

The ability of HIV to integrate into the host genome and establish latent reservoirs is the main hurdle preventing an HIV cure. LEDGINs are small-molecule integrase inhibitors that target the binding pocket of LEDGF/p75, a cellular cofactor that substantially contributes to HIV integration site selection.
KEYWORDS: HIV, LEDGIN, antiviral agents, block and lock, functional cure, integration
ABSTRACT
The ability of HIV to integrate into the host genome and establish latent reservoirs is the main hurdle preventing an HIV cure. LEDGINs are small-molecule integrase inhibitors that target the binding pocket of LEDGF/p75, a cellular cofactor that substantially contributes to HIV integration site selection. They are potent antivirals that inhibit HIV integration and maturation. In addition, they retarget residual integrants away from transcription units and toward a more repressive chromatin environment. As a result, treatment with the LEDGIN CX14442 yielded residual provirus that proved more latent and more refractory to reactivation, supporting the use of LEDGINs as research tools to study HIV latency and a functional cure strategy. In this study, we compared GS-9822, a potent, preclinical lead compound, with CX14442 with respect to antiviral potency, integration site selection, latency, and reactivation. GS-9822 was more potent than CX14442 in most assays. For the first time, the combined effects on viral replication, integrase-LEDGF/p75 interaction, integration sites, epigenetic landscape, immediate latency, and latency reversal were demonstrated at nanomolar concentrations achievable in the clinic. GS-9822 profiles as a preclinical candidate for future functional cure research.
INTRODUCTION
Present day combination antiretroviral therapy (cART) for HIV-1 infection is mainly based on four classes of antivirals targeting the HIV enzymes, nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), nonnucleotide reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), and integrase strand transfer inhibitors (INSTIs), though entry and fusion inhibitors have been developed as well (1). INSTIs such as dolutegravir and bictegravir are now part of preferred first-line cART cocktails (2–4). cART has been refined to once-daily regimens, and the extent of adverse effects has been greatly reduced. Still, there is no cure for the 37.9 million people living with HIV (5), as current antiretroviral treatments cannot clear HIV from the body. HIV integrates a provirus into the genome of the cells it infects, which can either lead to productive replication or remain in a latent state (6). Therefore, HIV can persist for decades in long-living memory cells, and this HIV persistence is a major focus in HIV research today.
In an attempt to eradicate HIV reservoirs, researchers have proposed the “shock and kill” or “kick and kill” strategy (7). Briefly, the goal is to reactivate latently infected cells so they are killed off by either viral cytopathic effects or the host immune system. Unfortunately, reactivating a sufficient fraction of latently infected cells and thus achieving a significant reduction of the latent reservoirs has proven difficult (8). An alternate approach is the “block and lock” strategy (9–11). In this approach to a “functional cure” or “HIV remission,” the virus is not eradicated, but a state of deep latency is induced, leading to sustained virological control in the absence of cART.
When HIV infects a cell, after virus entry, the reverse transcriptase copies the viral RNA into double-stranded viral DNA (vDNA). Along with other viral and cellular proteins, the vDNA and HIV integrase then form the so-called preintegration complex (PIC). Once inside the nucleus, integrase catalyzes both a 3′ processing and a strand transfer reaction, permanently inserting the vDNA into the host cell genome. The strand transfer reaction is impaired by INSTIs through competition with the vDNA for binding to the integrase catalytic core (12). HIV integrase has little resemblance to human proteins, resulting in a low risk for off-target effects and presents as an attractive therapeutic target (13). Next to INSTIs, a second class of integrase inhibitors exist, which target the interaction of HIV integrase with lens epithelium-derived growth factor (LEDGF/p75).
Albeit not sequence-specific, HIV integration is not random (14). HIV preferentially integrates in actively transcribed, gene-dense regions (14–20). One of the most important determinants of this selectivity is the interaction of HIV integrase with LEDGF/p75. LEDGF/p75 is ubiquitously expressed in human cells and functions as a transcriptional coactivator, tethering various proteins to active chromatin through recognition of the histone modification H3K36me3 by its N-terminal PWWP domain (21, 22). Next to PWWP, LEDGF/p75 is characterized by AT-hooks for chromatin interaction (23, 24) and a C-terminal domain containing the integrase binding domain (IBD) to which both HIV integrase and cellular partners bind (25–27). The dimeric catalytic core domain of all lentiviral integrases specifically interacts with this IBD (27–30). During HIV infection, LEDGF/p75 acts as a molecular tether linking the HIV PIC to the chromatin. In addition, it stimulates the integration reaction (25) and determines HIV-1 integration sites by targeting HIV toward active transcription units (18, 31–33).
An interhelical loop between the first two α-helices of the IBD (α1 and α2) of LEDGF/p75 fits in a tight cleft formed at the interface of the catalytic core dimer of integrase, outside the actual catalytic site which is targeted by INSTIs (29). Structure-based drug design led to the discovery of small molecules (LEDGINs) that mimic the IBD loop, disturb the integrase-LEDGF/p75 interaction, and inhibit HIV integration. This mechanism of action is now referred to as the early effect of LEDGINs (13, 34–36). As the binding site of LEDGINs differs from that of INSTIs, there is no cross-resistance between the two classes, and combination treatment even showed synergistic effects (37, 38). Interestingly, LEDGINs also affect HIV particle maturation (39–42), leading to less infectious viral particles, the so-called late effect. During assembly and through interaction with the LEDGF/p75 binding pocket in integrase, LEDGINs induce premature integrase multimerization which leads to a deformed viral capsid and displaces the viral genome outside the viral core, resulting in crippled viral progeny, with deficits in reverse transcription, nuclear import, and integration (39–43). Interestingly, the late effect occurs at lower LEDGIN concentrations and contributes to the potency of these compounds.
Various names have been used in literature to describe these molecules, referring to either the entire class or specific subsets—small molecules binding to the LEDGF/p75 binding site on integrase, (LEDGINs) (34); noncatalytic site integrase inhibitors or (NCINIs) (41), allosteric integrase inhibitors (ALLINIs) (44, 45), multimerization selective integrase inhibitors (MINIs) (46), and integrase-LEDGF allosteric inhibitors (INLAIs) (42). We prefer to call this novel class of antivirals LEDGINs since all chemotypes share the LEDGF/p75 binding site of HIV integrase that mediates both early and late effects. In addition, the first compounds belonging to this class were referred to as LEDGINs (35). Various LEDGINs with submicromolar and even nanomolar activity have been developed since (13, 35, 36, 47). After the discovery of the 3-quinoline acetic acid scaffold in a high-throughput 3′-processing assay, extensive lead optimization led to the development of BI224436 (48). In preclinical testing BI224436 showed potent in vitro antiviral activity against different HIV-1 laboratory strains in the low nanomolar range, no cross-resistance with common INSTI resistance mutations (including N155S, Q148H, and E92Q), and excellent pharmacokinetic profiles in rats, monkeys, and dogs (37). The compound was the first LEDGIN to advance into phase Ia clinical trials, showing adequate plasma concentrations at 100 mg and no safety concerns at concentrations up to 200 mg (49). A long-standing pursuit at Gilead for a novel antiretroviral agent with the potential for low-dose, unboosted once-daily oral dosing and a high barrier to resistance resulted in the identification of GS-9822 (Fig. 1d) (50). Screening of representatives from the LEDGIN library against a virus harboring the HIV-1 integrase polymorphisms A124T and A128T and the LEDGIN resistance mutation T174I revealed that the 5-indazolyl substitution of the 2-benzothiazole core gave a lower potency shift relative to wild-type virus. Subsequent optimization of the 5-indazole moiety yielded substantial improvements in potency while maintaining activity against LEDGIN binding-pocket variants. GS-9822 has high in vitro metabolic stability and favorable oral pharmacokinetic profiles with low systemic clearance in rats, dogs, and monkeys (50). However, a unique and difficult-to-monitor urothelial toxicity was observed in cynomolgus monkeys that poses a formidable challenge for further development of GS-9822.
FIG 1.
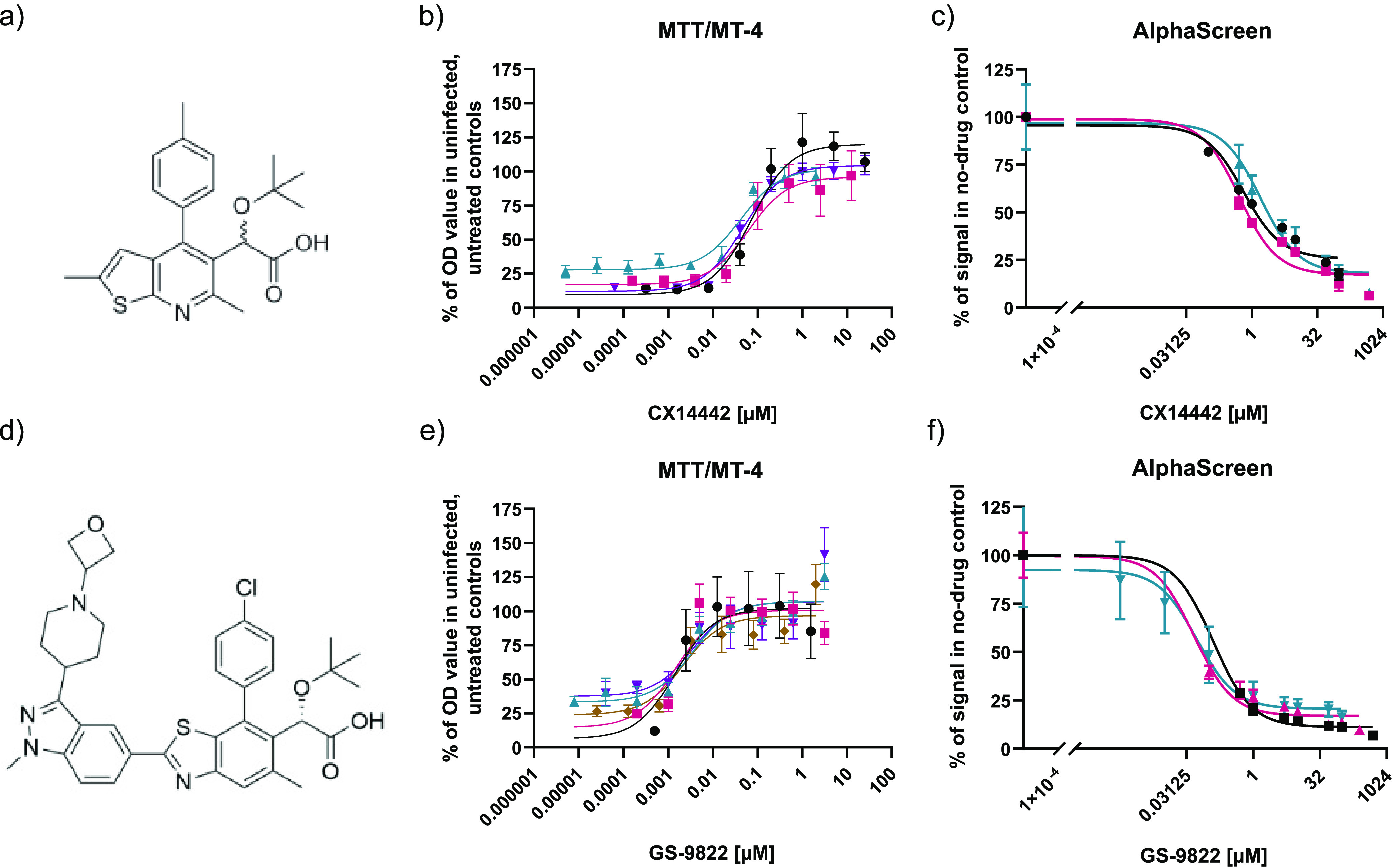
CX14442 and GS-9822 block HIV-1-induced cell toxicity and inhibit the interaction between HIV-1 integrase and LEDGF/p75. (a and d) Chemical structures of CX14442 and GS-9822. (b and e) Dose-response curves for MTT viability assays with CX14442 and GS-9822 in MT4-cells 5 days after infection with HIV-1 (strain IIIB). Cell viability, measured as optical density (OD) values, is greatly reduced upon HIV-1 infection but increases upon addition of the compounds. Results are plotted as a percentage of the OD values obtained in uninfected, untreated cells within the same experiment. Mean values and standard deviations (SD) are shown for CX14442 (n = 4) and GS-9822 (n = 5). Each sample was run in triplicate. (c and f) Dose-response curves of CX14442 and GS-9822 in AlphaScreen. Increasing concentrations of compound were added to 50 nM HIV-1 integrase (strain NL4.3) and 100 nM LEDGF/p75. Data shown are mean values and standard deviations for 3 experiments, with each analysis performed in duplicate. Data are plotted as a percentage of the signal in the no-drug control.
CX05045 and CX14442 are LEDGINs with submicromolar potency (38) used as research compounds to investigate the role of LEDGF/p75 in integration site selection (51). CX14442 (Fig. 1a) is a 2-(quinolin-3-yl)acetic acid with a tert-butyl ether as a hydrophobic bulk on the acetic acid 2-position, which aids in filling up the LEDGF/p75 binding pocket on HIV integrase, resulting in increased activity (38). As was previously shown for LEDGF/p75 depletion (31–33, 52, 53), treatment with LEDGIN CX14442 during transduction with a replication-deficient HIV-1-based vector reduced the number of integrated proviruses. In addition, LEDGIN treatment retargeted the site of residual integration away from active transcription units (51, 54). After treatment with CX05045 (38), the residual provirus was located away from the nuclear periphery toward the center of the nucleus (51). Importantly, by using the HIV-1 OGH double reporter vector (51, 55–58), the residual integrants after CX14442 treatment were shown to be more latent and more resistant to reactivation (51). Using barcoded HIV vectors (B-HIVE), the Fillion lab showed that integration in proximity to enhancers affects transcription and reactivation by different latency reversing agents (19). In a recent study, the same barcoded vector technology was used to show that CX14442 shifts integration away from H3K36me3, resulting in lower transcription levels (20). Residual transcription of retargeted provirus was most pronounced in the proximity of (super) enhancers. However, micromolar concentrations of CX14442 were necessary to achieve these results, as they are dependent on the early effect. The impact of the so-called late-effect of LEDGINs, leading to integrase multimerization and crippled progeny virions, on integration site selection in a subsequent round of infection was also studied (58). By treating only the producer cells with CX14442, residual integrants of a replication-deficient HIV vector were still retargeted toward less-gene-dense regions and a more repressive epigenetic environment, though the effect on transcription units and H3K36me3 was less clear (58). Interestingly, the residual integrants were more quiescent and resistant to reactivation. Lastly, the effect of treatment with CX14442 during multiple-round infection of primary cells with wild-type HIV was also investigated (58). Both retargeting out of transcription units and the creation of a more refractory reservoir were confirmed in this model that scores for both the early and the late effect (51, 58).
Here, we report on a side-by-side comparison of the preclinical lead compound GS-9822 with CX14442 with respect to their relative impacts on integration site selection, latency, and reactivation through the early effect of LEDGINs. GS-9822 displayed a block-and-lock phenotype at nanomolar concentrations, supporting a potential future clinical use of LEDGINs in a functional cure strategy.
RESULTS
GS-9822 has potent antiviral activity against wild-type HIV-1 viruses.
First, the 50% effective concentration (EC50) of CX14442 and GS-9822 against wild-type HIV-1 was determined by infecting MT-4 cells with wild-type HIV-1 subtype B viruses (Fig. 1b and e; Table 1). GS-9822 was 23-fold more potent against the IIIb strain and over 100-fold more potent against NL4.3 than CX14442, with an EC50 in the low nanomolar range for each strain. The 50% cytotoxic concentration (CC50) value of GS-9822 was about 15-fold lower than that of CX14442 in these cells. Due to its potency, the selectivity index (SI) of GS-9822 was still 2-fold higher than that of CX14442.
TABLE 1.
Potency of CX14442 and GS-9822 in antiviral and protein-protein interaction assays
| LEDGIN | AlphaScreen |
MTT/MT-4 assay |
||||||
|---|---|---|---|---|---|---|---|---|
| LEDGF/p75 - integrase IC50 (μM)a,f | CC50 (μM)b,f | Strain IIIb |
Strain NL4.3 |
|||||
| EC50 (μM)c,f | EC90 (μM)d,f | SIe,f | EC50c,g | EC90d,g | SIe,g | |||
| CX14442 | 0.92 ± 0.34 | 70.832 ± 7.022 | 0.051 ± 0.004 | 0.814 ± 0.594 | 1,725 | 0.296 ± 0.135 | 0.623 ± 0.345 | 239 |
| GS-9822 | 0.07 ± 0.02 | 4.7570 ± 0.5972 | 0.0022 ± 0.0003 | 0.0928 ± 0.0593 | 3,570 | 0.0025 ± 0.0008 | 0.0042 ± 0.0011 | 2,162 |
Inhibitory concentration (μM) required to inhibit the in vitro protein-protein interaction by 50%.
Cytotoxic concentration (μM) reducing cell viability by 50%.
Effective concentration (μM) required to reduce HIV-1-induced cytopathic effects by 50%.
Effective concentration (μM) required to reduce HIV-1-induced cytopathic effects by 90%.
SI, selectivity index: CC50/EC50.
Mean and SEM of at least 3 independent experiments.
Mean and SEM of 2 independent experiments.
GS-9822 potently inhibits the LEDGF/p75-integrase interaction in vitro.
Next, we evaluated if GS-9822 inhibits the integrase-LEDGF/p75 interaction, the mechanism of action required to affect integration site selection. Using X-ray crystallography, it was shown before that GS-9822 binds integrase at the LEDGF/p75 interaction interface (50). Therefore, AlphaScreen interaction assays with His6-tagged HIV-1 integrase and Flag-tagged LEDGF/p75 (34) were performed in the presence of increasing concentrations of CX14442 or GS-9822. A dose-dependent inhibition by each compound was evidenced (Fig. 1c and f; Table 1). GS-9822 inhibited the interaction at a 10-fold lower concentration (IC50 = 0.07 ± 0.02 μM) than CX14442 (50% inhibitory concentration [IC50], 0.92 ± 0.34 μM).
GS-9822 potently retargets HIV-1 integration.
To study the effect of GS-9822 on integration site selection, 100,000 SupT1 cells were transduced with 1.2 × 105 pg of the lentiviral vector CH-SFFV-eGFP-P2A-fLuc (Fig. 2a) and cultured for 3 days in the presence of GS-9822 or CX14442 or no drug. Cells were then washed and kept in culture for at least 10 days in the absence of compound to allow for the dilution of nonintegrated DNA. All samples were obtained from a single transduction experiment. Genomic DNA was extracted and prepared for Illumina Miseq integration site sequencing as well as Alu-long terminal repeat (Alu-LTR) quantitative PCR (qPCR) for proviral copy numbers. Sequencing data were analyzed using the INSPIIRED platform (59, 60). CX14442 and GS-9822 both induced a dose-dependent reduction in the number of integrated proviruses (see Fig. S1b in the supplemental material). Among the residual integrants, a reduction in the number of unique integration sites was detected for both inhibitors by the next-generation sequencing analysis (Fig. S1a). Thus, CX14442 and GS-9822 both inhibited HIV-1 OGH integration. In line with previous results (14–20), integration into genes was strongly favored in the no-drug control condition. Treatment with the compounds resulted in a proportion (up to 13%) of the residual integrants retargeting to regions outside genes (Table 2, Fig. S2), as was previously demonstrated for CX14442 (20, 51), though integration into genes was still preferred compared with the matched random controls. In addition, a dose-dependent reduction in the gene count surrounding residual integration sites was observed (Fig. 2b and d; Fig. S2 and S4). The effect was more pronounced with CX14442, with a maximal reduction (53.2%) at 25 μM (Fig. 2b), while a similar but less pronounced trend was also observed with GS-9822 (Fig. 2d). Under the no-drug control condition, the frequency of HIV-1 integration shows a positive correlation with the gene density per chromosome (Fig. 2c and e), as was shown previously (20). Treatment with GS-9822 significantly decreased the slope of this correlation for all the concentrations used. Taken together, these results point to LEDGINs retargeting residual integrants away from gene-dense regions as well.
FIG 2.
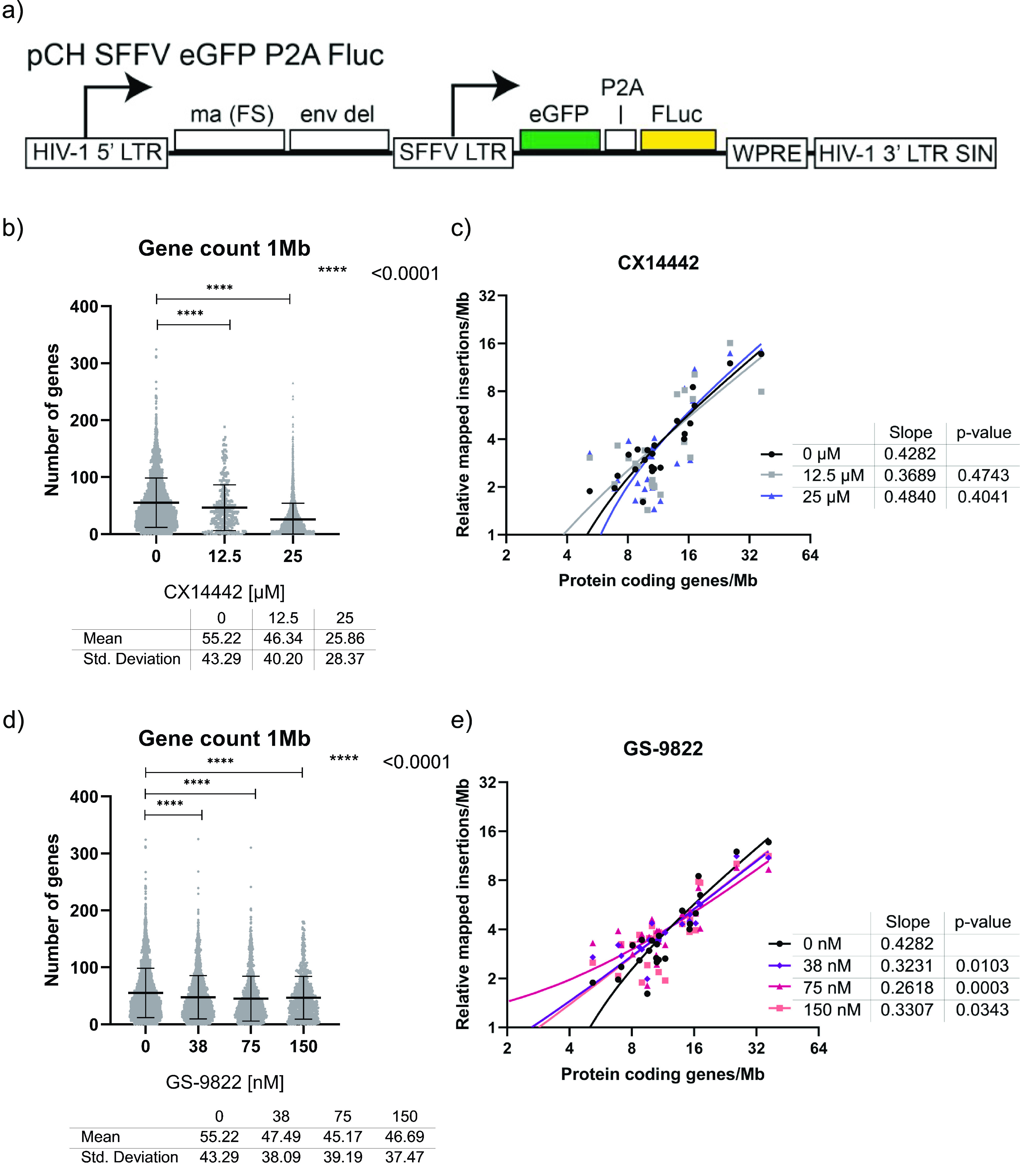
GS-9822 retargets integration away from gene-dense regions. SupT1 cells were transduced for 3 days with CH-SFFV-eGFP-P2A-fLuc in the presence or absence of various drug concentrations and kept in culture for at least 10 days. Next, genomic DNA was extracted for Illumina Miseq integration site sequencing, and data were analyzed via the INSPIIRED platform (59, 60). (a) Schematic representation of CH-SFFV-eGFP-P2A-fluc, a single round lentiviral vector containing an enhanced green fluorescent protein (eGFP) and firefly luciferase (fLuc) reporter gene used for integration site sequencing. (b and d) Graphs plotting the number of genes counted within a 1-Mb range of each integration site for samples treated with CX14442 and GS-9822, respectively. Annotated data were obtained using the University of California Santa Cruz (UCSC) Genome Browser website (http://genome.ucsc.edu, UCSC Known Genes) (90, 91). Mean values and standard deviations are plotted on top. Samples were compared to the no-compound condition using a Kruskall-Wallis test. (c and e) XY-plots showing the relative number of mapped insertions/Mb (y axis) over the UCSC protein coding gene density of each chromosome (x axis). For each sample, we calculated the insertions per Mb per chromosome, relative to the number of mapped insertions per condition to compensate for the differences in the number of integration sites between samples. For each condition, the total number of sites for all chromosomes is 100. Protein-coding genes were defined as gene entries containing a UniProt protein ID.
TABLE 2.
CX14442 and GS-9822 retarget integration away from gene-dense regions
| Type | CX14442 (μM) | No. of unique sites | Sites in genes (%)a,c | Sites in protein coding genes (%)b,c |
|---|---|---|---|---|
| Integration site | 0 | 11,624 | 76.49 | 72.76 |
| 12.5 | 350 | 70.57 (**) | 68.86 | |
| 25 | 427 | 71.66 (*) | 68.85 | |
| MRC | 0 | 34,872 | 41.75 | 37.28 |
| 12 | 1,050 | 39.52 | 36.38 | |
| 25 | 1,281 | 40.67 | 36.85 |
Percentage of integration sites within genes, calculated based on the University of Califorina Santa Cruz (UCSC) genes data set, which itself is based on data from RefSeq, GenBank, the Consensus Coding Sequence (CCDS) database, and UniProt (90, 91).
Percentage of integration sites within protein-coding genes, calculated based on the USCS genes data set. Protein-coding genes were defined as gene entries containing a UniProt protein ID.
Asterisks represent a statistically significant deviation from the control data set as calculated by the Chi-square test. *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
MRC, matched random control.
We analyzed other genomic markers to see if we could confirm these results (Fig. S1c to r and S2). DNase I hypersensitive sites, a marker for actively transcribed chromatin, were found slightly less frequently in proximity to residual integrants. As CpG islands occur mostly in proximity of promoters, the CpG count correlates with the number of genes in the region when examined at large ranges. With an increased concentration of GS-9822 (and, to a lesser extent, CX14442), a decrease in the CpG count at 1 Mb was seen. Gene-dense regions also tend to be more GC rich. As expected, the GC content surrounding residual integrants in a 1 kb range decreased with increasing concentrations of GS-9822 and CX14442, though the effect was most clear for GS-9822. Finally, the width of intergenic regions and genes is inversely correlated with gene density. We observed a trend toward an increase in the width of genes and intergenic regions with both GS-9822 and CX14442, implying once again a shift away from gene-dense regions, although this observation was not statistically significant. When looking at these genomic markers, results were consistent with a compound-induced retargeting away from gene-dense regions compared to the no-drug control, though gene-dense regions were still preferred compared with the matched random controls.
Overall, treatment with GS-9822 resulted in a similar retargeting pattern as previously observed for CX14442 (20, 51). However, nanomolar concentrations of the highly potent GS-9822 were sufficient to induce the retargeting, and for most parameters the effect of GS-9822 was more pronounced.
Retargeting of HIV-1 integration sites results in a different epigenetic landscape of the residual provirus.
The INSPIIRED platform was used to generate an epigenetic heat map, to analyze the effect of the retargeting on the epigenetic landscape surrounding the provirus (Fig. S3). Methylation marks that are associated with repressed chromatin, such as H4K20me3, H3K27me2, H3K27me3, H3K9me3, and H3K9me2 (61–64) are normally disfavored by HIV (51, 65–68). The presence of GS-9822 increased residual integration toward these methylation marks, even reaching levels comparable to the matched random controls for H4K20m3 under the 150-nM conditions. CX14442 produced a similar effect, though less pronounced, consistent with our previous observations (20, 51). Moreover, for GS-9822 in particular, integration was also shifted away from methylation marks usually highly favored by HIV, most notably H3K36me3, the methylation mark recognized by LEDGF/p75 (21, 22, 68–70). Like H3K36me3, H3K27me1, H2BK5me1, H4K20me1, and H3K9me1 are usually found along active gene bodies (61–64, 71–74) and favored by HIV (20, 51, 65–68). GS-9822 treatment shifted integration away from these marks, though the effect is less clear for H3K9me1, and integration near these marks was still favored compared to the matched random controls. These results further support our observation that LEDGINs and GS-9822 in particular retarget integration away from active transcription units (20, 51). Overall, a clear shift in the epigenetic features of the HIV integration sites was observed in the presence of the LEDGIN compounds, which was more pronounced with GS-9822 than with CX14442.
GS-9822 reduces infectivity and increases immediate latency of a single round, double reporter vector.
CX14442-mediated retargeting of HIV-1 integration to an altered chromatin landscape was associated with an increased immediate HIV-1 latency and less reactivation (20, 51). To study if treatment with GS-9822 is capable of similar effects at nanomolar concentrations, we used the HIV-1 OGH double reporter vector (51, 55, 56, 58, 75) (Fig. 3a). This previously described reporter vector combines LTR-dependent expression of enhanced green fluorescent protein (eGFP) replacing nef and a second reporter transcription unit immediately downstream, consisting of a constitutively active EF1α promoter, driving mutant Kusabira-Orange 2 (mKO2) reporter gene expression. This system allows us to distinguish cells containing productive and latent proviruses via flow cytometry (Fig. 3c). Other researchers have used this double reporter to show differences in integration sites between inducible and noninducible latent HIV-1 proviruses (75). SupT1 cells (3 × 105 cells) were transduced with 60 × 106 to 240 × 106 pg of the HIV-1 OGH double reporter in the presence of various concentrations of CX14442 or GS-9822. Raltegravir, an INSTI, was used as a control (Fig. 3b). We performed the first flow cytometry analysis 3 days after transduction. As expected, all three integrase inhibitors inhibited HIV-1 transduction as measured by the percentage of mKO2-positive cells with IC50 values of 0.002 μM, 0.007 μM, and 1.6 μM for RAL, GS-9822, and CX14442, respectively (Fig. 4a to c). For all compounds, the decrease in the number of eGFP-positive cells correlated with the decrease in proviral copy numbers per cell (Fig. 4a to c and e). When calculating the latent fraction (% single mKO2+ cells/[% single mKO2+ cells + % double positive cells] × 100 or C/[B + C] × 100 [Fig. 3c]), treatment with GS-9822 or CX14442 resulted in a dose-dependent increase in the latent fraction (Fig. 4d; Fig. S5a). For raltegravir, an increase in latency was only seen at concentrations of 17-fold the IC50 (Fig. 4d; Fig. S5a). Although the increase in latent fraction appears similar for GS-9822 and CX14442 when plotted with respect to fold IC50, the IC50 of GS-9822 is over 200-fold lower than that of CX14442, indicating that GS-9822 increases immediate HIV latency at nanomolar concentrations.
FIG 3.
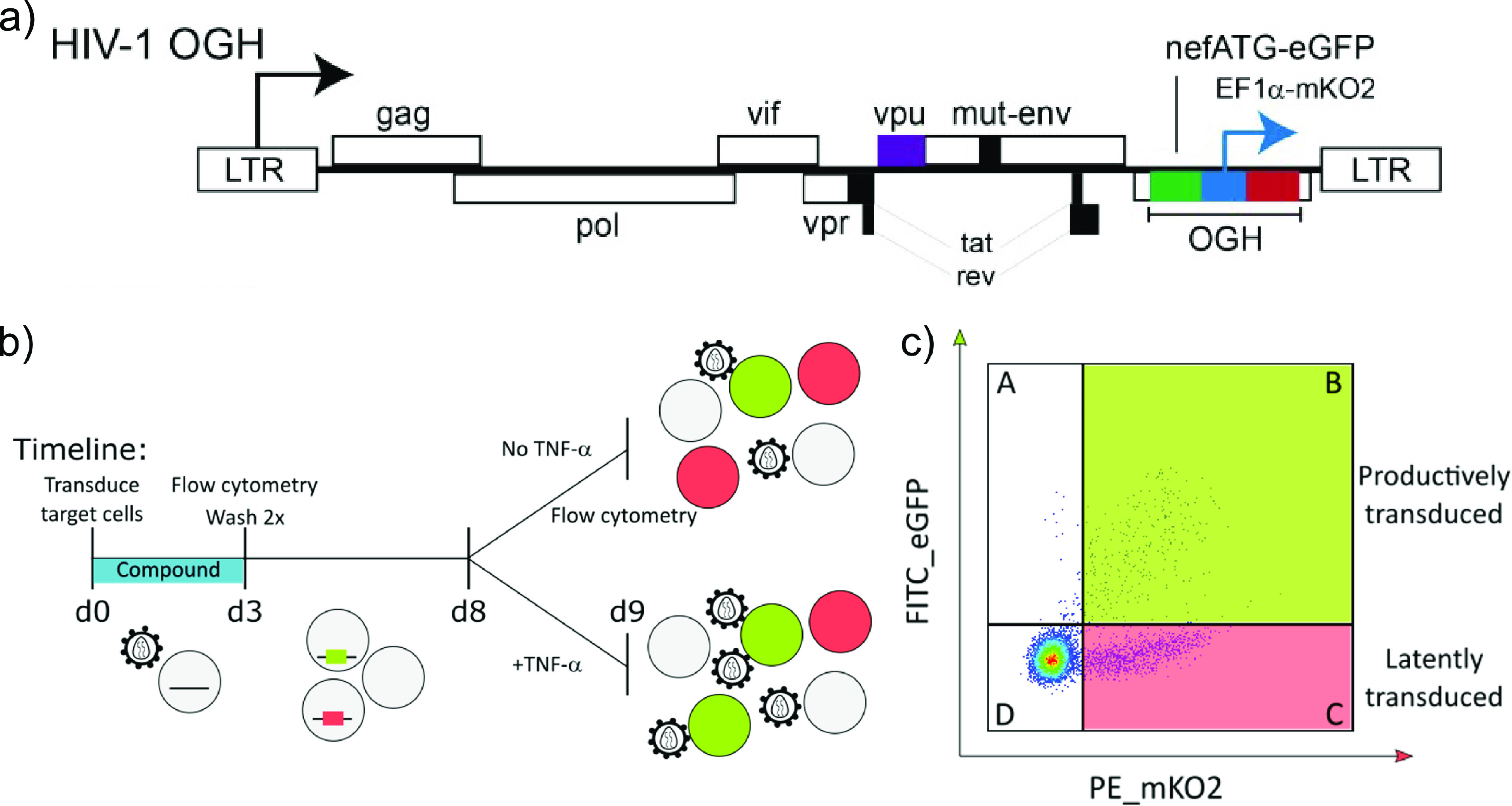
HIV-1 OGH: a double reporter construct used to analyze the impact of LEDGINs on immediate latency and reactivation. (a) Schematic representation of the HIV-1 OGH construct (51, 55, 56). HIV-1 OGH is a replication-deficient vector containing an eGFP gene under the control of the viral LTR promoter. HIV-1 OGH also carries a constitutively active transcriptional unit of an mKO2 reporter driven by an EF1α promoter. (b) Timeline of the transduction and reactivation experiments. SupT1 cells were transduced in the presence of CX14442 or GS-9822. Three days postransduction, vector and compounds were washed away, and flow cytometry analysis was performed. Eight days postransduction, cells were reactivated with 10 ng/ml TNF-α for 24 h. At day 9, 24 h postreactivation, cells were analyzed by flow cytometry. (c) Representative dot plot of SupT1 cells, transduced with 120 × 106 pg of HIV-1 OGH on day 9 (condition without TNF-α), showing how flow cytometry makes it possible to distinguish between different cell populations. Cells only expressing mKO2 from the constitutively active EF1α promoter have an inactive LTR and are thus considered to be latently transduced (quadrant C). If cells are productively transduced (quadrant B), the viral LTR promoter will drive eGFP expression as well, resulting in double positive cells.
FIG 4.

Treatment with CX14442 or GS-9822 but not raltegravir increases immediate latency of HIV-1 OGH double reporter construct. (a to c) Data of one representative experiment out of 4 plotting the average of duplicate measurements with standard deviation. Percentage of eGFP and mKO2-positive cells 3 days postransduction of SupT1 cells with a 1/20,000 dilution of HIV-1 OGH. Cells were treated with increasing concentrations of CX14442, GS-9822, or raltegravir. eGFP-positive cells are shown in green, and mKO2 positive cells are plotted in red. Mean IC50 values with standard error of the mean (SEM) for CX14442, GS-9822 and raltegravir are listed below the graphs. (d) The latent fraction (percentage of single mKO2-positive cells/[percentage of single mKO2-positive cells + percentage of double positive cells] · 100) or (quadrant C/[quadrant B + quadrant C]) as shown in panel c was calculated 3 days postransduction of SupT1 cells with a 1/20,000 dilution of HIV-1 OGH virus. Compound concentrations are plotted as a fold of the IC50. (e) Copies/million cells as calculated by Alu-LTR and CCR5 qPCR on day 3 postransduction of SupT1 cells with a 1/20,000 dilution of HIV-1 OGH virus. Mean values and SD are shown for one representative experiment out of 2. Compound concentrations are plotted as a fold of the IC50.
GS-9822 reduces HIV-1 reactivation in a single-round, double-reporter system.
Finally, we evaluated HIV-1 reactivation in SupT1 cells pretreated with CX14442 or GS-9822 or with raltegravir as a control. Cells were transduced with the HIV-1 OGH vector for 3 days in the presence of the compounds and then washed twice to remove compound and cell-free vector and kept in culture until day 8 in the absence of inhibitor. On day 8 postransduction, half of the cells were reactivated with 10 ng/ml tumor necrosis factor α (TNF-α), and the other half were left untreated. Thus, reporter gene expression and latency with and without reactivation could be compared (Fig. 3b). Upon reactivation, the percentage of mKO2-positive cells (transduced cells) remained unchanged, as expected, while an increase in eGFP-positive cells (reactivation of latent proviruses) was seen under all conditions (Figure 5a to c). The latent fraction decreased as transduced cells were reactivated from latency (Figure 5d to f). However, when cells had been treated with CX14442 or GS-9822 during initial transduction, a dose-dependent decrease in the reactivatable latent fraction was evidenced (Fig. 5d, e, and g; Fig. S5b). This effect was more pronounced for GS-9822 than for CX14442. Again, for raltegravir, a similar effect on reactivation was observed but only at concentrations of 17-fold the IC50 (Fig. 5f and g). Treatment with GS-9822 thus reduces reactivation from latency at nanomolar concentrations.
FIG 5.
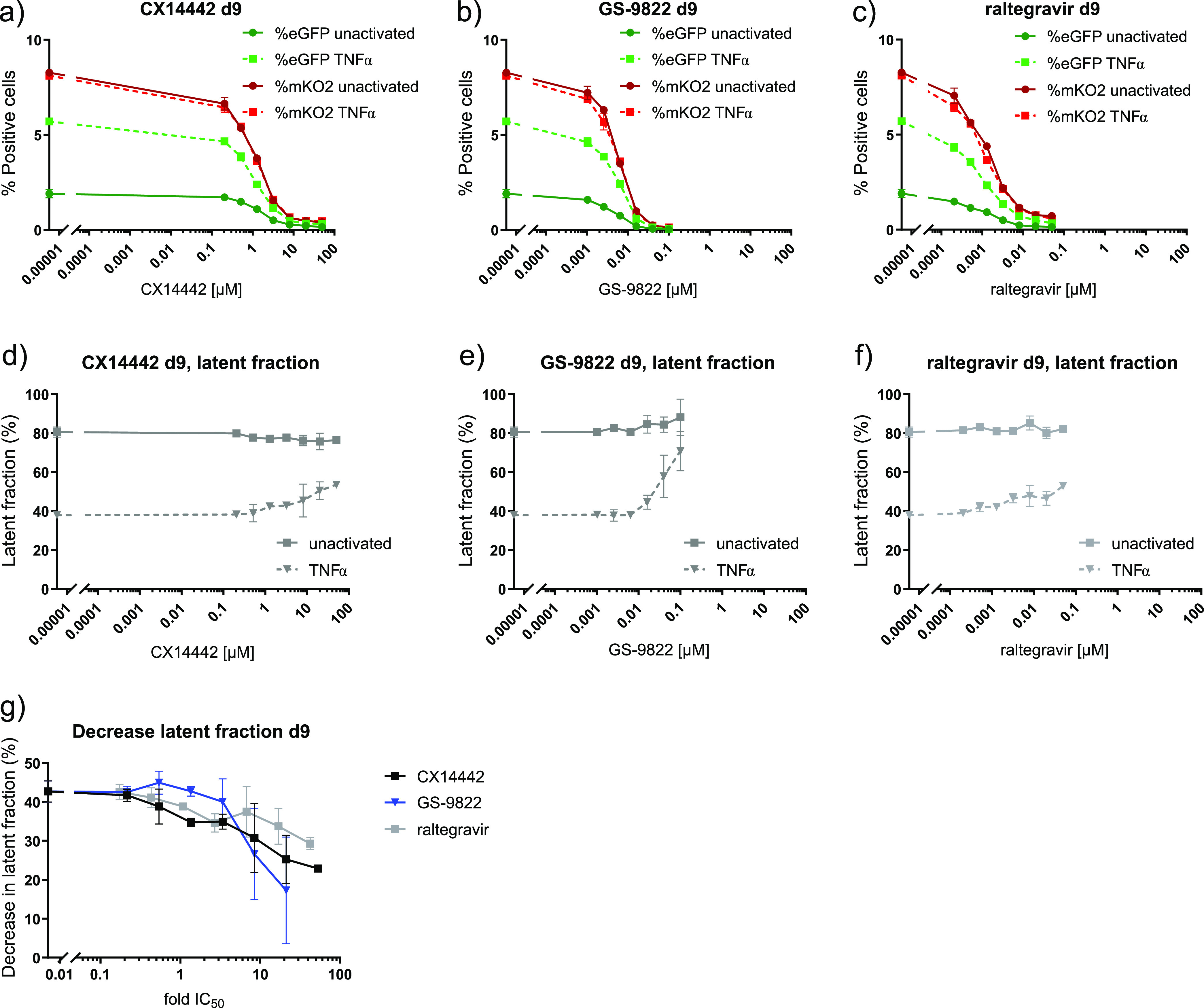
Treatment with CX14442 or GS-9822 but not raltegravir decreases reactivation from latency. Flow cytometry data on day 9 or 24 h after reactivation of HIV-1 OGH transduced SupT1 cells with 10 ng/ml TNF-α are shown for a 1/20,000 virus dilution. Full lines represent nonactivated cells, and dotted lines represent TNF-α-treated cells. Data show one representative experiment out of 4, and the average of duplicate measurements with standard deviation is plotted. (a to c) The percentages of mKO2- and eGFP-positive cells for cells pretreated with CX14442, GS-9822, or raltegravir are shown. eGFP-positive cells are shown in green, and mKO2-positive cells are plotted in red. (d to f) The latent fraction, calculated as described above, for cells pretreated with CX14442, GS-9822, or raltegravir is shown. (g) Upon reactivation, the latent fraction decreases, and the plotted decrease in latent fraction is calculated by subtracting the latent fraction under the TNF-α-treated condition from the latent fraction under the nontreated condition. Compound concentrations for this graph are plotted as a fold of the IC50.
DISCUSSION
GS-9822, a preclinical LEDGIN candidate, is a potent antiviral with nanomolar activity against wild-type HIV-1 viruses (Fig. 1b and e, Table 1). GS-9822 inhibits the LEDGF/p75-integrase interaction (Fig. 1c and f, Table 1) and reduces HIV-1 integration (Fig. S1a and b; Table 2, Fig. 4a to c and e). Much like CX14442 (20, 51), GS-9822 retargeted integration of residual proviruses away from genes and gene-dense regions (Table 2, Fig. 2b to e; Fig. S1c to r, S2, and S4), resulting in a more repressive epigenetic landscape (Fig. S3). Finally, when using a double reporter construct, CX14442 and GS-9822 were shown to reduce HIV-1 infectivity, increase immediate latency, and decrease the reactivation potential of residual integrants (Fig. 4 and 5). Remarkably, GS-9822 induced these effects at 200 to 300-fold lower concentrations than CX14442.
The present study is the first to show a combined effect on viral replication, integrase-LEDGF/p75 interaction, integration sites, chromatin landscape, immediate latency, and latency reversal after treatment with a LEDGIN at nanomolar concentrations. Compared to CX14442, GS-9822 was 20- to 100-fold more potent against wild-type HIV infection in MT-4 cells and inhibited the integrase-LEDGF/p75 interaction at a 10-fold lower concentration. When present during transduction with a single-round HIV-1-based lentiviral vector or the HIV-1 OGH double reporter, both GS-9822 and CX14442 hampered integration as shown by a reduction in copies per cell determined by qPCR (Fig. S1b; Fig. 4e), reporter gene expression via flow cytometry (Fig. 4a to d), and the number of unique sites obtained after integration site sequencing (Fig. S1a) (Table 2) (20, 51). Though HIV-1 normally favors integration in active genes and transcription units (14–20), both LEDGINs decreased integration within genes and gene-dense regions (Table 2 and Fig. 2b to e; Fig. S1c to r and S4) as was shown before for CX14442 (20, 51), CX05045 (51), and the BI/D compound (54). This shift in integration sites also coincided with differences in the epigenetic environment surrounding the integrated proviruses. Consistent with previous research on CX14442 (20, 51), GS-9822 reduced integrations in proximity to H3K36me3 (Fig. S3), a marker found along active gene bodies (61–64, 71–73), the histone modification targeted by LEDGF/p75 (21, 22, 69, 70), and according to an analysis by a deep learning neural network, the strongest predictor of HIV-1 integration of all epigenetic marks investigated (76). Recently, B-HIVE was used to show that upon retargeting by CX14442, proviruses integrated away from H3K36me3 had reduced RNA expression levels (20). GS-9822 clearly had a stronger effect than CX14442 (51) on other markers for active gene bodies (61, 63, 74), such as H3K27me1 and H2B5Kme1. H4K20me1 and H3K9me1, a marker usually located closest to the transcription start site (61, 73), were affected to a lesser degree. HIV usually disfavors integration near H3K27me2, H3K27me3, H3K9me2, H3K9me3, and H4K20me3 (20, 51, 65–68), all markers associated with silent genes and repressive chromatin (61–64, 74). As demonstrated previously for CX14442, retargeting of integration by GS-9822 resulted in relatively closer integration to these markers, at about the level of the generated matched random controls (MRCs) (20, 51). Overall, when comparing both compounds, GS-9822 showed stronger retargeting effects than CX14442 for most parameters and at nanomolar concentrations. Finally, GS-9822 and CX14442 both increased immediate latency (Fig. 4d; Fig. S5a) and reduced reactivation (Fig. 5, particularly Fig. 5g; Fig. S5b) from the residual integrated proviruses as was shown previously for CX14442 (20, 51). Here, the effect of both compounds seems comparable relative to their respective IC50s, though GS-9822 is about 200-fold more potent (GS-9822 IC50 = 6.929 nM versus CX14442 IC50 = 1.577 μM) and thus achieves the same effect at much lower concentrations. Based on these findings, we conclude that (i) the retargeting of integration, (ii) the increase in immediate latency, and (iii) the induction of a reservoir that is refractory to reactivation are all class effects related to the inhibition of the LEDGF-integrase interaction and are not specific for single compounds of the class. Notably, since MINIs may not exhibit this mechanism of action, this subtype of LEDGINs may not have this additional effect. Furthermore, highly potent LEDGIN compounds that inhibit the LEDGF-integrase interaction with good selectivity can induce these effects at low doses that could be achieved in the clinic, making LEDGINs an interesting candidate for functional HIV cure research.
LEDGINs are attractive antivirals given their multimodal effects, including blocking of viral maturation, integration, and induction of latency. Certain LEDGINs also reduce viral reactivation, though viral transcription is not fully blocked in each single cell. Whether the observed inhibition is enough to delay viral rebound or help to induce a functional cure phenotype will need to be investigated in animal models. On the other hand, research has shown that over time, intact proviruses located at integration sites suggestive of deeper latency accumulate in CD4+ T cells derived from patients on cART, pointing to selective pressures weeding out active proviruses (77). In addition, next-generation sequencing of proviruses and integration sites in elite controllers has shown that in these patients, proviruses are more frequently located in regions associated with heterochromatin features enriched in repressive chromatin marks (78). Thus, increasing the percentage of proviruses in the reservoir in such a latent state with LEDGINs could theoretically help some patients obtain viral control over time.
If used in the clinic with the specific goal of inducing a more latent reservoir, the timing of LEDGIN treatment will be important. Since we know that early treatment reduces the size of the HIV reservoir (79), treatment during primary-infection or at least early after infection is likely to have the most impact. Since even in the European Union/European Economic Area (EU/EAA), almost 50% of HIV patients are diagnosed in the late stages of infection (80), a relevant question requiring investigation is whether LEDGINs would have any impact on the existing latent reservoir of chronically infected patients. Recent research suggests that 60 to 70% of the HIV latent reservoir is formed in the year before cART initiation, although earlier strains do make up parts of the reservoir in certain patients (81, 82). Whether LEDGINs would exert a block-and-lock phenotype in chronically treated patients, perhaps after a treatment interruption, also remains to be investigated.
Since the effect of LEDGINs on latency has only been tested in cell culture, confirmation in animal models will be needed prior to clinical evaluation. GS-9822 has an excellent in vitro and pharmacokinetic profile, but an unmonitorable urothelial toxicity was seen in a monkey model, leading to a halt in further advancement (50). However, recently, a new LEDGIN compound, STP0404, was announced (47). STP0404 has favorable physicochemical and pharmacokinetic properties, nanomolar antiviral activity in different cell models, and a high therapeutic index. STP0404 was active against raltegravir-resistant HIV strains, while the typical LEDGIN resistance mutations Y99H and A128T conferred resistance to STP0404. No significant toxicities were reported in rat and dog. STP0404 was also shown to decrease reactivation from latency in primary resting CD4+ T cells. Given its high potency, STP0404 is being explored for long-acting antiretroviral therapy. Phase I clinical trials with this new LEDGIN were scheduled to start in 2020.
The present paper highlights the block-and-lock potential of LEDGINs in addition to their potent antiviral properties. Since we show that GS-9822 can affect HIV latency in vitro at clinically achievable doses, we believe that it should be investigated whether potent and safe LEDGIN compounds could act as modifiers and silencers of the functional HIV reservoir in vivo.
MATERIALS AND METHODS
Compounds.
CX14442 (38) was synthesized at Cistim/CD3 KU Leuven (courtesy of A. Marchand). GS-9822 (50) was provided by Gilead (courtesy of M. Balakrishnan). Raltegravir was purchased from the National Institute for Biological Standards and Control (NIBSC). All compounds were diluted in dimethyl sulfoxide (DMSO) to a stock concentration of 25 mM, which was then diluted in a 1/10 series and aliquoted for use in the different experiments.
Cell culture.
All cells were tested to be mycoplasma free (PlasmoTest, InvivoGen) and cultured in a humidified atmosphere at 5% CO2 and 37°C. MT-4 cells and SupT1 cells were provided by the National Institutes of Health (NIH) AIDS reagent program. They were cultured in RPMI 1640 (Gibco) supplemented with 10% (vol/vol) fetal bovine serum (FBS; Gibco) and 0.01% (vol/vol) gentamicin (Gibco). HEK293T cells were purchased from ATCC (293T/17 [HEK 293T/17; ATCC CRL-11268]) and cultured in Dulbecco modified Eagle medium (DMEM; Gibco) with 5% FBS (Gibco) and 0.01% gentamicin (Gibco).
Reporter viruses.
CH-SFFV-eGFP-P2A-fLuc. CH-SFFV-eGFP-P2A-fluc is a replication-deficient lentiviral vector containing an enhanced green fluorescent protein (eGFP) reporter and a firefly luciferase (fLuc) reporter, separated by a P2A sequence and driven by the spleen-focus-forming-virus (SFFV) promoter (58).
HIV-1 OGH. The dual-colored reporter vector, HIV-1 OGH, is an orange-green variant of a previously described LAI-based double reporter vector (55, 56, 75) used to study HIV latency (51, 58). It is replication deficient and contains an LTR-driven enhanced green fluorescent protein (eGFP) in the nef position followed by a constitutively active EF1α promoter driving a mutant Kusabira-Orange 2 (mKO2) reporter. The double reporter vector was a kind gift from the Verdin lab (Buck Institute for Research on Aging).
Viral strains.
The HIV-1 IIIB and NL4-3 strains were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH. HIV-1 IIIB virus was obtained from Robert Gallo (catalog number 398) and HIV-1 NL4-3 infectious molecular clone (pNL4-3) from Malcolm Martin (catalog number 114). HIV-1 IIIB was originally derived from concentrated culture fluids of peripheral blood or bone marrow from several patients with AIDS or AIDS-related diseases. The source of this chimeric molecular clone is the HIV-1 NY5 isolate (5′) and the HIV-1 LAV isolate (3′) cloned directly from genomic DNA (83–85). HIV-1 NL4-3 is a chimeric molecular clone based on the HIV-1 NY5 and HIV-1 LAV isolates (86).
Virus production.
HIV-1 OGH was produced in HEK293T cells by double transfection with pOGH plasmid and pVSVG, the VSV-G protein encoding plasmid, using linear polyethylenimine (PEI; Polysciences) in Optimem (Gibco). First, HEK293T cells were seeded overnight in DMEM (Gibco) at 5.7 million cells per petri dish. A DNA mixture was made consisting of 20 μg of transfer plasmid and 5 μg of pVSVG diluted in 700 μl of Optimem per petri dish. Next, a PEI mixture containing 68 μl of 10 μM PEI in 632 μl of 150 mM NaCl was added gently to the plasmid mixture. After incubation for 15 min at room temperature, 5 ml of Optimem was added, and the resulting medium was used to replace the seeding medium. After 6 h, the plates were washed twice with phosphate-buffered saline (PBS), and the medium was replaced with Optimem. After 72 h, the supernatant was collected for the first time, and the medium was replaced for a second collection after another 24 h. The supernatant was filtered through a 0.45-μm pore membrane (Merck), concentrated using a Vivaspin device with a 15- to 50-kDa cutoff (Merck), treated with 100 U/ml DNase (Roche Diagnostics) for 1 h at 37°C, and stored at –80°C until use.
CH-SFFV-eGFP-P2A-fluc was produced in HEK293T cells by triple transfection with 20 μg of the transfer plasmid pCH-SFFV-eGFP-P2A-fLuc, 10 μg of the packaging plasmid Δ8.91, and 5 μg of pVSV-G, using the protocol described above with 95 μl of PEI in 605 μl of 150 mM NaCl.
MTT assay.
In this assay, a yellow dye 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) is reduced by a mitochondrial dehydrogenase in metabolically active cells to a purple formazan derivative (87), which is measured spectrophotometrically. Here, we used the MTT assay to determine the inhibition of HIV-induced cytopathic effects (CPE) in MT-4 cells and thus the inhibition of HIV-1 infection. MT-4 cells were infected with wild-type HIV-1 subtype B viruses (IIIB and NL4.3) at a multiplicity of infection (MOI) of 0.01 in the presence of a dilution series of CX14442 and GS-9822 (1.28 nM to 100 μM for CX14442 and 0.064 nM to 5 μM for GS-9822). The MTT assay was performed after 5 days.
Expression and purification of recombinant proteins.
His6-tagged HIV-1 integrase and Flag-tagged LEDGF/p75 were purified for AlphaScreen applications as described previously over a HiTrap nickel affinity gel (Ni2+-NTA) and HiTrap heparin column, respectively (28, 88).
AlphaScreen assays.
We performed LEDGF/p75-integrase interaction AlphaScreen assays (Perkin Elmer, Waltham, MA) as described previously (34). The reaction buffer contained 25 mM Tris/HCl (pH 7.3), 150 mM NaCl, 1 mM MgCl2, 0.1% Tween 20, and 0.1% bovine serum albumin (BSA). All compounds and proteins were diluted to a 5× working solution in this buffer. We used a final volume of 25 μl in a 384-well microtiter plate (OptiPlate-384; Perkin Elmer). We used purified His6-tagged integrase (His-IN) protein from HIV-1, recognized by a nickel-chelate donor bead, and Flag-tagged LEDGF/p75 (Flag-p75), recognized by an anti-FLAG coated acceptor bead. First, AlphaScreen assays without compounds were run, with a range of concentrations (final concentrations, 10 to 200 μM) for both proteins to determine the optimal reaction conditions (data not shown). Then, 5 μl of His-IN was incubated for 1 h with 5 μl of Flag-p75 at 4°C. Next, 10 μl of a mix of nickel-chelate donor beads and anti-FLAG coated acceptor beads at a 1/100 dilution were added. The plate was then incubated for 3 h at 30°C while limiting exposure of the reaction mixture to light. The emission of light from the acceptor beads was analyzed in the Envision plate reader (Perkin Elmer) in AlphaScreen mode.
Next, we determined dose response curves in the presence of the compounds. First, 5 μl of His-IN at a final concentration of 50 nM was incubated for 30 min at 4°C in the presence of 5 μl of various concentrations of the compounds (final concentrations of 0.1 to 500 μM for CX14442 and 0.001 to 500 μM GS-9822). Next, 5 μl of Flag-tagged LEDGF/p75 at a final concentration of 100 nM was added for an additional 1 h at 4°C. Further steps were performed as described above. Data were analyzed using the Envision Manager software (Perkin Elmer) and GraphPad Prism.
Transduction and reactivation experiments with HIV-1 OGH.
SupT1 cells were transduced for 3 days in the presence of different concentrations of either CX14442 (38), GS-9822 (50), or raltegravir, after which the cells were washed twice with PBS. Eight days after infection, cells were reactivated in duplicate with 10 ng/ml tumor necrosis factor α (TNF-α; Immunosource). Flow cytometry analysis was performed 3 days postransduction and 24 h postreactivation (Fig. 5b). Drug concentrations are listed for each individual experiment. IC50 values were calculated using GraphPad Prism.
Flow cytometry.
Cells were fixed in 2% paraformaldehyde (PFA) for 15 min at room temperature and stored at 4°C. Cells were washed with and resuspended in PBS before flow cytometry analysis with a MACS Quant VYB analyzer (Mylteny Biotech). eGFP expression was measured using a 488-nm, 50-mW diode-pumped solid-state (DPSS) excitation laser, and the emitted signal was measured after a 525/50-nm band pass filter. mKO2 expression was measured using a 561-nm, 100-mW diode excitation laser and a 586/15-nm band pass filter. The gating strategy involved gating for lymphocytes using the forward and side scatter channel (FSC-H/SSC-H) and exclusion of doublets (FSC-A/FSC-H). At least 15,000 single live cells were counted in total, and each sample was measured in duplicate. Data were analyzed using FlowJo software.
Integration site analysis.
Integration sites were determined as previously described (60). SupT1 cells were seeded and transduced with a lentiviral vector (CH-SFFV-eGFP-P2A-fLuc) for 3 days in the presence of a range of concentrations of CX14442 or GS-9822. After 3 days, the cells were washed twice with PBS and kept in culture for at least 10 days to allow nonintegrated DNA to be diluted. Per condition, a single pellet of 5 to 10 million cells was used for further analysis. Next, genomic DNA was extracted using the GenElute mammalian genomic DNA miniprep kit (Sigma-Aldrich) and sheared by sonication with the Covaris M220 instrument. Linkers were added to the sheared DNA ends, and integration sites were amplified by nested PCR with primers complementary to the linker and the viral LTR. Finally, the PCR products were sequenced with Illumina Miseq (paired-end, 300 cycles). Sequencing data were analyzed using the INSPIIRED software (59) and Rstudio (BiocManager, GenomicRanges, and hiAnnotator packages). All samples were obtained from a single transduction experiment.
Integrated copies/cell qPCR.
Integrated HIV copies were quantified with a nested real-time Alu-LTR qPCR (58). For the first round, the PCR mix contained 5 μl of genomic DNA, 12.5 μl of iQ Supermix (Bio-Rad), 0.5 μl of 3 primers each at a 20 μM concentration (Alu forward [FW], TCCCAGCTACTGGGGAGGCTGAGG; Alu reverse [RV], TGCTGGGATTACAGGCGTGAG; HIV-1 LTR FWr1, GCTAACTAGGGAACCCACTGCTTA), and 6 μl of water. The PCR started with 10 min at 95°C, followed by 15 cycles of 95°C for 30 sec, 60°C for 40 sec, and 72°C for 3.5 min. The mix for the second round contained 12.5 μl of SuperiQ mix, 0.5 μl of 2 primers each at a 20 μM concentration (HIV-1 LTR FWr2, AGCTTGCCTTGAGTGCTTCAA; HIV-1 LTR RVr2, TGACTAAAAGGGTCTGAGGGATCT), 1 μl of probe at a 5 μM concentration (5′-FAM-TTACCAGAGTCACACAACAGACGGGCA-TAMRA-3′), and 5 μl of the first round PCR product. The PCR was performed in a LightCycler 480 device (Roche Life Science), starting with 5 min at 95°C, followed by 45 cycles of 95°C for 15 sec, 60°C for 30 sec, and 72°C for 1 min. To normalize for total input DNA, a CCR5 qPCR was performed on the same samples, as previously described (89). The PCR mix contained 5 μl of genomic DNA, 10 μl of Sybr Green (Invitrogen), 1 μl of 2 primers each at 20 μM concentration (LK46, GCTGTGTTTGCGTCTCTCCCAGGA; LK47, CTCACAGCCCTGTGCCTCTTCTTC), and 3 μl of water. The PCR was performed in a LightCycler 480 device (Roche Life Science). All samples were run at least in duplicate. Data were analyzed using the provided LightCycler 480 software.
Data availability.
All HIV-1 OGH integration sites with annotation, as obtained through analysis with the INSPIIRED software (59), are provided as supplemental material (Supplemental Data Sets S1 to S4).
Supplementary Material
ACKNOWLEDGMENTS
We thank Barbara Van Remoortel, Paulien Van de Velde, Jooke Van der Veeken, Siska Van Belle, and Jolien Blokken (Molecular Virology and Gene Therapy, KU Leuven) for their technical support. We are grateful to Kris Jacobs (Center for Cancer Bilogy, VIB-KU Leuven) for his assistance with flow cytometry measurements. Illumina MiSeq sequencing was performed by the KU Leuven Genomics core. We also thank Jonas Kaerts for helping to set up the INSPIIRED virtual machine.
A.B. and G.V. are doctoral fellows supported by the Flemish Fund for Scientific Research (FWO; Fonds voor Wetenschappelijk Onderzoek); Z.D. received financial support from the FWO (G0A5316N and SBO-Saphir) and the KU Leuven Research Council (C14/17/095-3M170311).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Z.D. and M.B. conceived the study. A.B., G.V., F.C., and Z.D. designed experiments. M.B. and M.L.M. provided GS-9822 that was first prepared by R.C. A.B., G.V., Paulien Van de Velde, Barbara Van Remoortel, and Jooke Van der Veeken conducted experiments. A.B. and G.V. performed bioinformatic analyses. A.B., G.V., F.C., and Z.D. analyzed the data. M.B. and M.L.M. critically reviewed the data and offered advice. Z.D. coordinated the study. A.B. and Z.D. prepared the manuscript. All authors read, corrected, and approved the final manuscript.
A.B., G.V., F.C., and Z.D. have no conflicts of interest to declare. M.B. and M.L.M. are employees and stockholders of Gilead Sciences. R.C. was an employee and stockholder of Gilead Sciences.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.National Institutes of Health. 2021. FDA-approved HIV medicines. Understanding HIV/AIDS. AIDSinfo. NIH, Bethesda, MD. https://hivinfo.nih.gov/understanding-hiv/fact-sheets/fda-approved-hiv-medicines. [Google Scholar]
- 2.EACS. 2019. European guidelines for the treatment of people living with HIV (PLWH) in Europe. Guidelines 122. EACS, Brussels, Belgium. https://www.eacsociety.org/guidelines/eacs-guidelines/eacs-guidelines.html. [Google Scholar]
- 3.DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents–A Working Group of the Office of AIDS Research Advisory Council (OARAC). 2018. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in adults and adolescents with HIV. Department of Health and Human Services, Washington, DC. https://clinicalinfo.hiv.gov/sites/default/files/inline-files/AdultandAdolescentGL.pdf. [Google Scholar]
- 4.World Health Organization (WHO). 2019. Policy brief: update of recommendations on first- and second-line antiretroviral regimens. WHO, Geneva, Switzerland. https://www.who.int/hiv/pub/arv/arv-update-2019-policy/en/. [Google Scholar]
- 5.Joint United Nations Programme on HIV/AIDS (UNAIDS). 2019. UNAIDS data 2019. UNAIDS, Geneva, Switzerland. https://www.unaids.org/en/resources/documents/2019/2019-UNAIDS-data. [Google Scholar]
- 6.Dahabieh MS, Battivelli E, Verdin E. 2015. Understanding HIV latency: the road to an HIV cure. Annu Rev Med 66:407–421. 10.1146/annurev-med-092112-152941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamer D. 2004. Can HIV be cured? Mechanisms of HIV persistence and strategies to combat it. Curr HIV Res 2:99–111. 10.2174/1570162043484915. [DOI] [PubMed] [Google Scholar]
- 8.Abner E, Jordan A. 2019. HIV “shock and kill” therapy: in need of revision. Antiviral Res 166:19–34. 10.1016/j.antiviral.2019.03.008. [DOI] [PubMed] [Google Scholar]
- 9.Darcis G, Van Driessche B, Van Lint C. 2017. HIV latency: should we shock or lock? Trends Immunol 38:217–228. 10.1016/j.it.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Vansant G, Bruggemans A, Janssens J, Debyser Z. 2020. Block-and-lock strategies to cure HIV infection. Viruses 12:84. 10.3390/v12010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahlenstiel CL, Symonds G, Kent SJ, Kelleher AD. 2020. Block and lock HIV cure strategies to control the latent reservoir. Front Cell Infect Microbiol 10.3389/fcimb.2020.00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lesbats P, Engelman AN, Cherepanov P. 2016. Retroviral DNA integration. Chem Rev 116:12730–12757. 10.1021/acs.chemrev.6b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi E, Mallareddy JR, Lu D, Kolluru S. 2018. Recent advances in the discovery of small-molecule inhibitors of HIV-1 integrase. Futur Sci OA 4:FSO338. 10.4155/fsoa-2018-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schroder ARW, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. 2002. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110:521–529. 10.1016/S0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 15.Lewinski MK, Bisgrove D, Shinn P, Chen H, Hoffmann C, Hannenhalli S, Verdin E, Berry CC, Ecker JR, Bushman FD. 2005. Genome-wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J Virol 79:6610–6619. 10.1128/JVI.79.11.6610-6619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewinski MK, Yamashita M, Emerman M, Ciuffi A, Marshall H, Crawford G, Collins F, Shinn P, Leipzig J, Hannenhalli S, Berry CC, Ecker JR, Bushman FD. 2006. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathog 2:e60. 10.1371/journal.ppat.0020060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brady T, Agosto LM, Malani N, Berry CC, O’Doherty U, Bushman F. 2009. HIV integration site distributions in resting and activated CD4+ T cells infected in culture. AIDS 23:1461–1471. 10.1097/QAD.0b013e32832caf28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Rijck J, Bartholomeeusen K, Ceulemans H, Debyser Z, Gijsbers R. 2010. High-resolution profiling of the LEDGF/p75 chromatin interaction in the ENCODE region. Nucleic Acids Res 38:6135–6147. 10.1093/nar/gkq410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H-CC, Martinez JP, Zorita E, Meyerhans A, Filion GJ. 2017. Position effects influence HIV latency reversal. Nat Struct Mol Biol 24:47–54. 10.1038/nsmb.3328. [DOI] [PubMed] [Google Scholar]
- 20.Vansant G, Chen H, Zorita E, Trejbalová K, Miklík D, Filion G, Debyser Z. 2020. The chromatin landscape at the HIV-1 provirus integration site determines viral expression. Nucleic Acids Res 48:7801–7817. 10.1093/nar/gkaa536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Nuland R, van Schaik FMAM, Simonis M, van Heesch S, Cuppen E, Boelens R, Timmers HMTM, van Ingen H. 2013. Nucleosomal DNA binding drives the recognition of H3K36-methylated nucleosomes by the PSIP1-PWWP domain. Epigenetics Chromatin 6:12. 10.1186/1756-8935-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eidahl JO, Crowe BL, North JA, McKee CJ, Shkriabai N, Feng L, Plumb M, Graham RL, Gorelick RJ, Hess S, Poirier MG, Foster MP, Kvaratskhelia M. 2013. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res 41:3924–3936. 10.1093/nar/gkt074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turlure F, Maertens G, Rahman S, Cherepanov P, Engelman A. 2006. A tripartite DNA-binding element, comprised of the nuclear localization signal and two AT-hook motifs, mediates the association of LEDGF/p75 with chromatin in vivo. Nucleic Acids Res 34:1653–1665. 10.1093/nar/gkl052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Llano M, Vanegas M, Hutchins N, Thompson D, Delgado S, Poeschla EM. 2006. Identification and characterization of the chromatin-binding domains of the HIV-1 integrase interactor LEDGF/p75. J Mol Biol 360:760–773. 10.1016/j.jmb.2006.04.073. [DOI] [PubMed] [Google Scholar]
- 25.Cherepanov P, Maertens G, Proost P, Devreese B, Van Beeumen J, Engelborghs Y, De Clercq E, Debyser Z. 2003. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J Biol Chem 278:372–381. 10.1074/jbc.M209278200. [DOI] [PubMed] [Google Scholar]
- 26.Ortiz-Hernandez GL, Sanchez-Hernandez ES, Casiano CA. 2020. Twenty years of research on the DFS70/LEDGF autoantibody-autoantigen system: many lessons learned but still many questions. Autoimmun Highlights 11:3. 10.1186/s13317-020-0126-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cherepanov P, Devroe E, Silver PA, Engelman A. 2004. Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase. J Biol Chem 279:48883–48892. 10.1074/jbc.M406307200. [DOI] [PubMed] [Google Scholar]
- 28.Maertens G, Cherepanov P, Pluymers W, Busschots K, De Clercq E, Debyser Z, Engelborghs Y. 2003. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J Biol Chem 278:33528–33539. 10.1074/jbc.M303594200. [DOI] [PubMed] [Google Scholar]
- 29.Cherepanov P, Sun ZYJ, Rahman S, Maertens G, Wagner G, Engelman A. 2005. Solution structure of the HIV-1 integrase-binding domain in LEDGF/p75. Nat Struct Mol Biol 12:526–532. 10.1038/nsmb937. [DOI] [PubMed] [Google Scholar]
- 30.Cherepanov P, Ambrosio ALB, Rahman S, Ellenberger T, Engelman A. 2005. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc Natl Acad Sci U S A 102:17308–17313. 10.1073/pnas.0506924102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, Bushman F. 2005. A role for LEDGF/p75 in targeting HIV DNA integration. Nat Med 11:1287–1289. 10.1038/nm1329. [DOI] [PubMed] [Google Scholar]
- 32.Shun M-C, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, Cherepanov P, Engelman A. 2007. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev 21:1767–1778. 10.1101/gad.1565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marshall HM, Ronen K, Berry C, Llano M, Sutherland H, Saenz D, Bickmore W, Poeschla E, Bushman FD. 2007. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One 2:e1340. 10.1371/journal.pone.0001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Van Remoortel B, Strelkov SV, De Maeyer M, Chaltin P, Debyser Z. 2010. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat Chem Biol 6:442–448. 10.1038/nchembio.370. [DOI] [PubMed] [Google Scholar]
- 35.Demeulemeester J, Chaltin P, Marchand A, De Maeyer M, Debyser Z, Christ F, Maeyer M, De Debyser Z, Christ F. 2014. LEDGINs, non-catalytic site inhibitors of HIV-1 integrase: a patent review (2006–2014.). Expert Opin Ther Pat 24:609–632. 10.1517/13543776.2014.898753. [DOI] [PubMed] [Google Scholar]
- 36.Engelman AN. 2019. Multifaceted HIV integrase functionalities and therapeutic strategies for their inhibition. J Biol Chem 294:15137–15157. 10.1074/jbc.REV119.006901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fenwick C, Amad M, Bailey MD, Bethell R, Bös M, Bonneau P, Cordingley M, Coulombe R, Duan J, Edwards P, Fader LD, Faucher A-M, Garneau M, Jakalian A, Kawai S, Lamorte L, LaPlante S, Luo L, Mason S, Poupart M-A, Rioux N, Schroeder P, Simoneau B, Tremblay S, Tsantrizos Y, Witvrouw M, Yoakim C. 2014. Preclinical profile of BI 224436, a novel HIV-1 non-catalytic-site integrase inhibitor. Antimicrob Agents Chemother 58:3233–3244. 10.1128/AAC.02719-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christ F, Shaw S, Demeulemeester J, Desimmie BA, Marchand A, Butler S, Smets W, Chaltin P, Westby M, Debyser Z, Pickford C, Marchan A, Butler S, Smets W, Chaltin P, Westby M, Debyser Z, Pickford C. 2012. Small-molecule inhibitors of the LEDGF/p75 binding site of integrase block HIV replication and modulate integrase multimerization. Antimicrob Agents Chemother 56:4365–4374. 10.1128/AAC.00717-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Desimmie BA, Schrijvers R, Demeulemeester J, Borrenberghs D, Weydert C, Thys W, Vets S, Van Remoortel B, Hofkens J, De Rijck J, Hendrix J, Bannert N, Gijsbers R, Christ F, Debyser Z. 2013. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology 10:57. 10.1186/1742-4690-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jurado KA, Wang H, Slaughter A, Feng L, Kessl JJ, Koh Y, Wang W, Ballandras-Colas A, Patel PA, Fuchs JR, Kvaratskhelia M, Engelman A. 2013. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc Natl Acad Sci U S A 110:8690–8695. 10.1073/pnas.1300703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balakrishnan M, Yant SR, Tsai L, O’Sullivan C, Bam RA, Tsai A, Niedziela-Majka A, Stray KM, Sakowicz R, Cihlar T. 2013. Non-catalytic site HIV-1 integrase inhibitors disrupt core maturation and induce a reverse transcription block in target cells. PLoS One 8:e74163. 10.1371/journal.pone.0074163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le Rouzic E, Bonnard D, Chasset S, Bruneau J-MM, Chevreuil F, Le Strat F, Nguyen J, Beauvoir R, Amadori C, Brias J, Vomscheid S, Eiler S, Lévy N, Delelis O, Deprez E, Saïb A, Zamborlini A, Emiliani S, Ruff M, Ledoussal B, Moreau F, Benarous R. 2013. Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology 10:144. 10.1186/1742-4690-10-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borrenberghs D, Dirix L, De Wit F, Rocha S, Blokken J, De Houwer S, Gijsbers R, Christ F, Hofkens J, Hendrix J, Debyser Z. 2016. Dynamic oligomerization of integrase orchestrates HIV nuclear entry. Sci Rep 6:36485. 10.1038/srep36485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kessl JJ, Jena N, Koh Y, Taskent-Sezgin H, Slaughter A, Feng L, de Silva S, Wu L, Le Grice SFJ, Engelman A, Fuchs JR, Kvaratskhelia M. 2012. Multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J Biol Chem 287:16801–16811. 10.1074/jbc.M112.354373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pozniak A, Arribas JR, Gathe J, Gupta SK, Post FA, Bloch M, Avihingsanon A, Crofoot G, Benson P, Lichtenstein K, Ramgopal M, Chetchotisakd P, Custodio JM, Abram ME, Wei X, Cheng A, McCallister S, SenGupta D, Fordyce MW, GS-US-292-0112 Study Team. 2016. Switching to tenofovir alafenamide, coformulated with elvitegravir, cobicistat, and emtricitabine, in HIV-infected patients with renal impairment: 48-week results from a single-arm, multicenter, open-label phase 3 study. J Acquir Immune Defic Syndr 71:530–537. 10.1097/QAI.0000000000000908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma A, Slaughter A, Jena N, Feng L, Kessl JJ, Fadel HJ, Malani N, Male F, Wu L, Poeschla E, Bushman FD, Fuchs JR, Kvaratskhelia M. 2014. A new class of multimerization selective inhibitors of HIV-1 integrase. PLoS Pathog 10:e1004171. 10.1371/journal.ppat.1004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahn S, Kim U-I, Seo WY, Choi S, Maehigashi T, Lindenberger J, Kvaratskhelia M, Kim B, Kim K. 2020. A highly potent and safe allosteric hiv-1 integrase inhibitor, STP0404, abstr 504. Conference on Retroviuses and Opportunistic Infections (CROI), 8 to 11 March 2020, Boston, MA. [Google Scholar]
- 48.Fader LD, Malenfant E, Parisien M, Carson R, Bilodeau F, Landry S, Pesant M, Brochu C, Morin S, Chabot C, Halmos T, Bousquet Y, Bailey MD, Kawai SH, Coulombe R, Laplante S, Jakalian A, Bhardwaj PK, Wernic D, Schroeder P, Amad M, Edwards P, Garneau M, Duan J, Cordingley M, Bethell R, Mason SW, Bös M, Bonneau P, Poupart MA, Faucher AM, Simoneau B, Fenwick C, Yoakim C, Tsantrizos Y. 2014. Discovery of BI 224436, a noncatalytic site integrase inhibitor (NCINI) of HIV-1. ACS Med Chem Lett 5:422–427. 10.1021/ml500002n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aslanyan S, Ballow C, Sabo JP, Habeck J, Roos D, MacGregor TR, Robinson P, Kort J. 2011. Safety and pharmacokinetics (PK) of single rising oral doses of a novel HIV integrase inhibitor in healthy volunteers. Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), 17 to 20 September 2011, Chicago, IL. https://www.natap.org/2011/ICAAC/ICAAC_35.htm. [Google Scholar]
- 50.Mitchell ML, Balakrishnan M, Brizgys G, Cai R, Landson E, Mulato A, Osier M, Wang J, Yu H, Sakowicz R. 2017. Novel non-catalytic site integrase inhibitor with improved resistance profile, abstr 434. Conference on Retroviuses and Opportunistic Infections (CROI), 13 to 16 February 2017, Seattle, WA. [Google Scholar]
- 51.Vranckx LS, Demeulemeester J, Saleh S, Boll A, Vansant G, Schrijvers R, Weydert C, Battivelli E, Verdin E, Cereseto A, Christ F, Gijsbers R, Debyser Z. 2016. LEDGIN-mediated inhibition of integrase–LEDGF/p75 interaction reduces reactivation of residual latent HIV. EBioMedicine 8:248–264. 10.1016/j.ebiom.2016.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schrijvers R, De Rijck J, Demeulemeester J, Adachi N, Vets S, Ronen K, Christ F, Bushman FD, Debyser Z, Gijsbers R. 2012. LEDGF/p75-independent HIV-1 replication demonstrates a role for HRP-2 and remains sensitive to inhibition by LEDGINs. PLoS Pathog 8:e1002558. 10.1371/journal.ppat.1002558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schrijvers R, Vets S, De Rijck J, Malani N, Bushman FD, Debyser Z, Gijsbers R. 2012. HRP-2 determines HIV-1 integration site selection in LEDGF/p75 depleted cells. Retrovirology 9:84. 10.1186/1742-4690-9-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feng L, Dharmarajan V, Serrao E, Hoyte A, Larue RC, Slaughter A, Sharma A, Plumb MR, Kessl JJ, Fuchs JR, Bushman FD, Engelman AN, Griffin PR, Kvaratskhelia M. 2016. The competitive interplay between allosteric HIV-1 integrase inhibitor BI/D and LEDGF/p75 during the early stage of HIV-1 replication adversely affects inhibitor potency. ACS Chem Biol 11:1313–1321. 10.1021/acschembio.6b00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Calvanese V, Chavez L, Laurent T, Ding S, Verdin E. 2013. Dual-color HIV reporters trace a population of latently infected cells and enable their purification. Virology 446:283–292. 10.1016/j.virol.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chavez L, Calvanese V, Verdin E. 2015. HIV latency is established directly and early in both resting and activated primary CD4 T cells. PLoS Pathog 11:e1004955. 10.1371/journal.ppat.1004955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Battivelli E, Dahabieh MS, Abdel-Mohsen M, Svensson JP, Tojal Da Silva I, Cohn LBLB, Gramatica A, Deeks S, Greene WC, Pillai SK, Verdin E, Silva IT, Da Cohn Lblb Gramatica A, Deeks S, Greene WC, Pillai SK, Verdin E, Da Silva IT, Cohn LBLB, Gramatica A, Deeks S, Greene WC, Pillai SK, Verdin E. 2018. Distinct chromatin functional states correlate with HIV latency reactivation in infected primary CD4+ T cells. Elife 7:e34655. 10.7554/eLife.34655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vansant G, Vranckx LS, Zurnic I, Van Looveren D, Van De Velde P, Nobles C, Gijsbers R, Christ F, Debyser Z. 2019. Impact of LEDGIN treatment during virus production on residual HIV-1 transcription. Retrovirology 16:8–17. 10.1186/s12977-019-0472-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berry CC, Nobles C, Six E, Wu Y, Malani N, Sherman E, Dryga A, Everett JK, Male F, Bailey A, Bittinger K, Drake MJ, Caccavelli L, Bates P, Hacein-Bey-Abina S, Cavazzana M, Bushman FD. 2017. INSPIIRED: quantification and visualization tools for analyzing integration site distributions. Mol Ther Methods Clin Dev 4:17–26. 10.1016/j.omtm.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sherman E, Nobles C, Berry CC, Six E, Wu Y, Dryga A, Malani N, Male F, Reddy S, Bailey A, Bittinger K, Everett JK, Caccavelli L, Drake MJ, Bates P, Hacein-Bey-Abina S, Cavazzana M, Bushman FD. 2017. INSPIIRED: a pipeline for quantitative analysis of sites of new DNA integration in cellular genomes. Mol Ther Methods Clin Dev 4:39–49. 10.1016/j.omtm.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. 2007. High-resolution profiling of histone methylations in the human genome. Cell 129:823–837. 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 62.Berger SL. 2007. The complex language of chromatin regulation during transcription. Nature 447:407–412. 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 63.Mellor J, Dudek P, Clynes D. 2008. A glimpse into the epigenetic landscape of gene regulation. Curr Opin Genet Dev 18:116–122. 10.1016/j.gde.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 64.Kimura H. 2013. Histone modifications for human epigenome analysis. J Hum Genet 58:439–445. 10.1038/jhg.2013.66. [DOI] [PubMed] [Google Scholar]
- 65.De Ravin SS, Su L, Theobald N, Choi U, Macpherson JL, Poidinger M, Symonds G, Pond SM, Ferris AL, Hughes SH, Malech HL, Wu X. 2014. Enhancers are major targets for murine leukemia virus vector integration. J Virol 88:4504–4513. 10.1128/JVI.00011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Larue RC, Plumb MR, Crowe BL, Shkriabai N, Sharma A, DiFiore J, Malani N, Aiyer SS, Roth MJ, Bushman FD, Foster MP, Kvaratskhelia M. 2014. Bimodal high-affinity association of Brd4 with murine leukemia virus integrase and mononucleosomes. Nucleic Acids Res 42:4861–4881. 10.1093/nar/gku135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roth SL, Malani N, Bushman FD. 2011. Gammaretroviral integration into nucleosomal target DNA in vivo. J Virol 85:7393–7401. 10.1128/JVI.00635-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marini B, Kertesz-Farkas A, Ali H, Lucic B, Lisek K, Manganaro L, Pongor S, Luzzati R, Recchia A, Mavilio F, Giacca M, Lusic M. 2015. Nuclear architecture dictates HIV-1 integration site selection. Nature 521:227–231. 10.1038/nature14226. [DOI] [PubMed] [Google Scholar]
- 69.Debyser Z, Christ F, De Rijck J, Gijsbers R. 2015. Host factors for retroviral integration site selection. Trends Biochem Sci 40:108–116. 10.1016/j.tibs.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 70.Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. 2012. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet 8:e1002717. 10.1371/journal.pgen.1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bannister AJ, Schneider R, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. 2005. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J Biol Chem 280:17732–17736. 10.1074/jbc.M500796200. [DOI] [PubMed] [Google Scholar]
- 72.Hon GC, Hawkins RD, Ren B. 2009. Predictive chromatin signatures in the mammalian genome. Hum Mol Genet 18:12–14. 10.1093/hmg/ddp409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huff JT, Plocik AM, Guthrie C, Yamamoto KR. 2010. Reciprocal intronic and exonic histone modification regions in humans. Nat Struct Mol Biol 17:1495–1499. 10.1038/nsmb.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. 2008. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40:897–903. 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Battivelli E, Dahabieh MS, Abdel-Mohsen M, Svensson JP, Da Silva IT, Cohn LB, Gramatica A, Deeks S, Greene WC, Pillai SK, Verdin E. 2018. Chromatin functional states correlate with HIV latency reversal in infected primary CD4+ T cells. bioRxiv 10.7554/elife.34655. [DOI] [PMC free article] [PubMed]
- 76.Hu H, Xiao A, Zhang S, Li Y, Shi X, Jiang T, Zhang L, Zhang L, Zeng J. 2019. DeepHINT: understanding HIV-1 integration via deep learning with attention. Bioinformatics 35:1660–1667. 10.1093/bioinformatics/bty842. [DOI] [PubMed] [Google Scholar]
- 77.Einkauf KB, Lee GQ, Gao C, Sharaf R, Sun X, Hua S, Chen SMY, Jiang C, Lian X, Chowdhury FZ, Rosenberg ES, Chun TW, Li JZ, Yu XG, Lichterfeld M. 2019. Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J Clin Invest 129:988–998. 10.1172/JCI124291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiang C, Lian X, Gao C, Sun X, Einkauf KB, Chevalier JM, Chen SMYY, Hua S, Rhee B, Chang K, Blackmer JE, Osborn M, Peluso MJ, Hoh R, Somsouk M, Milush J, Bertagnolli LN, Sweet SE, Varriale JA, Burbelo PD, Chun T-WW, Laird GM, Serrao E, Engelman AN, Carrington M, Siliciano RF, Siliciano JM, Deeks SG, Walker BD, Lichterfeld M, Yu XG. 2020. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature 585:261–267. 10.1038/s41586-020-2651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ananworanich J, Chomont N, Eller LA, Kroon E, Tovanabutra S, Bose M, Nau M, Fletcher JLK, Tipsuk S, Vandergeeten C, O’Connell RJ, Pinyakorn S, Michael N, Phanuphak N, Robb ML, RV217 and RV254/SEARCH010 Study Groups. 2016. HIV DNA set point is rapidly established in acute HIV infection and dramatically reduced by early ART. EBioMedicine 11:68–72. 10.1016/j.ebiom.2016.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.ECDC. 2017. Thematic report: HIV testing. ECDC, Solna, Sweden. https://www.ecdc.europa.eu/en/publications-data/thematic-report-hiv-testing. [Google Scholar]
- 81.Abrahams MR, Joseph SB, Garrett N, Tyers L, Moeser M, Archin N, Council OD, Matten D, Zhou S, Doolabh D, Anthony C, Goonetilleke N, Karim SA, Margolis DM, Pond SK, Williamson C, Swanstrom R. 2019. The replication-competent HIV-1 latent reservoir is primarily established near the time of therapy initiation. Sci Transl Med 11:eaaw5589. 10.1126/scitranslmed.aaw5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brodin J, Zanini F, Thebo L, Lanz C, Bratt G, Neher RA, Albert J. 2016. Establishment and stability of the latent HIV-1 DNA reservoir. Elife 5:e18889. 10.7554/eLife.18889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Popovic M, Read-Connole E, Gallo RC. 1984. T4 positive human neoplastic cell lines susceptible to and permissive for HTLV-III. Lancet 324:1472–1473. 10.1016/S0140-6736(84)91666-0. [DOI] [PubMed] [Google Scholar]
- 84.Popovic M, Sarngadharan MG, Read E, Gallo RC. 1984. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science 224:497–500. 10.1126/science.6200935. [DOI] [PubMed] [Google Scholar]
- 85.Ratner L, Haseltine W, Patarca R, Livak KJ, Starcich B, Josephs SF, Doran ER, Rafalski JA, Whitehorn EA, Baumeister K, Ivanoff L, Petteway SR, Pearson ML, Lautenberger JA, Papas TS, Ghrayeb J, Chang NT, Gallo RC, Wong-Staal F. 1985. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature 313:277–284. 10.1038/313277a0. [DOI] [PubMed] [Google Scholar]
- 86.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. 10.1128/JVI.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D, Herdewijn P, Desmyter J, De Clercq E. 1988. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J Virol Methods 20:309–321. 10.1016/0166-0934(88)90134-6. [DOI] [PubMed] [Google Scholar]
- 88.Bartholomeeusen K, De Rijck J, Busschots K, Desender L, Gijsbers R, Emiliani S, Benarous R, Debyser Z, Christ F. 2007. Differential interaction of HIV-1 integrase and JPO2 with the C terminus of LEDGF/p75. J Mol Biol 372:407–421. 10.1016/j.jmb.2007.06.090. [DOI] [PubMed] [Google Scholar]
- 89.Zhang L, Lewin SR, Markowitz M, Lin HH, Skulsky E, Karanicolas R, He Y, Jin X, Tuttleton S, Vesanen M, Spiegel H, Kost R, van Lunzen J, Stellbrink HJ, Wolinsky S, Borkowsky W, Palumbo P, Kostrikis LG, Ho DD. 1999. Measuring recent thymic emigrants in blood of normal and HIV-1-infected individuals before and after effective therapy. J Exp Med 190:725–732. 10.1084/jem.190.5.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haeussler M, Zweig AS, Tyner C, Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Hinrichs AS, Gonzalez JN, Gibson D, Diekhans M, Clawson H, Casper J, Barber GP, Haussler D, Kuhn RM, Kent WJ. 2019. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res 47:D853–D858. 10.1093/nar/gky1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hsu F, Kent JW, Clawson H, Kuhn RM, Diekhans M, Haussler D. 2006. The UCSC known genes. Bioinformatics 22:1036–1046. 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All HIV-1 OGH integration sites with annotation, as obtained through analysis with the INSPIIRED software (59), are provided as supplemental material (Supplemental Data Sets S1 to S4).