

Soils near living and decomposing roots form distinct niches that promote microorganisms with distinctive environmental preferences and interactions. Yet few studies have assessed the community-level cooccurrence of bacteria and fungi in these soil niches as plant roots grow and senesce.
KEYWORDS: arbuscular mycorrhizal fungi (AMF), metagenome-assembled genome (MAG), cooccurrence network, rhizosphere, detritusphere, hyphosphere, stable-isotope probing (SIP), spatial heterogeneity
ABSTRACT
Soil bacteria and fungi are known to form niche-specific communities that differ between actively growing and decaying roots. Yet almost nothing is known about the cross-kingdom interactions that frame these communities and the environmental filtering that defines these potentially friendly or competing neighbors. We explored the temporal and spatial patterns of soil fungal (mycorrhizal and nonmycorrhizal) and bacterial cooccurrence near roots of wild oat grass, Avena fatua, growing in its naturalized soil in a greenhouse experiment. Amplicon sequences of the fungal internal transcribed spacer (ITS) and bacterial 16S rRNA genes from rhizosphere and bulk soils collected at multiple plant growth stages were used to construct covariation-based networks as a step toward identifying fungal-bacterial associations. Corresponding stable-isotope-enabled metagenome-assembled genomes (MAGs) of bacteria identified in cooccurrence networks were used to inform potential mechanisms underlying the observed links. Bacterial-fungal networks were significantly different in rhizosphere versus bulk soils and between arbuscular mycorrhizal fungi (AMF) and nonmycorrhizal fungi. Over 12 weeks of plant growth, nonmycorrhizal fungi formed increasingly complex networks with bacteria in rhizosphere soils, while AMF more frequently formed networks with bacteria in bulk soils. Analysis of network-associated bacterial MAGs suggests that some of the fungal-bacterial links that we identified are potential indicators of bacterial breakdown and consumption of fungal biomass, while others intimate shared ecological niches.
INTRODUCTION
The biological communities and biogeochemical cycling in soil near roots versus distant from roots reflect distinct ecological niches. Rhizosphere soil (directly next to living roots) receives 10 to 40% of plant photosynthetic products as rhizodeposits (1) that include low-molecular-weight compounds easily consumed by microorganisms, metabolites related to plant defense and stress tolerance, and root debris resulting from death and sloughing off of root structures. These rhizodeposits frame the formation of a rhizomicrobiome occupying the rhizosphere niche (2). In soil distant from and less influenced by roots, litter-derived older organic materials become major resources. The decomposition of macromolecular organic compounds and mineralization of nutrients by saprotrophic organisms in bulk soil containing root litter (the detritusphere) are critical processes in terrestrial nutrient cycling (3). The distinct physicochemical environments within the rhizosphere and the detritusphere influence how associated soil microbial communities assemble and function. Despite rich research on the microbial composition of these soil niches, we know little about the covariation patterns of bacteria and fungi in these soil environments. It has been suggested that many bacteria depend on fungal hydrolysis of recalcitrant plant materials such as lignin and cellulose, forming intermediate products that are primary bacterial substrates in soil (4). In return, bacteria contribute to the resources needed by fungal decomposition processes by providing nutritional benefits, for example, through in situ N fixation (5). Exploring fungal-bacterial cooccurrence patterns in rhizosphere and detritusphere niches can identify evidence of niche sharing and potential interactions that collectively affect carbon (C) cycling and nutrient availability to plants.
Bacteria and fungi are the most diverse groups of soil inhabitants, and their versatile physiologies help regulate many soil processes (6). Cooperatively, saprotrophic fungi and bacteria can increase the efficiency of litter decomposition (4, 5, 7). Fungal hyphae can connect patches in unsaturated porous soil, acting as transport vectors or “fungal highways” for bacterial dispersal and access to substrates (8–11). Yet bacteria also consume fungal exudates and biomass released after hyphal damage or death and actively attack living hyphae through mycophagy, promoting the transfer of C from biotic forms to CO2, dissolved organic C, and mineral-associated C (12–14). In addition, antagonistic interactions between certain bacterial and fungal pathogens can promote plant pathogen resistance (15). As individual groups of organisms, fungi and bacteria are known to dynamically compete for space, nutrients, and essential minerals (16–18), especially under nutrient limitation (19). In aggregate, the location of these direct fungal-bacterial interactions is termed the hyphosphere (20). The hyphosphere is a hot spot for bacterial-fungal interactions with strong effects on bacterial niche development (13), the consequences of which are only beginning to be understood. Studies suggest that mycorrhizal and saprotrophic fungi modulate bacterial community composition (21–25), thereby having differential effects on C in rhizosphere, hyphosphere, and detritusphere soil habitats (26). Yet most of the existing bacterial-fungal interaction research has been carried out under highly controlled laboratory conditions (i.e., on artificial media), and much remains unclear about the distribution and dynamics of bacterial and fungal communities in natural root-soil-microbe systems.
The associations between arbuscular mycorrhizal fungi (AMF) and bacteria are particularly important to plant nutrient acquisition and the redistribution of the plant photosynthate into belowground C pools. AMF are strict biotrophs that depend on plant-derived C via the exchange of plant-fixed C for nutrients that AMF absorb from soil (23) and are estimated to colonize 71% of land plants (27). AMF-plant interactions occur in the rhizosphere during the establishment of symbiosis, but once established, they become conduits that redistribute C from the roots to areas beyond the rhizosphere (i.e., the “bulk” soil). Bacteria have been shown to inhabit the hyphosphere, the area around mycorrhizal hyphae (28, 29). Classical examples include helper bacteria that facilitate the establishment of plant-mycorrhizal fungus symbiosis (30), plant-growth-promoting rhizobacteria (PGPR) that work synergistically with AMF to enhance plant growth (31), and mycophagous bacteria that feed on live hyphae (32). While insightful research has been conducted on the influence of AMF-bacterial associations on soil and plants, little is known about the location of these hyphal-bacterial associations in soil and when they occur during the growth phases of plants.
Ecological networks have been increasingly reported as approximations of the complex relationships among members of diverse microbiomes (33–35), where nodes represent members and links depict characteristics of their relationships (directionality and weight, etc.). In plant and animal networks, links are based on prior knowledge of biotic relationships (e.g., food webs, pollination networks, and pathogenic networks). In macroorganisms, network behavioral studies have greatly expanded our understanding of community dynamics, stability, and evolution (36–38). In microbial ecology, however, networks are usually inferred based on abundance correlations between molecular markers (34), where the cooccurrence of microbial taxa can result from environmental filtering, resulting in niche partitioning or sharing (39) that is exceedingly difficult to document in the natural microbial world. Although these correlation-based networks have inevitable limitations in their capacity to distinguish different mechanisms underlying the links (39, 40), they are thought to capture predicted and novel patterns in microbial community organization, including bacterial competition following substrate injection into groundwater (41), fungal functional guild partitioning in agriculture soils (42), plant-microbe associations (43), disease suppression against plant pathogens (44), increases in soil bacterium cooccurrence in response to climate change (45), and microbial metabolic dependence (46). In a previous related study (47), network analysis revealed highly complex cooccurrence patterns among bacteria living in the rhizosphere during the progression of plant growth. These patterns contrasted with the more simple and static networks in nonrhizosphere soil and suggest that niche sharing is critical to rhizosphere community assembly. In that same study, a keystone species identified based on network topology was found to harbor quorum sensing ability, suggesting that these networks captured potentially interacting bacteria. Networks based on cooccurrence patterns can serve as an important starting point for exploring potential interactions in complex soil microbiomes by identifying possible biotic interactions that are worthy of further experimental validation (39). Here, we explore cross-kingdom networks among bacteria and fungi to provide a hypothesis-generating procedure to understand the interactions between these two consequential groups of soil organisms over space and time; the identified fungal-bacterial links can be further examined to assess possible interactions or niche sharing.
In this study, we investigated the cooccurrence patterns of bacterial and fungal communities over space and time, using amplicon sequences of bacterial 16S and fungal internal transcribed spacer (ITS) rRNA genes, in soil samples from a greenhouse experiment that has previously yielded information on bacterial succession and cooccurrence patterns (47, 48) as well as potential trophic interactions revealed by stable-isotope-informed genome-resolved metagenomics (49). This experiment used soil from a California annual grassland and the annual grass Avena fatua, which is common and naturalized to the area from which the soil was collected. Growth conditions approximated those of two consecutive growing seasons separated by a summer dry-down period characteristic of the Mediterranean climate. Both rhizosphere and bulk soils were harvested in a multipoint time series and analyzed to compare soil microbial communities adjacent to and away from roots, at multiple plant growth stages, and in response to the effect of summer dry-down. We constructed cooccurrence networks using a random matrix theory (RMT)-based approach (45, 50) and separately explored the cooccurrences of soil bacteria with both mycorrhizal and nonmycorrhizal fungi at the levels of communities, populations, and taxa in order to understand how soil niche and different life histories of organisms influence the dynamics of fungal-bacterial cooccurrence over space and time. We then used stable-isotope-enabled genome-centric metagenomics to investigate the links between bacterial and fungal taxa at different plant growth stages in soils from rhizosphere, bulk, and detritusphere zones. We sought to address the following: (i) What are the patterns of bacterial-fungal cooccurrence in rhizosphere versus bulk/detritusphere soil and over time through a plant growing season? (ii) How do cooccurrence patterns differ between bacteria and mycorrhizal versus nonmycorrhizal fungi? (iii) What taxa of bacteria and fungi cooccur, and what are the potential mechanisms behind these patterns? Compared to previous studies of fungal-bacterial interactions (primarily conducted under controlled conditions), our analysis is unique in its use of live soil and native fungal and bacterial communities.
RESULTS
Overall network topology.
Using 16S rRNA and ITS amplicons (see Materials and Methods for basic sequencing results), we created separate nonmycorrhizal (including saprotrophic, pathogenic, endophytic, and unclassifiable) and mycorrhizal (AMF) fungal-bacterial networks for five time points in each of two growing seasons, for both rhizosphere and bulk (or bulk and decaying root litter) soils (16 biological replicates were used to construct each network).
The nonmycorrhizal fungal-bacterial networks (Fig. 1; see also Table S1 in the supplemental material) have 310 to 783 nodes (operational taxonomic units [OTUs] whose relative abundance covaries with at least one other OTU) and 368 to 790 links (pairwise correlations of nodes), with an average degree (average number of links per node) ranging from 1.5 to 2.9. On average, 35% of the nodes are fungal, representing 12.5% of all detected nonmycorrhizal ITS OTUs at each sampling time point and soil location. On average, 85% of the links in the networks are positive (positive correlation between node relative abundances). There is no discernible pattern of node taxonomy changes over time at the phylum level (Fig. S2f).
FIG 1.

The succession of networks for bacteria and nonmycorrhizal fungi over different plant growth stages in two seasons. Weeks 0, 3, 6, 9, and 12 correspond to preplanted, seedling, vegetative growth, flowering, and senescent stages, respectively. Modules with more than 4 nodes are differentiated by color in each network. Smaller modules are in all gray.
Topological properties of networks between bacteria and nonmycorrhizal fungi over plant growth stages (a to e) and node taxonomic compositions of these networks at the phylum level (f). The linear regression line is shown as a solid line if the effect of time (in weeks after planting) is statistically significant (P ≤ 0.05). Otherwise, it is shown as a dashed line. Download FIG S2, PDF file, 0.3 MB (272.5KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Properties of nonmycorrhizal fungal-bacterial networks and AMF-bacterial networks based on Pearson and Spearman correlations. “−” shows where network topological properties do not apply because the network is empty or the network is too simple. Download Table S1, XLSX file, 0.01 MB (15.4KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
A total of 156 ITS OTUs were identified as Glomeromycota, covering a wide range of the AMF phylogeny. Among the 26 AMF OTUs included in network construction (those with frequencies of ≥10 in 16 replicates), all of which were detected in both rhizosphere and bulk soils, 22 had positive or negative links with bacterial OTUs. Notably, AMF-bacterial links were detected in bulk soils in both seasons but were detected in the rhizosphere only in season 2 (Fig. 2; Table S1). These AMF-based networks are relatively small, with 2 to 51 nodes and 1 to 41 links. Averaged across sampling times and locations, only 4% of the detected AMF OTUs were present in these networks, with averages of 6.3% for the bulk soil and only 1.1% for the rhizosphere. A total of 87% of the AMF network links are positive, and fungi make up 36% of all the nodes, similar to the nonmycorrhizal fungal-bacterial networks.
FIG 2.
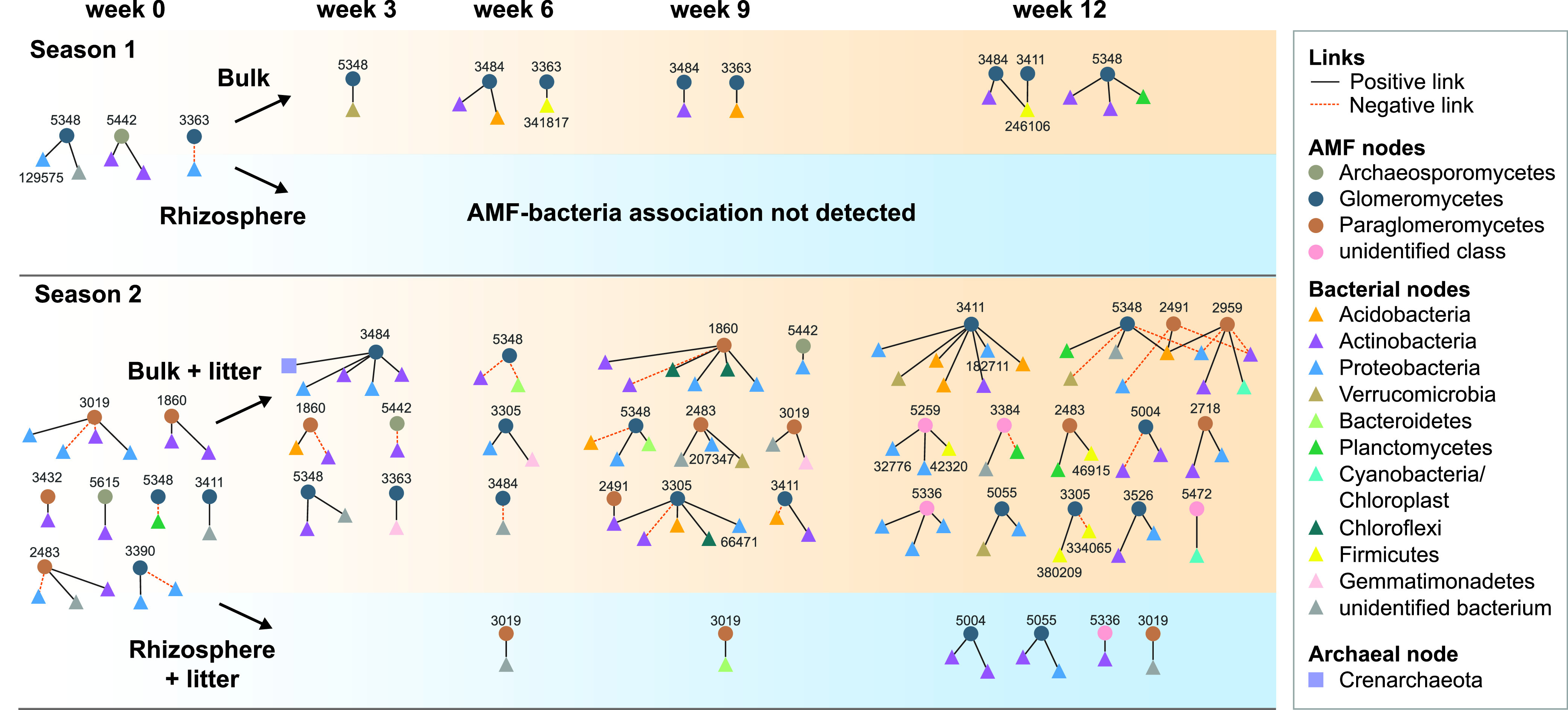
The succession of AMF-bacterial networks over different plant growth stages in two seasons. Node shapes represent AMF (circles), bacteria (triangles), or archaea (squares), and node colors differentiate phyla for bacteria/archaea and classes for AMF. The OTU identifier for each AMF node is marked and corresponds to those in Table S5 in the supplemental material. The bacterial nodes that are mentioned in the text are also marked with their OTU identifiers.
Taxonomy of AMF nodes in AMF-bacterial networks. Download Table S5, XLSX file, 0.01 MB (9.8KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Sampling location shapes network complexity and succession.
Sampling location had a strong impact on network topology, resulting in distinctly structured networks in rhizosphere and bulk soils. Although nonmycorrhizal fungal-bacterial networks (Fig. 1; Tables S1 and S2) have similar numbers of links between rhizosphere and bulk soils, the rhizosphere networks contained 24% fewer (F1,12 = 24.0; P < 0.001) nodes on average. Accordingly, the rhizosphere networks have a 30% higher average degree (F1,12 = 34.0; P < 0.001) and 85% higher connectance (F1,12 = 104.4; P < 0.001) than the bulk soil networks. Rhizosphere network modules are on average 47% larger (F1,12 = 16.72; P = 0.002) than the bulk soil networks. In addition, the proportion of positive links is 13% higher (F1,12 = 35.6; P < 0.001) in the rhizosphere, suggesting more cooccurrence than coexclusion of bacteria and nonmycorrhizal fungi.
Analysis of covariance (ANCOVA) on the topological parameters for the nonmycorrhizal fungal-bacterial networks and AMF-bacterial networks. Download Table S2, XLSX file, 0.01 MB (11.1KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
For the nonmycorrhizal fungal-bacterial networks, the rhizosphere networks illustrate a far stronger successional pattern with plant growth than the bulk soils. Sampling week had a significant effect on the network size (F1,12 = 6.2; P = 0.028), average degree (F1,12 = 10.9; P = 0.006), average module size (F1,12 = 5.99; P = 0.031), connectance (F1,12 = 46.9; P < 0.001), and proportion of positive links (F1,12 = 8.5; P = 0.013) in these networks (Table S2). General linear models (Fig. S2) show that the average degree (adjusted r2 = 0.81; P = 0.024), connectance (r2 = 0.78; P = 0.030), module size (r2 = 0.77; P = 0.031), and proportion of positive links (r2 = 0.72; P = 0.045) increased strongly and significantly over time for the rhizosphere networks in season 1. For the season 2 rhizosphere networks, only connectance showed a significant linear increase with time (r2 = 0.90; P = 0.008). Network size (r2 = 0.67; P = 0.056) decreases with time were marginally significant but with a meaningful effect size. In the rhizosphere networks, the average degree and average module size increased until week 9 but reversed in week 12 (plant senescence) in both seasons, suggesting that plant senescence decreased rhizosphere microbial network complexity. For both seasons, bulk soil networks remained static, with no topological parameter showing any significant change over time (Fig. S2).
Accounting for time points with no significant cooccurrence, the rhizosphere AMF-bacterial networks (Fig. 2; Tables S1 and S2) have on average 71% fewer nodes (F1,12 = 7.7; P = 0.017) and 75% fewer links (F1,12 = 8.1; P = 0.015) than the corresponding bulk soil networks. The differences in AMF-bacterial network size and complexity between rhizosphere and bulk soils are evident in Fig. 2.
For AMF-bacterial networks, due to a lack of data points in the rhizosphere networks, we did not detect any statistically significant change in network topological parameters over time with plant growth (Table S2). However, the bulk soil networks from season 2 (which contained decaying roots from the previous season) were significantly more complex than the season 1 bulk soils, where there was no root detritus. The numbers of nodes and links are also significantly different between these rhizosphere and bulk soil networks, with greater complexity in the bulk soils. In the season 2 bulk soils (which included root litter), we measured AMF-bacterial cooccurrence in the soil prior to A. fatua being planted (23 nodes and 15 links) (Fig. 2; Table S1).
Contrasting patterns of bacterial cooccurrence with AMF versus nonmycorrhizal fungi.
Bacterial cooccurrence with both AMF and nonmycorrhizal fungi was influenced by the presence of roots and plant growth timing but manifested quite differently between the AMF and nonmycorrhizal fungi. First, unlike nonmycorrhizal fungal-bacterial networks, AMF-bacterial cooccurrence was observed frequently and with increased complexity over time in bulk soil relative to rhizosphere soil, suggesting that although AMF are strict biotrophs, they tend to cooccur with bacteria away from roots. Second, growth season had a stronger impact on the cooccurrence of bacteria with AMF than with nonmycorrhizal fungi. There were over 5 times as many nodes (F1,12 = 10.8; P = 0.007) and 6 times as many links (F1,12 = 10.6; P = 0.007) in the AMF-bacterial networks for season 2 compared to season 1 (Fig. 2; Tables S1 and S2). For nonmycorrhizal fungal-bacterial networks, although their sizes were similar in the two seasons (F1,12 = 0.47; P = 0.507) (Table S2), the season 1 networks showed a slightly but significantly higher average degree (F1,12 = 6.6; P = 0.024), connectance (F1,12 = 12.8; P = 0.004), and proportion of positive links (F1,12 = 5.9; P = 0.032) than those in season 2 (Table S2 and Fig. S2).
The complexity of AMF-bacterial networks over time tracked with the increasing richness of the detected AMF taxa, while the complexity of nonmycorrhizal fungal-bacterial networks followed an opposing trend to the declining pattern of nonmycorrhizal fungal richness (Fig. S3). More AMF OTUs were detected in bulk soil than in the rhizosphere; bulk soil AMF OTUs increased throughout the two seasons, regardless of summer dry-down. In the rhizosphere, the richness of nonmycorrhizal fungi decreased with plant growth, yet the network complexity indicated by average degree and connectance increased (Fig. S2). In addition, the summer dry-down brought the richness and level of complexity of the nonmycorrhizal fungal-bacterial networks to levels similar to those at week 0 in season 1.
Richness (a and c) and relative abundances (b and d) of nonmycorrhizal fungi (a and b) and AMF (c and d) over time in bulk and rhizosphere soils. Richness was estimated by the number of OTUs. Relative abundance was estimated as a proportion of the number of sequences in total fungal sequences. There was a 3-month dry-down between week 12 of season 1 and week 0 of season 2. Download FIG S3, PDF file, 0.1 MB (152.1KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Fungi comprise the majority of keystone species, and fungal nodes tend to occur in multiple networks.
We identified keystone nodes (see Materials and Methods for criteria) in the fungal-bacterial networks (containing both nonmycorrhizal fungal-bacterial and AMF-bacterial links). Two to 18 module hubs were identified at each date and location, with a total of 172 module hubs belonging to 128 OTUs (Table S3). Among all the module hubs, 95% (163 out of 172) of the keystone nodes were fungi, mostly Ascomycota, while only 8 were bacteria. In contrast, for the composition of all the network nodes (including keystone nodes and those that were not), only 35% belonged to fungi. No connectors were identified in these networks.
Module hubs and their taxonomy in the fungal-bacterial networks. ITS OTUs that occurred in more than one network are in boldface type. Download Table S3, XLSX file, 0.01 MB (15.8KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
A higher percentage of fungal OTUs than bacterial OTUs occurred in multiple networks. In the 18 nonmycorrhizal fungal-bacterial networks, over 36% of the 541 unique fungal OTUs occurred in more than 9 of these networks (Fig. S4b), while only 0.2% of the 2,825 unique bacterial OTUs occurred in more than half of the networks (Fig. S4a). Similarly, all except one bacterial node in AMF-bacterial networks were present in only a single network (Fig. S4c), while one AMF node (OTU 5348) occurred in 8 of the 13 AMF-bacterial networks (Fig. S4d).
Numbers and proportions of nodes occurring in multiple networks. (a) Bacterial and archaeal nodes in nonmycorrhizal fungal-bacterial networks; (b) fungal nodes in nonmycorrhizal fungal-bacterial networks; (c) bacterial and archaeal nodes in AMF-bacterial networks; (d) AMF nodes in AMF-bacterial networks. On top of each bar, the number of nodes and the proportion are listed. Download FIG S4, PDF file, 0.5 MB (516.4KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Potential mechanisms underlying the observed fungal-bacterial cooccurrence as informed by genome-resolved metagenomics.
In a separate analysis of a subset of these soil samples (49), we resolved multiple bacterial metagenome-assembled genomes (MAGs) from samples collected from weeks 0, 6, and 9 of this study. In five of these MAGs, we discovered the corresponding V4 region of the 16S rRNA gene that matched with high confidence (>99% similarity for at least 200 bp) to the representative sequences of the 16S rRNA OTUs that we used to construct our networks. These bacterial genomes include two Acidobacteria (Acidobacteria_68_21 and Acidobacteria_60_12), one Actinobacteria (Actinobacteria_70_13), and two Alphaproteobacteria (Rhizobium_leguminosarum_59_9 and Mesorhizobium_63_15) genomes. A total of 38 links, including 3 negative and 35 positive ones, connected bacterial nodes matching these genomes to fungal nodes. The linked bacterial and fungal nodes belonged to 8 bacterial OTUs and 31 fungal OTUs. Half of these links were in rhizosphere networks, and the other half were in bulk soil networks (Fig. 3; Table S4).
FIG 3.
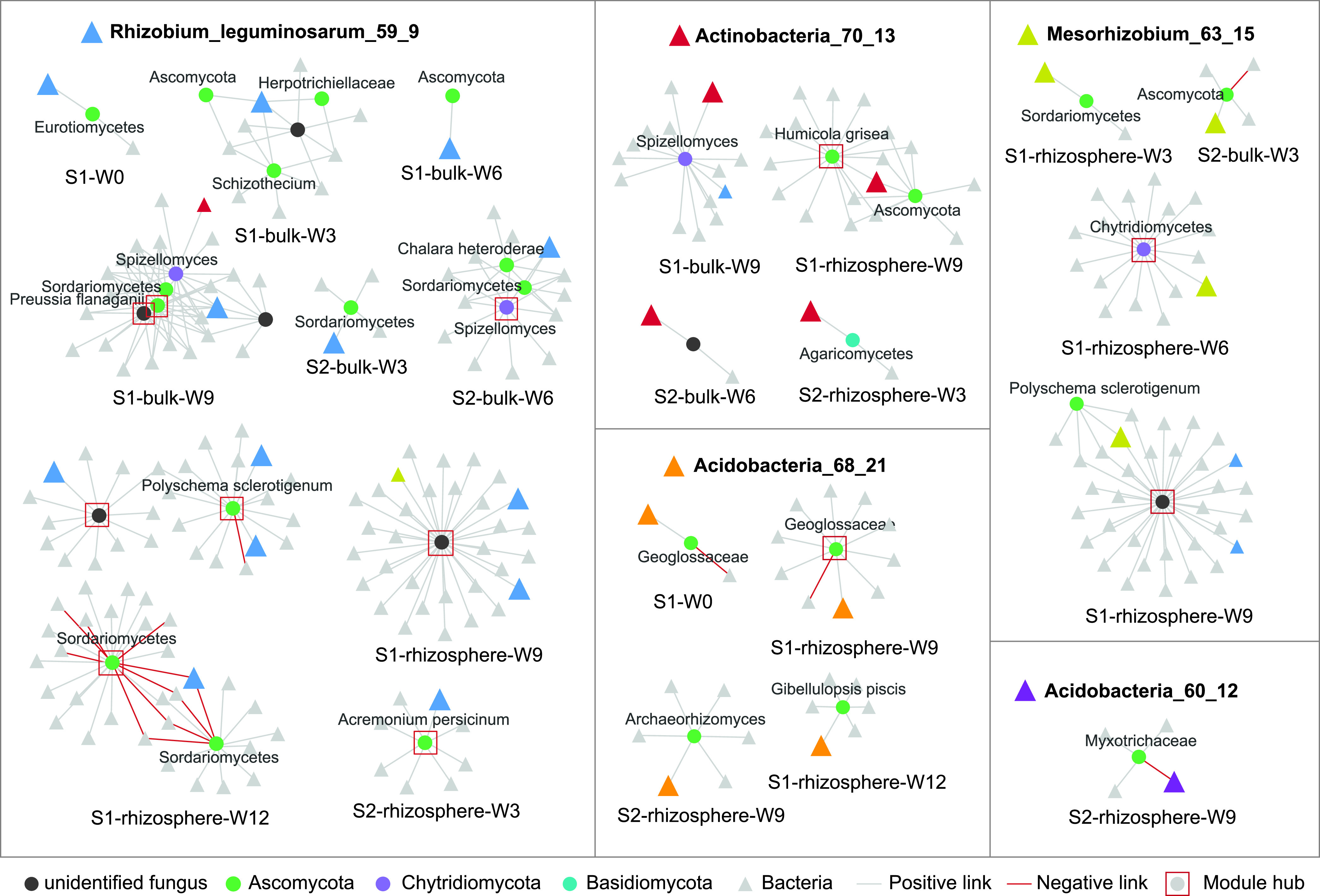
Subnetworks of the eight nodes matched to five metagenome-assembled bacterial genomes. Each frame contains one genome-matching bacterial node, fungal nodes directly linked to it, and its 2nd neighbors in the networks. The name of the genome is shown at the top within each frame, and the genome-matching nodes are represented by colored triangles in the networks. Fungal nodes directly linked to the genome-matching bacterial nodes are annotated and colored based on phylum.
Links connecting genome-resolved bacterial genomes and their fungal associates. Download Table S4, XLSX file, 0.01 MB (11.3KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The five MAGs corresponding to nodes in our networks harbored a number of genes that may have been involved in interorganismal interaction pathways (Table 1).
Based on their phylogeny and predicted ability to modulate plant hormones, Rhizobium_leguminosarum_59_9 and Mesorhizobium_63_15 may be plant symbionts. These two taxa were far more abundant in the rhizosphere than in bulk soils (Fig. S5a and b) and were also highly 13C enriched in the stable-isotope probing (SIP) analysis of the same samples (49). The 16S OTUs matching Rhizobium_leguminosarum_59_9 formed 21 positive and 2 negative links with nodes of Ascomycota, Chytridiomycota, and a few unclassified species in networks constructed for different plant growth stages. The OTU matching Mesorhizobium_63_15 formed five positive links with fungi (Table S4).
Acidobacteria_68_21 could be a bulk soil specialist based on its high relative abundance in bulk soil samples (Fig. S5c) and the fact that it was not labeled by plant-derived 13C. It harbors many defensive capabilities by carrying a large arsenal of biosynthetic gene clusters, a type IV secretion system, CRISPR-Cas systems, and, notably, insecticidal proteins, which could be used in defense or offense against other bacteria, nematodes, bacteriophages, and fungi. Its central metabolism is highly flexible, and its genome harbors a wide array of carbohydrate-active enzyme (CAZy) genes targeting different substrates. This MAG was the most abundant organism identified in the bulk soil in that related study (49). The OTU matching this genome had four positive links with fungal nodes (Table S4).
The bacterial node matching Acidobacteria_60_12 formed only one negative link, with a Myxotrichaceae node (Table S4). Acidobacteria_60_12 exhibits antifungal potential by carrying 18 CAZy genes targeting fungal biomass compounds (49, 51); a number of biosynthetic gene clusters, including one predicted to biosynthesize a phenazine-like compound, a documented antifungal compound (52); and antibiotic resistance modules and reactive oxygen species (ROS) protectants that could provide defense against fungi (49).
Finally, the Actinobacteria_70_13 genome carried a large number of CAZy CE14 genes, a chitin deacetylase, and some large genes likely used in adhesion to biological material (Table S4). The nodes matching this genome had five positive links with fungal nodes (Fig. 3).
TABLE 1.
Information regarding the genome-matching network nodesa
| Genome (matching network node[s]) |
Relevant gene(s)/pathway(s) |
13C enrichment of genome in the rhizosphere compared to bulk |
Atom% excess at wk 6 |
Atom% excess at wk 9 |
|---|---|---|---|---|
|
Rhizobium_leguminosarum_59_9 (OTUs 175932, 61972, 42846, 64784) |
Genes involved in modulating plant hormones | Yes | 58 | 39.2 |
|
Mesorhizobium_63_15 (OTU 13989) |
Genes involved in modulating plant hormones | Yes | 55.1 | 43.5 |
| Type VI secretion system | ||||
|
Acidobacteria_68_21 (OTU 2124) |
BGCs | No | 0 | 0 |
| Type IV secretion system | ||||
| CRISPR-Cas system | ||||
| Insecticidal proteins | ||||
| Wide array of CAZy genes targeting different substrates | ||||
| Highly versatile metabolism | ||||
|
Acidobacteria_60_12 (OTU 7765) |
18 CAZy genes targeting fungal biomass compounds | Yes | 42.5 | 29.9 |
| BGCs | ||||
| Antibiotic resistance modules | ||||
| Reactive oxygen species protectants | ||||
|
Actinobacteria_70_13 (OTU 2178) |
Chitin deacetylase | Yes | 62.4 | 24.4 |
| Genes likely used in adhesion to biological material | ||||
Data from reference 49. BGCs, biosynthetic gene clusters.
Relative abundances of the 16S OTUs matching the metagenome-resolved bacterial genomes. Bars represent averaged abundances of the OTUs across biological replicates in different locations, seasons, and weeks. Error bars show standard errors of the means (n = 16). Panels a to e separately show the abundances of the OTUs matching the genomes Rhizobium_leguminosarum_59_9, Mesorhizobium_63_15, Acidobacteria_68_21, Acidobacteria_60_12, and Actinobacteria_70_13, respectively. Download FIG S5, PDF file, 0.3 MB (347.3KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
DISCUSSION
By investigating fungal-bacterial networks in a native grassland soil, we found that different soil niches had distinctly different cooccurrence patterns for bacteria and AMF versus bacteria with nonmycorrhizal fungi. AMF were more frequently linked to bacteria in soils >2 to 4 mm from a root and in the presence of the previous season’s root litter, while nonmycorrhizal fungi formed more complex networks with bacteria in the rhizosphere. Cooccurrence of species can result from combined effects of environmental filtering related to niche sharing, biotic interactions, as well as dispersal limitation (53). However, it is likely that dispersal limitation is a less important driver for cooccurrence in our experiment because the soil was sieved and homogenized before planting. Our results indicate that distinct niches partitioned by the availability and forms of C substrates are likely fundamental drivers of how microorganisms of different life histories and ecologies cooccur and potentially interact in complex soil environments.
Cooccurrence between AMF and bacteria.
Known to be strictly biotrophic organisms that live on carbon derived from living roots, AMF had both higher richness and relative abundance in soils outside the rhizosphere, especially in the presence of root litter in our second growing season. This apparent contradiction may be explained by AMF’s life form in soils. First, as roots take up nutrients, a depletion zone is formed in their immediate vicinity (1 to 3 mm) (54–56). After colonizing roots, AMF are driven to extend the majority of their hyphae beyond this root-adjacent depletion zone to reach for nutrients that roots cannot access. In fact, it has been shown that AMF greatly extend the phosphorus depletion zone around clover roots (57, 58), demonstrating their search for nutrients away from roots; this could lead to a higher detectable AMF relative abundance in nonrhizosphere soils. Second, AMF spores are formed on hyphae outside roots, and previous evidence shows that the nucleic acid concentration is much higher in fungal spores than in vegetative hyphae (59–64). Thus, a combination of spores and hyphae may lead to the detection of more AMF using DNA-based molecular markers in nonrhizosphere soils than near roots. Finally, the second season’s root litter (derived from the first season’s root growth) likely included some AMF-colonized roots. These may have served as inocula, leading to AMF’s quick propagation in the second season and increased our ability to detect their presence. This explains the progressive increase of AMF richness and relative abundance over the two growth seasons despite the intervening summer dry-down as well as the high AMF diversity that we measured at week 0 in season 2, before seedlings had been planted. Our observations are consistent with another study that reports slightly higher richness of AMF in bulk soil than in the rhizosphere (65) in a sorghum cultivar, indicating that this pattern may persist across different plant species and soil types. Since only relative abundance is available for AMF versus nonmycorrhizal fungi in molecular marker-based studies, further tests such as hyphal and spore counts could be helpful to confirm the absolute abundance of AMF in rhizosphere versus bulk soil.
In our rhizosphere networks, a low AMF relative abundance, as mentioned above, together with a lack of common life history traits between AMF and bacteria, could have caused the underrepresentation of AMF-bacterial cooccurrence. Rhizodeposits, characterized by abundant low-molecular-weight C substrates, drive the formation of a rhizosphere niche shared by taxa that actively consume these substrates (2, 66). Bacteria in the rhizosphere form complex networks with each other because of their shared niche space (47). AMF, however, depend on C provided from inside the root rather than external rhizodeposits (67), representing a closer association with plants that is fundamentally different from the relationship between plant and rhizosphere bacterial communities. Also, the processes by which AMF sense and actively colonize roots (67) differ from the recruitment of rhizodeposit-consuming microbial communities (68), although AMF’s functional association with plants may overlap those of certain plant-growth-promoting rhizobacteria (PGPR), such as providing nutrients and alleviating drought stress (23, 69, 70). AMF perform these functions by transporting nutrients and water directly to internal root structures via hyphae (100), whereas PGPR function in soil outside roots to free nutrients as well as in the endodermis to regulate plant metabolites (70). In summary, although dependent on root C, AMF occupy a distinct ecological and spatial niche from rhizodeposit-dependent microorganisms.
Our detection of common AMF-bacterial cooccurrence in the detritusphere could also potentially result from functional associations between the two groups. AMF have no known saprotrophic capabilities but appear to be able to alter the rates of litter decomposition through their effects on saprotrophic organisms (7, 23). AMF exudates can provide C to bacteria in the detritusphere and promote bacterial litter decomposition. AMF then take up nutrients freed during decomposition. For example, Glomus geosporum and Glomus constrictum hyphae have been associated with bacteria of the genera Cellvibrio, Chondromyces, Flexibacter, Lysobacter, and Pseudomonas, which are known biopolymer degraders (71). Also, the phosphate-solubilizing bacterium Rahnella aquatilis can be induced to mineralize phytate when grown with the fructose-exuding AMF Rhizophagus irregularis (72). In our networks, we found AMF nodes linked to bacteria from the same orders (Myxococcales, Sphingobacteriales, and Xanthomonadales) that include the above-mentioned biopolymer degraders, although the genera of these nodes are unidentified. Material exchanges between AMF and bacteria in soil, which can foster cooccurrence, tend to be more active at extending hyphal tips, which reach far beyond the rhizosphere (67). In a separate study, using the same soil planted with Avena barbata, we found that the AMF Rhizophagus intraradices transferred a significant amount of recent photosynthetic C across a 3-mm air gap to a separate soil-containing hyphal chamber (A. Kakouridis, M. Yuan, J. A. Hagen, C. A. Fossum, M. L. Moore, D. J. Herman, P. Nico, P. Weber, J. Pett-Ridge, and M. K. Firestone, unpublished data), possibly releasing some of this carbon as hyphal exudates. Based on AMF transport of C to growing tips and where AMF and bacteria more frequently cooccur, we hypothesize that AMF-bacterial associations are most frequent at growing hyphal tips some distance from roots and that AMF hyphae can serve as connections between microbial activity hot spots in rhizosphere and detritusphere soils (73). This hypothesis may represent a critical characteristic of the hyphosphere and is worth further research. In addition, AMF spores are known to harbor intracellular bacterial microbiomes with two main groups of bacteria, Burkholderia-related Betaproteobacteria (74) and Mycoplasma-related Mollicutes (75). In our data, we found bacteria of the order Burkholderiales in networks linking to AMF OTUs, although further genetic information or culture experiments would be required to confirm these relationships in our system. Together, these observations imply that biological processes in the detritusphere could be important drivers of AMF-bacterial cooccurrence.
Cooccurrence between nonmycorrhizal fungi and bacteria.
The highly complex cooccurrence network of nonmycorrhizal fungi with bacteria in the rhizosphere could result in large part from niche sharing, a common occurrence among bacterial networks (47). As is typical in natural soil (76–78), the majority of nonmycorrhizal fungi detected in this study were either saprotrophic fungi or taxa that have saprotrophic capabilities during part of their life cycle or under certain abiotic conditions. The availability of organic C is a major driver of the distribution of both bacteria and saprotrophic fungi in soil. The C-rich rhizosphere could foster niche sharing between rhizodeposit-dependent bacteria and a diverse range of fungi that actively assimilate root exudates (79), debris, and litter, resulting in a pattern of cooccurrence between the two. Organic C availability in bulk soil, on the other hand, tends to be more limited; thus, cooccurrence is less likely to happen than in the rhizosphere. In our second growing season, the presence of litter coincided with a slight increase in the complexity of cooccurrence between nonmycorrhizal fungi and bacteria, possibly because of added amounts of C substrates. But the increase in network complexity with the presence of litter is less obvious than the large and obvious difference in network structures in rhizosphere versus bulk soil, implying that rhizosphere environmental filtering is the main driver of nonmycorrhizal fungal-bacterial cooccurrence.
Given the importance of the rhizosphere environment in driving the cooccurrence of nonmycorrhizal fungi with bacteria, biochemical changes in the rhizosphere associated with plant senescence are likely the cause of the consistent decrease in network complexity at week 12. Approaching senescence, Avena roots exude less, and the composition of exudates changes from sugars and amino acids to stress- and senescence-related molecules (66, 80). Soon thereafter, roots gradually become litter. The resulting altered substrate type can vastly change the environment and microbial assemblage. Root exudates during vegetative growth, like sugars and amino acids, are consumed by versatile microbial taxa, which may promote niche sharing and the cooccurrence of microorganisms; the decline of these microbially preferred exudates and the proportional increase of lignocellulose-rich root litter substrates could become unsuitable for exudate-dependent bacteria and fungi and dissipate shared niches.
Evidence from our nearly complete bacterial genomes, assembled from metagenomes derived from the same experiment, supports the hypothesis that some positive fungal-bacterial links may be explained by shared ecological niches. We traced the movement of plant-derived 13C into rhizosphere organisms’ genomes; the Rhizobium_leguminosarum_59_9 MAG became highly labeled, with 58 atom% excess 13C (49). This high level of isotope enrichment, in addition to plant symbiosis genetic systems present in the genome, suggests that the corresponding bacteria likely live in close association with the plant and actively take up exudates, thus representing an organism adapted to the rhizosphere. Here, we found that network nodes matching Rhizobium_leguminosarum_59_9 frequently cooccurred with phylogenetically diverse fungal nodes, including several module hubs. Many of the correlating fungal nodes, for example, Chalara heteroderae (81), Acremonium (82), Spizellomycetaceae (83), and Herpotrichiellaceae (83), come from clades with known plant pathogens or pathogens of plant pathogens that interact with plants frequently in the rhizosphere. The large number of fungal nodes that correlated with Rhizobium_leguminosarum_59_9 may represent a shared niche in the rhizosphere. In contrast, Acidobacteria_68_21 is likely a generalist adapted to living in bulk soil and decomposing a wide variety of compounds while protecting its own accumulated biomass against attack from other bacteria, microeukaryotes, and fungi. Based on the bacterium’s large metabolic potential, it may live in microhabitats devoid of any reliable C substrate. The fungi linked to Acidobacteria_68_21 may inhabit the same niche and may also be specialized for low-resource microhabitats.
Some other functional potentials harbored by the bacterial genomes that we have sequenced from this soil suggest that some of the fungal-bacterial links involve bacteria with the capacity to break down and consume fungal biomass as well as potential defense mechanisms. Acidobacteria_60_12 was one of the two genomes with a negative link to a fungus. This organism may be inhibitory toward soil fungi; its genome has a large number of antibiotic resistance modules and reactive oxygen species (ROS) protectants (which could shelter the bacterium from fungal defenses) and encodes the production of antifungals that may inhibit or kill organisms, including fungi. The large number of fungal biomass decomposition genes, including chitinases and glucanases (49, 51), suggests that this bacterium is involved in fungal carbon recycling, but it is unknown whether it actively parasitizes fungal hyphae.
Due to the harbored adhesin genes, Actinobacteria_70_13 may live physically attached to biological material, although whether the material is living or dead is unclear. It is also likely able to decompose fungal biomass based on its repertoire of CAZy genes that target fungal biopolymers (49, 51). Biomass decomposition is known to be a common process in the soil, but we are unable to draw further conclusions from genomic data.
The hyphosphere may be an important niche of fungal-bacterial cooccurrence in soil.
In heterogeneous soil habitats, the spatial scale over which organisms interact is an important factor that determines how communities assemble (Fig. 4a). This is reflected in the disproportionately higher number of fungal module hubs and the high frequency of occurrence of the same fungal nodes in multiple networks. Since fungal hyphae are much larger than individual bacterial cells, a single hypha or large AMF spores can occupy and influence a larger space and provide surface contact for a diverse group of bacterial cells, forming a hyphosphere in which diverse microbial processes can happen (84, 85). Physically attached hyphae and bacterial cells have been captured previously (74, 86, 87) and in a parallel study (Fig. 4b) (Kakouridis et al., unpublished). Our network analyses may have captured some of these hyphosphere “hot spots” where bacteria physically gather around fungal hyphae, with the fungi acting as module hubs, linking to a range of bacteria. In fact, different modular compartments may reflect hot spots of biotic interactions (88, 89), such as a leaky hyphal tip surrounded by bacteria or a particle of decomposing biomass. Along with the rhizosphere and detritusphere, the hyphosphere could represent a unique soil niche, especially in the context of fungal-bacterial interaction. The cooccurrence patterns between fungi and bacteria observed in this study imply that the hyphosphere could harbor processes involving cross-kingdom biotic interactions, whose importance to soil functioning is a currently murky area that demands future study.
FIG 4.

Illustration of the distribution of fungi in different soil niches and how such a distribution could foster fungal-bacterial cooccurrence. (a) Conceptual illustration showing the spatial heterogeneity of the soil environment around a root and how such heterogeneity affects bacterial-fungal cooccurrence. (1) Soil close to the root, representing the rhizosphere niche, is C rich because of root rhizodeposits but could be nutrient depleted due to plant uptake of N, P, and other essential elements. A wide range of nonmycorrhizal fungi that directly assimilate plant-derived C share this niche with many rhizosphere-adapted bacteria. AMF get C by colonizing the root itself and extend beyond the nutrient depletion zone to search for nutrients, showing a low relative abundance and a lack of niche sharing with rhizosphere bacteria. (2) Litter could serve as an AMF inoculum, and the detritusphere could be where AMF-exuded C primes bacterial decomposition of litter, possibly leading to cooccurrence between AMF and bacteria. (3) The higher relative abundance of AMF in bulk soil extends the space around the root where fresh photosynthetic C can be delivered and paves the way for the creation of a hyphosphere niche for bacteria living off of hyphal exudates. Nonmycorrhizal fungi also cooccur and interact with bacteria but more frequently in the rhizosphere, where substrates are abundant and microbial activity is higher. (4) Symbiosis with AMF spores in bulk soil. (5) Pathogenesis with bacteria consuming live hyphae. (6) Bacteria consuming dead fungal biomass. (b) Bacterial cells attached to the branching hyphae of Rhizophagus intraradices associated with A. barbata roots in a similar greenhouse experiment by our team, captured by scanning electron microscopy.
In summary, by constructing cooccurrence networks of soil bacteria and fungi associated with the growth of an annual grass, we found that plant growth greatly impacted the development of network structures over time, and we observed different cooccurrence patterns of AMF with bacteria versus nonmycorrhizal fungi with bacteria. Different ecological niches in the complex soil environment are likely major drivers of these cooccurrence patterns. In the rhizosphere, the complex and evolving networks between nonmycorrhizal fungi, mainly saprotrophic, and bacteria are probably the result of niche sharing driven by C availability. The rarity of AMF-bacterial cooccurrence in rhizosphere soil resulted from both a low relative abundance of AMF as well as a lack of common life history and potential niche requirements between AMF and rhizosphere-adapted bacteria. Instead, AMF and bacteria were more frequently linked in bulk soil, especially when it contained root litter, leading us to hypothesize that ecological functions involving the participation of both AMF and bacteria could be important in the detritusphere away from roots. SIP-enabled metagenomic analyses suggest that some links potentially involve bacterial symbiosis with eukaryotes, potentially plants or fungi, and their ability to break down fungal biomass, while others are more likely explained by the sharing of ecological niches.
It is possible that actual interactions caused no detectable covariation in the abundances of the partners at the spatial and temporal scales of our sampling regime, posing a strong argument to study biotic interactions using techniques beyond macro/mesoscale correlations in abundance. However, the mesoscale sampling (rhizosphere, bulk, and hyphosphere) applied in this study may be consistent with the scale of fungal niches.
Although caution needs to be used when interpreting molecular marker-based, reconstructed correlation networks (39, 40), the networks from our study highlight the unique characteristics of different soil niches in driving the cooccurrence of soil microorganisms and can serve as a useful first step in the exploration of potential associations within soil microbial communities.
MATERIALS AND METHODS
Greenhouse experiment setup and sampling.
The design and setup of the greenhouse experiment have been described previously (48) and are diagramed in Fig. S1a in the supplemental material. In brief, microcosms (11.5 by 2.9 by 25.5 cm) were packed with sieved (<2 mm) surface (0 to 10 cm) soils collected from the Little Buck watershed at the University of California’s Hopland Research and Extension Center, where the dominant type of vegetation was Avena species, a common type of Californian annual grass. Avena fatua was grown in these microcosms with controlled temperature, water, day length, and atmospheric CO2 concentration for two consecutive growing seasons in the Oxford Tract Greenhouse at the University of California, Berkeley. In each growth season, both rhizosphere and bulk soils were sampled at five time points, tracking the growth stages of Avena fatua at preplant (week 0), seedling (week 3), vegetative growth (week 6), flowering (week 9), and senescence (week 12). At the end of the first growing season, senescent shoots were cut and removed, and the dead roots were left with the soil inside the microcosms for a 3-month dry period, mimicking California’s summer drought, before the second season started and water was supplied again. Thus, dead roots from the first season remained in the microcosms at the beginning of the second season as litter. The rhizosphere soil was sampled as the soil and aggregates that were still attached to roots after gentle shaking. The bulk soil in the first season was collected from inside a 1-mm-mesh bag that was placed in every microcosm when packing, which contained sieved soils and was designed to prevent root penetration. In the second growing season, no “pure” bulk soil existed. After roots with rhizosphere soil were removed, the remaining soil in the microcosm was referred to as “bulk” soil but with the presence of litter. Thus, the rhizosphere and bulk soils in the two seasons represent four types of soil environment: rhizosphere, bulk soil, rhizosphere plus detritusphere, and detritusphere. About 1 to 2 g of soil was obtained per rhizosphere sample, and 5 g of soil was obtained per bulk soil sample. A total of 16 biological replicates were available for each of the combinations of growth season, growth stage, and soil location, resulting in 288 soil samples for analysis.
Diagram of the experimental design (a) and rarefaction curves of the sequences obtained from soil samples (b). In panel a, each microcosm shown represents one sampling time point, including 16 replicate microcosms (CO2 effect not detectable on the overall soil bacterial and fungal community structure [see Materials and Methods for details]). In panel b, the vertical line indicates the number of sequences (4,484) for rarefaction for each sample. Download FIG S1, PDF file, 1.4 MB (1.4MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Bacterial and fungal community analyses. (i) DNA extraction, library preparation, and sequencing.
Soil total DNA was extracted in a cetyltrimethylammonium bromide (CTAB) buffer using a phenol-chloroform purification protocol and checked for quality using PicoGreen. Bacterial and archaeal communities were surveyed using amplicon sequencing of the V4 region of 16S rRNA genes, amplified using the primer set 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). Fungal communities were investigated by sequencing the amplicons of the fungal ITS2 region using the primer set gITS7F (5′-GTGARTCATCGARTCTTTG-3′) and ITS4R (5′-TCCTCCGCTTATTGATATGC-3′). Amplification, library preparation, and sequencing were performed on an Illumina MiSeq 2.0 platform in 250-by-2 paired-end format at the Institute for Environmental Genomics, University of Oklahoma.
(ii) Sequence processing.
The data processing of 16S raw sequences was reported in detail previously (48). We inherited the use of operational taxonomic units (OTUs) from this published paper instead of using the higher-resolution amplicon sequencing variants (ASVs) so that the 16S nodes are comparable to the prokaryotic networks that have been discussed previously (48). The ITS raw sequences were processed through the pipeline on the Galaxy platform developed by the Institute for Environmental Genomics, University of Oklahoma. First, primers were trimmed with zero mismatch tolerance, followed by Btrim (90) to remove sequences with quality scores of <25 (window size of 5) or lengths of <60 bp. Next, forward and reverse sequences were combined using Flash (91), with a minimum required overlap of 10 bp and a maximum expected overlap length of 250 bp. The maximum allowed ratio of mismatch was set at 0.05 in the overlapping region. Afterward, the combined sequences were further cleaned by removing sequences containing ambiguous base N, deleting sequences with lengths of <100 bp or >500 bp, and deleting chimeras detected using UCHIME (92) with the UNITE (93) representative/reference sequences as the reference database. The remaining sequences were clustered into OTUs by UCLUST (94) at a similarity threshold of 0.97. The longest sequence in each OTU was chosen as the representative sequence of the OTU.
(iii) Plant sequence removal.
To identify and remove plant sequences, all the representative sequences were checked using BLASTN (95), nonfungal sequences (unclassified kingdom, plants, and animals) were discarded, and the remaining ITS representative sequences were all fungal sequences. After the removal of nonfungal sequences, the samples from bulk soil had much higher sequence numbers than those from the rhizosphere due to their close association with plant tissue, a known problem with ITS4 primers. Thus, the OTU table was rarified to 4,484 sequences/sample for all the samples to bring even the depth of fungal sequences. A total of 4,936 OTUs were obtained (Fig. S1b).
(iv) Fungal taxonomic assignment.
To assign taxonomic information to each OTU, we used the Training Feature Classifier in QIIME2 (96), trained a classifier with the UNITE database (93) v8 (2 February 2019), and classified the fungal ITS representative sequences afterward. One caveat of this classification method is that the confidence score of classification is given to each sequence only at the species level, while higher taxonomic information could be useful in identifying the ecological guild of the fungal taxa (e.g., AMF or nonmycorrhizal). We refined the output taxonomy from the above-described method using the following criteria. First, we accepted species-level assignment with a confidence score of ≥0.7. Second, we performed a BLAST comparison of our representative sequences to the UNITE v8 database (used to train the classifier) to acquire all the hits with ≥70% identity to each representative sequence. Third, for representative sequences with a species-level confidence score of ≥0.3, if more than 80% of the BLAST hits had the same phylum assignment with the classifier result, their phylum assignments were accepted. Fourth, for representative sequences with a species-level confidence score of ≥0.5, if more than 80% of the BLAST hits had the same class assignment with the classifier result, their phylum assignments were accepted. Taxonomic assignments that do not meet the above-described criteria are reported as “unidentified.” The refined results were further checked by (i) randomly picking representative sequences and performing a BLAST search against the NCBI database, which showed a high confidence of taxonomic assignment, and (ii) manually performing a BLAST search of all the Glomeromycota sequences against the NCBI database, which confirmed the assignments for all AMF sequences. In this way, 79% of the 4,936 OTUs were assigned to a phylum, 47% were assigned to a class, 28% were assigned to a genus, and 22% were assigned to a species.
(v) Fungal ecological guild.
To assess the ecological guilds of the fungal nodes in the networks, we applied FUNGuild (76) to the taxonomy assignments for the 563 ITS OTUs. A total of 30.9% (174) of these OTUs were assigned guilds at the confidence levels of “highly probable” and “probable.” Among them, 52.3% (91 OTUs) belonged to saprotrophs, 20.1% (35 OTUs) were symbiotrophs (including 22 AMF and 13 endophytes), 13.8% (24 OTUs) were pathogens, and the other 13.8% (24 OTUs) had combined guilds. A total of 65.5% (114) of these OTUs have saprotrophic capability. In addition, 62 OTUs were assigned guilds at the confidence level of “possible,” and 97% (60) of them have saprotrophic capability.
Cooccurrence bipartite fungal-bacterial network construction using RMT-based correlation cutoffs. (i) Network correlation cutoff.
We used the 16 biological replicates to construct a fungal-bacterial cooccurrence bipartite network for each sampling time point for either bulk or rhizosphere soil. To do this, we first combined the 16S and ITS OTU tables to represent one community and discarded the OTUs with <10 occurrences in 16 replicates to ensure the accuracy of the data association calculation. A total of 1,645 to 2,467 OTUs were included in the construction of each network. Among them, 84 to 90% were 16S OTUs, and 10 to 16% were ITS OTUs. Next, pairwise correlations of all the OTUs were calculated based on the Pearson correlation of their relative abundances in the 16 replicates. To determine the correlation cutoff for the network link, we applied a random matrix theory (RMT)-based approach (45, 50). The RMT method detects the transition of the correlation matrix from a noisy system to an organized nonrandom system as the correlation cutoff increases. The correlation cutoff detected ranged from 0.756 to 0.793 for the 18 networks. Pairs of nodes with a correlation above the cutoff are considered linked in the network. Positive correlations indicate positive links, and vice versa. To ensure that the constructed networks are not sensitive to correlation methods, we also used Spearman correlation to construct parallel networks, whose topology showed a trend similar to those from Pearson correlations (Table S1). In this study, we present only networks constructed based on Pearson correlation. The RMT-based correlation cutoff detection procedure is available through the Molecular Ecological Network Analysis Pipeline (MENAP) (http://ieg4.rccc.ou.edu/MENA/).
(ii) Bipartite subnetworks.
The 18 networks contained links of bacteria/archaea-bacteria/archaea, fungi-fungi, and fungi-bacteria/archaea. They were scale free, with the degree distribution following power law distributions with R2 values from 0.83 to 0.94. These networks had 1,096 to 2,020 nodes, with 1,688 to 3,627 links, and average degrees from 2.27 to 5.93. To specifically target fungal-bacterial/archaeal cross-kingdom cooccurrence, subnetworks were extracted to build bipartite networks that contain subsets of nodes and links specifically connecting fungi and bacteria/archaea. We observed only 14 archaeal nodes with 22 links to fungi across the 18 networks. Since most links are between fungi and bacteria, we refer to these bipartite networks as “fungal-bacterial” networks here. A total of 16% to 28% of the links in the full networks were between a bacterial/archaeal node and a fungal node and consist of the fungal-bacterial networks. A total of 29% to 44% of the nodes in the full networks remained in the bacterial-fungal networks.
(iii) AMF-bacterial versus nonmycorrhizal fungal-bacterial networks.
Mycorrhizal and nonmycorrhizal fungi represent different life strategies and ecological guilds, which are particularly important to differentiate in environments such as those near plant roots. Thus, we separated the fungal-bacterial networks into AMF-bacterial and nonmycorrhizal fungal-bacterial networks. AMF-bacterial networks contain all the AMF nodes and bacterial nodes linked to them; nonmycorrhizal fungal-bacterial networks contain all the non-AMF fungal nodes with bacterial nodes linked to them.
(iv) Spearman correlation for network construction.
In order to explore whether different correlation methods affect network construction, we also used Spearman correlation to construct networks. We show the topological properties of Spearman correlation-based fungal-bacterial networks in Table S1 to demonstrate that the successional trend of networks derived from Pearson and Spearman correlations over time and space are consistent. All the detailed analyses of nonmycorrhizal fungal-bacterial and AMF-bacterial networks are based on Pearson correlation.
(v) Network topological index calculation.
The following topological properties of the fungal-bacterial networks were analyzed using the R packages igraph (97) and bipartite (98).
Number of nodes and links. Each node represents an ITS or 16S OTU, whose relative abundance covaries with at least one other OTU at a correlation coefficient above the RMT-detected cutoff threshold. Each link represents one pairwise correlation between two nodes. The total node number (n) and link number (L) were calculated using the functions gorder and ecount.
R2 of the power law, which describes how well the distribution of node degrees (the number of links connected to each node) fits the power law model. A network possesses a scale-free feature if its degrees follow a power law distribution. The R script is available upon request. The nonmycorrhizal fungal-bacterial networks and AMF-bacterial networks constructed in this study are scale free, as the R2 of the power law model fit ranges from 0.89 to 1.
Average degree (avgK), the average number of links per node, equal to 2L/n. This was calculated using the degree function.
Average path length or geodesic distance (GD), the average length of the shortest path between nodes. This was calculated using the mean_distance function.
Number of modules and modularity. Nodes in a network can be separated into different components, within which nodes are closely linked and among which links are much looser. The fast greedy algorithm is used for module separation in this study. The number of modules indicates the modules that the nodes are separated into. Modularity is calculated based on Newman’s method to indicate how well a network is able to be separated into modules. Values are between 0 and 1, with 0 being not able to be separated at all and 1 being easily separated into components. These were calculated using the cluster_fast_greedy and modularity functions. The nonmycorrhizal fungal-bacterial networks are highly modular, with modularity ranging from 0.78 to 0.99. The 10 AMF-bacterial networks with >2 nodes all have modularity above 0.44 and show a basic modular structure with disconnected subcomponents.
Numbers and proportions of positive and negative links. Based on the sign of the Pearson correlation coefficient, a link is either positive or negative, indicating the pattern of covariation in the relative abundances of the two linked species.
Connectance, or graph density, the realized proportion of all possible links. For bipartite networks in this study, all possible links include only those between a bacterial node and a fungal node. Connectance and the following indices were all calculated using the network-level function.
(vi) Keystone node identification.
Based on network topology, nodes that are important in maintaining the network structure are identified as keystone nodes, including module hubs and connectors, as their removal could cause dramatic changes in the cooccurrence patterns of the involved organisms. Operationally, for modular networks (i.e., networks separable into closely interconnected subcomponents called modules), nodes with high within-module connectivity (zi ≥ 2.5 [highly connected to other nodes within the same module]) are identified as module hubs, while those with high among-module connectivity (pi ≥ 0.62 [tend to connect nodes in different modules]) are identified as connectors (89).
(vii) Random network generation.
To confirm that the observed network topology represents a nonrandom assembly of microorganisms, 100 random networks were generated by rewiring the links among the nodes and compared with the empirical ones for the 18 bipartite fungal-bacterial networks. A random network is generated according to the steps described below. First, for an empirical network with m bacterial nodes, n fungal nodes, and l total links, max(m,n) links were randomly placed between bacterial and fungal nodes so that no isolated node (node with zero degrees) exists. Second, the remaining links [l − max(m,n)] are randomly placed between bacterial and fungal nodes. In this way, m, n, and l are preserved, and the random networks are all bipartite. The average degree and connectance are thus preserved between the random and empirical networks. We calculated the means and standard deviations of the R2 power law and modularity for the 100 random networks. The R2 power law of the empirical networks was much higher than that of the random networks, and the empirical networks had much higher modularity than the random ones. The distinct topologies of empirical and random networks imply that the empirical networks that we constructed are not likely due to random wiring and can reflect microbial community assembly.
Metagenomic sequencing, bacterial genome resolving, and genome matching to network nodes.
For the same greenhouse experiment (48), the soil DNA was sequenced for metagenomes and analyzed in a separate study to construct metagenome-assembled genomes (MAGs) (49). Briefly, a total of 55 partial bacterial MAGs with >70% completeness and <10% contamination were reconstructed from these sequences, and high-quality 16S rRNA sequences for 13 of these MAGs were obtained (49). In order to match these MAGs to the network nodes, we performed a BLAST search of our representative sequences (16S V4 region) of the bacterial nodes in the network against the 14 high-quality 16S sequences obtained from the MAGs. Five MAGs were found matching with 9 bacterial nodes with >99% similarity of the 16S V4 regions for a length of at least 200 bp.
Statistical analysis.
Statistical analyses were performed using R version 3.5.3 (99). To compare the network topological indices between seasons, over plant growth, and between rhizosphere and bulk soils, an analysis of covariance (ANCOVA) model was applied to each network topological parameter using the aov function in the stats package. In the model “topological parameter ∼ Season*Week*Location,” season and location are considered categorical variables, and week is considered a continuous variable. To further determine the pattern that network topological indices change over time with plant growth, a general linear model, “topological parameter ∼ Week,” was fit for each index using the lm function in the stats package.
Data availability.
The 16S and ITS amplicon sequencing data are available through the NCBI SRA database under BioProject accession number PRJNA703382. R code for network construction and data analysis is available on GitHub at https://github.com/Mengting-Maggie-Yuan/Fungal-bacterial-network-construction-and-analysis.
ACKNOWLEDGMENTS
This research was supported by the U.S. Department of Energy Office of Science Office of Biological and Environmental Research Genomic Science program under awards DE-SC0020163 and DE-SC0010570 to M.F. at UC Berkeley and awards SCW1589 and SCW1678 to J.P.-R. at the Lawrence Livermore National Laboratory (LLNL). Work conducted at the LLNL was contributed under the auspices of the U.S. Department of Energy under contract DE-AC52-07NA27344.
We thank Donald J. Herman and Katerina Estera-Molina for the assistance in the greenhouse experiment; Liyou Wu, Qian Li, and Ying Fu for MiSeq sequencing; Daliang Ning and Zhou Shi for their valuable suggestions in data analysis; Kateryna Zhalnina for suggestions on data interpretation; and Christina Ramon for assistance in scanning electron microscopy imaging.
Experiment designed by S.S. and M.F.; experiment carried out by S.S., E.N., E.S., A.K., M.F., J.P.-R., and N.N.; analysis done by M.M.Y., A.K., E.S., S.S., and N.N.; and paper written by M.M.Y., A.K., M.F., and E.S. All authors contributed to writing and revision of the manuscript.
Footnotes
Citation Yuan MM, Kakouridis A, Starr E, Nguyen N, Shi S, Pett-Ridge J, Nuccio E, Zhou J, Firestone M. 2021. Fungal-bacterial cooccurrence patterns differ between arbuscular mycorrhizal fungi and nonmycorrhizal fungi across soil niches. mBio 12:e03509-20. https://doi.org/10.1128/mBio.03509-20.
REFERENCES
- 1.Newman EI. 1985. The rhizosphere: carbon sources and microbial populations, p 107–121. In Fitter AH, Atkinson D, Read DJ, Usher MB (ed), Ecological interactions in soil: plants, microbes and animals. CABI, Wallingford, United Kingdom. [Google Scholar]
- 2.Tian T, Reverdy A, She Q, Sun B, Chai Y. 2020. The role of rhizodeposits in shaping rhizomicrobiome. Environ Microbiol Rep 12:160–172. doi: 10.1111/1758-2229.12816. [DOI] [PubMed] [Google Scholar]
- 3.Gross CD, Harrison RB. 2019. The case for digging deeper: soil organic carbon storage, dynamics, and controls in our changing world. Soil Syst 3:28. doi: 10.3390/soilsystems3020028. [DOI] [Google Scholar]
- 4.Romaní AM, Fischer H, Mille-Lindblom C, Tranvik LJ. 2006. Interactions of bacteria and fungi on decomposing litter: differential extracellular enzyme activities. Ecology 87:2559–2569. doi: 10.1890/0012-9658(2006)87[2559:iobafo]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 5.Clausen CA. 1996. Bacterial associations with decaying wood: a review. Int Biodeterior Biodegradation 37:101–107. doi: 10.1016/0964-8305(95)00109-3. [DOI] [Google Scholar]
- 6.Frey-Klett P, Burlinson P, Deveau A, Barret M, Tarkka M, Sarniguet A. 2011. Bacterial-fungal interactions: hyphens between agricultural, clinical, environmental, and food microbiologists. Microbiol Mol Biol Rev 75:583–609. doi: 10.1128/MMBR.00020-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herman DJ, Firestone MK, Nuccio E, Hodge A. 2012. Interactions between an arbuscular mycorrhizal fungus and a soil microbial community mediating litter decomposition. FEMS Microbiol Ecol 80:236–247. doi: 10.1111/j.1574-6941.2011.01292.x. [DOI] [PubMed] [Google Scholar]
- 8.Kohlmeier S, Smits TH, Ford RM, Keel C, Harms H, Wick LY. 2005. Taking the fungal highway: mobilization of pollutant-degrading bacteria by fungi. Environ Sci Technol 39:4640–4646. doi: 10.1021/es047979z. [DOI] [PubMed] [Google Scholar]
- 9.Wick LY, Remer R, Würz B, Reichenbach J, Braun S, Schäfer F, Harms H. 2007. Effect of fungal hyphae on the access of bacteria to phenanthrene in soil. Environ Sci Technol 41:500–505. doi: 10.1021/es061407s. [DOI] [PubMed] [Google Scholar]
- 10.Warmink JA, Nazir R, Corten B, van Elsas JD. 2011. Hitchhikers on the fungal highway: the helper effect for bacterial migration via fungal hyphae. Soil Biol Biochem 43:760–765. doi: 10.1016/j.soilbio.2010.12.009. [DOI] [Google Scholar]
- 11.Banitz T, Johst K, Wick LY, Schamfuß S, Harms H, Frank K. 2013. Highways versus pipelines: contributions of two fungal transport mechanisms to efficient bioremediation. Environ Microbiol Rep 5:211–218. doi: 10.1111/1758-2229.12002. [DOI] [PubMed] [Google Scholar]
- 12.De Boer W, Gunnewiek PJK, Lafeber P, Janse JD, Spit BE, Woldendorp JW. 1998. Anti-fungal properties of chitinolytic dune soil bacteria. Soil Biol Biochem 30:193–203. doi: 10.1016/S0038-0717(97)00100-4. [DOI] [Google Scholar]
- 13.De Boer W, Folman LB, Summerbell RC, Boddy L. 2005. Living in a fungal world: impact of fungi on soil bacterial niche development. FEMS Microbiol Rev 29:795–811. doi: 10.1016/j.femsre.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 14.Kaiser K, Wemheuer B, Korolkow V, Wemheuer F, Nacke H, Schöning I, Schrumpf M, Daniel R. 2016. Driving forces of soil bacterial community structure, diversity, and function in temperate grasslands and forests. Sci Rep 6:33696. doi: 10.1038/srep33696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whipps JM. 2001. Microbial interactions and biocontrol in the rhizosphere. J Exp Bot 52:487–511. doi: 10.1093/jexbot/52.suppl_1.487. [DOI] [PubMed] [Google Scholar]
- 16.Fitter AH, Garbaye J. 1994. Interactions between mycorrhizal fungi and other soil organisms. Plant Soil 159:123–132. doi: 10.1007/BF00000101. [DOI] [Google Scholar]
- 17.Rousk J, Brookes PC, Baath E. 2010. Investigating the mechanisms for the opposing pH relationships of fungal and bacterial growth in soil. Soil Biol Biochem 42:926–934. doi: 10.1016/j.soilbio.2010.02.009. [DOI] [Google Scholar]
- 18.Leigh J, Fitter AH, Hodge A. 2011. Growth and symbiotic effectiveness of an arbuscular mycorrhizal fungus in organic matter in competition with soil bacteria. FEMS Microbiol Ecol 76:428–438. doi: 10.1111/j.1574-6941.2011.01066.x. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L, Xu M, Liu Y, Zhang F, Hodge A, Feng G. 2016. Carbon and phosphorus exchange may enable cooperation between an arbuscular mycorrhizal fungus and a phosphate-solubilizing bacterium. New Phytol 210:1022–1032. doi: 10.1111/nph.13838. [DOI] [PubMed] [Google Scholar]
- 20.Toljander JF, Lindahl BD, Paul LR, Elfstrand M, Finlay RD. 2007. Influence of arbuscular mycorrhizal mycelial exudates on soil bacterial growth and community structure. FEMS Microbiol Ecol 61:295–304. doi: 10.1111/j.1574-6941.2007.00337.x. [DOI] [PubMed] [Google Scholar]
- 21.Folman LB, Klein Gunnewiek PJA, Boddy L, de Boer W. 2008. Impact of white-rot fungi on numbers and community composition of bacteria colonizing beech wood from forest soil. FEMS Microbiol Ecol 63:181–191. doi: 10.1111/j.1574-6941.2007.00425.x. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen NH, Bruns TD. 2015. The microbiome of Pinus muricata ectomycorrhizae: community assemblages, fungal species effects, and Burkholderia as important bacteria in multipartnered symbioses. Microb Ecol 69:914–921. doi: 10.1007/s00248-015-0574-y. [DOI] [PubMed] [Google Scholar]
- 23.Nuccio EE, Hodge A, Pett-Ridge J, Herman DJ, Weber PK, Firestone MK. 2013. An arbuscular mycorrhizal fungus significantly modifies the soil bacterial community and nitrogen cycling during litter decomposition. Environ Microbiol 15:1870–1881. doi: 10.1111/1462-2920.12081. [DOI] [PubMed] [Google Scholar]
- 24.Warmink JA, Nazir R, Elsas JDV. 2009. Universal and species-specific bacterial ‘fungiphiles’ in the mycospheres of different basidiomycetous fungi. Environ Microbiol 11:300–312. doi: 10.1111/j.1462-2920.2008.01767.x. [DOI] [PubMed] [Google Scholar]
- 25.Nazir R, Semenov AV, Sarigul N, van Elsas JD. 2013. Bacterial community establishment in native and non-native soils and the effect of fungal colonization. Microbiol Discov 1:8. doi: 10.7243/2052-6180-1-8. [DOI] [Google Scholar]
- 26.Meier IC, Pritchard SG, Brzostek ER, McCormack ML, Phillips RP. 2015. The rhizosphere and hyphosphere differ in their impacts on carbon and nitrogen cycling in forests exposed to elevated CO2. New Phytol 205:1164–1174. doi: 10.1111/nph.13122. [DOI] [PubMed] [Google Scholar]
- 27.Brundrett MC, Tedersoo L. 2018. Evolutionary history of mycorrhizal symbioses and global host plant diversity. New Phytol 220:1108–1115. doi: 10.1111/nph.14976. [DOI] [PubMed] [Google Scholar]
- 28.Andrade G, Linderman RG, Bethlenfalvay GJ. 1998. Bacterial associations with the mycorrhizosphere and hyphosphere of the arbuscular mycorrhizal fungus Glomus mosseae. Plant Soil 202:79–87. doi: 10.1023/A:1004397222241. [DOI] [Google Scholar]
- 29.Koele N, Turpault M-P, Hildebrand EE, Uroz S, Frey-Klett P. 2009. Interactions between mycorrhizal fungi and mycorrhizosphere bacteria during mineral weathering: budget analysis and bacterial quantification. Soil Biol Biochem 41:1935–1942. doi: 10.1016/j.soilbio.2009.06.017. [DOI] [Google Scholar]
- 30.Garbaye J. 1994. Tansley review no. 76 helper bacteria: a new dimension to the mycorrhizal symbiosis. New Phytol 128:197–210. doi: 10.1111/j.1469-8137.1994.tb04003.x. [DOI] [PubMed] [Google Scholar]
- 31.Artursson V, Finlay RD, Jansson JK. 2006. Interactions between arbuscular mycorrhizal fungi and bacteria and their potential for stimulating plant growth. Environ Microbiol 8:1–10. doi: 10.1111/j.1462-2920.2005.00942.x. [DOI] [PubMed] [Google Scholar]
- 32.Lee Q, Widden P. 1996. Folsomia candida, a fungivorous collembolan, feeds preferentially on nematodes rather than soil fungi. Soil Biol Biochem 28:689–690. doi: 10.1016/0038-0717(95)00158-1. [DOI] [Google Scholar]
- 33.Barberán A, Bates ST, Casamayor EO, Fierer N. 2012. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J 6:343–351. doi: 10.1038/ismej.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faust K, Raes J. 2012. Microbial interactions: from networks to models. Nat Rev Microbiol 10:538–550. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 35.Lima-Mendez G, Faust K, Henry N, Decelle J, Colin S, Carcillo F, Chaffron S, Ignacio-Espinosa JC, Roux S, Vincent F, Bittner L, Darzi Y, Wang J, Audic S, Berline L, Bontempi G, Cabello AM, Coppola L, Cornejo-Castillo FM, d’Ovidio F, De Meester L, Ferrera I, Garet-Delmas M-J, Guidi L, Lara E, Pesant S, Royo-Llonch M, Salazar G, Sánchez P, Sebastian M, Souffreau C, Dimier C, Picheral M, Searson S, Kandels-Lewis S, Tara Oceans Coordinators, Gorsky G, Not F, Ogata H, Speich S, Stemmann L, Weissenbach J, Wincker P, Acinas SG, Sunagawa S, Bork P, Sullivan MB, Karsenti E, Bowler C, de Vargas C, Raes J. 2015. Determinants of community structure in the global plankton interactome. Science 348:1262073. doi: 10.1126/science.1262073. [DOI] [PubMed] [Google Scholar]
- 36.Dunne JA, Williams RJ, Martinez ND. 2002. Food-web structure and network theory: the role of connectance and size. Proc Natl Acad Sci U S A 99:12917–12922. doi: 10.1073/pnas.192407699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montoya JM, Pimm SL, Solé RV. 2006. Ecological networks and their fragility. Nature 442:259–264. doi: 10.1038/nature04927. [DOI] [PubMed] [Google Scholar]
- 38.Thébault E, Fontaine C. 2010. Stability of ecological communities and the architecture of mutualistic and trophic networks. Science 329:853–856. doi: 10.1126/science.1188321. [DOI] [PubMed] [Google Scholar]
- 39.Carr A, Diener C, Baliga NS, Gibbons SM. 2019. Use and abuse of correlation analyses in microbial ecology. ISME J 13:2647–2655. doi: 10.1038/s41396-019-0459-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blanchet FG, Cazelles K, Gravel D. 2020. Co-occurrence is not evidence of ecological interactions. Ecol Lett 23:1050–1063. doi: 10.1111/ele.13525. [DOI] [PubMed] [Google Scholar]
- 41.Deng Y, Zhang P, Qin Y, Tu Q, Yang Y, He Z, Schadt CW, Zhou J. 2016. Network succession reveals the importance of competition in response to emulsified vegetable oil amendment for uranium bioremediation. Environ Microbiol 18:205–218. doi: 10.1111/1462-2920.12981. [DOI] [PubMed] [Google Scholar]
- 42.Gao C, Montoya L, Xu L, Madera M, Hollingsworth J, Purdom E, Singan V, Vogel J, Hutmacher RB, Dahlberg JA, Coleman-Derr D, Lemaux PG, Taylor JW. 2020. Fungal community assembly in drought-stressed sorghum shows stochasticity, selection, and universal ecological dynamics. Nat Commun 11:34. doi: 10.1038/s41467-019-13913-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng K, Zhang Y, He Z, Ning D, Deng Y. 2019. Interdomain ecological networks between plants and microbes. Mol Ecol Resour 19:1565–1577. doi: 10.1111/1755-0998.13081. [DOI] [PubMed] [Google Scholar]
- 44.Ou Y, Penton CR, Geisen S, Shen Z, Sun Y, Lv N, Wang B, Ruan Y, Xiong W, Li R, Shen Q. 2019. Deciphering underlying drivers of disease suppressiveness against pathogenic Fusarium oxysporum. Front Microbiol 10:2535. doi: 10.3389/fmicb.2019.02535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou J, Deng Y, Luo F, He Z, Yang Y. 2011. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. mBio 2:e00122-11. doi: 10.1128/mBio.00122-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duran-Pinedo AE, Paster B, Teles R, Frias-Lopez J. 2011. Correlation network analysis applied to complex biofilm communities. PLoS One 6:e28438. doi: 10.1371/journal.pone.0028438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi S, Nuccio EE, Shi ZJ, He Z, Zhou J, Firestone MK. 2016. The interconnected rhizosphere: high network complexity dominates rhizosphere assemblages. Ecol Lett 19:926–936. doi: 10.1111/ele.12630. [DOI] [PubMed] [Google Scholar]
- 48.Shi S, Nuccio E, Herman DJ, Rijkers R, Estera K, Li J, da Rocha UN, He Z, Pett-Ridge J, Brodie EL, Zhou J, Firestone M. 2015. Successional trajectories of rhizosphere bacterial communities over consecutive seasons. mBio 6:e00746-15. doi: 10.1128/mBio.00746-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Starr EP, Shi S, Blazewicz SJ, Koch BJ, Probst AJ, Hungate BA, Pett-Ridge J, Firestone MK, Banfield JF. 2020. Stable isotope informed genome-resolved metagenomics uncovers potential trophic interactions in rhizosphere soil. bioRxiv doi: 10.1101/2020.08.21.262063. [DOI] [PMC free article] [PubMed]
- 50.Deng Y, Jiang Y-H, Yang Y, He Z, Luo F, Zhou J. 2012. Molecular ecological network analyses. BMC Bioinformatics 13:113. doi: 10.1186/1471-2105-13-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gruber S, Seidl-Seiboth V. 2012. Self versus non-self: fungal cell wall degradation in Trichoderma. Microbiology (Reading) 158:26–34. doi: 10.1099/mic.0.052613-0. [DOI] [PubMed] [Google Scholar]
- 52.Puopolo G, Masi M, Raio A, Andolfi A, Zoina A, Cimmino A, Evidente A. 2013. Insights on the susceptibility of plant pathogenic fungi to phenazine-1-carboxylic acid and its chemical derivatives. Nat Prod Res 27:956–966. doi: 10.1080/14786419.2012.696257. [DOI] [PubMed] [Google Scholar]
- 53.D’Amen M, Mod HK, Gotelli NJ, Guisan A. 2018. Disentangling biotic interactions, environmental filters, and dispersal limitation as drivers of species co-occurrence. Ecography 41:1233–1244. doi: 10.1111/ecog.03148. [DOI] [Google Scholar]
- 54.Jungk A, Claassen N. 1986. Availability of phosphate and potassium as the result of interactions between root and soil in the rhizosphere. J Plant Nutr Soil Sci 149:411–427. doi: 10.1002/jpln.19861490406. [DOI] [Google Scholar]
- 55.Syring KM, Claassen N. 1995. Estimation of the influx and the radius of the depletion zone developing around a root during nutrient uptake. Plant Soil 175:115–123. doi: 10.1007/BF02413016. [DOI] [Google Scholar]
- 56.York LM, Carminati A, Mooney SJ, Ritz K, Bennett MJ. 2016. The holistic rhizosphere: integrating zones, processes, and semantics in the soil influenced by roots. J Exp Bot 67:3629–3643. doi: 10.1093/jxb/erw108. [DOI] [PubMed] [Google Scholar]
- 57.Li X-L, George E, Marschner H. 1991. Phosphorus depletion and pH decrease at the root-soil and hyphae-soil interfaces of VA mycorrhizal white clover fertilized with ammonium. New Phytol 119:397–404. doi: 10.1111/j.1469-8137.1991.tb00039.x. [DOI] [Google Scholar]
- 58.Li X-L, George E, Marschner H. 1991. Extension of the phosphorus depletion zone in VA-mycorrhizal white clover in a calcareous soil. Plant Soil 136:41–48. doi: 10.1007/BF02465218. [DOI] [Google Scholar]
- 59.Cooke JC, Gemma JN, Koske RE. 1987. Observations of nuclei in vesicular-arbuscular mycorrhizal fungi. Mycologia 79:331–333. doi: 10.2307/3807669. [DOI] [Google Scholar]
- 60.Burggraaf AJP, Beringer JE. 1989. Absence of nuclear DNA synthesis in vesicular-arbuscular mycorrhizal fungi during in vitro development. New Phytol 111:25–33. doi: 10.1111/j.1469-8137.1989.tb04214.x. [DOI] [Google Scholar]
- 61.Viera A, Glenn MG. 1990. DNA content of vesicular-arbuscular mycorrhizal fungal spores. Mycologia 82:263–267. doi: 10.2307/3759857. [DOI] [Google Scholar]
- 62.Tommerup IC, Sivasithamparam K. 1990. Zygospores and asexual spores of Gigaspora decipiens, an arbuscular mycorrhizal fungus. Mycol Res 94:897–900. doi: 10.1016/S0953-7562(09)81303-8. [DOI] [Google Scholar]
- 63.Bécard G, Pfeffer PE. 1993. Status of nuclear division in arbuscular mycorrhizal fungi during in vitro development. Protoplasma 174:62–68. doi: 10.1007/BF01404043. [DOI] [Google Scholar]
- 64.Jany J, Pawlowska TE. 2010. Multinucleate spores contribute to evolutionary longevity of asexual Glomeromycota. Am Nat 175:424–435. doi: 10.1086/650725. [DOI] [PubMed] [Google Scholar]
- 65.Gao C, Montoya L, Xu L, Madera M, Hollingsworth J, Purdom E, Hutmacher RB, Dahlberg JA, Coleman-Derr D, Lemaux PG, Taylor JW. 2019. Strong succession in arbuscular mycorrhizal fungal communities. ISME J 13:214–226. doi: 10.1038/s41396-018-0264-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pett-Ridge J, Shi S, Estera-Molina K, Nuccio E, Yuan M, Rijkers R, Swenson T, Zhalnina K, Northen T, Zhou J, Firestone MK. 2021. Rhizosphere carbon turnover from cradle to grave: the role of microbe-plant interactions, p 51–73. In Gupta VVSR, Sharma AK (ed), Rhizosphere biology: interactions between microbes and plants. Springer, Singapore. [Google Scholar]
- 67.Smith SE, Read DJ. 2008. Mycorrhizal symbiosis, 3rd ed. Academic Press, San Diego, CA. [Google Scholar]
- 68.Berendsen RL, Pieterse CMJ, Bakker PAHM. 2012. The rhizosphere microbiome and plant health. Trends Plant Sci 17:478–486. doi: 10.1016/j.tplants.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 69.Wu Q-S, Xia R-X. 2006. Arbuscular mycorrhizal fungi influence growth, osmotic adjustment and photosynthesis of citrus under well-watered and water stress conditions. J Plant Physiol 163:417–425. doi: 10.1016/j.jplph.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 70.Lugtenberg B, Kamilova F. 2009. Plant-growth-promoting rhizobacteria. Annu Rev Microbiol 63:541–556. doi: 10.1146/annurev.micro.62.081307.162918. [DOI] [PubMed] [Google Scholar]
- 71.Roesti D, Ineichen K, Braissant O, Redecker D, Wiemken A, Aragno M. 2005. Bacteria associated with spores of the arbuscular mycorrhizal fungi Glomus geosporum and Glomus constrictum. Appl Environ Microbiol 71:6673–6679. doi: 10.1128/AEM.71.11.6673-6679.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang X, Ren B, Wu S, Sun Y, Chen B, Li R. 2018. Rhizophagus irregularis influences As and P uptake by alfafa [sic] and the neighboring non-host pepperweed growing in an As-contaminated soil. J Environ Sci (China) 67:36–44. doi: 10.1016/j.jes.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 73.Kuzyakov Y, Blagodatskaya E. 2015. Microbial hotspots and hot moments in soil: concept & review. Soil Biol Biochem 83:184–199. doi: 10.1016/j.soilbio.2015.01.025. [DOI] [Google Scholar]
- 74.Bianciotto V, Minerdi D, Perotto S, Bonfante P. 1996. Cellular interactions between arbuscular mycorrhizal fungi and rhizosphere bacteria. Protoplasma 193:123–131. doi: 10.1007/BF01276640. [DOI] [Google Scholar]
- 75.Naito M, Desirò A, González JB, Tao G, Morton JB, Bonfante P, Pawlowska TE. 2017. ‘Candidatus Moeniiplasma glomeromycotorum’, an endobacterium of arbuscular mycorrhizal fungi. Int J Syst Evol Microbiol 67:1177–1184. doi: 10.1099/ijsem.0.001785. [DOI] [PubMed] [Google Scholar]
- 76.Nguyen NH, Song Z, Bates ST, Branco S, Tedersoo L, Menke J, Schilling JS, Kennedy PG. 2016. FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol 20:241–248. doi: 10.1016/j.funeco.2015.06.006. [DOI] [Google Scholar]
- 77.Cline LC, Hobbie SE, Madritch MD, Buyarski CR, Tilman D, Cavender‐Bares JM. 2018. Resource availability underlies the plant-fungal diversity relationship in a grassland ecosystem. Ecology 99:204–216. doi: 10.1002/ecy.2075. [DOI] [PubMed] [Google Scholar]
- 78.Semchenko M, Leff JW, Lozano YM, Saar S, Davison J, Wilkinson A, Jackson BG, Pritchard WJ, Long JRD, Oakley S, Mason KE, Ostle NJ, Baggs EM, Johnson D, Fierer N, Bardgett RD. 2018. Fungal diversity regulates plant-soil feedbacks in temperate grassland. Sci Adv 4:eaau4578. doi: 10.1126/sciadv.aau4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hannula SE, Morriën E, van der Putten WH, de Boer W. 2020. Rhizosphere fungi actively assimilating plant-derived carbon in a grassland soil. Fungal Ecol 48:100988. doi: 10.1016/j.funeco.2020.100988. [DOI] [Google Scholar]
- 80.Zhalnina K, Louie KB, Hao Z, Mansoori N, da Rocha UN, Shi S, Cho H, Karaoz U, Loqué D, Bowen BP, Firestone MK, Northen TR, Brodie EL. 2018. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat Microbiol 3:470–480. doi: 10.1038/s41564-018-0129-3. [DOI] [PubMed] [Google Scholar]
- 81.Duczek LJ, White GP. 1986. Chalara heteroderae a fungal antagonist that parasitizes hyphae of Bipolaris sorokiniana. Soil Biol Biochem 18:655–659. doi: 10.1016/0038-0717(86)90090-8. [DOI] [Google Scholar]
- 82.Duncan CG, Eslyn WE. 1966. Wood-decaying ascomycetes and fungi imperfecti. Mycologia 58:642–645. doi: 10.2307/3757045. [DOI] [Google Scholar]
- 83.Cannon PF, Kirk PM. 2007. Fungal families of the world. CABI, Wallingford, United Kingdom. [Google Scholar]
- 84.Andrade G, Mihara KL, Linderman RG, Bethlenfalvay GJ. 1997. Bacteria from rhizosphere and hyphosphere soils of different arbuscular-mycorrhizal fungi. Plant Soil 192:71–79. doi: 10.1023/A:1004249629643. [DOI] [Google Scholar]
- 85.Liu Y, Sun Q, Li J, Lian B. 2018. Bacterial diversity among the fruit bodies of ectomycorrhizal and saprophytic fungi and their corresponding hyphosphere soils. Sci Rep 8:11672. doi: 10.1038/s41598-018-30120-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bonfante-Fasolo P, Vian B. 1984. Wall texture in the spore of a vesicular-arbuscular mycorrhizal fungus. Protoplasma 120:51–60. doi: 10.1007/BF01287617. [DOI] [Google Scholar]
- 87.Simon A, Bindschedler S, Job D, Wick LY, Filippidou S, Kooli WM, Verrecchia EP, Junier P. 2015. Exploiting the fungal highway: development of a novel tool for the in situ isolation of bacteria migrating along fungal mycelium. FEMS Microbiol Ecol 91:fiv116. doi: 10.1093/femsec/fiv116. [DOI] [PubMed] [Google Scholar]
- 88.Newman MEJ. 2006. Modularity and community structure in networks. Proc Natl Acad Sci U S A 103:8577–8582. doi: 10.1073/pnas.0601602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Olesen JM, Bascompte J, Dupont YL, Jordano P. 2007. The modularity of pollination networks. Proc Natl Acad Sci U S A 104:19891–19896. doi: 10.1073/pnas.0706375104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kong Y. 2011. Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics 98:152–153. doi: 10.1016/j.ygeno.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 91.Magoč T, Salzberg SL. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AFS, Bahram M, Bates ST, Bruns TD, Bengtsson‐Palme J, Callaghan TM, Douglas B, Drenkhan T, Eberhardt U, Dueñas M, Grebenc T, Griffith GW, Hartmann M, Kirk PM, Kohout P, Larsson E, Lindahl BD, Lücking R, Martín MP, Matheny PB, Nguyen NH, Niskanen T, Oja J, Peay KG, Peintner U, Peterson M, Põldmaa K, Saag L, Saar I, Schüßler A, Scott JA, Senés C, Smith ME, Suija A, Taylor DL, Telleria MT, Weiss M, Larsson K-H. 2013. Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol 22:5271–5277. doi: 10.1111/mec.12481. [DOI] [PubMed] [Google Scholar]
- 94.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 95.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 96.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Csárdi G, Nepusz T. 2006. The igraph software package for complex network research. Interjournal Complex Syst 2006:1695. [Google Scholar]
- 98.Dormann CF, Fründ J, Blüthgen N, Gruber B. 2009. Indices, graphs and null models: analyzing bipartite ecological networks. Open Environ Res J 2:7–24. doi: 10.2174/1874213000902010007. [DOI] [Google Scholar]
- 99.R Core Team. 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 100.Kakouridis A, Hagen JA, Kan MP, Mambelli S, Feldman LJ, Herman DJ, Pett-Ridge J, Firestone MK. 2020. Routes to roots: direct evidence of water transport by arbuscular mycorrhizal fungi to host plants. bioRxiv doi: 10.1101/2020.09.21.305409. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Topological properties of networks between bacteria and nonmycorrhizal fungi over plant growth stages (a to e) and node taxonomic compositions of these networks at the phylum level (f). The linear regression line is shown as a solid line if the effect of time (in weeks after planting) is statistically significant (P ≤ 0.05). Otherwise, it is shown as a dashed line. Download FIG S2, PDF file, 0.3 MB (272.5KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Properties of nonmycorrhizal fungal-bacterial networks and AMF-bacterial networks based on Pearson and Spearman correlations. “−” shows where network topological properties do not apply because the network is empty or the network is too simple. Download Table S1, XLSX file, 0.01 MB (15.4KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Taxonomy of AMF nodes in AMF-bacterial networks. Download Table S5, XLSX file, 0.01 MB (9.8KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Analysis of covariance (ANCOVA) on the topological parameters for the nonmycorrhizal fungal-bacterial networks and AMF-bacterial networks. Download Table S2, XLSX file, 0.01 MB (11.1KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Richness (a and c) and relative abundances (b and d) of nonmycorrhizal fungi (a and b) and AMF (c and d) over time in bulk and rhizosphere soils. Richness was estimated by the number of OTUs. Relative abundance was estimated as a proportion of the number of sequences in total fungal sequences. There was a 3-month dry-down between week 12 of season 1 and week 0 of season 2. Download FIG S3, PDF file, 0.1 MB (152.1KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Module hubs and their taxonomy in the fungal-bacterial networks. ITS OTUs that occurred in more than one network are in boldface type. Download Table S3, XLSX file, 0.01 MB (15.8KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Numbers and proportions of nodes occurring in multiple networks. (a) Bacterial and archaeal nodes in nonmycorrhizal fungal-bacterial networks; (b) fungal nodes in nonmycorrhizal fungal-bacterial networks; (c) bacterial and archaeal nodes in AMF-bacterial networks; (d) AMF nodes in AMF-bacterial networks. On top of each bar, the number of nodes and the proportion are listed. Download FIG S4, PDF file, 0.5 MB (516.4KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Links connecting genome-resolved bacterial genomes and their fungal associates. Download Table S4, XLSX file, 0.01 MB (11.3KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Relative abundances of the 16S OTUs matching the metagenome-resolved bacterial genomes. Bars represent averaged abundances of the OTUs across biological replicates in different locations, seasons, and weeks. Error bars show standard errors of the means (n = 16). Panels a to e separately show the abundances of the OTUs matching the genomes Rhizobium_leguminosarum_59_9, Mesorhizobium_63_15, Acidobacteria_68_21, Acidobacteria_60_12, and Actinobacteria_70_13, respectively. Download FIG S5, PDF file, 0.3 MB (347.3KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Diagram of the experimental design (a) and rarefaction curves of the sequences obtained from soil samples (b). In panel a, each microcosm shown represents one sampling time point, including 16 replicate microcosms (CO2 effect not detectable on the overall soil bacterial and fungal community structure [see Materials and Methods for details]). In panel b, the vertical line indicates the number of sequences (4,484) for rarefaction for each sample. Download FIG S1, PDF file, 1.4 MB (1.4MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Data Availability Statement
The 16S and ITS amplicon sequencing data are available through the NCBI SRA database under BioProject accession number PRJNA703382. R code for network construction and data analysis is available on GitHub at https://github.com/Mengting-Maggie-Yuan/Fungal-bacterial-network-construction-and-analysis.