

Abstract
With the aim of drawing comparisons to the highly reactive complex LCuOH (L = bis(2,6-diisopropylphenyl-carboxamido)pyridine), the complexes [Bu4N][LCuSR] (R = H or Ph) were prepared, characterized by spectroscopy and X-ray crystallography, and oxidized at low temperature to generate the species assigned as LCuSR on the basis of spectroscopy and theory. Consistent with the smaller electronegativity of S versus O, redox potentials for the LCuSR−/0 couples were ~50 mV lower than for LCuOH−/0, and the rates of the proton-coupled electron transfer reactions of LCuSR with anhydrous 1-hydroxy-2,2,6,6-tetramethylpiperidine at −80 °C were significantly slower (by more than 100 times) than the same reaction of LCuOH. Density functional theory (DFT) and time-dependent DFT calculations on LCuZ (Z = OH, SH, SPh) revealed subtle differences in structural and UV–visible parameters. Further comparison to complexes with Z = F, Cl, and Br using complete active space (CAS) self-consistent field and localized orbital CAS configuration interaction calculations along with a valence-bond-like interpretation of the wave functions showed differences with previously reported results (J. Am. Chem. Soc. 2020, 142, 8514), and argue for a consistent electronic structure across the entire series of complexes, rather than a change in the nature of the ligand field arrangement for Z = F.
Graphical Abstract
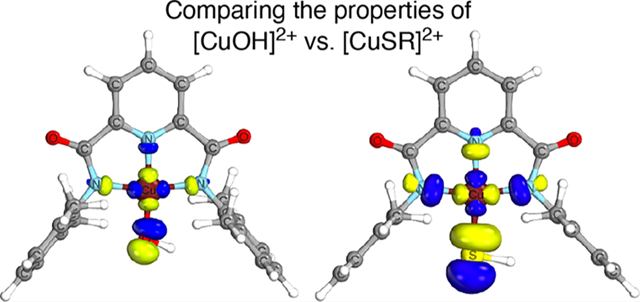
INTRODUCTION
A variety of novel copper–oxygen complexes1 have been prepared through research aimed at providing mechanistic insights into catalytic oxidations by synthetic2 and biological3 systems. Analogues that contain sulfur instead of oxygen also have been examined, in part to determine how the replacement of O by S influences the structure, properties, and function as well as to model copper–sulfur sites in enzymes.4–7 Such work has revealed new structural motifs and raised intriguing fundamental questions about chemical bonding.5b,g,8
Among the various copper–oxygen complexes that have been studied, those comprising the formally high-valent [CuOH]2+ core supported by sterically hindered, dianionic bis(carboxamido) ligands (Figure 1) have been shown to be highly reactive in a proton-coupled electron-transfer (PCET) involving substrate C–H and O–H bonds, processes that are relevant to catalytic oxidations.9 Comparative studies of analogues with [CuZ]2+ (Z = OOR′,10 halides,11 or carboxylates12) units also have been performed, leading to a greater understanding of electronic structural aspects and detailed PCET mechanisms. Intrigued by the insights provided by these studies and ones focused on copper–sulfur species,4–7 we asked: How would the properties of the heretofore unknown [CuSR]2+ core compare to those of [CuOH]2+ at parity of supporting ligand? Herein we report the results of synergistic experimental and theoretical work on the new species LCuSR (R = H or Ph) aimed at answering this question. As a part of this study, we were led to revisit the theoretical characterization of LCuZ (Z = F, Cl, Br) and found key similarities and differences with previous interpretations.11
Figure 1.
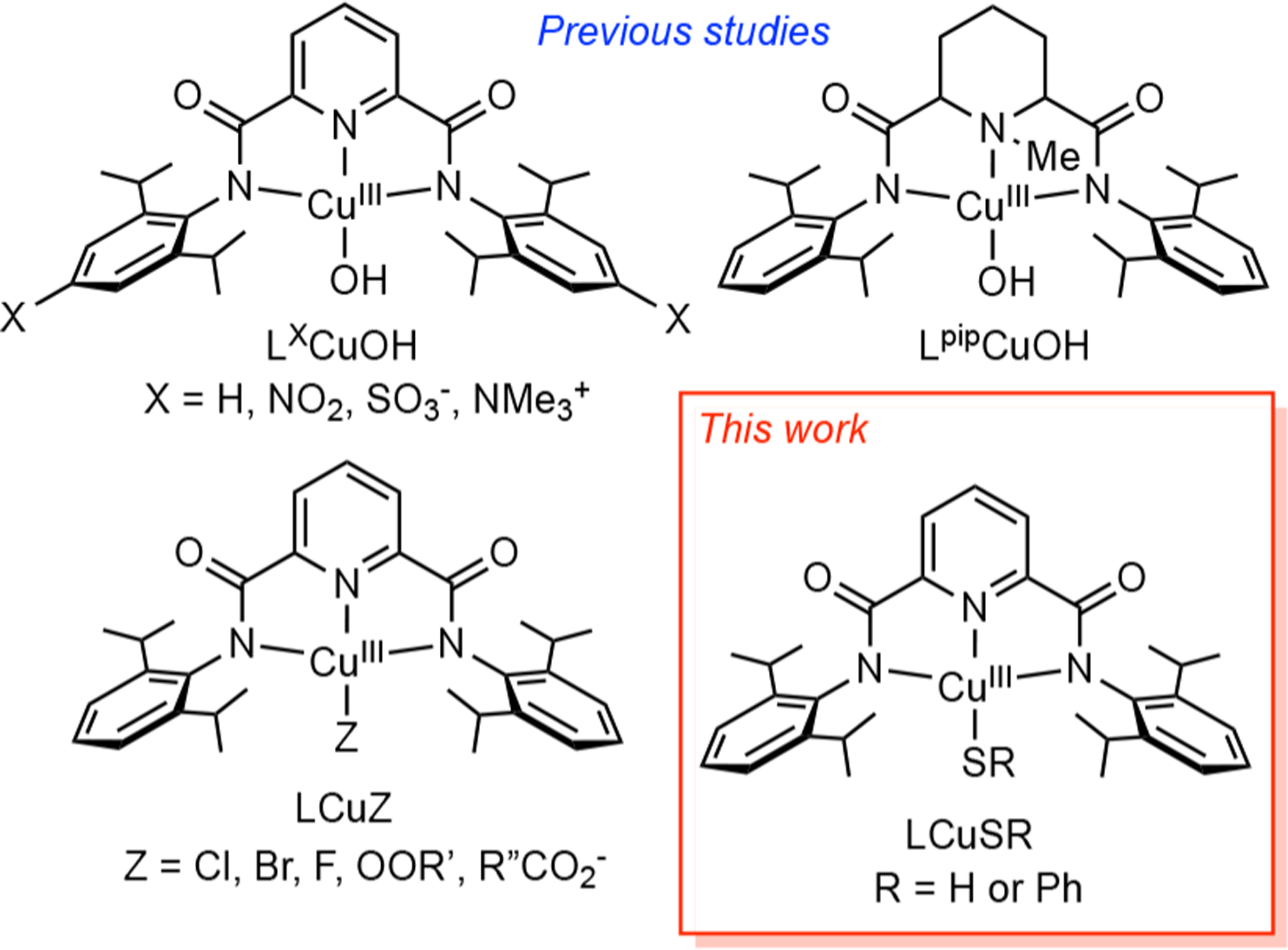
Previously studied complexes with [CuOH]2+ or [CuZ]2+ cores, and the analogues with [CuSR]2+ cores studied in this work. R′ = CMe2Ph or tBu; R″ = Me or aryl.
RESULTS AND DISCUSSION
Experiment.
Treatment of solutions of LCu(CH3CN) or LCuCl in tetrahydrofuran (THF) with Bu4NSH or NaSPh led to a color change from dark red to purple, from which purple crystalline products [Bu4N][LCuSH] ([Bu4N][1])13 or [Bu4N][LCuSPh] ([Bu4N][2]) were isolated in 73% or 63% yield, respectively. Both products were characterized by UV–vis and X-band electron paramagnetic resonance (EPR) spectroscopy, CHN analysis, and X-ray crystallography (Figure 2). Consistent with their analogous square-planar geometries evident in their solid-state structures, the Cu(II) complexes exhibit similar axial signals in their X-band EPR spectra (30 K) with Cu hyperfine (ACu(z) ≈ 520 MHz or 173 × 10−4 cm−1) and ligand N-hyperfine splitting patterns (AN ≈ 40–70 MHz or (13–23) × 10−4 cm−1) typical for Cu(II) complexes of L2− (Figure S1). A rare example of a terminal Cu(II)-SH species,7c,14 [Bu4N][1] exhibits a Cu–SH distance of 2.221(1) Å, similar to those seen in a few other Cu(II)-SH species (~2.25 Å), but longer than in a Cu(I)–SH complex (2.08 Å).
Figure 2.
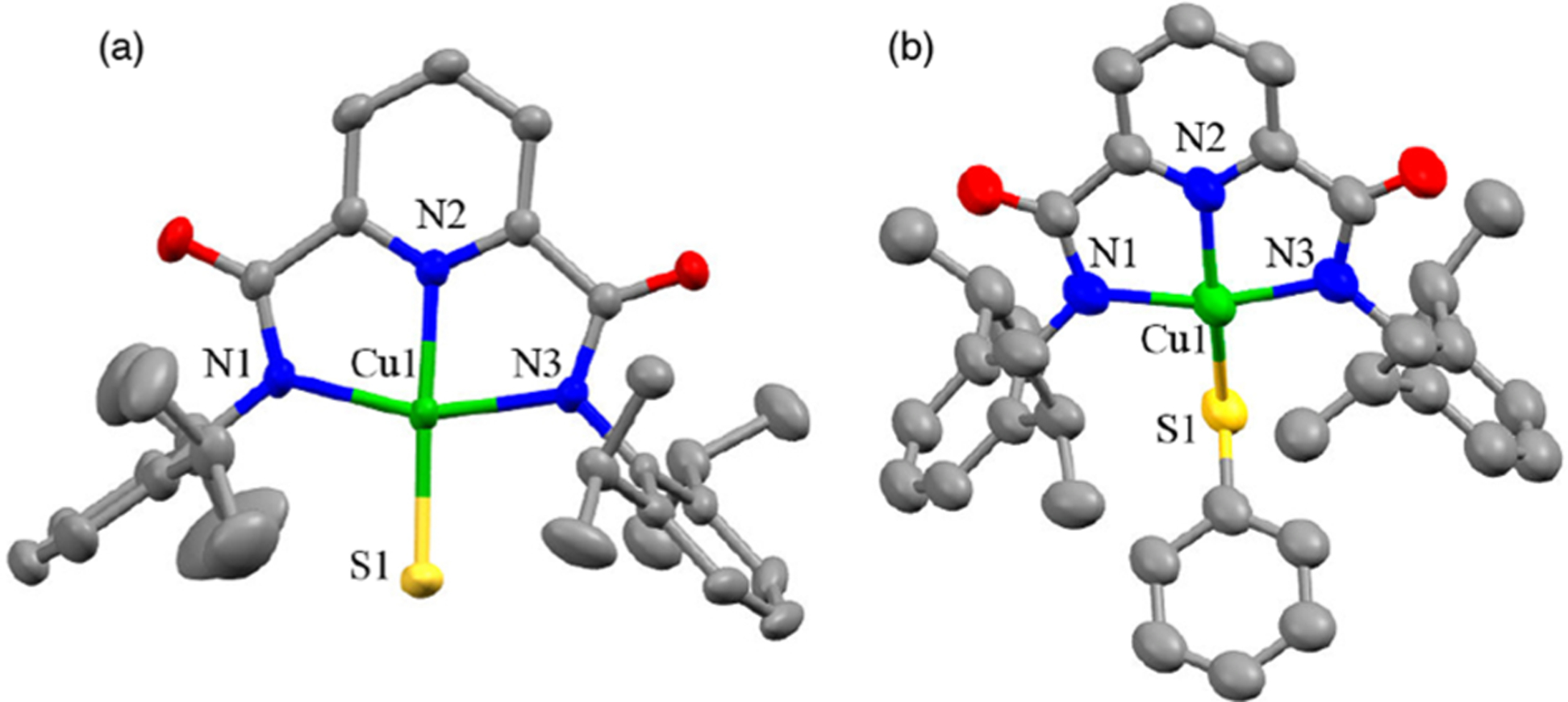
Representations of the anionic portions of the X-ray crystal structures of (a) [Bu4N][1] and (b) [Bu4N][2], showing all non-hydrogen atoms as 50% ellipsoids. Selected bond distances (Å) and angles (deg): [Bu4N][1]: Cu1–S1, 2.221(1); Cu1–N1, 2.021(2); Cu1–N2, 1.948(2); Cu1–N3, 2.021(2); N1–Cu1–S1, 98.30(6); N2–Cu1–S1, 175.58(6); N2–Cu1–N1, 79.26(8); N2–Cu1–N3, 79.41(8); N3–Cu1–S1, 103.23(6); N1–Cu1–N3, 158.35(8). [Bu4N][2]: Cu1–S1, 2.252(1); Cu1–N1, 2.002(4); Cu1–N2, 1.930(4); Cu1–N3, 2.004(5); N1–Cu1–S1, 103.1(1); N2–Cu1–S1, 161.1(1); N2–Cu1–N1, 79.5(1); N2–Cu1–N3, 79.6(1); N3–Cu1–S1, 100.0(2); N1–Cu1–N3, 156.5(1).
Cyclic voltammograms (THF, 0.1 M Bu4NPF6) showed pseudoreversible waves (Figure S2) with E1/2 values (vs Fc+/Fc) that are listed alongside values reported for other complexes LCuOH− and LCuZ− in Table 1. The value for LCuSH− (1−) is ~50 mV more negative than that for LCuOH−, consistent with the electronegativity order S < O. The 100 mV lower potential for LCuSPh− (2−) relative to LCuSH− (1−) implies greater electron donation by the thiophenolate, stabilizing the higher oxidation state. Both complexes exhibit redox potentials significantly lower than the carboxylate and halide complexes, consistent with poorer electron donation by their donor groups, and their values indicate that Fc+ would be a competent agent for chemical oxidation of the complexes.
Table 1.
Cyclic Voltammetry Dataa
| complex | R″ | E1/2 (V)b | ref |
|---|---|---|---|
| [CH3LCuCH2CN]− | −0.345 | 15 | |
| [LCuSPh]− (2 −) | −0.251 | this work | |
| [LCuOOtBu]− | −0.205 | 10 | |
| [LCuOOCMe2Ph]− | −0.154 | 10 | |
| [LCuSH]− (1−) | −0.209 | this work | |
| [LCuOH]− | −0.167 | 9i | |
| [LCuO2CR″]− | CH3 | 0.150 | 12a |
| C6H5 | 0.169 | 12a | |
| C6H4(NO2) | 0.239 | 12a | |
| C6F5 | 0.298 | 12a | |
| [LCuF]− | 0.605c | 11 | |
| [LCuCl]− | 0.665c | 11 | |
| [LCuBr]− | 0.665c | 11 |
Conditions: THF, Bu4NPF6 as supporting electrolyte, glassy carbon electrode.
Versus Fc+/Fc.
Values listed here versus Fc+/Fc were generated from the published values versus Ag/AgNO3 by using the correction +140 mV.16
Treatment of solutions of [Bu4N][1] or [Bu4N][2] at −80 °C with ferrocenium tetrakis(3,5-bis(trifluoromethyl)phenyl)-borate (FcBArF) led to an immediate color change to deep blue or purple, loss of the EPR signal, and the appearance of an intense UV–vis absorption peak with λmax = 582 nm (ε ≈ 6900 M−1cm−1) or 538 nm (ε ≈ 11 700 M−1 cm−1), respectively (Figure 3). These features are similar to those observed for LCuOH and LCuZ (Z = OOR, halide, O2CR),9 which were assigned as charge-transfer transitions (see theory results below). Repeated experiments with varying amounts of FcBArF (0.2–2 equiv) showed the maximum intensity of the product spectral features upon addition of 1 equiv (Figures S3 and S4). The starting spectrum is reformed upon addition of decamethylferrocene, and the product spectrum is regenerated upon subsequent addition of FcBArF (Figure S5). Together, these data show that the process involves a reversible, one-electron oxidation, consistent with the formation of the novel species LCuSR (R = H (1) or Ph (2)).
Figure 3.
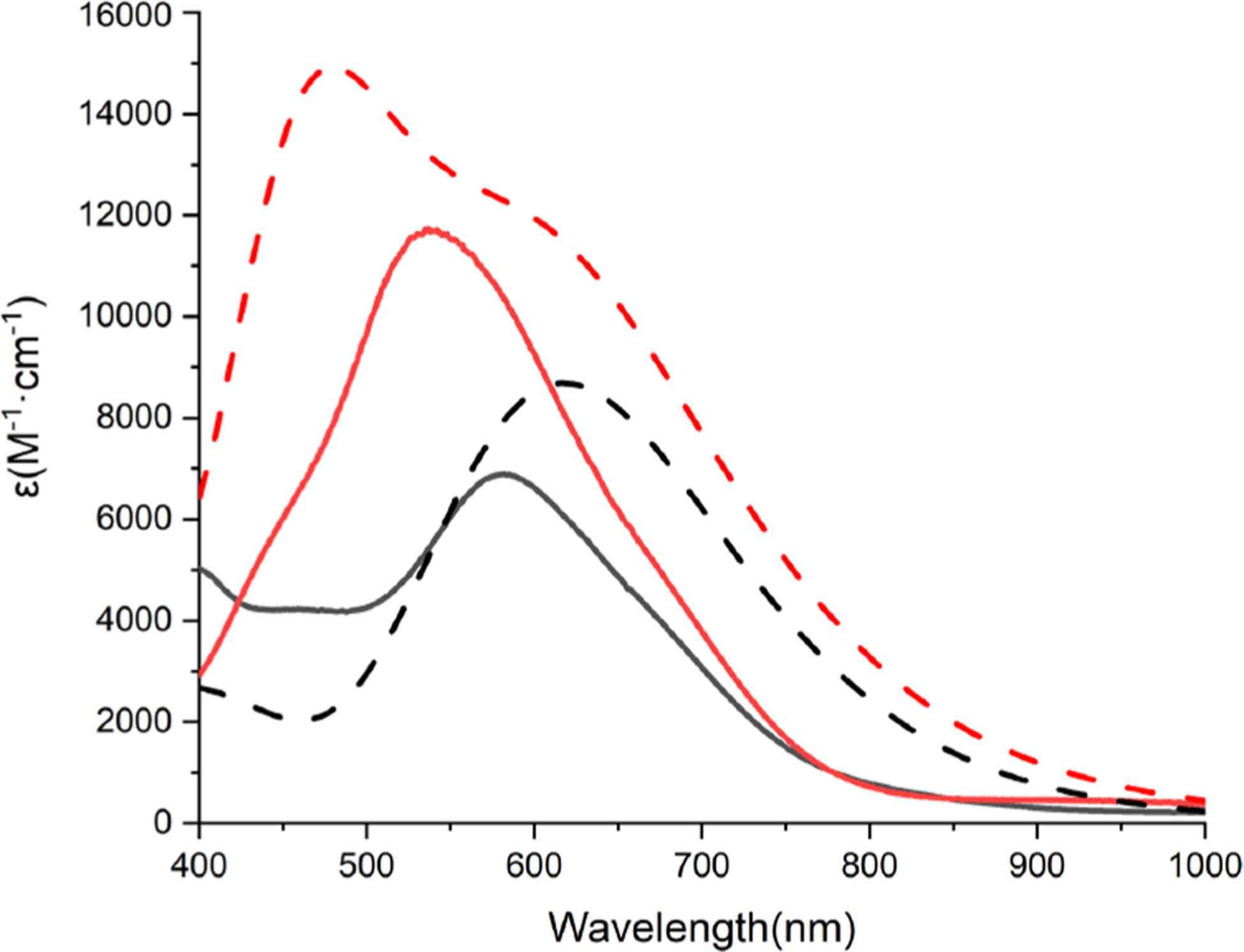
Overlay of experimental (solid lines) and calculated (TDDFT, dashed lines) UV–vis spectra of the products of the oxidation of [Bu4N][1] (black) and [Bu4N][2] (red). Experimental data measured for THF solutions at −80 °C.
The PCET reactivity of 1 and 2 was evaluated by monitoring of the decay of their diagnostic UV–vis absorbances upon addition of 1-hydroxy-2,2,6,6-tetramethylpiperidine (TEMPOH) (10–50 equiv) at −80 °C as a function of time. The formation of a TEMPO radical in these reactions was confirmed by EPR spectroscopy (Figure S6); a signal for an LCu(II) species also was observed that, on the basis of the UV–vis spectrum, we tentatively assign as LCu(THF)). Fits of the kinetic data provided pseudo-first-order rate constants (kobs), which when plotted versus [TEMPOH]0 yielded straight lines with zero intercepts (Figures S7 and S8), consistent with an overall second-order rate law with rate constants of k(1) = 4.2(2) M−1 s−1 and k(2) = 1.9 (1) M−1 s−1. The temperature dependencies (−40 °C to −80 °C) of the second-order rate constants were evaluated by the Eyring equation (Figure S9), yielding similar activation parameters for 1 (ΔH⧧ = 5.4(4) kcal/mol, ΔS⧧ = −27(2) eu) and 2 (ΔH⧧ = 4.7(2) kcal/mol, ΔS⧧ = −32(1) eu). The large negative ΔS⧧ values are consistent with a bimolecular process for the PCET reaction. It is particularly notable that the rates observed are much slower than those for the same reaction of LCuOH, which using 10 equiv at −80 °C is complete within 5 s (rate constant greater than 500 M−1 s−1, a more than 100 times faster reaction). This rate difference aligns with thermodynamic arguments based on the influences of redox potential and basicity on the product bond dissociation energy (greater driving force with increased redox potential and greater basicity). Accordingly, the PCET reactivity order LCuOH > LCuSR is consistent with lower redox potentials for 1− and 2− and the anticipated lower basicity of the hydrosulfido and thiophenolate ligands relative to hydroxide.17
Theory.
In order to understand the differences in properties between LCuOH and LCuSR (R = H or Ph), we performed a series of theoretical calculations aimed at comparing their electronic structures. Previous calculations on LCuOH serve as an important benchmark.9a,h,i We also were stimulated to revisit and draw comparisons to recently reported11 results of calculations on LCuZ (Z = F, Cl, Br), which were interpreted to indicate intriguing differences between the electronic structure for Z = F (an inverted ligand field) and X = Cl or Br (a classic ligand field) based on localized orbital CAS-CI calculations. We report density functional theory (DFT), complete active space self-consistent field (CASSCF), and localized orbital complete active space configuration interaction (CAS-CI) results that show a similar classic ligand field involving a dative covalent bond is present for all complexes.
As in previous work,9a,h,i DFT (mPWPW91) geometries for the S = 0 ground state for LCuZ (Z = OH, SH, SPh) are in excellent agreement with those of the experiment, and they also align with calculated results for analogues with the ligand truncated for convenience having methyl instead of isopropyl substituents, CH3LCuZ (Z = OH, SH, SPh, F, Cl, and Br; Table 2). All DFT calculations use the SDD basis set on Cu and 6-311+G(d,p) on the remaining atoms. With these geometries, UV–vis transitions were computed with B98 to determine the effect of the auxiliary ligand (Figure 3, Tables 3 and 4). Consistent with previous work,9a,h,i the transition corresponding to λmax is primarily from the ligand π highest occupied molecular orbital (HOMO) to the Cu lowest unoccupied molecular orbital (LUMO) for Z = OH, SH, F, Cl, and Br, while for Z = SPh the transitions from the −SPh π-system (HOMO–5) to the LUMO dominate (Figures S11–S19). As Z becomes less electron-withdrawing, this peak shifts to longer wavelengths as the HOMO–LUMO gap decreases due to the stabilization of the LUMO (Table 4). The DFT ground state is closed-shell S = 0, and the triplet state ranges from 16.0 to 31.3 kcal/mol higher in energy for all complexes (Table S4). All attempts to converge the broken-symmetry singlet solution converged to the closed-shell ground state suggesting the open-shell state is unstable or not accessible with DFT.
Table 2.
Bond Distances from Theory and Experiment (italics)a
| complex | Cu–Z | Cu–Npy | Cu-Nam | ref |
|---|---|---|---|---|
| LCuOH | 1.783 | 1.845 | 1.913 | 9i |
| 1.799(3) | 1.841(3) | 1.900(3) | 9ib | |
| LCuSH | 2.160 | 1.88 | 1.930 | c |
| LCuSPh | 2.206 | 1.907 | 1.974 | c |
| CH3LCuOH | 1.783 | 1.845 | 1.913 | c |
| CH3LCuSH | 2.160 | 1.880 | 1.930 | c |
| CH3LCuSPh | 2.20 | 1.901 | 1.959 | c |
| CH3LCuF | 1.770 | 1.842 | 1.924 | c |
| LCuF | 1.755(3) | 1.841(4) | 1.901(4) | 11 |
| CH3LCuCl | 2.130 | 1.865 | 1.940 | b |
| LCuCl | 2.1085(8) | 1.859(2) | 1.9132(16) | 11 |
| CH3LCuBr | 2.270 | 1.873 | 1.946 | c |
| LCuBr | 2.2562(4) | 1.8623(18) | 1.9159(13) | 11 |
Distances in angstroms; estimated standard deviations from experiment in parentheses; Cu–Npy = distance to ligand pyridine N atom; Cu–Nam = average distance to ligand carboxamide N atoms.
Experimental distance determined for the complex supported by a ligand with a methoxy group in the para position of the pyridine moiety.
This work.
Table 3.
UV–Vis Transitions and Oscillator Strengths Computed by TD-DFT (B98)
| complex | λmax (nm) | f |
|---|---|---|
| LCuOH | 546.1 | 0.3194 |
| LCuSH | 620.9 | 0.2063 |
| LCuSPh | 465.2, 619.8 | 0.2577, 0.1860 |
| CH3LCuOH | 552.7 | 0.2076 |
| CH3LCuSH | 613.1 | 0.2100 |
| CH3LCuSPh | 448.9 | 0.1867 |
| CH3LCuF | 676.6, 505.5 | 0.2492, 0.0902 |
| CH3LCuCl | 746.0, 553.1 | 0.1905, 0.1354 |
| CH3LCuBr | 861.9, 608.3, 274.0 | 0.0958, 0.1991, 0.2030 |
Table 4.
DFT (mPW1PW91) Orbital Energies (eV)
| complex | HOMO–LUMO gap | HOMO energy | LUMO energy |
|---|---|---|---|
| LCuOH | 2.90 | −6.6412 | −3.7386 |
| LCuSH | 2.73 | −6.6934 | −3.9674 |
| LCuSPh | 2.59 | −6.4962 | −3.9016 |
| CH3LCuOH | 2.83 | −6.5740 | −3.7443 |
| CH3LCuSH | 2.74 | −6.6817 | −3.9410 |
| CH3LCuSPh | 2.61 | −6.4647 | −3.8562 |
| CH3LCuF | 2.43 | −6.5128 | −4.0871 |
| CH3LCuCl | 2.30 | −6.5751 | −4.2795 |
| CH3LCuBr | 2.20 | −6.5430 | −4.3399 |
In light of recent work,11 a CASSCF study was undertaken for CH3LCuX (X = OH, SH, F, Cl, and Br). An active space with two electrons in two orbitals, denoted (2e,2o), was employed. The two orbitals are analogous to the DFT LUMO and its bonding counterpart and qualitatively similar for all species (Figure 4, see Figures S20–S24 for the validation of this choice). The CASSCF natural orbitals are labeled σ and σ* due to their covalent character with Cu contributing 50% on average to both orbitals (Table S9). The quantum theory of atoms in molecules (QTAIM) on the CASSCF density is also consistent with a dative bond (Table S10).
Figure 4.
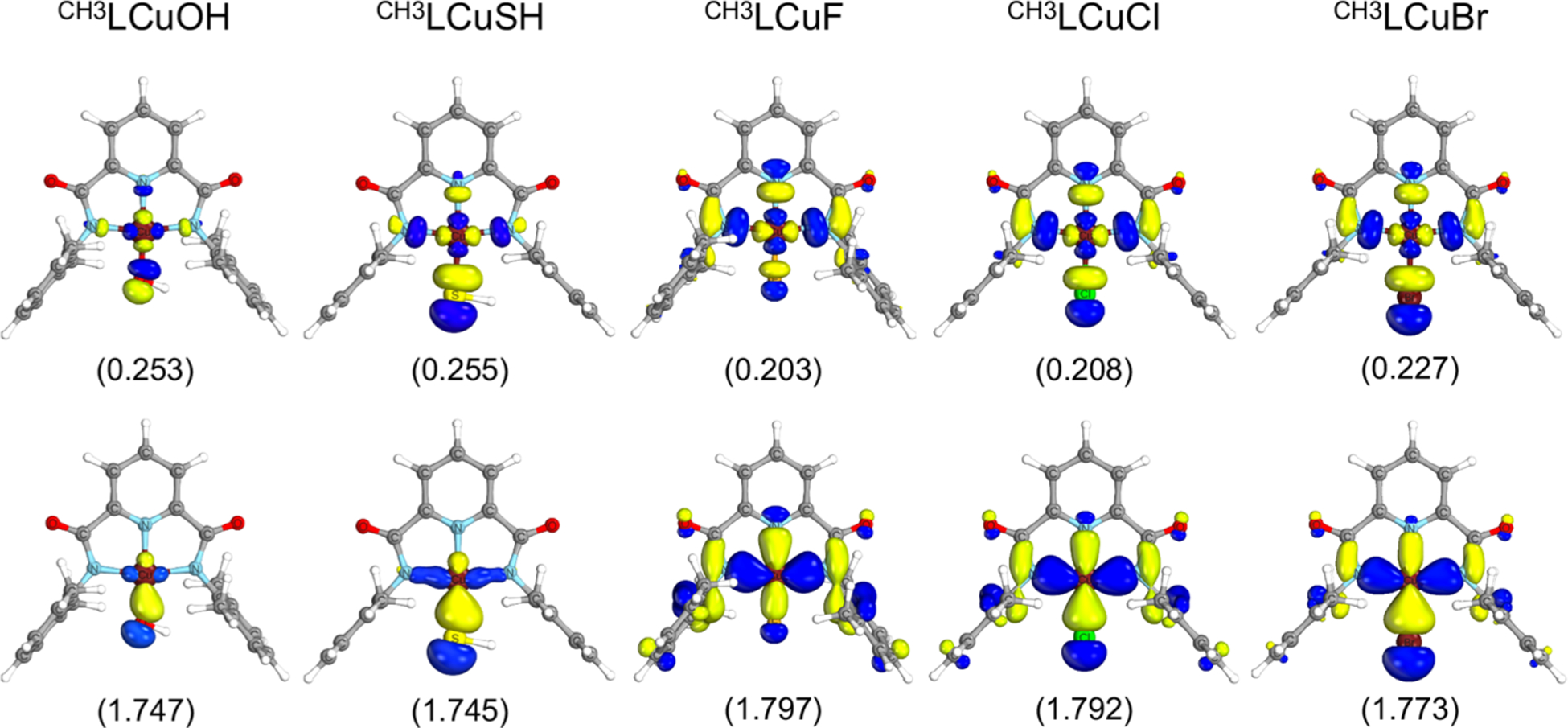
Active natural orbitals from the CASSCF (2e, 2o) calculations and corresponding occupation numbers. An isovalue of 0.04 is used.
Although the CASSCF natural orbitals do not have associated orbital energies, the σ* orbital generally has a higher partial occupation when DFT predicts a more stable LUMO. For the halide complexes, this is indeed observed. However, CASSCF and DFT differ in that σ* has a higher occupation for LCuOH and LCuSH compared to LCuF, LCuCl, and LCuBr, while the DFT LUMO is less stable. Perhaps this difference in the occupation numbers is due to differences in the σ* orbital, which is more localized on the Cu-RH bond compared to the other complexes.
Notwithstanding these subtle differences, the electronic structures are quite similar for all the complexes. There are three possible electron configurations: σ2σ*0, σ0σ*2, and σ1σ*1. The CASSCF wave function for all studied complexes involves only the σ2σ*0 and σ0σ*2 configurations with contributions of 88% and 12% on average, respectively (Table 5). No contribution from the σ1σ*1 electron configuration is observed. The bond itself can be further analyzed using the natural orbital occupation numbers (ONs) (Figure 4) to define an effective bond order (EBO).
Table 5.
Percent Contribution of Each Configuration to the Total CASSCF (2e,2o) Wavefunction Expressed in Terms of the Natural Orbitalsa
| complex | σ2σ*0 | σ0σ*2 | σ1σ*1 | EBO | % rad |
|---|---|---|---|---|---|
| CH3LCuOH | 87.2 | 12.8 | 0 | 0.745 | 25.5 |
| CH3LCuSH | 87.2 | 12.8 | 0 | 0.747 | 25.3 |
| CH3LCuF | 89.9 | 10.1 | 0 | 0.797 | 20.3 |
| LCuFb | 92.3 | 7.7 | 0 | 0.845 | 15.5 |
| CH3LCuCl | 89.3 | 10.7 | 0 | 0.792 | 20.8 |
| LCuClb | 91.4 | 8.6 | 0 | 0.828 | 17.2 |
| CH3LCuBr | 88.6 | 11.4 | 0 | 0.773 | 22.7 |
| LCuBrb | 90.5 | 9.5 | 0 | 0.811 | 18.9 |
The effective bond order (EBO) and the percentage of biradicaloid character (% rad) are also included.
Data from ref 11.
For a fully closed shell CuIIILX system, the EBO would be 1.0, while a fully CuII(LX)• would be 0.0. Therefore, the percentage of radical character (% rad) can be defined as
The complexes in this work have an effective bond order of 0.754 on average and a non-negligible biradicaloid contribution of 25% (Table 5). For comparison, a Cu corrole system was shown to have 10% and 50% biradical in the planar and saddled structures, respectively, and it was concluded that the former is closer to a formally CuIII center while the latter was indicative of corrole noninnocence.18 It was emphasized that the CuIIIL (pure dative bond) and CuIIL• (pure charge transfer) electronic structures represent extremes, while real systems fall along a spectrum. Notably, these results closely align with the CASSCF calculations reported previously for LCuX (X = F, Cl, Br) (Table 5). Thus, in agreement with our findings, this report describes an increase in the σ* occupation number from LCuF to LCuBr, as well as in the contributions of the electron configurations to the CASSCF wave function. Furthermore, the same trend in biradicaloid character emerges with an average EBO of 0.828 and a radical character of 17.2%. Therefore, LCuX complexes contain polar dative bonds where the bond strength as characterized by the EBO (or % rad) varies subtly with the ancillary ligand.
Multiconfigurational character is present in all complexes, yet the aforementioned DFT results are consistent with experiment. The type of electron correlation described here is the so-called left–right correlation, where the charge density is redistributed along the chemical bonds resulting in less charge near the atomic center.19 This type of correlation is frequently well-described by DFT as a result of its tendency to overdelocalize orbitals.20 Domain-based local pair natural orbital–coupled cluster singles doubles (DLPNO–CCSD) calculations were performed to compute the so-called T1 diagnostic. When this value is larger than 0.020, multiconfigurational character is likely present, and single-reference methods should not be used.21 The complexes in this work have values less than 0.019 (Table S11).
However, our work and that reported previously disagree with respect to the calculations in the localized orbital basis and the subsequent interpretation that LCuF has an inverted ligand field. We also performed localized orbital CAS-CI calculations in a localized orbital basis (see the Supporting Information), where the two localized orbitals are metal centered (d) and ligand-centered (LZ) (Figure S25). This approach has been used successfully in the literature to obtain a valence bond (VB)-like interpretation of the CASSCF wave function, since covalent contributions to bonding are removed via localization and the electron configurations can be related more easily to changes in oxidation states.18,22 The CASSCF wave function is invariant to the orbital choice within the active space; however, three different electron configurations are generated by occupying the localized orbitals instead of the natural orbitals, consisting of the configurations d2(LZ)0, d0(LZ)2, and d1(LZ)1 (see the Supporting Information for further discussion). The Cholesky localized orbitals d and LZ have Cu contributions of 94% and 6%, on average (Table S9).
The resulting wave functions are similar for the five complexes involving contributions from all three configurations (Table 6). The d1(LZ)1 configuration contributes 80.2%, on average, to the total wave function. This is followed by an average contribution from the d0(LZ)2 and d2(LZ)0 configurations of 13.2% and 6.5%, respectively. While these configurations were previously interpreted in terms of oxidation states, we argue that an alternative interpretation should be made.
Table 6.
Percent Contribution of Each Configuration to the Total Wavefunction in the Localized Orbital Basisa
| complex | d2(LZ)0(CuIZ+) | d0(LZ)2(CuIIIZ−) | d1(LZ)1(CuIIZ•) |
|---|---|---|---|
| CH3LCuOH | 4.5 | 13.6 | 81.9 |
| CH3LCuSH | 6.8 | 10.0 | 83.2 |
| CH3LCuF | 5.4 | 15.6 | 78.8 |
| LCuFb | 21.5 | 4.6 | 73.9 |
| CH3LCuCl | 6.9 | 12.7 | 80.4 |
| LCuClb | 0.6 | 34.0 | 65.4 |
| CH3LCuBr | 7.1 | 11.4 | 81.4 |
| LCuBrb | 2.2 | 24.5 | 73.3 |
Formal oxidation states for the indicated configurations are in parentheses.
Data are from ref 11.
For a simpler example of the proposed analysis, first consider the π-bond in ethene using this approach. The valence bond wave function ΨVB that represents the bond pair formed by two carbon p-orbitals and is the following
where
and
ψcovalent includes configurations in which each electron is occupying a p-orbital on a different carbon atom, while ψionic involves configurations where both electrons are on the same atom. The total wave function is a linear combination of the possible ways of occupying the two orbitals. The localized orbital CAS-CI results can be interpreted in the same way. The covalent contribution for ethene is 73.3%, and each ionic contribution is 13.3%. The fact that the p11p21 configuration has a large contribution does not suggest there is significant radical character in ethene. We argue that this is also the case for LCuZ and that the d1(LZ)1 configuration should be thought of as the covalent contribution and not as a radical contribution (i.e., d1(LZ)1 is not equivalent to σ1σ*1). The two ionic contributions are not symmetrically equivalent in this case, resulting in different contributions to the total wave function. On the one hand, as expected, CH3LCuSH has the largest covalent contribution, while CH3LCuF is the most ionic (Table 6). Furthermore, the d0(LZ)2 configuration can be assigned as CuIIIZ−, and its contribution decreases from F to Br, consistent with greater charge transfer from Z to Cu. On the other hand, the ionic contribution increases from Br to F. Moreover, differences between CH3LCuOH and CH3LCuSH are more apparent in the localized orbital basis than with CASSCF. CH3LCuSH has a slightly larger covalent contribution and a smaller CuIIIZ− contribution compared to LCuOH, which is consistent with a more polarizable and softer sulfur ligand.
While we see analogous trends from the localized orbital CAS-CI, CASSCF, and DFT calculations (Tables 4–6), in the previous work the results from LCuF were distinct, leading to the interpretation that the complex featured an inverted ligand field. Moreover, in the previous work the percent contributions for the two ionic contributions in LCuF are reversed: we find the “CuIII” d0(LZ)2 configuration to be larger, whereas it is smaller in the previous work. Recall that CASSCF and localized orbital CAS-CI are equivalent calculations; therefore, the trends related to the nature of the ligand should emerge in both methods. Although several localization approaches were tested (Tables S7 and S8) and the magnitude of the configurations does change, the trends described herein always emerge. At this juncture, we surmise that the previously reported localized orbital CAS-CI results are in error with respect to the interpretation of the nature of the ligand field trend, although the source of that error is unclear.
CONCLUSIONS
The complexes [Bu4N][LCuSH] ([Bu4N][1]) or [Bu4N]-[LCuSPh] ([Bu4N][2]), the former being a rare example of a terminal Cu(II)-SH species, were prepared and characterized by X-ray crystallography, UV–vis and EPR spectroscopy, and cyclic voltammetry. The latter showed pseudoreversible waves with E1/2 values that were ~50 mV lower than that of the LCuOH−/0 couple. Chemical oxidations at −80 °C with FcBArF led to a species identified as LCuSR (R = H or Ph) by diagnostic features in UV–vis spectra, EPR silence, titration data, and DFT/time-dependent (TD) DFT results. Comparisons of the latter across the series of LCuZ (Z = OH, SH, SPh, F, Cl, Br) showed the transition corresponding to the diagnostic UV–vis feature is primarily from the ligand π HOMO to the Cu LUMO (except for Z = SPh, where the transition from the −SPh π system (HOMO–5) to the LUMO is dominant) and that the HOMO–LUMO gap decreases due to the stabilization of the LUMO across the series, consistent with experimental observations. Kinetics studies of the reactions of LCuSR with TEMPOH revealed second-order rate constants ~100 times smaller than for LCuOH, consistent with the lower electronegativity of S and thermodynamic factors such as the lower redox potentials for the [CuSR]2+/+ couples and expected lower basicity of SR− versus OH−. DFT, CASSCF, and localized orbital CAS-CI results support the presence of dative covalent Cu-LZ bonds, with LCuF as the most ionic and LCuSH as the most covalent.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (GM47365). X-ray diffraction data were collected using a diffractometer acquired through NSF-MRI Award No. CHE-1827756. Computations supporting this project were performed on High Performance Computing systems at the University of South Dakota, funded by NSF Award No. OAC-1626516.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.1c00216.
Cartesian coordinates (XYZ)
Experimental and theoretical details, spectroscopic data, and data plots (PDF)
Accession Codes
CCDC 2058116 and 2058120 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.inorgchem.1c00216
Contributor Information
Wen Wu, Department of Chemistry, Washington University in St. Louis, St. Louis, Missouri 63130-4899, United States.
Jacqui Tehranchi De Hont, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Riffat Parveen, University of South Dakota, Vermillion, South Dakota 57069, United States.
Bess Vlaisavljevich, University of South Dakota, Vermillion, South Dakota 57069, United States.
William B. Tolman, Department of Chemistry, Washington University in St. Louis, St. Louis, Missouri 63130-4899, United States.
REFERENCES
- (1).(a) Selected reviews: Mirica LM; Ottenwaelder X; Stack TDP Sructure and Spectroscopy of Copper-Dioxygen Complexes. Chem. Rev 2004, 104, 1013–1045. [DOI] [PubMed] [Google Scholar]; (b) Lewis EA; Tolman WB Reactivity of Copper-Dioxygen Systems. Chem. Rev 2004, 104, 1047–1076. [DOI] [PubMed] [Google Scholar]; (c) Elwell CE; Gagnon NL; Neisen BD; Dhar D; Spaeth AD; Yee GM; Tolman WB Copper–Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev 2017, 117, 2059–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Quist DA; Diaz DE; Liu JJ; Karlin KD Activation of dioxygen by copper metalloproteins and insights from model complexes. JBIC, J. Biol. Inorg. Chem 2017, 22, 253–288. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liu JJ; Diaz DE; Quist DA; Karlin KD Copper(I)-Dioxygen Adducts and Copper Enzyme Mechanisms. Isr. J. Chem 2016, 56, 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Trammell R; Rajabimoghadam K; Garcia-Bosch I Copper-Promoted Functionalization of Organic Molecules: from Biologically Relevant Cu/O2 Model Systems to Organometallic Transformations. Chem. Rev 2019, 119, 2954–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Solomon EI; Heppner DE; Johnston EM; Ginsbach JW; Cirera J; Qayyum M; Kieber-Emmons MT; Kjaergaard CH; Hadt RG; Tian L Copper Active Sites in Biology. Chem. Rev 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Baeza Cinco MA; Hayton TW Progress Toward the Isolation of Late Metal Terminal Sulfides. Eur. J. Inorg. Chem 2020, 2020, 3613–3626. [Google Scholar]; (b) York JT; Bar-Nahum I; Tolman WB Copper-Sulfur Complexes Supported by N-Donor Ligands: Towards Models of the CuZ Site in Nitrous Oxide Reductase. Inorg. Chim. Acta 2008, 361, 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Bar-Nahum I; Gupta A; Huber S; Ertem M; Cramer C; Tolman W Reduction of Nitrous Oxide to Dinitrogen by a Mixed Valent Tricopper-Disulfido Cluster. J. Am. Chem. Soc 2009, 131, 2812–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sarangi R; York JT; Helton ME; Fujisawa K; Karlin KD; Tolman WB; Hodgson KO; Hedman B; Solomon EI X-Ray Absorption Spectroscopic and Theoretical Studies on (L)2 [Cu2 (S2)n]2+ Complexes: Disulfide versus Disulfide(1-) Bonding. J. Am. Chem. Soc 2008, 130, 676–686. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bar-Nahum I; York JT; Young VG; Tolman WB Novel Reactivity of Side-On (Disulfido)dicopper Complexes Supported by Bi- and Tridentate Nitrogen Donors: Impact of Axial Coordination. Angew. Chem., Int. Ed 2008, 47, 533–536. [DOI] [PubMed] [Google Scholar]; (d) York JT; Bar-Nahum I; Tolman WB Structural Diversity in Copper-Sulfur Chemistry: Synthesis of Novel Cu/S Clusters through Metathesis Reactions. Inorg. Chem 2007, 46, 8105–8107. [DOI] [PubMed] [Google Scholar]; (e) Brown EC; Bar-Nahum I; York JT; Aboelella NW; Tolman WB Ligand Structural Effects on Cu2S2 Bonding and Reactivity in Side-On Disulfido-Bridged Dicopper Complexes. Inorg. Chem 2007, 46, 486–496. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) York JT; Brown EC; Tolman WB Characterization of a Complex Comprising a {Cu2 (S2)2}2+ Core: Bis(μ-S22−)dicopper(III) or Bis(μ-S2−)dicopper-(II)? Angew. Chem., Int. Ed 2005, 44, 7745–7748. [DOI] [PubMed] [Google Scholar]; (g) Brown EC; York JT; Antholine WE; Ruiz E; Alvarez S; Tolman WB [Cu3 (μ-S)2]3+ Clusters Supported by N-Donor Ligands: Progress Towards a Synthetic Model of the Catalytic Site of Nitrous Oxide Reductase. J. Am. Chem. Soc 2005, 127, 13752–13753. [DOI] [PubMed] [Google Scholar]; (h) Brown EC; Aboelella NW; Reynolds AM; Aullón G; Alvarez S; Tolman WB A New Class of (μ-η2:η2-Disulfido)dicopper Complexes: Synthesis, Characterization, and Disulfide Exchange. Inorg. Chem 2004, 43, 3335–3337. [DOI] [PubMed] [Google Scholar]
- (6).(a) Johnson BJ; Lindeman SV; Mankad NP Assembly, Structure, and Reactivity of Cu4S and Cu3S Models for the Nitrous Oxide Reductase Active Site. Inorg. Chem 2014, 53, 10611–10619. [DOI] [PubMed] [Google Scholar]; (b) Johnson BJ; Antholine WE; Lindeman SV; Mankad NP A Cu4S Model for the Nitrous Oxide Reductase Active Sites Supported Only by Nitrogen Ligands. Chem. Commun 2015, 51, 11860–11863. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Johnson BJ; Antholine WE; Lindeman SV; Graham MJ; Mankad NP A One-Hole Cu4S Cluster with N2O Reductase Activity: A Structural and Functional Model for CuZ. J. Am. Chem. Soc 2016, 138, 13107–13110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hsu C-W; Rathnayaka SC; Islam SM; MacMillan SN; Mankad N N2O Reductase Activity of a [Cu4S] Cluster in the 4CuI Redox State Modulated by Hydrogen Bond Donors and Proton Relays in the Secondary Coordination Sphere. Angew. Chem., Int. Ed 2020, 59, 627. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Rathnayaka SC; Hsu CW; Johnson BJ; Iniguez SJ; Mankad NP Impact of Electronic and Steric Changes of Ligands on the Assembly, Stability, and Redox Activity of Cu4(μ4-S) Model Compounds of the CuZ Active Site of Nitrous Oxide Reductase (N2OR). Inorg. Chem 2020, 59, 6496–6507. [DOI] [PubMed] [Google Scholar]
- (7).(a) Wern M; Hoppe T; Becker J; Zahn S; Mollenhauer D; Schindler S Sulfur versus Dioxygen: Dinuclear (Trisulfido)copper Complexes. Eur. J. Inorg. Chem 2016, 2016, 3384–3388. [Google Scholar]; (b) Di Francesco GN; Gaillard A; Ghiviriga I; Abboud KA; Murray LJ Modeling Biological Copper Clusters: Synthesis of a Tricopper Complex, and Its Chloride- and Sulfide-Bridged Congeners. Inorg. Chem 2014, 53, 4647–4654. [DOI] [PubMed] [Google Scholar]; (c) Zhai J; Filatov AS; Hillhouse GL; Hopkins MD Synthesis, Structure, and Reactions of a Copper-Sulfido Cluster Comprised of the Parent Cu2S Unit: {(NHC)Cu}-2(μ-S). Chem. Sci 2016, 7, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Mealli C; Ienco A; Poduska A; Hoffmann R S4–2-Rings, Disulfides, and Sulfides in Transition-Metal Complexes: The Subtle Interplay of Oxidation and Structure. Angew. Chem., Int. Ed 2008, 47, 2864–2868. [DOI] [PubMed] [Google Scholar]; (b) Berry JF A Definitive Answer to a Bonding Quandary? The Role of One-Electron Resonance Structures in the Bonding of a {Cu3S2}3+ Core. Chem. - Eur. J 2010, 16, 2719–2724. [DOI] [PubMed] [Google Scholar]
- (9).(a) Donoghue PJ; Tehranchi J; Cramer CJ; Sarangi R; Solomon EI; Tolman WB Rapid C–H Bond Activation by a Monocopper(III)–Hydroxide Complex. J. Am. Chem. Soc 2011, 133, 17602–17605. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dhar D; Tolman WB Hydrogen Atom Abstraction from Hydrocarbons by a Copper(III)-Hydroxide Complex. J. Am. Chem. Soc 2015, 137, 1322–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gagnon N; Tolman WB [CuO]+ and [CuOH]2+ complexes: intermediates in oxidation catalysis? Acc. Chem. Res 2015, 48, 2126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Dhar D; Yee GM; Spaeth AD; Boyce DW; Zhang H; Dereli B; Cramer CJ; Tolman WB Perturbing the Copper(III)–Hydroxide Unit through Ligand Structural Variation. J. Am. Chem. Soc 2016, 138, 356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Dhar D; Yee GM; Markle TF; Mayer JM; Tolman WB Reactivity of the copper(III)-hydroxide unit with phenols. Chem. Sci 2017, 8, 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Spaeth AD; Gagnon NL; Dhar D; Yee GM; Tolman WB Determination of the Cu(III)–OH Bond Distance by Resonance Raman Spectroscopy Using a Normalized Version of Badger’s Rule. J. Am. Chem. Soc 2017, 139, 4477–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zerk TJ; Saouma CT; Mayer JM; Tolman WB Low Reorganization Energy for Electron Self-Exchange by a Formally Copper(III,II) Redox Couple . Inorg. Chem 2019, 58, 14151–14158. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Mandal M; Elwell CE; Bouchey CJ; Zerk TJ; Tolman WB; Cramer CJ Mechanisms for Hydrogen-Atom Abstraction by Mononuclear Copper(III) Cores: Hydrogen-Atom Transfer or Concerted Proton-Coupled Electron Transfer? J. Am. Chem. Soc 2019, 141, 17236–17244. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Krishnan VM; Shopov DY; Bouchey CJ; Bailey WD; Parveen R; Vlaisavljevich B; Tolman WB Structural Characterization of the [CuOR]2+ Core. J. Am. Chem. Soc 2021, 143, 3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Neisen BD; Gagnon NL; Dhar D; Spaeth AD; Tolman WB Formally Copper(III)-Alkylperoxo Complexes as Models of Possible Intermediates in Monooxygenase Enzymes. J. Am. Chem. Soc 2017, 139, 10220–10223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bower JK; Cypcar AD; Henriquez B; Stieber SCE; Zhang SC (sp3)-H Fluorination with a Copper(II)/(III) Redox Couple. J. Am. Chem. Soc 2020, 142, 8514–8521. [DOI] [PubMed] [Google Scholar]
- (12).(a) Elwell CE; Mandal M; Bouchey CJ; Que L Jr.; Cramer CJ; Tolman WB Carboxylate Structural Effects on the Properties and Proton-Coupled Electron Transfer Reactivity of [CuO2CR](2+) Cores. Inorg. Chem 2019, 58, 15872–15879. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Unjaroen D; Gericke R; Lovisari M; Nelis D; Mondal P; Pirovano P; Twamley B; Farquhar ER; McDonald AR High-Valent d(7) Ni(III) versus d(8) Cu(III) Oxidants in PCET. Inorg. Chem 2019, 58, 16838–16848. [DOI] [PubMed] [Google Scholar]
- (13).Tehranchi J Copper-Sulfur, Copper-Oxygen, and Copper-Alkyl Complexes Relevant to Copper Protein Active Site Intermediates and Catalysis, PhD Thesis, University of Minnesota, 2013. [Google Scholar]
- (14).(a) Gil-García R; Fraile R; Donnadieu B; Madariaga G; Januskaitis V; Rovira J; González L; Borrás J; Arnáiz FJ; García-Tojal J Desulfurization Processes of Thiosemicarbazonecopper(II) Derivatives in Acidic and Basic Aqueous Media. New J. Chem 2013, 37, 3568–3580. [Google Scholar]; (b) Strauch P; Dietzsch W; Golic L Photolytic Reactions of Binuclear Thiooxalate Complexes - Crystal and Molecular Structure of Hydrogensulfidopyridinebis-(triphenylphosphane)-copper(I. Z. Anorg. Allg. Chem 1997, 623, 129–134. [Google Scholar]; (c) Sreekanth A; Prathapachandra Kurup MR Structural and Spectral Studies on Four Coordinate Copper(Ii) Complexes of 2-Benzoylpyridine N(4),N(4)-(butane-1,4-diyl)thiosemicarbazone. Polyhedron 2003, 22, 3321–3332. [Google Scholar]; (d) Delgado S; Sanz Miguel PJ; Priego JL; Jiménez-Aparicio R; Gómez-García CJ; Zamora F A Conducting Coordination Polymer Based on Assembled Cu9 Cages. Inorg. Chem 2008, 47, 9128–9130. [DOI] [PubMed] [Google Scholar]; (e) Batsala GK; Dokorou V; Kourkoumelis N; Manos MJ; Tasiopoulos AJ; Mavromoustakos T; Simčič M; Golič-Grdadolnik S; Hadjikakou SK Copper(I)/(II) or Silver(I) Ions Towards 2-Mercaptopyrimidine: An Exploration of a Chemical Variability with Possible Biological Implication. Inorg. Chim. Acta 2012, 382, 146–157. [Google Scholar]; (f) Gupta G; Bhattacharya S Rich Synthetic and Structural Chemistry of Polynuclear Pb(II)–Cu(I)/Ag(I) Heterobimetallic Thiolate Clusters, Their Decomposition and Generation of a Cu(II) Hydrosulfide Variant. RSC Adv 2015, 5, 94486–94494. [Google Scholar]
- (15).Tehranchi J; Donoghue PJ; Cramer CJ; Tolman WB Reactivity of (Dicarboxamide)MII-OH (M = Cu, Ni) Complexes - Reaction with Acetonitrile to Yield MII-Cyanomethides. Eur. J. Inorg. Chem 2013, 2013, 4077–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).(a) Chowdhury DR; Spiccia L; Amritphale SS; Paul A; Singh A A Robust Iron Oxyhydroxide Water Oxidation Catalyst Operating under near Neutral and Alkaline Conditions. J. Mater. Chem. A 2016, 4, 3655–3660. [Google Scholar]; (b) Geiger WE Organometallic Electrochemistry: Origins, Development, And Future. Organometallics 2007, 26, 5738–5765. [Google Scholar]
- (17).Li Q; Lancaster JR Jr. Chemical Foundations of Hydrogen Sulfide Biology. Nitric Oxide 2013, 35, 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pierloot K; Zhao H; Vancoillie S Copper Corroles: the Question of Noninnocence. Inorg. Chem 2010, 49 (22), 10316–10329. [DOI] [PubMed] [Google Scholar]
- (19).Handy NC; Cohen AJ Left-Right Correlation Energy. Mol. Phys 2001, 99, 403–412. [Google Scholar]
- (20).(a) Polo V; Kraka E; Cremer D Some Thoughts about the Sta- bility and Reliability of Commonly Used Exchange-Correlation Functionals - Coverage of Dynamic and Nondynamic Correlation Effects. Theor. Chem. Acc 2002, 107, 291–303. [Google Scholar]; (b) Gräfenstein J; Kraka E; Cremer D The impact of the self-interaction error on the density functional theory description of dissociating radical cations: Ionic and covalent dissociation limits. J. Chem. Phys 2004, 120, 524–539. [DOI] [PubMed] [Google Scholar]; (c) Harvey JN On the accuracy of density functional theory in transition metal chemistry. Annu. Rep. Prog. Chem., Sect. C: Phys. Chem 2006, 102, 203–226. [Google Scholar]
- (21).Riplinger C; Neese F An Efficient and near Linear Scaling Pair Natural Orbital Based Local Coupled Cluster Method. J. Chem. Phys 2013, 138, No. 034106. [DOI] [PubMed] [Google Scholar]
- (22).Radon M; Broclawik E; Pierloot K Electronic Structure of Selected {FeNO}7 Complexes in Heme and Non-Heme Architectures: A Density Functional and Multireference ab Initio Study. J. Phys. Chem. B 2010, 114, 1518–1528. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.