

Abstract
Over the course of DNA replication, DNA lesions, transcriptional intermediates and protein-DNA complexes can impair the progression of replication forks, thus resulting in replication stress. Failure to maintain replication fork integrity in response to replication stress leads to genomic instability and predisposes to the development of cancer and other genetic disorders. Multiple DNA damage and repair pathways have evolved to allow completion of DNA replication following replication stress, thus preserving genomic integrity. One of the processes commonly induced in response to replication stress is fork reversal, which consists in the remodeling of stalled replication forks into four-way DNA junctions. In normal conditions, fork reversal slows down replication fork progression to ensure accurate repair of DNA lesions and facilitates replication fork restart once the DNA lesions have been removed. However, in certain pathological situations, such as the deficiency of DNA repair factors that protect regressed forks from nuclease-mediated degradation, fork reversal can cause genomic instability. In this review, we describe the complex molecular mechanisms regulating fork reversal, with a focus on the role of the SNF2-family fork remodelers SMARCAL1, ZRANB3 and HLTF, and highlight the implications of fork reversal for tumorigenesis and cancer therapy.
1. Introduction
DNA replication is the process by which genomic DNA is duplicated during cellular proliferation. Over the course of DNA replication, obstacles to replication fork progression cause replication stress, thus resulting in genomic instability, an enabling characteristic for cancer development [1–4]. Replication stress is induced by a variety of endogenous sources, including unbalanced nucleotide pools, abasic sites, modified DNA bases (e.g., oxidized or methylated bases, DNA crosslinks), DNA breaks, protein-DNA adducts, non-canonical DNA structures (e.g., hairpins, G-quadruplexes, and trinucleotide repeats), RNA-DNA hybrids (R-loops), replication slow zones (AT-rich regions or replication initiation poor sites), and collisions with transcription or non-histone DNA-bound proteins [5–7]. Replication stress can be further exacerbated by exposure to exogenous DNA damaging agents, including ultraviolet (UV) light, ionizing radiation (IR) and chemical compounds, such as DNA crosslinking and methylating agents (e.g., cisplatin, mitomycin C, methyl methanesulfonate), topoisomerase inhibitors (e.g., camptothecin) and inhibitors of nucleotide synthesis and DNA polymerase activity (e.g., hydroxyurea, aphidicolin) [8].
While replication obstacles on the lagging strand are bypassed by the synthesis of a new Okazaki fragment, impediments on the leading strand may result in the arrest of replication fork progression [9–11] (Figure 1, step I). DNA lesions that block the leading strand polymerase can induce the formation of RPA-coated ssDNA regions [11–13], which in turn results in the recruitment of the ATR kinase through its interacting partner ATRIP [14]. Following recruitment and activation, ATR phosphorylates a multitude of factors involved in cell cycle regulation, DNA replication and DNA repair in order to arrest cell cycle progression, suppress global origin firing, activate local dormant origins, slow replication fork movement, and promote replication fork stability [15–17]. Subsequently, several pathways become engaged to restore DNA synthesis and ensure successful completion of DNA replication by: 1) allowing the direct bypass of DNA lesions (Figure 1, steps II-III); 2) pausing and eventually resuming fork progression once the obstacles have been cleared (steps IV-V); or 3) repairing forks that have collapsed (steps VI-VIII) [18–20]. Additionally, completion of DNA synthesis at sites of replication fork stalling can also be achieved through convergence of replication forks originating from previously activated or newly fired origins [21]. The coordinated action of these pathways depends on the type of DNA lesion, cell cycle regulation, and protein post-translational modifications (e.g., PCNA ubiquitination) [17, 22]. Given the relevance of genome stability for human disease, understanding the crosstalk between the pathways that maintain replication fork integrity in physiological and pathological conditions is of utmost therapeutic relevance.
Figure 1. Simplified schematics of the mechanisms of replication fork restart upon replication stress.
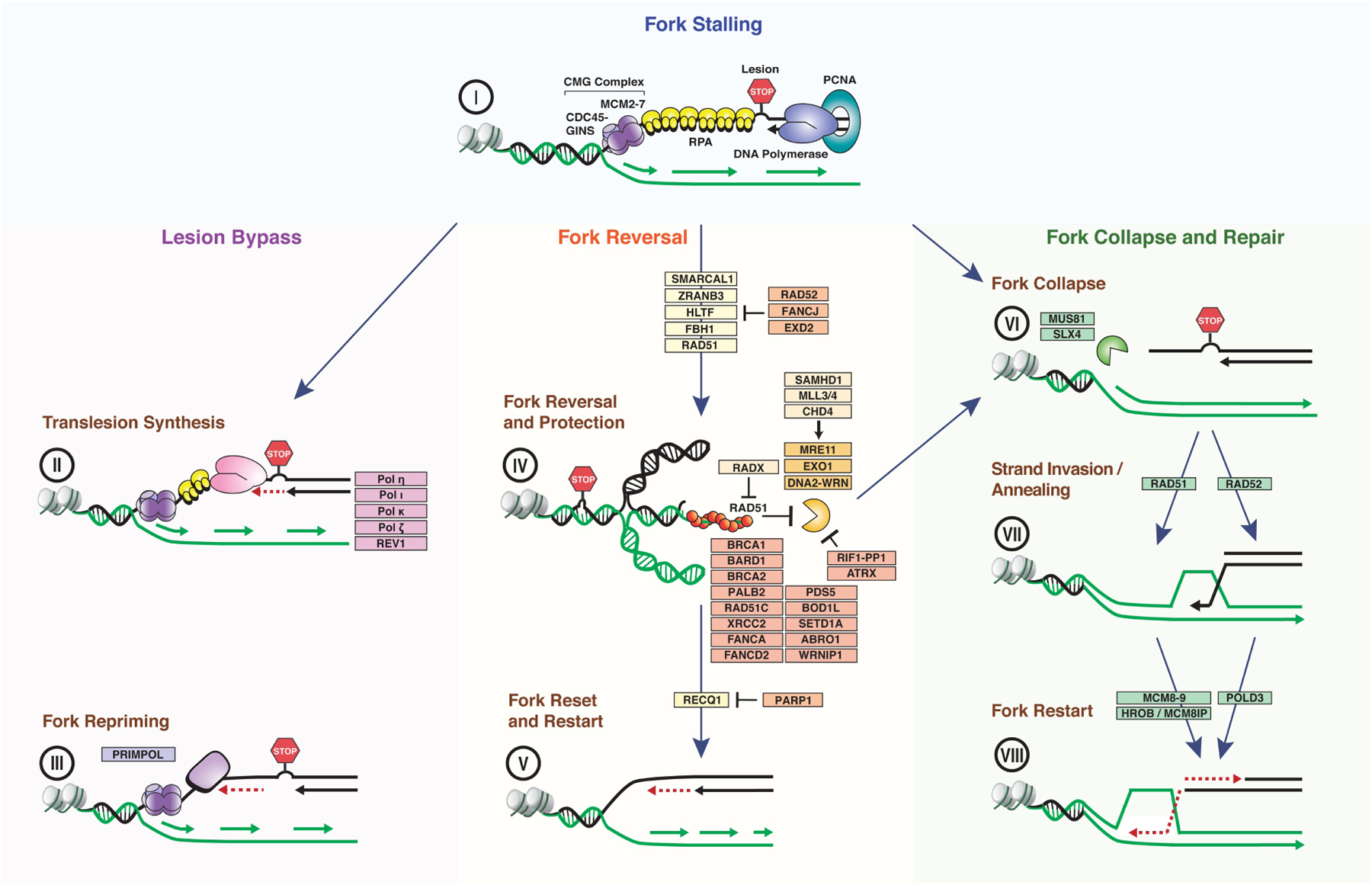
When the replication machinery encounters a replication block (stop sign), replication fork progression stalls, leading to the formation of single-stranded DNA (ssDNA) that is rapidly coated by the RPA trimer (I). Restart of stalled forks can occur through lesion bypass by translesion synthesis or fork repriming (II-III). Translesion synthesis allows the direct bypass of the DNA lesion (II), while fork repriming promotes the restart of DNA synthesis downstream of the lesion (III). Alternative to lesion bypass, stalled forks can undergo reversal mediated by fork remodelers, including the SNF2-family members SMARCAL1, ZRANB3 and HLTF (IV). Fork reversal slows down replication fork progression, thus allowing sufficient time for repair of the lesion. Reset of reversed forks by the RECQ1 helicase can then promote the restart of DNA synthesis once the lesion has been removed (V). Fork reversal could also directly promote the bypass of the DNA lesion by enabling DNA synthesis on the nascent DNA strand of the sister chromatid (green) through template switching (not shown). Following fork reversal, the exposed ends of the regressed fork are stabilized by RAD51 to limit fork resection by the nucleases MRE11, DNA2 and EXO1 and protect the integrity of the fork (IV). Limited fork resection can facilitate fork restart through the reset of reversed forks or HR-dependent strand invasion catalyzed by the 3’-end of the resected regressed arm (not shown). BRCA1/2, RAD51 paralogs and Fanconi anemia proteins cooperate with RAD51 to control fork resection and maintain fork protection. Defective fork protection causes extensive fork resection and processing of the regressed fork by the endonucleases SLX4 and MUS81, leading to fork collapse (VI). Fork collapse can also result from MUS81-mediated cleavage of persistently arrested replication forks or from forks encountering single-strand DNA breaks. Collapsed forks are repaired by HR-mediated fork restart pathways dependent on RAD51 and/or RAD52 (VI-VIII). Key factors involved in each pathway and described in the main text are indicated.
1.1. Lesion bypass
Certain DNA lesions, such as modified bases and DNA adducts, can be directly bypassed by translesion DNA synthesis (TLS) (Figure 1, step II). TLS is promoted by specialized DNA polymerases capable of DNA synthesis across DNA lesions. TLS polymerases are engaged at DNA damage sites through binding of monoubiquitinated PCNA mediated by their ubiquitin-binding motifs [23–26]. TLS requires the sequential action of two groups of DNA polymerases that insert the nucleotide across the lesion (Pol κ, Pol η, Pol ι, or REV1) and then extend the nascent DNA strand (Pol κ or Pol ζ) [19, 27–34]. Once the lesion has been bypassed, replicative polymerases can then resume DNA synthesis [35]. Distinct from the replicative polymerases, TLS polymerases are characterized by low processivity, efficiency and accuracy [32]. Lesion bypass can also occur by repriming of DNA synthesis downstream of DNA lesions (Figure 1, step III). Repriming is catalyzed by the DNA polymerase-primase PRIMPOL on templates containing DNA lesions (e.g., cyclobutane pyrimidine dimers and (6–4) T-T photoproducts), chain-terminating nucleotide analogs, and non-canonical DNA structures (e.g., G-quadruplexes) [36–43]. PRIMPOL presents weak affinity for DNA, thereby limiting its potentially error-prone repriming activity [39, 44].
Bypass of complex DNA lesions, such as DNA-protein crosslinks (DPCs) and interstrand crosslinks (ICLs), requires specialized machineries. DPCs and ICLs can be induced by exogenous sources, such as UV radiation and chemical agents (e.g., cisplatin, mitomycin C), or result from endogenous sources, like formaldehyde [45–48]. Bulky DPCs impair replication fork progression by delaying CDC45-MCM-GINS (CMG) helicase movement, while ICLs prevent the unwinding of the DNA double helix [49, 50]. RTEL1 facilitates the bypass of DPCs by the CMG helicase and is required for efficient DPC processing by the metalloprotease SPRTN [49]. MCM helicase complexes can also bypass ICLs at a single blocked replication fork through the actions of FANCD2, the BLM and FANCM helicases, and FANCM’s interacting partners MHF1 and MHF2 [51–54]. Subsequently, when two converging forks are arrested by an ICL, ubiquitination of the CMG helicase by the TRAIP E3 ligase controls whether the lesion is bypassed by TLS after ICL unhooking by the NEIL3 glycosylase [55–57] or is repaired by the Fanconi anemia pathway through ICL incision, DSB formation and homologous recombination (HR) [58–60].
1.2. Repair of collapsed forks
Forks irreversibly arrested by DNA lesions undergo MUS81/SLX4-dependent fork cleavage, which leads to DSB formation and fork collapse (Figure 1, step VI) [61, 62]. DSBs can be formed at single stalled forks or converging forks arrested by DNA lesions (e.g., ICLs) [59, 63]. HR then promotes DSB repair and fork restart through RAD51-dependent strand invasion, followed by DNA synthesis aided by the MCM8–9 helicase in complex with HROB/MCM8IP [64–67] (Figure 1, steps VI-VIII). If unrepaired DSBs persist into late G2 and mitosis, they can be resolved by RAD52-dependent mitotic DNA synthesis (MiDAS) (Figure 1, step VII) [68]. RAD52 facilitates MiDAS by promoting microhomology-mediated strand annealing, followed by DNA synthesis dependent on the POLD3 subunit of Pol δ (Figure 1, steps VI-VIII) [68–70]. Interestingly, in yeast a Rad51- and Rad52-dependent pathway called break-induced replication (BIR) proceeds through the invasion of the broken end into a homologous template, followed by conservative DNA synthesis until the end of the chromosome promoted by the Pif1 helicase [71–76]. While MiDAS and the repair of DSBs resulting from oncogene-induced replication stress, or induced during alternative lengthening of telomeres (ALT), have been suggested to be similar to the BIR pathway described in yeast [68, 69, 77, 78], the molecular mechanisms of BIR-like processes in mammals remain to be fully defined.
1.3. Fork reversal
Fork reversal occurs when the parental fork DNA strands re-anneal, thereby extruding the nascent DNA strands to form a four-way structure, also known as “chicken foot” [18] (Figure 1, step IV). Fork reversal has been proposed to limit excessive ssDNA accumulation at stalled forks, reposition DNA lesions into a dsDNA context to enable their repair, and prevent synthesis across DNA single-strand breaks (SSBs) to avoid their conversion into deleterious DSBs [18]. In addition, fork reversal could enable error-free bypass of DNA lesions by allowing the arrested leading strand to utilize the lagging strand as a template for DNA synthesis through template switching, a pathway suggested to depend on PCNA polyubiquitination [79]. While lesion bypass mediated by fork reversal has been demonstrated in vitro, evidence of the occurrence of such events in mammalian cells remains to be reported [20, 80, 81]. Nonetheless, recent studies using electron microscopy (EM) have enabled the visualization and quantification of reversed fork structures in replicating mammalian cells, demonstrating that exposure to endogenous and exogenous genotoxic stresses leads to an increase in fork reversal events [82–88]. Genetic and biochemical studies have shown that fork reversal is primarily catalyzed by the SNF2-family DNA translocases SMARCAL1, ZRANB3, and HLTF, along with the FBH1 helicase [86, 87, 89–96] (Figure 1, step IV). The RAD51 recombinase has also been proposed to have an HR-independent function in promoting fork reversal through mechanisms that are still unclear [83, 97]. In this review, we describe the functions of SNF2-family fork remodelers in the response to replication stress during physiological and pathological conditions.
2. SNF2-family DNA translocases and replication fork dynamics
The SNF2 family of proteins is structurally characterized by a SWI/SNF2 (switch/sucrose non-fermenting) translocase domain with a bi-lobed RecA-type ATPase configuration, which is present in the yeast protein Snf2 [98]. Functionally, SNF2-family members have been primarily involved in chromatin remodeling. The SNF2 family includes SNF2-like, INO80-like, SSO1653-like, RAD54-like, RAD5/16-like and SMARCAL1-like subfamilies [98, 99] (Figure 2). Below we describe the activities exhibited at the replication fork by the SMARCAL1-like and RAD5/16-like subfamily members SMARCAL1, ZRANB3 and HLTF.
Figure 2. The SNF2 family of proteins.
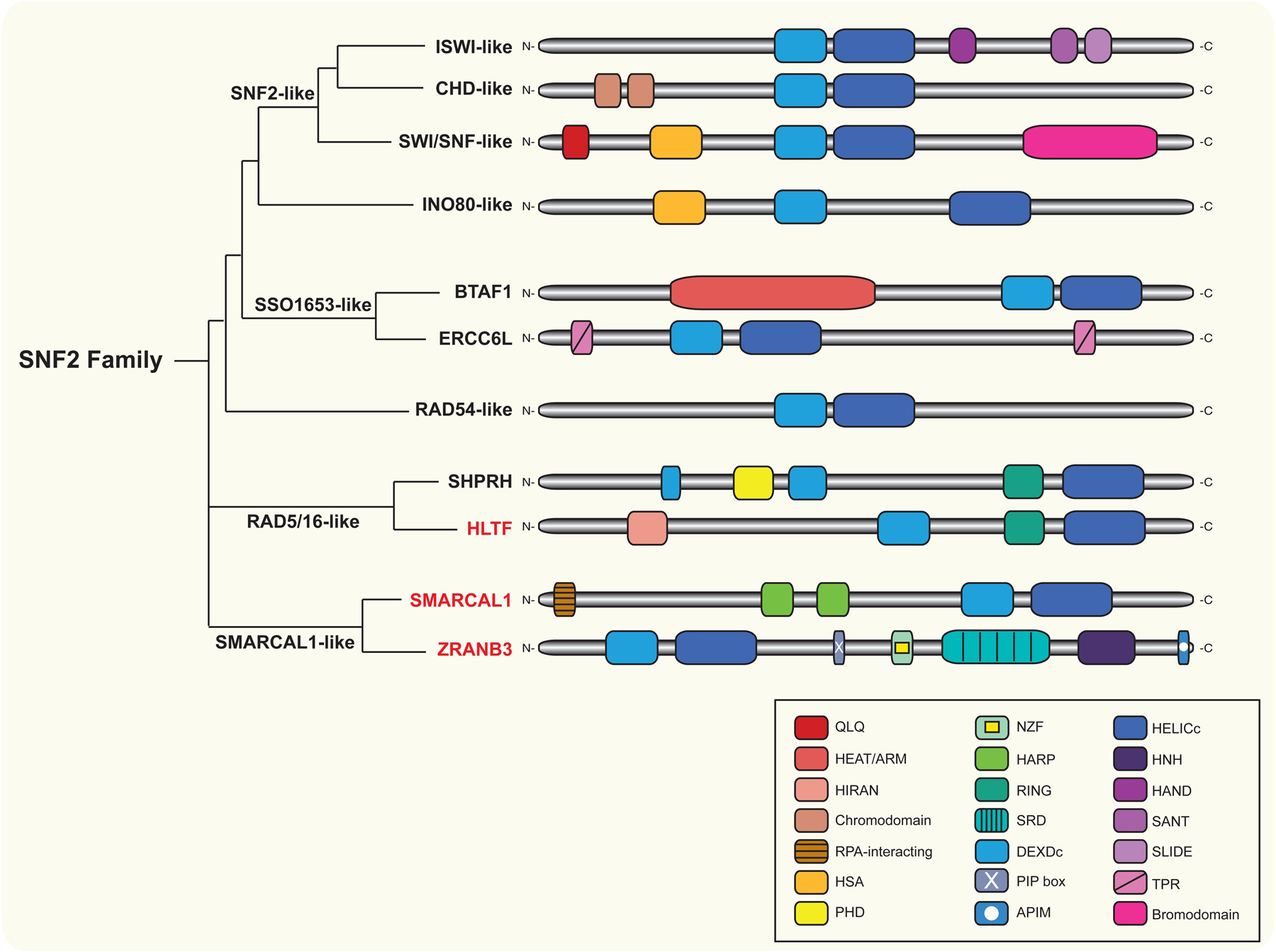
Depiction of SNF2-family group relationships, based on alignments of the DNA translocase domain (adapted from Flaus et al. [98]). Schematic representations (not to scale) of protein domains characteristic of each subfamily in humans are shown. All SNF2-family members contain a DNA translocase domain consisting of DEXDc and HELICc domains. The SNF2-like subfamily consists of ISWI-like, CHD-like and SWI/SNF-like groups. The ISWI-like group includes SMARCA1 (1054 aa) and SMARCA5 (1052 aa). The CHD-like group includes CHD1 (1710 aa), CHD2 (1828 aa), CHD3 (2000 aa), CHD4 (1912 aa), CHD5 (1954 aa), CHD6 (2715 aa), CHD7 (2997 aa), CHD8 (2581 aa), CHD9 (2897 aa) and CHD1L (897 aa). CHD3, CHD4 and CHD5 have N-terminal plant homeodomain (PHD) fingers not represented in the schematic, while CHD1L lacks the N-terminal chromodomains and contains a C-terminal macrodomain (not shown). The SWI/SNF-like group includes SMARCA2 (1590 aa) and SMARCA4 (1647 aa). HELLS (838 aa) is closely related to the SNF2-like subfamily, but it does not contain domains typical of the members of this subfamily (not shown). The INO80-like subfamily includes INO80 (1556 aa), SRCAP (3230 aa) and EP400 (3159 aa), which contain additional A/T hook and SANT domains, respectively, and SMARCAD1 (1026 aa), which lacks the HSA domain and contains two N-terminal coupling of ubiquitin conjugation to ER degradation (CUE) motifs not represented in the schematic. The SSO1653-like subfamily includes BTAF1 (1849 aa), which contains multiple HEAT/ARM repeats, and ERCC6L (1250 aa), which harbors two tetratricopeptide repeat (TPR) motifs and a PICH family domain (PFD) of unclear function not represented in the schematic. The RAD54-like subfamily includes RAD54 (747 aa) and ATRX (2492 aa), which contains an N-terminal ATRX-DNMT1-DNMT1L (ADD) domain not shown. The RAD5/16-like subfamily includes SHPRH (1683 aa), which contains a linker histone H1/H5 (H15) domain in addition to the domains represented in the figure, and HLTF (1009 aa). Finally, the SMARCAL1-like subfamily includes SMARCAL1 (954 aa) and ZRANB3 (1079 aa). SMARCAL1, ZRANB3 and HLTF (in red) are the only SNF2-family members that have been currently shown to regress stalled replication forks in vivo. HLTF contains a RING domain that mediates the interaction between HLTF and substrates to ubiquitinate. SMARCAL1 harbors an RPA2-binding motif to directly interact with RPA. ZRANB3 contains a PCNA-interacting protein (PIP) motif, an AlkB homolog 2 PCNA interacting motif (APIM) and a NPL4 zinc-finger (NZF) motif that cooperate to bind polyubiquitinated PCNA. The HIRAN, HARP and SRD (substrate recognition) domains of HLTF, SMARCAL1, and ZRANB3, respectively, enable structure specific DNA binding. The HNH motif of ZRANB3 is a nuclease domain.
2.1. SMARCAL1
SMARCAL1 (SWI/SNF-related, matrix-associated, actin-dependent, regulator of chromatin, subfamily A-like 1) is a distant member of the SNF2 family [98]. SMARCAL1 associates with the replication fork under unperturbed conditions, but becomes enriched at stalled forks upon replication stress through its direct binding to RPA mediated by an N-terminal RPA2 interaction motif [93, 100–105] (Figure 2). In addition to its RPA2-binding motif and SWI/SNF2 DNA translocase domain, SMARCAL1 contains two HepA-related protein (HARP) domains, which promote binding to synthetic fork-like structures [106]. Biochemical studies have shown that the HARP and DNA translocase domain of SMARCAL1 cooperate to promote ATP-dependent regression of fork-like structures through coordinated re-annealing of complementary strands [89, 91–93, 106] (Table 1). The fork remodeling activity of SMARCAL1 is stimulated by RPA, which targets SMARCAL1 to forks containing gaps on the leading strand [92, 107]. Besides remodeling fork-like structures, SMARCAL1 was also shown to promote Holliday junction branch migration and dissociation of D-loop recombination intermediates [91, 93]. The fork remodeling activity of SMARCAL1 has been confirmed in vivo by EM studies showing a 50% reduction in the number of reversed forks in SMARCAL1-deficient human cells [89]. In addition, loss of SMARCAL1 was shown to cause defective restart of stalled forks and delayed cell cycle progression following replication stress [100].
Table 1. List of functions of SNF2-family fork remodelers exhibited in biochemical assays, cellular and animal models, and human disease.
Citations of papers describing the indicated functions of SMARCAL1, ZRANB3 and HLTF are included in the table.
| HLTF | SMARCAL1 | ZRANB3 | |
|---|---|---|---|
| Biochemical Activities |
Activities • Fork reversal and restoration [90,94,114,116,117,120] • D-loop formation [121] • Ubiquitin ligase activity [112,113] Substrate preference • DNA binding: ssDNA and fork substrates with 3’-OH overhang [116–119] • Fork reversal: fork substrate with 3’-OH available in the vicinity of the fork branch point [116,117]; fork with gap on leading or lagging strand [117] • Fork restoration: fork with lagging strand gap (final product) [117] Inhibition by • FANCJ [135] |
Activities • Fork reversal and restoration [87,89,91–93,106,107,129] • D-loop dissolution [91] Substrate preference • DNA binding: ssDNA/dsDNA junction with 3’ or 5’ ssDNA overhangs, fork structures [93,293] • Fork reversal: fork substrate with a leading strand gap bound by RPA [92,107] • Fork restoration: fork with lagging strand gap bound by RPA (final product) [92] Association with • RPA2 [100,103,104] Stimulation by • RPA [92,107] Inhibition by • ATR [129] • MCM10 [294] • RAD52 [133] |
Activities • Fork reversal and restoration [89,91,92,108,111] • D-loop dissolution [91] • Inhibition of D-loop formation [91] • ATP-dependent endonuclease activity [108,110,111] Substrate preference • DNA binding: fork structures [108,110] • Fork reversal: fork with gap on leading or lagging strand [92] • Fork restoration: fork with lagging strand gap (final product) [92] • Endonuclease activity: splayed duplex [108] Association with • PCNA and poly-ubiquitinated PCNA [91,108,109] Stimulation by • PCNA [110] (endonuclease activity) Inhibition by • RPA [92] (fork remodeling activity) |
| Cellular Functions |
Functions • Fork reversal in response to replication stress [90] • Restart of stalled forks [94] • Suppression of PRIMPOL- and REV1-mediated restart of stalled forks [90] • Induction of PCNA polyubiquitination upon replication stress [112,113] • Stimulation of TLS [295,296] • Suppression of cellular resistance to HU [90,135] and MMC [90], promotion of cellular resistance to UV and MMS [112,113] • Suppression of replication stress-induced DSB formation [90] • Gene expression regulation [297,298] BRCA1/2-deficient cells • Promotion of replication stress-induced fork degradation and DSB formation [89] FANCJ-deficient cells • Promotion of replication stress-induced fork degradation and suppression of DSB formation [135] • Promotion of MMC sensitivity and HU resistance [135] |
Functions • Fork reversal in response to replication stress [87,89,93] • Restart of stalled forks [100] • Telomere maintenance in ALT [123,125,215] and non-ALT cells [124] and ALT suppression [124,187] • Stimulation of NHEJ [299] • Promotion of cellular resistance to HU, aphidicolin, MMC, IR and camptothecin [100,101,103,197,299] • Suppression of DNA damage formation [100,101,103,124,300] • Gene expression regulation in response to replication stress and heat shock [300–305] BRCA1/2-deficient cells • Promotion of replication stress-induced fork degradation [89] • Induction of DNA damage [159], replication stress-induced DSB formation and genomic rearrangements [89] • Suppression of cellular resistance to replication stress-inducing agents in breast cancer cells, but not in mammary epithelial cells [89] • Suppression of PRIMPOL-mediated restart of stalled forks [201] Myc-overexpressing cells • Promotion of replication fork progression and suppression of fork collapse [196] |
Functions • Fork reversal in response to replication stress [86] • Restart of stalled forks [91,109] • Recognition of poly-ubiquitinated PCNA [91,108] • Suppression of hyper-recombination [91] • Promotion of cellular resistance to camptothecin, HU, cisplatin, MMC [91,109] and MMS [108] BRCA1/2-deficient cells • Promotion of replication stress-induced fork degradation [89,96] • Induction of replication stress-induced DSBs and genomic rearrangements in mammary epithelial cells [89]; suppression of genomic rearrangements in U2OS cells [96] Myc-overexpressing cells • Promotion of replication fork progression and suppression of fork collapse [196] β-cells • Promotion of insulin secretion in response to glucose [179] |
| Mouse Models |
Hltf del/del • Semi-lethal / neonatal lethal (deletion of exons 11–12) [297,298,306] or exhibiting normal development, fertile, and normal life span (deletion of exons 1–5) [183] • Formation of carcinogen-induced colorectal cancer [306] Apcmin/+ • Hltf deletion increases intestinal adenocarcinoma invasion and malignancy [183] |
Smarcal1del/del • No developmental, growth, or physical abnormalities [305] • Reduced B-cell count [196,305] • Reduced growth and weight and albuminuria upon RNA pol II inhibition [305] • Hypersensitivity to campothecin, etoposide, and HU [301] • Increased survival, delayed T-cell lymphomagenesis, impaired T-lymphocyte reconstitution after IR [197] Eµ-myc transgenic mice • Smarcal1del/+, but not Smarcal1del/del, mice exhibit increased myc-induced B-cell lymphomagenesis and decreased survival [196] |
Zranb3−l− • No abnormalities [196] Eµ-myc transgenic mice • Loss of one or both Zranb3 alleles inhibits myc-induced B-cell lymphomagenesis [196] |
| Human Disease |
Cancer • Frequently silenced in colorectal and gastric cancers [181,182] • Overexpressed in esophageal, uterine, and squamous cell carcinoma [184] • Overexpression is associated with increased metastasis and poorer prognosis in non-small cell lung cancer [185] |
Genetic syndrome • Biallelic mutations cause Schimke immuno-osseous dysplasia (SIOD) [174,175,178,305,307] Cancer • Mutations identified in glioblastoma carrying wildtype TERT promoter and IDH1/2 genes, and ALT-positive [187] |
Genetic syndrome • Mutations associated with African-specifc type-2 diabetes [179] Cancer • Candidate tumor suppressor in endometrial cancer [186] • Mutations associated with carcinosarcoma [308] |
2.2. ZRANB3
ZRANB3 is related to SMARCAL1 and shares with it extensive similarity (44%) in the DNA translocase domain [91] (Figure 2). Distinct from SMARCAL1, ZRANB3 associates with PCNA via a PIP box and an APIM motif, which are required for ZRANB3’s recruitment to stalled replication forks [91, 108–110]. Moreover, ZRANB3 contains a NPL4 zinc-finger (NZF) motif that binds K63-linked polyubiquitin chains and cooperates with the PIP box and APIM motif to promote the association of ZRANB3 with polyubiquitinated PCNA [91, 108], suggesting a possible role for ZRANB3 in the template switching pathway. Biochemical studies have shown that ZRANB3 possesses ATP-dependent DNA translocase activity that promotes remodeling of synthetic fork-like structures and dissociation of D-loop intermediates [89, 91, 92, 108] (Table 1). In addition, ZRANB3 exhibits structure-specific endonuclease activity mediated by its HNH nuclease domain, which promotes ATP- and PCNA-dependent cleavage of splayed DNA duplex structures, generating 5’ overhangs in vitro [108, 110]. These observations have suggested the possibility that ZRANB3 could utilize its fork reversal activity to facilitate the excision of DNA lesions at stalled forks [108]. ZRANB3’s binding to DNA substrates relies on its sequence recognition domain (SRD), which is required for both fork remodeling and endonuclease activities [111]. Recent EM studies have shown that ZRANB3 requires its DNA translocase activity and its ability to associate with polyubiquitinated PCNA, but not its endonuclease activity, to catalyze fork reversal in vivo [86]. In line with its fork processing activities, ZRANB3 deficiency causes impaired fork slowing and defective fork restart in mammalian cells in response to replication stress [86, 91, 109].
2.3. HLTF
HLTF is a member of the RAD5/16-like subfamily (Figure 2). Analogous to the yeast Rad5 protein, HLTF promotes polyubiquitination of PCNA via its RING domain, suggesting a potential function in template switching [112, 113]. Similar to SMARCAL1 and ZRANB3, HLTF can also catalyze fork regression in vitro and in vivo through its DNA translocase activity [90, 94, 114–116] (Table 1). Besides HLTF’s ATPase domain, its HIP116/HLTF Rad5 N-terminus (HIRAN) domain plays an important role in fork reversal by recognizing 3’-ssDNA ends formed at stalled replication forks and properly orienting the motor activity of HLTF [116–120]. HLTF has also been shown to promote strand invasion and D-loop formation in vitro in an ATP-independent manner, a mechanism that could act as an alternative to fork remodeling for promoting fork restart [121]. In line with these findings, loss of HLTF results in impaired fork restart and defective fork slowing upon replication stress [90, 94, 116], as observed in the case of ZRANB3 deficiency [86, 91, 109].
2.4. Interplay between SMARCAL1, ZRANB3 and HLTF at the replication fork
Although SMARCAL1, ZRANB3 and HLTF share the same ability to reverse stalled forks, they play non-redundant functions during replication stress [122]. For instance, SMARCAL1, but not ZRANB3 and HLTF, protects telomeres from replication stress and is required for efficient telomere replication [123–125]. At a molecular level, the above DNA translocases exhibit differences in their mode of action (Table 1). As discussed above, SMARCAL1 is an RPA-associated factor and is recruited to stalled forks by RPA [100, 101, 103–105], while ZRANB3 associates to sites of replication stress in a manner dependent on PCNA and its polyubiquitination promoted by HLTF and UBC13 [91, 108, 112, 113]. In agreement with these observations, RPA stimulates the fork reversal activity of SMARCAL1, while it inhibits ZRANB3-mediated fork remodeling [92]. Furthermore, the fork reversal activity of ZRANB3 and HLTF, but not that of SMARCAL1, is thought to depend on PCNA ubiquitination [86, 126]. In addition to their different modes of activation, the above fork remodelers display distinct substrate specificities (Table 1). SMARCAL1 primarily acts on fork substrates containing leading strand gaps [92, 93]. Instead, ZRANB3 and HLTF remodel equally efficiently fork substrates with leading or lagging strand gaps, and HLTF has a strong preference for available 3’-OH groups near the fork junction [92, 116, 117, 119]. ZRANB3 may preferentially act upon forks stalled by damaged DNA bases that could be excised through its endonuclease activity [108]. These observations raise the possibility that SMARCAL1, ZRANB3 and HLTF might operate on distinct types of stalled forks induced in response to replication stress. These structures could arise independently from each other on distinct stalled forks or originate on the same fork during multiple remodeling attempts, especially under conditions where extensive fork remodeling is thought to occur (e.g., BRCA1/2-deficient cells). It has been proposed that, under those circumstances, SMARCAL1, ZRANB3, and HLTF may act cooperatively on their preferred substrates generated on the same fork, and that failure at any point in these sequential steps could impair fork reversal [89, 122]. SMARCAL1 could, for example, generate fork substrates amenable to HLTF-mediated fork remodeling by pulling the leading strand closer to the fork junction, and HLTF could control ZRANB3-dependent remodeling by regulating the levels of PCNA polyubiquitination. However, these models remain to be proven and the dynamics between fork remodelers elucidated. Understanding these processes will likely require the development of novel approaches to study replication fork dynamics that overcome the limitations of currently available methodologies, as discussed below (Section 4.1).
3. Roles of SNF2-family DNA translocases in maintaining genomic stability and preventing the development of cancer and genetic syndromes
3.1. Fork remodeling and genomic instability
Fork reversal is a double-edged sword, given that both failure to reverse stalled forks and excessive fork reversal can result in genomic instability due to enhanced nucleolytic processing of stalled forks [18, 81, 87–90, 96, 103, 127–131]. Therefore, fork remodelers need tight regulation to prevent unscheduled fork reversal activity. One such regulatory mechanism is provided by ATR, which phosphorylates SMARCAL1 on serine 652 to restrict its fork remodeling activity [129]. Moreover, RAD52 has recently been suggested to inhibit SMARCAL1-mediated fork reversal by directly blocking SMARCAL1 loading on stalled forks and possibly competing with SMARCAL1 for the same RPA2-binding site [132, 133]. In addition, the nuclease EXD2 has been suggested to counteract SMARCAL1-mediated fork reversal through mechanisms that need to be elucidated [134]. As previously mentioned, RPA impairs ZRANB3 fork remodeling activity in vitro, presumably by sterically hindering ZRANB3’s binding to DNA [92]. Furthermore, the helicase FANCJ restricts HLTF activity at the fork [135]. In addition to the above negative regulators of fork remodelers, the RECQ1 helicase counteracts fork reversal by resetting regressed forks once lesion repair or fork pausing has been completed, thus allowing fork restart (Figure 1, step V) [88]. RECQ1-mediated fork restoration is inhibited by PARP1, which promotes fork reversal [84, 88, 136].
An alternative mechanism restricts genomic instability caused by excessive fork reversal, namely fork protection. When a fork is reversed, the nascent DNA extruded from the “chicken foot” structure is susceptible to degradation by nucleases, such as MRE11, DNA2 and EXO1 [87, 89, 96, 131, 137–148]. EXO1-mediated processing of stalled forks could be aided by the SNF2-family member SMARCAD1, a chromatin remodeler that promotes DSB resection [149–152]. Interestingly, EM studies have shown that regressed forks contain nucleosomes, indicating that chromatin remodeling may play an important role in fork resection [153]. While limited fork resection by the above nucleases can assist in the removal of the regressed arm of the reversed fork and/or promote HR-mediated fork restart, uncontrolled fork degradation can lead to chromosomal instability [63, 81, 138]. To prevent fork degradation, RAD51 nucleofilaments are assembled on the regressed arm of reversed forks [87, 96, 97, 128, 141, 142, 154–156]. RAD51 nucleofilaments protect stalled forks in an HR-independent manner, as indicated by the observation that a RAD51 mutant deficient for strand invasion is capable of fork protection [97]. Several DDR factors regulate the assembly and stability of RAD51 nucleofilaments to protect stalled forks from degradation. Among those, BRCA1-BARD1 and BRCA2 play an HR-independent role in fork protection by assembling and stabilizing RAD51 nucleofilaments (Figure 1, step IV) [87, 89, 96, 141, 142, 144]. Recent studies have suggested that PIN1-dependent isomerization of the BRCA1-BARD1 complex allows enhanced binding of RAD51 and fork protection [157]. Interestingly, the BRCT domain of BARD1 exhibits an exclusive role in fork protection by recruiting the BRCA1-BARD1 heterodimer to stalled forks in a poly(ADP-ribose) (PAR)-dependent manner [144]. RPA phosphorylation, on the other hand, is thought to favor the recruitment of PALB2 and BRCA2 to stalled forks [158]. BRCA2 mediates fork protection through a RAD51 interaction site that, when mutated, renders BRCA2 unable to prevent fork degradation, despite being still competent for HR [142, 159]. Additionally, BRCA2 requires its DNA binding domain for fork protection and suppresses both MRE11- and DNA2-mediated fork degradation [142, 160]. Other DDR factors, such as FANCA, FANCD2, the RAD51 paralogs (XRCC2 and RAD51C), PDS5B, WRNIP1, ABRO1, and the SETD1A-BOD1L complex, also contribute to protecting stalled forks from nuclease-mediated degradation [141, 143, 145, 146, 161–164]. Distinct from these factors, the recently identified RADX protein has been shown to promote MRE11- and DNA2-mediated fork degradation by antagonizing RAD51 at stalled forks [127, 128].
Fork protection can also be achieved by nuclease inhibition or exclusion from nascent DNA. For instance, the SNF2-family member ATRX has been suggested to protect stalled forks from MRE11-dependent degradation through MRE11 sequestration, similar to its role at telomeres [165, 166]. Moreover, the phosphatase complex RIF1-PP1 negatively regulates the WRN-DNA2 complex to limit over-resection of stalled forks [139, 140]. Failure to protect forks from extensive degradation can lead to fork collapse and DSB formation induced by MUS81/SLX4-dependent fork cleavage [89, 131] (Figure 1, step VI). This observation is supported by the fact that MUS81-depletion prevents DSB formation after fork stalling similarly to deficiency of either MRE11 or EXO1, suggesting nucleolytic processing of reversed forks prior to cleavage [131]. MUS81-dependent fork processing has been shown to be promoted by the histone methyl-transferase EZH2 specifically in BRCA2-deficient cells, implying different roles for BRCA1- and BRCA2-mediated fork protection [167]. In addition, WRNIP1 protects stalled forks from SLX4-mediated processing to prevent unnecessary fork collapse and DSB formation [143, 168]. In the absence of fork protection mechanisms, as in BRCA1/2-deficient tumors, limitation of fork reversal becomes crucial to prevent extensive fork degradation and collapse [87, 89, 96], and consequent accumulation of DNA damage and chromosomal instability (Figure 3) [87, 89, 134, 141–143].
Figure 3. Cellular effects induced by replication stress.
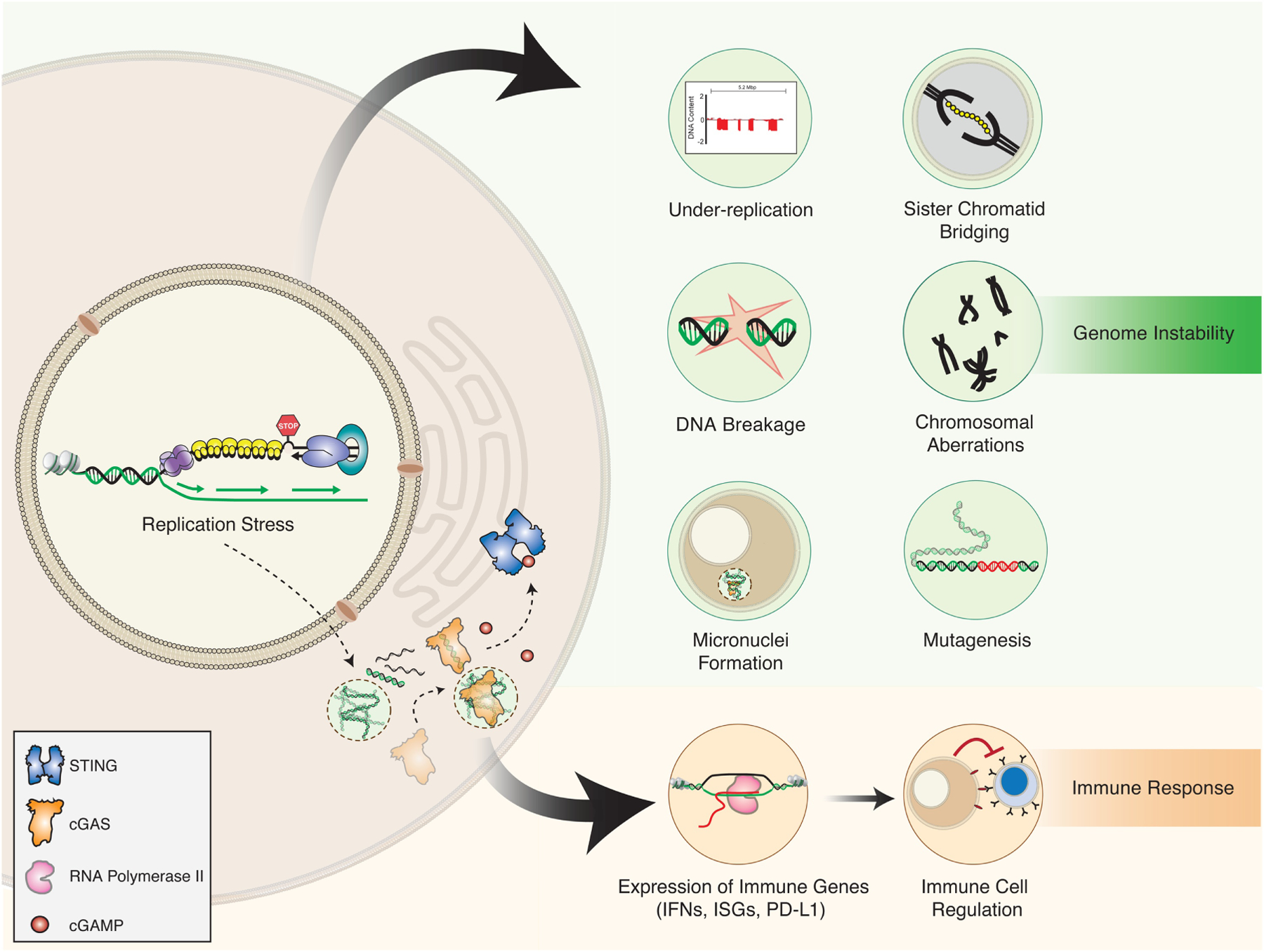
Failure to complete DNA replication due to replication stress results in under-replication and sister-chromatid bridge formation. If the bridges are not resolved during anaphase, the sister chromatids remain attached to each other, resulting in chromosome mis-segregation, lagging chromosome formation, or chromosome breakage. Chromosomal breakage and mis-segregation results in the formation of chromosomal aberrations. Chromosomal fragments or lagging chromosomes that fail to be incorporated into the daughter cell nucleus form micronuclei. Errors during DNA synthesis and DNA repair processes can result in the inaccurate duplication of the genome and mutagenesis. Cytoplasmic DNA fragments and micronuclei originating upon replication stress may be recognized by the cGAS-STING pathway, leading to the induction of interferons, immune stimulated genes (ISGs), and immune checkpoint factors (e.g., PD-L1). PD-L1 expression inhibits immune cell surveillance, leading to immune evasion and immunosuppression.
Failure to resume DNA synthesis after fork stalling/collapse leads to the accumulation of under-replicated regions and DNA breakage [169, 170] (Figure 3). Accordingly, difficult to replicate regions, such as common fragile sites, telomeres, and centromeres, are prone to the formation of sister chromatid bridges induced by replication stress [170]. The SNF2-family ERCC6L (PICH) DNA translocase is involved in the resolution of sister chromatid bridges due to under-replication, and its loss leads to micronuclei, chromosomal aberrations and cell death [171–173]. Overall, these findings indicate that proper processing of stalled/collapsed forks is critical for preventing genomic instability.
3.2. The role of fork remodeling in genetic disorders and cancer
Mutations in SNF2-family fork remodelers have been associated with human disease (Table 1). In particular, biallelic mutations in SMARCAL1 cause the autosomal recessive disorder Schimke immuno-osseous dysplasia (SIOD), which is characterized predominantly by spondyloepiphyseal dysplasia, growth retardation, T-cell immunodeficiency and renal failure [174–178]. More recently, mutations in ZRANB3 have been associated with an African-specific type-2 diabetes, implicating ZRANB3 in glucose response and insulin secretion in pancreatic β-cells [179]. It remains, however, to be determined whether these disorders result from deficiencies in fork remodeling or alterations of other biological processes (e.g., transcription) controlled by SMARCAL1 and ZRANB3 (Table 1).
Consistent with the role for DDR factors in suppressing tumor development [17, 180], loss-of-function mutations and down-regulation of SNF2-family members have been described in several human cancers (Table 1). HLTF is frequently silenced by promoter methylation in human colorectal and gastric cancers [181, 182]. Interestingly, while Hltf loss is insufficient to induce cancer development in mice, it cooperates with Apc deficiency to enhance colon cancer formation [183]. Paradoxically, HLTF is also commonly amplified and overexpressed in several cancer types, including esophageal, uterine and various types of squamous cell carcinoma [184]. HLTF overexpression is also associated with increased cancer metastatic potential and poor prognosis in non-small cell lung cancer patients [185]. While the precise function of HLTF amplification in cancer has not been fully investigated, studies conducted in yeast have shown that overexpression of Rad5, the HLTF yeast ortholog, induces replication fork instability [184], in line with the observation that alterations in the levels of fork remodelers cause DNA damage [103, 122]. Additional studies have reported a role for ZRANB3 as a putative tumor suppressor in endometrial cancer [186], whereas SMARCAL1 mutations have been described in a subtype of glioblastoma carrying wildtype TERT promoter and IDH1/2 genes and exhibiting alternative lengthening of telomeres (ALT) [187]. ATRX has also been found mutated in ALT-positive cancers, including gliomas, neuroendocrine tumors and various sarcomas [188–192]. Furthermore, CHD4 mutations are associated with serous endometrial carcinomas and ERCC6L is significantly mutated in breast and kidney cancer [193–195].
In agreement with their role in maintaining fork stability in response to replication stress, SMARCAL1 and ZRANB3 promote resistance to chemotherapeutic agents that induce replication stress (e.g., DNA crosslinking and methylating agents, topoisomerase inhibitors) in human and mouse cancer cell lines [91, 100, 101, 103, 108, 109] (Table 1). Distinct from SMARCAL1 or ZRANB3 loss, HLTF deficiency in the osteosarcoma U2OS cell line causes resistance to replication stress-inducing agents, such as HU and mitomycin C [90]. Accordingly, while loss of SMARCAL1 or ZRANB3 induces the accumulation of DNA damage and formation of DSBs after replication stress, HLTF deficiency results in decreased DSB induction and reduced DNA damage checkpoint signaling upon HU treatment, possibly contributing to the observed proliferative advantage of HLTF-deficient cells under replication stress [86, 90, 93, 100, 103, 109]. Based on these considerations, small-molecule inhibitors of SMARCAL1 and/or ZRANB3 could provide novel anti-cancer therapies. Inhibition of SMARCAL1 and/or ZRANB3 could be particularly effective in tumors with elevated replication stress induced by oncogenes. It was indeed shown that loss of Smarcal1 or Zranb3 increases replication stress and cell death induced by c-Myc overexpression and that Zranb3 deficiency reduces Myc-driven B-cell lymphomagenesis [196] (Table 1). In addition, loss of Smarcal1 was shown to reduce lymphomagenesis induced by ionizing radiation [197].
Distinct from the above observations, loss of SMARCAL1 or ZRANB3 in BRCA1/2-deficient mammary epithelial cells restores fork protection and reduces DNA damage and DNA break formation without re-establishing HR [89, 159] (Table 1). Similar findings were also obtained for HLTF depletion in BRCA1/2-deficient cells [89]. However, loss of SMARCAL1 does not restore cellular viability in BRCA1/2-deficient mammary epithelial cells upon treatment with replication stress-inducing agents [89], consistent with the observation that re-establishment of fork protection is not sufficient to suppress the growth defect exhibited by BRCA2-deficient mammary epithelial cells [159]. Notably, loss of fork protection is not sufficient to cause BRCA1/BARD1-deficient tumors [144]. It remains, however, to be investigated whether loss of fork protection is necessary for BRCA1/2-mutant tumor development. In such a scenario, re-establishment of fork protection through inhibition of SNF2-family fork remodelers might lead to a reduction of BRCA1/2-mutant tumor formation.
In contrast with the above findings in mammary epithelial cells, re-establishment of fork protection in BRCA1/2-deficient tumors has been associated with resistance to chemotherapeutic agents [89, 127, 167, 198]. In particular, loss of SMARCAL1 has been shown to restore chemoresistance in BRCA1-deficient breast cancer cells [89] (Table 1). Similar observations have been reported for CHD4, whose lower expression levels significantly correlate with poorer prognosis in patients with BRCA2-mutant tumors [198, 199]. Chemoresistance in BRCA1/2-deficient tumors can also occur as a result of the restoration of BRCA1/2 functionality, the re-establishment of HR-mediated DSB repair or the activation of compensatory DNA repair pathways in response to replication stress [200]. Recent studies have shown that the restart of stalled forks by PRIMPOL-mediated repriming is an alternative to SMARCAL1- or HLTF-dependent fork reversal [90, 201]. Remarkably, BRCA1-mutant ovarian cancer cells adapt to recurrent cisplatin treatments by activating fork repriming to avoid excessive nascent DNA degradation due to fork remodeling at the expense of ssDNA gap formation [201]. Besides fork reversal and repriming, TLS provides an additional mechanism to restart stalled forks. Notably, REV1-mediated TLS was recently shown to operate as an alternative pathway to HLTF-dependent fork reversal [90]. Furthermore, aberrant TLS activation induced by FANCJ deficiency was reported to reduce fork reversal and facilitate continuous DNA synthesis in the presence of HU, thus avoiding ssDNA gap formation due to repriming events [202]. ssDNA gaps formed following fork repriming can undergo resection and RAD51-mediated HR for post-replicative repair [203]. TLS can function alternatively to HR for post-replicative repair of ssDNA gaps generated during replication stress [156], and may therefore become essential in HR-deficient cells. Interestingly, genomic sequencing studies and analysis of mutational signatures upon treatment with genotoxic agents suggest that TLS is upregulated in BRCA1/2-deficient cells [204]. Understanding the complex interplay between fork reversal, repriming, TLS and HR in response to replication stress will be of paramount importance for developing novel cancer therapies.
3.3. Fork instability, innate immunity activation, and cancer immunotherapy
Innate immunity is an evolutionary conserved cell-intrinsic reaction to microbial pathogens, which involves multiple mechanisms, and sensor molecules. Cytosolic DNA sensing recognizes and reacts to foreign DNA found in the cytoplasm (e.g., viral DNA), leading to immune responses. The cGAMP synthase (cGAS)–stimulator of interferon genes (STING) pathway has been identified as a critical component of DNA sensing [205]. Briefly, cytosolic DNA sensed by cGAS activates STING, which triggers the expression of interferon-stimulated genes (ISGs) and induces the inflammatory response [205, 206] (Figure 3).
Recent studies have shown that unresolved DNA damage derived from replication fork instability, telomere fragility, common fragile site expression and chromosome mis-segregation often results in the formation of micronuclei upon mitotic progression, which can be detected by cGAS, thus leading to STING activation and the induction of innate immune signaling [207–209]. Accordingly, micronuclei formed as a result of deficiency in BRCA1/2 or RAD51 activate innate immune signaling, and this response is exacerbated by PARP inhibitor treatment [210–214]. Notably, loss of the SNF2-family members SMARCAL1 and ERCC6L has been associated with an increase of micronuclei formation [172, 215]. However, it remains to be determined whether deficiency of these factors leads to innate immunity induction.
In addition to micronuclei, short species of genomic DNA released into the cytoplasm during DNA replication and repair can also be recognized by cGAS, thereby triggering the activation of innate immune signaling. In particular, it was shown that cytoplasmic ssDNA derived from replication fork intermediates following HU treatment is degraded by TREX1 [216], a nuclease that suppresses innate immune signaling and maintains immune tolerance [217, 218], thus providing direct evidence that processing of stalled forks can generate cytoplasmic DNA and activate innate immunity. Additional studies have shown that the innate immune response is activated also by cytoplasmic ssDNA generated upon treatment with other replication stress-inducing agents, such as mitomycin C and cisplatin, or in response to IR [219]. Of note, depletion of nucleases such as DNA2 and EXO1, but not MRE11 and CtIP, restrains innate immune signaling, suggesting that ssDNA molecules that induce innate immunity require extensive DNA-end resection activities [219]. In addition, loss of SAMHD1, a factor that stimulates the exonuclease activity of MRE11 at stalled forks, was shown to prevent nascent DNA degradation and stimulate the helicase and endonuclease activities of RECQ1 and MRE11, respectively, thus resulting in the accumulation of cytosolic ssDNA fragments and the subsequent activation of the cGAS-STING pathway [220]. Cytoplasmic ssDNA fragments deriving from stalled replication intermediates and cGAS-STING-dependent innate immune signaling have also been observed in BRCA1-deficient breast cancer cells, although it remains unclear whether this phenotype is dependent on defective fork protection, as in the case of RAD51-depleted cells [212, 221]. Interestingly, it was reported that prostate cancer cells accumulate cytoplasmic dsDNA derived from nascent DNA in a manner dependent on MUS81, suggesting that it may originate from the cleavage of stalled forks [222]. Further studies are required to understand the relationship of fork remodeling with cytosolic DNA sensing and inflammation and evaluate whether inhibition of SMARCAL1, ZRANB3 and HLTF may stimulate the cGAS-STING pathway. Overall, the above findings demonstrate that replication fork instability leads to aberrant inflammatory response in both malignant and non-malignant cells (Figure 3).
cGAS-STING-dependent innate immune signaling enhances tumor immunogenicity, and consequently the response of tumors to immunotherapies, such as immune checkpoint blockade (ICB) treatments that use antibodies inhibiting the immune checkpoint factors PD-1, PD-L1 and CTLA-4 [223–228]. During oncogenesis, PD-L1 expressed on tumor cells restricts T cell-mediated anti-tumor immunity by binding to the PD-1 receptor on tumor-infiltrating T cells, thus promoting tumor development [229–231]. In the context of ICB therapy, higher levels of PD-L1 on tumor cells correlate with better response to PD-1/PD-L1 antibody-mediated immunotherapy [232]. Notably, the expression of PD-L1 depends upon STING activation and is induced in response to replication stress [221]. STING activation following replication stress can also result in the expression of CXCL10, a chemo-attractive cytokine that correlates with high tumor-infiltrating cytotoxic T cells [210], as well as type-I interferon signaling, which promotes anti-tumor immunity [233–235]. However, recent studies have also shown that activation of the cGAS-STING pathway in chromosomally unstable tumors drives metastasis [236]. Understanding the connection between replication fork instability and the cGAS-STING pathway will therefore be critical for defining underlying mechanisms of anti-tumor immunity and metastasis, as well as predicting the response to ICB therapy in cancer patients (Figure 3).
4. Technologies to define the function of SNF2-family members in replication fork metabolism, genome integrity and human disease
4.1. Methodologies to study replication fork transactions: current limitations and novel approaches
Our ability to study replication fork transactions is challenged by technological limitations. The current gold-standard assay used to study replication fork dynamics in response to replication stress relies on DNA fiber and combing techniques [237–244] (Figure 4). These techniques utilize thymidine analogs to label the progression of replication forks, followed by the stretching of the DNA onto a glass coverslip to obtain DNA fibers with a resolution limit of 1 μm, corresponding to 2–4 kb [238, 239, 243–245]. In addition to having resolution constraints that do not enable the visualization of fork structures, DNA fiber/combing techniques are unable to discriminate between leading and lagging strand-dependent DNA synthesis. Unlike DNA fiber techniques, EM enables the visualization and quantification of replication fork structures with a resolution of ~30–50 base pairs, thereby permitting the discrimination of ssDNA gaps, reversed forks and other fork intermediates [83, 155, 246–248]. Despite its high resolution, EM only provides a snapshot of replication intermediates captured at a single time point. Moreover, both EM-based approaches and DNA fiber/combing techniques lack the ability to visualize proteins associated with distinct replication fork intermediates. Current approaches to detect proteins associated with replication forks rely primarily on the iPOND technology, which enables the identification of proteins bound to nascent DNA by western blotting or mass spectrometry [102, 249] (Figure 4). Although highly sensitive for identifying fork-associated proteins, iPOND technologies do not provide spatial information on the location of the identified factors. Recent studies led to the development of proximity ligation assay (PLA)-based approaches that allow the detection of proteins on nascent DNA in response to replication stress using standard microscopy [89, 250, 251]. To enhance signal detection, PLA-based applications can be combined with super-resolution imaging technologies, which have been shown to enable the mapping of chromatin-associated proteins around a replicating area [136, 252]. Future developments of super-resolution imaging may allow the direct visualization of reversed forks and other replication fork transactions, thus enabling the study of replication fork dynamics through live imaging-based analyses. To gain additional mechanistic insights into replication fork transactions, cell-based assays can be coupled to biochemical assays that employ Xenopus egg extracts [50, 253–255] or purified replication factors [9, 256, 257] to study replication fork progression in vitro with and without the presence of fork barriers. Alternative biochemical assays can be utilized to monitor the enzymatic activities of recombinant DNA replication or repair factors on fork-like substrates [91, 93, 94] and define DNA transactions using single-molecule technologies, such as DNA curtains [258–260] or optical tweezers [261–263] (Figure 4). These approaches will contribute to further define the function of DNA replication and repair factors in replication fork metabolism.
Figure 4. Methods for studying replication fork transactions.
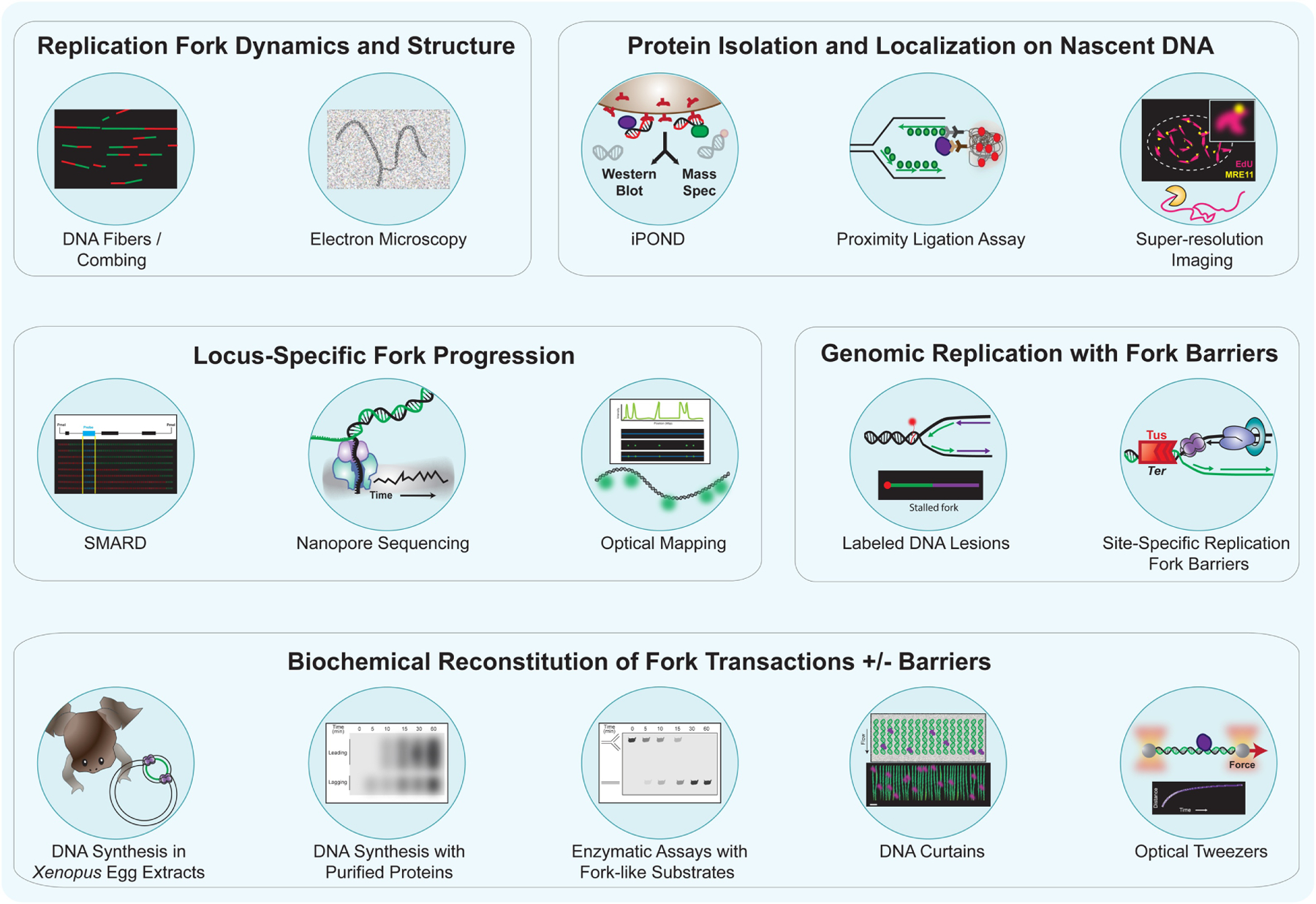
Overview of the current techniques used to (1) monitor replication fork dynamics and visualize replication fork structures, (2) identify proteins associated with and localized on nascent DNA, (3) analyze locus-specific replication fork progression, (4) monitor genomic replication at fork barriers, and (5) reconstitute in vitro replication fork transactions with and without the presence of barriers. DNA fiber and DNA combing techniques enable the analysis of the progression of individual replication forks, while electron microscopy allows high-resolution detection of DNA intermediates and replication fork structures, such as reversed forks and ssDNA gaps. Protein identification and localization on newly synthesized DNA can be obtained by iPOND, PLA-based approaches, and super-resolution microscopy methods, such as STORM. Techniques, such as SMARD, nanopore-based sequencing methods (D-NAscent, Fork-seq), and optical mapping enable the study of replication fork dynamics at genomic loci of interest. DNA lesions labeled with chemically modified adducts and/or antibodies or site-specific replication fork barriers, such as the E. coli Tus-Ter system, allow the analysis of events occurring when replication forks encounter obstacles. In vitro studies of replication fork transactions can be conducted using Xenopus egg extracts, which enable the study of replication fork progression, with and without fork barriers. Similar studies can be performed using purified replication factors that reconstitute DNA synthesis in vitro. Purified proteins can also be utilized to detect enzymatic activities on fork-like substrates or define DNA transactions at single-molecule level using DNA curtains or optical tweezers technologies combined with fluorescence microscopy.
Standard DNA fiber/combing assays can discern global changes in fork progression, but lack the ability to detect alterations caused by specific genomic contexts. To overcome this limitation, DNA combing techniques have been combined with fluorescence in situ hybridization to monitor replication fork progression at single loci using SMARD (single molecule analysis of the replicated DNA) [264–266] (Figure 4). SMARD allows the study of a limited number of genomic loci at a time. More recently, the use of optical mapping technologies have enabled the mapping of fluorescently-labeled replication tracts to genomic sites in human cells and Xenopus egg extracts [267, 268]. In addition, nanopore sequencing-based approaches, such as D-NAscent or FORK-seq, have been shown to discriminate thymidine analogue incorporation and identify DNA replication sites in yeast and mouse pluripotent stem cells [269–271]. We expect that further developments of optical mapping and nanopore sequencing technologies will enable genome-wide detection of site-specific replication events under normal or replication stress conditions in human cells.
Classical DNA fiber/combing assays are unable to discern the presence of DNA lesions on replication tracts, thus hindering the study of DNA lesion bypass and repair events. Recent studies have utilized digoxigenin-labeled psoralen to visualize DNA crosslinks on DNA fibers and study the bypass of those lesions [51] (Figure 4). Similar approaches might be possible using antibodies that detect DNA lesions (e.g., antibodies that recognize cisplatin-induced DNA lesions). Alternative methods employing the E.coli Tus-Ter system have been utilized in yeast and mammalian cells to study DNA repair events occurring at forks stalled by a single, site-specific replication fork barrier [272, 273]. More recently, a nuclease-dead Cas9 has been demonstrated to be a programmable barrier for replication fork progression [274]. Combining site-specific fork barriers with PLA and next-generation sequencing may enable the discrimination of proteins recruited to a single stalled fork and the detection of repair events induced by fork blockage. Ultimately, the development of new techniques to study replication dynamics will provide important insights into the mechanisms that control replication fork reversal and other fork transactions.
4.2. CRISPR-based technologies to investigate the contribution of SNF2-family members to genome stability and human disease
The mapping of genetic interactors of SNF2-family members can provide novel insight into their contribution to genome stability and other biological processes, as well as identify potential therapeutic options to treat SNF2-associated diseases. Epistatic relationships identified by genome-wide genetic interaction screens in SMARCAL1- or ZRANB3-deficient cells may potentially uncover new factors that promote fork reversal and/or identify biological pathways relevant to the suppression of multisystem developmental diseases, such as SIOD and diabetes. Furthermore, these same screens may also identify buffering interactions from which we could infer potential therapies to ameliorate disease symptoms, such as T-cell immunodeficiency in SIOD [275]. The identification of synthetic lethal partners of SNF2-family members may also provide supporting rationale for precision medicine in cancer treatment. For example, identifying synthetic lethal partners of ZRANB3 and HLTF may offer novel targets to treat endometrial and colorectal cancers, respectively [181, 186]. Moreover, identifying novel domains, separation-of-function mutations, and regulatory elements of the above SNF2-family fork remodelers may facilitate the classification of pathogenic mutations that contribute to human disease etiology.
The development of CRISPR-based technologies [276] has produced an expansive toolbox of genome editing reagents to define genetic interactions. Mapping of the genetic interactions of the above SNF2-family members can be achieved through combinatorial CRISPR-based screening in a pooled format [277–282]. Beyond cellular viability in response to replication stress-inducing agents as a readout of genomic stability, recent advances in the application of single-cell analyses to pooled CRISPR-Cas9-based screens have enabled the acquisition of richer content for deeper phenotypic characterization of DNA replication and repair processes and more refined definition of genetic interactions. For example, in situ sequencing of single guide RNAs (sgRNAs) or their corresponding barcodes in intact cells [283, 284] enables the use of cellular markers of replication stress and genome instability in pooled CRISPR screens. These markers can include replication stress-associated γH2AX and RPA foci, 53BP1 nuclear bodies, micronuclei, and DNA synthesis events in response to replication stress. Further advancements to CRISPR screening technologies may also allow the coupling of genetic screens with methods that monitor the association of proteins with nascent DNA, such as PLA-based assays [89, 250, 251]. As an alternative to these approaches, characterization of genetic interactions established by SNF2-family members can also be obtained by analyzing the transcriptional and chromatin state resulting from the deletion of SNF2-family members and their potential genetic interactors under different cellular conditions [285, 286]. These relationships can be elucidated by coupling CRISPR screening with single-cell RNA-seq (Perturb-seq, CROP-seq) and chromatin accessibility analyses (Perturb-ATAC) [285–288].
CRISPR-based technologies can also identify novel structure-function relationships of SNF2-family members beyond known domains that mediate nucleic acid-binding, protein-protein interactions or catalytic activity. To that end, the use of high-throughput saturation mutagenesis screens carries the potential to uncover novel functional domains, regulatory elements and separation-of-function mutations, as well as classify the pathogenicity of variants that occur in the human population. CRISPR-dependent base editing, and the recently developed prime editing are new technologies to study single nucleotide variants that have yet to be applied to systematic screening in human cells [289–291], but a saturation mutagenesis screen has already been conducted for BRCA1 using CRISPR-mediated HDR to introduce 4000 variants into 13 coding exons [292]. This proof-of-concept study identified hundreds of novel non-functional variants of BRCA1 and allowed for the potential to predict pathogenicity of variants that were previously of unknown significance. Application of these technologies to the SNF2-family members is similarly expected to produce a rich resource of novel structure-function relationships.
5. Conclusions and future perspectives
Members of the SNF2-family have been implicated in maintaining genomic stability in response to replication stress and regulating the development of human diseases. Future studies will better define the interplay among SNF2-family members at the replication fork and the interconnection between fork reversal and chromatin remodeling. The development of new techniques to study replication fork dynamics will help refine our understanding of these and other aspects of replication fork stability. Given the multiple roles of SNF2-family members in DNA metabolism, CRISPR screens will play an important role in defining the precise contributions of those factors to genomic stability, as well as their relationship with different DDR factors and pathways. Further studies will elucidate the contribution of SNF2-family fork remodelers to tumor development and the response to chemotherapy and immunotherapy. Ultimately, the development of small molecules able to efficiently inhibit the activity of specific SNF2-family fork remodelers may provide new cancer therapies and increase the therapeutic opportunities for cancer patients.
Acknowledgements
The authors would like to thank all the members of the Ciccia lab for their helpful advice and thoughtful discussions. We apologize for any work that could not be included or cited due to space constraints. This work was supported by the NIH grants R01CA197774, R01CA227450 and R01GM117064 to A.C., NSF GRFP Fellowship (DGE-16-44869) to S.A.J., NIH T32CA009503 to J.W.H., the Italian Association for Cancer Research (AIRC) post-doctoral research fellowship to G.L., and the European Molecular Biology Organisation (EMBO) Long-Term Fellowship (ALTF 366-2019) to R.C.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Fouad YA and Aanei C, Revisiting the hallmarks of cancer. Am J Cancer Res, 2017. 7(5): p. 1016–1036. [PMC free article] [PubMed] [Google Scholar]
- [2].Macheret M and Halazonetis TD, DNA replication stress as a hallmark of cancer. Annu Rev Pathol, 2015. 10: p. 425–48. [DOI] [PubMed] [Google Scholar]
- [3].Negrini S, Gorgoulis VG, and Halazonetis TD, Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol, 2010. 11(3): p. 220–8. [DOI] [PubMed] [Google Scholar]
- [4].Cortez D, Preventing replication fork collapse to maintain genome integrity. DNA Repair (Amst), 2015. 32: p. 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mirkin EV and Mirkin SM, Replication fork stalling at natural impediments. Microbiol Mol Biol Rev, 2007. 71(1): p. 13–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Magdalou I, et al. , The causes of replication stress and their consequences on genome stability and cell fate. Semin Cell Dev Biol, 2014. 30: p. 154–64. [DOI] [PubMed] [Google Scholar]
- [7].Lambert S and Carr AM, Impediments to replication fork movement: stabilisation, reactivation and genome instability. Chromosoma, 2013. 122(1–2): p. 33–45. [DOI] [PubMed] [Google Scholar]
- [8].Vesela E, et al. , Common Chemical Inductors of Replication Stress: Focus on Cell-Based Studies. Biomolecules, 2017. 7(1). [Google Scholar]
- [9].Taylor MRG and Yeeles JTP, The Initial Response of a Eukaryotic Replisome to DNA Damage. Mol Cell, 2018. 70(6): p. 1067–1080 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kose HB, et al. , Dynamics of the Eukaryotic Replicative Helicase at Lagging-Strand Protein Barriers Support the Steric Exclusion Model. Cell Rep, 2019. 26(8): p. 2113–2125 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Taylor MRG and Yeeles JTP, Dynamics of Replication Fork Progression Following Helicase-Polymerase Uncoupling in Eukaryotes. J Mol Biol, 2019. 431(10): p. 2040–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lopes M, Foiani M, and Sogo JM, Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell, 2006. 21(1): p. 15–27. [DOI] [PubMed] [Google Scholar]
- [13].Byun TS, et al. , Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev, 2005. 19(9): p. 1040–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zou L and Elledge SJ, Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science, 2003. 300: p. 1542–1548. [DOI] [PubMed] [Google Scholar]
- [15].Cimprich KA and Cortez D, ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol, 2008. 9(8): p. 616–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Flynn RL and Zou L, ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem Sci, 2011. 36(3): p. 133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ciccia A and Elledge SJ, The DNA damage response: making it safe to play with knives. Mol Cell, 2010. 40(2): p. 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Neelsen KJ and Lopes M, Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol, 2015. 16(4): p. 207–20. [DOI] [PubMed] [Google Scholar]
- [19].Goodman MF and Woodgate R, Translesion DNA polymerases. Cold Spring Harb Perspect Biol, 2013. 5(10): p. a010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cortez D, Replication-Coupled DNA Repair. Mol Cell, 2019. 74(5): p. 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Blow JJ, Ge XQ, and Jackson DA, How dormant origins promote complete genome replication. Trends Biochem Sci, 2011. 36(8): p. 405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ulrich HD and Walden H, Ubiquitin signalling in DNA replication and repair. Nat Rev Mol Cell Biol, 2010. 11(7): p. 479–89. [DOI] [PubMed] [Google Scholar]
- [23].Sale JE, Lehmann AR, and Woodgate R, Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol, 2012. 13(3): p. 141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bienko M, et al. , Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science, 2005. 310(5755): p. 1821–4. [DOI] [PubMed] [Google Scholar]
- [25].Tellier-Lebegue C, et al. , The translesion DNA polymerases Pol zeta and Rev1 are activated independently of PCNA ubiquitination upon UV radiation in mutants of DNA polymerase delta. PLoS Genet, 2017. 13(12): p. e1007119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kannouche PL, Wing J, and Lehmann AR, Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell, 2004. 14(4): p. 491–500. [DOI] [PubMed] [Google Scholar]
- [27].Haracska L, Prakash L, and Prakash S, Role of human DNA polymerase kappa as an extender in translesion synthesis. Proc Natl Acad Sci U S A, 2002. 99(25): p. 16000–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Johnson RE, et al. , Role of DNA polymerase eta in the bypass of a (6–4) TT photoproduct. Mol Cell Biol, 2001. 21(10): p. 3558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Garg P and Burgers PM, Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases eta and REV1. Proc Natl Acad Sci U S A, 2005. 102(51): p. 18361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Furrer A and van Loon B, Handling the 3-methylcytosine lesion by six human DNA polymerases members of the B-, X- and Y-families. Nucleic Acids Res, 2014. 42(1): p. 553–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shachar S, et al. , Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J, 2009. 28(4): p. 383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yang W and Gao Y, Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu Rev Biochem, 2018. 87: p. 239–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Johnson RE, et al. , Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature, 2000. 406: p. 1015–1019. [DOI] [PubMed] [Google Scholar]
- [34].Livneh Z, Ziv O, and Shachar S, Multiple two-polymerase mechanisms in mammalian translesion DNA synthesis. Cell Cycle, 2010. 9(4): p. 729–35. [DOI] [PubMed] [Google Scholar]
- [35].Guilliam TA and Yeeles JTP, Reconstitution of translesion synthesis reveals a mechanism of eukaryotic DNA replication restart. Nat Struct Mol Biol, 2020. 27(5): p. 450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mouron S, et al. , Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol, 2013. 20(12): p. 1383–9. [DOI] [PubMed] [Google Scholar]
- [37].Bianchi J, et al. , PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol Cell, 2013. 52(4): p. 566–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Garcia-Gomez S, et al. , PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell, 2013. 52(4): p. 541–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Guilliam TA, et al. , Human PrimPol is a highly error-prone polymerase regulated by single-stranded DNA binding proteins. Nucleic Acids Res, 2015. 43(2): p. 1056–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wan L, et al. , hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep, 2013. 14(12): p. 1104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Keen BA, et al. , Molecular dissection of the domain architecture and catalytic activities of human PrimPol. Nucleic Acids Res, 2014. 42(9): p. 5830–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schiavone D, et al. , PrimPol Is Required for Replicative Tolerance of G Quadruplexes in Vertebrate Cells. Mol Cell, 2016. 61(1): p. 161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kobayashi K, et al. , Repriming by PrimPol is critical for DNA replication restart downstream of lesions and chain-terminating nucleosides. Cell Cycle, 2016. 15(15): p. 1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Guilliam TA, et al. , PolDIP2 interacts with human PrimPol and enhances its DNA polymerase activities. Nucleic Acids Res, 2016. 44(7): p. 3317–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lai Y, et al. , Measurement of Endogenous versus Exogenous Formaldehyde-Induced DNA-Protein Crosslinks in Animal Tissues by Stable Isotope Labeling and Ultrasensitive Mass Spectrometry. Cancer Res, 2016. 76(9): p. 2652–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Molnar G, et al. , Quantification of DNA-protein interaction by UV crosslinking. Nucleic Acids Res, 1995. 23(16): p. 3318–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chvalova K, Brabec V, and Kasparkova J, Mechanism of the formation of DNA-protein cross-links by antitumor cisplatin. Nucleic Acids Res, 2007. 35(6): p. 1812–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Langevin F, et al. , Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature, 2011. 475(7354): p. 53–8. [DOI] [PubMed] [Google Scholar]
- [49].Sparks JL, et al. , The CMG Helicase Bypasses DNA-Protein Cross-Links to Facilitate Their Repair. Cell, 2019. 176(1–2): p. 167–181 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Raschle M, et al. , Mechanism of replication-coupled DNA interstrand crosslink repair. Cell, 2008. 134(6): p. 969–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Huang J, et al. , The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell, 2013. 52(3): p. 434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rohleder F, et al. , FANCM interacts with PCNA to promote replication traverse of DNA interstrand crosslinks. Nucleic Acids Res, 2016. 44(7): p. 3219–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ling C, et al. , Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discov, 2016. 2: p. 16047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Huang J, et al. , Remodeling of Interstrand Crosslink Proximal Replisomes Is Dependent on ATR, FANCM, and FANCD2. Cell Rep, 2019. 27(6): p. 1794–1808 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Li N, et al. , Cooperation of the NEIL3 and Fanconi anemia/BRCA pathways in interstrand crosslink repair. Nucleic Acids Res, 2020. 48(6): p. 3014–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wu RA, et al. , TRAIP is a master regulator of DNA interstrand crosslink repair. Nature, 2019. 567(7747): p. 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Semlow DR, et al. , Replication-Dependent Unhooking of DNA Interstrand Cross-Links by the NEIL3 Glycosylase. Cell, 2016. 167(2): p. 498–511 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Niraj J, Farkkila A, and D’Andrea AD, The Fanconi Anemia Pathway in Cancer. Annu Rev Cancer Biol, 2019. 3: p. 457–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhang J and Walter JC, Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair (Amst), 2014. 19: p. 135–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Knipscheer P, et al. , The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science, 2009. 326(5960): p. 1698–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ciccia A, McDonald N, and West SC, Structural and functional relationships of the XPF/MUS81 family of proteins. Annu Rev Biochem, 2008. 77: p. 259–87. [DOI] [PubMed] [Google Scholar]
- [62].Dehe PM and Gaillard PHL, Control of structure-specific endonucleases to maintain genome stability. Nat Rev Mol Cell Biol, 2017. 18(5): p. 315–330. [DOI] [PubMed] [Google Scholar]
- [63].Pasero P and Vindigni A, Nucleases Acting at Stalled Forks: How to Reboot the Replication Program with a Few Shortcuts. Annu Rev Genet, 2017. 51: p. 477–499. [DOI] [PubMed] [Google Scholar]
- [64].Natsume T, et al. , Acute inactivation of the replicative helicase in human cells triggers MCM8–9-dependent DNA synthesis. Genes Dev, 2017. 31(8): p. 816–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hustedt N, et al. , Control of homologous recombination by the HROB-MCM8-MCM9 pathway. Genes Dev, 2019. 33(19–20): p. 1397–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Long DT, et al. , Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science, 2011. 333(6038): p. 84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Huang J-W, et al. , MCM8IP activates the MCM8–9 helicase to promote DNA synthesis and homologous recombination upon DNA damage. Nature Communications, 2020. 11(1): p. 2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bhowmick R, Minocherhomji S, and Hickson ID, RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol Cell, 2016. 64(6): p. 1117–1126. [DOI] [PubMed] [Google Scholar]
- [69].Costantino L, et al. , Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science, 2014. 343(6166): p. 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ciccia A and Symington LS, Stressing Out About RAD52. Mol Cell, 2016. 64(6): p. 1017–1019. [DOI] [PubMed] [Google Scholar]
- [71].Donnianni RA and Symington LS, Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci U S A, 2013. 110(33): p. 13475–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Donnianni RA, et al. , DNA Polymerase Delta Synthesizes Both Strands during Break-Induced Replication. Mol Cell, 2019. 76(3): p. 371–381 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Elango R, et al. , Investigation of Break-Induced Replication in Yeast. Methods Enzymol, 2018. 601: p. 161–203. [DOI] [PubMed] [Google Scholar]
- [74].Anand RP, Lovett ST, and Haber JE, Break-induced DNA replication. Cold Spring Harb Perspect Biol, 2013. 5(12): p. a010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Llorente B, Smith CE, and Symington LS, Break-induced replication: what is it and what is it for? Cell Cycle, 2008. 7(7): p. 859–64. [DOI] [PubMed] [Google Scholar]
- [76].Wilson MA, et al. , Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature, 2013. 502(7471): p. 393–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Dilley RL, et al. , Break-induced telomere synthesis underlies alternative telomere maintenance. Nature, 2016. 539(7627): p. 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Sotiriou SK, et al. , Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol Cell, 2016. 64(6): p. 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ulrich HD, Timing and spacing of ubiquitin-dependent DNA damage bypass. FEBS Lett, 2011. 585(18): p. 2861–7. [DOI] [PubMed] [Google Scholar]
- [80].Bhat KP and Cortez D, RPA and RAD51: fork reversal, fork protection, and genome stability. Nat Struct Mol Biol, 2018. 25(6): p. 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Quinet A, Lemacon D, and Vindigni A, Replication Fork Reversal: Players and Guardians. Mol Cell, 2017. 68(5): p. 830–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Mutreja K, et al. , ATR-Mediated Global Fork Slowing and Reversal Assist Fork Traverse and Prevent Chromosomal Breakage at DNA Interstrand Cross-Links. Cell Rep, 2018. 24(10): p. 2629–2642 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zellweger R, et al. , Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol, 2015. 208(5): p. 563–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ray Chaudhuri A, et al. , Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol, 2012. 19(4): p. 417–23. [DOI] [PubMed] [Google Scholar]
- [85].Ahuja AK, et al. , A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nat Commun, 2016. 7: p. 10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Vujanovic M, et al. , Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol Cell, 2017. 67(5): p. 882–890 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kolinjivadi AM, et al. , Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell, 2017. 67(5): p. 867–881 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Berti M, et al. , Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol, 2013. 20(3): p. 347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Taglialatela A, et al. , Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell, 2017. 68(2): p. 414–430 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Bai G, et al. , HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis, Mol Cell 78 (6) (2020) 1237–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Ciccia A, et al. , Polyubiquitinated PCNA Recruits the ZRANB3 Translocase to Maintain Genomic Integrity after Replication Stress. Mol Cell, 2012. 47(3): p. 396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Betous R, et al. , Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep, 2013. 3(6): p. 1958–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Betous R, et al. , SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev, 2012. 26(2): p. 151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Blastyak A, et al. , Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol Cell Biol, 2010. 30(3): p. 684–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Fugger K, et al. , FBH1 Catalyzes Regression of Stalled Replication Forks, Cell Rep 10 (10) (2015) 1749–1757. [DOI] [PubMed] [Google Scholar]
- [96].Mijic S, et al. , Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat Commun, 2017. 8(1): p. 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Mason JM, et al. , Non-enzymatic roles of human RAD51 at stalled replication forks. Nat Commun, 2019. 10(1): p. 4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Flaus A, et al. , Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res, 2006. 34(10): p. 2887–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Prasad P, Lennartsson A, and Ekwall K, The roles of SNF2/SWI2 nucleosome remodeling enzymes in blood cell differentiation and leukemia. Biomed Res Int, 2015. 2015: p. 347571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Ciccia A, et al. , The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev, 2009. 23(20): p. 2415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Yuan J, Ghosal G, and Chen J, The annealing helicase HARP protects stalled replication forks. Genes Dev, 2009. 23(20): p. 2394–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Sirbu BM, et al. , Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J Biol Chem, 2013. 288(44): p. 31458–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Bansbach CE, et al. , The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev, 2009. 23(20): p. 2405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Yusufzai T, et al. , The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev, 2009. 23(20): p. 2400–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Postow L, et al. , Identification of SMARCAL1 as a component of the DNA damage response, J Biol Chem 284 (51) (2009) 35951–35961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Mason AC, et al. , A structure-specific nucleic acid-binding domain conserved among DNA repair proteins. Proc Natl Acad Sci U S A, 2014. 111(21): p. 7618–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Bhat KP, Betous R, and Cortez D, High-affinity DNA-binding domains of replication protein A (RPA) direct SMARCAL1-dependent replication fork remodeling. J Biol Chem, 2015. 290(7): p. 4110–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Weston R, Peeters H, and Ahel D, ZRANB3 is a structure-specific ATP-dependent endonuclease involved in replication stress response. Genes Dev, 2012. 26(14): p. 1558–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Yuan J, Ghosal G, and Chen J, The HARP-like Domain-Containing Protein AH2/ZRANB3 Binds to PCNA and Participates in Cellular Response to Replication Stress. Mol Cell, 2012. 47(3): p. 410–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Sebesta M, et al. , Structural insights into the function of ZRANB3 in replication stress response. Nat Commun, 2017. 8: p. 15847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Badu-Nkansah A, et al. , Identification of a Substrate Recognition Domain in the Replication Stress Response Protein Zinc Finger Ran-binding Domain-containing Protein 3 (ZRANB3). J Biol Chem, 2016. 291(15): p. 8251–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Unk I, et al. , Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc Natl Acad Sci U S A, 2008. 105(10): p. 3768–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Motegi A, et al. , Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc Natl Acad Sci U S A, 2008. 105(34): p. 12411–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Achar YJ, Balogh D, and Haracska L, Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc Natl Acad Sci U S A, 2011. 108(34): p. 14073–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Blastyak A, et al. , Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol Cell, 2007. 28(1): p. 167–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Kile AC, et al. , HLTF’s Ancient HIRAN Domain Binds 3’ DNA Ends to Drive Replication Fork Reversal. Mol Cell, 2015. 58(6): p. 1090–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Chavez DA, Greer BH, and Eichman BF, The HIRAN domain of helicase-like transcription factor positions the DNA translocase motor to drive efficient DNA fork regression. J Biol Chem, 2018. 293(22): p. 8484–8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Hishiki A, Sato M, Hashimoto H, Structure of HIRAN domain of human HLTF bound to duplex DNA provides structural basis for DNA unwinding to initiate replication fork regression, J Biochem 167 (6) (2020) 597–602. [DOI] [PubMed] [Google Scholar]
- [119].Hishiki A, et al. , Structure of a Novel DNA-binding Domain of Helicase-like Transcription Factor (HLTF) and Its Functional Implication in DNA Damage Tolerance. J Biol Chem, 2015. 290(21): p. 13215–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Achar YJ, et al. , Human HLTF mediates postreplication repair by its HIRAN domain-dependent replication fork remodelling. Nucleic Acids Res, 2015. 43(21): p. 10277–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Burkovics P, et al. , Strand invasion by HLTF as a mechanism for template switch in fork rescue. Nucleic Acids Res, 2014. 42(3): p. 1711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Poole LA and Cortez D, Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability. Crit Rev Biochem Mol Biol, 2017. 52(6): p. 696–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Cox KE, Marechal A, and Flynn RL, SMARCAL1 Resolves Replication Stress at ALT Telomeres. Cell Rep, 2016. 14(5): p. 1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Poole LA, et al. , SMARCAL1 maintains telomere integrity during DNA replication. Proc Natl Acad Sci U S A, 2015. 112(48): p. 14864–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Zhang T, et al. , Strand break-induced replication fork collapse leads to C-circles, C-overhangs and telomeric recombination. PLoS Genet, 2019. 15(2): p. e1007925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Thakar T, et al. , Ubiquitinated-PCNA protects replication forks from DNA2-mediated degradation by regulating Okazaki fragment maturation and chromatin assembly. Nat Commun, 2020. 11(1): p. 2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Dungrawala H, et al. , RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol Cell, 2017. 67(3): p. 374–386 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Bhat KP, et al. , RADX Modulates RAD51 Activity to Control Replication Fork Protection. Cell Rep, 2018. 24(3): p. 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Couch FB, et al. , ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev, 2013. 27(14): p. 1610–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Neelsen KJ, et al. , Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol, 2013. 200(6): p. 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Lemacon D, et al. , MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat Commun, 2017. 8(1): p. 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Grimme JM, et al. , Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes. Nucleic Acids Res, 2010. 38(9): p. 2917–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Malacaria E, et al. , Rad52 prevents excessive replication fork reversal and protects from nascent strand degradation. Nat Commun, 2019. 10(1): p. 1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Nieminuszczy J, et al. , EXD2 Protects Stressed Replication Forks and Is Required for Cell Viability in the Absence of BRCA1/2. Mol Cell, 2019. 75(3): p. 605–619 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Peng M, et al. , Opposing Roles of FANCJ and HLTF Protect Forks and Restrain Replication during Stress. Cell Rep, 2018. 24(12): p. 3251–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Whelan DR, et al. , Super-resolution visualization of distinct stalled and broken replication fork structures. bioRxiv, 2020: p. 2020.01.19.912014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Vallerga MB, et al. , Rad51 recombinase prevents Mre11 nuclease-dependent degradation and excessive PrimPol-mediated elongation of nascent DNA after UV irradiation. Proc Natl Acad Sci U S A, 2015. 112(48): p. E6624–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Thangavel S, et al. , DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol, 2015. 208(5): p. 545–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Garzon J, et al. , Human RIF1-Protein Phosphatase 1 Prevents Degradation and Breakage of Nascent DNA on Replication Stalling. Cell Rep, 2019. 27(9): p. 2558–2566 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Mukherjee C, et al. , RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat Commun, 2019. 10(1): p. 3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Schlacher K, Wu H, and Jasin M, A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell, 2012. 22(1): p. 106–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Schlacher K, et al. , Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell, 2011. 145(4): p. 529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Leuzzi G, et al. , WRNIP1 protects stalled forks from degradation and promotes fork restart after replication stress. EMBO J, 2016. 35(13): p. 1437–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Billing D, et al. , The BRCT Domains of the BRCA1 and BARD1 Tumor Suppressors Differentially Regulate Homology-Directed Repair and Stalled Fork Protection. Mol Cell, 2018. 72(1): p. 127–139 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Higgs MR, et al. , BOD1L Is Required to Suppress Deleterious Resection of Stressed Replication Forks. Mol Cell, 2015. 59(3): p. 462–77. [DOI] [PubMed] [Google Scholar]
- [146].Morales C, et al. , PDS5 proteins are required for proper cohesin dynamics and participate in replication fork protection. J Biol Chem, 2020. 295(1): p. 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Ding X, et al. , Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nat Commun, 2016. 7: p. 12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Przetocka S, et al. , CtIP-Mediated Fork Protection Synergizes with BRCA1 to Suppress Genomic Instability upon DNA Replication Stress. Mol Cell, 2018. 72(3): p. 568–582 e6. [DOI] [PubMed] [Google Scholar]
- [149].Bantele SCS and Pfander B, Nucleosome Remodeling by Fun30(SMARCAD1) in the DNA Damage Response. Front Mol Biosci, 2019. 6: p. 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Chakraborty S, et al. , SMARCAD1 Phosphorylation and Ubiquitination Are Required for Resection during DNA Double-Strand Break Repair. iScience, 2018. 2: p. 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Costelloe T, et al. , The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature, 2012. 489(7417): p. 581–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Densham RM, et al. , Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol, 2016. 23(7): p. 647–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Schmid JA, et al. , Histone Ubiquitination by the DNA Damage Response Is Required for Efficient DNA Replication in Unperturbed S Phase. Mol Cell, 2018. 71(6): p. 897–910 e8. [DOI] [PubMed] [Google Scholar]
- [154].Zadorozhny K, et al. , Fanconi-Anemia-Associated Mutations Destabilize RAD51 Filaments and Impair Replication Fork Protection. Cell Rep, 2017. 21(2): p. 333–340. [DOI] [PubMed] [Google Scholar]
- [155].Hashimoto Y, et al. , Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol, 2010. 17(11): p. 1305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Kolinjivadi AM, et al. , Moonlighting at replication forks: a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51, FEBS Lett 591 (8) (2017) 1083–1100. [DOI] [PubMed] [Google Scholar]
- [157].Daza-Martin M, et al. , Isomerization of BRCA1-BARD1 promotes replication fork protection. Nature, 2019. 571(7766): p. 521–527. [DOI] [PubMed] [Google Scholar]
- [158].Murphy AK, et al. , Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J Cell Biol, 2014. 206(4): p. 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Feng W and Jasin M, BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nat Commun, 2017. 8(1): p. 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Rickman KA, et al. , Distinct roles of BRCA2 in replication fork protection in response to hydroxyurea and DNA interstrand cross-links, Genes & Development 34 (11–12) (2020) 832–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Carvajal-Maldonado D, et al. , Perturbing cohesin dynamics drives MRE11 nuclease-dependent replication fork slowing. Nucleic Acids Res, 2019. 47(3): p. 1294–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Higgs MR, et al. , Histone Methylation by SETD1A Protects Nascent DNA through the Nucleosome Chaperone Activity of FANCD2. Mol Cell, 2018. 71(1): p. 25–41 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Xu S, et al. , Abro1 maintains genome stability and limits replication stress by protecting replication fork stability. Genes Dev, 2017. 31(14): p. 1469–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Somyajit K, et al. , Mammalian RAD51 paralogs protect nascent DNA at stalled forks and mediate replication restart. Nucleic Acids Res, 2015. 43(20): p. 9835–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Clynes D, et al. , Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat Commun, 2015. 6: p. 7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Huh MS, et al. , Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis, 2016. 7: p. e2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Rondinelli B, et al. , EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol, 2017. 19(11): p. 1371–1378. [DOI] [PubMed] [Google Scholar]
- [168].Porebski B, et al. , WRNIP1 Protects Reversed DNA Replication Forks from SLX4-Dependent Nucleolytic Cleavage. iScience, 2019. 21: p. 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Chan KL, North PS, and Hickson ID, BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J, 2007. 26(14): p. 3397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Chan KL, et al. , Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol, 2009. 11(6): p. 753–60. [DOI] [PubMed] [Google Scholar]
- [171].Chan YW, Fugger K, and West SC, Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat Cell Biol, 2018. 20(1): p. 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Albers E, et al. , Loss of PICH Results in Chromosomal Instability, p53 Activation, and Embryonic Lethality. Cell Rep, 2018. 24(12): p. 3274–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Nielsen CF, et al. , PICH promotes sister chromatid disjunction and co-operates with topoisomerase II in mitosis. Nat Commun, 2015. 6: p. 8962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Hunter KB, et al. , Schimke immunoosseous dysplasia: defining skeletal features. Eur J Pediatr, 2010. 169(7): p. 801–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Carroll C, et al. , Schimke Immunoosseous Dysplasia associated with undifferentiated carcinoma and a novel SMARCAL1 mutation in a child. Pediatr Blood Cancer, 2013. 60(9): p. E88–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Carroll C, et al. , A novel splice site mutation in SMARCAL1 results in aberrant exon definition in a child with Schimke immunoosseous dysplasia. Am J Med Genet A, 2015. 167A(10): p. 2260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Spranger J, et al. , Schimke immuno-osseous dysplasia: a newly recognized multisystem disease. J Pediatr, 1991. 119(1 Pt 1): p. 64–72. [DOI] [PubMed] [Google Scholar]
- [178].Boerkoel CF, et al. , Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet, 2002. 30(2): p. 215–20. [DOI] [PubMed] [Google Scholar]
- [179].Adeyemo AA, et al. , ZRANB3 is an African-specific type 2 diabetes locus associated with beta-cell mass and insulin response. Nat Commun, 2019. 10(1): p. 3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Jackson SP and Bartek J, The DNA-damage response in human biology and disease. Nature, 2009. 461(7267): p. 1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Moinova HR, et al. , HLTF gene silencing in human colon cancer. Proc Natl Acad Sci U S A, 2002. 99(7): p. 4562–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Leung WK, et al. , Inactivation of helicase-like transcription factor by promoter hypermethylation in human gastric cancer. Mol Carcinog, 2003. 37(2): p. 91–7. [DOI] [PubMed] [Google Scholar]
- [183].Sandhu S, et al. , Loss of HLTF function promotes intestinal carcinogenesis. Mol Cancer, 2012. 11: p. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Bryant EE, et al. , Rad5 dysregulation drives hyperactive recombination at replication forks resulting in cisplatin sensitivity and genome instability. Nucleic Acids Res, 2019. 47(17): p. 9144–9159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Li J, et al. , DCUN1D1 facilitates tumor metastasis by activating FAK signaling and up-regulates PD-L1 in non-small-cell lung cancer. Exp Cell Res, 2019. 374(2): p. 304–314. [DOI] [PubMed] [Google Scholar]
- [186].Lawrence MS, et al. , Discovery and saturation analysis of cancer genes across 21 tumour types. Nature, 2014. 505(7484): p. 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Diplas BH, et al. , The genomic landscape of TERT promoter wildtype-IDH wildtype glioblastoma. Nat Commun, 2018. 9(1): p. 2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Lee JC, et al. , Alternative lengthening of telomeres and loss of ATRX are frequent events in pleomorphic and dedifferentiated liposarcomas. Mod Pathol, 2015. 28(8): p. 1064–73. [DOI] [PubMed] [Google Scholar]
- [189].Singhi AD, et al. , Alternative Lengthening of Telomeres and Loss of DAXX/ATRX Expression Predicts Metastatic Disease and Poor Survival in Patients with Pancreatic Neuroendocrine Tumors. Clin Cancer Res, 2017. 23(2): p. 600–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Liau JY, et al. , Alternative lengthening of telomeres phenotype in malignant vascular tumors is highly associated with loss of ATRX expression and is frequently observed in hepatic angiosarcomas. Hum Pathol, 2015. 46(9): p. 1360–6. [DOI] [PubMed] [Google Scholar]
- [191].Brosnan-Cashman JA, et al. , ATRX loss induces multiple hallmarks of the alternative lengthening of telomeres (ALT) phenotype in human glioma cell lines in a cell line-specific manner. PLoS One, 2018. 13(9): p. e0204159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Ren X, et al. , Alternative lengthening of telomeres phenotype and loss of ATRX expression in sarcomas. Oncol Lett, 2018. 15(5): p. 7489–7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Zhao S, et al. , Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci U S A, 2013. 110(8): p. 2916–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Le Gallo M, et al. , Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet, 2012. 44(12): p. 1310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Pu SY, et al. , ERCC6L, a DNA helicase, is involved in cell proliferation and associated with survival and progress in breast and kidney cancers. Oncotarget, 2017. 8(26): p. 42116–42124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Puccetti MV, et al. , Smarcal1 and Zranb3 Protect Replication Forks from Myc-Induced DNA Replication Stress. Cancer Res, 2019. 79(7): p. 1612–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Puccetti MV, et al. , Defective replication stress response inhibits lymphomagenesis and impairs lymphocyte reconstitution. Oncogene, 2017. 36(18): p. 2553–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Ray Chaudhuri A, et al. , Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature, 2016. 535(7612): p. 382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Guillemette S, et al. , Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev, 2015. 29(5): p. 489–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Gogola E, Rottenberg S, and Jonkers J, Resistance to PARP Inhibitors: Lessons from Preclinical Models of BRCA-Associated Cancer. Annual Review of Cancer Biology, Vol 3, 2019. 3: p. 235–254. [Google Scholar]
- [201].Quinet A, et al. , PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol Cell, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Nayak S, et al. , Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Science Advances, 2020. 6(24): p. eaaz7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Piberger AL, et al. , PrimPol-dependent single-stranded gap formation mediates homologous recombination at bulky DNA adducts. bioRxiv, 2019: p. 773242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Zamborszky J, et al. , Loss of BRCA1 or BRCA2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene, 2017. 36(35): p. 5085–5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Sun L, et al. , Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science, 2013. 339(6121): p. 786–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Ishikawa H, Ma Z, and Barber GN, STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature, 2009. 461(7265): p. 788–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Luzhna L, Kathiria P, and Kovalchuk O, Micronuclei in genotoxicity assessment: from genetics to epigenetics and beyond. Front Genet, 2013. 4: p. 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Mackenzie KJ, et al. , cGAS surveillance of micronuclei links genome instability to innate immunity. Nature, 2017. 548(7668): p. 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Harding SM, et al. , Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature, 2017. 548(7668): p. 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Pantelidou C, et al. , PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov, 2019. 9(6): p. 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Reislander T, et al. , BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat Commun, 2019. 10(1): p. 3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Bhattacharya S, et al. , RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res, 2017. 45(8): p. 4590–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Ban S, et al. , Chromosomal instability in BRCA1- or BRCA2-defective human cancer cells detected by spontaneous micronucleus assay. Mutat Res, 2001. 474(1–2): p. 15–23. [DOI] [PubMed] [Google Scholar]
- [214].Heijink AM, et al. , BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat Commun, 2019. 10(1): p. 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Feng E, et al. , CSB cooperates with SMARCAL1 to maintain telomere stability in ALT cells. J Cell Sci, 2020. 133(4). [DOI] [PubMed] [Google Scholar]
- [216].Yang YG, Lindahl T, and Barnes DE, Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell, 2007. 131(5): p. 873–86. [DOI] [PubMed] [Google Scholar]
- [217].Kavanagh D, et al. , New roles for the major human 3’−5’ exonuclease TREX1 in human disease. Cell Cycle, 2008. 7(12): p. 1718–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Stetson DB, et al. , Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell, 2008. 134(4): p. 587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Erdal E, et al. , A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev, 2017. 31(4): p. 353–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Coquel F, et al. , SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature, 2018. 557(7703): p. 57–61. [DOI] [PubMed] [Google Scholar]
- [221].Parkes EE, et al. , Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J Natl Cancer Inst, 2017. 109(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Ho SS, et al. , The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity, 2016. 44(5): p. 1177–89. [DOI] [PubMed] [Google Scholar]
- [223].Wang H, et al. , cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A, 2017. 114(7): p. 1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Schadt L, et al. , Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep, 2019. 29(5): p. 1236–1248 e7. [DOI] [PubMed] [Google Scholar]
- [225].Callahan MK, Postow MA, and Wolchok JD, CTLA-4 and PD-1 Pathway Blockade: Combinations in the Clinic. Front Oncol, 2014. 4: p. 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Aerts JG and Hegmans JP, Tumor-specific cytotoxic T cells are crucial for efficacy of immunomodulatory antibodies in patients with lung cancer. Cancer Res, 2013. 73(8): p. 2381–8. [DOI] [PubMed] [Google Scholar]
- [227].Aguiar PN Jr., et al. , PD-L1 expression as a predictive biomarker in advanced non-small-cell lung cancer: updated survival data. Immunotherapy, 2017. 9(6): p. 499–506. [DOI] [PubMed] [Google Scholar]
- [228].Park YJ, Kuen DS, and Chung Y, Future prospects of immune checkpoint blockade in cancer: from response prediction to overcoming resistance. Exp Mol Med, 2018. 50(8): p. 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Dong H, et al. , Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med, 2002. 8(8): p. 793–800. [DOI] [PubMed] [Google Scholar]
- [230].Ahmadzadeh M, et al. , Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood, 2009. 114(8): p. 1537–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Akinleye A and Rasool Z, Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J Hematol Oncol, 2019. 12(1): p. 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Yi M, et al. , Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer, 2018. 17(1): p. 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Fuertes MB, et al. , Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med, 2011. 208(10): p. 2005–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Diamond MS, et al. , Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med, 2011. 208(10): p. 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Longhi MP, et al. , Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med, 2009. 206(7): p. 1589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Bakhoum SF, et al. , Chromosomal instability drives metastasis through a cytosolic DNA response. Nature, 2018. 553(7689): p. 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Nieminuszczy J, Schwab RA, and Niedzwiedz W, The DNA fibre technique - tracking helicases at work. Methods, 2016. 108: p. 92–8. [DOI] [PubMed] [Google Scholar]
- [238].Michalet X, et al. , Dynamic molecular combing: stretching the whole human genome for high-resolution studies. Science, 1997. 277(5331): p. 1518–23. [DOI] [PubMed] [Google Scholar]
- [239].Sidorova JM, et al. , Microfluidic-assisted analysis of replicating DNA molecules. Nat Protoc, 2009. 4(6): p. 849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Parra I and Windle B, High resolution visual mapping of stretched DNA by fluorescent hybridization. Nat Genet, 1993. 5(1): p. 17–21. [DOI] [PubMed] [Google Scholar]
- [241].Kaykov A, et al. , Molecular Combing of Single DNA Molecules on the 10 Megabase Scale. Sci Rep, 2016. 6: p. 19636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Bialic M, et al. , Analyzing the dynamics of DNA replication in Mammalian cells using DNA combing. Methods Mol Biol, 2015. 1300: p. 67–78. [DOI] [PubMed] [Google Scholar]
- [243].Petermann E, et al. , Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol Cell Biol, 2006. 26(8): p. 3319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Jackson DA and Pombo A, Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol, 1998. 140(6): p. 1285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Bensimon A, et al. , Alignment and sensitive detection of DNA by a moving interface. Science, 1994. 265(5181): p. 2096–8. [DOI] [PubMed] [Google Scholar]
- [246].Neelsen KJ, et al. , Visualization and interpretation of eukaryotic DNA replication intermediates in vivo by electron microscopy. Methods Mol Biol, 2014. 1094: p. 177–208. [DOI] [PubMed] [Google Scholar]
- [247].Vindigni A and Lopes M, Combining electron microscopy with single molecule DNA fiber approaches to study DNA replication dynamics. Biophys Chem, 2017. 225: p. 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Sogo JM, Lopes M, and Foiani M, Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science, 2002. 297: p. 599–602. [DOI] [PubMed] [Google Scholar]
- [249].Sirbu BM, et al. , Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev, 2011. 25(12): p. 1320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Roy S, Luzwick JW, and Schlacher K, SIRF: Quantitative in situ analysis of protein interactions at DNA replication forks. J Cell Biol, 2018. 217(4): p. 1521–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Petruk S, et al. , TrxG and PcG proteins but not methylated histones remain associated with DNA through replication. Cell, 2012. 150(5): p. 922–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Wooten M, et al. , Superresolution imaging of chromatin fibers to visualize epigenetic information on replicative DNA. Nat Protoc, 2020. 15(3): p. 1188–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Costanzo V, et al. , An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell, 2003. 11(1): p. 203–13. [DOI] [PubMed] [Google Scholar]
- [254].Walter J, Sun L, and Newport J, Regulated chromosomal DNA replication in the absence of a nucleus. Mol Cell, 1998. 1(4): p. 519–29. [DOI] [PubMed] [Google Scholar]
- [255].Blow JJ, et al. , Chromosome replication in cell-free systems from Xenopus eggs. Philos Trans R Soc Lond B Biol Sci, 1987. 317(1187): p. 483–94. [DOI] [PubMed] [Google Scholar]
- [256].Yeeles JT, et al. , Regulated eukaryotic DNA replication origin firing with purified proteins. Nature, 2015. 519(7544): p. 431–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Devbhandari S, et al. , Chromatin Constrains the Initiation and Elongation of DNA Replication. Mol Cell, 2017. 65(1): p. 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Greene EC, et al. , DNA curtains for high-throughput single-molecule optical imaging. Methods Enzymol, 2010. 472: p. 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Collins BE, et al. , DNA curtains: novel tools for imaging protein-nucleic acid interactions at the single-molecule level. Methods Cell Biol, 2014. 123: p. 217–34. [DOI] [PubMed] [Google Scholar]
- [260].Duzdevich D, et al. , The dynamics of eukaryotic replication initiation: origin specificity, licensing, and firing at the single-molecule level. Mol Cell, 2015. 58(3): p. 483–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Gross P, et al. , Combining optical tweezers, single-molecule fluorescence microscopy, and microfluidics for studies of DNA-protein interactions. Methods Enzymol, 2010. 475: p. 427–53. [DOI] [PubMed] [Google Scholar]
- [262].Brouwer I, et al. , Human RAD52 Captures and Holds DNA Strands, Increases DNA Flexibility, and Prevents Melting of Duplex DNA: Implications for DNA Recombination. Cell Rep, 2017. 18(12): p. 2845–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Wasserman MR, et al. , Replication Fork Activation Is Enabled by a Single-Stranded DNA Gate in CMG Helicase. Cell, 2019. 178(3): p. 600–611 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Norio P, et al. , Progressive activation of DNA replication initiation in large domains of the immunoglobulin heavy chain locus during B cell development. Mol Cell, 2005. 20(4): p. 575–87. [DOI] [PubMed] [Google Scholar]
- [265].Norio P and Schildkraut CL, Plasticity of DNA replication initiation in Epstein-Barr virus episomes. PLoS Biol, 2004. 2(6): p. e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Norio P and Schildkraut CL, Visualization of DNA replication on individual Epstein-Barr virus episomes. Science, 2001. 294(5550): p. 2361–4. [DOI] [PubMed] [Google Scholar]
- [267].De Carli F, et al. , High-Throughput Optical Mapping of Replicating DNA. Small Methods, 2018. 2(9): p. 1800146. [Google Scholar]
- [268].Klein K, et al. , Genome-Wide Identification of Early-Firing Human Replication Origins by Optical Replication Mapping. bioRxiv, 2017: p. 214841. [Google Scholar]
- [269].Hennion M, et al. , FORK-seq: replication landscape of the Saccharomyces cerevisiae genome by nanopore sequencing. bioRxiv, 2020: p. 2020.04.09.033720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Muller CA, et al. , Capturing the dynamics of genome replication on individual ultra-long nanopore sequence reads. Nat Methods, 2019. 16(5): p. 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Georgieva D, et al. , Detection of Base Analogs Incorporated During DNA Replication by Nanopore Sequencing. bioRxiv, 2019: p. 549220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Willis NA, et al. , BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature, 2014. 510(7506): p. 556–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Larsen NB, Hickson ID, and Mankouri HW, Tus-Ter as a tool to study site-specific DNA replication perturbation in eukaryotes. Cell Cycle, 2014. 13(19): p. 2994–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Whinn KS, et al. , Nuclease dead Cas9 is a programmable roadblock for DNA replication. Sci Rep, 2019. 9(1): p. 13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Clewing JM, et al. , Schimke immuno-osseous dysplasia: a clinicopathological correlation. J Med Genet, 2007. 44(2): p. 122–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Knott GJ and Doudna JA, CRISPR-Cas guides the future of genetic engineering. Science, 2018. 361(6405): p. 866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Shen JP, et al. , Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat Methods, 2017. 14(6): p. 573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Najm FJ, et al. , Orthologous CRISPR-Cas9 enzymes for combinatorial genetic screens. Nat Biotechnol, 2018. 36(2): p. 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Erard N, Knott SRV, and Hannon GJ, A CRISPR Resource for Individual, Combinatorial, or Multiplexed Gene Knockout. Mol Cell, 2017. 67(2): p. 348–354 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Du D, et al. , Genetic interaction mapping in mammalian cells using CRISPR interference. Nat Methods, 2017. 14(6): p. 577–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Norman TM, et al. , Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science, 2019. 365(6455): p. 786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Horlbeck MA, et al. , Mapping the Genetic Landscape of Human Cells. Cell, 2018. 174(4): p. 953–967 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Feldman D, et al. , Optical Pooled Screens in Human Cells. Cell, 2019. 179(3): p. 787–799 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Wang C, et al. , Imaging-based pooled CRISPR screening reveals regulators of lncRNA localization. Proc Natl Acad Sci U S A, 2019. 116(22): p. 10842–10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Adamson B, et al. , A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell, 2016. 167(7): p. 1867–1882 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Rubin AJ, et al. , Coupled Single-Cell CRISPR Screening and Epigenomic Profiling Reveals Causal Gene Regulatory Networks. Cell, 2019. 176(1–2): p. 361–376 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Datlinger P, et al. , Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods, 2017. 14(3): p. 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Dixit A, et al. , Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell, 2016. 167(7): p. 1853–1866 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Gaudelli NM, et al. , Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature, 2017. 551(7681): p. 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [290].Komor AC, et al. , Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature, 2016. 533(7603): p. 420–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Anzalone AV, et al. , Search-and-replace genome editing without double-strand breaks or donor DNA. Nature, 2019. 576(7785): p. 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [292].Findlay GM, et al. , Accurate classification of BRCA1 variants with saturation genome editing. Nature, 2018. 562(7726): p. 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Yusufzai T and Kadonaga JT, HARP is an ATP-driven annealing helicase. Science, 2008. 322(5902): p. 748–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Mayle R, et al. , Mcm10 has potent strand-annealing activity and limits translocase-mediated fork regression. Proc Natl Acad Sci U S A, 2019. 116(3): p. 798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].Seelinger M and Otterlei M, Helicase-Like Transcription Factor HLTF and E3 Ubiquitin Ligase SHPRH Confer DNA Damage Tolerance through Direct Interactions with Proliferating Cell Nuclear Antigen (PCNA). Int J Mol Sci, 2020. 21(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [296].Lin JR, et al. , SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol Cell, 2011. 42(2): p. 237–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].Helmer RA, et al. , Helicase-like transcription factor (Hltf) regulates G2/M transition, Wt1/Gata4/Hif-1a cardiac transcription networks, and collagen biogenesis. PLoS One, 2013. 8(11): p. e80461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Helmer RA, et al. , Role of helicase-like transcription factor (hltf) in the G2/m transition and apoptosis in brain. PLoS One, 2013. 8(6): p. e66799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [299].Keka IS, et al. , Smarcal1 promotes double-strand-break repair by nonhomologous end-joining. Nucleic Acids Res, 2015. 43(13): p. 6359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Pugliese GM, et al. , Inducible SMARCAL1 knockdown in iPSC reveals a link between replication stress and altered expression of master differentiation genes. Dis Model Mech, 2019. 12(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Baradaran-Heravi A, et al. , SMARCAL1 deficiency predisposes to non-Hodgkin lymphoma and hypersensitivity to genotoxic agents in vivo. Am J Med Genet A, 2012. 158A(9): p. 2204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Sharma T, et al. , SMARCAL1 Negatively Regulates C-Myc Transcription By Altering The Conformation Of The Promoter Region. Sci Rep, 2015. 5: p. 17910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [303].Haokip DT, et al. , Transcriptional Regulation of Atp-Dependent Chromatin Remodeling Factors: Smarcal1 and Brg1 Mutually Co-Regulate Each Other. Sci Rep, 2016. 6: p. 20532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [304].Patne K, et al. , BRG1 and SMARCAL1 transcriptionally co-regulate DROSHA, DGCR8 and DICER in response to doxorubicin-induced DNA damage. Biochim Biophys Acta Gene Regul Mech, 2017. 1860(9): p. 936–951. [DOI] [PubMed] [Google Scholar]
- [305].Baradaran-Heravi A, et al. , Penetrance of biallelic SMARCAL1 mutations is associated with environmental and genetic disturbances of gene expression. Hum Mol Genet, 2012. 21(11): p. 2572–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].Helmer RA, et al. , Helicase-like transcription factor (Hltf) gene-deletion promotes oxidative phosphorylation (OXPHOS) in colorectal tumors of AOM/DSS-treated mice. PLoS One, 2019. 14(8): p. e0221751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Elizondo LI, et al. , Schimke immuno-osseous dysplasia: SMARCAL1 loss-of-function and phenotypic correlation. J Med Genet, 2009. 46(1): p. 49–59. [DOI] [PubMed] [Google Scholar]
- [308].Jones S, et al. , Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat Commun, 2014. 5: p. 5006. [DOI] [PMC free article] [PubMed] [Google Scholar]