

Abstract
Structural abnormalities of the brain are increasingly recognized in patients that suffer from pharmacoresistant focal epilepsies by applying high‐resolution imaging techniques. In many of these patients, epilepsy surgery results in control of seizures. Neuropathologically, a broad spectrum of malformations of cortical development (MCD) is observed in respective surgical brain samples. These samples provide a unique basis to further understand underlying pathomechanisms by molecular approaches and develop improved diagnostics and entirely new therapeutic perspectives. Here we provide a comprehensive description of neuropathological findings, available classification systems as well as molecular mechanisms of MCDs. We emphasize the recently published ILEA classification system for focal cortical dysplasias (FCDs), which are now histopathologically distinguished as types I to III. However, this revised classification system represents a major challenge for molecular neuropathologists, as the underlying pathomechanisms in virtually all FCD entities will need to be specified in detail. The fact that only recently, the mammalian target of rapamycin (mTOR)‐antagonist Everolimus has been introduced as a treatment of epilepsies in the context of tuberous sclerosis‐associated brain lesions is a striking example of a successful translational “bedside to bench and back” approach. Hopefully, the exciting clinico‐pathological developments in the field of MCDs will in short term foster further therapeutic breakthroughs for the frequently associated medically refractory epilepsies.
Keywords: classification, mTOR, neuronal precursor, pathomechanisms.
INTRODUCTION
Malformations of cortical development (MCD) represent a wide range of cortical lesions resulting from derangements of normal intrauterine developmental processes and involving cells implicated in the formation of the cortical mantle. In the past, these malformations have been defined as “neuronal migration disorders”; however, as not all the cortical abnormalities have been proven to be due to migrational derangements, the general term of MCDs is preferred as a reflection of our improved knowledge on normal and pathological brain development. MCD are now recognized as a heterogeneous group of focal or diffuse anatomical abnormalities, determined by different causes, that develop during crucial periods of cortical ontogenesis and their pathological features depend thus largely on the timing of the defect in the developmental processes, as well as on its causes.
The etiology of malformative disorders often remains uncertain. Recent molecular biologic and genetic studies have, however, greatly expanded our knowledge on brain ontogenesis so that several disorders of cortical development have been recognized and, for some of them, specific causative genetic defects have been identified. Although brain malformations have been recognized by neuropathologists since the 19th century, improved imaging techniques, namely high resolution Magnetic resonance imaging (MRI), have a great diagnostic impact in this field, allowing in vivo diagnosis of various MCDs and providing correlations between imaging features, neurological deficits, developmental delays and electro‐clinical findings related to epilepsy (126) (Figure 1). Furthermore, new methodological techniques including quantitative image analysis, MR spectroscopy, functional MRI (fMRI), diffusion tensor imaging (DTI) and fiber tract reconstructions are increasingly available and useful in clinical practice, especially for the detection of small malformations. These novel technologies will further improve our understanding about the functional relevance of affected cortical areas.
Figure 1.
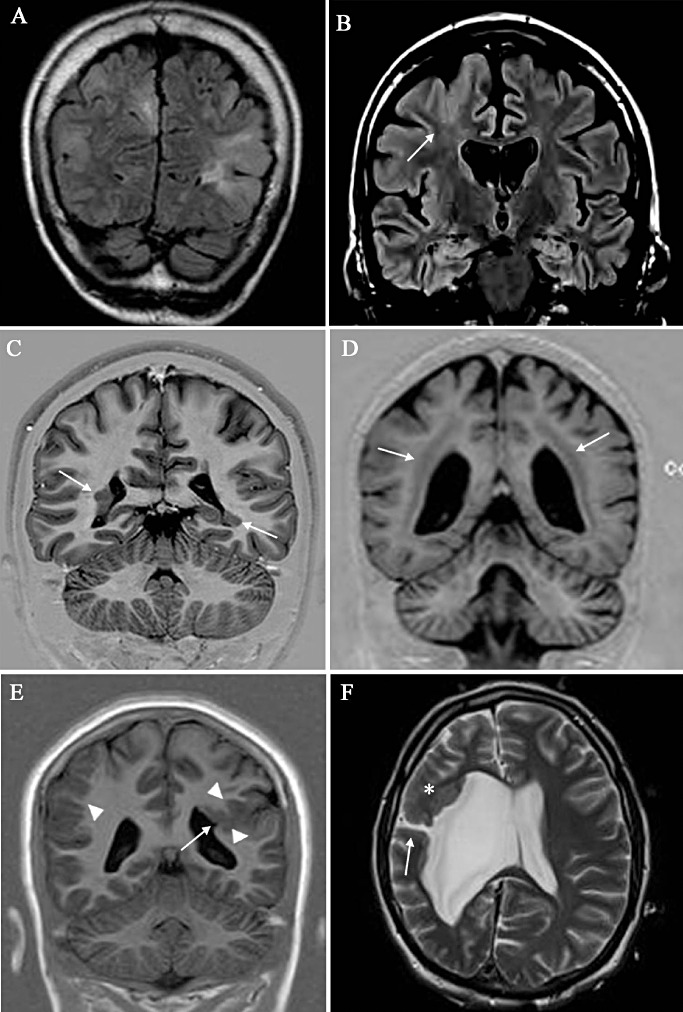
MRI revealing different types of MCD. A. Multiple bilateral nodules in a patient with TSC. B. FCD Type IIb (histologically proven) in the right frontal area; note the tapering from the sulcus toward the ventricle (arrow) considered a landmark for this type of dysplasia. C. Bilateral asymmetric periventricular nodular heterotopia (arrows). D. Bilateral symmetric band heterotopia (double cortex). E. Close‐lipped schizencephaly (arrow) associated with polymicrogyria (arrowheads); a polymicrogyric cortex is also evident in the contralateral cortex (arrowhead). F. Open‐lipped schizencephaly with communication with an enlarged right ventricle (arrow); a malformed cortex is also evident (asterisk) in the frontal cortex of the same side. FCD = focal cortical dysplasia; MCD = malformation of cortical development; MRI = magnetic resonance imaging; TSC = tuberous sclerosis complex.
Due to abnormal rearrangement of neuronal circuitries, MCDs can be intrinsically epileptogenic and this has been addressed by an increasing number of scientific publications during the last decade. The abnormal circuitry may be restricted to the malformed cortical area or may involve adjacent areas anatomically and functionally linked to the disorganized cortex. Due to the intrinsic epileptogenicity of these lesions, most of partial epilepsies previously defined as “cryptogenic” and frequently resistant to pharmacological therapy are now recognized to be associated with malformative cortical lesions and likely susceptible to surgical therapy. As a consequence, an increasing number of epilepsy patients are surgically treated. Increasing availability of surgical specimens also evokes the interest of neuropathologists. Their input not only covers a systematic microscopic workup for diagnosis, but also the possibility to answer basic questions about the pathogenesis of MCDs and related epilepsies. This has been recently made possible by the addition to traditional routine histopathological methods, “new” powerful techniques fruitfully applied to human surgical tissue samples.
A precise incidence of MCDs is not known, but it appears that they are more common, particularly in patients with epilepsy than was recognized in the pre‐MRI era. Furthermore, the increasing number of patients admitted to epilepsy surgery has further elucidated the neuropathological incidence of MCD in patients with otherwise unremarkable MRI. In large surgical series of patients with intractable epilepsy, the incidence of postsurgical diagnosis of different types of MCDs progressively increased from the early 1990s to the last few years 77, 132, 199, 219. Although the statistical incidence of MCD in surgically treated patients varies among different centers, biased by different factors such as methodological approaches, patient selection and MRI versus neuropathological diagnosis, it is estimated that 25%–40% of drug‐resistant childhood epilepsies are caused by MCD (96) and that 75% of patients with MCD will have epilepsy sometime in their life (133).
Classification systems
An increasing number of malformations are described in the current literature and many types and variants can be encountered in clinical practice. Some malformations have a clear pattern of inheritance, while others are almost exclusively sporadic; the genetic underpinnings of these malformations are increasingly being defined such that by now, more than 30 causative genes have been identified 98, 99. However, environmental causes such as in utero infections, trauma and vascular‐ischemic events could also be determinants for the generation of cortical malformations.
Abnormal cortical development represents a major cause of epilepsy and severe malformations manifest with profound developmental delay and early onset seizures. Mild malformations may be detected after seizure onset at various ages in otherwise healthy individuals 92, 97. There is no general consensus on these complex structural abnormalities and the current nomenclature is not uniform and, therefore, often misleading. Thus, formulating a classification scheme for MCD is difficult and despite many efforts, it is widely recognized that classifications utilized in the current literature are far from satisfactory. Indeed, a particular defect in corticogenesis may give rise to more than one morphological subcategory of MCD and conversely a morphological subtype of MCD may result from multiple different mechanisms. Due to the complexity of the central nervous system (CNS) and to continuous progress in different fields of neurosciences, a unified and comprehensive classification is far for being formulated and needs continued debate.
Neuropathology has had a great impact in this aspect and careful analysis of neuropathologic material, especially with the aid of new diagnostic methodologies, allowed the distinction and categorization of different forms of MCD. Differences among malformations, previously grouped together, have been revealed by high‐resolution MRI, while correlations between morphologically recognized malformations and electro‐clinical findings have provided the basis for new classification schemes 12, 148, 157, 166, 200, 204. Recent advances, particularly those derived from genetic analysis, highlight the need for more integrated and complex approaches to clarify MCD.
A combined morphological and genetic scheme referring to malformations of the entire brain has been proposed 75, 179, while Barkovich and coworkers (12) have developed a new classification, specifically addressed to MCD, based on major stages at which cortical development is affected. The categories are based on known developmental steps, pathologic features, genetics (when possible) and neuroimaging features. As focal cortical dysplasias (FCDs) are among the most frequent epileptogenic malformations, particularly susceptible to surgical treatment, a need for a specific classification of these abnormalities was envisaged. Based on combined neuroimaging, electro‐clinical and neuropathological findings, different classifications of FCD have been proposed 157, 200. In light of the latest data, including post‐surgical outcome, however, a new consensus classification has been proposed by an ad hoc task force of the International League Against Epilepsy (ILAE) Diagnostic Methods Commission (23).
FCD
Neuropathology and classification
FCD represents localized malformative brain lesions of unknown cause, frequently encountered in surgical resection specimens from patients with chronic medically intractable epilepsy 21, 190, 193.
Historical background
Taylor and colleagues (204) were first in 1971 to describe neuropathological features of FCD in surgical specimens of 10 epilepsy patients. They reported, as the most evident microscopic feature, the disruption of the normal cortical lamination by the presence of “large aberrant neurones,” as well as the presence of “grotesque cells” in both cortex and subcortical white matter (204). The authors suggested that these aberrant (“exotic”) populations of nerve cells may underlie the clinical manifestations of certain forms of focal epilepsy and also discuss the similarities with the neuropathological features of cortical lesions in patients with tuberous sclerosis complex (TSC), emphasizing the possibility of a forme fruste of TSC (204). Before the comprehensive description provided by Taylor and colleagues, few autopsy cases of focal cerebral gliosis with giant nerve cells in patients with intractable epilepsy had been reported and interpreted as a congenital condition of unknown etiology and pathogenesis (54). Already in 1896, Lombroso and Roncoroni reported focal cytoarchitectural cortical malformations reminiscent of the Taylor‐type FCD in patients with epilepsy [reviewed in (44)].
Pathological diagnosis and classification systems
Since the first reports, it has become clear that different histopathological features can be encountered in clinical practice and that the morphological spectrum of FCD is broad, including FCD variants with cortical dyslamination without significant cellular abnormalities. FCD may represent an isolated lesion, although cortical dyslamination can also be detected adjacent to hippocampal sclerosis, glio‐neuronal tumors, vascular malformations, as well as adjacent to a large variety of lesions acquired early during brain development (21). During the past 15 years, different FCD classification systems have been proposed 12, 148, 157, 200. The classification published by Palmini et al in 2004 was the consensus report of an international workshop and was mainly based on histopathological features, subclassifying the FCD into Type I and Type II categories. Palmini et al's FCD Type I referred to alterations in cortical lamination for Type Ia, with additional cytoarchitectural abnormalities found in Type Ib; Palmini's FCD Type II referred to FCD including both cortical dyslamination and cellular abnormalities (equivalent to the Taylor‐type FCD), such as the presence of dysplastic (dysmorphic) neurons (Type IIa) and balloon cells [Type IIb; (157) reviewed in (193)]. During the past 6 years, this classification system has been extensively used as a basis for several scientific reports [reviewed in 190, 193], but failed to provide consistent associations with clinical and neuroradiological features. Furthermore, the prediction of postsurgical seizure control remained ambiguous, particularly for FCD Type I 21, 121, 132, 193. This variability may possibly reflect inconsistent histological diagnoses, especially for Type I FCDs, which in the Palmini et al classification included different entities (ie, isolated and associated FCD variants). Accordingly, a recent study reported excellent interobserver concordance for the diagnosis of FCD Type II, but relatively poor neuropathologic inter‐ and intraobserver agreement for the Type I subtypes (41). These findings suggested the need to refine neuropathological diagnostic criteria of FCD subtypes to achieve improved reproducibility and enhanced insight into the clinico‐pathological correlations of FCD subtypes. A task force of the ILAE has recently generated a new consensus classification of FCD subtypes based on histopathological features and representing the basis for prospective clinical studies addressing the epileptogenicity of different FCD variants (23)
The ILAE classification system of FCD consists of a three‐tiered system, including both isolated and associated FCD variants [Table 1, (23)]. FCD Type I (Table 1 and Figure 2B,C) refers to isolated forms of FCD, characterized by an abnormal cortical layering, which may affect one or multiple lobes and is often observed in young patients with severe epilepsy and psychomotor retardation 22, 23, 122, 202. The new ILAE classification includes three FCD Type I subtypes, according to the pattern of cortical dyslamination (Table 1). FCD Type Ia is characterized by abnormal radial migration, as well as neuronal maturation with immature small diameter neurons organized in microcolumns, resembling the microcolumnar organization pattern described during the early stages of cortical development [(165), Table 1; Figure 2B]. However, the diagnosis of this FCD subtype requires particular attention when studying specimens of temporal polar cortex, including areas that tend to maintain a columnar appearance (64). In addition to the microcolumnar organization, increased numbers of heterotopic neurons are often observed and hypertrophic neurons can also be encountered outside layer 5 19, 23. FCD Type Ib (Table 1; Figure 2C) is characterized by abnormal cortical layering affecting the 6‐layered tangential organization of the neocortex and including a large spectrum of histopathological features ranging from alterations of the entire neocortical architecture to a more subtle abnormal layering involving specific layers, such as layer 2 or layer 4 or both (23). Also in this FCD subtypes, heterotopic neurons in white matter and hypertrophic neurons (outside layer 5) can be encountered, as well as normal neurons with disoriented dendrites 19, 23. FCD Type Ic refers to isolated lesions characterized by abnormal cortical layering affecting both radial and tangential cortical organization [Table 1; (23)]. Immunocytochemical analysis using antibodies directed against neuronal nuclear antigen (NeuN; Figure 2) can improve the visualization of the cortical layer abnormalities characteristic of the FCD Type I subtypes; heterotopic neurons within the white matter can be visualized using NeuN or microtubule‐associated protein 2 (MAP2) immunohistochemistry (19).
Table 1.
ILAE consensus classification system for FCD. Abbreviations: FCD = focal cortical dysplasia; ILAE = International League Against Epilepsy.
| FCD type I (isolated) | Ia: abnormal microcolumnar arrangement of neocortex | Ib: abnormal tangential cortical lamination | Ic: abnormal radial and tangential cortical lamination | |
| FCD type II (isolated) | IIa: FCD with dysmorphic neurons | IIb: FCD with dysmorphic neurons and balloon cells | ||
| FCD type III (associated with principal lesion) | IIIa: FCD in the temporal lobe associated with hippocampal sclerosis | IIIb: FCD adjacent to a glial or glioneuronal tumour | IIIc: FCD adjacent to a vascular malformation | IIId: FCD adjacent to any other lesion acquired during early life |
Three‐tiered ILAE classification system for FCDs. Modified from (23).
Figure 2.
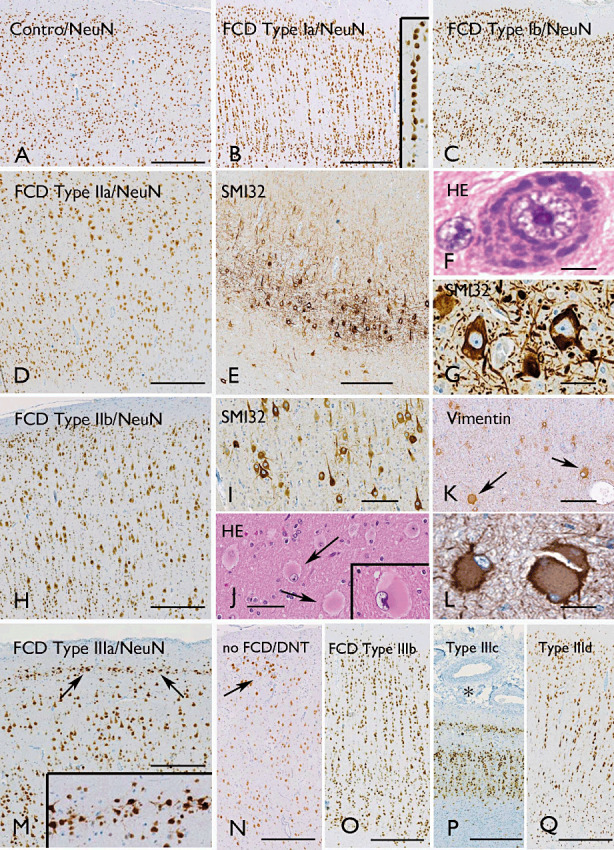
FCD variants. ILAE classification system 19, 23: A. Normal appearing neocortex (NeuN). B. Microcolumnar arrangements of small diameter neurons in FCD type Ia (NeuN); microcolumns are composed of more than eight neurons (insert in B). C. Abnormal cortical layering affecting the 6‐layered tangential organization of the neocortex in FCD Type Ib (NeuN). D–G. FCD Type IIA with cortical dyslamination (D, NeuN) and dysmorphic neurons with enlarged nuclei and aggregates of Nissl substance (F, H&E), as well as accumulation of nonphosphorylated neurofilaments (E, G; antibody SMI32). H–L. FCD Type IIb with cortical dyslamination (H, NeuN), dysmorphic neurons with accumulation of nonphosphorylated neurofilaments (I, antibody SMI32) and balloon cells (J, H&E, arrows and insert; K, L, vimentin, arrows in K). M. FCD Type IIIa with the characteristic abnormal Layer II (NeuN, arrows and insert in M). N. Neocortex adjacent to a dysembryoplastic neuroepithelial tumor (DNT) with clear cell tumor infiltrates that disrupt the cortical architecture (NeuN, no FCD); O. Neocortex adjacent to a DNT with microcolumnar arrangements, but without tumor infiltration (FCD Type IIIb, NeuN). P. Abnormal cortical architecture in a patient with meningioangiomatosis (FCD Type IIIc, NeuN). Q. Abnormal cortical architecture adjacent to a glial scar (NeuN). Scale bar in A: A–E, H, M–Q: 500 µm; I, K: 160 µm; J: 80 µm; G, L: 40 µm; F: 12 µm. FCD = focal cortical dysplasia; H&E = hematoxylin and eosin; ILAE = International League Against Epilepsy; NeuN = neuronal nuclear antigen.
FCD Type II refers to an isolated malformation characterized by disrupted cortical lamination and cytological abnormalities and [as in the previous classification system (157)] includes two subtypes, FCD Type IIa (with dysmorphic neurons, but without balloon cells) and FCD Type IIb (with dysmorphic neurons and balloon cells) (Table 1; Figure 2D–L; (23)). The cortical dyslamination in FCD Type II is often pronounced, without any recognizable layers (except for Layer I). In addition, blurring of gray/white matter junction is often observed, because of increased heterotopic neurons in white matter. Dysmorphic neurons in FCD Type II can be identified in routine hematoxylin and eosin (H&E) and cresyl violet (Nissl‐staining) staining. However, because they accumulate neurofilament proteins (phosphorylated and nonphosphorylated) in their cell bodies, antibodies directed against these proteins (2F11 or SMI32) may be useful to detect dysmorphic neurons, showing their abnormal shape, size and orientation (Figure 2E,G,I). Balloon cells (histologically identical to giant cells in tubers of TSC patients) can be detected in routine H&E; however, vimentin and the splice variant of GFAP, GFAP‐δ, may represent helpful markers for the demonstration of balloon cells in FCD specimens 30, 129, 142. In addition, some balloon cells also express the CD34 epitope (70) and the adhesion molecule on glia [AMOG; (31)]. Reduction of myelin staining, particularly evident in Type IIb, can be visualized using Luxol fast blue or Klüver–Barrera staining (19).
A major advance of the 2011 classification is represented by the FCD Type III category where FCD is combined with other potentially epileptogenic pathologies 20, 187. FCD Type III consists of four different subtypes: IIIa associated with hippocampal sclerosis (HS); IIIb associated with tumors; IIIc associated with vascular malformations; IIId associated with any other lesion acquired during early life [Table 1; Figure 2O–Q; (22)]. The histopathological features of FCD Type III subtypes may be similar to those detected in FCD Type I, with alterations in architectural (cortical dyslamination) and/or cytoarchitectural composition of the neocortex (hypertrophic neurons); however, distinct patterns can be identified in specific variants, such as FCD Type IIIa. In approximately 10% of surgical specimens of patients with HS, the temporal cortex shows an abnormal band of small and clustered “granular” neurons in the outer part of Layer II [81, 206; Figure 2M]. In addition, small “lentiform” nodular heterotopias have been reported in a subset of patients with HS (23). As discussed previously, immunocytochemical analysis using antibodies directed against NeuN, MAP2 and neurofilament proteins can improve the visualization of architectural and/or cytoarchitectural alterations of the cortex. Additional antibodies directed against CD34, p53, mutant IDH1 and Ki‐67 can also be helpful in the evaluation of FCD Type IIIb, to exclude neocortex infiltrated by neoplastic cells, which should not be diagnosed as FCD 19, 23. The new FCD classification system is based on histopathological criteria and its major aim is to improve the identification and homogeneity of different entities (ie, isolated and associated FCD subtypes). The next challenge is represented by the re‐evaluation of this new classification, involving different international centers and addressing the clinical, electrophysiological and imaging features, and the postsurgical seizure control. A collaboration between different neuropathology departments has already been established to examine both the interobserver and intraobserver variability associated with application of the ILAE classification system. In addition, we should now look forward to collaboration between clinicians and basic scientists to improve our still limited understanding of the underlying molecular mechanisms of FCD subtype formation (see below).
FCD—Molecular basis
With respect to molecular alterations in cortical malformations, major objectives aim at improving our understanding of gene defects and dysregulated signaling cascades, as well as molecular and cellular mechanisms mediating hyperexcitability. Given the revised classification of FCD, our current knowledge of the molecular pathology for distinct entities, particularly the FCDs Type I, is still limited. In contrast, molecular alterations of FCD type II are better understood. However, the delineation of cellular or molecular differences between these lesion types that refer to histological differences has the potential to become highly relevant with respect to the development of new diagnostic and therapeutic perspectives. Recent data suggest differential expression of several stem cell proteins and aberrant phosphorylation of mammalian target of rapamycin (mTOR) substrates, that is, S6 and S6 kinase 1 proteins, to distinguish Type II from Type I FCDs, that is, biomarkers for diagnostic pathology in resected epilepsy specimens (156). Epilepsy‐associated focal lesions and brain malformations in patients suffering from familial syndromes have served as an argument for overlapping molecular pathogenesis. Several clinico‐pathological studies have pointed to a role of specific genes and pathways in epilepsy‐associated glioneuronal lesions 15, 17, 150, generally underlying rare familial disorders such as tuberous sclerosis complex (TSC) 211, 212. TSC, as discussed in detail next, is caused by germline mutations of the TSC1 (hamartin) and TSC2 (tuberin) genes 67, 163. Generally, sporadic epilepsy patients with FCDs as well as benign glioneuronal tumors do not present with additional TSC‐associated stigmata (171). Nonetheless, FCD Type IIb brain specimens typically have sequence alterations in TSC1 (139). A base transition in exon 17 (2415C > T; His732Tyr) is increased in FCD Type IIb compared with controls. This base exchange is located in the interaction domain of hamartin with tuberin (212). Intriguingly, in FCD Type IIa, abundant genomic polymorphisms were detected in intron 4 of TSC2, but no allelic variants in exon 17 of TSC1 were observed (139). Are there potential functional consequences for allelic variants increased in FCD Type IIb? Co‐immunoprecipitation assays with tuberin revealed reduced tuberin binding of hamartin compared with wild‐type hamartin, similar to two TSC1 stop mutants. Furthermore, aberrant nuclear distribution of hamartin in vitro was observed, suggesting a fundamental functional impairment of hamartin (136). Several transgenic mouse studies have pointed to TSC1 with a major role for cellular abnormalities reflecting those in cortical malformations (144). Only recently, tuber/FCD Type IIb mimicking lesions in mice have been generated by using a sophisticated transgenic strategy (71). Hamartin and tuberin constitute a tumor suppressor mechanism, which plays a central role in the insulin/PI3K‐signaling pathway 107, 128. The PI3K pathway is critical for cell size‐ and growth‐control and cortical development (198). Binding of insulin to its membrane receptor activates the cascade components PI3K, Akt, TSC1/TSC2, mTOR (mammalian target of rapamycin) and the transcription factors p70S6kinase (S6K), as well as ribosomal S6 protein (S6) 68, 80. Inactivation of the TSC1/TSC2 complex by phosphorylation through Akt results in phosphorylation of mTOR and subsequent activation of the respective transcription factors interfering with cell size control; differential activation of distinct upstream elements have been demonstrated in FCD IIb 106, 143, 182, 183. Activation of individual PI3K‐pathway downstream components has also been reported in FCD Type IIb, that is, the eukaryotic translation initiation factor (eIF) 4G and phospho‐S6 15, 150. FCD Type II, but not FCD Type I cases exhibit activation of the mTOR cascade with strong neuronal expression of the phosphorylated isoform of S6 protein (108). Interfering with this cascade provides intriguing therapeutic perspectives in animal models of TSC‐associated lesions as well as in human TSC‐related malformations 5, 123, 145. Intriguingly, Everolimus has been established recently as the first mTOR inhibitor for the treatment of TSC patients with subependymal giant‐cell astrocytomas. Evidence supports reduction in tumor volume and improvement in epilepsy (123).
Recent data have pointed to a role of doublecortin‐like (DCL), a protein critically involved in neuronal division and radial migration during early corticogenesis in FCD as well as TSC. Balloon cells in FCD and giant cells in TSC, as well as dysmorphic neurons in both entities were observed to express strongly DCL. The prominent postnatal expression of DCL by cell elements of FCD and TSC supports an important role for this microtubule‐associated protein (28). Furthermore, in FCD Ia taken from patients up to 4 years of age, prominent DCX‐positive [DCX(+)] immature cells have been observed along the junction of layers I and II, with processes extending into the molecular layer. Such expression patterns in layer II of FCD Ia, but not in FCD II, may indicate delayed or abnormal cortical maturation rather than ongoing cytogenesis (194). Understanding mechanisms mediating increased excitability of respective lesions is for obvious reasons highly important. Recent analyses in experimental models of seizures and in temporal lobe epilepsy have pointed to a critical role of high‐mobility group box 1 and toll‐like receptor 4 signaling in mediating hyperexcitability and seizures (230). Although microglia reactivity is increased in FCD I compared with controls, the number of HLA‐DR‐positive cells is substantially denser in FCD II. There, a striking presence of perivascular and parenchymal CD3(+) T lymphocytes,a predominance of CD8(+) T‐cytotoxic/suppressor lymphocytes, a few dendritic cells, andexpression of components of the complement cascade, IL‐1β and MCP1 suggested the activity of both innate and adaptive immunities (108).
TUBEROUS SCLEROSIS
TSC is an autosomal dominant, multisystem disorder that results from mutations in the TSC1 or TSC2 genes 67, 211. CNS involvement is common in TSC and the neurological manifestations of TSC are the most disabling, including: developmental delay, neurobehavioral abnormalities such as autism, and severe epilepsy 33, 55. Although a clear genotype–phenotype correlation has not been established, the majority of studies suggest that patients with mutations in TSC2 have a more severe neurologic phenotype 9, 57, 154. Complex structural brain abnormalities are believed to underlie the neurological symptoms of TSC patients. Neuropathological examination of post‐mortem and surgical TSC brain specimens reveals three major lesions: subependymal nodules, subependymal giant cell tumors and cortical tubers 63, 151. However, recent advances in neuroimaging allow the detection of more subtle structural brain abnormalities present throughout the brain, which are believed to contribute to the complex neurological features of TSC 63, 89, 135.
Subependymal nodules (SENs) represent small (usually multiple) benign proliferative lesions lining the ventricular system that are believed to develop in fetal life and to be asymptomatic; microscopically, SENs often contain calcifications and are mainly composed of glial cells with variable morphology (plump or elongated glial cells), sometimes including small clusters of giant cells [26, 87, 151; Figure 3]. SENs may grow and develop into subependymal giant cell astrocytomas (SEGAs), which are low‐grade, slow‐growing tumors that arise from the periventricular region and can cause obstructive hydrocephalus 1, 87, 151.
Figure 3.
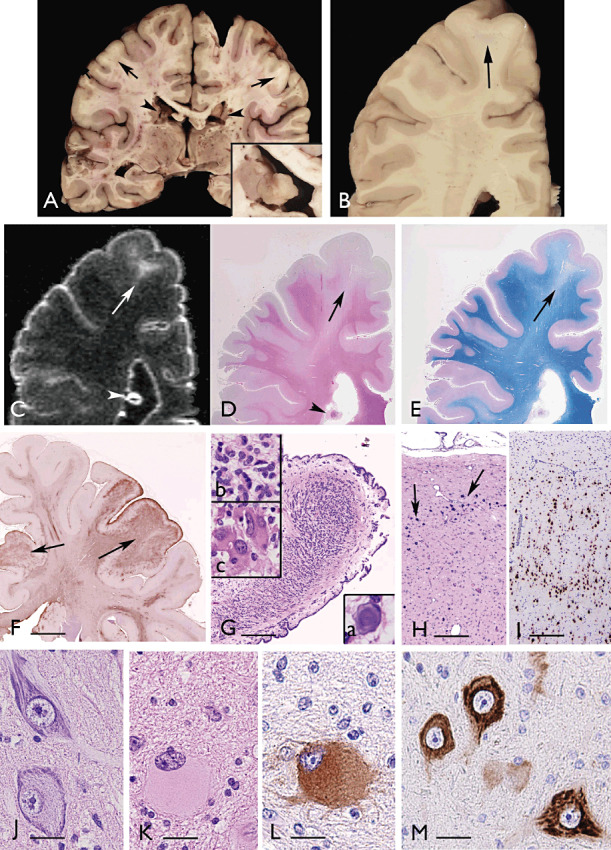
Tuberous sclerosis. Panel A,B. Coronal sections of the brain (32‐year‐old patient with a germline mutation in the TSC2 gene; (26)), showing several regions with blurring of the cortex/white matter junction (arrows) and subependymal nodules (SENs; arrowheads and insert in A). Panel C. Post‐mortem MRI; 3D‐FLAIR image, showing the hyperintense tuber (arrow), as well as SEN with calcifications (arrowhead). Panels D,E. Histologic stains [D: hematoxylin and eosin (H&E) and E: Luxol fast blue‐PAS‐staining]. Note the cortical tuber (arrows in D and E) with decreased density of myelinated fibers (E) and the calcified SEN (arrowhead in D). Panel F. Low magnification view showing strong GFAP immunoreactivity within cortical tubers (arrows). Panel G. H&E stain showing a low magnification image of a SEN appearing as an oval‐shaped compact and calcified lesion, with overlying ependyma, projecting into the lateral ventricle; insert a in G: calcification; inserts b–c: high‐magnification microphotographs of H&E stain showing the cellular components of a SEN, including relatively small, plump glial cells with eosinophilic cytoplasm (b) and giant aberrant cells with large nuclei displaying different size and shape (c). Panel H. H&E stain of cortical tubers showing the disorganized cortical cytoarchitecture with multiple punctate calcifications (arrows). Panel I. NeuN stain showing the disorganization of the neuronal component within the cortical tuber. Panel J. Dysmorphic neurons. Panel K. Giant cell with pale eosinophilic cytoplasm and balloon‐like appearance. Panels L,M. Microphotographs showing strong phospho‐S6 ribosomal protein (pS6) immunoreactivity in a giant cell within the subcortical white matter (L), and in dysmorphic neurons within the dysplastic cortex (M). Scale bars: F: 0.8 cm; G: 400 µm; H–I: 300 µm; J–M: 40 µm. MRI = magnetic resonance imaging; NeuN = neuronal nuclear antigen.
Cortical tubers are focal developmental malformations detected, as single or multiple lesions (Figure 3), in more than 80% of patients with TSC [for reviews, see 52, 155]. Several cases have been reported with prenatal detection of CNS lesions 43, 86, 222 and with histopathological confirmation of tubers as early as 20 weeks gestation (158), indicating that tubers form during embryonic brain development, probably between weeks 9 and 20 of human gestation. Cortical tubers display cortical dyslamination with different cell types, including dysmorphic neurons, reactive astrocytes and giant cells [26, 87, 151, 230; Figure 3]. There is no obvious cytological difference between the dysmorphic neurons observed in tubers of TSC patients and those detected in specimens of patients with FCD Type IIa/b. Similarly, the giant cells in TSC are histologically identical to balloon cells detected in FCD Type IIb [large cell body and opalescent glassy eosinophilic cytoplasm; (23)]. Such cells have been shown to express both neuronal and immature glial markers, suggesting a failure to differentiate prior to migration into the cortex [28, 51, 131, 197; for review, see (155)].
A growing body of evidence suggests that cortical tubers are not static lesions, but are instead highly dynamic, exhibiting evolving features over time. Recently, the cystic evolution of tubers has gained interest because of its strong association, in approximately 50% of patients, with a TSC2 gene mutation and with a more aggressive seizure phenotype 45, 46. Cyst‐like tubers possibly represent the end stage of a large spectrum of degenerative changes, affecting the white matter. An additional study suggests that astrogliosis in tubers is a dynamic process as well, with progression of astrocytes from “reactive” to “gliotic,” supporting the hypothesis that tubers are dynamic lesions (192).
The identification of the TSC1 and TSC2 genes has facilitated our understanding of the molecular mechanisms involved in the development of malformative and neoplastic lesions in TSC brain. Loss of function mutations results in a constitutive activation of the mTOR cascade, which plays a critical role in the regulation of cell growth and proliferation 42, 128, 156. Several studies have provided evidence of cell‐specific activation of the mTOR pathway in tubers, showing that dysmorphic neurons and giant cells are strongly immunoreactive for the phosphorylated isoform of p70S6 kinase1 and ribosomal protein S6 [pS6 15, 28, 150, 155, 197; Figure 3]. Expression of activated (phosphorylated) components of the mTOR pathway, such as pS6, now permits the analysis of the cellular components of tubers not simply on the basis of their morphology, but on the basis of the specific pathogenetic defect underlying the development of these lesions. Interestingly, recent observations in fetal brain indicate that mTOR hyperactivation represents an early event in the pathogenesis of tuber formation and is strictly linked to cytoarchitectural features characteristic of tubers (Crino and Aronica, unpub. obs.).
Many TSC‐associated tumors show loss of heterozygosity (LOH) at either the TSC1 or TSC2 locus 4, 42, 88. There is still debate about whether this “two‐hit” hypothesis explains tuber formation 115, 164. A recent study, by sequencing TSC1 and TSC2 in microdissected pS6–immunolabeled giant cells, identified in 5 out of 6 tuber specimens a somatic mutation in single giant cells that was not detected in whole tuber sections or leukocyte DNA, suggesting that a single somatic inactivating mutations may represent a mechanism underlying the formation of tubers adjacent to morphologically normal cortex (53).
Epilepsy is reported in over 80% of patients diagnosed with TSC and significantly affects psychomotor development and quality of life. Failure to respond to anticonvulsant drug treatment is particularly common in TSC patients 47, 55. Cortical tubers are believed to represent the neuropathological substrate of epilepsy in TSC patients. Recent advances in structural and functional imaging techniques have resulted in more accurate preoperative evaluation and improvements in surgical outcome 32, 217. However, the relationship between the tuber and the epileptogenic zone remains a challenge (32). Increasing evidence support the importance of the perituberal cortex in TSC, as well as in other malformations of cortical development [for review, see (220)]. Although epilepsy surgery, targeting tubers, often results in seizure freedom 120, 217, some patients continue to have seizures after target tuber resection and a few cases of TSC patients becoming seizure free after resection of nontuberal cortex have been reported [(216); for review, see (220)].
The cellular mechanism(s) underlying the epileptogenicity of cortical tubers remain largely unknown and possibly involved multiple mechanisms. Recent studies support the role of developmental alterations affecting the balance between excitation and inhibition in the epileptogenicity of cortical tubers [for review, see 102, 220]. Abnormal glutamate homeostasis, increased AMPA‐receptor‐mediated currents, impaired astrocytic gap junction coupling and potassium buffering, which would support seizure generation, have been described in mouse models of TSC 203, 216, 223, 228. Large‐scale gene expression studies indicate alterations in glutamatergic and GABAergic synaptic transmission within tubers in TSC patients, involving reduced expression of the glial glutamate transporter (GLT‐1 or EAAT2), reduced expression levels of GABAA receptor subunits and reduced expression of potassium channels (29). Selective alterations of ionotropic glutamate receptor subunit, as well as of metabotropic glutamate receptor (mGluR) subtypes, have been observed in the different cellular components of cortical tubers 27, 197, 218. Another recent development is represented by the demonstration in cortical tuber of a complex activation of pro‐inflammatory signaling pathways, including chemokines, complement, IL‐1β and Toll‐like receptor (TLR) mediated pathways 25, 29, 230, as well as evidence of activation of the plasminogen system (109); high expression of several factors involved in the regulation of angiogenesis has also been detected [(29); for review, see (7)]. Accordingly, epidermal growth factor (EGF), hepatocyte growth factor (HGF) and vascular endothelial growth factor (VEGF), which regulate angiogenesis and cell growth in the developing brain, have recently been shown to be up‐regulated in human SEGAs and tubers as well as in a mouse model of TSC, providing potential new target for therapy development in TSC (159).
HEMIMEGALENCEPHALY
Hemimegalencephaly (HMEG) is a rare malformation of cortical development characterized by enlargement and cytoarchitectural abnormalities of one cerebral hemisphere. In 1835, Sims in his article on “hypertrophy and atrophy of the brain”(186) discusses several autopsy cases of hypertrophy which have fallen under his observation, including one case of hypertrophy of one hemisphere. Since then, small series reports have been published, indicating that HMEG may occur as an isolated malformation or in syndromic form and that is associated with developmental delay, motor deficits and severe epilepsy with onset typically within the first few months of life 112, 176, 180, 207, 210. Epilepsy is often poorly controlled by antiepileptic drugs, requiring surgical therapy to remove or functionally disconnect the epileptogenic area within the affected hemisphere. However, epilepsy surgery in HMEG patients yields modest postsurgical seizure and cognitive outcomes (114). Radiologically, HMEG, in both isolated and syndromic forms, is typically characterized by an enlarged cerebral hemisphere with abnormal gyral pattern, thickened cortex and white matter abnormalities, with loss of gray‐white matter differentiation, on the enlarged side 74, 117, 153, 224. Structural changes have been also reported in structures outside the involved hemisphere, including ipsilateral olfactory nerve enlargement, cerebral vascular dilations, cerebellar enlargement with abnormal architecture of the cerebellar folia (181). The spectrum of CNS structural abnormalities includes cases variously defined as “hemi‐megalencephaly,”“focal megalencephaly” or “localized megalencephaly,” in which the abnormalities are detected only over a partial area of one hemisphere 58, 127, 153. In these cases, the detection of associated extracerebral abnormalities has been suggested to provide a clue for differentiating localized megalencephaly from multilobar cortical dysplasia (153).
HMEG has been reported in patients with various syndromes, such as Proteus syndrome (62), neurofibromatosis (56), hypomelanosis of Ito (195), Klippel–Weber–Trenauany syndrome (208), TSC (39) and linear sebaceous nevus syndromes 74, 174, 191. In both isolated and syndromic forms, a large spectrum of morphological alterations MCD can be observed [for review, see 74, 76, 141]. Macroscopic examination of the brain often reveals an abnormal gyral pattern (pachygyria, polygyria, polymicrogyria) and increased thickness of the cortex of the enlarged hemisphere (with blurring of the cortex‐white matter junction; Figure 4); microscopically, in the large majority of cases a severe dyslamination is observed throughout the cortex together with the presence of hypertrophic and dysmorphic neurons, as well as heterotopic neurons in the white matter and leptomeningeal glioneuronal heterotopia [6, 24, 61, 76, 172, 175, 226; for review, see 74, 76, 141; Figure 4]. Balloon cells have also been detected in HMEG specimens and have been considered a typical component of HMEG 11, 76. However, in a large series of surgery patients with HMEG, balloon cells were identified in less than 50% of the surgical specimens (175), with no detection in fetal examples (24). The observation that complex cytoarchitectural abnormalities are observed prenatally, also at a fetal age of 22 weeks (141), supports the hypothesis of an underlying primary genetic defect which affects the early stages of cortical development (between 10 and 20 weeks) involving the proliferation, migration and differentiation of precursor cells.
Figure 4.
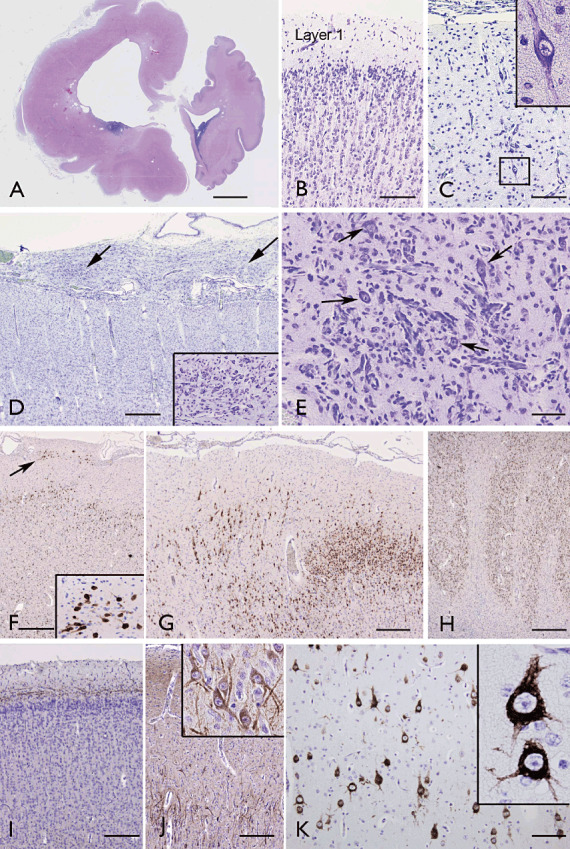
Hemimegalencephaly (HMEG). A. Coronal section showing diffuse and severe hyperthrophy of the left hemisphere with lateral ventricular dilatation, periventricular cysts, pachygyria and thickened cortex (32 weeks gestation; HMEG plus epidermal nevus (24). Panels B–D. Clear difference in the architecture of the cortex between the right side (“normal” side; B, cresyl violet) and the affected left side (HMEG; C, D, cresyl violet). HMEG side (C and D) shows severe cortical disorganization with loss of lamination and presence of cytomegalic neurons (insert in C) and thickened leptomeninges with clusters of heterotopic cells (arrows and insert in D). Panel E shows (arrows) a cluster of dysmorphic neurons within the affected cortex (cresyl violet). Panels F,G. (NeuN in HMEG cortex): loss of lamination with irregular distribution of neuronal cells and clusters of NeuN‐positive elements in Layer I (arrow and insert in F). H. NeuN staining in the occipital HMEG cortex with polymicrogyria. I,J. Neurofilament immunoreactivity in the unaffected (I) and affected (J) cortex, showing intense cytoplasmic staining in dysmorphic neurons (J). K. Phospho‐S6 ribosomal protein (pS6) staining; strong positivity is observed in a population of dysmorphic neurons within the affected cortex (K and insert). Scale bars: A: 1028 mm; B–C: 250 µm; D: 400 µm; E: 125 µm; F–H: 400 µm; I–J 250 µm; K: 200 µm. NeuN = neuronal nuclear antigen.
One unresolved question is whether syndromic HMEG is distinct from the sporadic forms or reflect a spectrum of hemispheric brain malformations, sharing common mechanisms of pathogenesis. The histopathology of HMEG with complex cytoarchitectural abnormalities, including in some cases the presence of dysmorphic neurons and balloon cells a [as in FCD Type IIb; (23)], has suggested a primary disorder of proliferation (196) or a disturbance of cellular lineage and cellular growth 74, 179. Barkovich et al (11) include HMEG among the non‐neoplastic disorders of proliferation (with abnormal cell types) together with tuberous sclerosis and cortical dysplasia with balloon cells (FCD Type IIb). Salamon et al (175) suggested that pathogenesis of HMEG probably involves an increased proliferation of the progenitor cells together with partial failure of postneurogenesis apoptosis in the molecular layer and subplate. Other authors (141) suggested the possibility of an accelerated neuronal differentiation as underlying pathogenetic mechanism, also emphasizing the importance of migration abnormalities. Excessive production and/or function of neuronal growth factors have been suggested to contribute to enhanced neuronal growth and/or differentiation. Attention has been focused on the potential role of EGF (119), nerve growth factor (NGF) and its high‐affinity receptor (6), as well as on the role of VEGF 24, 227.
Recent work from Crino and coworkers points to the role of new converging cell signaling pathways (Wnt/β‐catenin‐ and mammalian target of rapamycin, mTOR‐pathway) in the pathogenesis of HMEG [8, 24, 49, 227; Figure 2]. The identification of aberrant mTOR signaling in malformations of cortical development (such as FCD Type IIb, TSC and HMEG), characterized by cortical dyslamination, cytomegaly and intractable epilepsy, suggests a pathogenic link between these malformations, possibly representing a spectrum of disorders of mTOR signaling [so‐called “TORopathies”; 50, 221]. It has been suggested that HMEG could result from somatic mutations in genes encoding for proteins that regulate mTOR signaling pathway, occurring in progenitor cells in one hemisphere during the early stages of corticogenesis (50). A hemizygous deletion within chromosome 15q11.2–15q13.1 has recently been detected in the affected hemisphere in a single case of isolated HMEG (16).
Little is known about the epileptogenesis of HMEG. Using target cDNA array analysis and immunocytochemistry, the expression of multiple neurotransmitter receptors in surgery and autopsy HMEG specimens has been investigated 14, 24. These studies indicate that changes in expression and/or composition of ionotropic and metabotropic glutamate receptors could play a role in epileptogenesis in HMEG, as suggested for other types of focal malformations of cortical development [for review, see (220)]. In addition, the presence of activated glial cells in HMEG (24) may also be related to the epileptogenicity of this disorder through different mechanisms, including the production of inflammatory cytokines, such as IL‐1β; 184, 213. Accordingly, accumulating clinical evidence strongly supports the relevance of inflammation in the pathophysiology of human epilepsy 7, 214.
LISSENCEPHALY
Impaired cortical development can be caused by aberrant proliferation, differentiation or migration of neural precursor cells and neurons. Obviously, the latter defect leads to aberrant neuronal localization and lack of physiological cortical lamination (157). Migration of virtually all cortical neurons is severely affected in so‐called lissencephaly, that is, an abnormally smooth cerebral surface (215). Classic lissencephaly has a prevalence of 11.7 per million births (60). However, the frequency of milder forms of lissencephaly is not known. Hallmarks of the disorder are severe epilepsy and mental retardation, which in many patients go along with death at young ages. In normal brain development, the agyric cerebral surface, which is present until the 11th week of gestation, is replaced by gyri, which develop from the area of the sylvian fissure within the following weeks. Gyration is physiologically accomplished by approximately the 32nd fetal week. The critical time window for lissencephalic aberrations to initiate is between fetal weeks 11–13 (113). Lissencephaly/agyria and pachygyria, that is, cortical architecture somewhat more advanced, represent a spectrum of cortical aberrations.
Lissencephaly represents a genetic disorder with mutations particularly in two genes, that is, doublecortin (DCX) on the X chromosome (84) and LIS1 on chromosome 17 161, 169, 170. Mutations in DCX cause lissencephaly (XLIS) in hemizygous males and subcortical band heterotopia (SBH) in heterozygous females 65, 162. DCX is a microtubule‐associated phosphoprotein functionally important for cytoskeletal plasticity during axonal outgrowth, neuronal maturation and cell migration (104). Aberrant DCX function impairs the cellular shape and cytoskeletal function as well as migration of cells 84, 85, 178. Furthermore, DCX represents a candidate gene for women with cryptogenic epilepsy and retardation in the absence of male relatives or a known relevant family history (97). Intriguingly, in a rat model of subcortical band heterotopia (SBH) generated by in utero RNA interference of the Dcx gene, it was shown that aberrantly positioned neurons can be stimulated to migrate by re‐expressing Dcx after birth. Restarting migration in this way both reduced neocortical malformations and restored neuronal patterning. Furthermore, the capacity to reduce SBH continued into early postnatal development. Intervention after birth reduces the convulsant‐induced seizure threshold to a level similar to that in malformation‐free controls. For disorders of neuronal migration, these data provide an entirely novel perspective to reactivate developmental programs and, both, reducing the size of cortical malformations and seizure risk (140).
Heterozygous deletions of the chromosomal region 17p13 comprising Lissencephaly 1 (LIS1) causes the so‐called Miller–Dieker syndrome, which includes lissencephaly, craniofacial defects and substantial EEG abnormalities and is generally associated with a reduced lifespan. LIS1 encodes a subunit of the enzyme acetylhydroxylase and is particularly expressed by Cajal–Retzius cells 167, 185. Other genes located on chromosome 17p are likely to be involved in the phenotype of the Miller–Dieker syndrome such as the gene YWHAE encoding a protein termed 14‐3‐3e (209). It has been pointed out that the pachy‐ or agyral alterations associated with LIS1 are stronger in the posterior occipital area, whereas the structural abnormalities in XLIS particularly affect the anterior occipital brain (65).
In animal models, LIS1 is differentially activated by seizure activity (185). Nuclear distribution factor E‐homolog 1 (NDE1), LIS1 and NDE‐like 1 (NDEL1) together participate in essential neurodevelopmental processes, including neuronal precursor proliferation and differentiation, neuronal migration and neurite outgrowth (229). NDE1/LIS1/NDEL1 interacts with Disrupted in Schizophrenia 1 (DISC1) and the cAMP‐hydrolyzing enzyme phosphodiesterase 4 (PDE4). DISC1, PDE4, NDE1 and NDEL1 have each been raised as genetic risk factors for major mental disorders. Recently, it was shown that DISC1 and PDE4 modulate NDE1 phosphorylation by cAMP‐dependent protein kinase A (PKA). PKA‐dependent phosphorylation of the NDE1/LIS1/NDEL1 complex is DISC1‐PDE4 modulated and was suggested to regulate its neural functions (35). Interestingly, experimental approaches such as in utero knockdown of calpain by short hairpin RNA has shown potential to rescue defective cortical layering in Lis1(+/−) mice. Therefore, calpain inhibition can represent a potential therapeutic intervention for lissencephaly (225). DCX is expressed mainly in the developing brain. Epileptic seizures are generally regarded as a result of complex functional changes based on disruption of the normal development of the cortex and white matter and an impaired neuronal network structure.
A further variant of lissencephaly with autosomal‐recessive transmission is because of deletions in the reelin (RELN) gene (103). Generally, moderately severe pachygyria and extensive cerebellar hypoplasia coincide. In addition to seizures, severe hypotonia and developmental delay constitute clinical hallmarks of this syndrome. Type II lissencephaly's structural correlate is generally referred to as “pachygyric micropolygyria”(3). The cerebral surface is agyric but not entirely smooth. Histological assessment reveals abundant and confluent microgyri. In the cerebellum, cortical heterotopias occur in a multiple fashion. Type II lissencephaly is frequently associated with oculocerebellomuscular anomalies such as Walker–Warburg syndrome (177) as well as Fukuyama‐type congenital muscular dystrophy 79, 173. Macroscopic inspection of lissencephalic brains reveals absence (agyric) or decreased presence (pachygyric) of convolutions. In pachygyria, the gyri are plain and smooth. Microscopically, there is blurring of gray and white and impaired cortical structure. Two types of lissencephaly are distinguished (59): Type I is represented by a thick, rarely differentiated cortex under a smooth, largely agyric cerebral surface. There are only limited portions of white matter. The cortex generally comprises four layers:
-
•
a marginal (molecular) layer;
-
•
an outer neuronal layer;
-
•
a cell‐depleted layer with tangentially orientated myelinated fibers; and
-
•
an inner neuronal layer.
The respective architecture is frequently observed in the Miller–Dieker syndrome. The neurons with impaired migration can generate an additional cortical layer in the white matter, which underlies the double cortex formation. In contrast, Type II lissencephaly shows morphological CNS alterations with pachygyria, insufficient gyration of the cerebral surface and widened gyri (Figure 5). The claustrum is frequently absent and respective differences can be distinguished frequently by MRI (118). In the medulla and cerebellum, aberrant cerebellar cortex architecture and heterotopias of the nucleus olivaris can be observed (113). At the transition zone of the pachygyric to normal cortical areas, the outer neural layer continues into the normal cortex, whereas the inner neural layer does not continue in the normal cortex. This element can differentiate pachy‐ from polymicrogyria.
Figure 5.
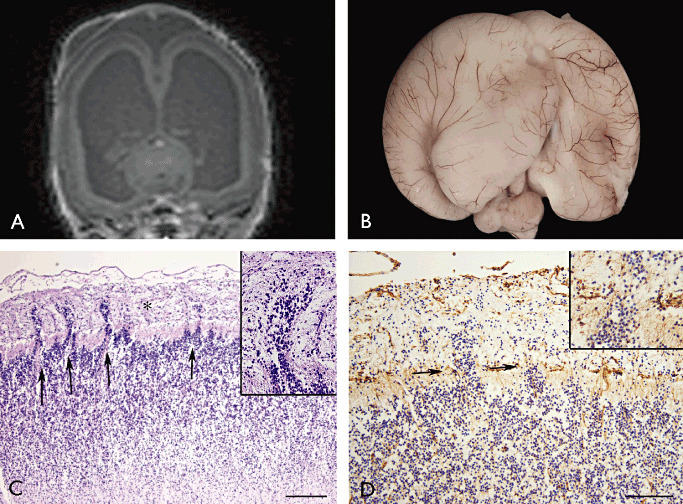
Type II lissencephaly. Fetal brain at 22 weeks of gestation with Walker–Warburg syndrome (POMT1 mutation). A. Post‐mortem MRI (hydrocephalus); B. External view of the brain. C. (H&E) cortical plate showing focal disruption of the pia‐glial limitans (arrows) with overmigration of cells into the leptomeninges (asterisk; high magnification in insert). D. Vimentin staining showing the multiple breaches of the pia‐glial limitans (arrows; high magnification in insert). Scale bars: A: 240 µM; B: 120 µM. H&E = hematoxylin and eosin; MRI = magnetic resonance imaging.
Only recently, tubulin alpha1A (TUBA1A), encoding a critical structural subunit of microtubules, has been implicated in lissencephaly. Novel and recurrent TUBA1A mutations are present in approximately 1% of children with classic lissencephaly and approximately 30% of children with lissencephaly with cerebellar hypoplasia (125). Mutations in the so‐called TUBA1A gene are found in patients with lissencephaly, pachygyria and polymicrogyria. Mutations in TUBA1A occur de novo and in the form of familial recurrence because of parental somatic mosaics in a parent (111). However, further genes such as the recently described GPR56, an orphan G protein‐coupled receptor (GPCR) from the family of adhesion GPCRs (137), may be linked to lissencephaly like maldevelopmental patterns in the future.
HETEROTOPIAS
Heterotopia comprises a group of MCD characterized by the presence of excessive neurons, either isolated or clustered, in the subcortical white matter (Figure 6). According with the location and distribution of heterotopic neurons, three groups are currently described:
Figure 6.
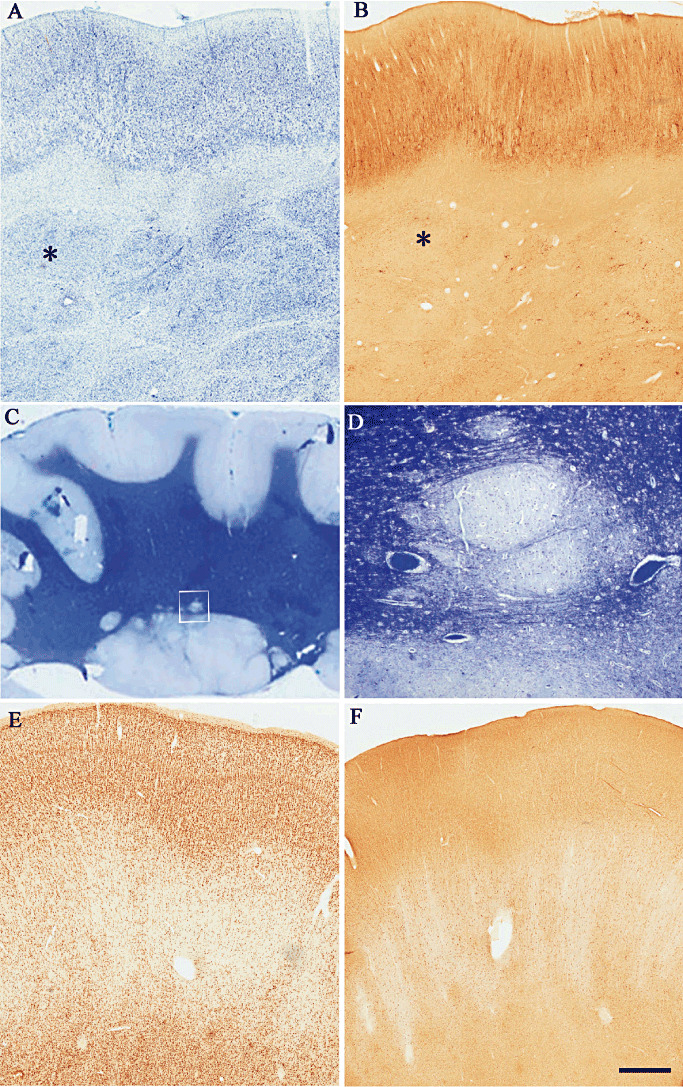
Heterotopia. A,B. Thionin stained and neurofilament immunostained coronal serial sections (asterisk is indicating the same nodule) from a surgical specimen showing multiple subcortical nodules. Note that the overlying cortex appears disorganized and no lamination can be detected. C. Low‐power photomicrograph of a Luxol fast blue‐stained section of occipital cortex, white matter, subependymal region with nodular formations around the ventricle and overlying cortex. D. High‐magnification detail of one of the nodules in C (square). E,F show two serial coronal sections immunostained with NeuN and MAP2 respectively from a patient with “double cortex.” The heterotopic band consists of unlayered cortex with haphazardly organized neurons. The white matter between the “outer” and “inner” cortex contains abundant heterotopic neurons arranged in columns. MAP2 = microtubule‐associated protein 2; NeuN = neuronal nuclear antigen.
-
•
neuronal heterotopia characterized by individual misplaced neurons in the white matter;
-
•
nodular heterotopia with nodules of gray matter within the white matter; and
-
•
band (or laminar) heterotopia (also defined as double cortex).
Although originally only subependymal nodular and band (laminar) forms were considered in the recent classification by Barkovich et al (12), the presence of abundant neurons in the white matter, frequently associated with different forms of MCD, is also considered. Gray matter heterotopia can be diffuse or localized. Diffuse forms include subcortical laminar heterotopia and extensive forms of bilateral periventricular nodular heterotopia. Localized forms can be subependymal, unilateral or bilateral, subcortical (nodular, laminar), or may extend from the subependymal region to the cortex unilaterally (transamantle). Among the different types of nodular heterotopia (NH), the number of bilateral and unilateral cases is approximately equal and bilateral symmetrical NH account for about one‐third of bilateral cases.
NH
The prevalence of NH has not been ascertained in the general population or patients with epilepsy 2, 66, but diagnoses of NH by MRI have been reported in 13%–20% of large series of epileptic patients with MCD [Figure 1C; 13, 66, 134, 166]. Two main groups of NH are currently recognized: (i) subependymal heterotopia (SHE) subsuming periventricular nodular heterotopias (PNH) and (ii) subcortical heterotopia (SCH), further subdivided on the basis of imaging and clinical data 10, 12, 13, 66, 201. Some PNH can be causally related to mutations of specific genes and chromosomal rearrangements [see (160) for references]. An X‐linked dominant inheritance has been established for familial bilateral periventricular nodular heterotopia (classical bilateral PNH) in females, a condition also characterized by a high incidence of spontaneous miscarriages especially in male fetuses and a 50% recurrence risk in the female offspring of affected women. Linkage analysis mapped this disorder to Xq28 and the gene responsible, filamin 1 (FLN1), has been identified. Sporadic cases of bilateral PNH both in females and males have also been described [see (94) for references.]. However, a genetic etiology has not always been found in other forms presenting single or multiple unilateral or bilateral nodules, sometimes associated with other brain malformations and thus suggesting that NH may represent a heterogeneous disorder 13, 152, 201.
Available surgical specimens were rare in the past as it was generally considered that epileptic patients with PNH did not respond to surgical treatment. However, recent reports indicate that in patients with unilateral heterotopia, surgery can be highly beneficial when epileptogenic zone is carefully identified (201). The surgical success also contributed to the neuropathological studies of these malformations providing new data on the intrinsic organization of the nodule and also suggesting possible etiological mechanisms
In all cases, the nodules consist of masses of neurons, with well‐defined boundaries, surrounded by white matter fibers, some of which infiltrate the nodules, suggesting a functional connection between the nodules and the overlying cortex (Figure 5). Neuropathologic investigations (147) reported that all of the nodules, regardless of their size and number (single or multiple), lobe and depth of location (subependymal and/or subcortical), have similar morphological and immunocytochemical characteristics. Similarly organized nodules have also been reported in patients with the filamin 1 gene mutation 116, 205. The reported data showed that the nodules contain pyramidal cells as well as all of the subtypes of inhibitory interneurons with apparent normal morphology (205). Interestingly, the presence of cell‐free zones with small vessels, reminiscent of the molecular layer, and glial cells, with radiating fibers similar to radial pattern of superficial gliosis, was described, suggesting a rudimentary laminar pattern within the nodules. In addition, small Reelin‐immunoreactive neurons within the cell‐free zone were identified, mirroring their normal location in the molecular layer of the overlying cortex (205). This peculiar organization was further confirmed by studies performed by analyzing the expression patterns of three layer‐specific genes (Ror β, ER81 and Nurr1) in tissues from patients with subcortical NH and demonstrating the prevalent expression of neurons belonging to layer VI and layer V in the external border of the nodules, with those belonging to layer IV in a more internal region (82).
The hypothesis that in subcortical NH, some of the Cajal–Retzius, Reelin‐secreting, cells remain displaced within the subplate during early stages of cortical development and that these cells, wrongly located in this transient layer, attract migrating neurons into a heterotopic position leading to the formation of a heterotopic malformation with a rudimentary laminar organization, was postulated (82). This hypothesis was indirectly confirmed by Kubo et al (124), demonstrating that ectopically expressed Reelin in developing mouse cortex is able to determine subcortical nodular aggregates with laminar organization similar to those observed in human pathology. Although MRI frequently suggests a polymicrogyric cortex overlying the NH, recent neuropathologic data reported abnormal gyration but without histological abnormalities consistent with the typical four‐layered or unlayered polymicrogyria. However, various degrees of cortical laminar disorganization similar to those observed in FCD has been described with reduced expression of calcium binding proteins, suggesting an impairment of the GABAergic system. In other cases, a normal cortical organization can be found although cortical disruption can be noted in areas where nodules invade the cortex 147, 201, 205. When NH involves the temporal lobe, hippocampal sclerosis may also be present.
Band heterotopia
This type of malformation, also defined as laminar heterotopia or double cortex, is included by Barkovich and coworkers (12) among those due to abnormal neuronal migration and incorporated in the subgroup defined as lissencephalies/subcortical band heterotopia spectrum. However, from the neuropathological aspect, this malformation is frequently considered within the broad group of “heterotopia.” Being a diffuse cortical malformation, frequently genetically determined, only a few neuropathological reports are available (138). Due to its frequent association with intractable epilepsy, focal surgical resections have been proposed in selected patients. However, despite the fact that in some surgically treated patients, satisfactory postsurgical outcome has been achieved, in general, surgery is considered to yield inadequate results in these cases. Despite the small number of surgical specimens, careful anatomical descriptions are available, thanks to the high‐resolution imaging also providing genotype/phenotype correlations of this malformation (93). The gross anatomical aspect observed on both MRI (Figure 1D) and available specimens shows a continuous bilateral, roughly symmetrical, band of gray matter located between the cortical mantle and the ventricles. The thickness of the band varies among patients and seems to correlate with the degree of associated mental retardation. A slight reduction in number of gyri and pachygyria can be observed in the “outer” cortex, but in general, it is of normal thickness and formed by the regular six layers with a normal distribution of neuronal elements with no cytological alterations. A zone of white matter of variable thickness containing abundant heterotopic neurons frequently arranged in columns is present between the “outer” and “inner” cortex (Figure 5E,F). The heterotopic band consists of unlayered cortex with small and medium‐sized rounded or pyramidal neurons. Although macroscopically the heterotopic band appears as a continuous aggregate of neurons, at the microscopic level it appears discontinuous and interrupted by radially oriented fibers. Within the white matter and in the heterotopic band, the pyramidal neurons are haphazardly oriented and nonpyramidal neurons express different calcium‐binding proteins, suggesting the presence of different subpopulations of GABAergic interneurons.
POLYMICROGYRIA
Polymicrogyria (PMG) is a cortical malformation characterized by an excessive number of small irregular gyri separated by shallow sulci leading to an irregular cortical surface with a lumpy aspect. PMG can be localized to a single gyrus or can involve a portion of one hemisphere; it can be bilateral and asymmetric, bilateral and symmetric, or diffuse. PMG may escape or be confused with “thickened cortex” at imaging investigations when CT scan or low‐field MRI are performed but, if appropriate age‐related protocols are used with high‐resolution MRI, it can be reliably differentiated from other forms of MCD and identified by the presence of numerous cortical gyration even in the absence of irregular gray/white matter junctions. The extent and location of PMG in different brain regions can determine a variety of specific syndromes with a wide range of clinical manifestations, from severe encephalopathy with intractable epilepsy to only selective impairment of cognitive function. Several malformation syndromes have been described such as bilateral perisylvian, bilateral parasagittal parieto‐occipital, bilateral frontal, and unilateral perisylvian or multilobar types 90, 95. However, the most common form of PMG is represented by the bilateral perisylvian PMG characterized by distinct clinico‐pathological features and consistent familial recurrence (93).
Various patterns of inheritance have been described for the different subtypes of polymicrogyria (90). No individual genes have been linked to any of the bilateral forms with isolated polymicrogyria, although a mutation in MECP2 was observed in a male patient with bilateral perisylvian disorder and severe neonatal encephalopathy (83). A recent hybrid genetic‐MRI approach led to the identification of the homeobox gene PAX6 as a factor, in which mutations can result in unilateral polymicrogyria (149). As observed frequently for other types of cortical malformations, polymicrogyria may be part of multiple congenital anomaly syndromes and is frequently observed in the vicinity of encephaloclastic lesions, such as schizencephaly [Figure 1E,F, (38)]. PMG can also be secondary to exogenic causes such as infections including cytomegaly, toxoplasmosis, rubeola, as well as to impaired hemodynamic disturbances, particularly referred to the perfusion area of the middle cerebral artery.
Although PMG is frequently associated with epileptic syndromes, the mechanisms of epileptogenesis related to PMG are not entirely understood. In an experimental model in which microgyri are generated by a freezing insult, it has been shown that functional abnormalities extend beyond the anatomic malformation 110, 168. These data corroborate the observations in humans revealing that the cortex surrounding the PMG is also involved in epileptogenesis and suggest that surgical resection restricted to the polymicrogyric area is not sufficient to substantially attenuate or abolish recurrent epileptic seizures 188, 189. Polymicrogyria is, however, not inevitably associated with epilepsy, and it may present as developmental delay or congenital hemiparesis.
In PMG, the gyri are atypically organized with abnormalities of the physiological laminar cytoarchitectural structure. Although a variety of intermediate neuropathological phenotypes exist, two main histological types are observed, the unlayered and the four‐layered types. In the unlayered PMG, the molecular layer is continuous and does not follow the convolution profiles. The neurons have a radial distribution without any laminar organization (72). This aspect suggests an early disruption of normal neuronal migration with subsequent cortical disorganization. By contrast, the four‐layered PMG shows a four‐layered laminar structure composed by a molecular layer, outer neuronal layer, nerve fiber layer and inner neuronal layer. Apparently, the third layer (nerve fiber) is a result of cellular necrosis. Occasionally, in the neuronal cell layers, granular as well as pyramidal neurons can be observed, resembling residuals of the normal six‐layer cortical architecture. This subtype of malformation is thought to be the result of perfusion failure, occurring between the 20th and 24th weeks of gestation, leading to intracortical laminar necrosis with consequent late migration disorder and postmigratory overturning of cortical organization 73, 78. These two histopathological subtypes do not necessarily have a distinct origin, as both may coexist in contiguous cortical regions 18, 93.
SCHIZENCEPHALY
Por‐/schizencephaly represent generally cystic lesions of cortical brain areas. Anatomical consequences are given by aberrant, immediate connections between the lateral ventricles and the subarachnoidal space. Schizencephaly is currently grouped with polymicrogyria among the malformations of cortical development (MCD; group IIIA 1,2) (11). The immediate pathological substrate of schizencephaly is given by a defect presenting with a transcortical cleft with open and partially closed parenchymal portions decorated by cortical tissue portions (Figure 1E,F).
For schizencephaly, several pathogenetic factors and etiological aspects exist, encountering regional lack of proliferating neurons and glial cells, which impairs cortical architecture. Furthermore, local impairment of neuronal migration or circumscribed ischemic necrosis comprising destruction of the radial glial fibers during early gestation has been claimed (91). Congenital cytomegaly virus infections have been linked with severe brain malformations including schizencephaly (34). Epidemiological data suggest that schizencephaly occurs more frequently in the fetuses of younger mothers and is associated with septo‐optic dysplasia, suggesting that the two conditions may be of common origin (105). The majority of schizencephaly patients are sporadic, but familial schizencephaly has been described. Recent data suggest molecular alterations in schizencephaly, encountering germline mutations in the EMX2 homeobox gene in approximately 70% of the cases 36, 37, 69. However, these data could not be confirmed by other studies (101). Sequencing analyses did not reveal genomic aberrations of LHX2, a gene with an important cortical patterning role; or HESX1 or SOX2, genes that have been associated with septo‐optic dysplasia in schizencephaly (146). As schizencephaly frequently occurs with additional cerebral malformations, such as hypoplasia or aplasia of the septum pellucidum or optic nerve, the involvement of genes important for the establishment of midline forebrain structures has been hypothesized and analyzed. These experiments resulted in observing heterozygous mutations in the SIX3 and SHH genes in a total of three unrelated patients and one fetus with schizencephaly; one of them without obvious associated malformations of midline forebrain structures. Three of these mutations have previously been reported in independent patients with holoprosencephaly. SIX3 operates immediately upstream of SHH, and the SHH pathway represents a key regulator of ventral forebrain patterning. These data suggest that schizencephaly may represent a pathogenetic aspect of a more complex aberration of ventral forebrain development in some patients, that is, belonging to the pathological pattern of holoprosencephaly related to mutations in SHH pathway components (101).
Schizencephaly frequently affects central brain regions as well as sites perfused by the middle cerebral artery. Hypoxic damage is of major importance during fetal development until the end of cortical maturation. Transitions of schizencephaly with polymicrogyria (72) (type 1) and co‐occurrence of schizencephaly with radial orientation of the gyri adjacent to the cortical defect (type 2) have been described 18, 40, 130. Schizencephaly often occurs bilaterally. However, the severity of the pathological manifestations frequently varies, that is, a porus is often present only on one side of the brain, whereas only abnormal gyration is present on the contralateral side. The inner glio‐ependymal surface can project to the external cortical surface with the structural correlate of a membranous, pale membrane. Clefts may be associated with a range of other malformations of septum, optic nerve, callosum or hippocampus (100). Epilepsy surgery represents a therapeutic option only in a minor fraction of schizencephalic patients with pharmacorefractory epilepsies (188). A major differential diagnosis of schizencephaly is given by cystic necrosis due to inappropriate perfusion at peri‐ and postnatal stages of development. However, these cystic lesions do not generally show communication between the inner and outer cerebrospinal fluid spaces.
ACKNOWLEDGMENTS
EA is supported by National Epilepsy Funds (NEF 09‐05) EU FP7 project NeuroGlia (Grant Agreement N° 202167); AJB is supported by the Deutsche Forschungsgemeinschaft (SFB TR3, KFO 177), BMBF NGFN EmiNET, the German Israeli Foundation, ERC EuroEpinomics and the Else Kröner Fresenius Foundation.
REFERENCES
- 1. Adriaensen ME, Schaefer‐Prokop CM, Stijnen T, Duyndam DA, Zonnenberg BA, Prokop M (2009) Prevalence of subependymal giant cell tumors in patients with tuberous sclerosis and a review of the literature. Eur J Neurol 16:691–696. [DOI] [PubMed] [Google Scholar]
- 2. Aghakhani Y, Kinay D, Gotman J, Soualmi L, Andermann F, Olivier A et al (2005) The role of periventricular nodular heterotopia in epileptogenesis. Brain 128:641–651. [DOI] [PubMed] [Google Scholar]
- 3. Aida N (1998) Fukuyama congenital muscular dystrophy: a neuroradiologic review. J Magn Reson Imaging 8:317–326. [DOI] [PubMed] [Google Scholar]
- 4. Al‐Saleem T, Wessner LL, Scheithauer BW, Patterson K, Roach ES, Dreyer SJ et al (1998) Malignant tumors of the kidney, brain, and soft tissues in children and young adults with the tuberous sclerosis complex. Cancer 83:2208–2216. [PubMed] [Google Scholar]
- 5. Anderl S, Freeland M, Kwiatkowski DJ, Goto J (2011) Therapeutic value of prenatal rapamycin treatment in a mouse brain model of tuberous sclerosis complex. Hum Mol Genet 20:4597–4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Antonelli A, Chiaretti A, Amendola T, Piastra M, Di Rocco C, Aloe L (2004) Nerve growth factor and brain‐derived neurotrophic factor in human paediatric hemimegalencephaly. Neuropediatrics 35:39–44. [DOI] [PubMed] [Google Scholar]
- 7. Aronica E, Crino PB (2011) Inflammation in epilepsy: clinical observations. Epilepsia 52:26–32. [DOI] [PubMed] [Google Scholar]
- 8. Aronica E, Boer K, Baybis M, Yu J, Crino P (2007) Co‐expression of cyclin D1 and phosphorylated ribosomal S6 proteins in hemimegalencephaly. Acta Neuropathol (Berl) 114:287–293. [DOI] [PubMed] [Google Scholar]
- 9. Au KS, Williams AT, Roach ES, Batchelor L, Sparagana SP, Delgado MR et al (2007) Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med 9:88–100. [DOI] [PubMed] [Google Scholar]
- 10. Barkovich AJ, Kjos BO (1992) Gray matter heterotopias: MR characteristics and correlation with developmental and neurologic manifestations. Radiology 182:493–499. [DOI] [PubMed] [Google Scholar]
- 11. Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB (2001) Classification system for malformations of cortical development: update 2001. Neurology 57:2168–2178. [DOI] [PubMed] [Google Scholar]
- 12. Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB (2005) A developmental and genetic classification for malformations of cortical development. Neurology 65:1873–1887. [DOI] [PubMed] [Google Scholar]
- 13. Battaglia G, Chiapparini L, Franceschetti S, Freri E, Tassi L, Bassanini S et al (2006) Periventricular nodular heterotopia: classification, epileptic history, and genesis of epileptic discharges. Epilepsia 47:86–97. [DOI] [PubMed] [Google Scholar]
- 14. Baybis M, Lynch D, Lee A, Patel A, McKhann G 2nd, Chugani D et al (2004) Altered expression of neurotransmitter‐receptor subunit and uptake site mRNAs in hemimegalencephaly. Epilepsia 45:1517–1524. [DOI] [PubMed] [Google Scholar]
- 15. Baybis M, Yu J, Lee A, Golden JA, Weiner H, McKhan G 2nd et al (2004) mTOR cascade activation distinguishes tubers from focal cortical dysplasias. Ann Neurol 56:478–487. [DOI] [PubMed] [Google Scholar]
- 16. Baybis M, Aronica E, Nathanson KL, Crino PB (2009) Deletion of 15q11.2‐15q13.1 in isolated human hemimegalencephaly. Acta Neuropathol (Berl) 118:821–823. [DOI] [PubMed] [Google Scholar]
- 17. Becker AJ, Urbach H, Scheffler B, Baden T, Normann S, Lahl R et al (2002) Focal cortical dysplasia of Taylor's balloon cell type: mutational analysis of the TSC1 gene indicates a pathogenic relationship to tuberous sclerosis. Ann Neurol 52:29–37. [DOI] [PubMed] [Google Scholar]
- 18. Bird CR, Gilles FH (1987) Type I schizencephaly: CT and neuropathologic findings. AJNR Am J Neuroradiol 8:451–454. [PMC free article] [PubMed] [Google Scholar]
- 19. Blümcke I, Muhlebner A (2011) Neuropathological work‐up of focal cortical dysplasias using the new ILAE consensus classification system—practical guideline article invited by the Euro‐CNS Research Committee. Clin Neuropathol 30:164–177. [DOI] [PubMed] [Google Scholar]
- 20. Blümcke I, Spreafico R (2011) An international consensus classification for focal cortical dysplasias. Lancet Neurol 10:26–27. [DOI] [PubMed] [Google Scholar]
- 21. Blümcke I, Vinters HV, Armstrong D, Aronica E, Thom M, Spreafico R (2009) Malformations of cortical development and epilepsies: neuropathological findings with emphasis on focal cortical dysplasia. Epileptic Disord 11:181–193. [DOI] [PubMed] [Google Scholar]
- 22. Blümcke I, Pieper T, Pauli E, Hildebrandt M, Kudernatsch M, Winkler P et al (2010) A distinct variant of focal cortical dysplasia type I characterised by magnetic resonance imaging and neuropathological examination in children with severe epilepsies. Epileptic Disord 12:172–180. [DOI] [PubMed] [Google Scholar]
- 23. Blümcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A et al (2011) The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 52:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boer K, Troost D, Spliet WG, Redeker S, Crino PB, Aronica E (2007) A neuropathological study of two autopsy cases of syndromic hemimegalencephaly. Neuropathol Appl Neurobiol 33:455–470. [DOI] [PubMed] [Google Scholar]
- 25. Boer K, Jansen F, Nellist M, Redeker S, van den Ouweland AM, Spliet WG et al (2008) Inflammatory processes in cortical tubers and subependymal giant cell tumors of tuberous sclerosis complex. Epilepsy Res 78:7–21. [DOI] [PubMed] [Google Scholar]
- 26. Boer K, Troost D, Jansen F, Nellist M, van den Ouweland AM, Geurts JJ et al (2008) Clinicopathological and immunohistochemical findings in an autopsy case of tuberous sclerosis complex. Neuropathology 28:577–590. [DOI] [PubMed] [Google Scholar]
- 27. Boer K, Troost D, Timmermans W, Gorter JA, Spliet WG, Nellist M et al (2008) Cellular localization of metabotropic glutamate receptors in cortical tubers and subependymal giant cell tumors of tuberous sclerosis complex. Neuroscience 156:203–215. [DOI] [PubMed] [Google Scholar]
- 28. Boer K, Lucassen PJ, Spliet WG, Vreugdenhil E, van Rijen PC, Troost D et al (2009) Doublecortin‐like (DCL) expression in focal cortical dysplasia and cortical tubers. Epilepsia 50:2629–2637. [DOI] [PubMed] [Google Scholar]
- 29. Boer K, Crino PB, Gorter JA, Nellist M, Jansen FE, Spliet WG et al (2010) Gene expression analysis of tuberous sclerosis complex cortical tubers reveals increased expression of adhesion and inflammatory factors. Brain Pathol 20:704–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boer K, Middeldorp J, Spliet WG, Razavi F, van Rijen PC, Baayen JC et al (2010) Immunohistochemical characterization of the out‐of frame splice variants GFAP Delta164/Deltaexon 6 in focal lesions associated with chronic epilepsy. Epilepsy Res 90:99–109. [DOI] [PubMed] [Google Scholar]
- 31. Boer K, Spliet WG, van Rijen PC, Jansen FE, Aronica E (2010) Expression patterns of AMOG in developing human cortex and malformations of cortical development. Epilepsy Res 91:84–93. [DOI] [PubMed] [Google Scholar]
- 32. Bollo RJ, Kalhorn SP, Carlson C, Haegeli V, Devinsky O, Weiner HL (2008) Epilepsy surgery and tuberous sclerosis complex: special considerations. Neurosurg Focus 25:E13. [DOI] [PubMed] [Google Scholar]
- 33. Bolton PF (2004) Neuroepileptic correlates of autistic symptomatology in tuberous sclerosis. Ment Retard Dev Disabil Res Rev 10:126–131. [DOI] [PubMed] [Google Scholar]
- 34. Bosnjak VM, Dakovic I, Duranovic V, Lujic L, Krakar G, Marn B (2011) Malformations of cortical development in children with congenital cytomegalovirus infection—a study of nine children with proven congenital cytomegalovirus infection. Coll Antropol 35:229–234. [PubMed] [Google Scholar]
- 35. Bradshaw NJ, Soares DC, Carlyle BC, Ogawa F, Davidson‐Smith H, Christie S et al (2011) PKA phosphorylation of NDE1 is DISC1/PDE4 dependent and modulates its interaction with LIS1 and NDEL1. J Neurosci 31:9043–9054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brunelli S, Faiella A, Capra V, Nigro V, Simeone A, Cama A, Boncinelli E (1996) Germline mutations in the homeobox gene EMX2 in patients with severe schizencephaly. Nat Genet 12:94–96. [DOI] [PubMed] [Google Scholar]
- 37. Capra V, De Marco P, Moroni A, Faiella A, Brunelli S, Tortori‐Donati P et al (1996) Schizencephaly: surgical features and new molecular genetic results. Eur J Pediatr Surg 6:27–29. [DOI] [PubMed] [Google Scholar]
- 38. Caraballo RH, Cersosimo RO, Fejerman N (2004) Unilateral closed‐lip schizencephaly and epilepsy: a comparison with cases of unilateral polymicrogyria. Brain Dev 26:151–157. [DOI] [PubMed] [Google Scholar]
- 39. Cartwright MS, McCarthy SC, Roach ES (2005) Hemimegalencephaly and tuberous sclerosis complex. Neurology 64:1634. [DOI] [PubMed] [Google Scholar]
- 40. Catala M, Bucourt M (2000) A destructive hemispheric cleft discovered in a female fetus. Clin Neuropathol 19:235–2357. [PubMed] [Google Scholar]
- 41. Chamberlain WA, Cohen ML, Gyure KA, Kleinschmidt‐DeMasters BK, Perry A, Powell SZ et al (2009) Interobserver and intraobserver reproducibility in focal cortical dysplasia (malformations of cortical development). Epilepsia 50:2593–2598. [DOI] [PubMed] [Google Scholar]
- 42. Chan JA, Zhang H, Roberts PS, Jozwiak S, Wieslawa G, Lewin‐Kowalik J et al (2004) Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J Neuropathol Exp Neurol 63:1236–1242. [DOI] [PubMed] [Google Scholar]
- 43. Chen CP, Su YN, Hung CC, Shih JC, Wang W (2006) Novel mutation in the TSC2 gene associated with prenatally diagnosed cardiac rhabdomyomas and cerebral tuberous sclerosis. J Formos Med Assoc 105:599–603. [DOI] [PubMed] [Google Scholar]
- 44. Chio A, Spreafico R, Avanzini G, Ghiglione P, Vercellino M, Mutani R (2003) Cesare Lombroso, cortical dysplasia, and epilepsy: keen findings and odd theories. Neurology 61:1412–1416. [DOI] [PubMed] [Google Scholar]
- 45. Chu‐Shore CJ, Frosch MP, Grant PE, Thiele EA (2009) Progressive multifocal cystlike cortical tubers in tuberous sclerosis complex: clinical and neuropathologic findings. Epilepsia 50:2648–2651.. [DOI] [PubMed] [Google Scholar]
- 46. Chu‐Shore CJ, Major P, Montenegro M, Thiele E (2009) Cyst‐like tubers are associated with TSC2 and epilepsy in tuberous sclerosis complex. Neurology 72:1165–1169. [DOI] [PubMed] [Google Scholar]
- 47. Connolly MB, Hendson G, Steinbok P (2006) Tuberous sclerosis complex: a review of the management of epilepsy with emphasis on surgical aspects. Childs Nerv Syst 22:896–908. [DOI] [PubMed] [Google Scholar]
- 48. Crino P, Cross JH, Delalande O, Dubeau F, Duncan J, Guerrini R et al (2011) The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 52:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Crino PB (2005) Molecular pathogenesis of focal cortical dysplasia and hemimegalencephaly. J Child Neurol 20:330–336. [DOI] [PubMed] [Google Scholar]
- 50. Crino PB (2009) Focal brain malformations: seizures, signaling, sequencing. Epilepsia 50:3–8. [DOI] [PubMed] [Google Scholar]
- 51. Crino PB, Trojanowski JQ, Dichter MA, Eberwine J (1996) Embryonic neuronal markers in tuberous sclerosis: single‐cell molecular pathology. Proc Natl Acad Sci U S A 93:14152–14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Crino PB, Nathanson KL, Henske EP (2006) The tuberous sclerosis complex. N Engl J Med 355:1345–1356. [DOI] [PubMed] [Google Scholar]
- 53. Crino PB, Aronica E, Baltuch G, Nathanson KL (2010) Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology 74:1716–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Crome L (1957) Infantile cerebral gliosis wtith giant nerve cells. J Neurol Neurosurg Psychiatry 20:117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Curatolo P, Verdecchia M, Bombardieri R (2002) Tuberous sclerosis complex: a review of neurological aspects. Eur J Paediatr Neurol 6:15–23. [DOI] [PubMed] [Google Scholar]
- 56. Cusmai R, Curatolo P, Mangano S, Cheminal R, Echenne B (1990) Hemimegalencephaly and neurofibromatosis. Neuropediatrics 21:179–182. [DOI] [PubMed] [Google Scholar]
- 57. Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J et al (2001) Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet 68:64–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. D'Agostino MD, Bastos A, Piras C, Bernasconi A, Grisar T, Tsur VG et al (2004) Posterior quadrantic dysplasia or hemi‐hemimegalencephaly: a characteristic brain malformation. Neurology 62:2214–2220. [DOI] [PubMed] [Google Scholar]
- 59. Dambska M, Wisniewski K, Sher JH (1983) Lissencephaly: two distinct clinico‐pathological types. Brain Dev 5:302–310. [DOI] [PubMed] [Google Scholar]
- 60. de Rijk‐van Andel JF, Arts WF, Hofman A, Staal A, Niermeijer MF (1991) Epidemiology of lissencephaly type I. Neuroepidemiology 10:200–204. [DOI] [PubMed] [Google Scholar]
- 61. De Rosa MJ, Secor DL, Barsom M, Fisher RS, Vinters HV (1992) Neuropathologic findings in surgically treated hemimegalencephaly: immunohistochemical, morphometric, and ultrastructural study. Acta Neuropathol (Berl) 84:250–260. [DOI] [PubMed] [Google Scholar]
- 62. DeLone DR, Brown WD, Gentry LR (1999) Proteus syndrome: craniofacial and cerebral MRI. Neuroradiology 41:840–843. [DOI] [PubMed] [Google Scholar]
- 63. DiMario FJ Jr (2004) Brain abnormalities in tuberous sclerosis complex. J Child Neurol 19:650–657. [DOI] [PubMed] [Google Scholar]
- 64. Ding SL, Van Hoesen GW, Cassell MD, Poremba A (2009) Parcellation of human temporal polar cortex: a combined analysis of multiple cytoarchitectonic, chemoarchitectonic, and pathological markers. J Comp Neurol 514:595–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dobyns WB, Truwit CL, Ross ME, Matsumoto N, Pilz DT, Ledbetter DH et al (1999) Differences in the gyral pattern distinguish chromosome 17‐linked and X‐linked lissencephaly. Neurology 53:270–277. [DOI] [PubMed] [Google Scholar]
- 66. Dubeau F, Tampieri D, Lee N, Andermann E, Carpenter S, Leblanc R et al (1995) Periventricular and subcortical nodular heterotopia. A study of 33 patients. Brain 118(Pt 5):1273–1287. [DOI] [PubMed] [Google Scholar]
- 67. ECTSC (1993) Identification and characterization of the tuberous sclerosis gene on chromosome 16. The European Chromosome 16 Tuberous Sclerosis Consortium. Cell 75:1305–1315. [DOI] [PubMed] [Google Scholar]
- 68. El‐Hashemite N, Zhang H, Henske EP, Kwiatkowski DJ (2003) Mutation in TSC2 and activation of mammalian target of rapamycin signalling pathway in renal angiomyolipoma. Lancet 361:1348–1349. [DOI] [PubMed] [Google Scholar]
- 69. Faiella A, Brunelli S, Granata T, D'Incerti L, Cardini R, Lenti C et al (1997) A number of schizencephaly patients including 2 brothers are heterozygous for germline mutations in the homeobox gene EMX2. Eur J Hum Genet 5:186–190. [PubMed] [Google Scholar]
- 70. Fauser S, Becker A, Schulze‐Bonhage A, Hildebrandt M, Tuxhorn I, Pannek HW et al (2004) CD34‐immunoreactive balloon cells in cortical malformations. Acta Neuropathol (Berl) 108:272–278. [DOI] [PubMed] [Google Scholar]
- 71. Feliciano DM, Su T, Lopez J, Platel JC, Bordey A (2011) Single‐cell Tsc1 knockout during corticogenesis generates tuber‐like lesions and reduces seizure threshold in mice. J Clin Invest 121:1596–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ferrer I (1984) A Golgi analysis of unlayered polymicrogyria. Acta Neuropathol (Berl) 65:69–76. [DOI] [PubMed] [Google Scholar]
- 73. Ferrer I, Catala I (1991) Unlayered polymicrogyria: structural and developmental aspects. Anat Embryol (Berl) 184:517–528. [DOI] [PubMed] [Google Scholar]
- 74. Flores‐Sarnat L (2002) Hemimegalencephaly: part 1. Genetic, clinical, and imaging aspects. J Child Neurol 17:373–384. [DOI] [PubMed] [Google Scholar]
- 75. Flores‐Sarnat L, Sarnat HB (2008) Axes and gradients of the neural tube for a morphological/molecular genetic classification of nervous system malformations. Handb Clin Neurol 87:1–11. [DOI] [PubMed] [Google Scholar]
- 76. Flores‐Sarnat L, Sarnat HB, Davila‐Gutierrez G, Alvarez A (2003) Hemimegalencephaly: part 2. Neuropathology suggests a disorder of cellular lineage. J Child Neurol 18:776–785. [DOI] [PubMed] [Google Scholar]
- 77. Frater JL, Prayson RA, Morris HH III, Bingaman WE (2000) Surgical pathologic findings of extratemporal‐based intractable epilepsy: a study of 133 consecutive resections. Arch Pathol Lab Med 124:545–549. [DOI] [PubMed] [Google Scholar]
- 78. Friede RL, Mikolasek J (1978) Postencephalitic porencephaly, hydranencephaly or polymicrogyria. A review. Acta Neuropathol (Berl) 43:161–168. [DOI] [PubMed] [Google Scholar]
- 79. Fukuyama Y, Osawa M, Suzuki H (1981) Congenital progressive muscular dystrophy of the Fukuyama type—clinical, genetic and pathological considerations. Brain Dev 3:1–29. [DOI] [PubMed] [Google Scholar]
- 80. Gao X, Zhang Y, Arrazola P, Hino O, Kobayashi T, Yeung RS et al (2002) Tsc tumour suppressor proteins antagonize amino‐acid‐TOR signalling. Nat Cell Biol 4:699–704. [DOI] [PubMed] [Google Scholar]
- 81. Garbelli R, Meroni A, Magnaghi G, Beolchi MS, Ferrario A, Tassi L et al (2006) Architectural (Type IA) focal cortical dysplasia and parvalbumin immunostaining in temporal lobe epilepsy. Epilepsia 47:1074–1078. [DOI] [PubMed] [Google Scholar]
- 82. Garbelli R, Rossini L, Moroni RF, Watakabe A, Yamamori T, Tassi L et al (2009) Layer‐specific genes reveal a rudimentary laminar pattern in human nodular heterotopia. Neurology 73:746–753. [DOI] [PubMed] [Google Scholar]
- 83. Geerdink N, Rotteveel JJ, Lammens M, Sistermans EA, Heikens GT, Gabreels FJ et al (2002) MECP2 mutation in a boy with severe neonatal encephalopathy: clinical, neuropathological and molecular findings. Neuropediatrics 33:33–36. [DOI] [PubMed] [Google Scholar]
- 84. Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I et al (1998) Doublecortin, a brain‐specific gene mutated in human X‐linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92:63–72. [DOI] [PubMed] [Google Scholar]
- 85. Gleeson JG, Lin PT, Flanagan LA, Walsh CA (1999) Doublecortin is a microtubule‐associated protein and is expressed widely by migrating neurons. Neuron 23:257–271. [DOI] [PubMed] [Google Scholar]
- 86. Glenn OA, Barkovich AJ (2006) Magnetic resonance imaging of the fetal brain and spine: an increasingly important tool in prenatal diagnosis, part 1. AJNR Am J Neuroradiol 27:1604–1611. [PMC free article] [PubMed] [Google Scholar]
- 87. Grajkowska W, Kotulska K, Jurkiewicz E, Matyja E (2010) Brain lesions in tuberous sclerosis complex. Review. Folia Neuropathol 48:139–149. [PubMed] [Google Scholar]
- 88. Green AJ, Smith M, Yates JR (1994) Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nat Genet 6:193–196. [DOI] [PubMed] [Google Scholar]
- 89. Griffiths PD, Batty R, Warren D, Hart A, Sharrard M, Mordekar SR et al (2011) The use of MR imaging and spectroscopy of the brain in children investigated for developmental delay: what is the most appropriate imaging strategy? Eur Radiol 21:1820–1830. [DOI] [PubMed] [Google Scholar]
- 90. Guerreiro MM, Andermann E, Guerrini R, Dobyns WB, Kuzniecky R, Silver K et al (2000) Familial perisylvian polymicrogyria: a new familial syndrome of cortical maldevelopment. Ann Neurol 48:39–48. [PubMed] [Google Scholar]
- 91. Guerrini R (2005) Genetic malformations of the cerebral cortex and epilepsy. Epilepsia 46:32–37. [DOI] [PubMed] [Google Scholar]
- 92. Guerrini R, Carrozzo R (2001) Epilepsy and genetic malformations of the cerebral cortex. Am J Med Genet 106:160–173. [DOI] [PubMed] [Google Scholar]
- 93. Guerrini R, Filippi T (2005) Neuronal migration disorders, genetics, and epileptogenesis. J Child Neurol 20:287–299. [DOI] [PubMed] [Google Scholar]
- 94. Guerrini R, Marini C (2006) Genetic malformations of cortical development. Exp Brain Res 173:322–333. [DOI] [PubMed] [Google Scholar]
- 95. Guerrini R, Barkovich AJ, Sztriha L, Dobyns WB (2000) Bilateral frontal polymicrogyria: a newly recognized brain malformation syndrome. Neurology 54:909–913. [DOI] [PubMed] [Google Scholar]
- 96. Guerrini R, Holthausen H, Parmeggiani L, Chiron C (2002) Epilepsy and malformations of the cerebral cortex. In: Epileptic Syndromes in Infancy, Childhood and Adolescence, 3rd edn. Roger J, Bureau M, Dravet C (eds), pp. 457–479. John Libbey: London. [Google Scholar]
- 97. Guerrini R, Moro F, Andermann E, Hughes E, D'Agostino D, Carrozzo R et al (2003) Nonsyndromic mental retardation and cryptogenic epilepsy in women with doublecortin gene mutations. Ann Neurol 54:30–37. [DOI] [PubMed] [Google Scholar]
- 98. Guerrini R, Sicca F, Parmeggiani L (2003) Epilepsy and malformations of the cerebral cortex. Epileptic Disord 5:9–26. [PubMed] [Google Scholar]
- 99. Guerrini R, Dobyns WB, Barkovich AJ (2008) Abnormal development of the human cerebral cortex: genetics, functional consequences and treatment options. Trends Neurosci 31:154–162. [DOI] [PubMed] [Google Scholar]
- 100. Hayashi N, Tsutsumi Y, Barkovich AJ (2002) Polymicrogyria without porencephaly/schizencephaly. MRI analysis of the spectrum and the prevalence of macroscopic findings in the clinical population. Neuroradiology 44:647–655. [DOI] [PubMed] [Google Scholar]
- 101. Hehr U, Pineda‐Alvarez DE, Uyanik G, Hu P, Zhou N, Hehr A et al (2010) Heterozygous mutations in SIX3 and SHH are associated with schizencephaly and further expand the clinical spectrum of holoprosencephaly. Hum Genet 127:555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Holmes GL, Stafstrom CE, Tuberous Sclerosis Study Group (2007) Tuberous sclerosis complex and epilepsy: recent developments and future challenges. Epilepsia 48:617–630. [DOI] [PubMed] [Google Scholar]
- 103. Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO et al (2000) Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet 26:93–96. [DOI] [PubMed] [Google Scholar]
- 104. Horesh D, Sapir T, Francis F, Wolf SG, Caspi M, Elbaum M et al (1999) Doublecortin, a stabilizer of microtubules. Hum Mol Genet 8:1599–1610. [DOI] [PubMed] [Google Scholar]
- 105. Howe DT, Rankin J, Draper ES (2011) Schizencephaly prevalence, prenatal diagnosis and clues to aetiology; a register based study. Ultrasound Obstet Gynecol 39:75–82. [DOI] [PubMed] [Google Scholar]
- 106. Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4:648–657. [DOI] [PubMed] [Google Scholar]
- 107. Ito N, Rubin GM (1999) Gigas, a Drosophila homolog of tuberous sclerosis gene product‐2, regulates the cell cycle. Cell 96:529–539. [DOI] [PubMed] [Google Scholar]
- 108. Iyer A, Zurolo E, Spliet WG, van Rijen PC, Baayen JC, Gorter JA et al (2010) Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia 51:1763–1773. [DOI] [PubMed] [Google Scholar]
- 109. Iyer AM, Zurolo E, Boer K, Baayen JC, Giangaspero F, Arcella A et al (2010) Tissue plasminogen activator and urokinase plasminogen activator in human epileptogenic pathologies. Neuroscience 167:929–945. [DOI] [PubMed] [Google Scholar]
- 110. Jacobs KM, Hwang BJ, Prince DA (1999) Focal epileptogenesis in a rat model of polymicrogyria. J Neurophysiol 81:159–173. [DOI] [PubMed] [Google Scholar]
- 111. Jansen AC, Oostra A, Desprechins B, De Vlaeminck Y, Verhelst H, Regal L et al (2011) TUBA1A mutations: from isolated lissencephaly to familial polymicrogyria. Neurology 76:988–992. [DOI] [PubMed] [Google Scholar]
- 112. Janszky J, Ebner A, Kruse B, Mertens M, Jokeit H, Seitz RJ et al (2003) Functional organization of the brain with malformations of cortical development. Ann Neurol 53:759–767. [DOI] [PubMed] [Google Scholar]
- 113. Jellinger K, Rett A (1976) Agyria‐pachygyria (lissencephaly syndrome). Neuropadiatrie 7:66–91. [DOI] [PubMed] [Google Scholar]
- 114. Jonas R, Nguyen S, Hu B, Asarnow RF, LoPresti C, Curtiss S et al (2004) Cerebral hemispherectomy: hospital course, seizure, developmental, language, and motor outcomes. Neurology 62:1712–1721. [DOI] [PubMed] [Google Scholar]
- 115. Jozwiak J, Jozwiak S (2005) Giant cells: contradiction to two‐hit model of tuber formation? Cell Mol Neurobiol 25:795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kakita A, Hayashi S, Moro F, Guerrini R, Ozawa T, Ono K et al (2002) Bilateral periventricular nodular heterotopia due to filamin 1 gene mutation: widespread glomeruloid microvascular anomaly and dysplastic cytoarchitecture in the cerebral cortex. Acta Neuropathol (Berl) 104:649–657. [DOI] [PubMed] [Google Scholar]
- 117. Kalifa GL, Chiron C, Sellier N, Demange P, Ponsot G, Lalande G, Robain O (1987) Hemimegalencephaly: MR imaging in five children. Radiology 165:29–33. [DOI] [PubMed] [Google Scholar]
- 118. Kara S, Jissendi‐Tchofo P, Barkovich AJ (2010) Developmental differences of the major forebrain commissures in lissencephalies. AJNR Am J Neuroradiol 31:1602–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kato M, Mizuguchi M, Sakuta R, Takashima S (1996) Hypertrophy of the cerebral white matter in hemimegalencephaly. Pediatr Neurol 14:335–338. [DOI] [PubMed] [Google Scholar]
- 120. Koh S, Jayakar P, Dunoyer C, Whiting SE, Resnick TJ, Alvarez LA et al (2000) Epilepsy surgery in children with tuberous sclerosis complex: presurgical evaluation and outcome. Epilepsia 41:1206–1213. [DOI] [PubMed] [Google Scholar]
- 121. Krsek P, Maton B, Korman B, Pacheco‐Jacome E, Jayakar P, Dunoyer C et al (2008) Different features of histopathological subtypes of pediatric focal cortical dysplasia. Ann Neurol 63:758–769. [DOI] [PubMed] [Google Scholar]
- 122. Krsek P, Pieper T, Karlmeier A, Hildebrandt M, Kolodziejczyk D, Winkler P et al (2009) Different presurgical characteristics and seizure outcomes in children with focal cortical dysplasia type I or II. Epilepsia 50:125–137. [DOI] [PubMed] [Google Scholar]
- 123. Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P et al (2010) Everolimus for subependymal giant‐cell astrocytomas in tuberous sclerosis. N Engl J Med 363:1801–1811. [DOI] [PubMed] [Google Scholar]
- 124. Kubo K, Honda T, Tomita K, Sekine K, Ishii K, Uto A et al (2010) Ectopic Reelin induces neuronal aggregation with a normal birthdate‐dependent “inside‐out” alignment in the developing neocortex. J Neurosci 30:10953–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kumar RA, Pilz DT, Babatz TD, Cushion TD, Harvey K, Topf M et al (2010) TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum Mol Genet 19:2817–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kuzniecky RI, Jackson GD (2005) Magnetic Resonance in Epilepsy. Neuroimaging Techniques, 2nd edn. Elsevier Academic Press: New York. [Google Scholar]
- 127. Kwa VI, Smitt JH, Verbeeten BW, Barth PG (1995) Epidermal nevus syndrome with isolated enlargement of one temporal lobe: a case report. Brain Dev 17:122–125. [DOI] [PubMed] [Google Scholar]
- 128. Kwiatkowski DJ (2003) Tuberous sclerosis: from tubers to mTOR. Ann Hum Genet 67(Pt 1):87–96. [DOI] [PubMed] [Google Scholar]
- 129. Lamparello P, Baybis M, Pollard J, Hol EM, Eisenstat DD, Aronica E, Crino PB (2007) Developmental lineage of cell types in cortical dysplasia with balloon cells. Brain 130(Pt 9):2267–2276. [DOI] [PubMed] [Google Scholar]
- 130. Landrieu P, Lacroix C (1994) Schizencephaly, consequence of a developmental vasculopathy? A clinicopathological report. Clin Neuropathol 13:192–196. [PubMed] [Google Scholar]
- 131. Lee A, Maldonado M, Baybis M, Walsh CA, Scheithauer B, Yeung R et al (2003) Markers of cellular proliferation are expressed in cortical tubers. Ann Neurol 53:668–673. [DOI] [PubMed] [Google Scholar]
- 132. Lerner JT, Salamon N, Hauptman JS, Velasco TR, Hemb M, Wu JY et al (2009) Assessment and surgical outcomes for mild type I and severe type II cortical dysplasia: a critical review and the UCLA experience. Epilepsia 50:1310–1335. [DOI] [PubMed] [Google Scholar]
- 133. Leventer RJ, Phelan EM, Coleman LT, Kean MJ, Jackson GD, Harvey AS (1999) Clinical and imaging features of cortical malformations in childhood. Neurology 53:715–722. [DOI] [PubMed] [Google Scholar]
- 134. Li LM, Dubeau F, Andermann F, Fish DR, Watson C, Cascino GD et al (1997) Periventricular nodular heterotopia and intractable temporal lobe epilepsy: poor outcome after temporal lobe resection. Ann Neurol 41:662–668. [DOI] [PubMed] [Google Scholar]
- 135. Luat AF, Makki M, Chugani HT (2007) Neuroimaging in tuberous sclerosis complex. Curr Opin Neurol 20:142–150. [DOI] [PubMed] [Google Scholar]
- 136. Lugnier C, Majores M, Fassunke J, Pernhorst K, Niehusmann P, Simon M et al (2009) Hamartin variants that are frequent in focal dysplasias and cortical tubers have reduced tuberin binding and aberrant subcellular distribution in vitro. J Neuropathol Exp Neurol 68:1136–1146. [DOI] [PubMed] [Google Scholar]
- 137. Luo R, Jeong SJ, Jin Z, Strokes N, Li S, Piao X (2011) G protein‐coupled receptor 56 and collagen III, a receptor‐ligand pair, regulates cortical development and lamination. Proc Natl Acad Sci U S A 108:12925–12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Mai R, Tassi L, Cossu M, Francione S, Lo Russo G, Garbelli R et al (2003) Aneuropathological, stereo‐EEG, and MRI study of subcortical band heterotopia. Neurology 60:1834–1838. [DOI] [PubMed] [Google Scholar]
- 139. Majores M, Blümcke I, Urbach H, Meroni A, Hans V, Holthausen H et al (2005) Distinct allelic variants of TSC1 and TSC2 in epilepsy‐associated cortical malformations without balloon cells. J Neuropathol Exp Neurol 64:629–637. [DOI] [PubMed] [Google Scholar]
- 140. Manent JB, Wang Y, Chang Y, Paramasivam M, LoTurco JJ (2009) Dcx reexpression reduces subcortical band heterotopia and seizure threshold in an animal model of neuronal migration disorder. Nat Med 15:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Manoranjan B, Provias JP (2010) Hemimegalencephaly: a fetal case with neuropathological confirmation and review of the literature. Acta Neuropathol (Berl) 120:117–130. [DOI] [PubMed] [Google Scholar]
- 142. Martinian L, Boer K, Middeldorp J, Hol EM, Sisodiya SM, Squier W et al (2009) Expression patterns of glial fibrillary acidic protein (GFAP)‐delta in epilepsy‐associated lesional pathologies. Neuropathol Appl Neurobiol 35:394–405. [DOI] [PubMed] [Google Scholar]
- 143. McManus EJ, Alessi DR (2002) TSC1‐TSC2: a complex tale of PKB‐mediated S6K regulation. Nat Cell Biol 4:E214–E216. [DOI] [PubMed] [Google Scholar]
- 144. Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M et al (2007) A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci 27:5546–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ (2008) Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci 28:5422–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Mellado C, Poduri A, Gleason D, Elhosary PC, Barry BJ, Partlow JN et al (2010) Candidate gene sequencing of LHX2, HESX1, and SOX2 in a large schizencephaly cohort. Am J Med Genet A 152A:2736–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Meroni A, Galli C, Bramerio M, Tassi L, Colombo N, Cossu M et al (2009) Nodular heterotopia: a neuropathological study of 24 patients undergoing surgery for drug‐resistant epilepsy. Epilepsia 50:116–124. [DOI] [PubMed] [Google Scholar]
- 148. Mischel PS, Nguyen LP, Vinters HV (1995) Cerebral cortical dysplasia associated with pediatric epilepsy. Review of neuropathologic features and proposal for a grading system. J Neuropathol Exp Neurol 54:137–153. [DOI] [PubMed] [Google Scholar]
- 149. Mitchell TN, Free SL, Williamson KA, Stevens JM, Churchill AJ, Hanson IM et al (2003) Polymicrogyria and absence of pineal gland due to PAX6 mutation. Ann Neurol 53:658–663. [DOI] [PubMed] [Google Scholar]
- 150. Miyata H, Chiang AC, Vinters HV (2004) Insulin signaling pathways in cortical dysplasia and TSC‐tubers: tissue microarray analysis. Ann Neurol 56:510–519. [DOI] [PubMed] [Google Scholar]
- 151. Mizuguchi M, Takashima S (2001) Neuropathology of tuberous sclerosis. Brain Dev 23:508–515. [DOI] [PubMed] [Google Scholar]
- 152. Montenegro MA, Li LM, Guerreiro MM, Cendes F (2002) Neuroimaging characteristics of pseudosubcortical laminar heterotopia. J Neuroimaging 12:52–54. [DOI] [PubMed] [Google Scholar]
- 153. Nakahashi M, Sato N, Yagishita A, Ota M, Saito Y, Sugai K et al (2009) Clinical and imaging characteristics of localized megalencephaly: a retrospective comparison of diffuse hemimegalencephaly and multilobar cortical dysplasia. Neuroradiology 51:821–830. [DOI] [PubMed] [Google Scholar]
- 154. Nellist M, Sancak O, Goedbloed MA, Rohe C, van Netten D, Mayer K et al (2005) Distinct effects of single amino‐acid changes to tuberin on the function of the tuberin‐hamartin complex. Eur J Hum Genet 13:59–68. [DOI] [PubMed] [Google Scholar]
- 155. Orlova KA, Crino PB (2010) The tuberous sclerosis complex. Ann N Y Acad Sci 1184:87–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Orlova KA, Tsai V, Baybis M, Heuer GG, Sisodiya S, Thom M et al (2010) Early progenitor cell marker expression distinguishes type II from type I focal cortical dysplasias. J Neuropathol Exp Neurol 69:850–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Palmini A, Najm I, Avanzini G, Babb T, Guerrini R, Foldvary‐Schaefer N et al (2004) Terminology and classification of the cortical dysplasias. Neurology 62:S2–S8. [DOI] [PubMed] [Google Scholar]
- 158. Park SH, Pepkowitz SH, Kerfoot C, De Rosa MJ, Poukens V, Wienecke R et al (1997) Tuberous sclerosis in a 20‐week gestation fetus: immunohistochemical study. Acta Neuropathol (Berl) 94:180–186. [DOI] [PubMed] [Google Scholar]
- 159. Parker WE, Orlova KA, Heuer GG, Baybis M, Aronica E, Frost M et al (2011) Enhanced epidermal growth factor, hepatocyte growth factor, and vascular endothelial growth factor expression in tuberous sclerosis complex. Am J Pathol 178:296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Parrini E, Ramazzotti A, Dobyns WB, Mei D, Moro F, Veggiotti P et al (2006) Periventricular heterotopia: phenotypic heterogeneity and correlation with filamin A mutations. Brain 129:1892–1906. [DOI] [PubMed] [Google Scholar]
- 161. Pilz DT, Matsumoto N, Minnerath S, Mills P, Gleeson JG, Allen KM et al (1998) LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Hum Mol Genet 7:2029–2037. [DOI] [PubMed] [Google Scholar]
- 162. Pilz DT, Kuc J, Matsumoto N, Bodurtha J, Bernadi B, Tassinari CA et al (1999) Subcortical band heterotopia in rare affected males can be caused by missense mutations in DCX (XLIS) or LIS1. Hum Mol Genet 8:1757–1760. [DOI] [PubMed] [Google Scholar]
- 163. Povey S, Burley MW, Attwood J, Benham F, Hunt D, Jeremiah SJ et al (1994) Two loci for tuberous sclerosis: one on 9q34 and one on 16p13. Ann Hum Genet 58:107–127. [DOI] [PubMed] [Google Scholar]
- 164. Qin W, Chan JA, Vinters HV, Mathern GW, Franz DN, Taillon BE et al (2010) Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second‐hit mutations in these genes are rare events. Brain Pathol 20:1096–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Rakic P, Lombroso PJ (1998) Development of the cerebral cortex: I. Forming the cortical structure. J Am Acad Child Adolesc Psychiatry 37:116–117. [DOI] [PubMed] [Google Scholar]
- 166. Raymond AA, Fish DR, Sisodiya SM, Alsanjari N, Stevens JM, Shorvon SD (1995) Abnormalities of gyration, heterotopias, tuberous sclerosis, focal cortical dysplasia, microdysgenesis, dysembryoplastic neuroepithelial tumour and dysgenesis of the archicortex in epilepsy. Clinical, EEG and neuroimaging features in 100 adult patients. Brain 118:629–660. [DOI] [PubMed] [Google Scholar]
- 167. Redecker C, Hagemann G, Gressens P, Evrard P, Witte OW (2000) Cortical dysgenesis. Current views on pathogenesis and pathophysiology. Nervenarzt 71:238–248. [DOI] [PubMed] [Google Scholar]
- 168. Redecker C, Luhmann HJ, Hagemann G, Fritschy JM, Witte OW (2000) Differential downregulation of GABAA receptor subunits in widespread brain regions in the freeze‐lesion model of focal cortical malformations. J Neurosci 20:5045–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Reiner O, Lombroso PJ (1998) Development of the cerebral cortex: II. Lissencephaly. J Am Acad Child Adolesc Psychiatry 37:231–232. [DOI] [PubMed] [Google Scholar]
- 170. Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB et al (1993) Isolation of a Miller‐Dieker lissencephaly gene containing G protein beta‐subunit‐like repeats. Nature 364:717–721. [DOI] [PubMed] [Google Scholar]
- 171. Roach ES, Gomez MR, Northrup H (1998) Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 13:624–628. [DOI] [PubMed] [Google Scholar]
- 172. Robain O, Floquet C, Heldt N, Rozenberg F (1988) Hemimegalencephaly: a clinicopathological study of four cases. Neuropathol Appl Neurobiol 14:125–135. [DOI] [PubMed] [Google Scholar]
- 173. Saito Y, Murayama S, Kawai M, Nakano I (1999) Breached cerebral glia limitans‐basal lamina complex in Fukuyama‐type congenital muscular dystrophy. Acta Neuropathol (Berl) 98:330–336. [DOI] [PubMed] [Google Scholar]
- 174. Sakuta R, Aikawa H, Takashima S, Ryo S (1991) Epidermal nevus syndrome with hemimegalencephaly: neuropathological study. Brain Dev 13:260–265. [DOI] [PubMed] [Google Scholar]
- 175. Salamon N, Andres M, Chute DJ, Nguyen ST, Chang JW, Huynh MN et al (2006) Contralateral hemimicrencephaly and clinical‐pathological correlations in children with hemimegalencephaly. Brain 129(Pt 2):352–365. [DOI] [PubMed] [Google Scholar]
- 176. Sanghvi JP, Rajadhyaksha SB, Ursekar M (2004) Spectrum of congenital CNS malformations in pediatric epilepsy. Indian Pediatr 41:831–838. [PubMed] [Google Scholar]
- 177. Santavuori P, Somer H, Sainio K, Rapola J, Kruus S, Nikitin T et al (1989) Muscle‐eye‐brain disease (MEB). Brain Dev 11:147–153. [DOI] [PubMed] [Google Scholar]
- 178. Sapir T, Horesh D, Caspi M, Atlas R, Burgess HA, Wolf SG et al (2000) Doublecortin mutations cluster in evolutionarily conserved functional domains. Hum Mol Genet 9:703–712. [DOI] [PubMed] [Google Scholar]
- 179. Sarnat HB, Flores‐Sarnat L (2004) Integrative classification of morphology and molecular genetics in central nervous system malformations. Am J Med Genet A 126:386–392. [DOI] [PubMed] [Google Scholar]
- 180. Sasaki M, Hashimoto T, Furushima W, Okada M, Kinoshita S, Fujikawa Y, Sugai K (2005) Clinical aspects of hemimegalencephaly by means of a nationwide survey. J Child Neurol 20:337–341. [DOI] [PubMed] [Google Scholar]
- 181. Sato N, Yagishita A, Oba H, Miki Y, Nakata Y, Yamashita F et al (2007) Hemimegalencephaly: a study of abnormalities occurring outside the involved hemisphere. AJNR Am J Neuroradiol 28:678–682. [PMC free article] [PubMed] [Google Scholar]
- 182. Schick V, Majores M, Engels G, Spitoni S, Koch A, Elger CE et al (2006) Activation of Akt independent of PTEN and CTMP tumor‐suppressor gene mutations in epilepsy‐associated Taylor‐type focal cortical dysplasias. Acta Neuropathol (Berl) 112:715–725. [DOI] [PubMed] [Google Scholar]
- 183. Schick V, Majores M, Engels G, Hartmann W, Elger CE, Schramm J et al (2007) Differential Pi3K‐pathway activation in cortical tubers and focal cortical dysplasias with balloon cells. Brain Pathol 17:165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Seifert G, Carmignoto G, Steinhauser C (2010) Astrocyte dysfunction in epilepsy. Brain Res Rev 63:212–221. [DOI] [PubMed] [Google Scholar]
- 185. Shmueli O, Cahana A, Reiner O (1999) Platelet‐activating factor (PAF) acetylhydrolase activity, LIS1 expression, and seizures. J Neurosci Res 57:176–184. [DOI] [PubMed] [Google Scholar]
- 186. Sims J (1835) On hypertrophy and atrophy of the brain. Med Chir Trans 19:315–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Sisodiya S (2011) Epilepsy: the new order‐classifying focal cortical dysplasias. Nat Rev Neurol 7:129–130. [DOI] [PubMed] [Google Scholar]
- 188. Sisodiya SM (2000) Surgery for malformations of cortical development causing epilepsy. Brain 123(Pt 6):1075–1091. [DOI] [PubMed] [Google Scholar]
- 189. Sisodiya SM (2004) Malformations of cortical development: burdens and insights from important causes of human epilepsy. Lancet Neurol 3:29–38. [DOI] [PubMed] [Google Scholar]
- 190. Sisodiya SM, Fauser S, Cross JH, Thom M (2009) Focal cortical dysplasia type II: biological features and clinical perspectives. Lancet Neurol 8:830–843. [DOI] [PubMed] [Google Scholar]
- 191. Solomon LM, Esterly NB (1975) Epidermal and other congenital organoid nevi. Curr Probl Pediatr 6:1–56. [DOI] [PubMed] [Google Scholar]
- 192. Sosunov AA, Wu X, Weiner HL, Mikell CB, Goodman RR, Crino PD, McKhann GM 2nd (2008) Tuberous sclerosis: a primary pathology of astrocytes? Epilepsia 49:53–62. [DOI] [PubMed] [Google Scholar]
- 193. Spreafico R, Blümcke I (2010) Focal cortical dysplasias: clinical implication of neuropathological classification systems. Acta Neuropathol (Berl) 120:359–367. [DOI] [PubMed] [Google Scholar]
- 194. Srikandarajah N, Martinian L, Sisodiya SM, Squier W, Blümcke I, Aronica E, Thom M (2009) Doublecortin expression in focal cortical dysplasia in epilepsy. Epilepsia 50:2619–2628. [DOI] [PubMed] [Google Scholar]
- 195. Tagawa T, Futagi Y, Arai H, Mushiake S, Nakayama M (1997) Hypomelanosis of Ito associated with hemimegalencephaly: a clinicopathological study. Pediatr Neurol 17:180–184. [DOI] [PubMed] [Google Scholar]
- 196. Takashima S, Chan F, Becker LE, Kuruta H (1991) Aberrant neuronal development in hemimegalencephaly: immunohistochemical and Golgi studies. Pediatr Neurol 7:275–280. [DOI] [PubMed] [Google Scholar]
- 197. Talos DM, Kwiatkowski DJ, Cordero K, Black PM, Jensen FE (2008) Cell‐specific alterations of glutamate receptor expression in tuberous sclerosis complex cortical tubers. Ann Neurol 63:454–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Tapon N, Ito N, Dickson BJ, Treisman JE, Hariharan IK (2001) The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell 105:345–355. [DOI] [PubMed] [Google Scholar]
- 199. Tassi L, Pasquier B, Minotti L, Garbelli R, Kahane P, Benabid AL et al (2001) Cortical dysplasia: electroclinical, imaging, and neuropathologic study of 13 patients. Epilepsia 42:1112–1123. [DOI] [PubMed] [Google Scholar]
- 200. Tassi L, Colombo N, Garbelli R, Francione S, Lo Russo G, Mai R et al (2002) Focal cortical dysplasia: neuropathological subtypes, EEG, neuroimaging and surgical outcome. Brain 125(Pt 8):1719–1732. [DOI] [PubMed] [Google Scholar]
- 201. Tassi L, Colombo N, Cossu M, Mai R, Francione S, Lo Russo G et al (2005) Electroclinical, MRI and neuropathological study of 10 patients with nodular heterotopia, with surgical outcomes. Brain 128:321–337. [DOI] [PubMed] [Google Scholar]
- 202. Tassi L, Garbelli R, Colombo N, Bramerio M, Lo Russo G, Deleo F et al (2010) Type I focal cortical dysplasia: surgical outcome is related to histopathology. Epileptic Disord 12:181–191. [DOI] [PubMed] [Google Scholar]
- 203. Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL (2005) Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 8:1727–1734. [DOI] [PubMed] [Google Scholar]
- 204. Taylor DC, Falconer MA, Bruton CJ, Corsellis JA (1971) Focal dysplasia of the cerebral cortex in epilepsy. J Neurol Neurosurg Psychiatry 34:369–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Thom M, Martinian L, Parnavelas JG, Sisodiya SM (2004) Distribution of cortical interneurons in grey matter heterotopia in patients with epilepsy. Epilepsia 45:916–923. [DOI] [PubMed] [Google Scholar]
- 206. Thom M, Eriksson S, Martinian L, Caboclo LO, McEvoy AW, Duncan JS, Sisodiya SM (2009) Temporal lobe sclerosis associated with hippocampal sclerosis in temporal lobe epilepsy: neuropathological features. J Neuropathol Exp Neurol 68:928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Tinkle BT, Schorry EK, Franz DN, Crone KR, Saal HM (2005) Epidemiology of hemimegalencephaly: a case series and review. Am J Med Genet A 139:204–211. [DOI] [PubMed] [Google Scholar]
- 208. Torregrosa A, Marti‐Bonmati L, Higueras V, Poyatos C, Sanchis A (2000) Klippel‐Trenaunay syndrome: frequency of cerebral and cerebellar hemihypertrophy on MRI. Neuroradiology 42:420–423. [DOI] [PubMed] [Google Scholar]
- 209. Toyo‐oka K, Shionoya A, Gambello MJ, Cardoso C, Leventer R, Ward HL et al (2003) 14‐3‐3epsilon is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller‐Dieker syndrome. Nat Genet 34:274–285. [DOI] [PubMed] [Google Scholar]
- 210. Trounce JQ, Rutter N, Mellor DH (1991) Hemimegalencephaly: diagnosis and treatment. Dev Med Child Neurol 33:261–266. [DOI] [PubMed] [Google Scholar]
- 211. van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S et al (1997) Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277:805–808. [DOI] [PubMed] [Google Scholar]
- 212. van Slegtenhorst M, Nellist M, Nagelkerken B, Cheadle J, Snell R, van den Ouweland A et al (1998) Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet 7:1053–1057. [DOI] [PubMed] [Google Scholar]
- 213. Vezzani A, Ravizza T, Balosso S, Aronica E (2008) Glia as a source of cytokines: implications for neuronal excitability and survival. Epilepsia 49:24–32. [DOI] [PubMed] [Google Scholar]
- 214. Vezzani A, French J, Bartfai T, Baram TZ (2011) The role of inflammation in epilepsy. Nat Rev Neurol 7:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Walsh CA (2000) Genetics of neuronal migration in the cerebral cortex. Ment Retard Dev Disabil Res Rev 6:34–40. [DOI] [PubMed] [Google Scholar]
- 216. Wang Y, Greenwood JS, Calcagnotto ME, Kirsch HE, Barbaro NM, Baraban SC (2007) Neocortical hyperexcitability in a human case of tuberous sclerosis complex and mice lacking neuronal expression of TSC1. Ann Neurol 61:139–152. [DOI] [PubMed] [Google Scholar]
- 217. Weiner HL, Carlson C, Ridgway EB, Zaroff CM, Miles D, LaJoie J, Devinsky O (2006) Epilepsy surgery in young children with tuberous sclerosis: results of a novel approach. Pediatrics 117:1494–1502. [DOI] [PubMed] [Google Scholar]
- 218. White R, Hua Y, Scheithauer B, Lynch DR, Henske EP, Crino PB (2001) Selective alterations in glutamate and GABA receptor subunit mRNA expression in dysplastic neurons and giant cells of cortical tubers. Ann Neurol 49:67–78. [DOI] [PubMed] [Google Scholar]
- 219. Wolf HK, Campos MG, Zentner J, Hufnagel A, Schramm J, Elger CE et al (1993) Surgical pathology of temporal lobe epilepsy. Experience with 216 cases. J Neuropathol Exp Neurol 52:499–506. [DOI] [PubMed] [Google Scholar]
- 220. Wong M (2008) Mechanisms of epileptogenesis in tuberous sclerosis complex and related malformations of cortical development with abnormal glioneuronal proliferation. Epilepsia 49:8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Wong M (2010) Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: from tuberous sclerosis to common acquired epilepsies. Epilepsia 51:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Wortmann SB, Reimer A, Creemers JW, Mullaart RA (2008) Prenatal diagnosis of cerebral lesions in Tuberous sclerosis complex (TSC). Case report and review of the literature. Eur J Paediatr Neurol 12:123–126. [DOI] [PubMed] [Google Scholar]
- 223. Xu L, Zeng LH, Wong M (2009) Impaired astrocytic gap junction coupling and potassium buffering in a mouse model of tuberous sclerosis complex. Neurobiol Dis 34:291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Yagishita A, Arai N, Tamagawa K, Oda M (1998) Hemimegalencephaly: signal changes suggesting abnormal myelination on MRI. Neuroradiology 40:734–738. [DOI] [PubMed] [Google Scholar]
- 225. Yamada M, Yoshida Y, Mori D, Takitoh T, Kengaku M, Umeshima H et al (2009) Inhibition of calpain increases LIS1 expression and partially rescues in vivo phenotypes in a mouse model of lissencephaly. Nat Med 15:1202–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Yasha TC, Santosh V, Das S, Shankar SK (1997) Hemimegalencephaly—morphological and immunocytochemical study. Clin Neuropathol 16:17–22. [PubMed] [Google Scholar]
- 227. Yu J, Baybis M, Lee A, McKhann G 2nd, Chugani D, Kupsky WJ et al (2005) Targeted gene expression analysis in hemimegalencephaly: activation of beta‐catenin signaling. Brain Pathol 15:179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Zeng LH, Ouyang Y, Gazit V, Cirrito JR, Jansen LA, Ess KC et al (2007) Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis complex. Neurobiol Dis 28:184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Zhu XJ, Liu X, Jin Q, Cai Y, Yang Y, Zhou T (2010) The L279P mutation of nuclear distribution gene C (NudC) influences its chaperone activity and lissencephaly protein 1 (LIS1) stability. J Biol Chem 285:29903–29910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Zurolo E, Iyer A, Maroso M, Carbonell C, Anink JJ, Ravizza T et al (2011) Activation of Toll‐like receptor, RAGE and HMGB1 signalling in malformations of cortical development. Brain 134(Pt 4):1015–1032. [DOI] [PubMed] [Google Scholar]