

Widespread antimicrobial resistance encourages repurposing/refining of nonantimicrobial drugs for antimicrobial indications. Gallium nitrate (GaNt), an FDA-approved medication for cancer-related hypercalcemia, recently showed good activity against several clinically significant bacteria.
KEYWORDS: gallium nitrate, tolerance, mechanisms of action, EvgS-EvgA acid stress response, reactive oxygen species, ROS detoxification, glyoxylate shunt
ABSTRACT
Widespread antimicrobial resistance encourages repurposing/refining of nonantimicrobial drugs for antimicrobial indications. Gallium nitrate (GaNt), an FDA-approved medication for cancer-related hypercalcemia, recently showed good activity against several clinically significant bacteria. However, the mechanism of GaNt antibacterial action is still poorly understood. In the present work, resistant and tolerant mutants of Escherichia coli were sought via multiple rounds of killing by GaNt. Multiround-enrichment yielded no resistant mutant; whole-genome sequencing of one representative GaNt-tolerant mutant uncovered mutations in three genes (evgS, arpA, and kdpD) potentially linked to protection from GaNt-mediated killing. Subsequent genetic analysis ruled out a role for arpA and kdpD, but two gain-of-function mutations in evgS conferred tolerance. The evgS mutation-mediated GaNt tolerance depended on EvgS-to-EvgA phosphotransfer; EvgA-mediated upregulation of GadE. YdeO, and SarfA also contributed to tolerance, the latter two likely through their regulation of GadE. GaNt-mediated killing of wild-type cells correlated with increased intracellular reactive oxygen species (ROS) accumulation that was abolished by the evgS-tolerant mutation. Moreover, GaNt-mediated killing was mitigated by dimethyl sulfoxide, and the evgS-tolerant mutation upregulated genes encoding enzymes involved in ROS detoxification and in the glyoxylate shunt of the tricarboxylic acid (TCA) cycle. Collectively, these findings indicate that GaNt kills bacteria through elevation of ROS; gain-of-function mutations in evgS confer tolerance by constitutively activating the EvgA-YdeO/GadE cascade of acid resistance pathways and by preventing GaNt-stimulated ROS accumulation by upregulating ROS detoxification and shifting TCA cycle carbon flux. The striking lethal activity of GaNt suggests that clinical use of the agent may not quickly lead to resistance.
INTRODUCTION
Antimicrobial resistance is one of the most significant challenges facing modern medicine. One example is the emergence and spread of pathogens, termed ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.), that resist most, if not all, currently available antimicrobials (1–3). A traditional way to address antimicrobial resistance is development of new compounds to treat pathogens already resistant to existing drugs. This approach is becoming increasingly difficult, as most of the feasible drug targets have been explored, and many derivatives of existing drug classes have been extensively refined (4, 5). Reinvestigating/refining an FDA-approved drug for new antibacterial indications is an attractive alternative, because pharmacological feasibility and toxicological concerns have already been addressed with these compounds (6). Gallium nitrate (GaNt) represents a candidate for reinvestigation, because it has been approved for treatment of a cancer-related hypercalcemia (7) and because it is active against many clinically significant multidrug-resistant bacterial pathogens in vitro (8–16), in animal models of infection (9, 11, 13, 17, 18), and in clinical trials (10, 18, 19). However, activity improvement is needed to protect from the emergence of resistance, to expand the spectrum of species covered, and to further reduce toxicity. A better understanding of action mechanism may facilitate these improvements.
Gallium-based antibacterial action has been thought to derive from interference with iron uptake (11, 15, 16), iron metabolism, and catalysis by iron-containing enzymes, because Ga3+ is similar to Fe3+ in terms of ionic radius, ionization potential, coordination chemistry, electronegativity, and electron affinity (7, 10). Since Fe3+ is redox active through reduction to a divalent state, while, under physiological conditions, Ga3+ is not, replacing Fe3+ with Ga3+ might disrupt the activity of many iron-containing respiratory enzymes that serve as redox centers. However, such an explanation for GaNt action is unsatisfactory, because mimicry of iron depletion and simple inhibition of enzyme activity usually confer bacteriostatic rather than bactericidal activity, and GaNt is strikingly lethal (8, 11, 14, 16). Events beyond enzyme inhibition likely occur that allow GaNt to rapidly kill bacteria.
Selection and characterization of resistant and/or tolerant mutants are traditional approaches for uncovering drug targets and action mechanisms. Unfortunately, mutants that are resistant to gallium nitrate, as indicated by an increase in MIC, are difficult to obtain (8), possibility due to gallium simultaneously interfering with multiple enzyme targets (10). The few weak resistance mutations, which evolved in vitro, are found in Pseudomonas aeruginosa and Escherichia coli genes that encode nonspecific transporters/pumps for iron citrate and other metals (20, 21) rather than in genes encoding gallium-specific targets. Gallium tolerance, defined as a reduced killing of bacteria with little or no change in MIC, has not been reported. Thus, the specific action mechanism of gallium nitrate is still largely unknown.
The present work explored gallium action by challenging E. coli with lethal concentrations of GaNt and then characterizing the resulting mutants. While no resistant mutant was obtained, several tolerant mutants were recovered. Whole-genome sequencing and subsequent genetic characterization identified two gain-of-function mutations in evgS, which encodes the sensor kinase of the EvgSA acid resistance two-component system. The mutations conferred gallium tolerance by (i) stimulating the EvgA-YdeO-GadE transcription regulatory cascade and (ii) suppressing a gallium-mediated surge of intracellular reactive oxygen species (ROS). We can now begin to think about ways to make GaNt even more lethal by enhancing ROS production.
RESULTS
MIC measurement and medium acidification determination.
We first measured MIC for GaNt with wild-type E. coli (strain BW25113) using LB broth lacking citrate. The MIC was 12.2 mM. At this concentration, GaNt acidified the LB medium to a pH of 4, a value that would inhibit bacterial growth and thus confound measurement of gallium nitrate-mediated antibacterial effects. To mitigate acidification by gallium nitrate from interfering with MIC measurements, we added 54.3 mM sodium citrate to LB medium. As shown in Tables S4 and S5 in the supplemental material, even with this citrate buffering, medium acidification occurred in a GaNt concentration-dependent manner. GaNt MICs determined with the citrate-buffered media were 18.4 and 12.2 mM, respectively, for LB and Mueller-Hinton (MH) media. At these GaNt concentrations, medium acidification was mild (e.g., pH > 5.6) (Tables S4 and S5); such acidification by itself did not inhibit bacterial growth during the 24-h incubation used for MIC determination. Thus, buffering with sodium citrate allowed determination of GaNt MIC without the confounding effect from GaNt-mediated medium acidification; citrate-buffered medium was used for all other experiments.
Since GaNt obtained from Sigma-Aldrich contains unspecified numbers of hydrates, we determined the number of the hydrates using a thermal decomposition method reported previously (22). The hydrate number was 9, which was used to adjust MICs estimated using anhydrous molecular weights supplied by the vendor. MICs in weight concentrations were 4.7 mg/ml and 3.1 mg/ml, respectively, for LB and MH media (Table S1).
Enrichment, isolation, and identification of mutants tolerant to gallium nitrate.
To gain insight into molecular details of gallium antibacterial action, we enriched E. coli mutants by challenging cultures with GaNt at lethal rather than sublethal concentrations, because previous in vitro evolution studies using sublethal concentrations generated mutants exhibiting only low to moderate loss of susceptibility arising from alterations in iron transporters or efflux pumps. Such mutations are uninformative for gallium action mechanism studies, because they nonspecifically reduce GaNt uptake rather than blocking the interaction of gallium with its cellular target (20, 21). Moreover, use of sublethal concentrations makes it difficult to enrich and study mutants that preferentially lack lethal activity (tolerant mutants).
Treatment of cultures with GaNt for 40 min at 2.5× MIC reduced survival by 5 orders of magnitude. After this treatment was applied for 4 and 5 rounds, survival rose to >80% (Fig. 1a). Surviving cells were purified as single colonies by plating on LB agar, and they were then screened for growth on agar containing 1× MIC of GaNt and survival in LB broth containing 2.5× MIC of GaNt. No colony grew on GaNt-containing agar, while hundreds of colonies formed on GaNt-free agar; gallium-resistant mutants were not enriched. When colonies recovered on GaNt-free agar were tested for killing and growth inhibition by GaNt, their MIC for GaNt was similar to that of the parental, wild-type strain, but survival of four mutants was much higher (>10,000-fold). These mutants were considered tolerant but not resistant to GaNt (no change in MIC). The most tolerant mutant, GaNt-3, showed >90% survival even after a 90-min treatment with 4× MIC of GaNt (Fig. 1b).
FIG 1.

Enrichment and characterization of gallium nitrate-tolerant mutants. (a) Mutant enrichment. Exponentially growing cultures of wild-type E. coli strain BW25113 were repeatedly challenged with 2.5× MIC of gallium nitrate for 40 min followed by determination of surviving CFU. (b) Reduced killing of one representative GaNt-tolerant mutant. Strain GaNt-3, obtained after 5 rounds of enrichment, and the wild-type (WT) control were grown to exponential phase and treated with 4× MIC of GaNt for the indicated times, and aliquots were assayed for survival by plating on drug-free agar. (c) Mutations recovered by enrichment that do not reduce GaNt-mediated killing. Mutations in arpA (E334A) and kdpD (P885T), found in strain GaNt-3 by whole-genome sequencing, were reconstructed in the parental strain and tested for survival to GaNt at 4× MIC for the indicated times. (d) Mutations in evgS reduced GaNt-mediated killing. Two mutations in evgS (E701G and S582P), as revealed by whole-genome sequencing of GaNt-3, were reconstructed in the parental strain and tested for survival of GaNt at 4× MIC for the indicated times. All experiments, except those whose results are shown in panel a (performed only once), were performed in triplicate; error bars indicate standard errors of the means.
Whole-genome sequencing of GaNt-3 revealed single-point mutations in three genes, arpA, kdpD, and evgS (Table 1). Reconstruction of these point mutations in each gene using the wild-type parental strain revealed that mutations in evgS, but not in arpA or kdpD, confer the tolerant phenotype (Fig. 1c and d). This conclusion was further solidified by PCR-mediated amplification and subsequent sequencing of the 3 genes identified above in the 3 other tolerant mutants recovered from the screen: mutations in evgS but not the other two genes were found (Table 1). Thus, two amino acid substitutions in EvgS (E701G and S582P) each drastically reduced killing by gallium nitrate without a change in MIC, a typical tolerance phenotype (23).
TABLE 1.
Mutational information of gallium nitrate-tolerant mutants
| Mutant | Gene name | Base change | Amino acid change | Gene product |
|---|---|---|---|---|
| GaNt-3 | evgS | A2102G | Glu701Gly | Sensory histidine kinase EvgS |
| arpA | A1001C | Glu334Ala | Regulator of acetyl-CoA synthetase | |
| kdpD | C2653A | Pro885Thr | Sensory histidine kinase KdpD | |
| GaNt-4 | evgS | T1744C | Ser582Pro | Sensory histidine kinase EvgS |
| GaNt-5 | evgS | A2102G | Glu701Gly | Sensory histidine kinase EvgS |
| GaNt-6 | evgS | T1744C | Ser582Pro | Sensory histidine kinase EvgS |
The extensive killing (e.g., >10,000-fold) of wild-type cells and the protection from killing by the mutations in evgS were mainly due to gallium nitrate itself rather than from gallium nitrate-mediated medium acidification. Control experiments using LB-citrate medium acidified by hydrochloric acid to pH 3.2, a value that is equal to acidification caused by GaNt at 4× MIC (Table S4), conferred only slight (e.g., <10-fold) lethality (Fig. S1). Addition of citrate, which helps distinguish killing by GaNt from that by medium acidification, did not interfere with the protection from killing by the evgS (E701G) mutation, because when low concentrations of GaNt (18.4 mM and 24.4 mM) were added to cultures grown in LB without citrate for longer incubation times, >100-fold protection from GaNt-mediated killing was observed; under the same conditions, medium acidification alone caused little killing (Fig. S2). Slow growth due to the mutations does not account for the tolerance phenotype, since the mutant and the wild-type strains exhibited similar growth curves (Fig. S3).
Contribution of the evgS-evgA two-component system and downstream regulatory cascade to gallium lethality.
Although evgS encodes the sensor kinase of the well-characterized EvgS-EvgA two-component-system response to acid stress, it was not obvious how gallium stress is related to EvgS-EvgA. The tolerance phenotype appears to derive from gain-of-function mutations, for the following reasons. (i) Deletion of evgS or evgA failed to confer tolerance (Fig. 2a). (ii) The EvgS E701G and S582P substitutions occur in the same PAS domain of EvgS in which other gain-of-function (constitutive activation) acid-resistant changes occur (24); these substitutions (e.g., N573K, S584F, and S600I) also conferred tolerance to GaNt-mediated killing (Fig. 2b). (iii) The EvgS E701G substitution upregulated three downstream genes, ydeO, gadE, and safA (Fig. 2c), that are controlled by EvgA and are involved in regulation of a response to acid stress. EvgS E701G-mediated tolerance likely depends on the EvgS-to-EvgA phosphorelay, because an amino acid substitution that abolishes the phosphorylation site of EvgA (D52A) eliminated gallium tolerance (Fig. 2d). As expected, killing was due mainly to GaNt itself rather than to GaNt-mediated medium acidification (Fig. S4). Since the E701G and S582P substitutions in EvgS conferred similar levels of gallium tolerance, subsequent characterization focused on the E701G substitution.
FIG 2.

Constitutive gain-of-function substitutions in EvgS reduce gallium nitrate-mediated killing via phosphorylation of EvgA. (a) Deficiency in evgS or evgA fails to reduce GaNt-mediated killing. Exponentially growing cultures of wild-type (WT), ΔevgS, or ΔevgA E. coli were grown and treated with 4× MIC of gallium nitrate for the indicated times followed by determination of survival. (b) Constitutive gain-of-function mutations in the PAS domain of EvgS, known to confer acid resistance, protect from GaNt-mediated killing. Cultures of the wild type and the indicated evgS mutants were grown, treated, and processed as for panel a. (c) The EvgS E701G gain-of-function mutation induces expression of several downstream regulators involved in acid stress response. Wild-type and evgS E701G mutant cultures were untreated or treated with 4× MIC of GaNt for 30 min. Samples were subjected to total RNA isolation followed by reverse transcription and PCR amplification of gadE, ydeO, and safA using 16S rRNA as an internal control. mRNA levels of all mutant samples were normalized to those of each gene in the untreated wild-type control. (d) The evgS E701G gain-of-function mutation confers GaNt tolerance through phosphotransfer from EvgS to EvgA. Wild-type, evgS E701G mutant, evgA phosphorylation site (D52A) mutant, and evgS-evgA double mutant cultures were grown, treated with GaNt, and processed as for panel a.
Since the EvgS-EvgA system controls several key downstream regulators (25, 26), we next investigated the contribution of three such regulators to EvgS E701G-mediated gallium tolerance. Deletion of gadE abolished tolerance, while deletion of ydeO or safA partially reduced it (Fig. 3a to c). SafA regulates another two-component system, PhoQ-PhoP, which ultimately controls RpoS degradation (27–29). Since RpoS is a master stress response regulator that affects gadE expression (29), we also asked whether EvgS E701G confers gallium tolerance via RpoS. Although a mutant deficient in rpoS was hypersensitive to killing by GaNt (Fig. 3d), introduction of EvgS E701G into the rpoS mutant still allowed drastic protection (e.g., 1,000-fold) from GaNt-mediated killing (Fig. 3d). These data indicate that EvgS E701G-mediated gallium tolerance is largely independent of RpoS. The YdeO effect may derive from its positive regulation of gadE, a gene whose expression is also regulated by EvgA and RpoS (25, 26). As described above, control experiments separated GaNt lethality from that caused by GaNt-mediated medium acidification, since reduced pH alone (without GaNt) showed a much lower (e.g., <100-fold) effect than treatment of samples with GaNt (Fig. S5).
FIG 3.

Contribution of downstream regulators of the EvgSA and RpoS systems in evgS gain-of-function-mediated protection from gallium nitrate lethality. (a) Deficiency of gadE abolishes evgS E701G-mediated protection from GaNt killing. Exponentially growing wild-type (WT), evgS (E701E) mutant, ΔgadE mutant, and evgS-ΔgadE double mutant E. coli cultures were grown and processed as for Fig. 1. (b) Deficiency of ydeO partially reverses evgS E701G-mediated protection from GaNt killing. Experimental conditions were as for panel a. (c) Deficiency of safA reduces evgS E701G-mediated protection from GaNt killing. Experimental conditions were as for panel a. (d) Deficiency of rpoS fails to eliminate evgS E701G-mediated protection from GaNt-mediated killing. Experimental conditions were as for panel a.
Partial overlap of acid and gallium tolerance pathways.
Since the EvgS-EvgA regulatory cascade is an acid resistance/tolerance system and since we found that mutations in evgS also confer tolerance to gallium-mediated killing, we next asked whether different responses to acid and gallium exist within this regulatory cascade. All mutants tested that have a mutation in the sequence encoding the PAS domain of EvgS exhibited tolerance to acid stress (Fig. 4a and c); all (Fig. 1d and 2b) but one (Fig. 4b) of these mutants also exhibited tolerance to GaNt. A deficiency in either gadE or rpoS conferred hyperlethality to acid and GaNt stress (Fig. 3a and d and 4c and d), but only a deficiency in gadE, not rpoS, abolished EvgS E701G-mediated protection from killing by GaNt (Fig. 3a and d). Neither a gadE nor an rpoS deficiency abolished protection by EvgS E701G to acid-mediated lethality (Fig. 4c and d). Collectively, these data indicate that gallium tolerance shares some regulatory circuits with acid resistance/tolerance centered on GadE, but not all acid resistance/tolerance factors play a role in gallium tolerance.
FIG 4.

Effect of mutations in evgS, gadE, and rpoS on protection from killing by acid and GaNt. (a) Constitutive gain-of-function mutations in the PAS domain of the EvgS all confer reduced killing from acid. Cultures of wild-type (WT) and five evgS mutants, indicated in the figure, were grown and processed as for Fig. 2b following acid treatment for the indicated times. (b) An evgS G658A mutation failed to protect from GaNt-mediated killing. Cultures were grown, treated, and processed as for Fig. 2b. (c) A deficiency of gadE failed to abolish evgS E701G-mediated protection from killing by low pH. Exponentially growing wild-type, evgS (E701E) mutant, ΔgadE mutant, and evgS-ΔgadE double-mutant E. coli cultures were grown and processed as for panel b following exposure to pH 2.50 for the indicated times. Survival of the ΔgadE mutant and the evgS-ΔgadE double mutant dropped below the detection limit (10−3%) after 20- and 40-min treatments, respectively. (d) Deficiency of rpoS fails to eliminate evgS E701G-mediated protection from acid. Experimental conditions were as for Fig. 3d except for treatment with acid (pH 2.50) for the indicated times.
Involvement of ROS in GaNt-mediated killing.
Due to its mimicry of ferric iron, gallium ion has long been thought to interfere with respiratory enzymes that rely on ferrous-ferric oscillation for electron transfer. Since ROS production is a by-product of respiration (30), gallium could affect ROS accumulation. However, previous work has not addressed whether gallium exposure stimulates intracellular accumulation of ROS in bacteria. We found that levels of ROS surged during GaNt treatment of wild-type E. coli and that the surge was absent in the evgS E701G mutant (Fig. 5a). Exogenous addition of ROS-mitigating chemicals, such as dimethyl sulfoxide (DMSO), protected E. coli from gallium-mediated killing (Fig. 5b). Moreover, GaNt treatment induced transcription of genes involved in ROS detoxification; such induction was more evident with the evgS E701G mutant than with wild-type cells (Fig. 5c).
FIG 5.

Involvement of intracellular ROS in gallium nitrate-mediated killing blocked by the EvgS E701G gain-of-function substitution. (a) GaNt-mediated surge in ROS accumulation abolished by the evgS E701G mutation. Exponentially growing cultures of wild-type (top) and evgS E701G (bottom) E. coli were incubated with 10 μM carboxy-H2DCFDA for 20 min before cells were treated with 4× MIC of GaNt for 1 h and subjected to flow cytometry for analysis of ROS accumulation. (b) The hydroxyl radical scavenger dimethyl sulfoxide (DMSO) protects against or delays GaNt-mediated killing. Exponentially growing cultures of wild-type E. coli were incubated with or without 5% (1/3 MIC) DMSO for 15 min before cells were treated with 4× MIC of GaNt for the indicated times. Samples were then processed as for Fig. 2b. (c) Induction of expression of genes involved in ROS detoxification by GaNt exposure stimulated by the evgS E701G gain-of-function mutation. Wild-type (WT) and mutant cultures were untreated or treated with 4× MIC of GaNt for 30 min; total cellular RNA was extracted, followed by reverse transcription and PCR amplification of the indicated genes, using 16S rRNA as an internal standard. mRNA levels were normalized to those of each gene with an untreated wild-type control. (d) Expression of genes involved in the TCA cycle by GaNt exposure as affected by the evgS E701G gain-of-function mutation. Cells were grown, treated, and processed as for panel c. (e) Preferential upregulation of genes involved in the glyoxylate shunt portion of the TCA cycle by the evgS E701G gain-of-function mutation during GaNt exposure. The ratio of evgS mutant to wild-type mRNA levels of each gene from GaNt-treated samples obtained in panel d are displayed in a scheme of the TCA cycle as fold changes (in parentheses) for each gene. Red font and positive numbers indicate upregulation. Blue font and negative numbers indicate downregulation. Black font indicates little change in transcription. PYR, pyruvate; AceCoA, acetyl coenzyme A; CIT, citrate; ICIT, isocitrate; OASUCC, oxalosuccinate; AKG, α-ketoglutarate; SuccCoA, succinyl-CoA; SUCC, succinate; FUM, fumarate; MAL, malate; OAA, oxaloacetate.
The evgS E701G mutant-mediated suppression of ROS accumulation may also derive from altered carbon flux from the tricarboxylic acid (TCA) cycle to the glyoxylate shunt, because GaNt treatment caused greater expression of genes (aceAB, acnAB, and fumABC) encoding key enzymes involved in the glyoxylate shunt with the mutant than with wild-type cells (Fig. 5d and e). These data are consistent with the TCA cycle fueling respiration to produce ROS, while the glyoxylate shunt helps mitigate oxidative stress by bypassing two decarboxylation steps that produce NADH (31), the fuel for ATP production and ROS generation. Taken together, these observations implicate ROS accumulation in gallium-mediated bacterial death.
DISCUSSION
The present work explored the antibacterial action of gallium nitrate by enriching and identifying mutants tolerant to gallium-mediated killing: the mutations had no effect on MIC, and their effects were therefore restricted to the death process. Two gain-of-function substitutions (E701G and S582P) in EvgS, a protein involved in the response to acid stress, mitigated gallium lethality. How EvgS may participate in gallium lethality is depicted in Fig. 6.
FIG 6.
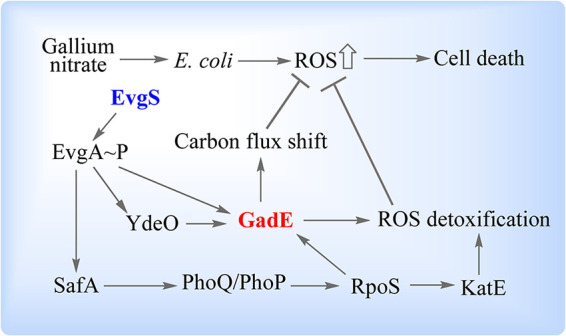
Schematic illustration of how an evgS gain-of-function mutation confers gallium nitrate tolerance. Treatment of E. coli with gallium nitrate causes a surge in intracellular ROS and subsequently results in cell death. EvgS gain-of-function substitutions constitutively stimulate EvgA phosphorylation. Phosphorylated EvgA either directly or indirectly through YdeO or SafA-PhoQ-PhoP-RpoS upregulates GadE, a central regulator of gallium tolerance. GadE shifts carbon flux from the full TCA cycle to the glyoxylate shunt, resulting in reduced NADH generation and downstream respiration that otherwise produces ROS as by-products. GadE also suppresses ROS accumulation by upregulating many genes involved in ROS detoxification, as does RpoS through upregulation of katE. Suppression of ROS accumulation protects from gallium-mediated killing.
Genetic analysis of the evgS-evgA regulatory cascade revealed that a constitutively activated gain-of-function tolerance mutation in evgS involves the phosphorelay from EvgS to EvgA. Phosphorylated EvgA then stimulates transcription of safA, ydeO, and gadE. After upregulation, YdeO can also stimulate the expression of gadE. Thus, GadE, which is a central regulator of the AR2 acid resistance pathway (24), is likely a key downstream factor in the EvgS-EvgA GaNt tolerance system. Indeed, deletion of gadE in the evgS E701G mutant background eliminated the gallium tolerance phenotype; deletion of ydeO or safA only partially reversed the tolerance phenotype, indicating that both YdeO and SafA may confer protection from GaNt-mediated killing through their partial regulation of GadE. SafA also regulates RpoS through the PhoQ-PhoP cascade, but that regulation appears to play little, if any, role in the protective effect of the EvgS E701G substitution, because protection occurs even in a ΔrpoS background despite RpoS also regulating GadE. As expected from gallium’s interference of iron-mediated redox reactions found in many respiratory enzymes, GaNt treatment causes a surge in intracellular ROS. That surge is reversed by the protective EvgS E701G substitution, thereby supporting a role for ROS in GaNt-mediated killing. To our knowledge, these observations are the first to probe the mechanism of gallium-mediated killing.
The relationship between the protective response of the EvgS-EvgA two-component system to both acid stress and gallium stress is not immediately obvious. We found that among all tested EvgS-EvgA-controlled proteins participating in acid stress, only some, apparently centering around GadE, are involved in gallium tolerance. We speculate that the EvgS-EvgA-GadE regulatory axis reduces proton gradients across the cell membrane (protecting from acid stress) and thereby downregulates ATP synthesis. That downregulation would reduce the generation of ROS, which are by-products of ATP synthesis, and would thereby reduce killing by gallium nitrate. Protection by the EvgS (E701G) substitution may also derive from its indirect upregulation of many genes that encode enzymes involved in ROS detoxification, in both the presence and absence of GaNt. The role of RpoS in GaNt lethality may derive from its regulation of katE (catalase) expression, which is highly induced during GaNt exposure, especially in the evgS E701G mutant.
We were unable to obtain resistant mutants by multiround in vitro enrichment using a highly lethal concentration of gallium nitrate (2.5× MIC). Previous work using successive sublethal (sub-MIC) concentrations obtained cells with mutations mapping in transporter genes that nonspecifically reduce gallium uptake (20, 21). The mutants we obtained specifically affected killing, not drug uptake or target interactions. These data emphasize the mechanistic distinction between blocking bacterial growth and lethal action (32). We speculate that our inability to recover resistant mutants may be due to multiple targets of gallium nitrate and to strong lethal action that kills resistant mutants before they are fixed in the population.
Refinement of GaNt for antimicrobial indications is considered feasible because FDA-approved doses achieve peak plasma concentration (28 μM) (33–36), which is above the MIC for Klebsiella pneumoniae, Mycobacterium tuberculosis, and many Acinetobacter baumannii and P. aeruginosa isolates (8–16). Toxicity issues, which occur mostly with high-bolus dosing (34, 37) or long-term, cumulative dosing of cancer patients (38), can be mitigated by mannitol diuresis (34). Moreover, toxicity may not be a widespread problem with antimicrobial applications, which are traditionally brief (39). We note that gallium induces the expression of virulence factors in P. aeruginosa at subinhibitory concentrations (40); however, this induction may not be problematic at lethal concentrations that rapidly lower bacterial numbers. Moreover, virulence factor induction can be mitigated by coadministration of brominated furanone C30 or zinc oxide nanoparticles (40, 41). Thus, approaches exist to address common clinical problems.
Although we expect that a better understanding of gallium action mechanism will lead to greater activity, lower toxicity, and an expanded spectrum of susceptible pathogens, we do not know whether GaNt will quickly succumb to resistance or whether it will be a broad-spectrum agent. Its extraordinary lethal activity may be sufficient to restrict the emergence of resistance while achieving patient cure.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and reagents.
Isogenic E. coli K-12 strains (Table S1) were acquired from the Yale University Coli Genetic Stock Center or constructed by bacteriophage P1-mediated transduction (42) or by a CRISPR (clustered regularly interspaced short palindromic repeat)-based allelic exchange (43). Plasmids and primers used for this work are listed in Tables S1 and S2 in the supplemental material. E. coli cultures were grown aerobically at 37°C in LB or MH liquid medium supplemented with 54.3 mM sodium citrate with rotary shaking at 250 rpm or as colonies on LB agar (44). Citrate was included in liquid media to mitigate GaNt-mediated medium acidification, which otherwise could inhibit growth of E. coli or even kill the bacterium, thereby confounding mechanism studies of GaNt antibacterial action. Citrate, which maintained a pH range between 3.2 and 6 after gallium nitrate addition, was chosen to reduce the possibility of high-pH-mediated gallium hydroxide formation (45), which is seen when the medium is buffered toward a more neutral pH by phosphate- or MOPS (morpholinepropanesulfonic acid)-based buffering systems. Gallium nitrate, sodium citrate, and other reagents, including antimicrobials, were obtained from Sigma-Aldrich Corp. (St. Louis, MO). Gallium nitrate stock solution (1.66 M) was prepared by dissolving the salt in 1.67 M sodium citrate. 5(6)-Carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA) was purchased from Thermo Fisher Scientific Corp. (Waltham, MA) and was dissolved in dimethyl sulfoxide (DMSO) as a 10 mM stock solution. PCR-mediated amplification and RNA isolation kits were purchased from TransGen Biotech Co. (Beijing, China). An RNA reverse transcription kit was purchased from Applied Biological Materials (Richmond, Canada).
Susceptibility determination.
The MIC was determined using a staggered 2-fold broth dilution method. Overnight cultures were first grown from frozen, single-colony seed lots; the resulting overnight cultures were diluted 200-fold and grown to exponential phase (i.e., to an optical density at 600 nm [OD600] of about 0.25), after which the cultures were diluted in LB or MH broth to ∼105 cells/ml, mixed with various amounts of drug, and incubated at 37°C for 24 h. The MIC (Table S1) was the lowest antimicrobial concentration that allowed no visible turbidity increase. Medium pH was determined using a pH meter (PB-10, Sartorius Scientific Instruments Co. Ltd.; Beijing, China) after addition of various concentrations of gallium nitrate into LB or MH broth before and after inoculation and incubation with bacterial cultures.
Determination of the hydrate amount in Ga(NO3)3·xH2O.
Since gallium nitrate purchased from Sigma-Aldrich (product no. 289892) contains an undefined amount of hydrate, we used a previously reported thermal decomposition method (22) to determine the hydrate number associated with each gallium nitrate molecule using a simultaneous thermo-analyzer (simultaneous DSC-TGA [SDT Q600 V20.9 Build 20]; TA Instruments, New Castle, DE). The MIC determined before hydrate number measurement using the anhydrous molecular weight provided in the product sheet was calibrated after the hydrate amount was experimentally determined.
Screening for gallium-resistant/tolerant mutants.
To seek mutants resistant or tolerant to GaNt, cultures of the wild-type E. coli strain BW25113 were grown to exponential phase (OD600 ∼ 0.25) in 30 ml LB medium at 37°C with shaking at 250 rpm. They were then treated with 2.5× MIC (46 mM, 11.8 mg/ml) GaNt for 40 min, which killed roughly 99.999% of the cells. Cells were concentrated by centrifugation (4,600 g, 4°C, 5 min) and resuspended in 20 ml LB medium. Centrifugation and culture resuspension were performed 2 more times to remove residual GaNt. Cultures were then diluted 100-fold, grown to exponential phase, and subjected to an additional round of enrichment. After 5 rounds of enrichment, treatment with 2.5× MIC of GaNt for 40 min killed less than 20% of the cells. These enriched cultures were washed as described above and plated on gallium-free agar and on agar containing 1× MIC of GaNt for subsequent recovery of tolerant or resistant mutants.
Whole-genome sequencing and genetic analysis for identification of tolerance mutations.
Chromosomal DNA was isolated from overnight cultures of wild-type and mutant strains using a bacterial chromosome DNA isolation kit (Tiangen Biotech Co., Beijing, China) according to the vendor’s technical manual. DNA samples were provided to Novogene Co., Ltd. (Beijing, China), for whole-genome sequencing and comparative sequence analysis against the E. coli BW25113 whole-genome sequence (GenBank accession number CP009273.1). Nonsynonymous mutations found in mutant strains were reconstructed by CRISPR-based allelic exchange (43) in the wild-type parental strain for verification of the tolerance phenotype. Additional mutants obtained during enrichment were surveyed for mutations in genes identified by whole-genome sequencing using PCR-mediated gene amplification and subsequent DNA sequence determination using primers listed in Table S2.
Bacterial killing assays.
Exponentially growing cultures (OD600 = ∼0.2 to 0.3), prepared as for MIC determination, were treated with 4× MIC GaNt at 37°C with rotary shaking at 250 rpm. At various times after treatment, samples were taken and subjected to 10-fold serial dilution in 0.9% NaCl. The diluted samples were plated (10 μl) in triplicate on drug-free LB agar. Agar plates were incubated for 24 h for CFU determination; percent survival was determined relative to an untreated culture sampled at the time of drug addition.
RNA extraction, reverse transcription, and RT-PCR measurement of gene expression.
Wild-type and evgS E701G mutant cultures were grown for 24 h and then diluted 100-fold in 25 ml LB liquid medium (containing 54.3 mM sodium citrate as indicated above) for subculturing at 37°C with 250 rpm rotary shaking until the OD600 was ∼0.3. Cultures were then treated with 4× MIC gallium nitrate. After treatment for 0 or 30 min, 5-ml bacterial samples were collected by centrifugation (4,600 × g for 10 min, 4°C), and total RNA was extracted using RNA extraction kit (TransGen Biotech Co., Beijing, China). Total RNA was reverse transcribed using random primers, and single-stranded cDNAs were synthesized according to protocols of the RNA reverse transcription kit (Applied Biological Materials, Richmond, Canada). The newly synthesized cDNA was diluted to 100 ng/ml and stored at −20°C for later use. Real-time PCR was performed using primers listed in Table S3 and the following thermal cycling profile: one cycle at 95°C for 30 s, then 40 cycles of 95°C for 5 s followed by 60°C for 30 s. Primers for 16S rRNA were used to obtain an internal reference for quantitative fluorescence RT-PCR determination of the relative transcription level for genes of interest.
Measurement of ROS by flow cytometry.
Bacterial fluorescence intensity was measured using fluorescence-based flow cytometry with a CytoFLEX A00-11102 flow cytometer (Beckman Coulter, Inc., Suzhou Xitogen Biotechnologies Co., Ltd., Suzhou, China). Carboxy-H2DCFDA (46, 47) was added to cultures at a final concentration of 10 μM for detection of intracellular ROS. A culture lacking carboxy-H2DCFDA served as a control for autofluorescence (none was observed). All culture-containing tubes were wrapped with aluminum foil to avoid light. Samples (200 μl), taken at various times, were chilled on ice, washed with 0.9% NaCl using centrifugation, and analyzed by flow cytometry. A total of 200,000 cells was analyzed at a rate of 35 μl/min for each sample to determine fluorescence. Detection parameters were 20 mV laser power and a 525/540 nm band pass filter (FITC channel). Data were analyzed using CytoFLEX software and FlowJo software.
Statistical analyses.
At least three biological replicates were used for every experiment. Each data point represents the mean of independent replicate experiments; plotted values are means and standard errors of the means (SEM).
Supplementary Material
ACKNOWLEDGMENTS
We thank Yaoqun Li and Lintao Xu for assistance in determining hydrate numbers of gallium nitrate and Yuzhi Hong for critical comments on the manuscript.
The work was supported by grants from National Natural Science Foundation of China (81673242 and 81661138005) and from a Public Health Research Institute internal bridging fund. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Rice LB. 2010. Progress and challenges in implementing the research on ESKAPE pathogens. Infect Control Hosp Epidemiol 31(Suppl 1):S7–S10. doi: 10.1086/655995. [DOI] [PubMed] [Google Scholar]
- 3.Falagas ME, Bliziotis IA. 2007. Pandrug-resistant Gram-negative bacteria: the dawn of the post-antibiotic era? Int J Antimicrob Agents 29:630–636. doi: 10.1016/j.ijantimicag.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Livermore DM, British Society for Antimicrobial Chemotherapy Working Party on The Urgent Need: Regenerating Antibacterial Drug Discovery and Development. 2011. Discovery research: the scientific challenge of finding new antibiotics. J Antimicrob Chemother 66:1941–1944. doi: 10.1093/jac/dkr262. [DOI] [PubMed] [Google Scholar]
- 5.Brown ED, Wright GD. 2016. Antibacterial drug discovery in the resistance era. Nature 529:336–343. doi: 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]
- 6.Savoia D. 2016. New antimicrobial approaches: reuse of old drugs. Curr Drug Targets 17:731–738. doi: 10.2174/1389450116666150806124110. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein LR. 1998. Mechanisms of therapeutic activity for gallium. Pharmacol Rev 50:665–682. [PubMed] [Google Scholar]
- 8.Arivett BA, Fiester SE, Ohneck EJ, Penwell WF, Kaufman CM, Relich RF, Actis LA. 2015. Antimicrobial activity of gallium protoporphyrin IX against Acinetobacter baumannii strains displaying different antibiotic resistance phenotypes. Antimicrob Agents Chemother 59:7657–7665. doi: 10.1128/AAC.01472-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banin E, Lozinski A, Brady KM, Berenshtein E, Butterfield PW, Moshe M, Chevion M, Greenberg EP, Banin E. 2008. The potential of desferrioxamine-gallium as an anti-Pseudomonas therapeutic agent. Proc Natl Acad Sci U S A 105:16761–16766. doi: 10.1073/pnas.0808608105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonchi C, Imperi F, Minandri F, Visca P, Frangipani E. 2014. Repurposing of gallium-based drugs for antibacterial therapy. Biofactors 40:303–312. doi: 10.1002/biof.1159. [DOI] [PubMed] [Google Scholar]
- 11.Kaneko Y, Thoendel M, Olakanmi O, Britigan BE, Singh PK. 2007. The transition metal gallium disrupts Pseudomonas aeruginosa iron metabolism and has antimicrobial and antibiofilm activity. J Clin Invest 117:877–888. doi: 10.1172/JCI30783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coleman M, Kuskie K, Liu M, Chaffin K, Libal M, Giguere S, Bernstein L, Cohen N. 2010. In vitro antimicrobial activity of gallium maltolate against virulent Rhodococcus equi. Vet Microbiol 146:175–178. doi: 10.1016/j.vetmic.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 13.Thompson MG, Truong-Le V, Alamneh YA, Black CC, Anderl J, Honnold CL, Pavlicek RL, Abu-Taleb R, Wise MC, Hall ER, Wagar EJ, Patzer E, Zurawski DV. 2015. Evaluation of gallium citrate formulations against a multidrug-resistant strain of Klebsiella pneumoniae in a murine wound model of infection. Antimicrob Agents Chemother 59:6484–6493. doi: 10.1128/AAC.00882-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdalla MY, Switzer BL, Goss CH, Aitken ML, Singh PK, Britigan BE. 2015. Gallium compounds exhibit potential as new therapeutic agents against Mycobacterium abscessus. Antimicrob Agents Chemother 59:4826–4834. doi: 10.1128/AAC.00331-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antunes LC, Imperi F, Minandri F, Visca P. 2012. In vitro and in vivo antimicrobial activities of gallium nitrate against multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 56:5961–5970. doi: 10.1128/AAC.01519-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olakanmi O, Britigan BE, Schlesinger LS. 2000. Gallium disrupts iron metabolism of mycobacteria residing within human macrophages. Infect Immun 68:5619–5627. doi: 10.1128/iai.68.10.5619-5627.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olakanmi O, Kesavalu B, Pasula R, Abdalla MY, Schlesinger LS, Britigan BE. 2013. Gallium nitrate is efficacious in murine models of tuberculosis and inhibits key bacterial Fe-dependent enzymes. Antimicrob Agents Chemother 57:6074–6080. doi: 10.1128/AAC.01543-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goss CH, Kaneko Y, Khuu L, Anderson GD, Ravishankar S, Aitken ML, Lechtzin N, Zhou G, Czyz DM, McLean K, Olakanmi O, Shuman HA, Teresi M, Wilhelm E, Caldwell E, Salipante SJ, Hornick DB, Siehnel RJ, Becker L, Britigan BE, Singh PK. 2018. Gallium disrupts bacterial iron metabolism and has therapeutic effects in mice and humans with lung infections. Sci Transl Med 10:eaat7520. doi: 10.1126/scitranslmed.aat7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goss CHH, Aitken ML, Caldwell E, Wilhelm E, Wolfstone A, Teresi M, Singh PK. 2012. Phase I pharmacokinetic and safety study of intravenous Ganite TM (gallium nitrate) in CF. Pediatric Pulmunol 47:303. [Google Scholar]
- 20.Garcia-Contreras R, Lira-Silva E, Jasso-Chavez R, Hernandez-Gonzalez IL, Maeda T, Hashimoto T, Boogerd FC, Sheng L, Wood TK, Moreno-Sanchez R. 2013. Isolation and characterization of gallium resistant Pseudomonas aeruginosa mutants. Int J Med Microbiol 303:574–582. doi: 10.1016/j.ijmm.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Graves JL, Jr, Ewunkem AJ, Ward J, Staley C, Thomas MD, Rhinehardt KL, Han J, Harrison SH. 2019. Experimental evolution of gallium resistance in Escherichia coli. Evol Med Public Health 2019:169–180. doi: 10.1093/emph/eoz025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melnikov P, Nascimento VA, Consolo LZZ. 2012. Thermal decomposition of gallium nitrate hydrate and modeling of thermolysis products. J Therm Anal Calorim 107:1117–1121. doi: 10.1007/s10973-011-1788-y. [DOI] [Google Scholar]
- 23.Brauner A, Fridman O, Gefen O, Balaban NQ. 2016. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat Rev Microbiol 14:320–330. doi: 10.1038/nrmicro.2016.34. [DOI] [PubMed] [Google Scholar]
- 24.Johnson MD, Bell J, Clarke K, Chandler R, Pathak P, Xia Y, Marshall RL, Weinstock GM, Loman NJ, Winn PJ, Lund PA. 2014. Characterization of mutations in the PAS domain of the EvgS sensor kinase selected by laboratory evolution for acid resistance in Escherichia coli. Mol Microbiol 93:911–927. doi: 10.1111/mmi.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itou J, Eguchi Y, Utsumi R. 2009. Molecular mechanism of transcriptional cascade initiated by the EvgS/EvgA system in Escherichia coli K-12. Biosci Biotechnol Biochem 73:870–878. doi: 10.1271/bbb.80795. [DOI] [PubMed] [Google Scholar]
- 26.Roggiani M, Yadavalli SS, Goulian M. 2017. Natural variation of a sensor kinase controlling a conserved stress response pathway in Escherichia coli. PLoS Genet 13:e1007101. doi: 10.1371/journal.pgen.1007101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bougdour A, Cunning C, Baptiste PJ, Elliott T, Gottesman S. 2008. Multiple pathways for regulation of sigmaS (RpoS) stability in Escherichia coli via the action of multiple anti-adaptors. Mol Microbiol 68:298–313. doi: 10.1111/j.1365-2958.2008.06146.x. [DOI] [PubMed] [Google Scholar]
- 28.Eguchi Y, Ishii E, Yamane M, Utsumi R. 2012. The connector SafA interacts with the multi-sensing domain of PhoQ in Escherichia coli. Mol Microbiol 85:299–313. doi: 10.1111/j.1365-2958.2012.08114.x. [DOI] [PubMed] [Google Scholar]
- 29.Eguchi Y, Utsumi R. 2014. Alkali metals in addition to acidic pH activate the EvgS histidine kinase sensor in Escherichia coli. J Bacteriol 196:3140–3149. doi: 10.1128/JB.01742-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zorov DB, Juhaszova M, Sollott SJ. 2014. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94:909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahn S, Jung J, Jang IA, Madsen EL, Park W. 2016. Role of glyoxylate shunt in oxidative stress response. J Biol Chem 291:11928–11938. doi: 10.1074/jbc.M115.708149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao X, Hong Y, Drlica K. 2015. Moving forward with reactive oxygen species involvement in antimicrobial lethality. J Antimicrob Chemother 70:639–642. doi: 10.1093/jac/dku463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelsen DP, Alcock N, Yeh S, Brown J, Young C. 1980. Pharmacokinetics of gallium nitrate in man. Cancer 46:2009–2013. doi:. [DOI] [PubMed] [Google Scholar]
- 34.Krakoff IH, Newman RA, Goldberg RS. 1979. Clinical toxicologic and pharmacologic studies of gallium nitrate. Cancer 44:1722–1727. doi:. [DOI] [PubMed] [Google Scholar]
- 35.Seligman PA, Moran PL, Schleicher RB, Crawford ED. 1992. Treatment with gallium nitrate: evidence for interference with iron metabolism in vivo. Am J Hematol 41:232–240. doi: 10.1002/ajh.2830410403. [DOI] [PubMed] [Google Scholar]
- 36.Todd PA, Fitton A. 1991. Gallium nitrate. A review of its pharmacological properties and therapeutic potential in cancer related hypercalcaemia. Drugs 42:261–273. doi: 10.2165/00003495-199142020-00007. [DOI] [PubMed] [Google Scholar]
- 37.Bedikian AY, Valdivieso M, Bodey GP, Burgess MA, Benjamin RS, Hall S, Freireich EJ. 1978. Phase I clinical studies with gallium nitrate. Cancer Treat Rep 62:1449–1453. [PubMed] [Google Scholar]
- 38.Senderowicz AM, Reid R, Headlee D, Abornathy T, Horti J, Lush RM, Reed E, Figg WD, Sausville EA. 1999. A phase II trial of gallium nitrate in patients with androgen-metastatic prostate cancer. Urol Int 63:120–125. doi: 10.1159/000030430. [DOI] [PubMed] [Google Scholar]
- 39.Bernstein LR. 2005. Therapeutic gallium compounds, p 259–277. In Tiekink G (ed), Metallotherapeutic drugs and metal-based diagnostic agents: the use of metals in medicine. John Wiley & Sons, Ltd., Oxford, United Kingdom. [Google Scholar]
- 40.Garcia-Contreras R, Perez-Eretza B, Lira-Silva E, Jasso-Chavez R, Coria-Jimenez R, Rangel-Vega A, Maeda T, Wood TK. 2014. Gallium induces the production of virulence factors in Pseudomonas aeruginosa. Pathog Dis 70:95–98. doi: 10.1111/2049-632X.12105. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Lara B, Saucedo-Mora MA, Roldan-Sanchez JA, Perez-Eretza B, Ramasamy M, Lee J, Coria-Jimenez R, Tapia M, Varela-Guerrero V, Garcia-Contreras R. 2015. Inhibition of quorum-sensing-dependent virulence factors and biofilm formation of clinical and environmental Pseudomonas aeruginosa strains by ZnO nanoparticles. Lett Appl Microbiol 61:299–305. doi: 10.1111/lam.12456. [DOI] [PubMed] [Google Scholar]
- 42.Wall JD, Harriman PD. 1974. Phage P1 mutants with altered transducing abilities for Escherichia coli. Virology 59:532–544. doi: 10.1016/0042-6822(74)90463-2. [DOI] [PubMed] [Google Scholar]
- 43.Jiang Y, Chen B, Duan C, Sun B, Yang J, Yang S. 2015. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl Environ Microbiol 81:2506–2514. doi: 10.1128/AEM.04023-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller J. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 45.Hacht B. 2008. Gallium(III) ion hydrolysis under physiological conditions. Bull Korean Chem Soc 29:372–376. [Google Scholar]
- 46.Bass DA, Parce JW, Dechatelet LR, Szejda P, Seeds MC, Thomas M. 1983. Flow cytometric studies of oxidative product formation by neutrophils: a graded response to membrane stimulation. J Immunol 130:1910–1917. [PubMed] [Google Scholar]
- 47.Brandt R, Keston AS. 1965. Synthesis of diacetyldichlorofluorescin: a stable reagent for fluorometric analysis. Anal Biochem 11:6–9. doi: 10.1016/0003-2697(65)90035-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.