

Abstract
Background
Depressive disorders are the most common psychiatric comorbidity in people with epilepsy, affecting around one‐third, with a significant negative impact on quality of life. There is concern that people may not be receiving appropriate treatment for their depression because of uncertainty regarding which antidepressant or class works best, and the perceived risk of exacerbating seizures. This review aimed to address these issues, and inform clinical practice and future research.
This is an updated version of the original Cochrane Review published in Issue 12, 2014.
Objectives
To evaluate the efficacy and safety of antidepressants in treating depressive symptoms and the effect on seizure recurrence, in people with epilepsy and depression.
Search methods
For this update, we searched CRS Web, MEDLINE, SCOPUS, PsycINFO, and ClinicalTrials.gov (February 2021). We searched the World Health Organization Clinical Trials Registry in October 2019, but were unable to update it because it was inaccessible. There were no language restrictions.
Selection criteria
We included randomised controlled trials (RCTs) and prospective non‐randomised studies of interventions (NRSIs), investigating children or adults with epilepsy, who were treated with an antidepressant and compared to placebo, comparative antidepressant, psychotherapy, or no treatment for depressive symptoms.
Data collection and analysis
The primary outcomes were changes in depression scores (proportion with a greater than 50% improvement, mean difference, and proportion who achieved complete remission) and change in seizure frequency (mean difference, proportion with a seizure recurrence, or episode of status epilepticus). Secondary outcomes included the number of participants who withdrew from the study and reasons for withdrawal, quality of life, cognitive functioning, and adverse events.
Two review authors independently extracted data for each included study. We then cross‐checked the data extraction. We assessed risk of bias using the Cochrane tool for RCTs, and the ROBINS‐I for NRSIs. We presented binary outcomes as risk ratios (RRs) with 95% confidence intervals (CIs) or 99% CIs for specific adverse events. We presented continuous outcomes as standardised mean differences (SMDs) with 95% CIs, and mean differences (MDs) with 95% CIs.
Main results
We included 10 studies in the review (four RCTs and six NRSIs), with 626 participants with epilepsy and depression, examining the effects of antidepressants. One RCT was a multi‐centre study comparing an antidepressant with cognitive behavioural therapy (CBT). The other three RCTs were single‐centre studies comparing an antidepressant with an active control, placebo, or no treatment. The NRSIs reported on outcomes mainly in participants with focal epilepsy before and after treatment for depression with a selective serotonin reuptake inhibitor (SSRI); one NRSI compared SSRIs to CBT.
We rated one RCT at low risk of bias, three RCTs at unclear risk of bias, and all six NRSIs at serious risk of bias. We were unable to conduct any meta‐analysis of RCT data due to heterogeneity of treatment comparisons. We judged the certainty of evidence to be moderate to very low across comparisons, because single studies contributed limited outcome data, and because of risk of bias, particularly for NRSIs, which did not adjust for confounding variables.
More than 50% improvement in depressive symptoms ranged from 43% to 82% in RCTs, and from 24% to 97% in NRSIs, depending on the antidepressant given. Venlafaxine improved depressive symptoms by more than 50% compared to no treatment (mean difference (MD) ‐7.59 (95% confidence interval (CI) ‐11.52 to ‐3.66; 1 study, 64 participants; low‐certainty evidence); the results between other comparisons were inconclusive. Two studies comparing SSRIs to CBT reported inconclusive results for the proportion of participants who achieved complete remission of depressive symptoms.
Seizure frequency data did not suggest an increased risk of seizures with antidepressants compared to control treatments or baseline. Two studies measured quality of life; antidepressants did not appear to improve quality of life over control. No studies reported on cognitive functioning.
Two RCTs and one NRSI reported comparative data on adverse events; antidepressants did not appear to increase the severity or number of adverse events compared to controls. The NSRIs reported higher rates of withdrawals due to adverse events than lack of efficacy. Reported adverse events for antidepressants included nausea, dizziness, sedation, headache, gastrointestinal disturbance, insomnia, and sexual dysfunction.
Authors' conclusions
Existing evidence on the effectiveness of antidepressants in treating depressive symptoms associated with epilepsy is still very limited. Rates of response to antidepressants were highly variable. There is low certainty evidence from one small RCT (64 participants) that venlafaxine may improve depressive symptoms more than no treatment; this evidence is limited to treatment between 8 and 16 weeks, and does not inform longer‐term effects. Moderate to low evidence suggests neither an increase nor exacerbation of seizures with SSRIs.
There are no available comparative data to inform the choice of antidepressant drug or classes of drug for efficacy or safety for treating people with epilepsy and depression.
RCTs of antidepressants utilising interventions from other treatment classes besides SSRIs, in large samples of patients with epilepsy and depression, are needed to better inform treatment policy. Future studies should assess interventions across a longer treatment duration to account for delayed onset of action, sustainability of treatment responses, and to provide a better understanding of the impact on seizure control.
Plain language summary
Antidepressants for people with epilepsy and depression
Background
Depressive disorders occur in approximately one‐third of people with epilepsy, often requiring antidepressant treatment. However, depression often goes untreated in people with epilepsy, partly due to fear that antidepressants might cause seizures. There are different classes of antidepressants, however they all aim to increase key nerve chemicals in the brain, thereby alleviating depressive symptoms.
Characteristics of studies
We found ten studies that included 626 patients with epilepsy and depression treated with an antidepressant. Four were randomised controlled trials, and six were non‐randomised prospective cohort studies. The studies observed the effect of different antidepressants, mainly a class of antidepressant called a selective serotonin reuptake inhibitor (SSRI). One randomised controlled trial and one prospective study also observed the effect of cognitive behavioural therapy on depression.
Results
Taking all the evidence into account, the review found that there is very limited evidence that antidepressants decrease depressive symptoms more than other treatments, placebo, or no treatment in epilepsy. There was limited information on the effect of antidepressants on seizure control, however in the studies reporting this outcome there did not appear to be any significant worsening of seizures. The evidence is current to February 2021.
Quality of the studies
We assessed the studies with regard to bias and quality. Overall, the quality of the evidence was rated as moderate to low for the clinical trials and low to very low for the non‐randomised prospective cohort studies. Large, high quality trials of antidepressants are needed to examine how different classes of antidepressant compare, and what impact they are likely to have on seizure control.
Summary of findings
Background
This is an updated version of a Cochrane Review published in Issue 12, 2014 (Maguire 2014).
Description of the condition
Depressive disorders are the most common psychiatric comorbidity in people with epilepsy (Tellez‐Zenteno 2007), and they are the strongest predictor of poor quality of life (Boylan 2004). Symptoms of depression include low mood, tiredness, and apathy. Sleep and cognitive functioning may also be affected. Depressive disorders occur in approximately one‐third of the people with epilepsy (Baker 1996; Indaco 1992; Jacoby 1996; Mendez 1986). These disorders are broadly divided into unipolar (depression only) and bipolar disorders (depression associated with mania or hypomania; (APA 2000)). Depressive disorders in epilepsy may be mediated via the interplay of neurobiological, psychosocial, and iatrogenic factors (Lambert 1999). Depressive symptoms or episodes may occur inter‐ictally (i.e. they appear unrelated to seizures) or peri‐ictally (preceding, during, or following seizures). This is an important distinction, as a person may require modification of his or her antiepileptic drug regimen, commencement of antidepressant drug therapy, or both. In some people, the depressive symptoms may follow a significant period of seizure remission in previously uncontrolled epilepsy, thought to occur via neuro‐biochemical changes, and termed 'forced normalisation' (Trimble 1998). Studies examining clinical predictors of risk for depression in people with epilepsy have produced inconsistent results (Lin 2012). There is a perceived greater risk of depression in people with temporal lobe epilepsy, although elevated rates of depression have been found in generalised and extra‐temporal focal epilepsy (Adams 2008). Epilepsy‐related factors as predictors of risk for depression are inconsistent. Psychosocial factors, such as life stress, coping style, social support, perceived stigma, and personality are more consistent predictors of depression in people with epilepsy (Hermann 2000).
In 2008, the Food and Drug Administration issued a health alert about an increased risk of suicidal ideation in people taking antiepileptic drugs (Hesdorffer 2009). This alert was based on a meta‐analysis of approximately 28,000 participants who had participated in randomised controlled trials (RCTs) investigating 11 antiepileptic drugs. There were four completed suicides, all of whom had taken antiepileptic drugs, compared to no suicides in the placebo groups (odds ratio (OR) 1.8; 95% confidence interval (CI) 1.24 to 2.66). Since this alert, a number of observational studies have investigated the association, reporting conflicting results, and the International League Against Epilepsy (ILAE) Commission on Neuropsychobiology a has published a consensus statement on the risk of suicide with antiepileptic drugs (Mula 2013). Whilst the exact risk of suicide with antiepileptic drugs is unknown, depression, as a treatment emergent adverse effect, is associated with some antiepileptic drugs (GABAergic antiepileptic drugs: benzodiazepines, vigabatrin, gabapentin; and also topiramate, levetiracetam, and zonisamide; (Mula 2009)). Other antiepileptic drugs appear to have mood‐stabilising properties (valproic acid, lamotrigine, carbamazepine, oxcarbazepine), which may benefit people with epilepsy and depression. Enzyme‐inducing antiepileptic drugs (i.e. carbamazepine) may lower plasma levels of antidepressants, thus impacting on their effectiveness.
Case control studies have shown that participants with depression have a two‐ to seven‐fold higher risk of developing epilepsy, implying a bi‐directional relationship (Hesdorffer 2000; Hesdorffer 2006; Hesdorffer 2012). A number of factors may explain this, for example shared pathophysiology involving disturbances in several key neurotransmitter systems (Bagdy 2007), structural lesions (frontal lobe tumours), or a genetic susceptibility. However, there is also the possibility that the use of antidepressants may trigger seizures. This is a common concern for healthcare professionals, and may influence their decisions to start antidepressant treatment (Cotterman‐Hart 2010).
Description of the intervention
Antidepressants are a heterogeneous class of drugs that have been the mainstay of pharmacological treatment for depressive disorders. There are 10 classes of antidepressants used to treat depressive disorders, with 60% to 70% of depressive episodes responding to current treatment (Klerman 1990; Sackeim 2006). These are:
tricyclic antidepressants;
selective serotonin reuptake inhibitors;
serotonin‐norepinephrine reuptake inhibitors;
monoamine oxidase inhibitors;
serotonin/antagonist reuptake inhibitors (i.e. trazodone);
dopamine and norepinephrine reuptake inhibitors (i.e. bupropion);
a‐2 antagonists (i.e. mirtazapine);
norepinephrine reuptake inhibitors (i.e. reboxetine);
selective serotonin reuptake enhancers (i.e. tianeptine); and
serotonin 5HT₂C receptor antagonists (i.e. agomelatine).
These drugs work by targeting serotonergic, or noradrenergic, or dopaminergic neurotransmission, or a combination, with the aim of increasing their synaptic concentrations (Stahl 2000). Glutamate antagonists represent a novel class of drug currently being tested in refractory depression (Zarate 2006).
The risk of seizures with antidepressants was reported in early studies of the first generation antidepressants, notably tricyclic antidepressants (Preskorn 1992; Wroblewski 1990). Alper 2007 reviewed the incidence of seizures in 75,000 non‐epileptic participants in phase II and phase III trials of antidepressant treatment. They reported lower incidence rates of seizures in those randomised to an antidepressant versus placebo (standardised incidence ratio 0.48; 95% CI 0.36 to 0.61). Coupland 2011 examined 60,746 primary care participants aged 65 and over, treated for depression with antidepressants, between 1996 and 2007, and showed increased risks of epilepsy or seizures for selective serotonin reuptake inhibitors (hazard ratio (HR) 1.80; 95% CI 1.32 to 2.43), and other antidepressant classes (HR 2.20; 95% CI 1.46 to 3.30) versus tricyclic antidepressants. Venlafaxine was associated with the highest risk of seizures.
How the intervention might work
There appears to be a significant relationship between epilepsy and depression. From studies, it is emerging that they share common neurobiological substrates that involve hyperactivity of the hypothalamic pituitary adrenal axis, and the disturbance of different neurotransmitter systems, mainly serotonin and norepinephrine (Dell'osso 2013). The density of serotonin receptors is high in the mesial temporal and prefrontal areas (Gilliam 2005b). In critical brain regions, such as the limbic system and prefrontal areas, enforced serotonergic circuits seem to be responsible for increasing seizure threshold (Kondziella 2009).
Antidepressants of the selective serotonin reuptake inhibitor family have been reported to be safe in treating depression in people with epilepsy, and to possess antiepileptic properties in animal models of epilepsy (Hamid 2013). Based on clinical data, it has been suggested that selective serotonin reuptake inhibitors can decrease the seizure frequency in refractory epilepsy (Kondziella 2009). This is believed to be due to the increase in the concentration of serotonin. The study shows that the concentration of endogenous serotonin (5‐HT) and the activity of its receptor subtypes, 5–HT(1A), 5‐HT(2C), 5‐HT(3), and 5–HT(7), play a significant role in the pathogenesis of epilepsies (Bagdy 2007). Therefore, medications with serotonin agonist and antagonist properties can play a significant role in the pathogenesis of epilepsies.
Why it is important to do this review
Depression is common in people with epilepsy, and has a significant negative impact on quality of life (Gilliam 2005b; Kondziella 2009). There is concern that they may not be receiving appropriate treatment for their depression because of uncertainty around which antidepressant, or class, works best, and the perceived risk of exacerbating seizures. This review aims to address these issues, and to inform clinical practice and future research.
Objectives
To evaluate the efficacy and safety of antidepressants in treating depressive symptoms and the effect on seizure recurrence, in people with epilepsy and depression.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs)
-
Prospective non‐randomised cohort controlled and uncontrolled studies (NRSI; with a control group including participants acting as their own control group (i.e. before‐after studies)).
We considered prospective non‐randomised cohort studies in this review because of the known delayed effect of antidepressants on depressive symptoms, which may not be effectively detected in short‐term randomised trials. Similarly, prospective non‐randomised studies are more likely to recruit populations of participants who better reflect clinical practice, since depression can affect any person with epilepsy.
Types of participants
We considered participants who satisfied all of the following criteria:
any age;
diagnosis of epilepsy (any type);
treated with antidepressants for co‐existing depression (including participants with major depressive disorder, adjustment disorder, and dysthymic disorder), based on standardised criteria, according to participant scores on validated tools, or both (e.g. Hamilton Rating Scale for Depression).
Types of interventions
Intervention group: participants who received an antidepressant drug in addition to an existing antiepileptic drug regimen
Control group(s): participants who received a placebo, comparative antidepressant, psychotherapy, or no treatment in addition to an existing antiepileptic drug regimen
Types of outcome measures
Primary outcomes
-
Depression scores
The proportion of participants with a greater than 50% improvement in depressive symptoms (defined as a 'response') compared to baseline
Mean difference in depression scores following treatment (compared to baseline or between‐group comparison)
The proportion of participants achieving complete remission of depressive symptoms
If the data allowed, we planned to analyse outcomes at ≤ 12 weeks (short‐term), 13 to 26 weeks (medium‐term), and ≥ 26 weeks (long‐term); however, we were unable to perform these analyses.
-
Change in seizure frequency
The mean difference in seizure frequency
The proportion of participants with a seizure recurrence
The proportion of participants with an episode of status epilepticus
Secondary outcomes
-
Withdrawals
For specific reasons
For any reasons
-
Global state
Clinically important change in global state (as defined by the individual studies)
Relapse (as defined by the individual studies)
-
Mental state
Clinically important change in general mental state score
General mental state score (average and end point)
Clinically important change in specific symptoms (sleep, anhedonia, suicidal ideas)
Specific symptom score (average and end point)
-
General functioning
Clinically important change in general functioning
General functioning score (average and end point)
-
Cognitive functioning
Clinically important change in overall cognitive functioning
Overall cognitive functioning score (end point and average)
Clinically important change in specific cognitive functioning (attention, concentration, memory, language, executive functioning)
Specific cognitive score (average and end point)
-
Quality of life
Clinically important change in quality of life
Any change in quality of life score (average and end point)
-
Behaviour
Clinically important change in general behaviour
Any important change in general behaviour (average and end point)
Clinically important change in specific aspects of behaviour
Any important change in specific aspects of behaviour score (average and end point)
-
Adverse effects
Death
Any non‐serious general adverse effects (e.g. gastrointestinal effects, anorexia, dizziness, dry mouth, insomnia, sexual dysfunction, hypotension)
Any serious, specific adverse effects (hypersensitivity reaction)
Any change in general adverse effect score (average and end point)
Clinically important change in specific adverse effects
Any change in specific adverse effects score (average and end point)
Search methods for identification of studies
Electronic searches
We ran searches for the original review in March 2013. We ran subsequent searches in May 2014, October 2016, July 2018, and October 2019. For the latest update, we searched the following databases.
Cochrane Central Register of Controlled Trials (CENTRAL), in CRS Web (searched 1 February 2021), using the search strategy shown in Appendix 1;
MEDLINE Ovid (1946 to 29 January 2021), using the search strategy shown in Appendix 2;
SCOPUS (1823 to 1 February 2021), using the search strategy shown in Appendix 3;
PsycINFO EBSCOhost (1887 to 1 February 2021), using the search strategy shown in Appendix 4;
ClinicalTrials.gov (searched 1 February 2021), using the search strategy shown in Appendix 5;
WHO International Clinical Trials Registry Platform (ICTRP; searched 22 October 2019), using the search strategy shown in Appendix 6. We were unable to update this search because the ICTRP website was inaccessible.
CRS Web includes randomised or quasi‐randomised, controlled trials from the Specialized Registers of Cochrane Review Groups, including Epilepsy, CENTRAL, PubMed, Embase, ClinicalTrials.gov, and ICTRP.
There were no language restrictions.
Searching other resources
We checked the reference lists of retrieved studies for additional reports of relevant studies.
We also contacted lead study authors for any relevant unpublished material.
We identified duplicate studies by screening reports according to title, authors’ names, location, and medical institute, omitting any duplicated studies.
We identified any grey literature studies published in the last five years by searching: 1. Zetoc database; 2. ISI Proceedings; 3. International Bureau for Epilepsy (IBE) congress proceedings database; 4. International League Against Epilepsy (ILAE) congress proceedings database; 5. Abstract books of symposia and congresses, meeting abstracts, and research reports
Data collection and analysis
Selection of studies
Two authors (MM, SN) independently assessed all citations generated from the searches for inclusion. Where disputes arose, we acquired the full report for more detailed scrutiny.
Data extraction and management
Two authors (MM, SJN) undertook separate data extraction for each included study. We then cross‐checked the data extraction. We extracted data using pre‐standardised data extraction forms. We discussed any disagreement, documented decisions, and if necessary, contacted trialists for clarification.
We extracted the following information from the included studies.
Methodological and trial design
Year of publication
Number of study centres
Language
Industry funding
Study design (RCT, prospective cohort study, retrospective cohort study)
Blinding
Type of control group (placebo, comparative antidepressant, no treatment)
Sample size
Follow‐up period
Class of antidepressant as intervention
Dose range of intervention
Inclusion and exclusion criteria
Participant demographic information
Age range
Number of male/female participants
Duration of epilepsy
Previous number of antiepileptic drugs
Epilepsy type (focal, generalised, unclassified)
Location of epilepsy (temporal, extra‐temporal)
Baseline mean depression score or severity
Baseline mean seizure frequency/month
Outcomes
The number of participants experiencing each outcome recorded per treatment group
Number of dropouts
Assessment of risk of bias in included studies
Two review authors (SJN, MJM) independently assessed the risk of bias for the included studies.
Due to the non‐randomised design of some studies, we assessed risk of bias for non‐randomised studies using the ROBINS‐I tool (Sterne 2016). This tool considers seven domains of bias: two domains of bias pre‐intervention (bias due to confounding and bias in selection of participants into the study); one domain of bias at intervention (bias in the measurement of interventions); and four domains of bias post‐intervention (bias due to departures from intended interventions, bias due to missing data, bias in measurement of outcomes, and bias in selection of the reported result). We planned to perform a separate 'Risk of bias' assessment for each outcome of interest in the study.
Important confounders of interest in this Cochrane Review included:
mean age;
epilepsy type (focal or generalised);
mean duration of epilepsy;
location of epilepsy;
mean baseline seizure frequency;
mean baseline depression score.
Each domain of bias contained signalling questions to facilitate judgements of risk of bias. The response options for the signalling questions were: yes; probably yes; probably no; no; and no information. We specified the signalling questions for each domain in Appendix 7.
The 'Risk of bias' judgement options for each domain were:
low risk of bias: the study is comparable to a well‐performed randomised trial with regard to this domain;
moderate risk of bias: the study is sound for a non‐randomised study with regard to this domain, but cannot be considered comparable to a well‐performed randomised trial;
serious risk of bias: the study has some important problems in this domain;
critical risk of bias: the study is too problematic in this domain to provide any useful evidence on the effects of the intervention;
no information on which to base a judgement about risk of bias for this domain.
We presented guidance for an overall risk of bias for a study, based on outcomes from 'Risk of bias' judgements of each domain, in Table 7.
1. Criteria for overall risk of bias judgements from ROBINS‐I.
| Risk of bias judgement | Criteria based on seven risk of bias domains |
| Low risk of bias: the study is comparable to a well‐performed randomised trial | The study is judged to be at low risk of bias for all domains |
| Moderate risk of bias: the study appears to provide sound evidence for a non‐randomised study but cannot be considered comparable to a well‐performed randomised trial | The study is judged to be at low or moderate risk of bias for all domains |
| Serious risk of bias: the study has some important problems | The study is judged to be at serious risk of bias in at least 1 domain, but not at critical risk of bias in any domain |
| Critical risk of bias: the study is too problematic to provide any useful evidence on the effects of intervention | The study is judged to be at critical risk of bias in at least 1 domain |
| No information on which to base a judgement about risk of bias | There is no clear indication that the study is at serious or critical risk of bias, and there is a lack of information in 1 or more key domains of bias (a judgement is required for this) |
For RCTs, we assessed all domains of the Cochrane tool for assessing risk of bias (Higgins 2011). We rated each of the following six domains as low, high, or unclear risk of bias: method of generating random sequence, allocation concealment, blinding methods, incomplete outcome data, selective outcome reporting, and other sources of bias.
The two review authors resolved any discrepancies in the 'Risk of bias' judgements by discussion.
Measures of treatment effect
For binary outcomes (50% or greater improvement in depressive symptoms, complete remission of depressive symptoms, and % treatment withdrawal), we presented results as risk ratios (RR) with 95% confidence interval (CI). To allow for multiple statistical testing, we presented RRs with 99% CIs for specific adverse events.
For continuous outcomes (mean change in depression score), we presented results as mean differences (MD) or standardised mean differences (SMD) with 95% CIs.
Unit of analysis issues
Studies using a variety of depression measures created issues when we wanted to combine results in a meta‐analysis. Where appropriate, we used the SMD to allow for these variances.
Dealing with missing data
We sought missing statistics from studies through contact with the study authors. We sought reasons for missing data to determine whether the data were missing at random or not. We found no data missing at random.
Assessment of heterogeneity
We assessed clinical heterogeneity by comparing the distribution of important participant factors between studies (age, epilepsy type, duration of epilepsy, baseline depression score, baseline seizure frequency) and trial factors (study design, type of control group, antidepressant drug class, type of depression disorder). We assessed statistical heterogeneity by using the I² statistic. We considered an I² value of 75% or more indicated considerable heterogeneity, 50% to 90% indicated substantial heterogeneity, and 30% to 60% indicated moderate heterogeneity. If the I² value was 75% or more, we made an a priori decision not to carry out meta‐analysis; instead, we used a narrative form for the review, and discussed all comparisons according to the findings presented within the studies. We planned meta‐regression techniques, where possible, to investigate possible sources of heterogeneity, however, we were unable to investigate this within this review.
Assessment of reporting biases
1. Protocol versus full study
We investigated outcome reporting bias using the ORBIT classification system, allocating studies a letter from A to I if we suspected the presence of selective outcome reporting bias (Kirkham 2010).
2. Funnel plot
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Higgins 2020; Sterne 2000). Funnel plots can be used to investigate reporting biases, but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes for which there were 10 or fewer studies, or when all studies were of similar sizes.
Data synthesis
We synthesised data using the RR, the MD, or the SMD, depending on the measures used in both the controlled and uncontrolled studies. We carried out a sensitivity analysis to check for differences between a random‐effects model and fixed‐effect model in influencing conclusions. If differences between the models existed, we intended to report outcomes based on the random‐effects model, which incorporates an assumption that the different studies are estimating different, yet related, intervention effects.
For controlled studies, we intended to carry out meta‐analysis using the Mantel‐Haenszel method for dichotomous outcomes, and the inverse variance method for continuous outcomes. For before‐after studies, we used the inverse variance methods for continuous outcomes in meta‐analysis.
We did not combine data for outcomes measured in both randomised and non‐randomised studies. We reported combined data on outcomes for randomised and non‐randomised studies separately.
We stratified each comparison by type of control group, study design, study characteristics, or a combination, to ensure appropriate combination of study data.
Subgroup analysis and investigation of heterogeneity
Where possible, we planned to stratify subgroup analysis by antidepressant drug class, epilepsy type, and age. For investigation of heterogeneity, please see Assessment of heterogeneity.
Sensitivity analysis
We intended to carry out sensitivity analysis if peculiarities in study quality were found (Assessment of risk of bias in included studies). We planned to report the analysis for all studies, and then compare this to an analysis of only studies at low risk of bias. However, as we could not combine RCTs in meta‐analysis, and we judged all NRSIs to be at serious risk of bias, we did not perform any sensitivity analyses.
Summary of findings and assessment of the certainty of the evidence
We made an overall summary judgement of risk of bias for each study per outcome, followed by an overall judgement per outcome across studies. We had planned to incorporate the 'Risk of bias' judgements into the analysis using a sensitivity analysis, so that a secondary analysis of the data included only studies rated as low risk of bias. However, we were unable to do this due to the small amount of studies and lack of data. We presented both results in the Results section of the review. Where applicable, we created 'Summary of findings' tables for outcomes, and graded each outcome using the GRADE approach (Guyatt 2008). Outcomes reported in 'Summary of findings' tables include: depression scores, seizure frequency, withdrawals, cognitive functioning, quality of life, and adverse effects.
Results
Description of studies
Results of the search
We identified 18 potentially eligible studies. We made our final assessment of eligibility by checking the full text of the reports. Figure 1 outlines the flow diagram of search results, eligible records, and study exclusions.
1.
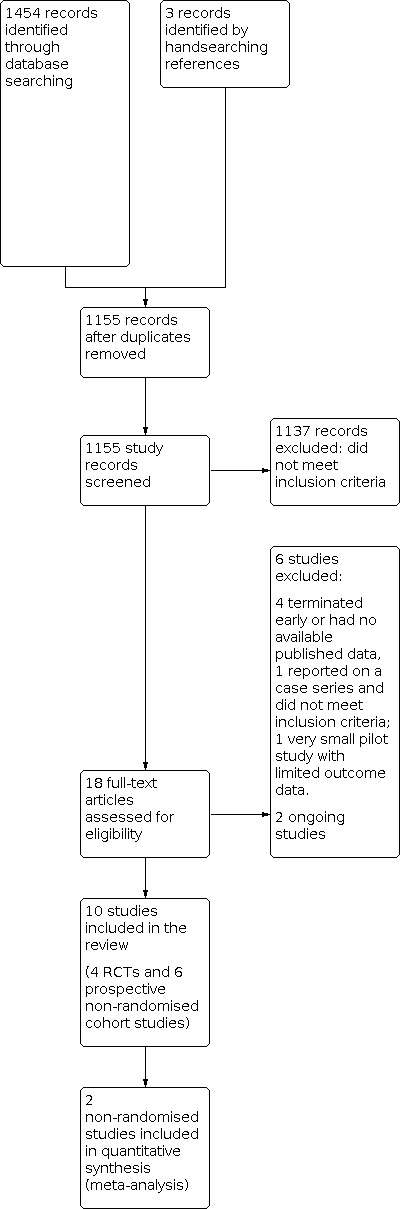
Study flow diagram
We excluded six studies; three studies did not meet the inclusion criteria, and a further three studies met the inclusion criteria but did not report any results for any of the primary and secondary outcomes. We attempted to contact trial authors of these three studies, but received no response.
Two studies are currently ongoing (EUCTR2017‐000990‐35‐IT; EUCTR2018‐003464‐32‐HU).
Included studies
Of the ten remaining studies, four were randomised controlled trials and six non‐randomised prospective cohort studies examining the effect of antidepressant drugs.
We found four randomised trials of antidepressant versus cognitive behavioural therapy, active drug control, placebo, or no treatment, which reported on the primary efficacy outcome (Gilliam 2019; Li 2005; Robertson 1985; Zhu 2004). One was a multi‐centre study (Gilliam 2019), the rest were single centre. A total of 313 participants were randomised in these studies; 199 participants had focal epilepsy. The remaining six non‐randomised prospective cohort studies reported on a total of 313 participants treated with an antidepressant, reported on the primary efficacy outcome (Hovorka 2000; Kanner 2000; Kuhn 2003; Orjuela‐Rojas 2015; Specchio 2004; Thome‐Souza 2007). Two hundred and ninety seven participants had focal epilepsy; 290 were treated with a selective serotonin reuptake inhibitor, and seven with cognitive behavioural therapy.
Seven studies reported outcomes for adults participants only; three studies reported outcomes for adults and children (Kanner 2000; Thome‐Souza 2007; Zhu 2004).
Eight studies included participants with focal onset epilepsy; two studies included participants with generalised onset epilepsy (Li 2005; Zhu 2004). In all trials, there was a larger or equal number of female participants.
Six studies evaluated the efficacy of selective serotonin reuptake inhibitors (citalopram, sertraline, fluoxetine) versus no treatment, a tricyclic antidepressant (doxepin), a norepinephrine reuptake inhibitor (reboxetine), or an alpha‐2 antagonists (mirtazapine; (Hovorka 2000; Kanner 2000; Kuhn 2003; Li 2005; Specchio 2004; Thome‐Souza 2007)). One study evaluated a serotonin‐norepinephrine reuptake inhibitor (venlafaxine) versus no treatment (Zhu 2004); another, a tricyclic antidepressant (amitriptyline) versus dopamine and a norepinephrine reuptake inhibitor (nomifensine; (Robertson 1985)). Two studies evaluated the efficacy of selective serotonin reuptake inhibitors (sertraline, citalopram) versus cognitive behavioural therapy (Gilliam 2019; Orjuela‐Rojas 2015).
Five studies used the Hamilton Rating Scale for Depression (HAMD; (Hovorka 2000; Kuhn 2003; Li 2005; Robertson 1985; Zhu 2004)). Two studies used both the Mini International Neuropsychiatric Interview (MINI) and Beck Depression Inventory (BDI; (Gilliam 2019; Orjuela‐Rojas 2015)). One study used the Montgomery‐Åsberg Depression Rating Scale (MADRS; Specchio 2004)), one study used the Kiddie SADS depression score (Thome‐Souza 2007), and one study did not report the use of a specific depression rating scale (Kanner 2000).
Randomised Controlled Trials
Gilliam 2019 was a published multi‐centre, randomised controlled trial, conducted in the USA, with 140 participants. Participants had a mean age of 39.6 years, 77 participants were female, and 56% of participants had focal epilepsy. Participants were randomised to receive sertraline (50 mg to 200 mg/day) or cognitive behavioural therapy (CBT). Changes in the MINI score, CES‐D, Beck Depression Inventory (BDI), QOLIE‐89 score, and adverse event profile (AEP) scores were compared from baseline to 8 weeks and 16 weeks. Changes in monthly seizure rates were compared from a retrospective 3‐month baseline, to 8 weeks and 16 weeks. Recurrence of a GTCS during the study was compared in those subjects who had not had a GTCS in a retrospective six‐month period prior to study enrolment. Of the 140 participants, 42 did not complete treatment as assigned, 15 of whom were lost to follow‐up. All participants were included in the reported analysis.
Li 2005 was a published, single‐centre, randomised controlled trial, conducted in China, with 67 participants. Forty‐two participants had generalised onset epilepsy. The participants were aged between 14 and 62 years, and 35 participants were female. Thirty‐three participants were randomised to paroxetine, which was started at 10 mg/day and titrated up to 40 mg/day, depending on response. Thirty‐four participants were randomised to doxepin, which was started at 25 mg/day and titrated up, according to response (mean dose 100 mg/day). The Hamilton Rating Scale for Depression (HAMD) score was measured at eight weeks and compared to the baseline score. Seizure frequency was not assessed. Three participants in the doxepin treatment arm dropped out and were not included in the primary analysis.
Robertson 1985 was a published, single‐centre, randomised, placebo‐controlled trial, conducted in the UK, with 42 participants. The majority had focal onset epilepsy. The participants were aged between 18 and 60 years, and 26 were female. Participants were randomised to amitriptyline, nomifensine, or placebo. All treatment arms completed a six‐week phase, and then both active treatment arms continued the study for a further six weeks. At 12 weeks of treatment, Hamilton Rating Scale for Depression (HAMD) scores were compared to baseline. Three participants withdrew from the study. Twenty‐eight participants in the active treatment arms were included in the primary outcome analysis at 12 weeks.
Zhu 2004 was a published, single‐centre, randomised trial of venlafaxine versus no treatment, conducted in China, with 64 participants. The participants were aged between 7 and 60 years. Thirty‐two participants were randomised to venlafaxine 25 mg/day to 75 mg/day; 32 participants received no treatment. Depression scores were measured at eight weeks of treatment, using the Hamilton Rating Scale for Depression (HAMD), and compared to baseline. Seizure frequency was not reported. There were no dropouts, and all participants were included in the primary outcome analysis.
Non‐Randomised Prospective Cohort Studies
Hovorka 2000 was a published, single‐centre, prospective cohort study, conducted in the Czech Republic, with 43 participants. Two‐thirds of the participants had focal epilepsy. Participants were between the ages of 12 and 49 years, and 35 participants were female. . All participants received citalopram (mean daily dose 22.6 mg ± 8.3 mg) for eight weeks. At four and eight weeks, Hamilton Rating Scale for Depression (HAMD) depression scores and seizure frequencies were measured and compared to an unspecified baseline period. There were no treatment withdrawals; all 43 participants were included in the reported analysis.
Kanner 2000 was a published, single‐centre, prospective cohort study, conducted in the USA, with 100 participants. Participants were aged between 6 and 62 years, 95% had focal onset epilepsy, and 49 participants were female. All participants received sertraline (25 mg/day to 200 mg/day; mean dose of 108 mg/day ± 56.9 mg/day), and were followed up for 0.2 to 38 months. Monthly seizure frequencies were compared during the treatment period, and to a 3‐ and 12‐month retrospective baseline period. No changes in depression scores were reported. Of the 100 participants, 18 withdrew from the study. All participants were included in the primary efficacy analysis.
Kuhn 2003 was a published, single‐centre, prospective cohort study, conducted in Germany, with 75 participants. All had focal onset epilepsy (temporal lobe). The participants were aged between 19 and 68 years, and 45 participants were female. Twenty‐seven participants received mirtazepine (mean daily dose 32.2 mg), 33 participants received citalopram (mean daily dose 24.2 mg), and 15 participants received reboxetine (mean daily dose 6.9 mg). Changes in Hamilton Rating Scale for Depression (HAMD) depression scores and treatment responders were measured at four weeks and 20 to 30 weeks, and compared to baseline scores. Changes in seizure frequency were not measured. Forty‐two participants dropped out; eight dropped out between baseline and week four, 34 dropped out between week four and weeks 20 to 30. The last observation carried forward method was used, and all participants were included in the primary efficacy outcomes.
Orjuela‐Rojas 2015 was a published, single‐centre, prospective study, conducted in Mexico, with 15 participants. All participants had temporal lobe epilepsy, and 11 participants were female. Seven participants received 12 sessions of cognitive behavioural therapy, and 8 participants received an SSRI (either sertraline (200 mg/day to 400 mg/day or citalopram 20 mg/day) over a 12‐week period. The Beck Depression Inventory (BDI) score, HADS score, QOLIE‐31, MINI, and monthly seizure frequency were compared at 6 and 12 weeks to baseline scores. There were two dropouts in the CBT group, and one participant was lost to follow‐up in the SSRI group. All participants were included in the reported analyses.
Specchio 2004 was a published, multi‐centre, prospective cohort study, conducted in Italy, with 45 participants. Forty‐four participants had focal onset epilepsy. The participants had a mean age of 42.7 years, and 31 were female. All participants received citalopram for four months. Montgomery–Åsberg Depression Rating Scale (MADRS) depression scores and seizure frequency were measured at two and four months on citalopram, and compared to baseline measures. Six participants withdrew from the study and were omitted from the primary outcome analysis.
Thome‐Souza 2007 was a published, single‐centre, prospective cohort study, conducted in Brazil, with 36 participants with focal onset epilepsy. The participants were aged between six and 18 years, and 19 were female. Twenty‐eight participants received sertraline (50 mg/day to 200 mg/day), and eight participants received fluoxetine (20 mg/day to 80 mg/day) for 12 to 78 months. Change in Kiddie SADS score was measured during the treatment phase, and compared to a six‐month baseline score. Seizure exacerbation was also observed during the treatment phase. One participant dropped out of the study. All participants were included in the primary outcome analysis.
Excluded studies
We excluded six studies. One was a clinical trial comparing escitalopram and no treatment (NCT01244724). This trial was terminated early due to problems with recruitment, and there were no available published data. The second study was a very small pilot trial comparing escitalopram and referral to psychiatry (NCT03464383). Only three participants were recruited to each study arm, and limited outcome data were available. The third study reported on a small case series of participants with epilepsy, who were taking a combined tricyclic antidepressant and SSRI for depression, and did not fulfil the inclusion criteria (Blumer 1997).
A further three studies were excluded because they did not report any primary or secondary outcome data and trial authors could not be contacted (Harmant 1990; Machado 2010; NCT00595699).
See Characteristics of excluded studies for more details of the studies.
Risk of bias in included studies
We rated risk of bias across each domain for each study, and then made an overall judgement on risk of bias for each study, using the Cochrane 'Risk of bias' tool for the RCTs, and the ROBINS‐I tool for the NRSIs.
See Characteristics of included studies for review authors' judgements about each 'Risk of bias' item for each included RCT, and Table 8 for review authors' judgements about each 'Risk of bias' item for each included non‐randomised study.
2. Risk of bias judgements for non‐randomised studies (ROBINS‐I).
| Domain and risk of bias judgement | Study | |||||
| Hovorka 2000 | Kanner 2000 | Kuhn 2003 | Orjuela‐Rojas 2015 | Specchio 2004 | Thome‐Souza 2007 | |
| Bias due to confounding | Serious | Moderate | Serious | Moderate | Serious | Serious |
| Bias in selection of participants into the study | Moderate | Low | Moderate | Low | Serious | Moderate |
| Bias in classification of interventions | Low | Low | Moderate | Low | Low | Low |
| Bias due to deviations from intended interventions | Low | Low | Moderate | Moderate | Moderate | Low |
| Bias due to missing data | Low | Low | Serious | Serious | Serious | Low |
| Bias in measurement of outcomes | Moderate | Serious | Moderate | Moderate | Moderate | Moderate |
| Bias in selection of the reported result | Low | Moderate | Low | Moderate | Low | Low |
| Overall judgement | Serious | Serious | Serious | Serious | Serious | Serious |
| Support for judgement | No adjustment for confounding; unclear if participants were recruited consecutively; and by design, blinding was not possible, which may have influenced subjectively‐assessed outcomes | Some analyses to investigate prognostic variables, but not a complete analysis of confounders; unclear which variables were of interest in advance, and if other characteristics were tested and analysed; and by design, blinding was not possible, which may have influenced subjectively‐assessed outcomes. The measure of depression was not an accurate or reliable measure | No adjustment for confounding; many discontinuations due to adverse events and non‐compliance, with outcome data analysed by last observation carried forward; unclear if participants were already receiving the intervention on entry into the study, and exactly how groups were assigned. Lack of blinding may have influenced some participant‐reported outcomes. | Some analyses to investigate prognostic variables, but not a complete analysis of confounders; unclear which variables were of interest in advance, and if other characteristics were tested and analysed; small groups and dropouts, with outcome data analysed by last observation carried forward, Lack of blinding may have influenced some participant‐reported outcomes. | No adjustment for confounding; outcome data included only for those who completed analysis (6 participants excluded); and by design, blinding was not possible, which may have influenced subjectively‐assessed outcomes | No adjustment for confounding; unclear if participants were recruited consecutively; and by design, blinding was not possible, which may have influenced subjectively‐assessed outcomes. |
Overall, we rated one RCT at low risk of bias (Gilliam 2019), three RCTs at unclear risk of bias (Li 2005; Robertson 1985; Zhu 2004), and all six NRSIs at serious risk of bias.
Allocation
Generation of random sequence and allocation concealment (RCTs)
We rated three RCTs at low risk of bias for sequence generation, as they used adequate methods (Gilliam 2019; Li 2005; Robertson 1985). For allocation concealment, we rated Li 2005 at unclear risk of bias, Robertson 1985 at low risk of bias, and Gilliam 2019 at low risk of bias, as it was not possible to conceal allocation due to different treatment types (CBT versus sertraline). We rated the fourth RCT at unclear risk of bias for both sequence generation and allocation concealment (Zhu 2004). (See the 'Characteristics of included studies' for more detailed information on methodology).
Selection of participants into the study (NRSIs)
We judged two studies, which recruited consecutive participants over a specified time frame, to be at low risk of bias in the selection of participants into the study (Kanner 2000; Orjuela‐Rojas 2015); three studies to be at moderate risk of bias, as we were unclear if participants were consecutive, the time frame of recruitment was unclear, or we were unclear if participants were already taking an antidepressant at recruitment (Hovorka 2000; Kuhn 2003; Thome‐Souza 2007); and one study to be at serious risk of bias, as although consecutive participants were recruited, only those who completed the intervention were included in the study results (Specchio 2004).
Classification of interventions (NRSIs)
We judged five studies to be at low risk of bias in classification of interventions: four studies had a before‐after design, measuring outcomes before and after treatment with antidepressant medications (Hovorka 2000; Kanner 2000, Specchio 2004; Thome‐Souza 2007), and one study allocated participants to antidepressants or CBT according to the feasibility of participants travelling to attend CBT (Orjuela‐Rojas 2015). We judged one study, with three treatment groups receiving different antidepressant medications, to be at moderate risk of bias, as it was unclear exactly how participants were assigned to these groups, and if they were already taking the treatment at recruitment into the study (Kuhn 2003).
Deviations from intended interventions (NSRIs)
All studies aimed to assess the effect of starting and adhering to an intervention, and in all studies, all included participants received an intervention plus any anti‐seizure medications needed to maintain stability during the study. We judged three studies to be at low risk of bias, as all participants were included in the study and analysis (Hovorka 2000; Kanner 2000; Thome‐Souza 2007), and three studies to be at moderate risk of bias, since participants who discontinued the intervention or the study were not included in analysis (Kuhn 2003; Orjuela‐Rojas 2015; Specchio 2004).
Blinding
Blinding of participants, personnel, and outcome assessors (RCTs)
We rated two RCTs at low risk of bias, as study personnel, participants, and outcome assessors were blinded (Li 2005; Robertson 1985). One RCT did not report any clear methods of blinding, therefore, we rated this study at unclear risk of bias (Zhu 2004). One RCT reported an absence of blinding for study personnel or participants due to different treatment interventions (CBT versus sertraline), but did report blinding of study investigators. Therefore, we rated this RCT at low risk of bias (Gilliam 2019).
Measurement of outcomes (NSRIs)
By design, participants were not blinded in any of the six NRSIs, since they completed their own seizure diaries, in an un‐blinded manner. In one study, with three treatment groups receiving different antidepressant medications, a blinded outcome assessor assessed depression; in the other studies, depression was measured before and after the intervention in an un‐blinded manner. As lack of blinding may have influenced some participant‐reported or subjectively assessed outcomes, we judged five studies to be at moderate risk of bias due to measurement of outcomes (Hovorka 2000; Kuhn 2003; Orjuela‐Rojas 2015; Specchio 2004; Thome‐Souza 2007). We judged one study to be at serious risk of bias, because they used an insufficient measure of depression, by looking for 'complete resolution of identified target psychiatric symptoms' as a measure of response to treatment (Kanner 2000).
Incomplete outcome data
For the RCTs, two reported missing data and did not perform an intention‐to‐treat analysis, but they did report both numerator and denominator data (Li 2005; Robertson 1985). We rated these as unclear risk of bias. The third RCT did not report any missing data and carried out an intention‐to‐treat analysis, therefore, we rated this study at low risk of bias (Zhu 2004). The fourth RCT reported missing data, with 15 participants lost to follow‐up, but reported an intention‐to‐treat analysis. We rated this study at low risk of bias (Gilliam 2019).
We judged three NRSIs to be at low risk of bias, as they included all participants in the study; none of the participants discontinued the study (Hovorka 2000; Thome‐Souza 2007), or all participants were included in an intention‐to‐treat analysis (Kanner 2000). We judged three studies to be at serious risk of bias, since participants who discontinued the intervention or the study were not included in the analysis, or data were imputed using the simple method 'last observation carried forward', which assumes no change in outcomes, so this may have introduced bias into the results (Kuhn 2003; Orjuela‐Rojas 2015; Specchio 2004).
Selective reporting
We rated the four RCTs at low risk of bias, as they reported outcomes that were clearly stated in their methods section (Gilliam 2019; Li 2005; Robertson 1985; Zhu 2004).
We judged four NSRIs to be at low risk of bias in their selection of the reported results when summary statistics were presented for all outcomes defined in the methods, and no formal statistical analyses were conducted (Hovorka 2000; Thome‐Souza 2007), or all outcomes and analyses defined in the methods were reported in the results (Kuhn 2003; Specchio 2004). We judged two studies to be at moderate risk of bias, when some participant characteristics were considered in the analyses, but the methods contained no details about which participant characteristics were of interest, and how many characteristics were examined and tested (Kanner 2000; Orjuela‐Rojas 2015).
Other potential sources of bias
For one RCT, seizure data at baseline was collected retrospectively, so we judged this trial to be at high risk of recall bias (Gilliam 2019). For the other RCTs, it was unclear whether there were any other potential sources of bias, so we rated all the RCTs as unclear risk of bias for this domain.
Confounding variables (NSRIs)
We judged four studies to be at serious risk of bias due to confounding, as they conducted no adjustments for any confounding variables (Hovorka 2000; Kuhn 2003; Specchio 2004; Thome‐Souza 2007); and we judged two studies to be at moderate risk of bias due to confounding, as they conducted some analyses to investigate differences in participant subgroups, but they did not consider all the important, prespecified confounders for this review, and it was unclear if the analyses were conducted specifically to investigate confounding (Kanner 2000; Orjuela‐Rojas 2015; see Assessment of risk of bias in included studies).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6
Summary of findings 1. Paroxetine compared to doxepin for people with epilepsy and depression.
| Paroxetine compared to doxepin for people with epilepsy and depression | ||||||
| Patient or population: people with epilepsy and depression Settings: outpatients Intervention: paroxetine Comparison: doxepin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| doxepin | paroxetine | |||||
|
> 50% reduction in depressive symptoms Follow‐up: 8 weeks |
706 per 1000 | 819 per 1000 (621 to 1000) | RR 1.16 (0.88 to 1.52) | 67 (1 RCT) | ⊕⊕⊕⊝ moderatea | |
|
Mean depression scores (HAMD scores; lower = better) Follow‐up: 8 weeks |
NA | The mean HAMD depression score in the intervention groups was 0.65 higher (2.15 lower to 3.45 higher) | NA | 67 (1 RCT) | ⊕⊕⊕⊝ moderatea | |
|
Seizure frequency Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
|
Withdrawals Follow‐up: 8 weeks |
88 per 1000 | 13 per 1000 (1 to 242) | RR 0.15 (0.01 to 2.74) | 67 (1 RCT) | ⊕⊕⊕⊝ moderatea |
doxepin: 3 withdrew paroxetine: 0 withdrew |
|
Cognitive functioning Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
|
Quality of life Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
|
Adverse effects Follow‐up: 8 weeks |
Reported adverse events: blurred vision, dizziness, dry mouth, sleep disorders, and urinary retention | Reported adverse events: blurred vision, dizziness, dry mouth, and sleep disorders | NA | 67 (1 RCT) |
⊕⊕⊕⊝ moderatea |
There were no significant differences between treatment groups for any reported adverse events |
| *The basis for the assumed risk is the event rate in the doxepin group. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HAMD: Hamilton Rating Scale for Depression; NA: not applicable; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty. Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty, Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty. Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty. We are very uncertain about the estimate. | ||||||
aCertainty of the evidence downgraded for imprecision, because only one small study contributed to the outcomes.
Summary of findings 2. Amitriptyline compared to nomifensine for people with epilepsy and depression.
| Amitriptyline compared to nomifensine for people with epilepsy and depression | ||||||
| Patient or population: people with epilepsy and depression Settings: outpatients Intervention: amitriptyline Comparison: nomifensine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| nomifensine | amitriptyline | |||||
|
> 50% reduction in depressive symptoms Follow‐up: 12 weeks |
786 per 1000 | 432 per 1000 (220 to 833) | RR 0.55 (0.28 to 1.06) | 28 (1 RCT) | ⊕⊕⊝⊝ lowa | |
|
Mean depression scores Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
|
Seizure frequency Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
|
Withdrawals Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) | ‐ | |
|
Cognitive functioning Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
|
Quality of life Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
|
Adverse effects Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
| *The basis for the assumed risk is the event rate in the nomifensine group. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; NA: not applicable; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty. Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty, Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty. Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty. We are very uncertain about the estimate. | ||||||
aCertainty of the evidence downgraded twice for imprecision, because only very small study contributed limited outcome data.
Summary of findings 3. Venlafaxine compared to no treatment for people with epilepsy and depression.
| Venlafaxine compared to no treatment for people with epilepsy and depression | ||||||
| Patient or population: people with epilepsy and depression Settings: outpatients Intervention: venlafaxine Comparison: no treatment | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| no treatment | venlafaxine | |||||
|
> 50% reduction in depressive symptoms Follow‐up: 8 weeks |
125 per 1000 | 406 per 1000 (149 to 1000) | RR 3.25 (1.19 to 8.9) | 64 (1 RCT) | ⊕⊕⊝⊝ lowa,b |
|
|
Mean depression scores (HAMD scores; lower = better) Follow‐up: 8 weeks |
NA | The mean HAMD depression score in the intervention group was 7.59 lower (11.52 lower to 3.66 lower) | NA | 64 (1 RCT) | ⊕⊕⊝⊝ lowa,b |
|
|
Seizure frequency Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
|
Withdrawals Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) | ‐ | |
|
Cognitive functioning Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
|
Quality of life Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
|
Adverse effects Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
| *The basis for the assumed risk is the event rate in the no treatment group. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HAMD: Hamilton Rating Scale for Depression; NA: not applicable; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty. Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty, Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty. Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty. We are very uncertain about the estimate. | ||||||
aCertainty of the evidence downgraded for imprecision, because only one small study contributed to the outcomes. bCertainty of the evidence downgraded once due to risk of bias; unclear methodological information provided regarding randomisation and allocation concealment.
Summary of findings 4. Sertraline compared to cognitive behavioural therapy for people with epilepsy and depression.
| Sertraline compared to cognitive behavioural therapy for people with epilepsy and depression | ||||||
| Patient or population: people with epilepsy and depression Settings: outpatients Intervention: sertraline Comparison: cognitive behavioural therapy (CBT) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| CBT | sertraline | |||||
|
> 50% reduction in depressive symptoms Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
|
Mean depression scores (BDI scores; lower = better) Follow‐up: 16 weeks |
NA | The mean BDI depression score in the intervention group was 0.50 lower (4.47 lower to 3.47 higher) | NA | 117 (1 RCT) | ⊕⊕⊕⊝ moderatea |
At 8 weeks: MD ‐2.50 (95% CI ‐6.28 to 1.28; 104 participants) |
|
Seizure frequency Follow‐up: 16 weeks |
NA | The mean frequency of GTCS per month in the intervention group was 0 lower (‐0.10 lower to 0.10 higher) The mean frequency of focal seizures with impaired awareness per month in the intervention group was 3.00 lower (7.81 lower to 1.81 higher) |
NA | 96 with GTCS plus 75 with focal seizures (1 RCT) |
⊕⊕⊝⊝ lowb |
At 8 weeks: GTCS per month: MD ‐0.10 (95% CI ‐0.26 to 0.06; 86 participants) focal seizures with impaired awareness per month: MD ‐2.60 (95% CI ‐6.52 to 1.32; 75 participants) |
|
Withdrawals Follow‐up: 16 weeks |
176 per 1000 |
222 per 1000 (113 to 434 per 1000) |
RR 1.26 (0.64 to 2.46) | 140 (1 RCT) | ⊕⊕⊕⊝ moderatea |
CBT: 6 withdrew, 6 lost to follow‐up sertraline: 7 withdrew, 9 lost to follow‐up |
|
Cognitive functioning Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
|
Quality of life (QOLIE‐89 scale; lower = better) Follow‐up: 16 weeks |
NA | The mean QOLIE‐89 score in the intervention group was 3.10 higher (3.41 lower to 9.61 higher) | NA | 118 (1 RCT) |
⊕⊕⊕⊝ moderatea |
at 8 weeks: MD 6.10 (95% CI ‐0.28 to 12.48; 104 participants) |
|
Adverse effects Follow‐up: 16 weeks |
NA | The mean adverse event profile score in the intervention group was 2.10 lower (6.21 lower to 2.01 higher) | NA | 118 (1 RCT) |
⊕⊕⊕⊝ lowa,c |
Sertraline resulted in more cases of tiredness than CBT (RR 3.54, 99% CI 1.40 to 8.96; 140 participants) Sertraline did not result in more cases of any other adverse effects than CBT. |
| *The basis for the assumed risk is the event rate in the CBT group. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). BDI: Beck Depression Inventory; CBT: cognitive behavioural therapy; CI: confidence interval; MD: mean difference; NA: not applicable; QOLIE: Quality of life in Epilepsy; RCT: randomised controlled trial; RR: risk ratio; GTCS: generalised tonic‐clonic seizures | ||||||
| GRADE Working Group grades of evidence High certainty. Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty, Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty. Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty. We are very uncertain about the estimate. | ||||||
aCertainty of the evidence downgraded once due to risk of bias: participants and personnel not blinded, and lack of blinding may have influenced outcome bCertainty of the evidence downgraded twice due to risk of bias and imprecision: risk of recall bias as seizure frequency data at baseline was collected retrospectively, and data not available for all participants
cCertainty of the evidence downgraded once due to imprecision: adverse event data not available for all participants who received an intervention
Summary of findings 5. Citalopram (before and after treatment) for people with epilepsy and depression.
| Citalopram (before and after treatment) for people with epilepsy and depression | ||||
| Patient or population: people with epilepsy and depression Settings: outpatients Intervention: citalopram Control: before citalopram treatment | ||||
| Outcomes | Illustrative comparative risks* (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments |
| Citalopram (before and after) | ||||
| > 50% reduction in depressive symptoms Follow‐up: 4 months |
11 out of 45 participants (24%) showed a 50% or more improvement in depression scores after treatment compared to baseline. | 45 (1 NRSI) |
⊕⊕⊝⊝ lowa |
|
|
Mean depression scores (HAMD scores; lower = better) Follow‐up: 8 weeks to 4 months |
Improved depression scores were shown after citalopram compared to before (see comment) | 88 (2 NRSI) | ⊕⊕⊝⊝ lowa,b,c | SMD in HAMD score was 1.17 (95% CI 0.96 to 1.38), indicating improved outcomes and a large treatment effect. |
|
Seizure frequency Follow‐up: 8 weeks to 4 months |
See comment | 88 (2 NRSI) | ⊕⊝⊝⊝ very lowa,c | Results were mixed between studies; due to very high heterogeneity (I² = 81%), we did not present the overall effect estimate. |
|
Withdrawals Follow‐up: 8 weeks to 4 months |
6/45 participants (13%) withdrew from one study; 0/43 from the other study | 88 (2 NRSI) |
⊕⊕⊝⊝ lowa |
|
|
Cognitive functioning Follow‐up: NA |
‐ | 0 (0 studies) |
‐ | |
|
Quality of life Follow‐up: NA |
‐ | 0 (0 studies) |
‐ | |
|
Adverse effects Follow‐up: 8 weeks to 4 months |
22/45 participants (56%) experienced adverse events in one study; 5/43 (12%) in the other study | 88 (2 NRSI) |
⊕⊕⊝⊝ lowa |
Specific adverse events reported: nausea, sexual dysfunction, headache, dizziness, drowsiness, and fatigue |
| CI: confidence interval; HAMD: Hamilton Rating Scale for Depression; NRSI: non‐randomised studies of interventions | ||||
| GRADE Working Group grades of evidence High certainty. Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty, Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty. Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty. We are very uncertain about the estimate. | ||||
aCertainty of the evidence downgraded twice as studies were judged to be at serious risk of bias due to lack of blinding, which may have influenced participant‐recorded outcomes, and lack of adjustment for confounding variables. bCertainty of the evidence upgraded once as large effect found. cCertainty of the evidence downgraded due to inconsistency: substantial statistical heterogeneity was present (I² > 50%).
Summary of findings 6. Selective serotonin reuptake inhibitors compared to cognitive behavioural therapy for people with epilepsy and depression.
| Selective serotonin reuptake inhibitorscompared to cognitive behavioural therapy for people with epilepsy and depression | ||||||
| Patient or population: people with epilepsy and depression Settings: outpatients Intervention: selective serotonin reuptake inhibitors (SSRIs; sertraline or citalopram) Comparison: cognitive behavioural therapy (CBT) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| CBT | SSRIs | |||||
|
> 50% reduction in depressive symptoms Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
|
Mean depression scores (BDI scores; lower = better) Follow‐up: 12 weeks |
NA | The mean BDI depression score in the intervention group was 4.90 lower (14.90 lower to 4.80 higher) |
NA | 15 (1 NRSI) | ⊕⊝⊝⊝ very lowa,b |
at 6 weeks: MD ‐2.60 (95% CI ‐11.58 to 6.38; 15 participants) |
|
Seizure frequency Follow‐up: 12 weeks |
NA | The mean frequency of seizures per month in the intervention group was 1.60 lower (5.63 lower to 2.43 higher) | NA | 15 (1 NRSI) | ⊕⊝⊝⊝ very lowa,b |
|
|
Withdrawals Follow‐up: 12 weeks |
286 per 1000 |
126 per 1000 (14 to 1000 per 1000) |
RR 0.44 (0.05 to 3.85) |
15 (1 NRSI) | ⊕⊝⊝⊝ very lowa,b |
CBT: 2 lost to follow‐up SSRI: 1 lost to follow‐up in the |
|
Cognitive functioning Follow‐up: NA |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
|
Quality of life ‐ QOLIE‐31 scale Follow‐up: 12 weeks |
NA | The mean QOLIE‐31 score in the intervention group was 0.50 lower (19.67 lower to 18.67 higher) | NA | 15 (1 NRSI) | ⊕⊝⊝⊝1,2 very low |
|
|
Adverse effects ‐ adverse event profile Follow‐up: 16 weeks |
‐ | ‐ | ‐ | 0 (0 studies) |
‐ | |
| *The basis for the assumed risk is the event rate in the CBT group. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). BDI: Beck Depression Inventory; CBT: cognitive behavioural therapy; CI: confidence interval; MD: mean difference; NA: not applicable; QOLIE: Quality of life in Epilepsy; RCT: randomised controlled trial; RR: risk ratio; SSRI: selective serotonin reuptake inhibitors | ||||||
| GRADE Working Group grades of evidence High certainty. Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty, Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty. Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty. We are very uncertain about the estimate. | ||||||
1. Certainty of the evidence downgraded twice as the study was judged to be at serious risk of bias with regards to lack of blinding which may have influenced participant recorded outcomes and lack of adjustment for confounding variables.
2. Certainty of the evidence downgraded once due to imprecision: very small study of 15 participants, confidence intervals around effect estimates wide
We evaluated six comparisons in this review:
Paroxetine compared to doxepin for people with epilepsy and depression (Table 1). We judged the certainty of evidence for this comparison to be moderate; evidence was downgraded due to imprecision, as only one small RCT contributed data.
Amitriptyline compared to nomifensine for people with epilepsy and depression (Table 2). We judged the certainty of evidence for this comparison to be low; we downgraded the evidence due to serious imprecision, as only one very small RCT contributed data.
Venlafaxine compared to no treatment for people with epilepsy and depression (Table 3). We judged the certainty of evidence for this comparison to be low; we downgraded the evidence due to risk of bias and imprecision, as only one small RCT contributed data.
Sertraline compared to cognitive behavioural therapy (CBT) for people with epilepsy and depression (Table 4). We judged the certainty of evidence for this comparison to be moderate to low; we downgraded the evidence due to risk of bias (lack of blinding, and retrospective collection of seizure frequency data at baseline).
Citalopram (before and after) for people with epilepsy and depression (Table 5). We judged the certainty of evidence for this comparison to be low to very low; we downgraded the evidence from two NRSIs due to serious risk of bias, and substantial statistical heterogeneity was present where data could be pooled.
SSRIs (sertraline or citalopram) compared to cognitive behavioural therapy (CBT) for people with epilepsy and depression (Table 6). We judged the certainty of evidence for this comparison to be very low; we downgraded the evidence due to serious risk of bias and imprecision, as only one very small NRSI of 15 participants contributed evidence, without any adjustment for confounding variables
Primary outcomes
Depression scores
1. Proportion with a greater than 50% improvement
Three of four RCTs reported on the proportion with a 50% or more improvement in depression scores. The RCTs analysed different treatment comparisons, and we were unable to combine the data in meta‐analysis.
Li 2005 (N = 67) compared paroxetine (20 mg/day to 40 mg/day) to doxepin (mean dose 100 mg/day). There were 27/33 (82%) responders in the paroxetine group and 24/34 (71%) in the doxepin group. The risk ratio (RR) for the proportion with a 50% of more improvement in depression scores for paroxetine versus doxepin was 1.16 (95% confidence interval (CI) 0.88 to 1.52; P > 0.05; Analysis 1.1).
1.1. Analysis.

Comparison 1: RCT: paroxetine versus doxepin, Outcome 1: > 50% reduction in depressive symptoms
Robertson 1985 (N = 42) compared amitriptyline (75 mg/day), nomifensine (75 mg/day), and placebo. At 12 weeks, there were 6/14 (43%) responders in the amitriptyline group and 11/14 (79%) in the nomifensine group. The risk ratio for the proportion with a 50% or more improvement in depression scores for amitriptyline versus nomifensine was 0.55 (95% CI 0.28 to 1.06; P > 0.05; Analysis 2.1).
2.1. Analysis.

Comparison 2: RCT: amitriptyline versus nomifensine, Outcome 1: > 50% reduction in depressive symptoms
Zhu 2004 (N = 64) compared venlafaxine (25 mg/day to 75 mg/day) to no treatment. There were 22/32 (69%) responders in the venlafaxine group and 6/32 (19%) in the no treatment group. More participants had a 50% or more improvement in depression scores in the venlafaxine group than in the no treatment group (RR 3.25, 95% CI 1.19 to 8.90; P < 0.05; Analysis 3.1).
3.1. Analysis.

Comparison 3: RCT: venlafaxine versus no treatment controls, Outcome 1: > 50% reduction in depressive symptoms
Four of the six NRSIs reported on the proportion of participants with a 50% or more improvement in depression scores. One study did not use a validated depression scale (Kanner 2000).
Hovorka 2000 observed 28/43 (65%) participants with a 50% or more improvement in depression scores following eight weeks of treatment with citalopram (mean dose 23 mg/day) compared to baseline.
Kuhn 2003 observed 17/27 participants (52%) in the mirtazepine group (mean dose 32 mg/day), 12/33 participants (36%) in the citalopram group (mean dose 24 mg/day), and 8/15 participants (53%) in the reboxetine group (mean dose 7 mg/day) with a 50% or more improvement in depression scores following 20 to 30 weeks of treatment, compared to baseline.
Specchio 2004 observed 11/45 participants (24%) with a 50% or more improvement in depression scores following four months of treatment with citalopram (20 mg/day), compared to baseline.
Thome‐Souza 2007 observed 35/36 participants (97%) with a 50% or more improvement in depression scores following one year of treatment with sertraline (mean dose 111 mg/day), or fluoxetine (mean 46 mg/day), compared to baseline.
2. Mean difference
Three of the four RCTS reported on the mean difference in depression scores. Two RCTs used the HAMD scale (Li 2005; Zhu 2004), and one used the BDI‐II (Gilliam 2019). The RCTs compared different treatment groups, and we were unable to combine the data in any meta‐analysis.
Gilliam 2019 (N = 140) compared sertraline (50 mg/day to 200 mg/day) to cognitive behavioural therapy (CBT). The mean difference (MD) in depression scores for sertraline versus CBT was ‐2.50 (95% CI ‐6.28 to 1.28) at 8 weeks, and ‐0.50 (95% CI ‐4.47 to 3.47) at 16 weeks (Analysis 4.1).
4.1. Analysis.

Comparison 4: RCT: sertraline versus cognitive behavioural therapy (CBT), Outcome 1: Mean depression scores (BDI)
Li 2005 (N = 67) compared paroxetine (20 mg/day to 40 mg/day) and doxepin (mean dose 100 mg/day). The mean difference in depression scores following treatment for paroxetine versus doxepin was 0.65 (95% CI ‐2.15 to 3.45; Analysis 1.2).
1.2. Analysis.

Comparison 1: RCT: paroxetine versus doxepin, Outcome 2: Mean depression scores
Zhu 2004 (N = 64) compared venlafaxine (25 mg to 75 mg/day) versus no treatment. Depression scores following treatment with venlafaxine were better than in the no treatment group (MD ‐7.59, 95% CI ‐11.52 to ‐3.66; Analysis 3.2).
3.2. Analysis.

Comparison 3: RCT: venlafaxine versus no treatment controls, Outcome 2: Mean depression scores ‐ HAMD
Four out of the six NSRIs reported on the mean difference in depression scores; either the mean difference before and after citalopram treatment (Hovorka 2000; Specchio 2004), or the mean difference between treatment groups following treatment (Kuhn 2003; Orjuela‐Rojas 2015).
We were able to meta‐analyse data from two before‐after studies of citalopram. The average dose of citalopram was 22 mg/day in Hovorka 2000, and 20 mg/day in Specchio 2004. The standardised mean difference (SMD) in depression scores indicated a large effect, with an estimate of 1.17 (95% CI 0.96 to 1.38; 2 studies, 88 participants; Analysis 5.1). The I² was 53%, which may reflect differences between the treatment periods. Specchio 2004 reported outcomes following four months of treatment; Hovorka 2000 after two months of treatment. Due to the level of statistical heterogeneity, we conducted a random‐effects analysis, which showed similar results (SMD 1.17; 95% CI 0.86 to 1.47).
5.1. Analysis.

Comparison 5: NRSI: citalopram (before and after), Outcome 1: Mean depression scores HAMD‐21
Kuhn 2003 reported mean depression scores before and after treatment with mirtazepine, citalopram, and reboxetine. From baseline to 20 to 30 weeks of treatment, the mean depression scores on the HAMD decreased with mirtazepine from 23 to 13.5, citalopram from 22.5 to 14, and reboxetine from 23 to 13.5.
Orjuela‐Rojas 2015 reported mean depression scores before and after treatment with an SSRI (sertraline or citalopram) versus CBT. The mean difference in depression scores for SSRIs versus CBT was ‐2.60 (95% CI ‐11.58 to 6.38) following six weeks of treatment; and ‐4.90 (95% CI ‐14.60 to 4.80; Analysis 6.1) following 12 weeks of treatment.
6.1. Analysis.

Comparison 6: NRSI: SSRIs (sertraline or citalopram) versus CBT, Outcome 1: Mean depression scores (BDI)
3. Remission
One of the four RCTs reported the proportion of participants who achieved a remission in depressive symptoms.
Gilliam 2019 reported 38/72 (53%) participants in the sertraline group and 41/68 (60%) in the CBT group who achieved a remission in depressive symptoms, measured on the MINI at 16 weeks of treatment (RR 0.88, 95% CI 0.65 to 1.17; 140 participants; Analysis 4.2).
4.2. Analysis.

Comparison 4: RCT: sertraline versus cognitive behavioural therapy (CBT), Outcome 2: Remission in depressive symptoms
One of the six NRSIs reported the proportion of participants who achieved a remission in depressive symptoms.
Orjuela‐Rojas 2015 observed 7/8 (87%) in the SSRI group and 4/7 (57%) in the CBT group who achieved a remission in depressive symptoms, measured on the BDI (BDI < 14) at 12 weeks of treatment (RR 1.53, 95% CI 0.77 to 3.06; 15 participants; Analysis 6.2).
6.2. Analysis.

Comparison 6: NRSI: SSRIs (sertraline or citalopram) versus CBT, Outcome 2: Remission in depressive symptoms
Seizure frequency
1. Mean difference
One RCT reported mean difference in seizure frequency between baseline and end of treatment period (Gilliam 2019). One RCT reported no change in seizure frequency in either the paroxetine or the doxepin treatment groups (Li 2005).
Gilliam 2019 reported inconclusive results for a change in the average number of generalised tonic‐clonic seizure (GTCS) per month at eight weeks (MD ‐0.10, 95% CI ‐0.26 to 0.06), and 16 weeks (MD 0.00, 95% CI ‐0.10 to 0.10) of treatment with either sertraline or CBT; and for focal seizures with impaired awareness at eight weeks (MD ‐2.60, 95% CI ‐6.52 to 1.32), and 16 weeks (MD ‐3.00, 95% CI ‐7.81 to 1.81; Analysis 4.3) of treatment with either sertraline or CBT.
4.3. Analysis.

Comparison 4: RCT: sertraline versus cognitive behavioural therapy (CBT), Outcome 3: Seizure frequency
Four of the six NRSIs reported on changes in mean seizure frequency; either the mean difference before and after citalopram treatment (Hovorka 2000; Specchio 2004), or the mean difference between treatment groups following treatment (Kuhn 2003; Orjuela‐Rojas 2015).
We conducted a meta‐analysis with data from the two before‐after studies of citalopram (Hovorka 2000; Specchio 2004). Calculating SMD, and using both a fixed‐effect and random‐effects model, the I² was 81%. Therefore, we did not report the pooled effect estimate. Individually, Hovorka 2000 showed inconclusive results between groups (MD 0.03, 95% CI ‐0.23 to 0.30; 43 participants), while Specchio 2004 showed possible improvement after treatment (MD 0.50, 95% CI 0.21 to 0.78; 45 participants; Analysis 5.2). Possible reasons for a high I² value are differences in mean baseline seizure frequency, treatment duration, and mean age of participants.
5.2. Analysis.

Comparison 5: NRSI: citalopram (before and after), Outcome 2: Mean monthly seizure frequency
Kanner 2000 reported no statistically significant difference in seizure frequency between baseline and post‐treatment for the 100 participants treated with sertraline (mean dose 108 mg/day).
The results for a change in seizure frequency between baseline and 12 weeks of treatment, between the eight participants receiving an SSRI (sertraline or citalopram) and the seven participants receiving CBT, were inconclusive (MD ‐1.60, 95% CI ‐5.63 to 2.43; 15 participants; Analysis 6.3; Orjuela‐Rojas 2015).
6.3. Analysis.

Comparison 6: NRSI: SSRIs (sertraline or citalopram) versus CBT, Outcome 3: Seizure frequency per month at 12 weeks
2. Seizure recurrence
One RCT reported on the recurrent of GTCS during the study in participants who had not experienced a GTCS in the six months prior to enrolment (Gilliam 2019). It reported a recurrence in 4/51 people in the sertraline group versus 4/53 in the CBT (RR 1.04, 95% CI 0.27 to 3.94; 104 participants; Analysis 4.4).
4.4. Analysis.

Comparison 4: RCT: sertraline versus cognitive behavioural therapy (CBT), Outcome 4: Seizure recurrence
One NRSI reported that one participant experienced a recurrence of seizures (Specchio 2004).
3. Episode of status epilepticus
None of the 10 studies reported episodes of status epilepticus.
Secondary outcomes
Withdrawals for any reason
Three of the RCTs clearly reported the number of participants who withdrew for any reason (Gilliam 2019; Li 2005; Robertson 1985).
Gilliam 2019 reported 7/72 withdrawals in the sertraline group (three withdrew consent; four left the study due to worsening depression), and 6/68 in the CBT group (two withdrew consent; four left the study due to worsening depression). There were also 9/72 participants lost to follow‐up from the sertraline group, and 6/68 from the CBT group (RR 1.26, 95% CI 0.64 to 2.46; 140 participants; Analysis 4.5).
4.5. Analysis.

Comparison 4: RCT: sertraline versus cognitive behavioural therapy (CBT), Outcome 5: Withdrawals (any reason)
Li 2005 reported that 3/34 participants (9%) withdrew from the doxepin group versus 0/33 from the paroxetine group (RR 0.15, 95% CI 0.01 to 2.74; P = 0.20; 67 participants; Analysis 1.3). The specific reasons for withdrawal were not reported.
1.3. Analysis.

Comparison 1: RCT: paroxetine versus doxepin, Outcome 3: Withdrawals (any reason)
Robertson 1985 reported that 1/14 participants (7%) withdrew from the amitriptyline group, 1/14 from the nomifensine group, and 1/14 from the placebo group. The participant withdrew from the nomifensine group because of increased seizures; the other reasons were not reported.
All six prospective non‐randomised studies reported on the number of participants withdrawing from the study.
Hovorka 2000 reported no treatment withdrawals from the study.
Kanner 2000 reported that 18/100 participants (18%) withdrew due to adverse events from the sertraline.
Kuhn 2003 reported that 20/27 participants (74%) withdrew from the mirtazepine group, 16/33 participants (48%) from the citalopram group, and 6/15 participants (40%) from the reboxetine group withdrew after 20 to 30 weeks of treatment. Adverse events accounted for 8/20 withdrawals from the mirtazepine group, 6/16 from the citalopram group, and 3/6 from the reboxetine group. Three participants (two from mirtazepine and one from citalopram) withdrew due to inefficacy. The remaining 22 participants were lost to follow‐up.
Orjuela‐Rojas 2015 reported that 2/7 withdrew from the CBT group (1 due to health problems unrelated to epilepsy, and 1 due to severe psychosocial situation deemed unrelated to either epilepsy or depression). There were no withdrawals from the SSRI group, but one was lost to follow‐up due to a road traffic accident. There were inconclusive results between groups (RR 0.44, 95% CI 0.05 to 3.85; 15 participants; Analysis 6.4).
6.4. Analysis.

Comparison 6: NRSI: SSRIs (sertraline or citalopram) versus CBT, Outcome 4: Withdrawals (any reason)
Specchio 2004 reported that 6/45 participants (13%) withdrew from the study (four because of adverse events from the citalopram, one due to poor compliance, and one due to concurrent illness).
Thome‐Souza 2007 reported that 1/36 participant, treated with sertraline (SSRI) withdrew because of an exacerbation of seizures.
Global State
None of the included studies reported on global state outcomes.
Mental State
None of the included studies reported on mental state outcomes.
General Functioning
None of the included studies reported on general functioning outcomes.
Cognitive Functioning
None of the included studies reported on cognitive functioning outcomes.
Quality of life
One of the four RCTs assessed quality of life, using the 89‐item Quality of Life in Epilepsy Inventory (QOLIE‐89; (Gilliam 2019)).
There were inconclusive results in improved quality of life between the sertraline and CBT groups at 8 weeks (MD 6.10, 95% CI ‐0.28 to 12.48; 104 participants), and at 16 weeks (MD 3.10, 95% CI ‐3.41 to 9.61; 118 participants Analysis 4.6).
4.6. Analysis.

Comparison 4: RCT: sertraline versus cognitive behavioural therapy (CBT), Outcome 6: Quality of life (QOLIE‐89)
One of the six NRSIs assessed quality of life with the QOLIE‐31 (Orjuela‐Rojas 2015).
There were inconclusive results between groups (MD ‐0.50, 95% CI ‐19.67 to 18.67; 15 participants; Analysis 6.5)
6.5. Analysis.

Comparison 6: NRSI: SSRIs (sertraline or citalopram) versus CBT, Outcome 5: Quality of life (QOLIE‐31 overall score)
Behaviour
None of the included studies reported on behaviour outcomes.
Adverse effects
Two RCTs reported the number of participants who experienced specific side effects (Gilliam 2019; Li 2005).
Li 2005 compared paroxetine and doxepin.
The results were inconclusive between groups for blurred vision (RR 0.34, 99% CI 0.09 to 1.32; 67 participants), dizziness (RR 0.21, 99% CI 0.03 to 1.37; 67 participants), dry mouth (RR 0.26, 99% CI 0.06 to 1.20; 67 participants), sleep disorders (RR 0.32, 99% CI 0.08 to 1.20; 67 participants), and urinary retention (RR 0.34, 99% CI 0.01 to 21.99; 67 participants; Analysis 1.4).
1.4. Analysis.

Comparison 1: RCT: paroxetine versus doxepin, Outcome 4: Adverse effects
Gilliam 2019 compared sertraline and CBT.
There were 10 serious adverse events in the sertraline group, three of which were possibly related to the study (one mania and psychosis, two worsening depression requiring hospitalisation). There were 12 serious adverse events in the CBT group, three of which were possibly related to the study (worsening depression and suicidal). One participant in the CBT group died of sudden unexpected death in epilepsy (SUDEP).
The difference in adverse events profile (AEP) scores was inconclusive between the groups at 8 weeks (MD ‐2.70, 95% CI ‐6.62 to 1.22; 106 participants), and 16 weeks (MD ‐2.10, 95% CI ‐6.21 to 2.01; 118 participants; Analysis 4.7).
4.7. Analysis.

Comparison 4: RCT: sertraline versus cognitive behavioural therapy (CBT), Outcome 7: Adverse events profile
There were more cases of tiredness reported by the participants on sertraline compared to those receiving CBT (RR 3.54, 99% CI 1.40 to 8.96; 140 participants; Analysis 4.8)
4.8. Analysis.

Comparison 4: RCT: sertraline versus cognitive behavioural therapy (CBT), Outcome 8: Adverse events
Results were inconclusive between the groups for the other five most commonly reported adverse events: headache (RR 1.48, 99% CI 0.69 to 3.19), insomnia (RR 2.16, 99% CI 0.73 to 6.38), shakiness (RR 12.28, 99% CI 0.88 to 171.59), nausea (RR 2.60, 99% CI 0.62 to 10.96), and diarrhoea (RR 19.85, 99% CI 0.49 to 805.53; 140 participants; Analysis 4.8).
Five of six NRSIs reported on adverse events. The studies analysed different treatment comparisons, so we were unable to combine the data in meta‐analysis.
Hovorka 2000 reported that 3/43 participants (7%) experienced nausea, and 2/43 (5%) experienced sexual dysfunction following eight weeks of treatment with citalopram.
Kanner 2000 reported that 9/100 participants (9%) experienced sedation, 7/100 (7%) experienced hypomanic symptoms, 1/100 (1%) experienced rheumatic pain, and 1/100 participants (1%) experienced myoclonus following an average of 10 months on sertraline.
Kuhn 2003 reported that 13/75 participants experienced side effects. The most common were; weight gain (5/75), sedation (2/75), and sexual dysfunction (2/75).
Specchio 2004 reported that 22/45 participants (56%) experienced side effects. The most common were; headache (15%), nausea (11%), dizziness (9%), drowsiness (7%), and fatigue (7%).
Thome‐Souza 2007 reported that 1/36 participants (3%) experienced facial rash, and 1/36 (3%) experienced gastrointestinal disorders.
Discussion
Summary of main results
We identified four randomised controlled trials (RCTs; N = 313) of antidepressant treatment for people with epilepsy, three of which had small sample sizes (42 to 67 participants). One trial compared antidepressant medication with cognitive behavioural therapy (CBT). Venlafaxine improved depressive symptoms when compared to no treatment; amitriptyline was not better than nomifensine in improving depressive symptoms; depressive symptoms were improved with both sertraline or CBT, but one was not better than the other. The RCTs did not allow meaningful comparisons among the different classes of antidepressants. Therefore, we do not know which antidepressant or class of antidepressant is most effective.
Only one RCT reported on changes in seizure frequency. The results were inconclusive for a change in frequency of generalised tonic clonic seizures for sertraline or CBT. Only one RCT reported on adverse events. The results were inconclusive between groups for adverse events including suicidal risk except tiredness which occurred more often in the sertraline group. The top reported side effects were; sedation, headache, insomnia and gastrointestinal symptoms.
The six prospective cohort studies (N = 313) were of serious risk of bias examining small numbers of participants treated predominantly with selective serotonin reuptake inhibitors (SSRIs). One study compared SSRIs to CBT. The combined meta‐analysis of two studies examining citalopram at 20 mg/day showed that citalopram improved depressive symptoms. We could not combine the data on changes in seizure frequency due to marked heterogeneity. However, in the three studies reporting changes in seizure frequency, the results were inconclusive. Only one study reported on one participant on antidepressant treatment (citalopram) who experienced seizure recurrence. Whilst the data are of low certainty in terms of impact on seizures, there is broad agreement across the prospective cohort studies of limited or no impact on seizures with selective serotonin reuptake inhibitors (SSRIs).
Participants withdrawing from antidepressants were more likely to do so because of adverse events rather than lack of efficacy. Reported adverse events for SSRIs included nausea, dizziness, sedation, gastrointestinal disturbance, and sexual dysfunction. We have no reliable information on the comparative risk of adverse events with different classes of antidepressant treatment.
Overall completeness and applicability of evidence
This review has ascertained that there is very limited evidence of an effect of antidepressants on depressive symptoms in participants with epilepsy. Depressive symptoms were improved with both sertraline or CBT, but one was not better than the other. We do not have any reliable high certainty evidence to inform on the best choice of antidepressant drug or class of drug for treating depression with the lowest risk of seizure exacerbation.
Quality of the evidence
Overall, we rated one study at low risk of bias (Gilliam 2019), five prospective cohort studies at high risk of bias (Hovorka 2000; Kanner 2000; Kuhn 2003; Orjuela‐Rojas 2015; Thome‐Souza 2007) and four studies (three RCTs and one prospective cohort study) at unclear risk of bias (Li 2005; Robertson 1985; Specchio 2004; Zhu 2004).
The 'Summary of findings' tables for each comparison examined shows that the certainty of the evidence for the outcomes ranged from moderate certainty to low certainty. For comparisons where RCTs were available, we downgraded for imprecision as for these comparisons, only one small study contributed to the outcomes. For comparisons where only NRSIs were available, we rated the certainty of evidence as low to very low and evidence was downgraded due to risk of bias, imprecision and substantial heterogeneity for some outcomes.
Potential biases in the review process
It is possible that despite the exhaustive searches carried out in this review, we did not identify other sources of unpublished data. This can be more of an issue for reviews including non‐randomised study designs, such as this review.
Including non‐randomised evidence, which is inherently at risk of additional biases to RCT evidences, may have also introduced bias into the review. However, as stated in Types of studies, prospective non‐randomised studies may be more reflective of clinical practice in terms of the populations recruited, and because of the known delayed effect of antidepressants on depressive symptoms, which may not be effectively detected in short‐term randomised trials.
We carried out a detailed quality assessment of the non‐randomised studies included in this review, using an appropriate tool (ROBINS‐I), and interpreted evidence from the non‐randomised studies taking into account the likely biases present within these studies.
Agreements and disagreements with other studies or reviews
We are not aware of any other reviews on this topic.
Authors' conclusions
Implications for practice.
Existing evidence on the effectiveness of antidepressants in treating depressive symptoms associated with epilepsy is still very limited. There is low to moderate certainty evidence from two RCTs that venlafaxine, sertraline, and CBT may reduce depressive symptoms. Sertraline and CBT may improve quality of life, but moderate certainty evidence did not find one superior to the other. We have no high certainty evidence to inform the choice of antidepressant drug or class of drug for treating depression in people with epilepsy. None of the treatments appeared to increase seizure activity, but there are no available comparative data on antidepressant classes and safety in relation to seizures.
Implications for research.
Randomised controlled trials of antidepressants, utilising interventions from other treatment classes besides SSRIs, in large cohorts of participants with epilepsy and depression, are needed to better inform treatment policy in the future. The studies need to be of longer duration to assess efficacy of treatment responses and provide better understanding on the impact of seizure control.
What's new
| Date | Event | Description |
|---|---|---|
| 1 February 2021 | New search has been performed | Searches updated 1 February 2021; two new studies were included (Gilliam 2019; Orjuela‐Rojas 2015). |
| 1 February 2021 | New citation required but conclusions have not changed | Conclusions are unchanged. |
History
Protocol first published: Issue 7, 2013 Review first published: Issue 12, 2014
Acknowledgements
This review update was supported by the National Institute for Health Research via Cochrane Infrastructure funding to the Epilepsy Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS, or the Department of Health and Social Care.
We acknowledge Jennifer Pulman (Weston) and Jasvinder Singh for contributions to the original review protocol and to previous versions of the review.
Appendices
Appendix 1. CRS Web search strategy
1. (antidepressant* or antidepressive* or "af 1161" or "ba 34276" or "bc 105" or "brl 29060" or "brl 29060" or "cl 67772" or "cp 15467 61" or "du 23000" or "fg 7051" or "ici 58834" or "l deprenyl" or "leo 640" or "lilly 110140" or "lu 10 171" or "ma 1291" or "nsc 16895" or "org gb 94" or "r 55667" or "ro 11 1163" or "trans 2 phenylcyclopropylamine" or "ym 35 995" or "ym 992" or "zk 62711" or abilify or adapin or adaptol or adderall or agomelatin* or aiglonyl or allegron or altrulin* or amfebutamon* or amineptin* or amineurin or amisulprid* or amitrip* or amitrol or amizol or amoxapin* or amphetamin* or anafranil or anapsique or aponal or ardeydorm or ardeytropin or aremis or arima or aripiprazol* or arminol or aropax or asenapin* or asendin or astyl or atomoxetin* or aurorix or aventyl or axiomin or azepin* or benactyzin* or benzeneacetic acid or besitran or bolvidon or bosnyl or brofaramin* or bupropion or buspar or buspiron* or butriptylin* or carbamazepin* or celexa or chlomipramin* or chlorgylin* or cipralex or cipramil or citalopram* or clomipramin* or clorgilin* or clorgylin* or concerta or cymbalta or cytalopram* or dalcipran or damilen or deanol or defanyl or deftan or demanyl or demolox or depakote or deponerton or deprax or deprenorm or deprilept or deptran or desidox or desiflu or desipramin* or desisulpid or desitriptylin* or desmethylamitriptylin* or desmethylimipramin* or desmethylloxapin* or desvenlafaxin* or desyrel or dexedrin* or dexmethylphenidat* or dextroamphetamin* or dibencycladin* or digton or dilithium carbonate or dimethylaminoethanol or dimethylethanolamin* or dmi or dogmatil or dolmatil or domical or doneurin or dosulepin or dothiepin or doxepia or doxepin* or duloxetin* or dumirox or edronax or effexor or eglonyl or ekilid or elavil or eldepryl or eldoral or emovit or emsam or endep or escitalopram or eskalith or espadox or espiride or etonin or etoperidone or evaden* or favarin or fenelzin or feprapax or feraken or fevarin or floxyfral or fluoxetin* or fluvoxadura or fluvoxamin* or focalin or gamanil or gladem or guastil or herphonal or hydiphen or imidobenzyl* or imipramin* or imizin or insidon or iprazid or iprindol* or iproniazid or isocarboxazid or ixel or janimin* or jatrosom or lamictal or lamotrigin* or laroxyl or leboprid* or lentizol or lerivon or lexapro or lisdexamfetamin* or lithan* or lithium or lithobid or lofepramin* or lomont or lopramin* or lubazodon* or lucidil or ludiomil or lustral or luvox or lyphan or manerix or maprolu or maprotilin* or mareen or marplan or melitracen or meresa or meridia or methylphenidat* or mianserin or micalith or midalcipran or milnacepra* or mirpan or mirtazapin* or moclamin* or moclix or moclobemid* or moclobeta or moclodura or moclonorm or modal or molipaxin or nardelzin* or nardil or naturruhe or nefadar or nefazodon* or neogama or nialamid* or norfenazin or noritren or norpramin* or nortrilen or nortriptylin* or norval or novoprotect or olanzapin* or opipramol or optimax or oxitriptan* or pamelor or parnate or paroxetin* or paxil or paxtibi or pertofran* or pertrofran or petylyl or phenelzin* or phenethylhydrazin* or phenylethylhydrazin* or pirazidol or pirlindol* or pizotifen or pizotylin* or polomigran or pontirid* or pramolan or priadel or pristiq or prondol or prothiaden or protriptylin* or prozac or prudoxin or pryleugan or psicocen or psymion or quetiapin* or quilinorm* or quipazin* or quitaxon or quomen or r55667 or reboxetin* or reductil or remeron or rhotrimin* or rimoc or ritalin or ritanserin or rolipram or sandomigran or saphris or sarafem or saroten or sarotex or savella or sealdin or sediel or selegilin* or sendis or seroquel or seroxat or sertralin* or serzone or sibutramin* or sinequan or solian or stangyl or strattera or sulp or sulpirid* or sulpitil or sulpivert or sulpor or surmontil or sycrest or symbyax or synedil or syneudon or tandospiron* or tegretol or tepavil or thombran or tianeptin* or tofranil or toledomin or tolvon or tonibral or tradozon* or tramadol or tramal or transamine or tranylcypromin* or trazodon* or trimepr* or trimidura or trimineurin or trimipr* or tripramin* or triptafen or trittico or trofan or tryptacin or tryptan or tryptanol or tryptin* or tryptizol or tryptophan* or tyrima or ultram or valdoxan or valpro* or venlafaxin* or viibryd or vilazodon* or viloxazin* or vivactil or vivalan or vyvanse or wellbutrin or xepin or yentreve or zelapar or zimelidin* or zispin or zoloft or zonalon or zyban or zyntabac):AB,KW,MC,MH,TI AND INSEGMENT
2. MeSH DESCRIPTOR Antidepressive Agents Explode All AND INSEGMENT
3. #1 OR #2 AND INSEGMENT
4. MeSH DESCRIPTOR Depression Explode All AND INSEGMENT
5. MeSH DESCRIPTOR Depressive Disorder Explode All AND INSEGMENT
6. MeSH DESCRIPTOR Dysthymic Disorder Explode All AND INSEGMENT
7. (depression* or depressive*):AB,KW,MC,MH,TI AND INSEGMENT
8. "respiratory depression":AB,KW,MC,MH,TI AND INSEGMENT
9. (#4 OR #5 OR #6 OR #7) NOT #8 AND INSEGMENT
10. #3 AND #9 AND INSEGMENT
11. MESH DESCRIPTOR Epilepsy EXPLODE ALL AND INSEGMENT
12. MESH DESCRIPTOR Seizures EXPLODE ALL AND INSEGMENT
13. (epilep* OR seizure* OR convuls*):AB,KW,MC,MH,TI AND INSEGMENT
14. #11 OR #12 OR #13 AND INSEGMENT
15. #10 AND #14 AND INSEGMENT
16. MESH DESCRIPTOR Electroconvulsive Therapy EXPLODE ALL AND INSEGMENT
17. #15 NOT #16 AND INSEGMENT
18. (antidepressant* or antidepressive* or "af 1161" or "ba 34276" or "bc 105" or "brl 29060" or "brl 29060" or "cl 67772" or "cp 15467 61" or "du 23000" or "fg 7051" or "ici 58834" or "l deprenyl" or "leo 640" or "lilly 110140" or "lu 10 171" or "ma 1291" or "nsc 16895" or "org gb 94" or "r 55667" or "ro 11 1163" or "trans 2 phenylcyclopropylamine" or "ym 35 995" or "ym 992" or "zk 62711" or abilify or adapin or adaptol or adderall or agomelatin* or aiglonyl or allegron or altrulin* or amfebutamon* or amineptin* or amineurin or amisulprid* or amitrip* or amitrol or amizol or amoxapin* or amphetamin* or anafranil or anapsique or aponal or ardeydorm or ardeytropin or aremis or arima or aripiprazol* or arminol or aropax or asenapin* or asendin or astyl or atomoxetin* or aurorix or aventyl or axiomin or azepin* or benactyzin* or benzeneacetic acid or besitran or bolvidon or bosnyl or brofaramin* or bupropion or buspar or buspiron* or butriptylin* or carbamazepin* or celexa or chlomipramin* or chlorgylin* or cipralex or cipramil or citalopram* or clomipramin* or clorgilin* or clorgylin* or concerta or cymbalta or cytalopram* or dalcipran or damilen or deanol or defanyl or deftan or demanyl or demolox or depakote or deponerton or deprax or deprenorm or deprilept or deptran or desidox or desiflu or desipramin* or desisulpid or desitriptylin* or desmethylamitriptylin* or desmethylimipramin* or desmethylloxapin* or desvenlafaxin* or desyrel or dexedrin* or dexmethylphenidat* or dextroamphetamin* or dibencycladin* or digton or dilithium carbonate or dimethylaminoethanol or dimethylethanolamin* or dmi or dogmatil or dolmatil or domical or doneurin or dosulepin or dothiepin or doxepia or doxepin* or duloxetin* or dumirox or edronax or effexor or eglonyl or ekilid or elavil or eldepryl or eldoral or emovit or emsam or endep or escitalopram or eskalith or espadox or espiride or etonin or etoperidone or evaden* or favarin or fenelzin or feprapax or feraken or fevarin or floxyfral or fluoxetin* or fluvoxadura or fluvoxamin* or focalin or gamanil or gladem or guastil or herphonal or hydiphen or imidobenzyl* or imipramin* or imizin or insidon or iprazid or iprindol* or iproniazid or isocarboxazid or ixel or janimin* or jatrosom or lamictal or lamotrigin* or laroxyl or leboprid* or lentizol or lerivon or lexapro or lisdexamfetamin* or lithan* or lithium or lithobid or lofepramin* or lomont or lopramin* or lubazodon* or lucidil or ludiomil or lustral or luvox or lyphan or manerix or maprolu or maprotilin* or mareen or marplan or melitracen or meresa or meridia or methylphenidat* or mianserin or micalith or midalcipran or milnacepra* or mirpan or mirtazapin* or moclamin* or moclix or moclobemid* or moclobeta or moclodura or moclonorm or modal or molipaxin or nardelzin* or nardil or naturruhe or nefadar or nefazodon* or neogama or nialamid* or norfenazin or noritren or norpramin* or nortrilen or nortriptylin* or norval or novoprotect or olanzapin* or opipramol or optimax or oxitriptan* or pamelor or parnate or paroxetin* or paxil or paxtibi or pertofran* or pertrofran or petylyl or phenelzin* or phenethylhydrazin* or phenylethylhydrazin* or pirazidol or pirlindol* or pizotifen or pizotylin* or polomigran or pontirid* or pramolan or priadel or pristiq or prondol or prothiaden or protriptylin* or prozac or prudoxin or pryleugan or psicocen or psymion or quetiapin* or quilinorm* or quipazin* or quitaxon or quomen or r55667 or reboxetin* or reductil or remeron or rhotrimin* or rimoc or ritalin or ritanserin or rolipram or sandomigran or saphris or sarafem or saroten or sarotex or savella or sealdin or sediel or selegilin* or sendis or seroquel or seroxat or sertralin* or serzone or sibutramin* or sinequan or solian or stangyl or strattera or sulp or sulpirid* or sulpitil or sulpivert or sulpor or surmontil or sycrest or symbyax or synedil or syneudon or tandospiron* or tegretol or tepavil or thombran or tianeptin* or tofranil or toledomin or tolvon or tonibral or tradozon* or tramadol or tramal or transamine or tranylcypromin* or trazodon* or trimepr* or trimidura or trimineurin or trimipr* or tripramin* or triptafen or trittico or trofan or tryptacin or tryptan or tryptanol or tryptin* or tryptizol or tryptophan* or tyrima or ultram or valdoxan or valpro* or venlafaxin* or viibryd or vilazodon* or viloxazin* or vivactil or vivalan or vyvanse or wellbutrin or xepin or yentreve or zelapar or zimelidin* or zispin or zoloft or zonalon or zyban or zyntabac):AB,KW,MC,MH,TI AND CENTRAL:TARGET
19. MeSH DESCRIPTOR Antidepressive Agents Explode All AND CENTRAL:TARGET
20. #18 OR #19 AND CENTRAL:TARGET
21. MeSH DESCRIPTOR Depression Explode All AND CENTRAL:TARGET
22. MeSH DESCRIPTOR Depressive Disorder Explode All AND CENTRAL:TARGET
23. MeSH DESCRIPTOR Dysthymic Disorder Explode All AND CENTRAL:TARGET
24. (depression* or depressive*):AB,KW,MC,MH,TI AND CENTRAL:TARGET
25. "respiratory depression":AB,KW,MC,MH,TI AND CENTRAL:TARGET
26. (#21 OR #22 OR #23 OR #24) NOT #25 AND CENTRAL:TARGET
27. #20 AND #26 AND CENTRAL:TARGET
28. MESH DESCRIPTOR Epilepsy EXPLODE ALL AND CENTRAL:TARGET
29. MESH DESCRIPTOR Seizures EXPLODE ALL AND CENTRAL:TARGET
30. (epilep* OR seizure* OR convuls*):AB,KW,MC,MH,TI AND CENTRAL:TARGET
31. #28 OR #29 OR #30 AND CENTRAL:TARGET
32. #27 AND #31 AND CENTRAL:TARGET
33. MESH DESCRIPTOR Electroconvulsive Therapy EXPLODE ALL AND CENTRAL:TARGET
34. #32 NOT #33 AND CENTRAL:TARGET
35. #17 OR #34
Appendix 2. MEDLINE Ovid search strategy
1. exp Depression/ or exp Depressive Disorder/ or exp Dysthymic Disorder/ or (depression$ or depressive$).tw.
2. "respiratory depression".tw.
3. 1 not 2
4. exp Antidepressive Agents/ or anti?depressant$.tw. or anti?depressiv$.tw.
5. ("af 1161" or "bc 105" or "brl 29060" or "cl 67772" or "cp 15467 61" or "du 23000" or "ici 58834" or "leo 640" or "lilly 110140" or "ma 1291" or "nsc 16895" or "org gb 94" or "r 55667" or "ro 11‐1163" or "trans 2 phenylcyclopropylamine" or "ym‐35,995" or "zk 62711" or abilify or adapin or adaptol or adderall or af?1161 or agomelatine or aiglonyl or allegron or altruline or amfebutamone or amineptine or amineurin or amisulpride or amitrip or amitriptylin$ or amitrol or amiz?l or amoxapine or amphetamine or anafranil or anapsique or apo?doxepin or apo?moclob?mide or apo?nortriptyline or apo?sertraline or apo?trimip or apoamitriptyline or aponal or ardeydorm or ardeytropin or aremis or arima or aripiprazole or arminol or aropax or asenapine or asendin or astyl or atomoxetine or auror?x or aventyl or axiomin or ba?34276 or bc?105 or benactyzine or benzeneacetic acid or besitran or beta?phenylethylhydrazine or bolvidon or bosnyl or brl?29060 or brofaramine or bupropion or buspar or buspirone or butriptyline or carbamazepine or celexa or chlomipramine or chlorgyline or cipralex or cipramil or citalopram or cl?67772 or clomipramine or clorgilin$ or clorgyline or concerta or cp?15467?61 or cymbalta or cytalopram or dalcipran or damilen or de?methylimipramine or deanol or defanyl or deftan or deman?l or demolox or depakote or deponerton or deprax or deprenorm or deprilept or deptran or desidox or desiflu or desipramine or desisulpid or desitriptyline or desmethylamitriptylin or desmethylloxapine or desvenlafaxine or desyrel or dexedrine or dexmethylphenidate or dextroamphetamine or dibencycladine or digton or dilithium carbonate or dimethylaminoethanol or dimethylethanolamine or dogmatil or dolmatil or domical or doneurin or dosulepin or dothiepin or doxepia or doxepin$ or du?23000 or duloxetine or dumirox or edronax or ef?exor or eglonyl or ekilid or elavil or eldepryl or eldoral or emovit or emsam or endep or escitalopram or eskalith or espadox or espiride or etonin or etoperidone or evadene or favarin or fenelzin or feprapax or feraken or fevarin or fg?7051 or floxyfral or fluoxetin$ or fluvoxadura or fluvoxamin$ or focalin or gam?nil or gen?nortriptyline or gen?sertraline or gladem or guastil or herphonal or hydiphen or ici?58834 or imidobenzyle or imipramine or imizin or insidon or iprazid or iprindole or iproniazid or isocarboxazid or ixel or janimine or jatrosom or lamictal or lamotrigine or laroxyl or l‐deprenyl or lebopride or lentizol or lerivon or levo?tryptophan or lexapro or lilly?110140 or lisdexamfetamine or lithane or lithium or lithobid or lofepramine or lomont or lopramine or l‐tryptophan or lu?10?171 or lubazodone or lucidil or ludiomil or lustral or luvox or lyphan or ma?1291 or manerix or maprolu or maprotilin$ or mareen or marplan or melitracen or meresa or meridia or methylphenidate or mianserin or micalith or midalcipran or milnacepram or milnacipra? or mirpan or mirtazapine or moclamine or moclix or moclob?mide or moclobemid$ or moclobeta or moclodura or moclonorm or modal or molipaxin or nardelzine or nardil or naturruhe or nefadar or nefazodone or neogama or nialamide or nor?nortriptyline or norfenazin or norpramin or nortrilen or nortriptyline or norval or novo?doxepin or novo?moclob?mide or novo?nortriptyline or novo?sertraline or novo?tripramine or novoprotect or nsc?16895 or nu?moclob?mide or nu?nortriptyline or nu?trimipramine or nu?tripramine or numo?moclob?mide or olanzapine or opipramol or optimax or oxitriptan or pamelor or parnate or paroxetine or paxil or paxtibi or pert?ofran$ or petylyl or phenelzine or phenethylhydrazine or phenylethylhydrazine or pirazidol or pirlindole or pizotifen or pizotyline or pms?moclob?mide or pms?nortriptyline or pms?tryptophan or polomigran or pontiride or pramolan or priadel or pristiq or prondol or prothiaden or protriptyline or prozac or prudoxin or pryleugan or psicocen or psymion or quetiapine or quilinorm?retard or quipazine or quitaxon or quomen or r55667 or r‐55667 or ratio?nortriptyline or ratio?sertraline or ratio?tryptophan or reboxetine or reductil or remeron or rhotrimine or rhoxal?sertraline or rimoc or ritalin or ritanserin or ro‐11‐1163 or rolipram or sandomigran or saphris or sarafem or saroten or sarotex or savella or sealdin or sediel or selegiline or sendis or seroquel or seroxat or sertraline or serzone or sibutramine or sin?quan or solian or stangyl or strattera or sulp or sulpiride or sulpitil or sulpivert or sulpor or surmontil or sycrest or symbyax or synedil or syneudon or tandospirone or tegretol or tepavil or thombran or tianeptine or tofranil or toledomin or tolvon or tonibral or tradozone or tramadol or tramal or trans‐2‐phenylcyclopropylamine or transamine or tranylcypromine or trazodon$ or trim?pr?min$ or trimidura or trimineurin or trimip or tripramine or triptafen or trittico or trofan or tryptacin or tryptan or tryptanol or tryptine or tryptizol or tryptophan or tyrima or ultram or valdoxan or valproic acid or venlafaxine or viibryd or vilazodone or viloxazine or vivactil or vivalan or vyvanse or wellbutrin or xepin or yentreve or ym‐992 or zelapar or zimelidine or zispin or zk?62711 or zoloft or zonalon or zyban or zyntabac).mp.
6. 4 or 5
7. exp Epilepsy/
8. exp Seizures/
9. (epilep$ or seizure$ or convuls$).tw.
10. 7 or 8 or 9
11. exp Pre‐Eclampsia/ or exp Eclampsia/
12. 10 not 11
13. 3 and 6 and 12
14. exp *Electroconvulsive Therapy/
15. 13 not 14
16. exp controlled clinical trial/ or (randomi?ed or placebo or randomly).ab.
17. clinical trials as topic.sh.
18. trial.ti.
19. 16 or 17 or 18
20. exp cohort studies/ or cohort$.tw,hw.
21. exp epidemiologic methods/ or exp follow‐up studies/ or exp prospective studies/
22. limit 21 to yr=1966‐1989
23. exp controlled before‐after studies/ or ("before and after" or "before‐and‐after").tw,hw.
24. 19 or 20 or 22 or 23
25. exp animals/ not humans.sh.
26. 24 not 25
27. 26 not case reports.pt.
28. 15 and 27
29. remove duplicates from 28
Appendix 3. SCOPUS search strategy
(((((((TITLE‐ABS‐KEY(antidepressant* OR antidepressiv*)) OR (TITLE‐ABS‐KEY("af 1161" OR "ba 34276" OR "bc 105" OR "brl 29060" OR "brl 29060" OR "cl 67772" OR "cp 15467 61" OR "du 23000" OR "fg 7051" OR "ici 58834" OR "l deprenyl" OR "leo 640" OR "lilly 110140" OR "lu 10 171" OR "ma 1291" OR "nsc 16895" OR "org gb 94" OR "r 55667" OR "ro 11 1163" OR "trans 2 phenylcyclopropylamine" OR "ym 35 995" OR "ym 992" OR "zk 62711" OR *amitriptyline OR *doxepin OR *moclobemide OR *nortriptyline OR *phenylethylhydrazine OR *sertraline OR *trimip OR *trimipramine OR *tripramine OR *tryptophan)) OR (TITLE‐ABS‐KEY(abilify OR adapin OR adaptol OR adderall OR agomelatine OR aiglonyl OR allegron OR altruline OR amfebutamone OR amineptine OR amineurin OR amisulpride OR amitrip OR amitriptylin* OR amitrol OR amiz?l OR amoxapine OR amphetamine OR anafranil OR anapsique OR aponal OR ardeydorm OR ardeytropin OR aremis OR arima OR aripiprazole OR arminol OR aropax OR asenapine OR asendin OR astyl OR atomoxetine OR auror?x OR aventyl OR axiomin)) OR (TITLE‐ABS‐KEY(benactyzine OR benzeneacetic acid OR besitran OR bolvidon OR bosnyl OR brofaramine OR bupropion OR buspar OR buspirone OR butriptyline OR carbamazepine OR celexa OR chlomipramine OR chlorgyline OR cipralex OR cipramil OR citalopram OR clomipramine OR clorgilin* OR clorgyline OR concerta OR cymbalta OR cytalopram)) OR (TITLE‐ABS‐KEY(dalcipran OR damilen OR de*methylimipramine OR deanol OR defanyl OR deftan OR deman?l OR demolox OR depakote OR deponerton OR deprax OR deprenorm OR deprilept OR deptran OR desidox OR desiflu OR desipramine OR desisulpid OR desitriptyline OR desmethylamitriptylin OR desmethylloxapine OR desvenlafaxine OR desyrel OR dexedrine OR dexmethylphenidate OR dextroamphetamine OR dibencycladine OR digton OR dilithium carbonate OR dimethylaminoethanol OR dimethylethanolamine OR dogmatil OR dolmatil OR domical OR doneurin OR dosulepin OR dothiepin OR doxepia OR doxepin* OR duloxetine OR dumirox))) OR ((TITLE‐ABS‐KEY(edronax OR ef*exor OR eglonyl OR ekilid OR elavil OR eldepryl OR eldoral OR emovit OR emsam OR endep OR escitalopram OR eskalith OR espadox OR espiride OR etonin OR etoperidone OR evadene OR favarin OR fenelzin OR feprapax OR feraken OR fevarin OR floxyfral OR fluoxetin* OR fluvoxadura OR fluvoxamin* OR focalin OR gam?nil OR gladem OR guastil OR herphonal OR hydiphen)) OR (TITLE‐ABS‐KEY(imidobenzyle OR imipramine OR imizin OR insidon OR iprazid OR iprindole OR iproniazid OR isocarboxazid OR ixel OR janimine OR jatrosom OR lamictal OR lamotrigine OR laroxyl OR lebopride OR lentizol OR lerivon OR lexapro OR lisdexamfetamine OR lithane OR lithium OR lithobid OR lofepramine OR lomont OR lopramine OR lubazodone OR lucidil OR ludiomil OR lustral OR luvox OR lyphan)) OR (TITLE‐ABS‐KEY(manerix OR maprolu OR maprotilin* OR mareen OR marplan OR melitracen OR meresa OR meridia OR methylphenidate OR mianserin OR micalith OR midalcipran OR milnacepra* OR mirpan OR mirtazapine OR moclamine OR moclix OR moclob?mide OR moclobemid* OR moclobeta OR moclodura OR moclonorm OR modal OR molipaxin OR nardelzine OR nardil OR naturruhe OR nefadar OR nefazodone OR neogama OR nialamide OR norfenazin OR norpramin OR nortrilen OR norval OR novoprotect OR olanzapine OR opipramol OR optimax OR oxitriptan)) OR (TITLE‐ABS‐KEY(pamelor OR parnate OR paroxetine OR paxil OR paxtibi OR pertofran* OR pertrofran OR petylyl OR phenelzine OR phenethylhydrazine OR pirazidol OR pirlindole OR pizotifen OR pizotyline OR polomigran OR pontiride OR pramolan OR priadel OR pristiq OR prondol OR prothiaden OR protriptyline OR prozac OR prudoxin OR pryleugan OR psicocen OR psymion OR quetiapine OR quilinorm* OR quipazine OR quitaxon OR quomen)))) OR (TITLE‐ABS‐KEY(r55667 OR reboxetine OR reductil OR remeron OR rhotrimine OR rimoc OR ritalin OR ritanserin OR rolipram OR sandomigran OR saphris OR sarafem OR saroten OR sarotex OR savella OR sealdin OR sediel OR selegiline OR sendis OR seroquel OR seroxat OR serzone OR sibutramine OR sin*quan OR solian OR stangyl OR strattera OR sulp OR sulpiride OR sulpitil OR sulpivert OR sulpor OR surmontil OR sycrest OR symbyax OR synedil OR syneudon)) OR (TITLE‐ABS‐KEY(tandospirone OR tegretol OR tepavil OR thombran OR tianeptine OR tofranil OR toledomin OR tolvon OR tonibral OR tradozone OR tramadol OR tramal OR transamine OR tranylcypromine OR trazodon* OR trimeprimin* OR trimidura OR trimineurin OR triptafen OR trittico OR trofan OR tryptacin OR tryptan OR tryptanol OR tryptine OR tryptizol OR tyrima OR ultram OR valdoxan OR valproic acid OR venlafaxine OR viibryd OR vilazodone OR viloxazine OR vivactil OR vivalan OR vyvanse OR wellbutrin OR xepin OR yentreve OR zelapar OR zimelidine OR zispin OR zoloft OR zonalon OR zyban OR zyntabac))) AND (TITLE‐ABS‐KEY(dysthymic OR depression* OR depressive*) AND NOT TITLE‐ABS‐KEY("respiratory depression"))) AND ((TITLE‐ABS‐KEY(epilep* OR "infantile spasm" OR "ring chromosome 20" OR "R20" OR "myoclonic encephalopathy" OR "pyridoxine dependency") OR (TITLE‐ABS‐KEY(syndrome) W/2 (aicardi OR angelman OR doose OR dravet OR janz OR jeavons OR "landau kleffner" OR "lennox gastaut" OR ohtahara OR panayiotopoulos OR rasmussen OR rett OR "sturge weber" OR tassinari OR "unverricht lundborg" OR west)) OR TITLE(seizure OR convuls*) OR (TITLE‐ABS‐KEY(lafora*) W/4 (disease OR epilep*) AND NOT (TITLE(dog OR canine) OR INDEXTERMS(dog OR canine)))) AND NOT (TITLE(*eclampsia) OR INDEXTERMS(*eclampsia)) AND NOT INDEX(medline))) AND NOT (TITLE(electroconvulsive OR ECT))) AND ((((TITLE((randomiz* OR randomis* OR controlled OR placebo OR blind* OR unblind* OR "parallel group" OR crossover OR "cross over" OR cluster OR "head to head") PRE/2 (analy* OR method OR procedure OR study OR studies OR trial))) OR (ABS((randomiz* OR randomis* OR controlled OR placebo OR blind* OR unblind* OR "parallel group" OR crossover OR "cross over" OR cluster OR "head to head") PRE/2 (analy* OR method OR procedure OR study OR studies OR trial)))) OR (TITLE(("before and after" OR cohort OR prospective) PRE/2 (trial OR method OR procedure OR study))) OR (ABS(("before and after" OR cohort OR prospective) PRE/2 (trial OR method OR procedure OR study)))) AND NOT (INDEXTERMS("case report") OR TITLE ("case report") OR DOCTYPE(re)))
Appendix 4. PsycINFO EBSCOhost search strategy
S11 S6 AND S10
S10 S7 OR S8 OR S9
S9 TI (("before and after" OR cohort OR prospective) W2 (analy* OR method OR procedure OR study OR studies OR trial))
S8 AB (("before and after" OR cohort OR prospective) W2 (analy* OR method OR procedure OR study OR studies OR trial))
S7 TI ( (randomiz* OR randomis* OR controlled OR placebo OR blind* OR unblind* OR "parallel group" OR crossover OR cross‐over OR cluster OR "head to head") N2 (analy* OR method OR procedure OR study OR studies OR trial) ) OR AB ( (randomiz* OR randomis* OR controlled OR placebo OR blind* OR unblind* OR "parallel group" OR crossover OR cross‐over OR cluster OR "head to head") N2 (analy* OR method OR procedure OR study OR studies OR trial) )
S6 S5 NOT DE "Electroconvulsive Shock Therapy"
S5 (S1 OR S2) AND S3 AND S4
S4 DE "Epilepsy" OR DE "Seizures" OR DE "Epileptic Seizures" OR DE "Grand Mal Seizures" OR DE "Petit Mal Seizures" OR DE "Status Epilepticus" OR epilep* OR seizure* OR convuls*
S3 (DE "Depression (Emotion)" OR DE "Major Depression" OR DE "Anaclitic Depression" OR DE "Dysthymic Disorder" OR DE "Endogenous Depression" OR DE "Postpartum Depression" OR DE "Reactive Depression" OR DE "Recurrent Depression" OR DE "Treatment Resistant Depression" OR depression* OR depressive*) NOT "respiratory depression"
S2 DE "Serotonin Reuptake Inhibitors" OR DE "Chlorimipramine" OR DE "Citalopram" OR DE "Fluoxetine" OR DE "Fluvoxamine" OR DE "Paroxetine" OR DE "Zimeldine" OR DE "Serotonin Agonists" OR DE "Triptans" OR DE "Serotonin Antagonists" OR DE "Dihydroxytryptamine" OR DE "Lysergic Acid Diethylamide" OR DE "Mianserin" OR DE "Molindone" OR DE "Parachlorophenylalanine" OR DE "Ritanserin" OR DE "Tetrabenazine" OR DE "Tryptophan" OR DE "Hydroxytryptophan (5‐)" OR DE "Serotonin Precursors" OR DE "Tryptophan" OR DE "Antidepressant Drugs" OR DE "Bupropion" OR DE "Citalopram" OR DE "Fluoxetine" OR DE "Fluvoxamine" OR DE "Iproniazid" OR DE "Isocarboxazid" OR DE "Lithium Carbonate" OR DE "Methylphenidate" OR DE "Mianserin" OR DE "Moclobemide" OR DE "Molindone" OR DE "Nefazodone" OR DE "Nialamide" OR DE "Nomifensine" OR DE "Paroxetine" OR DE "Phenelzine" OR DE "Pheniprazine" OR DE "Pipradrol" OR DE "Serotonin Norepinephrine Reuptake Inhibitors" OR DE "Sertraline" OR DE "Sulpiride" OR DE "Tranylcypromine" OR DE "Trazodone" OR DE "Tricyclic Antidepressant Drugs" OR DE "Venlafaxine" OR DE "Zimeldine" OR DE "Serotonin Norepinephrine Reuptake Inhibitors" OR DE "Venlafaxine" OR DE "Tricyclic Antidepressant Drugs" OR DE "Amitriptyline" OR DE "Chlorimipramine" OR DE "Desipramine" OR DE "Doxepin" OR DE "Imipramine" OR DE "Maprotiline" OR DE "Nortriptyline" OR DE "Lithium" OR DE "Lithium Carbonate" OR DE "Monoamine Oxidase Inhibitors" OR DE "Iproniazid" OR DE "Isocarboxazid" OR DE "Moclobemide" OR DE "Nialamide" OR DE "Pargyline" OR DE "Phenelzine" OR DE "Pheniprazine" OR DE "Tranylcypromine" OR antidepressant OR antidepressive
S1 "af 1161" OR "ba 34276" OR "bc 105" OR "brl 29060" OR "brl 29060" OR "cl 67772" OR "cp 15467 61" OR "du 23000" OR "fg 7051" OR "ici 58834" OR "l deprenyl" OR "leo 640" OR "lilly 110140" OR "lu 10 171" OR "ma 1291" OR "nsc 16895" OR "org gb 94" OR "r 55667" OR "ro 11 1163" OR "trans 2 phenylcyclopropylamine" OR "ym 35 995" OR "ym 992" OR "zk 62711" OR abilify or adapin or adaptol or adderall or agomelatine or aiglonyl or allegron or altruline or amfebutamone or amineptine or amineurin or amisulpride or amitrip or amitriptylin* or amitrol or amiz?l or amoxapine or amphetamine or anafranil or anapsique or apo#doxepin or apo#moclob?mide or apo#nortriptyline or apo#sertraline or apo#trimip or apoamitriptyline or aponal or ardeydorm or ardeytropin or aremis or arima or aripiprazole or arminol or aropax or asenapine or asendin or astyl or atomoxetine or auror?x or aventyl or axiomin or benactyzine or benzeneacetic acid or besitran or beta#phenylethylhydrazine or bolvidon or bosnyl or brofaramine or bupropion or buspar or buspirone or butriptyline or carbamazepine or celexa or chlomipramine or chlorgyline or cipralex or cipramil or citalopram or clomipramine or clorgilin* or clorgyline or concerta or cymbalta or cytalopram or dalcipran or damilen or de#methylimipramine or deanol or defanyl or deftan or deman?l or demolox or depakote or deponerton or deprax or deprenorm or deprilept or deptran or desidox or desiflu or desipramine or desisulpid or desitriptyline or desmethylamitriptylin or desmethylloxapine or desvenlafaxine or desyrel or dexedrine or dexmethylphenidate or dextroamphetamine or dibencycladine or digton or dilithium carbonate or dimethylaminoethanol or dimethylethanolamine or dogmatil or dolmatil or domical or doneurin or dosulepin or dothiepin or doxepia or doxepin* or duloxetine or dumirox or edronax or ef#exor or eglonyl or ekilid or elavil or eldepryl or eldoral or emovit or emsam or endep or escitalopram or eskalith or espadox or espiride or etonin or etoperidone or evadene or favarin or fenelzin or feprapax or feraken or fevarin or floxyfral or fluoxetin* or fluvoxadura or fluvoxamin* or focalin or gam?nil or gen#nortriptyline or gen#sertraline or gladem or guastil or herphonal or hydiphen or imidobenzyle or imipramine or imizin or insidon or iprazid or iprindole or iproniazid or isocarboxazid or ixel or janimine or jatrosom or lamictal or lamotrigine or laroxyl or lebopride or lentizol or lerivon or levo#tryptophan or lexapro or lisdexamfetamine or lithane or lithium or lithobid or lofepramine or lomont or lopramine or l#tryptophan or lubazodone or lucidil or ludiomil or lustral or luvox or lyphan or manerix or maprolu or maprotilin* or mareen or marplan or melitracen or meresa or meridia or methylphenidate or mianserin or micalith or midalcipran or milnacepram or milnacipra? or mirpan or mirtazapine or moclamine or moclix or moclob?mide or moclobemid* or moclobeta or moclodura or moclonorm or modal or molipaxin or nardelzine or nardil or naturruhe or nefadar or nefazodone or neogama or nialamide or nor#nortriptyline or norfenazin or norpramin or nortrilen or nortriptyline or norval or novo#doxepin or novo#moclob?mide or novo#nortriptyline or novo#sertraline or novo#tripramine or novoprotect or nu#moclob?mide or nu#nortriptyline or nu#trimipramine or nu#tripramine or numo#moclob?mide or olanzapine or opipramol or optimax or oxitriptan or pamelor or parnate or paroxetine or paxil or paxtibi or pert#ofran* or petylyl or phenelzine or phenethylhydrazine or phenylethylhydrazine or pirazidol or pirlindole or pizotifen or pizotyline or pms#moclob?mide or pms#nortriptyline or pms#tryptophan or polomigran or pontiride or pramolan or priadel or pristiq or prondol or prothiaden or protriptyline or prozac or prudoxin or pryleugan or psicocen or psymion or quetiapine or quilinorm#retard or quipazine or quitaxon or quomen or r55667 or ratio#nortriptyline or ratio#sertraline or ratio#tryptophan or reboxetine or reductil or remeron or rhotrimine or rhoxal#sertraline or rimoc or ritalin or ritanserin or rolipram or sandomigran or saphris or sarafem or saroten or sarotex or savella or sealdin or sediel or selegiline or sendis or seroquel or seroxat or sertraline or serzone or sibutramine or sin#quan or solian or stangyl or strattera or sulp or sulpiride or sulpitil or sulpivert or sulpor or surmontil or sycrest or symbyax or synedil or syneudon or tandospirone or tegretol or tepavil or thombran or tianeptine or tofranil or toledomin or tolvon or tonibral or tradozone or tramadol or tramal or transamine or tranylcypromine or trazodon* or trim?pr?min* or trimidura or trimineurin or trimip or tripramine or triptafen or trittico or trofan or tryptacin or tryptan or tryptanol or tryptine or tryptizol or tryptophan or tyrima or ultram or valdoxan or valproic acid or venlafaxine or viibryd or vilazodone or viloxazine or vivactil or vivalan or vyvanse or wellbutrin or xepin or yentreve or zelapar or zimelidine or zispin or zoloft or zonalon or zyban or zyntabac
Appendix 5. ClinicalTrials.gov search strategy
Interventional Studies | Epilepsy | Antidepressant
Appendix 6. ICTRP search strategy
Condition: epilepsy
Intervention: antidepressant
Recruitment status: All
Phases: All
Appendix 7. Signalling questions for the seven 'Risk of bias' domains of the ROBINS‐I tool
| Bias due to confounding | |
| 1.1 Is there potential for confounding of the effect of intervention in this study? If N/PN to 1.1: the study can be considered to be at low risk of bias due to confounding and no further signalling questions need be considered If Y/PY to 1.1: determine whether there is a need to assess time‐varying confounding |
Y/PY/PN/N |
| 1.2. If Y/PY to 1.1: Was the analysis based on splitting participants’ follow‐up time according to intervention received? If N/PN to 1.2, answer questions relating to baseline confounding (1.4 to 1.6) If Y/PY to 1.2, go to question 1.3 |
Y/PY/PN/N/NI/NA |
| 1.3 Were intervention discontinuations or switches likely to be related to factors that are prognostic for the outcome? If N/PN to 1.3, answer questions relating to baseline confounding (1.4 to 1.6) If Y/PY to 1.3, answer questions relating to both baseline and time‐varying confounding (1.7 and 1.8) |
Y/PY/PN/N/NI/NA |
| 1.4. Did the authors use an appropriate analysis method that controlled for all the important confounding domains? | Y/PY/PN/N/NI/NA |
| 1.5. If Y/PY to 1.4, were confounding domains that were controlled for measured validly and reliably by the variables available in this study? | Y/PY/PN/N/NI/NA |
| 1.6. Did the authors control for any post‐intervention variables that could have been affected by the intervention? | Y/PY/PN/N/NI/NA |
| 1.7. Did the authors use an appropriate analysis method that controlled for all the important confounding domains and for time‐varying confounding? | Y/PY/PN/N/NI/NA |
| 1.8. If Y or PY to 1.7, were confounding domains that were controlled for measured validly and reliably by the variables available in this study? | Y/PY/PN/N/NI/NA |
| 'Risk of bias' judgement | Low/Moderate/Serious/Critical/NI |
| Bias in selection of participants into the study | |
| 2.1. Was selection of participants into the study (or into the analysis) based on participant characteristics observed after the start of intervention? If N/PN to 2.1, go to 2.4 |
Y/PY/PN/N/NI |
| 2.2. If Y/PY to 2.1, were the post‐intervention variables that influenced selection likely to be associated with intervention? | Y/PY/PN/N/NI/NA |
| 2.3. If Y/PY to 2.2, were the post‐intervention variables that influenced selection likely to be influenced by the outcome or a cause of the outcome? | Y/PY/PN/N/NI/NA |
| 2.4. Do start of follow‐up and start of intervention coincide for most participants? | Y/PY/PN/N/NI |
| 2.5. If Y/PY to 2.2 and 2.3, or N/PN to 2.4, were adjustment techniques used that are likely to correct for the presence of selection biases? | Y/PY/PN/N/NI/NA |
| 'Risk of bias' judgement | Low/Moderate/Serious/Critical/NI |
| Bias in classification of interventions | |
| 3.1 Were intervention groups clearly defined? | Y/PY/PN/N/NI |
| 3.2 Was the information used to define intervention groups recorded at the start of the intervention? | Y/PY/PN/N/NI |
| 3.3 Could classification of intervention status have been affected by knowledge of the outcome or risk of the outcome? | Y/PY/PN/N/NI |
| 'Risk of bias' judgement | Low/Moderate/Serious/Critical/NI |
|
Bias due to deviations from intended interventions If your aim for this study is to assess the effect of assignment to intervention, answer questions 4.1 and 4.2 If your aim for this study is to assess the effect of starting and adhering to intervention, answer questions 4.3 to 4.6 | |
| 4.1. Were there deviations from the intended intervention beyond what would be expected in usual practice? | Y/PY/PN/N/NI |
| 4.2. If Y/PY to 4.1, were these deviations from intended intervention unbalanced between groups and likely to have affected the outcome? | Y/PY/PN/N/NI/NA |
| 4.3. Were important co‐interventions balanced across intervention groups? | Y/PY/PN/N/NI |
| 4.4. Was the intervention implemented successfully for most participants? | Y/PY/PN/N/NI |
| 4.5. Did study participants adhere to the assigned intervention regimen? | Y/PY/PN/N/NI |
| 4.6. If N/PN to 4.3, 4.4, or 4.5, was an appropriate analysis used to estimate the effect of starting and adhering to the intervention? | Y/PY/PN/N/NI/NA |
| 'Risk of bias' judgement | Low/Moderate/Serious/Critical/NI |
| Bias due to missing data | |
| 5.1 Were outcome data available for all, or nearly all, participants? | Y/PY/PN/N/NI |
| 5.2 Were participants excluded due to missing data on intervention status? | Y/PY/PN/N/NI |
| 5.3 Were participants excluded due to missing data on other variables needed for the analysis? | Y/PY/PN/N/NI |
| 5.4 If PN/N to 5.1, or Y/PY to 5.2 or 5.3, are the proportion of participants and reasons for missing data similar across interventions? | Y/PY/PN/N/NI/NA |
| 5.5 If PN/N to 5.1, or Y/PY to 5.2 or 5.3, is there evidence that results were robust to the presence of missing data? | Y/PY/PN/N/NI/NA |
| 'Risk of bias' judgement | Low/Moderate/Serious/Critical/NI |
| Bias in measurement of outcomes | |
| 6.1 Could the outcome measure have been influenced by knowledge of the intervention received? | Y/PY/PN/N/NI |
| 6.2 Were outcome assessors unaware of the intervention received by study participants? | Y/PY/PN/N/NI |
| 6.3 Were the methods of outcome assessment comparable across intervention groups? | Y/PY/PN/N/NI |
| 6.4 Were any systematic errors in measurement of the outcome unrelated to intervention received? | Y/PY/PN/N/NI |
| 'Risk of bias' judgement | Low/Moderate/Serious/Critical/NI |
|
Bias in selection of the reported result Is the reported effect estimate likely to be selected, on the basis of the results, from... | |
| 7.1. ... multiple outcome measurements within the outcome domain? | Y/PY/PN/N/NI |
| 7.2 ...multiple analyses of the intervention‐outcome relationship? | Y/PY/PN/N/NI |
| 7.3 ...different subgroups? | Y/PY/PN/N/NI |
| 'Risk of bias' judgement | Low/Moderate/Serious/Critical/NI |
| Abbreviations: Y: yes; PY: probably yes; PN: probably no; N: no; NI: no information; NA: not applicable | |
Data and analyses
Comparison 1. RCT: paroxetine versus doxepin.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 > 50% reduction in depressive symptoms | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.88, 1.52] |
| 1.2 Mean depression scores | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.2.1 HAMD scores | 1 | 67 | Mean Difference (IV, Fixed, 95% CI) | 0.65 [‐2.15, 3.45] |
| 1.3 Withdrawals (any reason) | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.01, 2.74] |
| 1.4 Adverse effects | 1 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 1.4.1 Blurred vision | 1 | 67 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.34 [0.09, 1.32] |
| 1.4.2 Dizziness | 1 | 67 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.21 [0.03, 1.37] |
| 1.4.3 Dry mouth | 1 | 67 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.26 [0.06, 1.20] |
| 1.4.4 Sleep disorders | 1 | 67 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.32 [0.08, 1.20] |
| 1.4.5 Urinary retention | 1 | 67 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.34 [0.01, 21.99] |
Comparison 2. RCT: amitriptyline versus nomifensine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 > 50% reduction in depressive symptoms | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.28, 1.06] |
Comparison 3. RCT: venlafaxine versus no treatment controls.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 > 50% reduction in depressive symptoms | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.25 [1.19, 8.90] |
| 3.2 Mean depression scores ‐ HAMD | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | ‐7.59 [‐11.52, ‐3.66] |
Comparison 4. RCT: sertraline versus cognitive behavioural therapy (CBT).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Mean depression scores (BDI) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1.1 BDI‐II scores at 8 weeks | 1 | 104 | Mean Difference (IV, Fixed, 95% CI) | ‐2.50 [‐6.28, 1.28] |
| 4.1.2 BDI‐II scores at 16 weeks | 1 | 117 | Mean Difference (IV, Fixed, 95% CI) | ‐0.50 [‐4.47, 3.47] |
| 4.2 Remission in depressive symptoms | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.65, 1.17] |
| 4.3 Seizure frequency | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.3.1 Generalised tonic‐clonic seizures per month at 8 weeks | 1 | 86 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.26, 0.06] |
| 4.3.2 Generalised tonic‐clonic seizures per month at 16 weeks | 1 | 96 | Mean Difference (IV, Fixed, 95% CI) | 0.00 [‐0.10, 0.10] |
| 4.3.3 Focal seizures with impaired awareness per month at 8 weeks | 1 | 75 | Mean Difference (IV, Fixed, 95% CI) | ‐2.60 [‐6.52, 1.32] |
| 4.3.4 Focal seizures with impaired awareness per month at 16 weeks | 1 | 73 | Mean Difference (IV, Fixed, 95% CI) | ‐3.00 [‐7.81, 1.81] |
| 4.4 Seizure recurrence | 1 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.27, 3.94] |
| 4.5 Withdrawals (any reason) | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.64, 2.46] |
| 4.6 Quality of life (QOLIE‐89) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.6.1 QOLIE‐89 score at 8 weeks | 1 | 104 | Mean Difference (IV, Fixed, 95% CI) | 6.10 [‐0.28, 12.48] |
| 4.6.2 QOLIE‐89 score at 16 weeks | 1 | 118 | Mean Difference (IV, Fixed, 95% CI) | 3.10 [‐3.41, 9.61] |
| 4.7 Adverse events profile | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.7.1 Adverse event profile at 8 weeks | 1 | 106 | Mean Difference (IV, Fixed, 95% CI) | ‐2.70 [‐6.62, 1.22] |
| 4.7.2 Adverse event profile at 16 weeks | 1 | 118 | Mean Difference (IV, Fixed, 95% CI) | ‐2.10 [‐6.21, 2.01] |
| 4.8 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 4.8.1 Anxiety | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 8.51 [0.19, 386.16] |
| 4.8.2 Chest pain | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.47 [0.05, 4.21] |
| 4.8.3 Cold | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.63 [0.13, 3.13] |
| 4.8.4 Diarrhoea | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 19.85 [0.49, 805.53] |
| 4.8.5 Dizziness | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.51 [0.37, 6.14] |
| 4.8.6 Dry mouth | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.78 [0.22, 65.10] |
| 4.8.7 Headache | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.48 [0.69, 3.19] |
| 4.8.8 Insomnia | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.16 [0.73, 6.38] |
| 4.8.9 Memory difficulty | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.71 [0.10, 4.82] |
| 4.8.10 Muscle strain or pain | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.21 [0.36, 4.12] |
| 4.8.11 Nausea | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.60 [0.62, 10.96] |
| 4.8.12 Rash | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 4.72 [0.29, 76.72] |
| 4.8.13 Sexual dysfunction | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 8.51 [0.19, 386.16] |
| 4.8.14 Shakiness | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 12.28 [0.88, 171.59] |
| 4.8.15 Tiredness | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.54 [1.40, 8.96] |
| 4.8.16 Unsteadiness | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.57 [0.25, 9.81] |
| 4.8.17 Worsening depression | 1 | 140 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.13 [0.25, 5.07] |
Comparison 5. NRSI: citalopram (before and after).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Mean depression scores HAMD‐21 | 2 | 176 | Std. Mean Difference (IV, Fixed, 95% CI) | 1.17 [0.96, 1.38] |
| 5.2 Mean monthly seizure frequency | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only |
Comparison 6. NRSI: SSRIs (sertraline or citalopram) versus CBT.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Mean depression scores (BDI) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1.1 BDI at 6 weeks | 1 | 15 | Mean Difference (IV, Fixed, 95% CI) | ‐2.60 [‐11.58, 6.38] |
| 6.1.2 BDI at 12 weeks | 1 | 15 | Mean Difference (IV, Fixed, 95% CI) | ‐4.90 [‐14.60, 4.80] |
| 6.2 Remission in depressive symptoms | 1 | 15 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.53 [0.77, 3.06] |
| 6.3 Seizure frequency per month at 12 weeks | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐1.60 [‐5.63, 2.43] | |
| 6.4 Withdrawals (any reason) | 1 | 15 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.44 [0.05, 3.85] |
| 6.5 Quality of life (QOLIE‐31 overall score) | 1 | 15 | Mean Difference (IV, Fixed, 95% CI) | ‐0.50 [‐19.67, 18.67] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Gilliam 2019.
| Study characteristics | ||
| Methods | Multi‐centre, randomised controlled trial conducted in the USA Baseline period: 3 month retrospective baseline (seizures) Treatment period:16 weeks |
|
| Participants | 140 participants; 77 female 21 to 75 years old 58% focal epilepsy CES‐D score >14 |
|
| Interventions | Sertraline 50 mg/day to 200 mg/day versus CBT | |
| Outcomes | BDI, CES‐D, seizure recurrence and monthly frequency, AEP, adverse events | |
| Notes | ITT analysis, 49/72 in sertraline group completed assigned treatment, 49/68 in CBT group completed assigned treatment | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Simple non‐stratified computerised randomisation code generated by an investigator not otherwise involved in the study |
| Allocation concealment (selection bias) | Low risk | Treatment assignment obtained by telephone communication (centralised randomisation) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding of participants and personnel not possible by design (sertraline or CBT) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Site investigators blinded to outcome assessment but research assistants implementing study procedures not blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 23/72 participants (32%) in the sertraline group and 19/68 participants (28%) in the CBT group did not complete treatment. Intention‐to‐treat analysis imputed missing data by multiple imputation |
| Selective reporting (reporting bias) | Low risk | Outcomes stated in methods section of report are present in the results. No protocol available |
| Other bias | High risk | High risk of recall bias as seizure rates were collected retrospectively at baseline |
Hovorka 2000.
| Study characteristics | ||
| Methods | A single‐centre, non‐randomised, uncontrolled, prospective before and after study (Prague) Baseline period: 2 months Treatment period: 8 weeks | |
| Participants | 43 people with focal epilepsy exceeding 15 points on the Hamilton Rating Scale for Depression (HAMD) 21 scale for depression 35 females and 8 males Aged 21 to 49 years: mean 33.2 years | |
| Interventions | Citalopram at a flexible dose; the average dose was 19.3 mg ± 2.6 mg at the end of the first month, 22.62 mg ± 8.3 mg at the end of the second month | |
| Outcomes | 1) Seizure frequency 2) Depressive symptoms measured by the HAMD‐21 3) Adverse effects |
|
| Notes | No dropouts and no exclusions from the analysis | |
Kanner 2000.
| Study characteristics | ||
| Methods | A single‐centre, non‐randomised, uncontrolled, prospective before and after study (USA) Baseline period: not reported Treatment period: mean 10.3 months (0.2 to 38 months) |
|
| Participants | 100 people with focal epilepsy, with depressive or obsessive compulsive disorder 51 males and 49 females Aged 6 to 62 years: mean 29.9 years | |
| Interventions | Sertraline, mean dose of 108 mg ± 56.9 mg per day | |
| Outcomes | 1) Improvement in depressive symptoms 2) Seizure frequency 3) Adverse effects |
|
| Notes | 18 people withdrew due to adverse effects; all included in analysis | |
Kuhn 2003.
| Study characteristics | ||
| Methods | A single‐centre, non‐randomised, prospective study (Germany) Baseline period: 4 days Observation period: 20 to 30 weeks | |
| Participants | 75 people with temporal lobe epilepsy exceeding 15 points on the HAMD‐21 scale for depression 45 females and 30 males Aged 19 to 68 years: mean 40.1 years | |
| Interventions | Citalopram (N = 33), dose at endpoint: 24.2 mg Mirtazapine (N = 27), dose at endpoint: 32.2 mg Reboxetine (N = 15), dose at endpoint: 6.9 mg |
|
| Outcomes | 1) Improvement in depressive symptoms 2) Seizure frequency and severity 3) Adverse effects |
|
| Notes | Large amount of withdrawals from week 4 to weeks 20 to 30. Last observation carried forward approach used | |
Li 2005.
| Study characteristics | ||
| Methods | A single‐centre, randomised controlled trial (China) Baseline period: unclear Treatment phase: 8 weeks | |
| Participants | 67 participants with epilepsy and depression (meeting CCMD‐3 criteria for depression and HAMD‐21 score > 18) | |
| Interventions | Paroxetine (N = 33): 17 males, 16 females aged 14 to 62 years, dose 10 mg/day to 40 mg/day Doxepin (N = 34): 15 males, 19 females, aged 16 to 59 years, dose 25 mg/day titrated up according to response (mean dose 100 mg) |
|
| Outcomes | 1) Change in depression scores (HAMD‐21) from baseline 2) Adverse events |
|
| Notes | 3 participants discontinued study in doxepin arm because of adverse events, with 31 participants analysed for this treatment arm | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation carried out by flipping of a coin |
| Allocation concealment (selection bias) | Unclear risk | No details available regarding methods of allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessor blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Missing data reported, ITT not used |
| Selective reporting (reporting bias) | Low risk | Outcomes stated in methods section of report were present in the results. No protocol available |
| Other bias | Unclear risk | Insufficient details in report to judge the influence of other bias |
Orjuela‐Rojas 2015.
| Study characteristics | ||
| Methods | Single centre, prospective cohort study in Mexico Baseline period; not reported Treatment period; 12 weeks |
|
| Participants | 15 participants, 11 female, > 18 years old, all with temporal lobe epilepsy | |
| Interventions | SSRI (sertraline or citalopram) vs CBT | |
| Outcomes | BDI HADS‐D HADS‐A QOLIE‐31 MINI Seizures per month |
|
| Notes | 2 dropouts reported in CBTgroup, 1 lost to follow‐up | |
Robertson 1985.
| Study characteristics | ||
| Methods | A single‐centre, randomised, double‐blind, controlled trial (UK) Baseline period: unclear Treatment period: 12 weeks (6 weeks for all 3 arms of trial, then 6 weeks for the 2 antidepressants only) | |
| Participants | 42 people with epilepsy exceeding 15 points on the HAMD‐21 scale for depression 26 females and 13 males Aged 18 to 60 years | |
| Interventions | Amitriptyline (N = 14) 25 mg TDS Nomifensine (N = 14) 25mg TDS Placebo (N = 14) |
|
| Outcomes | 1) Improvement in depressive symptoms 2) Seizure frequency 3) Adverse effects |
|
| Notes | 39 people included in the analysis. At 6 weeks, non‐responders in the active drug arms had dose doubled, and those in the placebo arm were withdrawn from the study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number codes used, however generation of this randomisation sequence is unclear |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study personnel and participants blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessor blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Missing data detected and attrition reported |
| Selective reporting (reporting bias) | Low risk | Outcomes stated in methods section of report are present in the results. No protocol available |
| Other bias | Unclear risk | After 6 weeks, placebo group removed from trial; only active antidepressant treatment groups continued in the trial |
Specchio 2004.
| Study characteristics | ||
| Methods | A multi‐centre, non‐randomised, uncontrolled, prospective before‐after study (Italy)
Baseline period: not reported Treatment period: 4 months |
|
| Participants | 45 people with focal epilepsy, and exceeding or equal to 20 on the MADRS 31 females and 14 males Mean age of 42.7 years | |
| Interventions | Citalopram 20 mg per day | |
| Outcomes | 1) Seizure frequency 2) Improvement in depression measured by MADRS and Zung‐SDS 3) Adverse effects |
|
| Notes | 39 participants received intended treatment and analysed | |
Thome‐Souza 2007.
| Study characteristics | ||
| Methods | A single‐centre, non‐randomised, uncontrolled, prospective before‐after study (Brazil) Baseline period: not reported Treatment period: mean 25.8 months (range 12 months to 78 months) |
|
| Participants | 36 children and adolescents with focal epilepsy and diagnosis of depression 19 females and 17 males Aged 5 to 18 years, mean: 12.7 years | |
| Interventions | Sertraline up to 200 mg/day, mean dose 111.5 mg/day (50 mg/day to 200 mg/day) Fluoxetine up to 80 mg/day, mean dose 45.7 mg/day (20 mg/day to 80 mg/day) |
|
| Outcomes | 1) Seizure severity 2) Improvement in depressive symptoms 3) Adverse effects |
|
| Notes | No dropouts | |
Zhu 2004.
| Study characteristics | ||
| Methods | Single‐centre, randomised trial of venlafaxine versus no treatment (China)
Baseline period: not reported Treatment period: 8 weeks |
|
| Participants | 64 people with epilepsy (presumed genetic or cause unknown) and depression 39 males and 25 females Aged 7 to 60 years (mean 27 years) | |
| Interventions | Venlafaxine 25 mg to 75 mg/day (N = 32) No treatment (N = 32) |
|
| Outcomes | 1) Change in HAMD‐21 scores 2) Adverse events |
|
| Notes | No dropouts | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Methods for generation of random sequence were not detailed in the report |
| Allocation concealment (selection bias) | Unclear risk | Methods for allocation were not detailed in the report |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details of blinding methods in the report |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear whether outcome assessor blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing data |
| Selective reporting (reporting bias) | Low risk | Outcomes stated in methods section of report were present in the results. No protocol available |
| Other bias | Unclear risk | Insufficient details in report to judge the influence of other bias |
AEP: Adverse Events Profile BDI: Beck Depression Inventory CCMD‐3: Chinese Classification of Mental Disorders CBT: Cognitive Behavioral Therapy CES‐D: Center for Epidemiologic Studies Depression HADS‐D: Hospital Anxiety and Depression Scale‐Depression HADS‐A: Hospital Anxiety and Depression Scale‐Anxiety HAMD: Hamilton Rating Scale for Depression ITT: Intention‐To‐Treat MADRS: Montgomery–Åsberg Depression Rating Scale MINI: Mini International Neuropsychiatric Interview NA: Not Applicable SSRI: Selective serotonin Reuptake Inhibitor TDS: Three times a day QOLIE‐31: Quality of Life in Epilepsy Inventory ‐31 Zung‐SDS: Zung Self‐Rating Depression Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Blumer 1997 | Case series; study not meeting the inclusion criteria |
| Harmant 1990 | Did not report any results for any of the primary and secondary outcomes. We attempted to contact trial author of the study but received no response. |
| Machado 2010 | Did not report any results for any of the primary and secondary outcomes. We attempted to contact trial author of the study but received no response. |
| NCT00595699 | Did not report any results for any of the primary and secondary outcomes. We attempted to contact trial author of the study but received no response. |
| NCT01244724 | Trial listed on ClinicalTrials.gov and recorded as terminated |
| NCT03464383 | Very small pilot study with only 3 participants recruited to antidepressant (escitalopram 10 mg) and referral to psychiatry treatment arms |
Characteristics of ongoing studies [ordered by study ID]
EUCTR2017‐000990‐35‐IT.
| Study name | Effects of the antidepressive therapy with agomelatin and escitalopram in people with depression and epilepsy |
| Methods | A double‐blind randomised study with active control (escitalopram) with parallel groups |
| Participants | 222 |
| Interventions | escitalopram 10 mg daily vs agomelatine 25 mg daily for 6 months |
| Outcomes | Primary outcome: efficacy (depression BDI‐II) Secondary outcomes: depression (HDRS), quality of life (QOLIE‐31P, quality of life in epilepsy II version), sleep quality (PSQI), daytime sleepiness (ESS), and cognition (MDB) |
| Starting date | 2019 |
| Contact information | Prof. Fabio Placidi |
| Notes |
EUCTR2018‐003464‐32‐HU.
| Study name | Effect of mirtazapine on seizure frequency in epileptic patients with vagal nerve stimulation device |
| Methods | Double‐blind, randomised, placebo controlled trial |
| Participants | Target sample size 30 participants Adults (18 to 65 years ) with drug resistant epilepsy with focal seizures, with or without loss of consciousness, and a vagal nerve stimulation device implanted and activated > 6 months prior to enrolment |
| Interventions | Mirtazapine (30 mg) compared to placebo |
| Outcomes | Change in seizure frequency at weeks 12 and 27 Quality of Life: using the self‐administered Quality of Life in Epilepsy 89 (QOLIE‐89) at weeks 12, 15, and 27 Depression: using Beck Depression Inventory (BDI), and Hamilton Depression Rating Scale (HAM‐D) at weeks 12, 15, and 27 |
| Starting date | 08 /11/2019 |
| Contact information | None provided |
| Notes |
Differences between protocol and review
Update 2021:
Wording of the type of eligible studies was clarified as "Prospective non‐randomised cohort controlled and uncontrolled studies (with a control group including participants acting as their own control group (i.e. before and after studies))."
Wording of outcomes was clarified, and sub‐outcome 'The proportion of people achieving complete remission of depressive symptoms' was added for the outcome 'Depression.'
Risk of bias approach for NRSIs was updated to use the ROBINS‐I tool
Text was added to clarify that "For specific adverse events, to allow for multiple statistical testing, we presented RRs with 99% CIs."
The additional primary outcome 'The proportion of people achieving complete remission of depressive symptoms'
Contributions of authors
MM, JS wrote the review protocol.
MM holds responsibility for managing the review process.
SN oversaw the statistical analyses, and contributed to the writing of the updated review.
AM provided supervision and comments throughout the development of the protocol, review and review updates.
Sources of support
Internal sources
No sources of support supplied
External sources
National Institute for Health Research, UK
Declarations of interest
MM: none known AGM is partly funded by the National Institute for Health Research Applied Research Collaboration North West Coast (NIHR ARC NWC). A consortium of pharmaceutical companies (GSK, EISAI, UCB Pharma) funded the National Audit of Seizure Management in Hospitals (NASH) through grants paid to the University of Liverpool. SN: none known
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Gilliam 2019 {published data only}
- Gilliam FG, Black KJ, Carter J, Freedland KE, Sheline YI, Tsai W-Y, et al. A trial of sertraline or cognitive behavior therapy for depression in epilepsy. Annals of Neurology 2019;86(4):552-60. [DOI: ] [ISSN: 1531-8249] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hovorka 2000 {published data only}
- Hovorka J, Herman E, Nemcova I. Treatment of interictal depression with citalopram in patients with epilepsy. Epilepsy and Behavior 2000;1:444-7. [DOI] [PubMed] [Google Scholar]
Kanner 2000 {published data only}
- Kanner A, Kozak A, Frey M. The use of sertraline in patients with epilepsy: is it safe? Epilepsy and Behavior 2000;1:100-5. [DOI] [PubMed] [Google Scholar]
Kuhn 2003 {published data only}
- Kuhn K, Quednow B, Thiel M, Falkai P, Maier W, Elger C. Antidepressive treatment in patients with temporal lobe epilepsy and major depression: a prospective study with three different antidepressants. Epilepsy and Behavior 2003;4:674-9. [DOI] [PubMed] [Google Scholar]
Li 2005 {published data only}
- Li W, Ma D. A randomized controlled trial to evaluate the efficacy of paroxetine and doxepin in treating epileptic patients with depression. Chinese Journal of Clinical Rehabilitation 2005;9(12):20-1. [Google Scholar]
Orjuela‐Rojas 2015 {published data only}
- Orjuela-Rojas JM, Martinez-Juarez IE, Ruiz-Chow A, Crail-Melendez D. Treatment of depression in patients with temporal lobe epilepsy: a pilot study of cognitive behavioral therapy vs. selective serotonin reuptake inhibitors. Epilepsy & Behavior 2015;51:176-81. [DOI: 10.1016/j.yebeh.2015.07.033] [PMID: ] [DOI] [PubMed] [Google Scholar]
Robertson 1985 {published data only}
- Robertson M, Trimble M. The treatment of depression in patients with epilepsy: a double blind trial. Journal of Affective Disorders 1985;9:127-36. [DOI] [PubMed] [Google Scholar]
Specchio 2004 {published data only}
- Specchio L, Iudice A, Specchio N, La Neve A, Spinelli A, Galli R, et al. Citalopram as treatment of depression in patients with epilepsy. Clinical Neuropharmacology 2004;27(3):133-6. [DOI] [PubMed] [Google Scholar]
Thome‐Souza 2007 {published data only}
- Thome-Souza M, Kuczynski E, Valente K. Sertraline and fluoxetine: safe treatments for children and adolescents with epilepsy and depression. Epilepsy & Behavior 2007;10:417-25. [DOI] [PubMed] [Google Scholar]
Zhu 2004 {published data only}
- Zhu S, Luo L, Gui Y. Short-term efficacy of venlafaxine treating the depression in epilepsy patients. Chinese Journal of Rehabilitation 2004;19(2):101. [Google Scholar]
References to studies excluded from this review
Blumer 1997 {published data only}
- Blumer D. Antidepressant and double antidepressant treatment for the affective disorder of epilepsy. Journal of Clinical Psychiatry 1997;58(1):3-11. [DOI: 10.4088/JCP.v58n0101] [DOI] [PubMed] [Google Scholar]
Harmant 1990 {published data only}
- Harmant J, Van Rijckevorsel-Harmant K, De Barsy T, Hendrickx B. Fluvoxamine: an antidepressant with low (or no) epileptogenic effect. Lancet 1990;336(8711):386. [DOI] [PubMed] [Google Scholar]
Machado 2010 {published data only}
- Machado R, Espinosa A, Montoto A. Cholesterol concentrations and clinical response to sertraline in patients with epilepsy: preliminary results. Epilepsy & Behavior 2010;19:509-12. [DOI] [PubMed] [Google Scholar]
NCT00595699 {unpublished data only}
- NCT00595699. Escitalopram treatment of major depression in patients with temporal lobe epilepsy. clinicaltrials.gov/ct/show/NCT00595699 (first posted 16 January 2008).
NCT01244724 {published data only}
- NCT01244724. Lexapro for major depression in patients with epilepsy. clinicaltrials.gov/show/NCT01244724 (first posted 19 November 2010).
NCT03464383 {published data only}
- NCT03464383. Anxiety and depression in epilepsy: a treatment study [Anxiety and depression in epilepsy: a pilot epileptologist-driven treatment study]. clinicaltrials.gov/ct2/show/record/NCT03464383 (first received 7 March 2018).
References to ongoing studies
EUCTR2017‐000990‐35‐IT {published data only}
- EUCTR2017-000990-35-IT. Effects of the antidepressive therapy with agomelatin and escitalopram in people with depression and epilepsy [Effects of antidepressant treatment with agomelatine on patients affected by depression and epilepsy. A double blind randomized study with active control (escitalopram) with parallel groups]. www.clinicaltrialsregister.eu/ctr-search/search?query=2017-000990-35 (first posted 6 February 2019).
EUCTR2018‐003464‐32‐HU {published data only}
- EUCTR2018-003464-32-HU. Effect of mirtazapine on seizure frequency in epileptic patients with vagal nerve stimulation device. www.clinicaltrialsregister.eu/ctr-search/search?query=2018-003464-32 (first posted 5 November 2019).
Additional references
Adams 2008
- Adams SJ, O'Brien TJ, Lloyd J, Kilpatrick CJ, Salzberg MR, Velakoulis D. Neuropsychiatric morbidity in focal epilepsy. British Journal of Psychiatry 2008;192(6):464-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Alper 2007
- Alper K, Schwartz KA, Kolts RL, Khan A. Seizure incidence in psychopharmacological clinical trials: an analysis of Food and Drug Administration (FDA) summary basis of approval reports. Biological Psychiatry 2007;62(4):345-54. [PMID: ] [DOI] [PubMed] [Google Scholar]
APA 2000
- American Psychiatric Association. Diagnostic and Statistical Manual. 4th edition. Washington DC: American Psychiatric Association, 2000. [Google Scholar]
Bagdy 2007
- Bagdy G, Kecskemeti V, Riba P, Jakus R. Serotonin and epilepsy. Journal of Neurochemistry 2007;100(4):857-73. [PMID: ] [DOI] [PubMed] [Google Scholar]
Baker 1996
- Baker G, Jacoby A, Chadwick D. The associations of psychopathology in epilepsy: a community study. Epilepsy Research 1996;25:29-39. [DOI] [PubMed] [Google Scholar]
Boylan 2004
- Boylan LS, Flint LA, Labovitz DL, Jackson SC, Starner K, Devinsky O. Depression but not seizure frequency predicts quality of life in treatment-resistant epilepsy. Neurology 2004;62(2):258-61. [DOI] [PubMed] [Google Scholar]
Cotterman‐Hart 2010
- Cotterman-Hart S. Depression in epilepsy: why aren't we treating? Epilepsy & Behavior 2010;19(3):419-21. [PMID: ] [DOI] [PubMed] [Google Scholar]
Coupland 2011
- Coupland C, Dhiman P, Morriss R, Arthur A, Barton G, Hippisley-Cox J. Antidepressant use and risk of adverse outcomes in older people: population based cohort study. BMJ 2011;343:d4551. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dell'osso 2013
Gilliam 2005b
- Gilliam FG. Diagnosis and treatment of mood disorders in persons with epilepsy. Current Opinion in Neurology 2005;18(2):129-33. [PMID: ] [DOI] [PubMed] [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist G, Kunz R, Falck-Ytter Y, Alonso-Coello P, et al, for the GRADE Working Group. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hamid 2013
- Hamid H, Kanner AM. Should antidepressant drugs of the selective serotonin reuptake inhibitor family be tested as antiepileptic drugs? Epilepsy & Behavior 2013;26(3):261-5. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hermann 2000
- Hermann BP, Seidenberg M, Bell B. Psychiatric comorbidity in chronic epilepsy: identification, consequences, and treatment of major depression. Epilepsia 2000;41(Suppl 2):S31-41. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hesdorffer 2000
- Hesdorffer DC, Hauser WA, Annegers JF, Cascino G. Major depression is a risk factor for seizures in older adults. Annals of Neurology 2000;47(2):246-9. [PubMed] [Google Scholar]
Hesdorffer 2006
- Hesdorffer DC, Hauser WA, Olafsson E, Ludvigsson P, Kjartansson O. Depression and suicide attempt as risk factors for incident unprovoked seizures. Annals of Neurology 2006;59(1):35-41. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hesdorffer 2009
- Hesdorffer DC, Kanner AM. The FDA alert on suicidality and antiepileptic drugs: fire or false alarm? Epilepsia 2009;50(5):978-86. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hesdorffer 2012
- Hesdorffer DC, Ishihara L, Mynepalli L, Webb DJ, Weil J, Hauser WA. Epilepsy, suicidality, and psychiatric disorders: a bidirectional association. Annals of Neurology 2012;72(2):184-91. [PMID: ] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from training.cochrane.org/handbook/archive/v5.1/.
Higgins 2020
- Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). Cochrane, 2020. Available from www.training.cochrane.org/handbook.
Indaco 1992
- Indaco A, Carrieri B, Naapi C, Gentile S, Striano S. Interictal depression in epilepsy. Epilepsy Research 1992;12:45-50. [DOI] [PubMed] [Google Scholar]
Jacoby 1996
- Jacoby A, Baker G, Steen N, Potts P, Chadwick D. The clinical course of epilepsy and its psychological correlates: finding from a U.K. community study. Epilepsia 1996;37:148-61. [DOI] [PubMed] [Google Scholar]
Kirkham 2010
- Kirkham JJ, Dwan KM, Altman DG, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340:c365. [PMID: ] [DOI] [PubMed] [Google Scholar]
Klerman 1990
- Klerman G. Treatment of recurrent unipolar major depressive disorder. Commentary on the Pittsburgh study. Archives of General Psychiatry 1990;47(12):1158-61. [DOI] [PubMed] [Google Scholar]
Kondziella 2009
- Kondziella D, Asztely F. Don't be afraid to treat depression in patients with epilepsy! Acta Neurologica Scandinavica 2009;119(2):75-80. [PMID: ] [DOI] [PubMed] [Google Scholar]
Lambert 1999
- Lambert M, Robertson M. Depression in epilepsy: etiology, phenomenology, and treatment. Epilepsia 1999;40(Suppl 10):s21-47. [DOI] [PubMed] [Google Scholar]
Lin 2012
- Lin JJ, Mula M, Hermann BP. Uncovering the neurobehavioural comorbidities of epilepsy over the lifespan. Lancet 2012;380(9848):1180-92. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mendez 1986
- Mendez M, Cummings J, Benson D. Depression in epilepsy: significance and phenomenology. Archives of Neurology 1986;43:766-70. [DOI] [PubMed] [Google Scholar]
Mula 2009
- Mula M, Schmitz B. Depression in epilepsy: mechanisms and therapeutic approach. Therapeutic Advances in Neurological Disorders 2009;2(5):337-44. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mula 2013
- Mula M, Kanner AM, Schmitz B, Schachter S. Antiepileptic drugs and suicidality: an expert consensus statement from the Task Force on Therapeutic Strategies of the ILAE Commission on Neuropsychobiology. Epilepsia 2013;54(1):199-203. [PMID: ] [DOI] [PubMed] [Google Scholar]
Preskorn 1992
- Preskorn SH, Fast GA. Tricyclic antidepressant-induced seizures and plasma drug concentration. Journal of Clinical Psychiatry 1992;53(5):160-2. [PMID: ] [PubMed] [Google Scholar]
Sackeim 2006
- Sackeim HA, Roose SP, Lavori PW. Determining the duration of antidepressant treatment: application of signal detection methodology and the need for duration adaptive designs (DAD). Biological Psychiatry 2006;59(6):483-92. [PMID: ] [DOI] [PubMed] [Google Scholar]
Stahl 2000
- Stahl SM. Blue genes and the mechanism of action of antidepressants. Journal of Clinical Psychiatry 2000;61(3):164-5. [PMID: ] [DOI] [PubMed] [Google Scholar]
Sterne 2000
- Sterne JA, Gavaghan D, Egger M. Publication and related bias in meta-analysis: power of statistical tests and prevalence in the literature. Journal of Clinical Epidemiology 2000;53(11):1119-29. [PMID: ] [DOI] [PubMed] [Google Scholar]
Tellez‐Zenteno 2007
- Tellez-Zenteno JF, Patten SB, Jette N, Williams J, Wiebe S. Psychiatric comorbidity in epilepsy: a population-based analysis. Epilepsia 2007;48(12):2336-44. [PMID: ] [DOI] [PubMed] [Google Scholar]
Trimble 1998
- Trimble MR. New antiepileptic drugs and psychopathology. Neuropsychobiology 1998;38(3):149-51. [PMID: ] [DOI] [PubMed] [Google Scholar]
Wroblewski 1990
- Wroblewski BA, McColgan K, Smith K, Whyte J, Singer WD. The incidence of seizures during tricyclic antidepressant drug treatment in a brain-injured population. Journal of Clinical Psychopharmacology 1990;10(2):124-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Zarate 2006
- Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Archives of General Psychiatry 2006;63(8):856-64. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Maguire 2013
- Maguire MJ, Pulman J, Singh J, Marson AG. Antidepressants for people with epilepsy and depression. Cochrane Database of Systematic Reviews 2013, Issue 7. Art. No: CD010682. [DOI: 10.1002/14651858.CD010682] [DOI] [PMC free article] [PubMed] [Google Scholar]
Maguire 2014
- Maguire MJ, Weston J, Singh J, Marson AG. Antidepressants for people with epilepsy and depression. Cochrane Database of Systematic Reviews 2014, Issue 12. Art. No: CD010682. [DOI: 10.1002/14651858.CD010682.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]