘driver’ role for histone variants in tumorigenesis, and these mutated variants have been coined ‘oncohistones’125. This includes H3.3 K27M, K36M and G34R/W substitutions in paediatric gliomas and skeletal tumours, which appear to be the most frequent and most well studied, and constitute our primary focus in the following sections.
These oncohistone mutations function in large part by blocking histone modifier enzymatic activity (see later); however, such enzymes do not necessarily display preference for H3 family members. Therefore, whether mutation in one of the H3.3 genes impacts the oncohistone pathology differently than mutation in a canonical H3 gene remains unresolved. Besides mutation, H3.3 may also contribute to tumour progression through changes in its levels. For example, the balance between H3.3 and replicative H3 deposition could modulate metastatic behaviour through transcriptional reprogramming126. Identifying the defining features of H3.3 that contribute to tumour progression, including genomic distribution by virtue of its specific chaperones as well as its relationship to the developmental time window and proliferative state of the cell, remains of great interest to the field.
H3K27M in paediatric high-grade gliomas.
The H3.3 K27M mutation was first identified in diffuse intrinsic pontine glioma (DIPG), glioblastoma, thalamic high-grade glioma (HGG) and spinal HGG in children and adolescent patients127–136 (FIG. 2). These disorders are molecularly similar and share a devastating prognosis due to their undifferentiated character and diffuse spread, leading the WHO to classify them under a single disease, ‘diffuse midline glioma K27M-mutant’137. K27M mutations are infrequent in paediatric HGGs affecting the cerebral hemispheres and in adults127,128,130,131,135,138.
The K27M mutation mainly affects H3F3A127,128,130,131,135 (FIG. 2a), with less frequent mutations in H3.1 paralogues HIST1H3B129 and HIST1H3C131,133 or the H3.2 gene HIST2H3C139. The mutation occurs as an AAG>ATG transition in H3F3A and HIST1H3B (cBioPortal for Cancer Genomics52), and would require a much less likely dinucleotide substitution to generate the same outcome in H3F3B-encoded K27, which is is encoded by an AAA codon. The organization of canonical histone genes in multiple, highly similar copies complicates efforts to map mutations, which may contribute to an underestimation of their prevalence in cancer140.
EZH2, the catalytic subunit of PRC2, trimethylates H3K27 to maintain gene silencing and cellular identity141. Despite representing a small fraction of the total H3 pool, K27M decreases global levels of H3K27me3, suggestive of a gain-of-function EZH2 inhibitor142–145. Exactly how this occurs remains a matter of debate. Together with increased interaction of PRC2 with the mutant due to stronger binding of the K27M peptide in the active site, the hypothesis that K27M sequesters PRC2 and prevents K27 methylation of non-mutant H3 in trans was proposed142,146. Genome-wide profiling of H3K27me3 in K27M-mutant tumours and neural progenitor cells also showed H3K27me3 gains at numerous loci142,144,147, inconsistent with complete PRC2 inhibition. Indeed, sharp peaks of H3K27me3 and the PRC2 subunit SUZ12 are present in K27M-mutant gliomas, notably at unmethylated CpG islands, which are high-affinity recruitment sites for PRC2 (REF.148). Such retained H3K27me3 peaks also display lower levels of H3.3K27M-mutant incorporation, potentially explaining this localized residual PRC2 activity149. However, the modification is unable to spread to form characteristic PRC2-mediated large repressive domains148,150, which may be due to stable conformational changes of PRC2 induced by K27M. Indeed, H3K27M-mediated inhibition preferentially affects the PRC2 complex once it binds to nucleosomes and becomes allosterically activated by existing H3K27me3 (REF.150). This mode of action would predict PRC2’s capacity to propagate existing K27me3 to neighbouring nucleosomes is impaired by K27M.
The K27M mutation appears to occur early, as it is present in all primary and metastatic tumour cells of K27M-mutant gliomas and can be traced to the cell of origin through evolutionary reconstruction125. However, K27M is often accompanied by other driver mutations affecting canonical oncogenic and tumour suppressor pathways131,133,135,136. Whereas the dominant effect of K27M on H3K27me3 levels can be recapitulated across multiple unrelated cell types143,148,150, its contribution to tumorigenesis is highly context dependent. First, K27M is almost exclusively found in paediatric and adolescent tumours, which suggests it preferentially impacts a vulnerable developmental time window, consistent with the role of PRC2 in differentiation (FIG. 2; TABLE 1). Second, efforts to model the oncogenic effects of K27M in cell culture or in vivo revealed that it induced senescence in human fibroblasts and astrocytes147 and demonstrated embryonic lethality in mice151, while targeted expression of the H3.3K27M mutant in neural progenitor cells did not induce tumours on its own151–153. Third, K27M does induce proliferation in neural progenitor cells147 or p53-deficient nestin-positive progenitors of the neonatal mouse brainstem143, but generation of undifferentiated, invasive K27M-mutant midline gliomas in mice required cooperating oncogenic events during central nervous system development151–153. Importantly, animal models recapitulate global H3K27me3 loss, as well as instances of local enrichment, and gene expression profiles found in human K27M HGGs. Among PRC2 target genes, there is upregulation of developmental factors associated with neurogenesis, such as WNT and HOX family members, but also repression of CDKN2A, indicating enhanced self-renewal and proliferation151–153. Furthermore, many genes upregulated in K27M mouse tumours have bivalent promoters in K27 wild-type controls153. Although it remains unclear which of these transcriptional changes are necessary and sufficient for tumorigenesis, they serve to emphasize the developmental nature of K27M-associated pathology. K27M, and thus PRC2 inhibition, likely maintains HGG cells in an undifferentiated state. Deletion of the mutant H3f3a gene in mouse DIPG xenograft models rescues H3K27me3 spreading and abolishes tumour formation148, and knockdown significantly decreases proliferation and restores a repressive chromatin state at bivalent genes, pushing cell identity towards the oligodendrocyte lineage154.
H3K36M in chondroblastoma.
Strikingly, a K36M mutation of H3.3 was identified in 73 of 77 cases of chondroblastoma155, a benign tumour of the cartilage of bone growth plates in adolescents and young adults (FIG. 2b). The mutation appears diagnostic as it can distinguish chondroblastoma from histologic mimics156. The high frequency of mutation, coupled with the lack of other recurring mutations and genomic aberrations, implies a driver role for H3K36M. H3F3B was preferentially mutated; only 7% of H3.3 mutations occurred in H3F3A155. Subsequent studies identified K36M in the H3.1 genes in paediatric soft tissue sarcomas and adult head and neck squamous cell carcinomas157,158, albeit at much lower frequencies.
Studies based on transgenic expression of K36M in various cell types143,157, knock-in K36M immortalized human chondrocytes and chromatin immunoprecipitation followed by sequencing of primary tumours159 revealed that the mutation decreased overall levels of H3K36 dimethylation and trimethylation, suggesting a dominant-negative effect similar to K27M. Accordingly, K36M inhibits deposition of both the dimethyl mark by NSD2 (but not other K36 methyltransferases) and the trimethyl mark by SETD2 (REFS157,159). Given that SETD2 binds with stronger affinity to the mutant peptide and nucleosome, as proposed for PRC2 at K27M, K36M likely sequesters methyltransferase activity157,159. Importantly, the mutation not only affects modification of K36 but also enhances K27 methylation by reducing the antagonistic effect of K36 methylation on EZH2 (REF160). Loss of K36me3 instructs the acquisition of K27me3 at nearby intergenic regions and leads to a redistribution of PRC1 (REF.157). PRC1-catalysed repression is therefore lost at its target genes, several of which promote self-renewal and lineage specification of mesenchymal stem cells, thereby impairing their differentiation into chondrocytes. Accordingly, exogenous expression of K36M in mesenchymal progenitor cells drives sarcoma growth in mouse allografts157.
H3.3 G34 mutations in gliomas and skeletal tumours.
G34R, and less frequently G34V, mutations were identified in more than 10% of HGGs (FIG. 2a), and in contrast to K27M, occur more frequently in tumours of the cerebral hemispheres in teenagers and young adults127,129,130. In more than 90% of giant cell tumours of the bone (GCTB), G34 is mutated to W and less so to L155, and like K36M in chondroblastoma, it is highly specific for GCTB while absent in histologically similar tumours156. Nearly all of these mutations affect H3F3A, which cannot be explained by codon usage alone — the two H3.3 genes and three H3.2 genes share the same codon at this position, and the same R and V substitutions could arise in H3.1-encoding genes through either an identical or an adjacent point mutation. In gliomas, the G34 substitutions are consistently accompanied by mutations in ATRX or DAXX, which may promote alternative lengthening of telomeres (ALT; see later)127,130,131,135. Nevertheless, it is unclear whether the G34 mutation impacts ATRX–DAXX function in these tumours and would select for the concomitant inactivation of this complex. Similarly to K27M, G34R/V mutations often co-occur with established cancer drivers127,131,135.
While G34 is not a substrate for modification, it lies close to K36, and is mechanistically distinct from K-to-M mutations. It was reported that G34R/V mutants blocked K36 methylation only in cis and therefore did not have a dominant-negative effect on K36me3 (REFS143,161). This can be explained by the structure of the SETD2 K36 methyltransferase, whose narrow substrate channel can accommodate only the minimal side chains of G34 within the H3 tail, thereby preventing G34-mutant histones carrying invariably bulkier side chains from accessing its active site162,163. Intriguingly, a targeted G34R mutation of a single copy of H3f3a in mouse ES cells led to local gains of K36me3 at lysine-specific demethylase 4 (KDM4)-enriched loci. G34R displayed increased affinity for KDM4 and inhibited its demethylase activity towards its K36me3 substrate, leading to accumulation of its other substrate, H3K9me3 (REF164). The importance of the K36me3 axis for gliomagenesis is additionally underscored by the fact that SETD2 itself is frequently mutated in paediatric HGGs165, and driver mutations in the isocitrate dehydrogenase genes IDH1 and IDH2, which occur in a distinct set of gliomas lacking H3.3 mutations166, promote tumorigenesis through abnormal production of an oncometabolite that inhibits DNA and histone demethylases, including KDM4 (REFS167,168). GCTB mutant G34W/L similarly blocks K36 methylation in cis when overexpressed, through a mechanism involving SETD2 inhibition169. The same in vitro study showed impaired K27 methylation and binding of HIRA to G34-mutant histones. G34W-mutant tumour cells isolated from GCTB displayed increased proliferation and invasive capacity, which could be recapitulated by overexpression of the mutant in an osteosarcoma cell line170. Furthermore, selective depletion of the mutant in GCTB cell lines revealed it was necessary to sustain their proliferation and migration in vitro and in vivo171.
Therapeutic opportunities for H3.3-mutant tumours.
The presence of the K27M mutation predicts shorter survival in DIPG128 and other HGGs172,173. Detection of K27M either by sequencing, immunohistochemistry (using H3K27M antibodies) or consequent decrease in H3K27me3 is equivalent139,173, carrying a uniformly fatal prognosis independent of the anatomic site or histologic grade172. Small-molecule inhibitors targeting histone-modifying enzymes allow direct modulation of K27 methylation, or indirect modulation by targeting complementary histone PTMs to reverse the mutant gene expression profile. Therefore, K27M mutation provides a therapeutic opportunity in HGGs. For example, inhibition of the K27 demethylase JMJD3 in K27M-mutant brainstem glioma cell lines174 or menin, a subunit of the MLL–SET1 methyltransferase complex175, in an ES cell-derived model147 decreased viability and proliferation of tumour cells both in vitro and in murine xenografts. Importantly, antitumour activity was dependent on the presence of mutated K27M, and efficiently targeted the brainstem, highlighting the capacity of these drugs to cross the blood—brain barrier147,174. Even though global loss of K27me3 is found in DIPG, the use of EZH2 inhibitors impaired proliferation and induced senescence in primary patient-derived K27M DIPG cell cultures176. Given that several tumour suppressor genes (for example, CDKN2A) retain residual H3K27me3 in DIPG cells149,152,176,177, it is likely that EZH2 inhibition is synthetically lethal in the presence of K27M owing to re-expression of these tumour suppressors.
Concurrent with global H3K27me3 loss, K27M tumours may harbour an increase in H3K27 acteylation (H3K27ac) at genomic regions enriched in the mutant histone177. Given that H3K27ac loci are co-occupied by BRD2 and BRD4, therapeutic use of BETi was investigated in this context. In one study, BETi use led to impaired proliferation of DIPG cells in vitro and orthotopically by downregulating the sonic hedgehog pathway177. This was challenged in another study where the presence of K27M in DIPG-derived isogenic cell systems led not to BETi sensitivity but to pervasive H3K27ac deposition across the genome, including at repetitive elements178. The resulting derepression of endogenous retroviruses is a therapeutic vulnerability that was further amplified using DNA demethylating agents and histone deacetylase inhibitors, increasing the immunogenicity of these tumours through viral mimicry178. Finally, a K27M-derived tumour neoantigen has been identified which elicits a CD8+ T cell response in patients with DIPG. Engineered antigen-specific T cells directed at this epitope caused glioma cell killing in vitro and controlled tumour growth in vivo179, providing a proof of principle that engineered T cell approaches may be feasible in the future. While efforts to target H3.3 mutations have focused on K27M glioma, epigenetic drugs that counterbalance the consequences of K36 or G34 mutations at the PTM level are promising candidates for future research.
DAXX and ATRX mutations
Mutations in the H3.3 chaperone complex DAXX—ATRX are common in tumours of neural or neural crest origin, such as pancreatic neuroendocrine tumours, glioma and neuroblastoma127,180–182 (TABLE 1). In addition, DAXX is upregulated in The Cancer Genome Atlas (TCGA) datasets compared with normal tissue, and in metastatic versus primary tumour samples183. Upregulation of DAXX carries a negative prognostic value in prostate and ovarian cancer184–186.
DAXX is enriched in promyelocytic leukaemia bodies and has been reported to interact with various nuclear proteins besides ATRX, suggesting additional functions beyond H3.3 deposition183. ATRX is a chromatin remodeller that contains an ADD domain, a DAXX-interacting region and an SNF2 helicase domain, and also has DAXX-independent functions187. ATRX interacts with macroH2A to exclude it from subtelomeric regions71 and contributes to silencing of the inactive X chromosome and PRC2 target genes188.
To attain replicative immortality, a prerequisite for tumour growth, aberrantly proliferating cells prevent telomere shortening either by activating mutations of the telomerase reverse transcriptase gene (TERT) or via the process of ALT. ALT is observed, for example, in pancreatic neuroendocrine tumours, glioblastoma, oligodendrogliomas, neuroblastoma, and osteosarcomas, and is highly correlated with loss or mutations in ATRX or DAXX180,181,189 (TABLE 1). ATRX disruption occurs in a mutually exclusive manner with TERT activation190 and is associated with larger tumour size, higher risk of metastasis and higher tumour grade191. Impaired ATRX or DAXX function was required to sustain ALT in a cancer cell line, as reintroduction of either protein potently suppressed telomere lengthening189,192. The involvement of ATRX in this process likely stems from its enrichment at G-quadruplex structures, whose replication and transcription it facilitates192–194. At telomeres, where these structures are enriched, absence of ATRX leads to collapse of stalled replication forks, which would trigger homology-directed DNA repair and ultimately ALT192. Depletion of another H3 chaperone, ASF1 (BOX 1), induces hallmarks of ALT in immortalized and transformed human cell lines, suggesting that improper H3 incorporation at telomeres plays a causal role in ALT195.
Mutually exclusive mutations of ATRX and DAXX were identified in pancreatic neuroendocrine tumours, the second most lethal type of pancreatic cancer181, where they correlate with shorter overall survival191,196–199. In midline gliomas, ATRX mutations occur alone or together with H3.3 mutation, particularly G34R/V127,131,135. In neuroblastoma, point mutations, indels and large in-frame fusions were identified in ATRX, but not DAXX or the genes encoding H3.3 (REFS182,200,201), raising the possibility that ATRX mutation in neuroblastoma may be independent of H3.3 deposition. In both midline glioma and neuroblastoma, ATRX mutations are more common in older children (8 years old, up to adolescent age) and are associated with significantly shorter overall survival compared with patients with wild-type ATRX128,182,202,203.
Therapeutic opportunities for ATRX and DAXX-mutated cancers.
Given the strong correlation between DAXX and ATRX mutations and ALT, loss of function of the complex could serve as a useful prognostic marker191. Telomere instability associated with replication stress in the absence of ATRX—DAXX could also constitute a vulnerability, which could be exploited by disabling homology-directed DNA repair. This principle has been demonstrated in vitro using inhibitors of the DNA damage response kinase ATR, which display selective cytotoxicity in ALT-positive osteosarcoma cell lines204. Similarly, inhibition of PARP1, whose activation was induced by fork stalling in the absence of ATRX in the developing brain, exacerbated DNA damage and attenuated proliferation in ATRX-deleted HeLa cells205. Further support for this exploitable vulnerability came from a recent, high-throughput compound screen which identified an increased susceptibility to ATM inhibitors and PARP inhibitors in ATRX-altered neuroblastoma models206. Furthermore, a novel therapeutically tractable connection has been established between ATRX in-frame fusions and EZH2 inhibitor sensitivity in neuroblastoma; we demonstrated that these gain-of-function ATRX mutations promote silencing of neurogenesis genes through REST activation and thus suppression of differentiation programmes, which can be reversed by EZH2 inhibition201.
CENP-A
Essential for cellular proliferation.
CENP-A (encoded by the gene CENPA) is a divergent H3 variant found at a subset of nucleosomes associated with centromeric satellite repeat sequences207 (FIG. 1). CENP-A has a dual role, directing the assembly of the kinetochore208 and propagating centromere identity across mitosis and meiosis in an epigenetic manner209. In mammalian cells, CENP-A dynamics are tightly linked to cell cycle progression. Unlike canonical H3 or variant H3.3, CENP-A expression and translation peak in G2 phase210,211, and deposition at centromeres is restricted to early G1 phase by the inhibitory activity of CDK1 and CDK2 (REFS212,213). Holliday junction recognition protein (HJURP) was identified as a CENP-A chaperone214,215 that relies on structural differences in H3 variants to specifically recognize CENP-A216. HJURP activity is limited during the cell cycle by cyclin A, which inhibits its association with centromeres outside G1 phase217. CENP-A is indispensable for embryogenesis218, and together with HJURP, depletion in vertebrate cell lines results in failure of kinetochore assembly and impaired chromosome segregation214,215,219,220, illustrating their essential role in maintaining centromere function.
CENP-A and HJURP accumulate during transformation.
HJURP and CENP-A are upregulated in a plethora of solid tumours (FIG. 2b; TABLE 1). Increased CENPA and HJURP mRNA levels may distinguish tumour versus normal cells or late versus early lesions regardless of the cell of origin, as differential expression reaches significance in most solid tumours221. This, along with their mechanistic connection to cell cycle progression and correlation with Ki67 (REFS222–225), suggests high CENP-A and HJURP levels are indicators of aberrant cellular proliferation.
Survey of TCGA data across cancer types reveals very low rates of somatic CENPA (FIG. 2a) and HJURP mutation (cBioPortal for Cancer Genomics52). Thus, the accumulation of the two centromeric factors in tumours is likely a result of transcriptional deregulation or increased protein stability. The increased efficiency of centromere propagation when both the variant and the chaperone are abundant could contribute to the fitness of frequently dividing cancer cells226. Tumours harbouring deletion or gain-of-function point mutations in TP53 have increased CENPA and HJURP expression227. This may be explained at least in part by the presence of binding motifs in CENPA and HJURP promoters, which restrict the peak of their expression to G2/M phase228,229 by recruiting the p53–DREAM complex227. This can be recapitulated ex vivo, as upregulation of HJURP and CENP-A occurs in p53-deficient mouse embryonic fibroblasts and, conversely, p53 activation leads to downregulation of these proteins in human cancer cell lines. In addition to transcriptional regulation, when one partner is exogenously overexpressed or depleted, the other follows227, suggesting reciprocal stabilization.
High levels of CENP-A can induce its promiscuous mislocalization into chromosome arms when exogenously overexpressed in HeLa cells230,231, but the chaperone responsible for this is not HJURP. Excess CENP-A hijacks DAXX, which deposits an abnormal CENP-A–H3.3 hybrid nucleosome at loci where H3.3 is normally present231. However, in such exogenous experimental models, CENP-A expression is uncoupled from cell cycle control of its chaperone machinery, which could in turn contribute to its aberrant deposition. Moreover, in colorectal cancer cell lines displaying ectopic incorporation, increased CENP-A levels correlate with the upregulation of DAXX and ATRX, but not HJURP232. This imbalance appears to determine CENP-A mis-targeting, as overexpression of HJURP decreases the number of CENP-A extracentromeric peaks and, conversely, HJURP depletion can allow ectopic CENP-A accumulation even in cancer cell lines where the variant is not overexpressed233. However, in TCGA datasets for breast invasive carcinoma, lung adenocarcinoma and lower-grade glioma, CENPA and HJURP display a remarkable level of co-expression (Spearman correlation coefficients of more than 0.90; cBioPortal for Cancer Genomics52), likely reflecting their common mode of transcriptional regulation. Thus, studying their contribution to cancer progression requires identification of tumours where CENP-A and HJURP levels are uncoupled. In these instances, promiscuously incorporated CENP-A could contribute to tumorigenesis on the one hand by seeding the formation of neocentromeres, which promote genomic instability and rescue broken chromosome fragments234. On the other hand, accumulation of CENP-A at gene regulatory regions and transcription factor-binding sites231,232 could interfere with gene expression, but both hypotheses are yet to be confirmed in cancer models.
Therapeutic opportunities for targeting CENP-A deposition.
High levels of HJURP and CENP-A correlate with tumour progression and carry a negative prognosis (TABLE 1); however, they also predict increased tumour sensitivity to adjuvant chemotherapy or radiotherapy221,222, likely as a consequence of CENP-A involvement in cellular proliferation. Moreover, genomic instability, a potential consequence of perturbed CENP-A deposition, can either promote or hamper tumour progression235. CENP-A deposition may represent a vulnerability in the context of p53 loss. In a mouse embryonic fibroblast model, the effect of Hjurp knockout was limited to proliferative arrest in wild-type Trp53 cells, whereas in Trp53-null transformed cells and tumours it led to altered ploidy and cell death227. Highly proliferating cancer cells rapidly lose CENP-A from centromeres without active deposition220,236, and loss of p53 will allow centromere-challenged cells to continue proliferating and accumulate catastrophic levels of aneuploidy237. Therefore, inhibiting CENP-A deposition might preferentially target transformed cells while sparing p53-proficient normal cells. Furthermore, aneuploidy resulting from perturbed centromere or kinetochore function generates replication-coupled DNA damage, proteotoxic stress and a senescence-associated secretory phenotype in human cells238,239 (BOX 2), which can enhance the immune response elicited by the death of transformed cells240.
No existing pharmacological compounds specifically target CENP-A deposition; however, multiple enzymatic activities are important for the propagation of centromere identity241. Unlike transcriptional regulatory PTMs found on other histones, CENP-A is modified to modulate its deposition and mitotic role. CDK1 and CDK2 complexes phosphorylate CENP-A and components of its deposition machinery to inhibit loading of the variant outside G1 phase213,217,242–246, while phosphorylation of HJURP by the M-phase kinase PLK1 promotes CENP-A deposition247. Furthermore, ubiquitylation of CENP-A K124 facilitates its interaction with HJURP and proper centromeric targeting248. Perturbing the balance of these modifications could provide a chemical means to block CENP-A deposition. Importantly, PLK1 inhibitors have progressed into clinical trials, but their efficacy in cancer cells is impaired by a functional p53 response249. This suggests that p53-deficient tumours with a high degree of dependency on CENP-A incorporation might be vulnerable to such a therapeutic approach.
Perspectives
As described throughout this Review, histone variants contribute to various processes of the ‘hallmarks of cancer’250, which has led us to adapt these to histone variant biology (FIG. 3a). For example, metabolic rewiring is an essential feature of cancer cells, and the ADP ribose-binding properties of macroH2A1.1 are emerging as a connection between metabolites and the epigenome that warrants further investigation in vivo. Given that the stages of tumorigenesis from initiation to growth, metastasis, dormancy, and treatment resistance are driven by distinct cellular pathways, we suspect that deregulation of histone variant functions is co-opted by transformed cells with distinct consequences along the evolutionary trajectory of a tumour (FIG. 3b). However, we lack knowledge of histone variant biology in important processes such as tumour cell dormancy and drug resistance.
Fig. 3 |. Histone variant hallmarks in tumorigenesis.
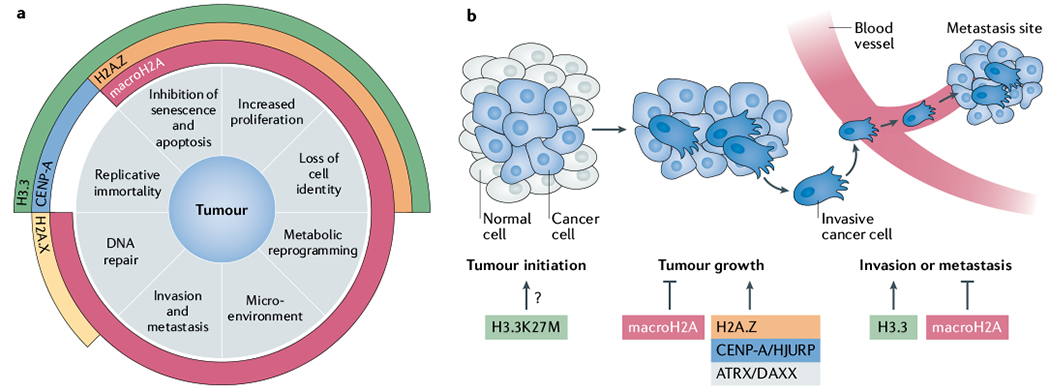
a | The role of altered histone variants in tumorigenesis can be categorized into eight major hallmarks as shown. MacroH2A is associated with the highest number of hallmarks, underlying its highly versatile role in tumour development. Centromeric protein A (CENP-A) contributes to replicative immortality via its function in chromosome segregation. H3.3 alteration affects pathways involving replicative immortality due to its role in telomere maintenance, senescence, increased proliferation and loss of cell identity. Lastly, H2A.Z alterations affect mainly proliferation-associated pathways. b | Contribution of altered histone variants and their chaperones to the stages of tumour development. A large number of studies have addressed the impact of altered histone variants on proliferation, as shown in the ‘tumour growth’ phase. Although H3.3K27M cannot initiate transformation on its own, this mutation occurs very early during tumorigenesis. Few reports have linked histone variants to invasion or metastasis mechanistically; however, there is evidence that macroH2A may reduce tumour invasiveness and metastatic potential, while deposition of H3.3 in favour of H3.1 sustains induction of genes that promote epithelial–mesenchymal transition. ATRX, α-thalassaemia/mental retardation syndrome X-linked; DAXX, death domain-associated protein 6, HJURP, Holliday junction recognition protein.
We anticipate increased efforts for the foreseeable future to characterize the diversity of the histone mutational spectrum across cancer types, as well the impact of emerging histone point mutations on the transcriptome and epigenome, in particular by leveraging large cancer datasets. For example, while most H3 mutations have been found in solid tumours, such mutations are also emerging in haematological cancers, albeit at lower frequencies251–255. Moreover, a study of more than 3,000 unique patient samples from common cancer types revealed that all core histones are recurrently mutated in ~4% of tumours, but the functional contribution of these mostly novel substitutions remains to be characterized140.
Immunotherapies have brought about remarkable increases in survival in otherwise highly lethal cancers; however, ongoing efforts strive to understand tumour response or resistance and predict patient outcome. A handful of reports have linked macroH2A to regulation of cytokine and immune effector expression95,256,257, leading us to hypothesize that variant-driven gene expression changes intrinsic to tumour cells or tumour-associated cells might shape the immune microenvironment. Therefore, it is important to assess the potential impact of histone variant dysfunction on genes involved in immune evasion, and ultimately on response to immunotherapy.
Our capacity to exploit histone variant dysfunction in cancers will rely on our developing novel, pharmaceutically viable ways to target histone variant deposition. As yet, this has proven exceptionally challenging, given that most histone chaperones lack enzymatic activities, and binding to their respective substrates occurs through large surfaces devoid of deep pockets where small-molecule inhibitors might intervene. Alternatively, given the roles of histone variants and chaperones in regulating cell identity, it is reasonable to consider whether loss of cell fate associated with their deregulation could constitute a vulnerability to epigenetic reprogramming agents in poorly differentiated tumours147,174,201. Awaiting these discoveries, histone variants could still find use as prognostic markers or predictors of therapy response, contributing to more personalized treatments and ultimately adding to overall patient survival and quality of life.
Supplementary Material
Acknowledgements
The authors thank members of the Bernstein laboratory for discussions relevant to this Review. This work was supported by NIH/NCI grant R01CA154683, NIH/NCI grant R01CA218024, NIH/NINDS grant R01NS110837 (E.B.) and the American Skin Association Ping Y. Tai Foundation Research Grant in Skin Cancer/Melanoma (D.F). The authors apologize to those whose work was not cited due to space constraints.
Glossary
- Oncohistones
Missense mutations in a histone variant, most commonly H3.3, often altering a key post-translational modification site that leads to a tumorigenic gene expression programme driving specific malignancies in children or young adults.
- YEATS domain
Histone post-translational modification reader module found in transcriptional regulator proteins that selectively binds to acetylated and crotonylated lysine residues.
- Pericentric heterochromatin
Constitutively condensed chromatin localized adjacent to the centromere, formed on repetitive DNA satellite sequences repressed by DNA methylation and histone H3 lysine 9 trimethylation (H3K9me3).
- Bivalent chromatin domains
Genomic regions that harbor both the repressive mark histone H3 lysine 27 trimethylation (H3K27me3) and the activating mark histone H3 lysine 4 trimethylation (H3K4me3) and poise a subset of genes silenced in embryonic stem cells for activation during subsequent cell fate decisions to drive normal development.
- PRC2
Polycomb repressive complex 2 (PRC2) catalyses trimethylation of histone H3 lysine 27 to silence developmental genes during differentiation, and contributes to facultative heterochromatin formation at the inactive X chromosome and imprinted genes.
- PRC1
Polycomb repressive complex 1 (PRC1) monoubiquitylates histone H2A lysine 119 to induce gene repression, in coordination with histone H3 lysine 27 trimethylation (H3K27me3) deposited by PRC2.
- Poly(ADP-ribose)
polymerase An enzyme that attaches chains of ADP-ribose from an NAD+ donor molecule to acceptor proteins (including poly(ADP-ribose) polymerases themselves) to regulate DNA repair and carbohydrate and lipid metabolism.
- Triple-negative breast cancer
Breast cancer subtype that does not express oestrogen and progesterone receptors or HER2, and therefore is not suited to hormone therapy or HER2 inhibition.
- Euchromatin
The loosely compacted, transcriptionally active and gene-enriched fraction of chromatin.
- Alternative lengthening of telomeres
(ALT). Telomerase-independent mechanism that exploits homologous recombination of repetitive telomere sequences to promote telomere length maintenance in certain immortalized cell lines and tumours.
- Promyelocytic leukaemia bodies
Membraneless nuclear compartments formed on a scaffold of promyelocytic leukaemia protein involved in telomere lengthening and DNA damage response.
- ADD domain
Protein domain consisting of an amino-terminal GATA-like zinc-finger, a plant homeodomain finger and a long carboxy-terminal α-helix which binds histone H3 unmethylated at lysine 4 and trimethylated at lysine 9.
- SNF2 helicase domain
Protein domain that uses the energy of ATP hydrolysis to apply torsional strain to DNA in order to remodel nucleosomes and other DNA—protein complexes.
- G-quadruplex structures
Stable secondary DNA structures present in G-rich DNA held together by G-G base pairs formed within the same DNA strand.
- ATM
Ataxia telangiectasia mutated (ATM) is a DNA damage kinase that phosphorylates itself and downstream effectors in response to DNA double-strand breaks to coordinate the cellular DNA damage response.
- Kinetochore
Multicomplex protein structure localized at the centromere during mitosis to attach, orient and move sister chromatids along the mitotic spindle, ensuring accurate chromosome segregation.
- Neocentromeres
Abnormal centromeres formed de novo at a location distinct from the primary constriction of a chromosome.
Footnotes
Competing interests
The authors declare no competing interests.
Peer review information
Nature Reviews Cancer thanks M. Buschbeck and Z. Zhang for their contribution to the peer review of this work.
Supplementary information is available for this paper at https://doi.org/10.1038/s41568-020-00330-0.
References
- 1.Mendiratta S, Gatto A & Almouzni G Histone supply: multitiered regulation ensures chromatin dynamics throughout the cell cycle. J. Cell Biol. 218, 39–54 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buschbeck M & Hake SB Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Bio 18, 299–314(2017). [DOI] [PubMed] [Google Scholar]
- 3.Talbert PB & Henikoff S Histone variants on the move: substrates for chromatin dynamics. Nat. Rev. Mol. Cell Bio 18, 115–126 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Mattiroli F, D’Arcy S & Luger K The right place at the right time: chaperoning core histone variants. EMBO Rep. 16, 1454–1466 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filipescu D, Müller S & Almouzni G Histone H3 variants and their chaperones during development and disease: contributing to epigenetic control. Annu. Rev. Cell Dev. Biol. 30, 615–646 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Corujo D & Buschbeck M Post-translational modifications of H2A histone variants and their role in cancer. Cancers 10, 59 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao Z & Shilatifard A Epigenetic modifications of histones in cancer. Genome Biol. 20, 245 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang T et al. Histone variants: critical determinants in tumour heterogeneity. Front. Med. 13, 289–297 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Faast R et al. Histone variant H2A.Z is required for early mammalian development. Curr. Biol. 11, 1183–1187 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Eirín-López JM, González-Romero R, Dryhurst D, Ishibashi T & Ausió J The evolutionary differentiation of two histone H2A.Z variants in chordates (H2A.Z-1 and H2A.Z-2) is mediated by a stepwise mutation process that affects three amino acid residues. BMC Evol. Biol. 9, 31 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dryhurst D et al. Characterization of the histone H2A.Z-1 and H2A.Z-2 isoforms in vertebrates. BMC Biol. 7, 86 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horikoshi N et al. Structural polymorphism in the L1 loop regions of human H2A.Z.1 and H2A.Z.2. Acta Crystallogr. D Biol. Crystallogr. 69, 2431–2439 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vardabasso C et al. Histone variant H2A.Z.2 mediates proliferation and drug sensitivity of malignant melanoma. Mol. Cell 59, 75–88 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that H2A.Z.2 together with BRD2 promotes expression of E2F target genes and proposes BETi as a therapeutic strategy for melanoma.
- 14.Dunn CJ et al. Histone hypervariants H2A.Z.1 and H2A.Z.2 play independent and context-specific roles in neuronal activity-induced transcription of Arc/Arg3.1 and other immediate early genes. eNeuro 10.1523/ENEURO.0040-17.2017(2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenberg RS, Long HK, Swigut T & Wysocka J Single amino acid change underlies distinct roles of H2A.Z subtypes in human syndrome. Cell 178, 1421–1436.e24 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamaa A et al. Integrated analysis of H2A.Z isoforms function reveals a complex interplay in gene regulation. eELife 9, e53375 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan JY, Gordon F, Luger K, Hansen JC & Tremethick DJ The essential histone variant H2A.Z regulates the equilibrium between different chromatin conformational states. Nat. Struct. Biol. 9, 172–176 (2002) [DOI] [PubMed] [Google Scholar]
- 18.Jin C & Felsenfeld G Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Gene Dev. 21, 1519–1529 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin C et al. H3.3/H2A.Z double variant–containing nucleosomes mark “nucleosome-free regions” of active promoters and other regulatory regions. Nat. Genet. 41, 941–945 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ku M et al. H2A.Z landscapes and dual modifications in pluripotent and multipotent stem cells underlie complex genome regulatory functions. Genome Biol. 13, R85 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perell GT et al. Specific acetylation patterns of H2A.Z form transient interactions with the BPTF bromodomain. Biochemistry 56, 4607–4615 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giaimo BD, Ferrante F, Herchenröther A, Hake SB & Borggrefe T The histone variant H2A.Z in gene regulation. Epigenet. Chromatin 12, 37 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dalvai M et al. H2A.Z-dependent crosstalk between enhancer and promoter regulates Cyclin D1 expression. Oncogene 32, 4243–4251 (2013) [DOI] [PubMed] [Google Scholar]
- 24.Giaimo BD et al. Histone variant H2A.Z deposition and acetylation directs the canonical Notch signaling response. Nucleic Acids Res. 46, gky551 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi J, Heo K & An W Cooperative action of TIP48 and TIP49 in H2A.Z exchange catalyzed by acetylation of nucleosomal H2A. Nucleic Acids Res. 37, 5993–6007 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu C-C et al. Gas41 links histone acetylation to H2A.Z deposition and maintenance of embryonic stem cell identity. Cell Discov. 4, 28 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho HJ et al. GAS41 recognizes diacetylated histone H3 through a bivalent binding mode. ACS Chem. Biol. 13, 2739–2746 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Obri A et al. ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature 505, 648–653 (2014) [DOI] [PubMed] [Google Scholar]
- 29.Alatwi HE & Downs JA Removal of H2A.Z by INO80 promotes homologous recombination. EMBO Rep. 16, 986–994 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gursoy-Yuzugullu O, Ayrapetov MK & Price BD Histone chaperone Anp32e removes H2A.Z from DNA double-strand breaks and promotes nucleosome reorganization and DNA repair. Proc. Natl Acad. Sci. USA 112, 7507–7512 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greaves IK, Rangasamy D, Ridgway P & Tremethick DJ H2A.Z contributes to the unique 3D structure of the centromere. Proc. Natl Acad. Sci. USA 104, 525–530 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Y et al. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell 48, 723–733 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boyarchuk E, Filipescu D, Vassias I, Cantaloube S & Almouzni G The histone variant composition of centromeres is controlled by the pericentric heterochromatin state during the cell cycle. J. Cell Sci. 127, 3347–3359 (2014) [DOI] [PubMed] [Google Scholar]
- 34.Creyghton MP et al. H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell 135, 649–661 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subramanian V et al. H2A.Z acidic patch couples chromatin dynamics to regulation of gene expression programs during ESC differentiation. PLoS Genet. 9, e1003725 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang HD et al. Oncogenic potential of histone-variant H2A.Z.1 and its regulatory role in cell cycle and epithelial-mesenchymal transition in liver cancer. Oncotarget 7, 11412–11423 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rispal J et al. The H2A.Z histone variant integrates Wnt signaling in intestinal epithelial homeostasis. Nat. Commun. 10, 1827 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frank SR et al. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 4, 575–580 (2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeong KW et al. Recognition of enhancer element–specific histone methylation by TIP60 in transcriptional activation. Nat. Struct. Mol. Biol. 18, 1358–1365 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Slupianek A, Yerrum S, Safadi FF & Monroy MA The chromatin remodeling factor SRCAP modulates expression of prostate specific antigen and cellular proliferation in prostate cancer cells. J. Cell Physiol. 224, 369–375 (2010) [DOI] [PubMed] [Google Scholar]
- 41.Hua S et al. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression. Mol. Syst. Biol. 4, 188 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]; This article identifies E-boxes at H2A.Z promoters and shows that oestrogen synergizes with MYC to induce H2A.Z expression.
- 42.Svotelis A, Gévry N, Grondin G & Gaudreau L H2A.Z overexpression promotes cellular proliferation of breast cancer cells. Cell Cycle 9, 364–370 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Gévry N et al. Histone H2A.Z is essential for estrogen receptor signaling. Gene Dev. 23, 1522–1533 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsai C-H et al. SMYD3-mediated H2A.Z.1 methylation promotes cell cycle and cancer proliferation. Cancer Res. 76, 6043–6053 (2016). [DOI] [PubMed] [Google Scholar]
- 45.He HH et al. Nucleosome dynamics define transcriptional enhancers. Nat. Genet. 42, 343–347 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valdés-Mora F et al. Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer. Nat. Commun. 8, 1346 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dryhurst D, McMullen B, Fazli L, Rennie PS & Ausió J Histone H2A.Z prepares the prostate specific antigen (PSA) gene for androgen receptor-mediated transcription and is upregulated in a model of prostate cancer progression. Cancer Lett. 315, 38–47 (2012) [DOI] [PubMed] [Google Scholar]
- 48.Ito S et al. MRGBP promotes AR-mediated transactivation of KLK3 and TMPRSS2 via acetylation of histone H2A.Z in prostate cancer cells. Biochim. Biophys. Acta Gene Regul. Mech. 1861, 794–802 (2018) [DOI] [PubMed] [Google Scholar]
- 49.Valdés-Mora F et al. Acetylation of H2A.Z is a key epigenetic modification associated with gene deregulation and epigenetic remodeling in cancer. Genome Res. 22, 307–321 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sarcinella E, Zuzarte PC, Lau PNI, Draker R & Cheung P Monoubiquitylation of H2A.Z distinguishes its association with euchromatin or facultative heterochromatin. Mol. Cell Biol. 27, 6457–6468 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Draker R, Sarcinella E & Cheung P USP10 deubiquitylates the histone variant H2A.Z and both are required for androgen receptor-mediated gene activation. Nucleic Acids Res. 39, 3529–3542 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cerami E et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hsu C-C et al. Recognition of histone acetylation by the GAS41 YEATS domain promotes H2A.Z deposition in non-small cell lung cancer. Gene Dev. 32, 58–69 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mattera L et al. The p400/Tip60 ratio is critical for colorectal cancer cell proliferation through DNA damage response pathways. Oncogene 28, 1506–1517 (2009) [DOI] [PubMed] [Google Scholar]
- 55.Chevillard-Briet M et al. Interplay between chromatin-modifying enzymes controls colon cancer progression through Wnt signaling. Hum. Mol. Genet. 23, 2120–2131 (2014) [DOI] [PubMed] [Google Scholar]
- 56.Draker R et al. A combination of H2A.Z and H4 acetylation eecruits Brd2 to chromatin during transcriptional activation. PLoS Genet. 8, e1003047 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Filippakopoulos P et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang B et al. H2A.Z regulates tumorigenesis, metastasis and sensitivity to cisplatin in intrahepatic cholangiocarcinoma. Int. J. Oncol. 52, 1235–1245 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Long H et al. H2A.Z facilitates licensing and activation of early replication origins. Nature 577, 1–18 (2020) [DOI] [PubMed] [Google Scholar]
- 60.Latrick CM et al. Molecular basis and specificity of H2A.Z–H2B recognition and deposition by the histone chaperone YL1. Nat. Struct. Mol. Biol. 23, 309–316 (2016) [DOI] [PubMed] [Google Scholar]
- 61.Liang X et al. Structural basis of H2A.Z recognition by SRCAP chromatin-remodeling subunit YL1. Nat. Struct. Mol. Biol. 23, 317–323 (2016) [DOI] [PubMed] [Google Scholar]
- 62.Pehrson & Fried V MacroH2A, a core histone containing a large nonhistone region. Science 257, 1398–1400 (1992) [DOI] [PubMed] [Google Scholar]
- 63.Chakravarthy S et al. Structural characterization of the histone variant macroH2A. Mol. Cell Biol. 25, 7616–7624 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pehrson JR, Costanzi C & Dharia C Developmental and tissue expression patterns of histone macroH2A1 subtypes. J. Cell. Biochem. 65, 107–113 (1997) [DOI] [PubMed] [Google Scholar]
- 65.Costanzi C & Pehrson JR Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 393, 599–601 (1998) [DOI] [PubMed] [Google Scholar]
- 66.Costanzi C & Pehrson JR MACROH2A2, a new member of the MACROH2A core histone family. J. Biol. Chem. 276, 21776–21784 (2001) [DOI] [PubMed] [Google Scholar]
- 67.Zhang R et al. Formation of macroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 8, 19–30 (2005) [DOI] [PubMed] [Google Scholar]
- 68.Buschbeck M et al. The histone variant macroH2A is an epigenetic regulator of key developmental genes. Nat. Struct. Mol. Biol. 16, 1074–1079 (2009) [DOI] [PubMed] [Google Scholar]
- 69.Changolkar LN et al. Genome-wide distribution of macroH2A1 histone variants in mouse liver chromatin. Mol. Cell Biol. 30, 5473–5483 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gamble MJ, Frizzell KM, Yang C, Krishnakumar R & Kraus WL The histone variant macroH2A1 marks repressed autosomal chromatin, but protects a subset of its target genes from silencing. Gene Dev. 24, 21–32 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ratnakumar K et al. ATRX-mediated chromatin association of histone variant macroH2A1 regulates α-globin expression. Gene Dev. 26, 433–438 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gaspar-Maia A et al. MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat. Commun. 4, 1565 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Douet J et al. MacroH2A histone variants maintain nuclear organization and heterochromatin architecture. J. Cell Sci. 130, jcs.199216 (2017). [DOI] [PubMed] [Google Scholar]
- 74.Sun Z et al. Transcription-associated histone pruning demarcates macroH2A chromatin domains. Nat. Struct. Mol. Biol. 25, 958–970 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kustatscher G, Hothorn M, Pugieux C, Scheffzek K & Ladurner AG Splicing regulates NAD metabolite binding to histone macroH2A. Nat. Struct. Mol. Biol. 12, 624–625 (2005) [DOI] [PubMed] [Google Scholar]
- 76.Kozlowski M et al. MacroH2A histone variants limit chromatin plasticity through two distinct mechanisms. EMBO Rep. 19, e44445 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Simonet NG et al. SirT7 auto-ADP-ribosylation regulates glucose starvation response through mH2A1. Sci. Adv. 6, eaaz2590 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ouararhni K et al. The histone variant mH2A1.1 interferes with transcription by down-regulating PARP-1 enzymatic activity. Gene Dev. 20, 3324–3336 (2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Timinszky G et al. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 16, 923–929 (2009) [DOI] [PubMed] [Google Scholar]
- 80.Xu C, Xu Y, Gursoy-Yuzugullu O & Price BD The histone variant macroH2A1.1 is recruited to DSBs through a mechanism involving PARP1. Febs Lett. 586, 3920–3925 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marjanović MP et al. MacroH2A1.1 regulates mitochondrial respiration by limiting nuclear NAD+ consumption. Nat. Struct. Mol. Biol. 24, 902–910 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tong L & Denu JM Function and metabolism of sirtuin metabolite O-acetyl-ADP-ribose. Biochim. Biophys. Acta Proteins Proteom. 1804, 1617–1625 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nusinow DA et al. Poly(ADP-ribose) polymerase 1 is inhibited by a histone H2A variant, macroH2A, and contributes to silencing of the inactive X chromosome. J. Biol. Chem. 282, 12851–12859 (2007) [DOI] [PubMed] [Google Scholar]
- 84.Pasque V et al. Histone variant macroH2A marks embryonic differentiation in vivo and acts as an epigenetic barrier to induced pluripotency. J. Cell Sci. 125, 6094–6104 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pasque V, Gillich A, Garrett N & Gurdon JB Histone variant macroH2A confers resistance to nuclear reprogramming. EMBO J. 30, 2373–2387 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barrero MJ et al. Macrohistone variants preserve cell identity by preventing the gain of H3K4me2 during reprogramming to pluripotency. Cell Rep. 3, 1005–1011 (2013) [DOI] [PubMed] [Google Scholar]
- 87.Sporn JC et al. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene 28, 3423–3428 (2009) [DOI] [PubMed] [Google Scholar]
- 88.Novikov L et al. QKI-mediated alternative splicing of the histone variant macroH2A1 regulates cancer cell proliferation. Mol. Cell Biol. 31, 4244–4255 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first article to identify a regulator of macroH2A1 alternative splicing, and a differential impact of splice variants on cell proliferation.
- 89.Li F et al. QKI5-mediated alternative splicing of the histone variant macroH2A1 regulates gastric carcinogenesis. Oncotarget 7, 32821–32834 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 90.Vieira-Silva TS et al. Histone variant macroH2A1 is downregulated in prostate cancer and influences malignant cell phenotype. Cancer Cell Int. 19, 112 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dardenne E et al. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat. Struct. Mol. Biol. 19, 1139–1146 (2012) [DOI] [PubMed] [Google Scholar]; This study shows that splicing factors regulate macroH2A1 splice variant switching, contributing to breast cancer invasiveness.
- 92.Yip BH et al. The U2AF1S34F mutation induces lineage-specific splicing alterations in myelodysplastic syndromes. J. Clin. Invest. 127, 2206–2221 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu D et al. Skp2–MacroH2A1–CDK8 axis orchestrates G2/M transition and tumorigenesis. Nat. Commun. 6, 6641 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hodge DQ, Cui J, Gamble MJ & Guo W Histone variant macroH2A1 plays an isoform-specific role in suppressing epithelial-mesenchymal transition. Sci. Rep. 8, 841 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim J-M et al. MacroH2A1.2 inhibits prostate cancer-induced osteoclastogenesis through cooperation with HP1α and H1.2. Oncogene 37, 5749–5765 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim J et al. Regulation of breast cancer-induced osteoclastogenesis by macroH2A1.2 involving EZH2-mediated H3K27me3. Cell Rep. 24, 224–237 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kapoor A et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 468, 1105–1109 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first report of a functional role for macroH2A variants in cancer.
- 98.Boulard M et al. Histone variant macroH2A1 deletion in mice causes female-specific steatosis. Epigenet. Chromatin 3, 8 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Changolkar LN et al. Developmental changes in histone macroH2A1-mediated gene regulation. Mol. Cell Biol. 27, 2758–2764 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sheedfar F et al. Genetic ablation of macrohistone H2A1 leads to increased leanness, glucose tolerance and energy expenditure in mice fed a high-fat diet. Int. J. Obes. 39, 331–338 (2015) [DOI] [PubMed] [Google Scholar]
- 101.Pehrson JR, Changolkar LN, Costanzi C & Leu NA Mice without macroH2A histone variants. Mol. Cell Biol. 34, 4523–4533 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rappa F et al. Immunopositivity for histone macroH2A1 isoforms marks steatosis-associated hepatocellular carcinoma. PLoS ONE 8, e54458 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Borghesan M et al. DNA hypomethylation and histone variant macroH2A1 synergistically attenuate chemotherapy-induced senescence to promote hepatocellular carcinoma progression. Cancer Res. 76, 594–606 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Re OL et al. Histone variant macroH2A1 rewires carbohydrate and lipid metabolism of hepatocellular carcinoma cells towards cancer stem cells. Epigenetics 13, 829–845 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hu WH et al. Loss of histone variant macroH2A2 expression associates with progression of anal neoplasm. J. Clin. Pathol. 69, 627 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Garcia H et al. Facilitates chromatin transcription complex is an “accelerator” of tumor transformation and potential marker and target of aggressive cancers. Cell Rep. 4, 159–173 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cedeno RJ et al. The histone variant macroH2A confers functional robustness to the intestinal stem cell compartment. PLoS ONE 12, e0185196 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Curtin NJ & Szabo C Poly(ADP-ribose) polymerase inhibition: past, present and future. Nat. Rev. Drug Discov. 19, 711–736 (2020) [DOI] [PubMed] [Google Scholar]
- 109.Wu RS, Tsai S & Bonner WM Patterns of histone variant synthesis can distinguish go from G1 cells. Cell 31, 367–374 (1982) [DOI] [PubMed] [Google Scholar]
- 110.Piña B & Suau P Changes in histones H2A and H3 variant composition in differentiating and mature rat brain cortical neurons. Dev. Biol. 123, 51–58 (1987) [DOI] [PubMed] [Google Scholar]
- 111.Hake SB et al. Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J. Biol. Chem. 281, 559–568 (2006) [DOI] [PubMed] [Google Scholar]
- 112.Goldberg AD et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140, 678–691 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ray-Gallet D et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol. Cell 44, 928–941 (2011) [DOI] [PubMed] [Google Scholar]
- 114.Zhang H et al. RPA interacts with HIRA and regulates H3.3 deposition at gene regulatory elements in mammalian cells. Mol. Cell 65, 272–284 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Adam S, Polo SE & Almouzni G Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell 155, 94–106 (2013) [DOI] [PubMed] [Google Scholar]
- 116.Banaszynski LA et al. Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells. Cell 155, 107–120 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Filipescu D, Szenker E & Almouzni G Developmental roles of histone H3 variants and their chaperones. Trends Genet. 29, 630–640 (2013) [DOI] [PubMed] [Google Scholar]
- 118.Chen P et al. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Gene Dev. 27, 2109–2124 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Armache A et al. Histone H3.3 phosphorylation amplifies stimulation-induced transcription. Nature 583, 852–857 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lewis PW, Elsaesser SJ, Noh K-M, Stadler SC & Allis CD Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl Acad. Sci. USA 107, 14075–14080 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Elsässer SJ, Noh K-M, Diaz N, Allis CD & Banaszynski LA Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 522, 240–244 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Voon HPJ et al. ATRX plays a key role in maintaining silencing at interstitial heterochromatic loci and imprinted genes. Cell Rep. 11, 405–418 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.He Q et al. The Daxx/Atrx complex protects tandem repetitive elements during DNA hypomethylation by promoting H3K9 trimethylation. Cell Stem Cell 17, 273–286 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Udugama M et al. Histone variant H3.3 provides the heterochromatic H3 lysine 9 tri-methylation mark at telomeres. Nucleic Acids Res. 43, 10227–10237 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nikbakht H et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat. Commun. 7, 11185 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gomes AP et al. Dynamic incorporation of histone H3 variants into chromatin is essential for acquisition of aggressive traits and metastatic colonization. Cancer Cell 36, 402–417.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schwartzentruber J et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012) [DOI] [PubMed] [Google Scholar]; This study is among the first, along with Wu et al. (2012), to identify ‘oncohistone’ H3.3, ATRX and DAXX mutations in paediatric midline gliomas.
- 128.Khuong-Quang D-A et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 124, 439–447 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wu G et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is among the first, along with Schwartzentruber et al. (2012), to identify ‘oncohistone’ H3.3 and H3 mutations in paediatric midline gliomas.
- 130.Sturm D et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22, 425–437 (2012) [DOI] [PubMed] [Google Scholar]
- 131.Fontebasso AM et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 46, 462–466 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Aihara K et al. H3F3A K27M mutations in thalamic gliomas from young adult patients. Neuro Oncol. 16, 140–146 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Buczkowicz P et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 46, 451–456 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bechet D et al. Specific detection of methionine 27 mutation in histone 3 variants (H3K27M) in fixed tissue from high-grade astrocytomas. Acta Neuropathol. 128, 733–741 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wu G et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 46, 444–450 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Taylor KR et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 46, 457–461 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Louis DN et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820 (2016) [DOI] [PubMed] [Google Scholar]
- 138.Parsons DW et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Castel D et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 130, 815–827 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nacev BA et al. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 567, 473–478 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]; This study analyses an extensive dataset across human malignancies and reveals that histone mutations are widespread.
- 141.Holoch D & Margueron R Mechanisms regulating PRC2 recruitment and enzymatic activity. Trends Biochem. Sci. 42, 531–542 (2017) [DOI] [PubMed] [Google Scholar]
- 142.Bender S et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672 (2013) [DOI] [PubMed] [Google Scholar]; This study identifies a dominant-negative effect of K27M on H3K27 trimethylation via PRC2 inhibition.
- 143.Lewis PW et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates a common mechanism by which H3 methionine for lysine substitutions inhibit SET domain-containing lysine methyltransferases.
- 144.Chan K-M et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Gene Dev. 27, 985–990 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]; This article describes a global decrease and a local redistribution of H3K27me3 as a consequence of K27M with consequent gene expression changes.
- 145.Venneti S et al. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol. 23, 558–564 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Justin N et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 7, 11316 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Funato K, Major T, Lewis PW, Allis CD & Tabar V Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346, 1529–1533 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]; This study models differentiation using the H3.3K27M mutation and proposes an epigenetic therapy for H3-mutant gliomas.
- 148.Harutyunyan AS et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 10, 1262 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fang D et al. H3.3K27M mutant proteins reprogram epigenome by sequestering the PRC2 complex to poised enhancers. eLife 7, e36696 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Stafford JM et al. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci. Adv. 4, eaau5935 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pathania M et al. H3.3K27M cooperates with Trp53 loss and PDGFRA gain in mouse embryonic neural progenitor cells to induce invasive high-grade gliomas. Cancer Cell 32, 684–700.e9 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cordero FJ et al. Histone H3.3K27M represses p16 to accelerate gliomagenesis in a murine model of DIPG. Mol. Cancer Res. 15, 1243–1254 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Larson JD et al. Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell 35, 140–155.e7 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Silveira AB et al. H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathol. 137, 637–655 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Behjati S et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 45, 1479–1482 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]; This article identifies highly prevalent H3 mutations in rare cancer types affecting cartilage and growth plates of bones in children and young adults.
- 156.Schaefer I et al. Immunohistochemistry for histone H3G34W and H3K36M is highly specific for giant cell tumor of bone and chondroblastoma, respectively, in FNA and core needle biopsy. Cancer Cytopathol. 126, 552–566 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lu C et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352, 844–849 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]; This article characterizes the mechanism of action of K36M mutations, which affect H3K27me3 distribution and inhibit mesenchymal differentiation.
- 158.Papillon-Cavanagh S et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat. Genet. 49, 180–185 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fang D et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 352, 1344–1348 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]; This article characterizes the mechanism of action of K36M mutations by inhibiting H3K36 lysine methyltransferases.
- 160.Schmitges FW et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 42, 330–341 (2011) [DOI] [PubMed] [Google Scholar]
- 161.Bjerke L et al. Histone H3.3 mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 3, 512–519 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yang S et al. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Gene Dev. 30, 1611–1616 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fang J et al. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3–MutSα interaction. Proc. Natl Acad. Sci. USA 115, 201806355 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Voon HPJ et al. Inhibition of a K9/K36 demethylase by an H3.3 point mutation found in paediatric glioblastoma. Nat. Commun. 9, 3142 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fontebasso AM et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 125, 659–669 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Williams MJ, Singleton WGB, Lowis SP, Malik K & Kurian KM Therapeutic targeting of histone modifications in adult and pediatric high-grade glioma. Front. Oncol. 7, 45 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Xu W et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chowdhury R et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 12, 463–469 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Shi L, Shi J, Shi X, Li W & Wen H Histone H3.3 G34 mutations alter histone H3K36 and H3K27 methylation in cis. J. Mol. Biol. 430, 1562–1565 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lim J et al. The histone variant H3.3 G34W substitution in giant cell tumor of the bone link chromatin and RNA processing. Sci. Rep. 7, 13459 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fellenberg J et al. Knock-down of oncohistone H3F3A-G34W counteracts the neoplastic phenotype of giant cell tumor of bone derived stromal cells. Cancer Lett. 448, 61–69 (2019) [DOI] [PubMed] [Google Scholar]
- 172.Karremann M et al. Diffuse high-grade gliomas with H3 K27M mutations carry a dismal prognosis independent of tumor location. Neuro Oncol. 20, 123–131 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Huang T et al. Detection of histone H3 K27M mutation and post-translational modifications in pediatric diffuse midline glioma via tissue immunohistochemistry informs diagnosis and clinical outcomes. Oncotarget 9, 37112–37124 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hashizume R et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 20, 1394–1396 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]; This is one of the first studies to propose an epigenetic therapy for H3-mutant gliomas.
- 175.Yokoyama A et al. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 123, 207–218 (2005) [DOI] [PubMed] [Google Scholar]
- 176.Mohammad F et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 23, 483–492 (2017) [DOI] [PubMed] [Google Scholar]
- 177.Piunti A et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 23, 493–500 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Krug B et al. Pervasive H3K27 acetylation leads to ERV expression and a therapeutic vulnerability in H3K27M gliomas. Cancer Cell 35, 782–797.e8 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chheda ZS et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J. Exp. Med. 215, 141–157 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Heaphy CM et al. Altered Telomeres in Tumors with ATRX and DAXX Mutations. Science 333, 425–425 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]; This article identifies a connection between ATRX/ DAXX inactivation and acquisition of the ALT phenotype.
- 181.Jiao Y et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331, 1199–1203 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identifies ATRX/DAXX mutations in a poorly understood pancreatic cancer subtype.
- 182.Cheung N-KV et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. Jama 307, 1062–1071 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mahmud I & Liao D DAXX in cancer: phenomena, processes, mechanisms and regulation. Nucleic Acids Res. 47, 7734–7752 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kwan PS et al. Daxx regulates mitotic progression and prostate cancer predisposition. Carcinogenesis 34, 750–759 (2013) [DOI] [PubMed] [Google Scholar]
- 185.Tsourlakis MC et al. Overexpression of the chromatin remodeler death-domain–associated protein in prostate cancer is an independent predictor of early prostate-specific antigen recurrence. Hum. Pathol. 44, 1789–1796 (2013) [DOI] [PubMed] [Google Scholar]
- 186.Pan WW et al. Death domain-associated protein DAXX promotes ovarian cancer development and chemoresistance. J. Biol. Chem. 288, 13620–13630 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Dyer MA, Qadeer ZA, Valle-Garcia D & Bernstein E ATRX and DAXX: mechanisms and mutations. Cold Spring Harb. Perspect. Med. 7, a026567 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sarma K et al. ATRX directs binding of PRC2 to Xist RNA and polycomb targets. Cell 159, 869–883 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yost KE et al. Rapid and reversible suppression of ALT by DAXX in osteosarcoma cells. Sci. Rep. 9, 4544 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Killela PJ et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl Acad. Sci. USA 110, 6021–6026 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Singhi AD et al. Alternative lengthening of telomeres and loss of DAXX/ATRX expression predicts metastatic disease and poor survival in patients with pancreatic neuroendocrine tumors. Clin. Cancer Res. 23, 600–609 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Clynes D et al. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 6, 7538 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Law MJ et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 143, 367–378 (2010) [DOI] [PubMed] [Google Scholar]
- 194.Levy MA, Kernohan KD, Jiang Y & Bérubé NG ATRX promotes gene expression by facilitating transcriptional elongation through guanine-rich coding regions. Hum. Mol. Genet. 24, 1824–1835 (2015) [DOI] [PubMed] [Google Scholar]
- 195.O’Sullivan RJ et al. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol. 21, 167–174 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Marinoni I et al. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology 146, 453–460.e5 (2014) [DOI] [PubMed] [Google Scholar]
- 197.Park JK et al. DAXX/ATRX and MEN1 genes are strong prognostic markers in pancreatic neuroendocrine tumor. Oncotarget 5, 49796–49806 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Chou A et al. ATRX loss is an independent predictor of poor survival in pancreatic neuroendocrine tumors. Hum. Pathol. 82, 249–257 (2018) [DOI] [PubMed] [Google Scholar]
- 199.Chan CS et al. ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat. Commun. 9, 4158 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Molenaar JJ et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483, 589–593 (2012). [DOI] [PubMed] [Google Scholar]
- 201.Qadeer ZA et al. ATRX in-frame fusion neuroblastoma is sensitive to EZH2 inhibition via modulation of neuronal gene signatures. Cancer Cell 36, 512–527.e9 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Pugh TJ et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 45, 279–284 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kurihara S, Hiyama E, Onitake Y, Yamaoka E & Hiyama K Clinical features of ATRX or DAXX mutated neuroblastoma. J. Pediatr. Surg. 49, 1835–1838 (2014) [DOI] [PubMed] [Google Scholar]
- 204.Flynn RL et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347, 273–277 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Huh MS et al. Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis. 7, e2220 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.George SL et al. Therapeutic vulnerabilities in the DNA damage response for the treatment of ATRX mutant neuroblastoma. Ebiomedicine 59, 102971 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sullivan KF, Hechenberger M & Masri K Human CENP-A contains a histone H3 related histone fold domain that is required for targeting to the centromere. J. Cell Biol. 127, 581–592 (1994) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Musacchio A & Desai A A molecular view of kinetochore assembly and function. Biology 6, 5 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Black BE & Cleveland DW Epigenetic centromere propagation and the nature of CENP-A nucleosomes. Cell 144, 471–479 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Shelby RD, Vafa O & Sullivan KF Assembly of CENP-A into centromeric chromatin requires a cooperative array of nucleosomal DNA contact sites. J. Cell Biol. 136, 501–513 (1997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Shelby RD, Monier K & Sullivan KF Chromatin assembly at kinetochores is uncoupled from DNA replication. J. Cell Biol. 151, 1113–1118 (2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Jansen LET, Black BE, Foltz DR & Cleveland DW Propagation of centromeric chromatin requires exit from mitosis. J. Cell Biol. 176, 795–805 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Silva MCC et al. Cdk activity couples epigenetic centromere inheritance to cell cycle progression. Dev. Cell 22, 52–63 (2012) [DOI] [PubMed] [Google Scholar]
- 214.Dunleavy EM et al. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 137, 485–497 (2009) [DOI] [PubMed] [Google Scholar]
- 215.Foltz DR et al. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell 137, 472–484 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Hu H et al. Structure of a CENP-A–histone H4 heterodimer in complex with chaperone HJURP. Gene Dev. 25, 901–906 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Stankovic A et al. A dual inhibitory mechanism sufficient to maintain cell-cycle-restricted CENP-A assembly. Mol. Cell 65, 231–246 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Howman EV et al. Early disruption of centromeric chromatin organization in centromere protein A (Cenpa) null mice. Proc. Natl Acad. Sci. USA 97, 1148–1153 (2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Régnier V et al. CENP-A is required for accurate chromosome segregation and sustained kinetochore association of BubR1. Mol. Cell Biol. 25, 3967–3981 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Fachinetti D et al. A two-step mechanism for epigenetic specification of centromere identity and function. Nat. Cell Biol. 15, 1056–1066 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zhang W et al. Centromere and kinetochore gene misexpression predicts cancer patient survival and response to radiotherapy and chemotherapy. Nat. Commun. 7, 12619 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Hu Z et al. The expression level of HJURP has an independent prognostic impact and predicts the sensitivity to radiotherapy in breast cancer. Breast Cancer Res. 12, R18 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Li Y et al. ShRNA-targeted centromere protein a inhibits hepatocellular carcinoma growth. PLoS ONE 6, e17794 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.McGovern SL, Qi Y, Pusztai L, Symmans WF & Buchholz TA Centromere protein-A, an essential centromere protein, is a prognostic marker for relapse in estrogen receptor-positive breast cancer. Breast Cancer Res. 14, R72 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Gu X-M et al. Expression and prognostic relevance of centromere protein A in primary osteosarcoma. Pathol. Res. Pract. 210, 228–233 (2014) [DOI] [PubMed] [Google Scholar]
- 226.Luo J, Solimini NL & Elledge SJ Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Filipescu D et al. Essential role for centromeric factors following p53 loss and oncogenic transformation. Gene Dev. 31, 463–480 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]; This work identifies a dependency on increased levels of CENP-A and HJURP for aberrant proliferation of p53-deficient cells.
- 228.Müller GA et al. The CHR promoter element controls cell cycle-dependent gene transcription and binds the DREAM and MMB complexes. Nucleic Acids Res. 40, 1561–1578 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Müller GA et al. The CHR site: definition and genome-wide identification of a cell cycle transcriptional element. Nucleic Acids Res. 42, 10331–10350 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hooser AAV et al. Specification of kinetochoreforming chromatin by the histone H3 variant CENP-A. J. Cell Sci. 114, 3529–3542 (2001). [DOI] [PubMed] [Google Scholar]
- 231.Lacoste N et al. Mislocalization of the centromeric histone variant CenH3/CENP-A in human cells depends on the chaperone DAXX. Mol. Cell 53, 631–644 (2014) [DOI] [PubMed] [Google Scholar]
- 232.Athwal RK et al. CENP-A nucleosomes localize to transcription factor hotspots and subtelomeric sites in human cancer cells. Epigenet Chromatin 8, 2 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Nye J, Sturgill D, Athwal R & Dalal Y HJURP antagonizes CENP-A mislocalization driven by the H3.3 chaperones HIRA and DAXX. PLoS ONE 13, e0205948 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Barra V & Fachinetti D The dark side of centromeres: types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun. 9, 4340 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Santaguida S & Amon A Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat. Rev. Mol. Cell Biol. 16, 473–485 (2015) [DOI] [PubMed] [Google Scholar]
- 236.Zasadzińska E et al. Inheritance of CENP-A nucleosomes during DNA replication requires HJURP. Dev. Cell 47, 348–362.e7 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Thompson SL & Compton DA Proliferation of aneuploid human cells is limited by a p53-dependent mechanismp53 suppresses aneuploidy. J. Cell Biol. 188, 369–381 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Ohashi A et al. Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53-mediated post-mitotic apoptosis in SAC-impaired cells. Nat. Commun. 6, 7668 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Santaguida S et al. Chromosome mis-segregation generates cell-cycle-arrested cells with complex karyotypes that are eliminated by the immune system. Dev. Cell 41, 638–651.e5 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Galluzzi L, Buqué A, Kepp O, Zitvogel L & Kroemer G Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111 (2017) [DOI] [PubMed] [Google Scholar]
- 241.Srivastava S, Zasadzińska E & Foltz DR Posttranslational mechanisms controlling centromere function and assembly. Curr. Opin. Cell Biol. 52, 126–135 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Müller S et al. Phosphorylation and DNA binding of HJURP determine its centromeric recruitment and function in CenH3CENP-A loading. Cell Rep. 8, 190–203 (2014) [DOI] [PubMed] [Google Scholar]
- 243.Yu Z et al. Dynamic phosphorylation of CENP-A at Ser68 orchestrates its cell-cycle-dependent deposition at centromeres. Dev. Cell 32, 68–81 (2015) [DOI] [PubMed] [Google Scholar]
- 244.Pan D et al. CDK-regulated dimerization of M18BP1 on a Mis18 hexamer is necessary for CENP-A loading. eLife 6, e23352 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Spiller F et al. Molecular basis for Cdk1-regulated timing of Mis18 complex assembly and CENP-A deposition. EMBO Rep. 18, 894–905 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Takada M et al. FBW7 loss promotes chromosomal instability and tumorigenesis via cyclin E1/ CDK2–mediated phosphorylation of CENP-A. Cancer Res. 77, 4881–4893 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.McKinley KL & Cheeseman IM Polo-like kinase 1 licenses CENP-A deposition at centromeres. Cell 158, 397–411 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Niikura Y et al. CENP-A K124 ubiquitylation is required for CENP-A deposition at the centromere. Dev. Cell 32, 589–603 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Smith L et al. The responses of cancer cells to PLK1 inhibitors reveal a novel protective role for p53 in maintaining centrosome separation. Sci. Rep. 7, 16115 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011) [DOI] [PubMed] [Google Scholar]
- 251.Atak ZK et al. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in T-cell acute lymphoblastic leukemia. PLoS Genet. 9, e1003997 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Huether R et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat. Commun. 5, 3630 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lehnertz B et al. H3 K27M/I mutations promote context-dependent transformation in acute myeloid leukemia with RUNX1 alterations. Blood 130, 2204–2214 (2017) [DOI] [PubMed] [Google Scholar]
- 254.Collord G et al. Recurrent histone mutations in T-cell acute lymphoblastic leukaemia. Brit J. Haematol. 184, 676–679 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Boileau M et al. Mutant H3 histones drive human pre-leukemic hematopoietic stem cell expansion and promote leukemic aggressiveness. Nat. Commun. 10, 2891 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Hussey KM et al. The histone variant macroH2A1 regulates target gene expression in part by recruiting the transcriptional coregulator PELP1. Mol. Cell Biol. 34, 2437–2449 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Chen H et al. MacroH2A1 and ATM play opposing roles in paracrine senescence and the senescence-associated secretory phenotype. Mol. Cell 59, 719–731 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Rhodes DR et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc. Natl Acad. Sci. USA 101, 9309–9314 (2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Hardy S et al. The euchromatic and heterochromatic landscapes are shaped by antagonizing effects of transcription on H2A.Z deposition. PLoS Genet. 5, e1000687 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Segala G, Bennesch MA, Pandey DP, Hulo N & Picard D Monoubiquitination of histone H2B blocks eviction of histone variant H2A.Z from inducible enhancers. Mol. Cell 64, 334–346 (2016) [DOI] [PubMed] [Google Scholar]
- 261.Ma X-J et al. Gene expression profiles of human breast cancer progression. Proc. Natl Acad. Sci. 100, 5974–5979 (2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Oca R. M. de et al. The histone chaperone HJURP is a new independent prognostic marker for luminal A breast carcinoma. Mol. Oncol. 9, 657–674 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Lei S, Long J & Li J MacroH2A suppresses the proliferation of the B16 melanoma cell line. Mol. Med. Rep. 10, 1845–1850 (2014) [DOI] [PubMed] [Google Scholar]
- 264.Hu B et al. Holliday junction–recognizing protein promotes cell proliferation and correlates with unfavorable clinical outcome of hepatocellular carcinoma. Onco. Targets Ther. 10, 2601–2607 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Wu Q et al. Expression and prognostic significance of centromere protein A in human lung adenocarcinoma. Lung Cancer 77, 407–414 (2012) [DOI] [PubMed] [Google Scholar]
- 266.Kato T et al. Activation of Holliday junction–recognizing protein involved in the chromosomal stability and immortality of cancer cells. Cancer Res. 67, 8544–8553 (2007) [DOI] [PubMed] [Google Scholar]
- 267.Barrios O. de et al. ZEB1-induced tumourigenesis requires senescence inhibition via activation of DKK1/mutant p53/Mdm2/CtBP and repression of macroH2A1. Gut 66, 666 (2017) [DOI] [PubMed] [Google Scholar]
- 268.Tomonaga T et al. Overexpression and mistargeting of centromere protein-A in human primary colorectal cancer. Cancer Res. 63, 3511–3516 (2003) [PubMed] [Google Scholar]
- 269.Sang Y et al. CDK5-dependent phosphorylation and nuclear translocation of TRIM59 promotes macroH2A1 ubiquitination and tumorigenicity. Nat. Commun. 10, 4013 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Stangeland B et al. Combined expressional analysis, bioinformatics and targeted proteomics identify new potential therapeutic targets in glioblastoma stem cells. Oncotarget 6, 26192–26215 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Tayrac M. de et al. Prognostic significance of EDN/RB, HJURP, p60/CAF-1 and PDLI4, four new markers in high-grade gliomas. PLoS ONE 8, e73332 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Valente V et al. Modulation of HJURP (Holliday junction-recognizing protein) levels is correlated with glioblastoma cells survival. PLoS ONE 8, e62200 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Qiu J-J et al. Prognostic value of centromere protein-A expression in patients with epithelial ovarian cancer. Tumor Biol. 34, 2971–2975 (2013) [DOI] [PubMed] [Google Scholar]
- 274.Li L, Li X, Meng Q, Khan AQ & Chen X Increased expression of Holliday junction-recognizing protein (HJURP) as an independent prognostic biomarker in advanced-stage serous ovarian carcinoma. Med. Sci. Monit. 24, 3050–3055 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Liu Y, Hua T, Chi S & Wang H Identification of key pathways and genes in endometrial cancer using bioinformatics analyses. Oncol. Lett. 17, 897–906 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Chen X et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 7, 104–112 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Mason-Osann E et al. Identification of a novel gene fusion in ALT positive osteosarcoma. Oncotarget 9, 32868–32880 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Lee PJ et al. Spectrum of mutations in leiomyosarcomas identified by clinical targeted next-generation sequencing. Exp. Mol. Pathol. 102, 156–161 (2017) [DOI] [PubMed] [Google Scholar]
- 279.Mäkinen N et al. Exome sequencing of uterine leiomyosarcomas identifies frequent mutations in TP53, ATRX, and MED12. PLoS Genet. 12, e1005850 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Liau J-Y et al. Leiomyosarcoma with alternative lengthening of telomeres is associated with aggressive histologic features, loss of ATRX expression, and poor clinical outcome. Am. J. Surg. Pathol. 39, 236–244 (2015) [DOI] [PubMed] [Google Scholar]
- 281.Liau J-Y et al. Comprehensive screening of alternative lengthening of telomeres phenotype and loss of ATRX expression in sarcomas. Mod. Pathol. 28, 1545–1554 (2015) [DOI] [PubMed] [Google Scholar]
- 282.Yang C-Y et al. Targeted next-generation sequencing of cancer genes identified frequent TP53 and ATRX mutations in leiomyosarcoma. Am. J. Transl. Res. 7, 2072–2081 (2015) [PMC free article] [PubMed] [Google Scholar]
- 283.Tachiwana H et al. Structures of human nucleosomes containing major histone H3 variants. Acta Crystallogr. D Biol. Crystallogr. 67, 578–583 (2011) [DOI] [PubMed] [Google Scholar]
- 284.Tachiwana H et al. Crystal structure of the human centromeric nucleosome containing CENP-A. Nature 476, 232–235 (2011) [DOI] [PubMed] [Google Scholar]
- 285.Chakravarthy S, Luger K PDB Entry - 2F8N. Worldwide Protein Data Bank. www.wwpdb.org/pdb?id=pdb_00002f8n (2011) [Google Scholar]
- 286.Reynolds CR, Islam SA & Sternberg MJE EzMol: a web server wizard for the rapid visualization and image production of protein and nucleic acid structures. J. Mol. Biol. 10.1016/j.jmb.2018.01.013 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Luger K, Mäder AW, Richmond RK, Sargent DF & Richmond TJ Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 (1997) [DOI] [PubMed] [Google Scholar]
- 288.Tagami H, Ray-Gallet D, Almouzni G & Nakatani Y Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116, 51–61 (2004) [DOI] [PubMed] [Google Scholar]
- 289.Latreille D, Bluy L, Benkirane M & Kiernan RE Identification of histone 3 variant 2 interacting factors. Nucleic Acids Res. 42, 3542–3550 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Shibahara K & Stillman B Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 96, 575–585 (1999) [DOI] [PubMed] [Google Scholar]
- 291.Moggs JG et al. A CAF-1–PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol. Cell Biol. 20, 1206–1218 (2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Mello JA et al. Human Asf1 and CAF-1 interact and synergize in a repair-coupled nucleosome assembly pathway. EMBO Rep. 3, 329–334 (2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Tang Y et al. Structure of a human ASF1a–HIRA complex and insights into specificity of histone chaperone complex assembly. Nat. Struct. Mol. Biol. 13, 921–929 (2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Huang H et al. A unique binding mode enables MCM2 to chaperone histones H3–H4 at replication forks. Nat. Struct. Mol. Biol. 22, 618–626 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Groth A et al. Regulation of replication fork progression through histone supply and demand. Science 318, 1928–1931 (2007) [DOI] [PubMed] [Google Scholar]
- 296.Herranz N & Gil J Mechanisms and functions of cellular senescence. J. Clin. Invest. 128, 1238–1246 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Chandra T et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol. Cell 47, 203–214 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Lee K, Lau ZZ, Meredith C & Park JH Decrease of p400 ATPase complex and loss of H2A.Z within the p21 promoter occur in senescent IMR-90 human fibroblasts. Mech. Ageing Dev. 133, 686–694 (2012) [DOI] [PubMed] [Google Scholar]
- 299.Duarte LF et al. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat. Commun. 5, 5210 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates a novel tumour-suppressive role for H3.3 proteolytic processing.
- 300.Chicas A et al. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc. Natl Acad. Sci.USA 109, 8971–8976 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Corpet A, Olbrich T, Gwerder M, Fink D & Stucki M Dynamics of histone H3.3 deposition in proliferating and senescent cells reveals a DAXX-dependent targeting to PML-NBs important for pericentromeric heterochromatin organization. Cell Cycle 13, 249–267 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Rai TS et al. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Gene Dev. 28, 2712–2725 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]; This article identifies a novel tumour-suppressive role for the histone chaperone HIRA.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.