

Abstract
The mechanism underlying neurofibrillary tangles (NFTs) in Alzheimer's disease (AD) and other neurodegenerative disorders remains elusive. Niemann–Pick disease type C (NPC) is a kind of genetic neurovisceral disorder in which the intracellular sequestration of cholesterol and other lipids in neurons, NFT formation and neuronal degeneration in brain are the neuropathology hallmarks. The age of onset and progression of the disease vary dramatically. We have analyzed the hippocampus from 17 NPC cases, aged from 7 months to 55 years, to depict the temporal characteristics of NFT formation. Unexpectedly, classic NFT was observed in about 4‐year‐old NPC brain, suggesting that NFT is not aging dependent, and that juvenile brain neurons satisfy the requirements for NFT formation. NFT in the hippocampus of NPC was significantly increased in number with the advance of age. More importantly, multiple mitotic phase markers, which are not usually found in normal mature neurons, were abundant in the affected neurons and incorporated into NFT. The unusual activation of cdc2/cyclin B kinase and downstream mitotic indices are closely associated with the age‐dependent NFT formation, signifying the contribution of abortive cell cycle to neurodegeneration. The cdc2 inhibitors may be therapeutically used for early intervention of neurodegeneration and NFT formation in NPC.
Keywords: cdc2, cyclin B1, mitotic marker, neurodegeneration, tau phosphorylation
INTRODUCTION
The neurofibrillary tangles (NFTs) were considered as a diagnostically pathological marker for Alzheimer's disease (AD); however, the lesions have been observed in several different neurodegenerative diseases, such as Down syndrome (17) or frontotemporal dementia (25). Because most of these disorders affect aged individuals, NFTs are generally considered to be the aging‐associated lesions. Subsequently, NFTs were discovered to be present in a variety of juvenile neurologic diseases, such as Niemann–Pick disease type C (NPC) (1), Cockayne syndrome (37) or Hallervorden–Spatz disease (13). Despite the seemingly distinct etiologies and pathologies of these various diseases, the NFTs may represent a marker for neuronal degeneration, and the presence in a wide range of neurologic diseases implies a common final pathway leading to their formation.
NPC is a kind of rare and fatal neurovisceral lipidoses, caused by mutations in NPC1 or NPC2 gene (32). Various mutations in the NPC genes produce the clinical forms of neonatal, infantile, childhood, adolescent or adult onset NPC, presenting a protean constellation of neuropsychiatric manifestations and premature death (35). The overwhelming majority of NPC cases have been described in children and adolescents, but there is a prolonged progression of pathology leading to death in adulthood 27, 36. Neuropathologically, neurons distended with lipid storage material, NFT formation and neuronal death characterize NPC (36). Although the biological significance of tau phosphorylation and NFT formation has not been fully ascertained 12, 18, the mechanisms for NFT formation have drawn much attention from neuroscientists based on the statement that the number and the extent of NFT in AD brain are paralleled with the degree of dementia of AD patients other than senile plaques (5). The NFTs in NPC are found to be composed of the paired helical filaments that are similar to those in AD (1), but the NFT pathology in NPC has not been characterized in detail, and the mechanisms leading to NFT formation are not known yet. A few kinases such as cycle division kinase cdk5/p25, glycogen synthese kinase ‐3β (GSK‐3β), mitogen‐activated protein kinase (MAPK), which have been found to get involved in abnormal phosphorylation and NFT formation in AD, have been investigated in NPC as well. In our previous studies, we had initially described that the cdk5/p25 activation may contribute to the cytoskeletal pathology in murine NPC 7, 47. However, the possibility has been suspected by one of our subsequent studies showing that the cytoskeletal pathology was persistent after a significant reduction of cdk5 activity in p35 knockout murine NPC (14). Another kinase, the MAPK, once described to be related to tau phosphorylation in NPC pathology (34), has been recently found not to contribute to NPC pathology in our work (48). GSK‐3β, a kinase convinced to play an important role in tau hyperphosphorylation and NFT formation in AD, had been demonstrated by us not to be involved in NPC pathology in a murine NPC model (47). Interestingly, in the cerebellum of human NPC where no visible NFT was found, the abnormal phosphorylation of tau similar to that in AD pattern has been accumulated relative to control cerebellum (6). In addition, we have described that a cohort of mitotic markers were abnormally enriched in the degenerating Purkinje cells, suggesting the potential involvement of mitotic kinase cdc2/cyclin B1 in the neuronal degeneration in NPC (6). The belief that the cell cycle activation is contributed to NPC cytoskeletal pathology was further substantiated by our observation that the intraventricular introduction of cell cycle division kinases (cdk) inhibitors such as roscovitine and olomoucine has significantly ameliorated the Purkinje cell loss, reduced the number of axonal spheroids (a kind of cytoskeletal pathology described in NPC) and improved the motor ability in a murine NPC model (47). Based on the observations above, we raised a hypothesis that the abnormal activation of the cell cycle in post‐mitotic neurons plays a key role in NFT formation in human NPC, just like the presence of a spectrum of cell cycle regulators in NFT‐bearing neurons in several neurodegenerative disorders 16, 40.
We have taken an advantage of a group of NPC cases having an unusually wide range of onset and progression, to delineate the temporal and qualitative characteristics of NFT formation in human NPC with the advance of age, and to chart out the possible early steps in the NFT‐forming cascade in NPC.
EXPERIMENTAL PROCEDURES
All these procedures were approved by Institutional Review Board of the Huazhong University of Science and Technology in Wuhan, China.
Antibodies
Tau antibodies
PHF‐1 monoclonal antibody was generously provided by Dr. Peter Davies (Albert Einstein College of Medicine, Bronx, N.Y.) and has been epitope‐mapped to phosphoserines 396 and 404 of human tau. The MC1 antibody recognizes an early pathological conformation in tau (44), and the TG‐5 antibody is directed at primary sequence in human tau.
Cell cycle antibodies
The TG‐3, MPM‐2, H5 and H14 antibodies recognize mitotic phosphoepitopes produced by the mitotic cdc2/cyclin B1 kinase (15). The TG‐3 antibody recognizes a conformation‐dependent phosphothreonine 231 of AD tau, and a phosphoepitope in nucleolin (20). The MPM‐2 mitotic protein monoclonal #2 antibody recognizing a mitotic phosphorylated epitope was obtained from Upstate Biotechnologies (Lake Placid, N.Y.). The H5 and H14 mouse monoclonal IgM antibodies were provided by Dr. David Bregman (Albert Einstein College of Medicine). The two antibodies detect phosphorylated serine numbers 2 and 5 respectively, of the carboxyl terminal domain heptapeptide repeat region of RNA polymerase II (RNAP II) (43). The cdc2 polyclonal antibody recognizing cdc2 phosphorylated at Thr161 was purchased from Cell Signaling Technology (Danvers, Mass.). Another cdc2 polyclonal antibody recognizing C‐terminus of cdc2 was obtained from Upstate Biotechnologies. The human cyclin B1 monoclonal antibody was purchased from Santa Cruz Biotechnologies (Santa Cruz, Calif.).
Human brain tissues
Post‐mortem human brain from 17 cases of clinically and biochemically confirmed NPC (aged from 7 months to 55 years), two cases of AD and five controls (aged 7 days to 23 years) were collected worldwide (see Table 1). All of the NPC cases were neuropathologically evaluated. The controls were free of neurofibrillary changes or any other neuropathological features. Hippocampal tissue blocks were fixed in 10% buffered formalin for at least 1 week, and embedded in paraffin. The tissues were sectioned at 6 µm.
Table 1.
Pathological changes in Niemann–Pick disease type C cases. Abbreviation: NFTs = neurofibrillary tangles.
| Case number | Age at death | NFTs* | Purkinje cell loss | Phospho‐tau | Mitotic markers† | Neuronal storage |
|---|---|---|---|---|---|---|
| 914 | 7 Months | − | − | +− | +− | +− |
| 64534 | 4.5 Years | + | + | +− | + | |
| 381‐72 | 6 Years | − | + | + | + | +++ |
| 98‐28 | 10 Years | + | + | + | + | + |
| 96‐195 | 10 Years | ++ | + | + | +− | + |
| A11 | 11 Years | ++ | ++ | + | + | + |
| 200‐88 | 17 Years | ++ | ++ | + | + | +++ |
| 7476 | 17 Years | ++ | + | ++ | ++ | +++ |
| 88‐84 | 19 Years | ++ | ++ | ++ | ++ | ++ |
| 94A109 | 19 Years | +++ | ++ | +++ | +++ | ++ |
| 26651 | 20 Years | +++ | +++ | +++ | +++ | ++ |
| 930537 | 25 Years | ++++ | +++ | +++ | +++ | ++ |
| A31 | 31 Years | ++++ | ++ | +++ | +++ | ++ |
| A97‐124 | 32 Years | ++ | +++ | ++ | ++ | ++ |
| A94‐74 | 43 Years | ++++ | +++ | +++ | +++ | ++ |
| 63771 | 46 Years | ++++ | +++ | +++ | +++ | ++ |
| A97‐123 | 55 Years | ++++ | +++ | +++ | +++ | ++ |
NFTs: −, no tangles; +, a few tangles in CA region; ++, many tangles in CA region; +++, widespread tangles in CA region and subiculum; ++++, extended to other regions such as the thalamus and colliculi.
Mitotic markers: recognized by monoclonal antibodies TG‐3, MPM‐2, H5, H14, cdc2 or cyclin B1. Not all cases displayed all positive markers.
Bielschowsky silver impregnation
Modified Bielschowsky silver impregnation staining was carried out on paraffin‐embedded sections as described (28).
Immunocytochemistry and double immunofluorescence labeling
The immunocytochemical staining was performed on paraffin‐embedded sections as indicated, using a library of antibodies specific for AD tau and mitotic markers. The paraffin‐embedded sections were deparaffinized and rehydrated in xylene and graded ethanol. Deparaffinized and vibratome sections were permeabilized by incubating with 0.25% Triton X and 3% hydrogen peroxidase for 30 minutes at room temperature. After blocking the nonspecific binding for 30 minutes in 5% blocking milk, the sections were incubated with primary antibodies in blocking milk overnight at 4°C. The TG‐3, CP13 and PHF‐1 antibodies were used at a 1:10 dilution in blocking milk, H5 and H14 1:2, MPM‐2 and cdc2 1:100 and cyclin B1 3 ug/mL, respectively. For detecting the specific binding of primary antibodies, the horseradish peroxidase‐conjugated appropriate secondary antibodies (purchased from Southern Biotechnology Associates, Alabaster. Ala.) were applied to the tissue at a dilution of 1:500 for 1 h. Antibody binding was visualized by reaction with diaminobenzidine (Sigma, St. Louis, Mo.). The sections were counterstained with hematoxylin for 10 s. The immunofluorescence labeling was carried out according to the previously described method (41).
Photography
Light and fluorescent micrographs were collected using a Nikon® microscope (Nikon, Tokyo, Japan) connected with a computerized SPOT™ digital camera (Diagnostic Instruments Inc., Sterling Heights, Mich.), and processed for generation of the figures with Adobe® Photoshop®.
Preparation of brain lysates
The hippocampal tissue obtained at autopsy from the 11‐ and 31‐year‐old NPC cases, a 25‐year‐old control and a typical AD case used for biochemical studies were quickly frozen at –80°C. The frozen tissues were homogenized with a polytron in buffer containing10 mM Tris–HCl, 150 mM NaCl, 20 mM NaF, 1 mM sodium vanadate, 25 µM leupeptin, 2 mM ethylene glycol tetraacetic acid, 0.5% Triton X‐100, 2 mM phenylmethylsulfonyl fluoride and 0.1% sodium dodecylsulfate (SDS). The homogenates were aliquoted and stored at –80°C.
Western blot analysis
Just before use, the aliquots were thawed and centrifuged at 12 000 × g for 5 minutes, and the soluble fraction was used as extract for immunoblotting. The protein contents in supernatants were determined using Bio‐Rad Protein Assay kit (Hercules, Calif.) and a microplate reader (Molecular Devices, Calif.). The proteins were separated on SDS–polyacrylamide gel electrophoresis after boiling with sample buffer for 5 minutes. For the detection of tau, 30 µg of protein was loaded per well. The separated proteins were electrophoretically transferred to nitrocellulose membrane. Following transfer, the nonspecific binding sites of proteins were blocked with 5% fat‐free milk in Tris‐buffered saline for 1 h at room temperature. The blots were then incubated with primary antibodies on a shaker overnight at 4°C. Enhanced chemiluminescence was used to visualize bound antibody.
RESULTS
All the cases studied here had evidence of lipid storage as indicated by foamy deposits in the neuronal soma or dendrites that were negatively stained with silver reagent. Neuronal loss was also evident to varying degrees in each case. We first compared immunoreactivity with PHF‐1, an antibody widely used for highlighting NFT in AD, with that of TG‐3, a reagent specific for the early cell cycle changes associated with NFT formation. In addition, other antibodies specific for mitotic antigens that are aberrantly expressed in degenerating AD neurons were also used (15). We have previously published that over 25 control cases ranging in age from 27 to 91 years did not display immunoreactivity with any of the mitotic antibodies 40, 42. For the first time here, we have extrapolated these findings to juvenile control cases, and so we only show these new data. Overall, the results with the mitotic antibodies were similar to those with TG‐3 and PHF‐1. Thus, for the most part, the figures concentrate on the results with PHF‐1 and TG‐3, and some examples with the mitotic antibodies are also presented. A summary of all the data for each case including NFT and other pathological changes is presented in Table 1. The details with respect to age dependence and other qualitative changes are discussed in the ensuing part of the Results section.
Age‐dependent NFT formation in NPC
The hippocampus is the most vulnerable to NFT in AD, and is commonly affected in NPC as well. To examine the dependence on age of NFT in NPC, we have conducted immunohistochemical analysis of the hippocampus from NPC cases aged 7 months to 55 years using the spectrum of NFT antibodies recognizing phospho‐tau (PHF‐1) or mitotic antigens (TG‐3). Representative light micrographs from the CA1 region of Ammon's horn show a direct increase in the number of PHF‐1‐ and TG‐3‐positive NFT with increasing age (Figure 1). The earliest detection of a few NFT was in the CA1 region in the 4.5‐year‐old case (arbitrarily assigned a “+” in Table 1). The NFT in the 4.5‐year‐old case visualized with either PHF‐1 or TG‐3 looked pretty mature (Figure 1, the inlets). The number of NFT in the entire Ammon's horn region in cases aged up to 10 years (unfortunately, no hippocampal tissue was available in the 6‐year‐old NPC case) did not increase dramatically (Figure 1, 4.5‐ and 10‐year cases), but in cases aged from 14 to 18 years, the number was increased to 10–20, and similar numbers were seen scattered within the entorhinal cortex and parahippocampal gyrus (++, the representative case at 17). In cases between 19 and 25 years of age, the number of NFT in the entire hippocampal region was exponentially increased (+++, Table 1) and even further in cases older than 25 years (++++, the representative cases aged 55 years.), at which point the distribution approached that of a classic stage III AD case (Figure 1, AD case). The NFT visualized with PHF‐1 and TG‐3 antibodies displayed a similar pattern in distribution in each case (Figure 1). However, extracellular neuritic plaques, a characteristic of AD pathological changes, were not observed in any NPC case.
Figure 1.
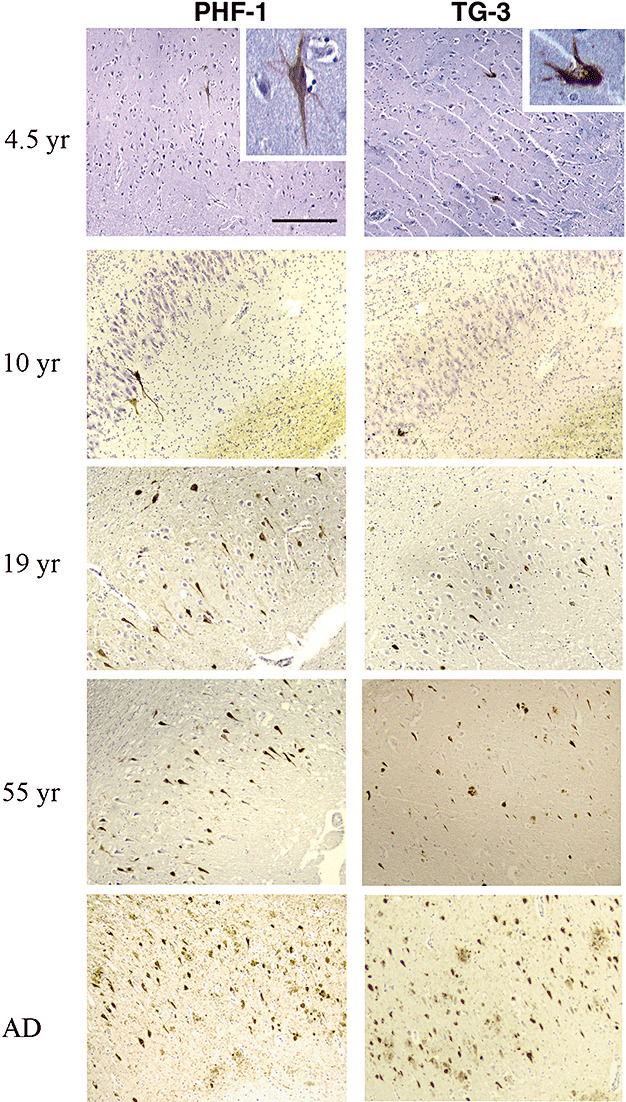
Progression of NFT formation with age in NPC. Paraffin‐embedded sections from hippocampus (for the 6‐year‐old case only the temporal cortex was available) were immunostained with the PHF‐1 and TG‐3 antibodies (brown), and each section was counter‐stained with Hematoxylin to visualize the nuclei (violet) of all cells in a given field. Light micrographs from NPC cases with different ages show the relative numbers and distribution of NFT in CA1 region of hippocampus, which look similar to those in a typical AD case (20×, scale bar = 80 microns).
Typical NFTs were detected in the children NPC cases
In NPC cases aged 4.5 to 10 years, a few NFTs were detected (Figure 2B–F), two to five NFTs in the deepest layer of the CA1/subiculum (Figure 2B,C) and 8–15 NFTs in the CA4 subregion (Figure 2E,F). Similar numbers of scattered NFTs were also observed in the entorhinal cortex and parahippocampal gyrus of these cases. This distribution of NFTs is different from the pattern in AD, characteristic of initial appearance in the entorhinal cortex, followed by the CA1 and subiculum, and then the parahippocampal gyrus and/or CA4 region (3). Most of the NFTs were contained in neurons with elaborate process staining (Figure 2C), suggesting that they were relatively “young,” that is, of the group 2‐type described by Braak et al (2). Scattered group I neurons lacking NFTs were also noted (Figure 2, affected neurons in CA2, arrowhead in C, and CA4, D). Some neurons containing foamy lipid accumulation were stained with both TG‐3 and PHF‐1 antibodies, demonstrating the incorporation of mitotic antigens into NFT (Figure 2F, only TG‐3 shown, arrow). But no frank NFT was visualized with PHF‐1 or TG‐3 staining in hippocampus (Figure 2A). In addition, scattered neuropils were easily observed in the hippocampus in all children cases except the 7‐month‐old case.
Figure 2.
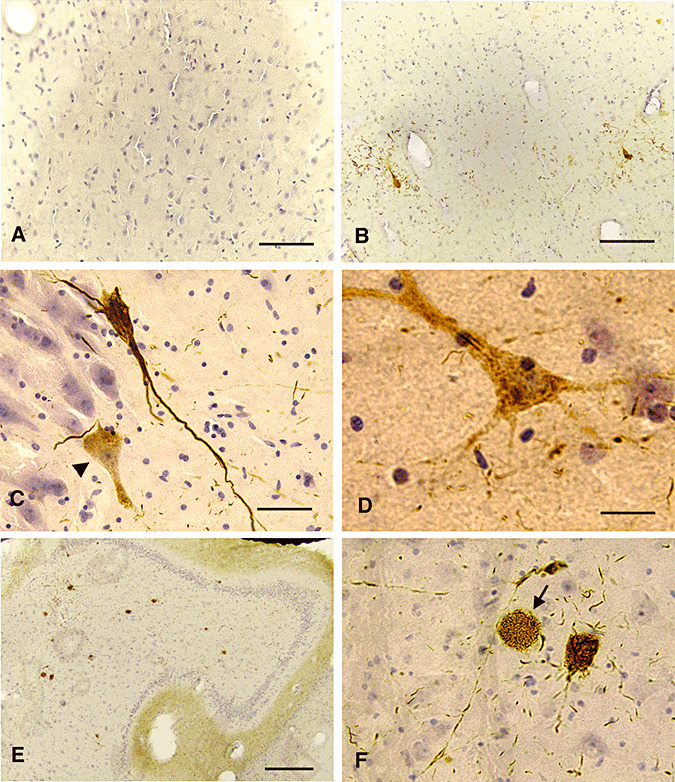
NFT pathology in juvenile NPC. Sections from the hippocampus of the 7‐month‐old (A), the 4.5‐year‐old (B) and an 10‐year‐old NPC cases (C–F) were stained with PHF‐1 (C–D) or TG‐3 (A, B, E, F) and counter‐stained with hematoxylin (A, B, F, bar = 80 microns; C, D, bar = 40 microns; E, bar = 160 microns). PHF‐1 or TG‐3 did not stain the neurons in the 7‐month‐old NPC case (A). A mature NFT was developed in the CA1 region of the 4.5‐year‐old NPC case (B). Similarly, fully‐developed NFT were stained with PHF‐1 in the 10‐year‐old NPC case (C) and some neurons with diffuse PHF‐1 immunoreactivity were seen (arrowhead in C, and D). Increased numbers of NFT were common in the CA4 hippocampal region in the 10‐year‐old NPC case (E), and some neurons with only lipid storage were also noted (F).
The number of NFTs were markedly increased in NPC cases aged 11–25 years
In NPC cases aged 11–25 years, NFT pathology was advanced relative to that observed in the children cases described earlier. NFT and neurons involved in early stages of NFT formation were prominent in the CA1 and CA4 regions (Figure 3B), or even the parahippocampus (Figure 3A,C), and also concentrated in the deepest layers of the CA1/subiculum (Figure 3D). The number of TG‐3‐ and PHF‐1‐positive group 1 neurons, that is, without obvious NFT, was greater than in children cases (Figure 3C,D, respectively). Several NFT‐bearing neurons were contended with lipid storage that was either immunoreactive with TG‐3 (Figure 3C, arrows) or with PHF‐1 (Figure 3D, arrow). However, a significant number of lipid‐containing neurons devoid of TG‐3 or PHF‐1 immunoreactivity (Figure 3C,D, arrowheads).
Figure 3.
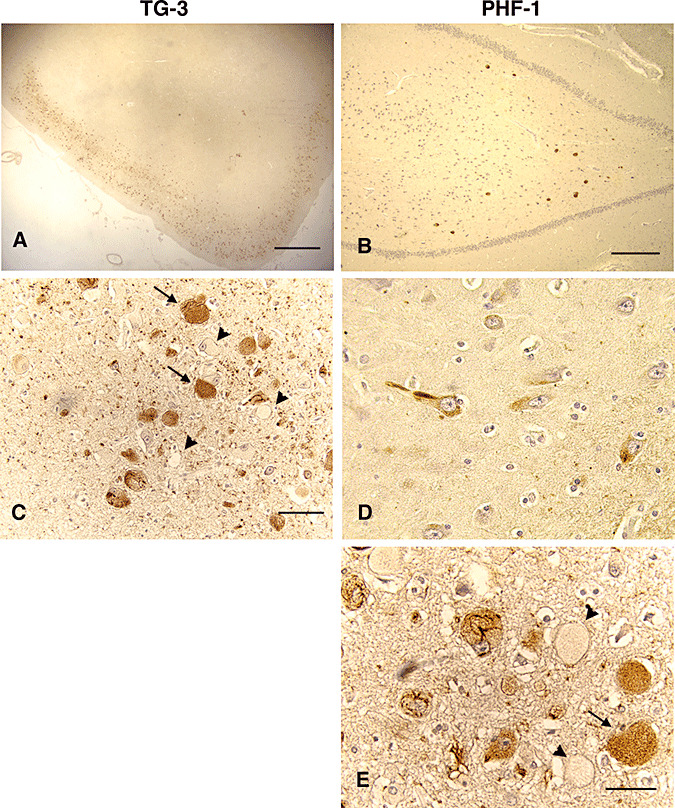
NFT pathology in young adult NPC. Sections from the hippocampus of a 17‐year‐old NPC case were stained with PHF‐1 and TG‐3 and counter‐stained with hematoxylin. Numerous NFT and storage containing neurons were detected in the cortical layer of the parahipocampal cortex (Panels A, scale = 800 microns, and C, bar = 80 microns, both for TG‐3), and some NFT were present in the CA4 region (panel B, bar = 400 microns, PHF‐1). Deeper layers of the CA1/subiculum had scattered NFT and neuronal staining resembling pre‐tangle stages of pathology (panel D, PHF‐1, bar = 40 microns). Both antibodies reacted with NFT and storage material (arrows, panel C for TG‐3, and panel D for PHF‐1), although some storage‐containing neurons were also unstained (arrowheads, panels C and D).
NFT pathology in adult NPC resembled to that in AD
In the adult NPC cases aged over 25 years, the number of NFTs was considerably increased, and NFTs occupied every hippocampal subregion in addition to the CA4 and CA1/subiculum (Figure 4, Bielschowsky silver impregnation, A; TG‐3 staining, B). Interestingly, even the neurons in the dentate granule layer that are somewhat more refractory to AD pathology (38) were visibly involved with NFT (Figure 4B,I). In the adult cases, many unaffected neurons were also apparent, but larger numbers of cells throughout Ammon's horn contained NFTs (Figure 4A, silver impregnation; B, TG‐3 staining). Both classic AD‐type NFT in which the filaments were ordered in the soma along the axis of the apical dendrites (Figure 4C,D, arrows), and large numbers of a more reticular NFT, lacking any orientation within the soma (Figure 4D, arrowheads), were visualized. Neurons distended with storage material in cytoplasm negatively stained with Bielschowsky reagent were common (Figure 4C, arrowheads). Immunohistochemical analysis of the adult NPC cases with TG‐3 enabled visualization of both classic (Figure 4E,F) and reticular (Figure 4G) NFTs as well. Some lipid‐containing neurons were immunostained with TG‐3 (Figure 4E,H, arrow, and H inset), whereas others were not (Figure 4E, arrowhead). Some cells were essentially bulged with TG‐3 immunoreactivity and lipid which displaced the nucleus to one end of the soma (Figure 4H, the inlet), and some neurons contained the beginning of a tangle in the middle (Figure 4H, arrow).
Figure 4.
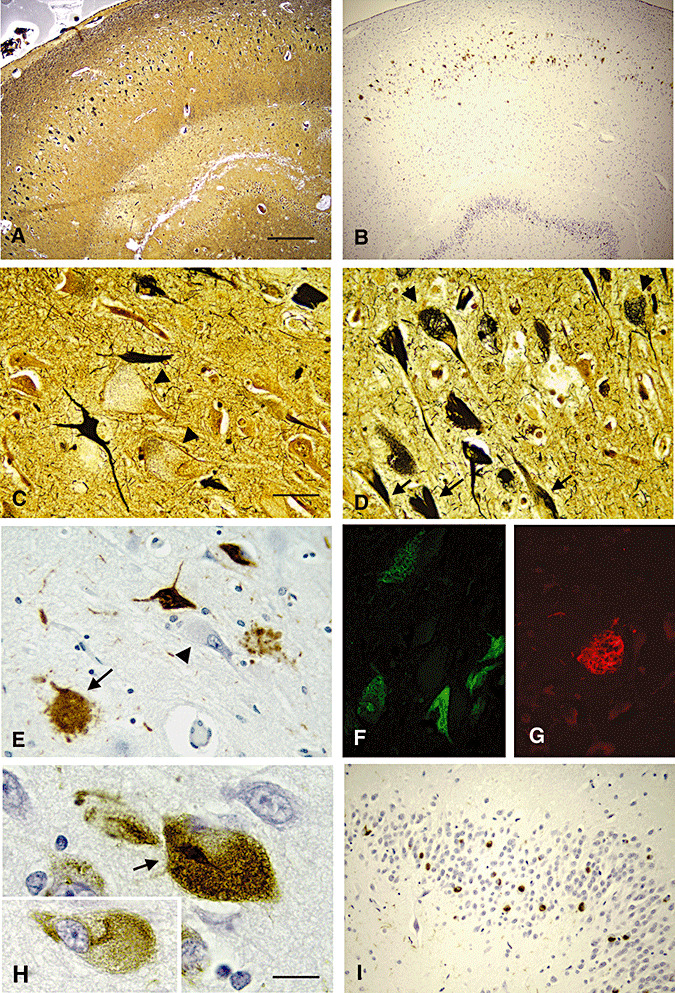
NFT pathology in adult NPC. Hippocampal sections from a 32‐year‐old adult case of NPC were stained either with Bielschowsky silver reagent (panels A, C and D) or TG‐3 (panels B, E–I) (A, B, bar = 160 microns; C–G, bar = 40 microns; H, bar = 15 microns; I, bar = 80 microns). The number and distribution of NFT as visualized with Bielschowsky reagent appeared similar to those with TG‐3 (Panels A and B). Apart from the classic AD‐type NFT (panels C & D, arrows), reticular type NFT were also seen (panel D, arrowheads). A lot of distended neurons with storage were negatively stained by silver impregnation (arrowheads, panel C). Both NFT types were stained with TG‐3 (panels E & F, brown and green respectively for AD‐type; G, red for reticular type). Neurons with lipid storage were prominently stained with TG‐3 (panels E and H arrows), and although most had no visible NFT (panel H, inset), some rare neurons contained storage and small bundles of PHF (panel H, arrow). NFT were also frequent in the dentate granule layer of the hippocampus (panel I).
Mitotic markers were incorporated into NFT
The antibodies specific for mitotic antigens which displayed characteristic fibrillar pathology in AD brain also stained similar changes in NPC brain (Figure 5). The MPM‐2‐recognized mitotic phosphoepitopes were immunoreactive in classic NFT (Figure 5A,D). Phosphorylated RNAP II visualized with the H14 (Figure 5B) and H5 antibodies (Figure 5C,E) was also evident in the classic NFT. Notably, the phosphorylated RNAP II was relocated from its functional compartment in the nuclei of normal cells (Figure 5C, arrowhead) to the cytoplasm of affected neurons (Figure 5C, arrow and inset, and E), as in AD brain (16). The various mitotic antigens were colocalized in affected neurons, as shown for MPM‐2 (Figure 5D) and H5 (Figure 5E). Concomitant with the appearance of various mitotic phosphoepitopes in NPC was the presence of cdc2 kinase (Figure 5F) and cyclin B1 (Figure 5G), which are convinced to generate the mitotic antigens in post‐mitotic neurons. Similarly, the active form of cdc2 was mislocated in the cytoplasm, instead in the nuclei where it will be activated by cyclin B1 and exerts normal functions.
Figure 5.
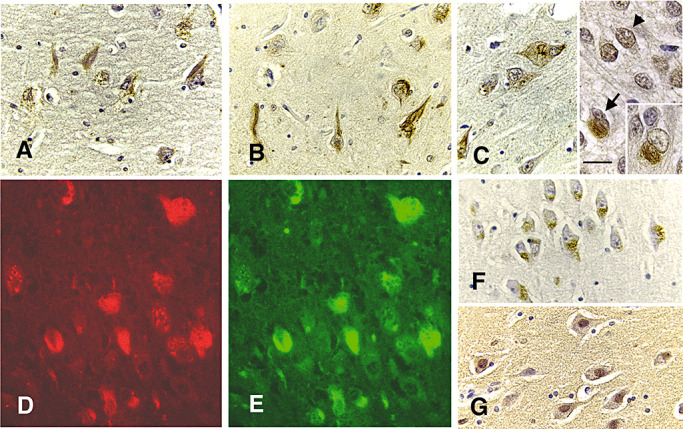
Mitotic epitopes demarcating NFT. Hippocampal sections from a 31‐year‐old case were immunostained with MPM‐2 (panels A, D), H14 (panel B), H5 (panels C, E), cdc2 (panel F) and cyclin B1 (G) and counter‐stained with hematoxylin (all panels, bar = 40 microns). Classic NFT were stained with every antibody (panels A and D for MPM‐2, and panel B for H14, and C and E, H‐5), but cdc2 and cyclin B1 staining of some neurons lacking obvious NFT was more prominent (panels F and G, respectively). The H5 staining shows a shift in distribution of the enzyme from the nucleus (panel C, arrowhead) to the cytoplasm (panel C, arrow, and inset). MPM‐2 (panel D, red) and H5 (panel E, green) colocalize in the same NFT.
The nature of the antigens in NPC NFT immunohistochemically detected above was further confirmed by analyzing the lysates from the hippocampus of two NPC cases and an AD patient using immunoblotting (Figure 6). The PHF‐1‐recognized triplet of tau bands resembling the typical PHF pattern in AD was apparently accumulated in the adult NPC case (adt), but not in the juvenile NPC case (juv) or in the control (C). The phosphorylated tau, labeled by the MC1 and Alz‐50 antibodies recognizing the same primary sequence and conformational‐dependent tau epitope, appeared to be enriched in both NPC cases when compared with the control (Figure 6, shown for MC1 only). Interestingly, in the adult case, the tau bands also appeared elevated in molecular weight, consistent with increased phosphorylation of tau (Figure 6, MC1). However, the total tau, recognized by TG‐5, a primary sequence‐dependent tau antibody, did not present any dramatic difference in the amount in the NPC cases relative to the control, and all the expected tau isoforms were present in both NPC cases and the control (Figure 6, TG‐5).
Figure 6.
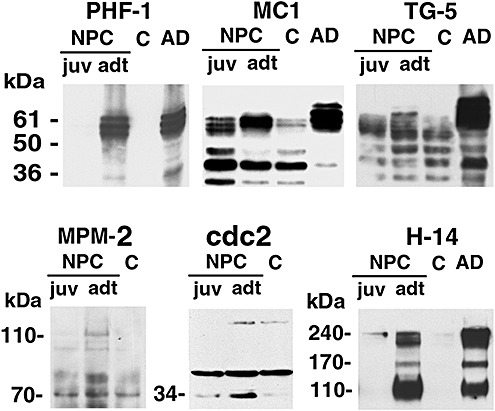
Immunoblot analysis of NFT antigens. Lysates from frozen pieces of hippocampus taken from the 11 year and 31 year old NPC‐1 cases (NPC, juv and adt, respectively), a 23 year old control (C) and an AD case (AD) were subjected to immunoblotting with the indicated antibodies. The PHF‐1 antibody showed the predominant presence of PHF‐tau in the adult NPC case as in AD, but not in the juvenile case. Consistent with this increase in phosphorylation, the MC1 antibody displayed an upward shift in apparent molecular weight of tau in the adult case. The total tau‐immunoreactive protein as detected with TG‐5 antibody appeared no significant change NPC cases when compare to that in the control. A mitotic marker, MPM‐2 detected an expecting band of 110 kD in both NPC cases but not in control. Cdc2 PISTAIRE antibody stained a 34 band accumulated in NPC cases, in which the non‐specific binding band is same in all the samples. H‐14 immunolabeled bands in the adult case similar to those in AD.
In harmony with the immunohistochemical staining, multiple mitotic markers in NPC seemed to be enriched when compared to the control (Figure 6, MPM‐2, cdc2 and H‐14), although no statistical data can be obtained because of the limited cases available. MPM‐2 antibody, which often revealed mitotic phosphoepitopes in different molecular sizes in AD brain (41), showed an enrichment in about 110 kD mitotic phosphoepitopes. H‐14‐recognized bands were accumulated in the adult NPC case, similar to those in the AD brain. Unfortunately, cyclin B1 was not detected in all the samples (not shown).
DISCUSSION
It has been widely accepted that NFTs are lesions associated with the aged brain, although the literature is dotted with reports of NFTs in juvenile cases with rare neurologic conditions, like NPC 27, 39. We have had the opportunity to amass a cohort of NPC cases with age of death ranging from 7 months to 55 years, which has enabled us to examine the relationship between NFT pathology and age of onset and duration of disease. The earliest age at which frank NFT of the AD‐type was detected was 4.5 years, and every case in the childhood category exhibited small number of NFT. These findings reaffirm that advanced age is not a prerequisite for NFT formation, an idea purported previously by Braak and Braak (4). With progression of NPC disease into adulthood, the number of NFT increases exponentially, eventually resembling definitive stage III AD cases (3). The NFTs found in NPC brain include both the classic AD type and the reticular type. With respect to the isoform composition of tau in NPC, we have found that all six isoforms are present and incorporated into PHF (6). Thus, NPC NFTs are more similar to AD NFTs than to other types of filamentous lesions, that is, straight filaments and pick bodies, that result from disproportionate increases of certain tau isoforms 10, 19. With respect to posttranslational modifications, PHF tau incorporated into NPC NFT is also hyperphosphorylated in a similar manner as that from AD NFT. Auer et al previously reported similarities in phosphorylation of serines 198/199, 202, 235, 262 and 396 recognized by the AT270, AT8, AT180, 12E8 and PHF‐1 antibodies respectively (1). Qualitatively, we did not note any differences in the biochemical, structural and immunological profiles of NFT in juvenile and adult cases of NPC. However, we have found that the increase in tau phosphorylation in juvenile NPC cases is too trivial to be detected by Western blotting. In adult cases with more widespread NFT pathology, PHF protein approaches levels of a classic AD case.
The appearance of NFT in children NPC, decades earlier to that in AD, suggests that there are some special cofactors to greatly accelerate the process of NFT formation in NPC brain. The cofactors are unlikely to be tau, because it has been demonstrated that the switch in alternative splicing leading to mature tau occurs in human neurons by 2 years of age 22, 29. Tau is a cytosolic protein and one of the most highly soluble proteins known. In physiological buffer conditions in vitro and at the typical concentrations of tau in neurons (µM or less), the polymerization of tau into PHF is exceedingly slow (9). The rate‐limiting steps in PHF assembly are dimerization and cooperative nucleation of tau, both of which are accelerated by high tau concentrations, increased cellular reducing potential or optimal ratios of tau to cytosolic polyanions. The interplay of these factors or perhaps others may account for the protracted nature of PHF formation. However, it may be possible that NPC neurons having a particular genotypic trait overcome the thermodynamic hurdles for PHF assembly a lot sooner than others. In this respect, we suggest that NPC gene mutations have a more potent effect on tau assembly process than mutations in APP or presenilin genes, trisomy 21 or tau mutations. This would insinuate that cholesterol dysmetabolism exerts a stronger influence on NFT formation than amyloid deposition, which is considered the inducing factor for NFT in AD (23). It has been reported that the content of free cholesterol as measured by semiquantitative filipin fluorescence microscopy in AD and NPC is higher in tangle‐bearing neurons than in tangle‐free neurons (8). We did not directly correlate cholesterol levels with the distribution of NFT, because nearly all of the tissues we obtained for study were paraffin embedded, and deparaffinization of sectioned tissues will dissolve cholesterol. The presence of numerous neurons swollen with lipid storage contained no tangles in contrast to the tangle‐bearing neurons in the same region may insinuate the elusive correlation between the cytoplasmic accumulation of cholesterol and NFT formation (3, 4). Regardless, the connection between NFT formation and disordered cholesterol metabolism is indeed intriguing and has obviously triggered the search for amyloid deposits in NPC. These results show the abnormal deposition of β‐amyloid in late endosomes of NPC fibroblasts and neurons (21), but no frank plaques were obviously observed in the 17 human cases examined here, nor in other papers devoted to the pathology of NPC 27, 36.
Apart from these tau changes, the multiple mitotic markers including the TG‐3, MPM2, H5 and H14 phosphoepitopes, and the mitotic kinase complex composed of the cdc2/cyclin B1 are evidently associated with NFT formation. The mitotic antigens do not normally appear in post‐mitotic neurons, but they are accumulated in AD neurons at risk for developing NFT and eventually incorporated into NFT 16, 40. To date, we have established that mitotic kinase and its phosphorylated substrates are common to other diseases involving NFT, such as Down syndrome, progressive supranuclear palsy and frontotemporal dementia linked to chromosome 17 (15). Despite the array of genetic and nongenetic factors causing these diseases, activation of cdc2/cyclin B1 appears to be a convergent mechanism leading to protein phosphorylation, NFT formation and neuronal death in all of them. The involvement of the activated mitotic kinase does not exclude the possibility that other kinds of kinases will be superimposed in the phosphorylation of the cytoskeletal proteins and the formation of NFT, although the cdk5/25, MAPK and GSK‐3β have been found not to contribute to the cytoskeletal pathology in a murine model of NPC 47, 48.
The increase in mitotic antigens appeared comparable in both juvenile and adult NPC cases, although the accumulation of phosphorylated tau shows a sharp difference at two different ages (Figure 6). We have made similar observations in the cerebellum of human NPC, a region affected early in the disease, and in npc‐1 mutant mice (6). The possible explanation for this discrepancy is that the mitotic antigens in degenerating neurons may appear much earlier than phospho‐tau does, especially in young NPC cases. Unfortunately, we have not obtained enough brain tissue from infantile cases to demonstrate that the appearance of cell cycle markers precedes NFT formation. This possibility could be tested in young npc mice in future studies. It is consistent with previous suggestions that NFTs are not an absolute requirement for neuronal death 11, 31, 45, or may help neurons escape form apoptosis (26). The errant activation of the mitotic cdc2/cyclin B kinase in post‐mitotic neurons wreaks metabolic changes that are sufficient to cause neurodegeneration even in the absence of NFT (15). The possibility was at least partially demonstrated by three independent reports 24, 30, 33. These studies showed that the neurons are destined for neurodegeneration after they are forced to reenter the cell cycle using proto‐oncogens. Especially, the transgenic mice expressing neuronal MYC have developed cognitive deficits, neuronal cell death and gliosis (24). Based on those observations, we suggest that the mitotic cdc2/cyclin B1 kinase may be the major culprit in NPC degenerative process because a large number of neurons will be killed before NFTs develop in both juvenile and adult NPC cases. If it is confirmed that inappropriate cell cycle activation is triggered before cytoskeletal changes and neuronal death, targeting at cdc2/cyclin B1 kinase for abrogating progressive neuronal degeneration could be therapeutically significant in NPC, as well as in AD. The potential therapeutical significance of our findings is also suggested in Woods et al's review, in which developing new agents abrogating the cell cycle to protect the neurons that reentered the cell cycle is advocated (46).
However, we still do not know how the adult neurons in NPC reenter the cell cycle. In somatic cells of NPC patients and animal models, accumulation of unesterified cholesterol is considered as the major biochemical change, but in neurons, multiple neutral lipids are sequestrated. If the reactivation of the cell cycle in the neurons is associated with the alteration in neutral lipids, or directly mediated by mutations in NPC1 or NPC2 gene, more studies are needed.
ACKNOWLEDGMENTS
We are indebted to Drs. M. Elleder, H. Budka, S. Love, W. Pendlebury, S. Shahriar, K. Suzuki, S. Salmat and C. Fligner, and the Kathleen Price Bryan Brain Bank for generously providing us with NPC tissue for the study. We also thank J. Hussemann for his technical assistance. We also appreciate the kind gifts of antibodies from Drs. Peter Davies, David Bregman and Harish C. Pant. This project was supported by a grant from the Education Ministry of China to B.B. (20071108), and the grants from NSFC to B.B. (30670735) and W.W. (30725019) respectively, and a grant from the Seattle Jim Lambright Medical Research Foundation to I.V.
REFERENCES
- 1. Auer IA, Schmidt ML, Lee VM, Curry B, Suzuki K, Shin RW et al (1995) Paired helical filament tau (PHFtau) in Niemann–Pick type C disease is similar to PHFtau in Alzheimer's disease. Acta Neuropathol 90:547–551. [DOI] [PubMed] [Google Scholar]
- 2. Braak E, Braak H, Mandelkow EM (1994) A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol 87:554–567. [DOI] [PubMed] [Google Scholar]
- 3. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 4. Braak H, Braak E (1997) Frequency of stages of Alzheimer‐related lesions in different age categories. Neurobiol Aging 18:351–357. [DOI] [PubMed] [Google Scholar]
- 5. Braak H, Braak E, Bohl J (1993) Staging of Alzheimer‐related cortical destruction. Eur Neurol 33:403–408. [DOI] [PubMed] [Google Scholar]
- 6. Bu B, Klunemann H, Suzuki K, Li J, Bird T, Jin LW, Vincent I (2002) Niemann–Pick disease type C yields possible clue for why cerebellar neurons do not form neurofibrillary tangles. Neurobiol Dis 11:285–297. [DOI] [PubMed] [Google Scholar]
- 7. Bu B, Li J, Davies P, Vincent I (2002) Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann–Pick type C murine model. J Neurosci 22:6515–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Distl R, Meske V, Ohm TG (2001) Tangle‐bearing neurons contain more free cholesterol than adjacent tangle‐free neurons. Acta Neuropathol 101:547–554. [DOI] [PubMed] [Google Scholar]
- 9. Friedhoff P, Von Bergen M, Mandelkow EM, Davies P, Mandelkow E (1998) A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc Natl Acad Sci U S A 95:15712–15717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ghetti B, Murrell JR, Zolo P, Spillantini MG, Goedert M (2000) Progress in hereditary tauopathies: a mutation in the tau gene (G389R) causes a Pick disease‐like syndrome. Ann N Y Acad Sci 920:52–62. [DOI] [PubMed] [Google Scholar]
- 11. Gomez‐Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC et al (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol 41:17–24. [DOI] [PubMed] [Google Scholar]
- 12. Grundke‐Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI (1986) Abnormal phosphorylation of the microtubule‐associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83:4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Halliday W (1995) The nosology of Hallervorden–Spatz disease. J Neurol Sci 134:84–91. [DOI] [PubMed] [Google Scholar]
- 14. Hallows KR (2005) Emerging role of AMP‐activated protein kinase in coupling membrane transport to cellular metabolism. Curr Opin Nephrol Hypertens 14:464–471. [DOI] [PubMed] [Google Scholar]
- 15. Husseman JW, Nochlin D, Vincent I (2000) Mitotic activation: a convergent mechanism for a cohort of neurodegenerative diseases. Neurobiol Aging 21:815–828. [DOI] [PubMed] [Google Scholar]
- 16. Husseman JW, Hallows JL, Bregman DB, Leverenz JB, Nochlin D, Jin LW, Vincent I (2001) Hyperphosphorylation of RNA polymerase II and reduced neuronal RNA levels precede neurofibrillary tangles in Alzheimer disease. J Neuropathol Exp Neurol 60:1219–1232. [DOI] [PubMed] [Google Scholar]
- 17. Hyman BT (1992) Down syndrome and Alzheimer disease. Prog Clin Biol Res 379:123–142. [PubMed] [Google Scholar]
- 18. Iqbal K, Grundke‐Iqbal I (2008) Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics and prevention. J Cell Mol Med 12:38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ishizawa K, Ksiezak‐Reding H, Davies P, Delacourte A, Tiseo P, Yen SH, Dickson DW (2000) A double‐labeling immunohistochemical study of tau exon 10 in Alzheimer's disease, progressive supranuclear palsy and Pick's disease. Acta Neuropathol 100:235–244. [DOI] [PubMed] [Google Scholar]
- 20. Jicha GA, Lane E, Vincent I, Otvos L Jr, Hoffmann R, Davies P (1997) A conformation‐ and phosphorylation‐dependent antibody recognizing the paired helical filaments of Alzheimer's disease. J Neurochem 69:2087–2095. [DOI] [PubMed] [Google Scholar]
- 21. Jin LW, Shie FS, Maezawa I, Vincent I, Bird T (2004) Intracellular accumulation of amyloidogenic fragments of amyloid‐beta precursor protein in neurons with Niemann–Pick type C defects is associated with endosomal abnormalities. Am J Pathol 164:975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kosik KS (1990) Tau protein and Alzheimer's disease. Curr Opin Cell Biol 2:101–104. [DOI] [PubMed] [Google Scholar]
- 23. Launer LJ, White LR, Petrovitch H, Ross GW, Curb JD (2001) Cholesterol and neuropathologic markers of AD: a population‐based autopsy study. Neurology 57:1447–1452. [DOI] [PubMed] [Google Scholar]
- 24. Lee HG, Casadesus G, Nunomura A, Zhu X, Castellani RJ, Richardson SL et al (2009) The neuronal expression of MYC causes a neurodegenerative phenotype in a novel transgenic mouse. Am J Pathol 174:891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee VM (2001) Biomedicine. Tauists and beta‐aptists united—well almost! Science 293:1446–1447. [DOI] [PubMed] [Google Scholar]
- 26. Li HL, Wang HH, Liu SJ, Deng YQ, Zhang YJ, Tian Q et al (2007) Phosphorylation of tau antagonizes apoptosis by stabilizing beta‐catenin, a mechanism involved in Alzheimer's neurodegeneration. Proc Natl Acad Sci U S A 104:3591–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Love S, Bridges LR, Case CP (1995) Neurofibrillary tangles in Niemann–Pick disease type C. Brain 118:119–129. [DOI] [PubMed] [Google Scholar]
- 28. Luna LG (1968) Manual of Histologic Staining Methods of the Armed Forces Institute of Pathology. McGraw‐Hill: New York. [Google Scholar]
- 29. Matus A (1990) Microtubule‐associated proteins and the determination of neuronal form. J Physiol 84:134–137. [PubMed] [Google Scholar]
- 30. McShea A, Lee HG, Petersen RB, Casadesus G, Vincent I, Linford NJ et al (2007) Neuronal cell cycle re‐entry mediates Alzheimer disease‐type changes. Biochim Biophys Acta 1772:467–472. [DOI] [PubMed] [Google Scholar]
- 31. Morsch R, Simon W, Coleman PD (1999) Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol 58:188–197. [DOI] [PubMed] [Google Scholar]
- 32. Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R et al (2000) Identification of HE1 as the second gene of Niemann–Pick C disease. Science 290:2298–2301. [DOI] [PubMed] [Google Scholar]
- 33. Park KH, Hallows JL, Chakrabarty P, Davies P, Vincent I (2007) Conditional neuronal simian virus 40 T antigen expression induces Alzheimer‐like tau and amyloid pathology in mice. J Neurosci 27:2969–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sawamura N, Gong JS, Garver WS, Heidenreich RA, Ninomiya H, Ohno K et al (2001) Site‐specific phosphorylation of tau accompanied by activation of mitogen‐activated protein kinase (MAPK) in brains of Niemann–Pick type C mice. J Biol Chem 276:10314–10319. [DOI] [PubMed] [Google Scholar]
- 35. Scriver CR (2001) The Metabolic and Molecular Bases of Inherited Disease, 8th edn. McGraw‐Hill: New York. [Google Scholar]
- 36. Suzuki K, Parker CC, Pentchev PG, Katz D, Ghetti B, D’Agostino AN, Carstea ED (1995) Neurofibrillary tangles in Niemann–Pick disease type C. Acta Neuropathol 89:227–238. [DOI] [PubMed] [Google Scholar]
- 37. Takada K, Becker LE (1986) Cockayne's syndrome: report of two autopsy cases associated with neurofibrillary tangles. Clin Neuropathol 5:64–68. [PubMed] [Google Scholar]
- 38. Tolnay M, Spillantini MG, Goedert M, Ulrich J, Langui D, Probst A (1997) Argyrophilic grain disease: widespread hyperphosphorylation of tau protein in limbic neurons. Acta Neuropathol 93:477–484. [DOI] [PubMed] [Google Scholar]
- 39. Vanier MT, Suzuki K (1998) Recent advances in elucidating Niemann–Pick C disease. Brain Pathol 8:163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vincent I, Jicha G, Rosado M, Dickson DW (1997) Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci 17:3588–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vincent I, Zheng JH, Dickson DW, Kress Y, Davies P (1998) Mitotic phosphoepitopes precede paired helical filaments in Alzheimer's disease. Neurobiol Aging 19:287–296. [DOI] [PubMed] [Google Scholar]
- 42. Vincent I, Bu B, Hudson K, Husseman J, Nochlin D, Jin L (2001) Constitutive Cdc25B tyrosine phosphatase activity in adult brain neurons with M phase‐type alterations in Alzheimer's disease. Neuroscience 105:639–650. [DOI] [PubMed] [Google Scholar]
- 43. Warren SL, Landolfi AS, Curtis C, Morrow JS (1992) Cytostellin: a novel, highly conserved protein that undergoes continuous redistribution during the cell cycle. J Cell Sci 103:381–388. [DOI] [PubMed] [Google Scholar]
- 44. Weaver CL, Espinoza M, Kress Y, Davies P (2000) Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging 21:719–727. [DOI] [PubMed] [Google Scholar]
- 45. Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293:711–714. [DOI] [PubMed] [Google Scholar]
- 46. Woods J, Snape M, Smith MA (2007) The cell cycle hypothesis of Alzheimer's disease: suggestions for drug development. Biochim Biophys Acta 1772:503–508. [DOI] [PubMed] [Google Scholar]
- 47. Zhang M, Li J, Chakrabarty P, Bu B, Vincent I (2004) Cyclin‐dependent kinase inhibitors attenuate protein hyperphosphorylation, cytoskeletal lesion formation, and motor defects in Niemann–Pick type C mice. Am J Pathol 165:843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang M, Hallows JL, Wang X, Bu B, Wang W, Vincent I (2008) Mitogen‐activated protein kinase activity may not be necessary for the neuropathology of Niemann–Pick type C mice. J Neurochem 107:814–822. [DOI] [PubMed] [Google Scholar]