

Abstract
A key part of the optimization of small molecules in pharmaceutical inhibitor development is to vary the molecular design to enhance complementarity of chemical features of the compound with the positioning of amino acids in the active site of a target enzyme. Typically this involves iterations of synthesis, to modify the compound, and biophysical assay, to assess the outcomes. Selective targeting of the anti‐cancer carbonic anhydrase isoform XII (CA XII), this process is challenging because the overall fold is very similar across the twelve CA isoforms. To enhance drug development for CA XII we used a reverse engineering approach where mutation of the key six amino acids in the active site of human CA XII into the CA II isoform was performed to provide a protein chimera (chCA XII) which is amenable to structure‐based compound optimization. Through determination of structural detail and affinity measurement of the interaction with over 60 compounds we observed that the compounds that bound CA XII more strongly than CA II, switched their preference and bound more strongly to the engineered chimera, chCA XII, based on CA II, but containing the 6 key amino acids from CA XII, behaved as CA XII in its compound recognition profile. The structures of the compounds in the chimeric active site also resembled those determined for complexes with CA XII, hence validating this protein engineering approach in the development of new inhibitors.
Keywords: drug design, enzyme models, fluorescence spectroscopy, sulfonamides, X-ray diffraction
Chimeric carbonic anhydrase XII (chCA XII), based on CA II with six key amino acids in the active site mutated according to CA XII, recognized ligands as CA XII and not CA II. The compounds that bound CA XII more strongly than CA II switched their preference and interacted with higher affinity with chCA XII.

1. Introduction
The development of selective inhibitors targeted at structurally highly similar enzyme isoforms is a challenging task in drug discovery. Enzyme‐inhibitor binding interactions occur via key amino acids at defined positions, usually in the active site of the enzyme. Identifying the bonding patterns of the residues participating in the intermolecular interactions provides insight into the catalytic mechanism and the opportunity for inhibitor recognition. [1] In the case of inhibition of protein isoforms, detecting the distinguishing amino acid interactions is crucial for the design of isoform‐selective inhibitors.
Carbonic anhydrases (CAs, EC 4.2.1.1) catalyze reversible hydration of carbon dioxide to bicarbonate and protons. Their isoforms play roles in numerous physiological and pathological processes by regulating the pH and bicarbonate balance.[ 2 , 3 , 4 , 5 ] CA inhibitors have been used as therapeutic options in a number of disease areas including anti‐glaucoma agents,[ 6 , 7 ] diuretics [8] and antiepileptics.[ 9 , 10 , 11 , 12 ] However, most of the clinically used inhibitors lack selectivity towards targeted CA isoforms and exhibit various side effects. There is thus, a demand for selective inhibitors that target only the disease‐associated CA isoform, for instance, isoforms IX or XII, which regulate the pH of a tumor microenvironment. [13]
Design and development of CA isoform‐selective inhibitors is challenging due to the high amino acid sequence homology between isoforms. Humans have twelve catalytically active CA isoforms: CA I, CA II, CA III, CA VII, CA XIII (cytosolic), CA VA and CA VB (mitochondrial), CA VI (secreted), CA IV, CA IX, CA XII, CA XIV (membrane‐bound). Active sites of CA isoforms are cone‐shaped and differ from each other by several residues.
CA XII is an important isoform since it controls the extracellular pH of cancer cells under physiological conditions and acts as a hypoxia‐induced tumor marker. CA XII is regulated by estrogen receptor in breast cancer. [14] High expression of CA XII correlates with favorable survival of patients having invasive breast [15] or resectable non‐small cell lung cancers. [16] However, it is associated with poor prognosis in patients with colorectal, [17] hepatocellular [18] carcinomas, and diffuse astrocytomas. [19] Furthermore, CA XII is considered as a pharmacological target to overcome chemoresistance in cancer cells. [20] The lower wide‐spread expression of CA XII has also been detected in human eye, [21] gut, [22] kidney, [23] pancreas, [24] gastric mucosa, [25] epithelial components of skin, [26] endometrial epithelium [27] and male excurrent ducts. [28] Additional roles of CA XII in the human body include glaucoma progression, [21] salt uptake in sweat glands, [29] and pH regulation of nucleus pulposus cells. [30] CA XII function in disease is reviewed in detail by Waheed and Sly. [31]
CA XII is an important but highly challenging drug target. Traditional screening and drug design protocols are limited because it is a membrane‐associated dimeric enzyme containing N‐terminal extracellular catalytic domain, a membrane‐spanning α‐helix, and a 30‐residue small intracellular C‐terminal region. CA XII has been the focus of numerous studies to determine its structure and function down to the atomic level. The first crystallographic structure of CA XII at 1.55 Å resolution was solved in 2001. [32] The protein has GXXXG and GXXXS dimerization motifs in the transmembrane segment, two glycosylation sites, Asn52 and Asn136, exposed on the surface and a single disulfide linkage between Cys23 and Cys203. The active site composition is analogous to the most studied CA II: the zinc ion is coordinated by His94, His96, His119 and a water molecule. The pK a of the zinc‐bound water molecule at 25 °C is 7.0–7.1.[ 33 , 34 ] A large body of work has resulted in the deposition of structural data for 24 crystal structures of chlorinated/fluorinated mono‐/di‐substituted benzenesulfonamides bound to CA XII complexes[ 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 ] to the PDB. This forms a valuable resource for CA XII inhibitor design.
The high‐resolution structural data is complemented by a wealth of thermodynamic, kinetic and structural analysis of interactions between more than 400 inhibitors and 12 CA isoforms. These reveal that minor structural differences between CA isoforms can cause unpredictable effects to the changes of selective recognition between a ligand and target isoform. [43] Most conserved residues are found deep within the active site (close to the zinc ion), [44] whilst the top of the binding site is more variable.
Here we use a protein engineering approach to produce a chimeric version based on the CA II isoform which is soluble and highly amenable to drug development strategies. In an earlier study we showed that CA II‐based mutant with six CA XII‐specific residues (A65S, N67K, I91T, F130A, V134S, and L203N), resembled the active site of CA XII. [39] We, therefore, produced a chimeric CA XII protein (chCA XII) which recapitulates the CA XII enzyme activity and function. We have characterized the catalytic activity and thermal stability of chCA XII, as well as conducted ligand binding and inhibition studies with 63 small molecule compounds by enzymatic and biophysical assays. For the design of novel and selective anticancer drug candidates we have analyzed the structural differences between the targeted CA XII and off‐target, ubiquitously expressed, CA II, which share 34 % sequence identity. [45] We have obtained nine crystal structures of inhibitors bound to chCA XII and compared the binding modes together with binding affinities of inhibitors to CA II and CA XII. The detailed analysis validates the protein engineering a route to selective inhibitors of the CA XII isoform.
2. Results
The principal hypothesis and approach in constructing chCA XII is shown in Figure 1. The chimeric protein produced is essentially the same CA II, except that it would recognize inhibitors that would be normally recognized by CA XII. The sequences of CA II and CA XII were aligned (supplementary Figure S1) and six amino acids (A65S, N67K, I91T, F130A, V134S, and L203N) in the vicinity of the ligand binding site that differ between CA II and CA XII were mutated resulting in a chimeric form of CA II (chCA XII) whose ligand recognition mirrored that of CA XII.
Figure 1.
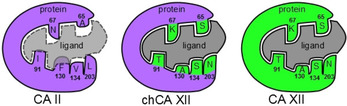
The general scheme of engineering the chimeric CA XII (chCA XII). The CA XII‐selective inhibitor is shown in gray. The hypothetical inhibitor is unable to bind to CA II (violet) but is supposed to bind well to chCA XII (violet‐green) and CA XII (green). To obtain chCA XII, six residues in the active site of CA II were replaced by the ones present in CA XII at the same locations of the active site (N67→K67, A65→S65, I91→T91, F130→A130, V134→S134, and L203→N203).
2.1. Catalytic Properties of chCA XII Closely Mimic CA XII
To ascertain the role of key amino acids in the active site of CA XII, the thermal stability and catalytic activity of the chCA XII was measured in a wide pH range and compared to the properties of CA II and CA XII.[ 46 , 47 ] Six mutations (A65S, N67K, I91T, F130A, V134S and L203N) in the active site of CA II resulted in increased thermal stability by 0.5 °C in the pH range 4.5–10.0 (Figure 2A). The CA II and chCA XII were most stable at pH 6.0–8.0 with melting temperature T m 55.6±0.1 °C and 56.1±0.1 °C, respectively. The CA XII had different stability profile with the maximum T m=53.6±0.2 °C at pH 6.0. In addition, the chCA XII had similar maximal catalytic activity as CA II (k cat=6.0 × 105 s−1), but larger pK a value of His64 by 0.6 units (pK a=7.9±0.2 for chCA XII and pK a=7.3±0.2 for CA II) as shown in Figure 2B. The determined maximal catalytic constant of CA XII was (4.1±0.9) × 105 s−1 with pK a of 7.6±0.2. Interestingly, the Michaelis‐Menten constant of chCA XII (K M=3.6±2.0 mM) was similar to CA XII (K M=3.6±1.5 mM) and differed from CA II (K M=4.7±1.0 mM) in the pH range 6.8–9.0 (Figure S2). The site‐directed mutagenesis of CA II slightly stabilized the enzyme and retained the catalytic efficiency (k cat has not changed), but the pK a of a proton shuttle His64 has increased while the K M value decreased to approach the CA XII.
Figure 2.

Experimental data of stability, catalytic activity and inhibitor binding of CA II (violet squares), CA XII (dark green circles) and chCA XII (light green squares) by FTSA (panels A, C, E, H), ITC (panels D, F) and SFA (panels G, I) techniques. (A) Thermal stability profiles of CA's at pH 4.5–10.0. (B) Catalytic constant of CA dependence on pH in the range 6.0–9.0. (C) Unfolding curves of the proteins at 0, 25 and 200 μM inhibitor 24 concentrations. (D) Raw curves of CA titrations with inhibitor 13. (E) Thermal shifts of CA melting temperature at increasing inhibitor 24 concentrations up to 200 μM. (F) Integrated titration curves for inhibitor 13 binding to CA. (G) Acidification curves of spontaneous and catalyzed CO2 hydration reaction by CA at several inhibitor 34 concentrations. (H) Correlations of observed pK d’s between chCA XII and CA XII (R2=0.90) or CA II (R2=0.24). The theoretically perfect positive correlation is represented as a black dashed line and linear regression models are shown as green (y=1.03x–0.31) and violet (y=0.52x+3.20) lines. (I) Dose‐response curves of inhibition of CA enzymatic activity by 34. Overall, all shown figures here confirm that the chCA XII protein is recognized by compounds similarly to CA XII, but different from CA II.
2.2. chCA XII Structure‐Affinity Data Recapitulate Wild Type Interactions
To understand the reasons for affinity and selectivity of inhibitors targeting CA II and CA XII, we measured interactions between a series of 63 sulfonamide‐based inhibitors and chCA XII by fluorescent thermal shift assay (FTSA, also called Thermofluor® or differential scanning fluorimetry) and compared the binding constants to those of CA II and CA XII. [43] We also used stopped‐flow CO2 hydration assay (SFA) to confirm the inhibitory properties of chemical compounds and isothermal titration calorimetry (ITC) to determine the thermodynamic parameters of binding. As control compounds, we used commercially available inhibitors ‐ ethoxzolamide (EZA), acetazolamide (AZM), methazolamide (MZM), naphthalene‐2‐sulfonamide (2‐NSA) and 1‐(4‐fluorophenyl)‐3‐(4‐sulfamoylphenyl)urea (U‐104/SLC‐0111). Figure 2 shows representative experimental data by FTSA (panels C, E), ITC (panels D, F) and SFA (panels G, I).
X‐ray crystallographic structures of chCA XII were solved in complex with nine compounds (6, 9, 11, 13, 43, 44, 48, 50, and 52). Electron densities of compounds are shown in Figure S3 and data collection and refinement statistics are presented in the Table S1. Several compounds bound to CA II and CA XII have been characterized previously, so we compared the binding of inhibitors by chCA XII with parental CA II and CA XII isoforms. Inhibitors used in the study could be arranged to 3 groups according to binding affinities: first that possessed more than 10‐fold selectivity towards CA II over CA XII, second that possessed more than 10‐fold selectivity for CA XII over CA II, and third that bound CA II and CA XII with similar dissociation constants. The binding affinities determined by FTSA were in good agreement with ITC and SFA assays as previously discussed.[ 43 , 46 , 48 ] As shown there, we use the K d values obtained by FTSA method since this technique provides the most accurate results.
2.3. CA II‐Selective Compounds
The compounds analyzed formed three broad groups. The first group comprised drug ethoxzolamide (1), non‐halogenated, chlorinated, or fluorinated benzensulfonamides with substituents at para‐ (2–16), ortho‐ (17) or meta‐ (18–22) positions (Figure 3, upper part) that bind to CA II more strongly than CA XII. The binding affinities are provided in Table 1. The majority of chemical compounds 1–22 (with exception of 2, 4 and 17) are strong nanomolar inhibitors of CA II (K d 0.65–67 nM) possessing up to 200‐fold selectivity over CA XII (K d 37–1100 nM) and chCA XII (K d 25–2900 nM). Ureidosulfonamide 2 (U104/SLC‐0111) has been previously published as a strong inhibitor of CA XII with K i=4.5 nM and a moderate inhibitor of CA II with K i=96 nM. [49] However, we have observed that the binding affinities of compound 2 were stronger for CA II (K d 330 nM by FTSA, 150 nM by SFA, 200 nM by ITC) than for CA XII (K d 5500 nM by FTSA, 1900 nM by SFA, unobservable K d by ITC due to weak binding). According to our results, this compound inhibits CA II, chCA XII and CA XII weaker than other para‐substituted compounds 3–16.
Figure 3.

Chemical structures of compounds 1–34 used in this study arranged according to their selectivity. Benzenesulfonamides 1–22 bound more than 10‐fold stronger to CA II than to CA XII, while compounds 23–34 were more than 10‐fold stronger binders of CA XII.
Table 1.
The observed dissociation constants (K d, nM) of CA II‐selective compounds 1–22 and CA XII‐selective compound 23–34 binding to recombinant human CA II, chCA XII and CA XII, determined by fluorescent thermal shift assay (pH 7.0, 37 °C) and confirmed by isothermal titration calorimetry (shown in the parentheses, pH 7.0, 37 °C) and stopped‐flow CO2 hydration assay (shown in the parentheses as second value in italic font, pH 7.5, 24 °C).
|
Compound |
Carbonic anhydrase K d [nM][a] |
K d ratio |
|||
|---|---|---|---|---|---|
|
No. |
Lab. name |
CA II |
chCA XII |
CA XII |
CA XII/CA II |
|
>10‐fold selectivity for CA II over CA XII | |||||
|
1 |
EZA |
1.3 |
25 |
71 |
55 |
|
2 |
U‐104/ SLC‐0111 |
330 (200, 150) |
1800 (780, 300) |
5500 (NA, 1900) |
17 |
|
3 |
VD12‐10 |
25 |
330 |
330 |
13 |
|
4 |
VD11‐31 |
140 (100) |
2200 (330) |
2900 (5000) |
21 |
|
5 |
E79 |
4.0 |
770 |
910 |
228 |
|
6 |
E11‐8 |
11 (38) |
910 (920) |
830 (600) |
75 |
|
7 |
E11‐18 |
28 |
770 |
330 |
12 |
|
8 |
E53 |
67 |
2860 |
1100 |
16 |
|
9 |
VD12‐05 |
2.2 (≤12) |
86 (44) |
37 (47) |
17 |
|
10 |
VD10‐13 |
11 |
310 |
220 |
20 |
|
11 |
VD10‐35 |
17 (≤12) |
250 (180) |
250 (140) |
15 |
|
12 |
VD10‐12 |
18 |
670 |
500 |
28 |
|
13 |
VD10‐50 |
9.6 (≤12) |
300 (150) |
280 (230) |
29 |
|
14 |
VD10‐49 |
0.65 |
84 |
120 |
185 |
|
15 |
VD11‐9 |
1.7 |
75 |
76 |
45 |
|
16 |
VR16‐05 |
2.0 |
175 |
83 |
42 |
|
17 |
VD11‐67 |
5900 |
100000 |
100000 |
17 |
|
18 |
VD11‐26 |
5.9 |
290 |
290 |
49 |
|
19 |
VD11‐25 |
1.7 |
77 |
67 |
39 |
|
20 |
VD11‐15 |
1.2 |
450 |
150 |
125 |
|
21 |
VD12‐42 |
2.9 |
27 |
41 |
14 |
|
22 |
EA4‐1 |
10 |
140 |
170 |
17 |
|
>10‐fold selectivity for CA XII over CA II CA II/ CA XII | |||||
|---|---|---|---|---|---|
|
23 |
E13‐4 |
5000 |
2000 |
420 |
12 |
|
24 |
EA1A‐2 |
31 |
0.4 |
1.9 |
16 |
|
25 |
LJ15‐32 |
6.3 |
0.77 |
0.53 |
12 |
|
26 |
EA12‐3 |
13 |
1.6 |
0.4 |
33 |
|
27 |
EA3‐7 |
420 |
50 (≤12) |
24 (≤12) |
17 |
|
28 |
EA8‐7 |
330 (120) |
7.2 (≤12) |
17 (19) |
20 |
|
29 |
EA12‐7 |
3000 (500) |
59 (21, <25) |
19 (≤12, <25) |
160 |
|
30 |
EA11‐7 |
530 |
17 (25) |
20 (56) |
27 |
|
31 |
VD11‐4‐2 |
56 |
6.7 |
2.9 |
19 |
|
32 |
VD12‐39 |
710 |
25 |
65 |
11 |
|
33 |
VD12‐65 |
>200000 |
220 |
500 |
>400 |
|
34 |
VD16‐22 |
58000 (200000) |
3330 (910) |
830 (500) |
70 |
The observed binding affinities of nonfluorinated compounds 3 and 4 were approximately ten‐fold lower than analogous fluorinated compounds 9 and 10 due higher pK a value of sulfonamide group of inhibitors 3 and 4. [41] Modification of methyl group in 3 and 9 to hydroxy group (compounds 4 and 10) of para‐substituent weakened the binding by several times. The additional morpholine substituent at ortho‐ position (compound 17) decreased the binding affinity for CA II by over 1000‐fold (K d 5.9 μM) and chCA XII, CA XII (K d 100 μM). However, additional substituents at meta‐ position (compounds 18–21) did not have notable change of binding affinity or selectivity. Dichloro meta‐substituted compound 22 also bound CA II 14–17‐fold stronger than chCA XII or CA XII. The binding affinities of compounds 1–22 representing the first group were similar for chCA XII and CA XII, thus chCA XII closely mimicked the CA XII.
Crystal structures of four para‐substituted compounds (6, 9, 11, and 13) bound to chCA XII were solved and compared to the complexes of CA II and CA XII with these inhibitors. The superimposed binding modes of compounds 6, 9, 11 and 13 in the active sites of CA II, CA XII and chCA XII are shown in Figure 4. This group of compounds bound CA II stronger than CA XII (as well as chCA XII): 6–75‐fold (11 vs 830 nM), 9–17‐fold (2.2 vs 37 nM), 11–15‐fold (17 vs 250 nM), and 13–29‐fold (9.6 vs 280 nM). The fluorinated ring of para‐substituted benzenesulfonamides (9, 11, 13) could be found in two orientations in the active site of CA II, chCA XII and CA XII. For example, compound 13 (Figure 4D) had identical orientation of fluorinated ring in CA II and chCA XII active sites, whereas in CA XII we found two alternative positions of fluorinated ring. Despite such differences in the binding mode of compound 13 to CA XII and chCA XII, the binding affinities were similar. The stronger binding to CA II could be due to dimethyl pyrimidine interaction with the phenylalanine side chain Phe131, whereas in chCA XII and CA XII the position of dimethyl pyrimidine varied. The electron density of the dimethyl pyrimidine in the crystal structure of chCA XII was rather weak, suggesting mobility. Compound 11 had two alternative positions of fluorinated ring in active site of chCA XII, whereas in the crystal structures of CA II and CA XII it had a single but different position of fluorinated ring (Figure 4C). In CA II, the side chain of Phe130 reduced the mobility of the flexible tail. In chCA XII, the electron density of the tail was also weak. The electron density of 9 bound in chCA XII was well‐defined and compound was modeled in a single orientation. In the crystal structures of CA II, the position of the ligand was different. Compound bound in all four subunits of asymmetric unit of CA XII is found in multiple conformations with variable positions of the tail (Figure 4B). The position of the chlorinated benzene ring of compound 6 as in the case of other chlorosubstituted benzenesulfonamide derivatives was fixed.[ 38 , 50 ]
Figure 4.

X‐ray crystallographic structures of CA II‐selective compounds bound to CA II, chCA XII and CA XII. Zn(II) is shown as magenta sphere. Black line denotes the shape of the active site, whereas blue transparent ellipse denotes the hydrophilic part of active site which contains water molecules. Protein side chains and ligands bound to CA II are colored magenta, chCA XII‐light green, CA XII‐dark green. (A) Compound 6 bound in the active site of CA II (PDB: 4KNJ), chCA XII (PDB: 6YH5) and CA XII (PDB: 4KP5). (B) Compound 9 bound in the active site of CA II (PDB: 4WW6), chCA XII (PDB: 6YHA) and CA XII (PDB: 5MSA). (C) Compound 11 bound in the active site of CA II (PDB: 4PZH), chCA XII (PDB: 6YH4) and CA XII (PDB: 5MSB). (D) Compound 13 bound in the active site of CA II (PDB: 4HT0), chCA XII (PDB: 6YHB) and CA XII (PDB: 4HT2).
There were differences of binding mode in the position of para‐substituent. The positions of the pyrimidine ring in CA XII differed from that in CA II and chCA XII (Figure 4A). Despite the differences in the position of para‐substituent, compound 6 showed the same binding affinities towards chCA XII and CA XII. Note that the affinities of compound 6 for CA II and CA XII differed 75‐fold. The reason for this was that pyrimidine substituent of compound 6 in CA II is located in the hydrophobic cavity formed by side chains of Phe130, Val134, Leu203 and conservative between CA II and CA XII‐Pro201 and Leu197. The para‐substituents of compounds 9 and 13 also had close interactions with this hydrophobic cavity in active site of CA II. Active site of CA XII does not contain such hydrophobic cavity and has alanine instead Phe130 (in CA II), serine instead Val134 and asparagine instead Leu203.
2.4. CA XII‐Selective Compounds
The second group of compounds included halogenated benzensulfonamides 23–34 that possess higher than ten‐fold selectivity for anticancer target CA XII over off‐target CA II (Figure 3 lower part). The chemical compounds are nanomolar inhibitors of CA XII (as well as chCA XII) as listed in Table 1. Inhibitors 23–29 have Cl− or Br− atom at the ortho‐ position, cyclohexylamino or cyclohexylsulfanyl tail at para‐ position and varying tail with amide linkage (except 23) at meta‐ position. Compound 23 with methyl substituent at meta‐ position bound CAs with K d 420–5000 nM while compounds 24–29 with amide linkage at the same position strengthens the binding up to 5000‐fold. The strongest binders 24–26 (K d 0.4–1.9 nM for CA XII, K d 0.4‐1.6 for chCA XII, and K d 6.3–31 nM for CA II) bear phenylsulfanyl or cyclohexylsulfanyl substituent at para position. Replacement of cyclohexylsulfanyl to cyclohexylamino tail decreased the binding affinities approximately ten‐fold. In this study we have synthesized two new inhibitors ‐ 29 (EA12‐7) and 30 (EA11‐7) that were analogues to lead compound 26 (EA12‐3). [36] We have measured binding affinities towards all 12 catalytically active CAs (Figure 5, Table S2) and showed that compound 29 (EA12‐7) bound all CAs with decreased affinity compared to 26 (EA12‐3), but significantly increased selectivity. It has 155‐fold selectivity towards CA XII (K d=19 nM) over CA II (K d=3000 nM) and bound very weakly or did not bind CAs I, III, VA, VB, VI, VII and CA XII (K d≥10000–200000 nM). Methylation of carboxy group at meta‐substituent (compound 30) did not increase affinity or selectivity in comparison to 26 and 29.
Figure 5.

CA XII‐selective inhibitors: 26 (EA12‐3), 29 (EA12‐7) and 30 (EA11‐7) and their affinities for all twelve human CA isoforms determined by fluorescent thermal shift assay. The white line on the bars represents the limit of affinity determination (K d 200 μM). Substitution of sulfur atom to nitrogen atom in the para position decreased the binding affinity for all 12 CAs, but increased inhibitor selectivity for CA XII over CA II from 33‐fold to 155‐fold and up to 10000‐fold over remaining 10 CA isoforms. However, additional methylation of carboxy group at meta substituent increased the binding affinity for all CAs up to 25‐fold, but reduced selectivity towards CA XII.
In our previous study we showed that fluorinated compounds with bulky substituents on benzensulfonamide ring have greater affinity for CA XII than CA II. [40] The lead compound 31 (VD11‐4‐2) of anticancer target CA IX (K d=50 pM) [39] bound CA XII and chCA XII stronger than CA II (K d=2.9, 6.7 and 56 nM, respectively). Replacement of cyclooctylamino group to cyclododecylamino substituent at meta‐ position and addition of phenyl ring to para‐ position (32) decreased the affinity for all tested CAs. However, the same displacement of hydrophobic ring and addition of 2‐(4‐hydroxyphenyl)ethylamino group at second meta‐ position with SO2CH2CH2OH tail (33) increased the size of inhibitor so that it could not fit in the active site of CA II, but bound to CA XII and chCA XII (K d 500 and 220 nM). Another di‐meta‐substituted benzenesulfonamide bearing phenyl groups 34 also very weakly bound CA II (K d 58000 nM) but stronger to CA XII and chCA XII (K d=830 and 3300 nM). Overall, the chCA XII was a suitable model for screening of CA XII‐selective inhibitors. It bound halogenated benzenesulfonamides 23–34 with similar affinities as CA XII and accommodated large compounds with bulky substituents in the active site that hardly or did not fit in the active site of CA II.
2.5. Nonselective Inhibitors
The third group incorporated inhibitors 35–63 that bound to CA II, chCA XII and CA XII with affinities differing by up to ten‐fold (Figure 6, Table 2). Drugs acetazolamide (35) and methazolamide (36) were nanomolar inhibitors of CA II (K d 46 and 100 nM) and bound up to 9‐fold stronger than chCA XII or CA XII, while naphthalene‐2‐sulfonamide (37) similarly bound CA II stronger than chCA XII and CA XII (K d for CA II was 770 nM, while for chCA XII and CA XII ‐ 6700 and 6300 nM, respectively). The chlorinated benzensulfonamides bearing thiazole (38), benzimidazole (39) or pyrimidine (40, 41) fragments at para‐ position were weaker inhibitors of CA II, chCA XII and CA XII than acetazolamide (35) and 2,3,5,6‐tetrafluoro‐4‐(phenylthio)benzensulfonamide (42). The weak binding for CA XII and chCA XII also shown by ortho‐substituted with bulky hydrophobic groups fluorinated benzenesulfonamides 43 (K d 10000 and 12500 nM), 44 (K d 720 and 4000 nM).
Figure 6.

Chemical structures of compounds 35–63, that bound to CA II and CA XII with similar binding affinities (<10‐fold differences). Their binding affinities were also close for chCA XII and thus were uninformative about the switch of affinities in the chimeric protein.
Table 2.
The observed dissociation constants (K d, nM) of nonselective compound 35–63 binding to recombinant human CA II, chCA XII and CA XII, determined by fluorescent thermal shift assay (pH 7.0, 37 °C).
|
Compound |
Carbonic anhydrase K d [nM][a] |
K d ratio |
|||
|---|---|---|---|---|---|
|
No. |
Lab. name |
CA II |
chCA XII |
CA XII |
CA XII/CA II |
|
Similar binding (<10‐fold selectivity) | |||||
|
35 |
AZM |
46 |
415 |
130 |
2.8 |
|
36 |
MZM |
100 |
550 |
500 |
5.0 |
|
37 |
2‐NSA |
770 |
6700 |
6300 |
8.2 |
|
38 |
E11‐64 |
100 |
625 |
500 |
5.0 |
|
39 |
E2 |
1600 |
5000 |
3100 |
1.9 |
|
40 |
E50 |
140 |
2600 |
1200 |
8.6 |
|
41 |
E45 |
120 |
2900 |
1100 |
9.2 |
|
42 |
VD10‐18 |
3.4 |
50 |
18 |
5.3 |
|
43 |
VD12‐29‐2 |
2200 |
12500 |
10000 |
4.5 |
|
44 |
VD12‐36 |
230 |
4000 |
720 |
3.1 |
|
45 |
AZ13‐2‐2 |
2200 |
2000 |
250 |
0.1 |
|
46 |
VD12‐09 |
1000 |
500 |
240 |
0.2 |
|
47 |
VD12‐14 |
10000 |
12500 |
1400 |
7.0 |
|
48 |
VD12‐25‐2 |
64 |
46 |
16 |
0.3 |
|
49 |
VD12‐30 |
42 |
1.25 |
5.0 |
0.1 |
|
50 |
VD12‐34 |
17 |
12.5 |
8.3 |
0.5 |
|
51 |
VD11‐28 |
6.7 |
33 |
40 |
6.0 |
|
52 |
VD10‐39b |
83 |
40 |
25 |
0.3 |
|
53 |
EA3‐3 |
10 |
1.7 |
2.0 |
0.2 |
|
54 |
EA11‐3 |
10 |
2.0 |
1.3 |
8.0 |
|
55 |
EA4‐2 |
5.0 |
0.25 |
0.83 |
0.2 |
|
56 |
LJ15‐22 |
13 |
4.0 |
1.7 |
7.6 |
|
57 |
LJ14‐9 |
10 |
0.29 |
0.8 |
0.1 |
|
58 |
LJ15‐33 |
5.3 |
6.7 |
4.0 |
0.8 |
|
59 |
EA4‐8 |
20 |
28 |
130 |
6.5 |
|
60 |
EA10‐8 |
40 |
31 |
330 |
8.3 |
|
61 |
LJ15‐37 |
31 |
26 |
9.5 |
0.3 |
|
62 |
EA4‐2o |
3.3 |
3.3 |
3.1 |
0.9 |
|
63 |
EA12‐4 |
15 |
10 |
6.4 |
0.4 |
Structural arrangement of 43 and 44 in the active site of chCA XII is compared in Figure 7A. Compounds differed in the position of the sulfonamide group at Zn(II). Compound 43 contains relatively rigid hydrophobic group in the ortho‐position. Due to its geometry, 43 could not occupy the canonical position of sulfonamide group in the active site. Only the positions of the sulfonamide nitrogen atoms of both compounds coincided. The sulfonamide nitrogen atom of 43 also formed the hydrogen bond with hydroxyl of Thr199 and the coordination bond with Zn(II). Compound 44 contains in the ortho‐position bulky but flexible hydrophobic cyclooctylamino group. The sulfonamide group of this compound occupies the canonical position of sulfonamide‐based inhibitors in the active site of human CA isoforms (sulfonamide made two hydrogen bonds with Thr199 and coordinated Zn(II)).
Figure 7.
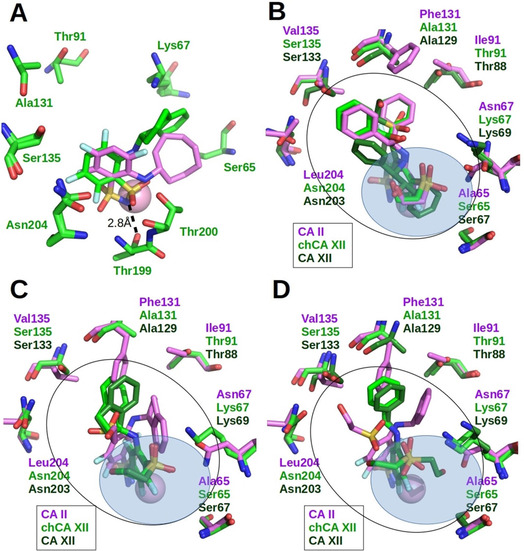
X‐ray crystallographic structures of several nonselective compounds bound to CA II, chCA XII and CA XII. Zn(II) is shown as magenta sphere. Black line denotes the shape of active site, whereas blue transparent area marked the hydrophilic part of active site which contains water molecules. Protein side chains and ligands bound to CA II are colored magenta, chCA XII‐light green, CA XII‐dark green. (A) Compounds 43 (PDB: 6YH8) and 44 (magenta, PDB: 6YH6) bound in chCA XII. (B) Compound 48 bound in CA II (PDB: 4QIY), chCA XII (PDB: 6YH9) and CA XII (PDB: 4QJ0, main orientation (from 4 protein subunits: 3 vs 1)). (C) Compound 50 bound in CA II (PDB: 5DRS), chCA XII (PDB: 6YH7) and CA XII (PDB: 5LLO). (D) Compound 52 bound in CA II (PDB: 5EHE), chCA XII (PDB: 6YHC) and CA XII (PDB: 4QJW).
Fluorinated benzensulfonamides 45–47 with cyclooctylamino substituents at ortho‐ position and varying tails at para‐ position also exhibited moderate affinity towards tested CAs (K d 240–12500 nM). Stronger binders of CA II, chCA XII and CA XII were meta‐, para‐ di‐substituted tri‐fluorinated, or chlorinated/brominated at ortho‐ position benzensulfonamides 48–63. They showed similar binding affinities for all three proteins (K d 3.3–88 nM for CA II, 0.25–46 nM for chCA XII, and K d=0.8–330 nM for CA XII). The crystal structures of compounds 48, 50, 52 bound to chCA XII were determined.
The compound 50 was synthesized by modifying 11: the hydrophobic substituent was added to the meta‐ position of fluorinated ring. Compounds 50 and 11 have similar binding affinities to CA II (K d 17 nM) so the impact of additional surface was not detected. The additional hydrophobic surface at the meta‐position of fluorinated ring has improved the binding affinity (K d) of the 50 for CA XII and chCA XII by 20‐fold and 30‐fold, respectively. The binding mode of 50 was the same in the active sites of CA XII and chCA XII, whereas in CA II the position of the compound was different (Figure 7C). The binding affinities of 48 and 52 for CA XII were 5–6‐fold higher compared to 11 (10–16‐fold for chCA XII). Despite the similar binding affinities of the compound 52 for CA II, CA XII and chCA XII, the binding modes also differed as in case of 50. The binding mode of compound 52 was similar only in the active sites of CA XII and chCA XII (Figure 7D). The binding mode of 48 was the same in the active sites of CA II and chCA XII and in one protein chain out of four in CA XII. In the other three protein chains of CA XII, the meta‐substituent of the compound was located on the opposite side (Figure 7B). The bulky 48 molecule fully occupied the middle part of the active site cavity closing the access of water molecules to the deeper parts of active site. The consequence of this could be a decreased dissociation rate. [37] The possible reasons of similar binding affinities of 11 and 50 to CA II have been analyzed, [37] and the enthalpy‐entropy compensation was observed for this pair of compounds. Compounds 11 and 50 bound to CA XII differently (compare Figure 4C and Figure 7C). Compound 50 efficiently filled the active site of CA XII. Thus, the meta‐substituent of 50 was located in the hydrophobic environment and sterically displaced the remaining part of the compound into the hydrophilic part of CA XII active site, where the formation of hydrogen bonds with protein surface is more likely. Such explanation could be also applicable to the comparison of 11 and 52.
The binding, inhibition and structural studies revealed that the engineered chCA XII protein mimicked the active site of CA XII. The experimental dosing/titration curves of chCA XII were closer to CA XII, but not CA II (Figure 2E, F, I). Figure 2H summarizes the binding data and shows the correlations between observed pK d’s of chCA XII and CA II or CA XII. There is a strong correlation between pK d’s of chCA XII and CA XII with coefficient of determination R 2=0.90, and nearly absent correlation between binding affinities of chCA XII and CA II with R 2=0.24. The correlations of intrinsic values were also similar to observed ones and were provided in Figure S4. The binding affinities of several compounds were closer between CA II and chCA XII, than between chCA XII and CA XII, but all of them belonged to the group of nonselective inhibitors.
3. Discussion
This study has used a reverse engineering approach to provide an opportunity to develop selective inhibitors to the difficult target CA XII. In achieving this it has shed light on outstanding questions surrounding protein‐ligand recognition. Despite decades of effort on drug development aimed at the carbonic anhydrases even the apparently most basic questions remain open. For example, is carbonic anhydrase behaving as rather rigid protein that does not undergo significant conformational or other structural changes upon ligand binding, or is the protein rather flexible where motions of substantial parts are essential for ligand recognition? Recent studies indicated that motion is important even for an allegedly “rigid protein”, a prototypical textbook example such as CA II. [51] In our opinion, carbonic anhydrase active sites are rather rigid and recognize ligands primarily via a Lock‐and‐Key mechanism. [52] However, the question is largely open and requires further studies to understand the principles of recognition and enable truly rational design of high‐affinity and high‐selectivity compounds.
Several studies[ 44 , 45 , 53 ] have distinguished the active site residues that differ between CA II and CA XII and could be exploited for selective inhibitor design. The first active site mutants of CA II were generated in the early 1990 s.[ 54 , 55 ] CA II was used as a model isoform due to simple expression and purification, ultra‐high catalytic activity (k cat=0.6–1.4×106 s−1), moderate thermal stability (T m 55–59 °C)[ 56 , 57 ] and off‐target function ‐ it is found in erythrocytes and regulates blood ion concentrations. [58]
By engineering chCA XII we have shown that six mutations in the active site of CA II (A65S, N67K, I91T, F130A, V134S, and L203N) were sufficient to switch its inhibitor recognition profile to the CA XII. The kinetic studies showed that these mutations did not affect the catalytic activity (k cat of CA II and chCA XII is 6.0×105 s−1) but increased the pK a value of the proton shuttle His64 from 7.3 to 7.9. The observed maximal catalytic constant of CA XII was (4.1±0.9)×105 s−1 and pK a of His64 was 7.6±0.1. The parameters closely matched the previously published k cat value of (4.0±1.7)×105 s−1 and pK a of 8.0±0.2 by stopped‐flow spectrophotometry and 7.5±0.1 by 18O‐mass spectrometry. [34] Our previous study showed, that A65T, N67Q, F130Y, V134Q, and L203T mutations did not affect the pK a value of His64 in CA II. [59] For this reason, we assumed that I91T mutation increased the pK a of mutant enzyme and Thr88 (CA XII numbering) was a key member in catalytic mechanism of CA XII.
The chCA XII recognized the substrate with similar K M constant as CA XII (K M=3.6 mM) as well as observed pK d’s of 63 inhibitors binding to chCA XII were significantly more similar to CA XII (R 2=0.90) than to CA II (R 2=0.24). Compounds 9, 11, 13, 48 and 50 occupied similar positions in the active sites of CA XII and chCA XII, and binding modes differed from CA II. These crystal structures supported the similar binding by CA XII and chCA XII, as suggested by binding data. However, several cases demonstrated different binding modes of the ligand in chCA XII and CA XII, and the binding modes in chCA XII resembled CA II. Despite similar position, the key residues interacting with ligand in CA II and chCA XII differed and contacts between ligand and protein differed. Detailed interaction analysis of multiple binding modes as in CA XII could be helpful here.
In this study we have compared inhibitor binding properties of CA II, chCA XII and CA XII using the observed parameters, because both isoforms have similar pK a value of zinc‐bound water equal to 7.0–7.1±0.2 at 25 °C.[ 33 , 34 , 60 ] The binding of sulfonamide compound to CA occurs only when sulfonamide group is in the deprotonated form and zinc‐bound water in the active site is protonated.[ 43 , 61 ] For this reason, attention should be paid to the pK a’s of zinc‐bound water molecule and sulfonamide group of an inhibitor when comparing binding affinities of several CAs or inhibitors. Structure‐affinity correlation analysis should be performed using intrinsic parameters of binding[ 41 , 50 , 62 , 63 , 64 , 65 , 66 ] that are independent on experimental condition (pH, buffer, etc.). Our previous studies with chimeric carbonic anhydrase VI revealed that intrinsic binding affinities had a better linear correlation between chCA VI and CA VI than the observed affinities. [59]
However, in the case of chimeric chCA XII, the pK a of the Zn(II)‐bound water molecule did not differ from CA II or CA XII. The pK a values were 6.9 for CA II, 6.9 for chCA XII and 6.8 for CA XII at 37 °C. Therefore, in the calculation of intrinsic binding constants, the only reason for the differences from the observed constants remained the pK a of the compound sulfonamide amino group. Furthermore, when comparing binding to these three proteins, the relative ratio of observed or intrinsic values remained essentially the same and thus Figure 2H compares the switch of the observed values, while the corresponding intrinsic constants and their switch is provided in the supplementary materials. The switch was equally well observed in both approaches.
When studying correlations between compound's chemical structure and observed binding affinity for CA II or CA XII, the studied para‐substituted benzenesulfonamides bound to CA II with more than ten‐fold higher affinity than to CA XII. The data were consistent with other published studies.[ 42 , 48 , 67 , 68 , 69 ] Despite the fact that 4‐(4‐fluorophenylureido)benzenesulfonamide U‐104/SLC‐0111 [49] (2 in this study) has been tested in patients with advanced solid tumors and has finished Phase 1 clinical trial, [70] our study indicates, that as confirmed by three different assays (FTSA, ITC and SFA), the U‐104/SLC‐0111 exhibits rather weak, micromolar, affinity for anticancer target CA XII and binds it 17‐fold weaker than off‐target CA II. Recently, Jonsson and Liljas have expressed doubts about proper experimental data for promoting the U‐104/SLC‐0111 compound into preclinical studies. [71] Our results show that lead compound EA12‐3 (26) [36] is a significantly stronger inhibitor of CA XII (K d=0.4 nM) and its newly synthesized analogue EA12‐7 (29) possessed significant selectivity for CA XII. Further studies are needed, by as many techniques as possible, to confirm the validity of the affinities, especially because the enzyme inhibition assay is limited by the concentration of the enzyme and is therefore difficult to apply it for determination of sub‐nanomolar affinities.
4. Conclusions
The detailed thermodynamic and kinetic profiling combined with X‐ray structural studies revealed that CA II‐based chimeric protein chCA XII resembled the active site of CA XII. Amino acids S67, K69, T88, A129, S133, and N203 were confirmed as key residues of CA XII for inhibitor recognition, while T88 played a major role in the catalytic mechanism. A series of 63 sulfonamide compounds bound chCA XII with similar affinities to CA XII, but not CA II. Structurally, the switch from CA II to CA XII was very sensitive and therefore both alternative inhibitor binding positions were observed in the crystal structures of chCA XII. As a result, chCA XII was shown as a suitable model for designing CA XII‐selective inhibitors, such as compound EA12‐3 and a newly synthesized analogue EA12‐7. Our study provided additional insight to the basis for isoform selectivity of sulfonamides binding to CA XII, over CA II. The strategy could potentially be applied to other drug design projects in medicinal chemistry if the protein was sufficiently rigid.
Experimental Section
Protein Preparation
Carbonic anhydrase protein isoforms I–XIV, including chCA XII were expressed and purified as described previously. [39] The molecular weight of enzymes was confirmed by mass‐spectrometry, purity was analyzed by SDS‐PAGE and concentrations were determined by UV‐Vis spectrophotometry.
Synthesis of Chemical Compounds
Ethoxzolamide (EZA), acetazolamide (AZM), methazolamide (MZM), naphthalene‐2‐sulfonamide (2‐NSA), and all starting materials and reagents were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Compounds 2, 23, 29 and 30 were newly designed and synthesis is reported in electronic supplementary information. Synthesis of other chemical compounds was described earlier.[ 35 , 36 , 38 , 39 , 40 , 42 , 52 , 67 , 72 , 73 , 74 ] All compounds were dissolved in dimethyl sulfoxide (DMSO) at 10–30 mM concentration and diluted in aqueous buffers just prior to the FTSA, ITC or SFA experiments.
Crystallization
The chCA XII protein was concentrated by ultrafiltration to 28 mg/mL. Crystallization buffer contained 0.1 M sodium bicine (pH 9.0), 0.2 M ammonium sulfate and 2 M sodium malonate (pH 7.0). The ligand solutions for crystal soaking were made by mixing of 50 μL of corresponding reservoir solution and 3 μL of 50 mM ligand solution (in DMSO).
Data Collection and Structure Determination
All datasets of X‐ray diffraction were collected at the EMBL beamline P13. The datasets were processed by XDS program [75] and scaled with SCALA. [76] The molecular replacement was made using MOLREP program. [77] The initial phases were received from PDB entry 4HT0. The 3D models of compounds were built by AVOGADRO program. [78] The library files with complete chemical and geometric descriptions of compounds were generated using LIBCHECK program.[ 79 , 80 ] The models were built using COOT [81] and refined by REFMAC.[ 82 , 83 ] Coordinates and structure factors have been deposited to the RCSB Protein Data Bank (PDB). The PDB access codes, data collection and refinement statistics are listed in supporting information Table S1.
Fluorescent Thermal Shift Assay
Thermal stability of chCA XII and inhibitor binding affinities were determined using fluorescent thermal shift assay, also known as ThermoFluor® or differential scanning fluorimetry (DSF). QIAGEN Rotor‐Gene Q (Corbett Rotor‐Gene 6000) cycler with the blue channel (365±20 nm excitation and 460±15 nm detection) was used to monitor the fluorescence of 1‐anilino‐8‐naphthalene sulfonate (ANS) upon carbonic anhydrase unfolding. The temperature of samples was elevated from 25 to 99 °C at a heating rate of 1 °C/min.
Thermal stability experiments at different pH's (from pH 4.5 to 10.0) were performed using 10 μM chCA XII in universal buffer (50 mM sodium acetate, 25 mM sodium borate, 50 mM sodium phosphate and 50 mM sodium chloride) containing 2 % DMSO compound and 50 μM ANS. The melting temperature T m was determined using a model for fluorescence yield upon protein unfolding.[ 84 , 85 ]
Inhibitor binding affinities were determined upon addition of 0–200 μM (in 2 % DMSO) to the 5 μM CA (except 10 μM for CA IV) solution in 50 mM sodium phosphate, 100 mM sodium chloride buffer (pH 7.0). The thermal shift data were fitted using a quantitative model[ 85 , 86 ] yielding the K d values.
Isothermal Titration Calorimetry
Isothermal titration calorimetry experiments were carried out at 37 °C using VP‐ITC and iTC‐200 calorimeters. The solution containing 6–20 μM carbonic anhydrase in the cell was titrated with 60–200 μM inhibitor solution in the syringe. Experiments were performed in 50 mM sodium phosphate buffer containing 100 mM sodium chloride at pH 7.0 with a final DMSO concentration of 2 %. CAs were dialyzed in the reaction buffer before the titrations. Experimental setup used for VP‐ITC instrument: 25 injections of 10 μL every 180 s at reference power of 4 μcal/s and stirring speed of 260 rpm; iTC200 instrument: 19 injections of 2 μL every 180 s at reference power of 5 μcal/s and stirring speed of 750 rpm. The thermodynamic parameters of binding were determined using NITPIC, [87] SEDPHAT [88] and/or MicroCal Origin software. The first injection point was deleted in the integrated data and a single binding model was applied to fit the data.
Stopped‐Flow CO2 Hydration Assay
Catalytic properties of chCA XII, CA XII and inhibition constants were determined using the stopped‐flow CO2 hydration assay. Experiments were performed using Applied Photophysics SX.18 MV‐R Stopped‐Flow Spectrometer at 24 °C temperature. Saturated substrate solution was prepared by bubbling CO2 gas into milli‐Q water for 1 hour at room temperature.
The catalytic constant k cat and Michaelis‐Menten constant K M of chCA XII and CA XII in pH range 6.0–9.0 was determined using 25 mM buffer and 30–100 μM indicator pairs with similar pK a values: Mes and Bromocresol purple (λ 590 nm, pH 6.0–6.4), Mops and Bromothymol blue (λ 615 nm, pH 6.8–7.1), Hepes and Phenol red (λ 557 nm, pH 7.2–7.8), Tris and m‐Cresol purple (λ 575 nm, pH 8.1–8.4), Ches and Thymol blue (λ 595 nm, pH 8.6–9.0). Solutions also contained 0.2 M sodium sulfate to maintain ionic strength. Enzyme concentration was 150–200 nM for chCA XII and 50–150 nM for CA XII. The saturated substrate solution was diluted with milli‐Q water and final carbon dioxide concentrations were determined according to Ref. [46] The kinetic parameters were obtained using Lineweaver‐Burk and Michaelis‐Menten plots. The pK a values of His64 were determined by fitting k cat dependence on pH data using a single ionization event model:
| (1) |
Inhibition of enzymatic activity experiments were performed in 25 mM Hepes buffer, containing 30 μM Phenol red indicator, 0.2 M sodium sulfate, pH 7.5. The samples consisted of 30–100 nM CA II, 50–200 nM chCA XII, 50–100 nM CA XII and 0–100 μM inhibitor (in <1 % DMSO). The raw curves of time‐dependent absorbance change were fitted using a single exponential model. The rates of spontaneous CO2 hydration reaction were subtracted from the observed catalyzed reaction rates. The K d values were calculated by fitting dose‐response curves to a Morrison equation:[ 89 , 90 ]
| (2) |
Supporting information
Synthesis of chemical compounds 2, 23, 29 and 30; sequence alignment of CA II and CA XII; the dependence of Michelis‐Menten constant on pH; the electron densities of inhibitors bound to CAs of the crystal structures described in this study; correlations of intrinsic pK d’s between chCA XII and either CA XII or CA II; data collection and refinement statistics of chimeric CA XII complexes with inhibitors 6, 9, 11, 13, 43, 44, 48, 50, and 52; the observed dissociation constants (K d, nM) of compounds 26, 29 and 30 binding to recombinant human carbonic anhydrases I–XIV; the pK a values of compound's sulfonamide group and the intrinsic dissociation constants of compounds 1–63 binding to recombinant human CA II, chCA XII and CA XII.
Contributor Roles
D. M., J. S., L. B., J. E. L.‐designed and supervised the project; J. S., L.B., A. Zu.‐performed the enzymatic and biophysical experiments; J. S., V. P., L. B., A. Zu.‐derived the models and analyzed the data; A.S., E. M., S. G.‐determined the crystal structures of CA‐inhibitor complexes, wrote the crystallography part; A. M., V. M.‐expressed and purified the CA proteins; A. Za., E. Č.‐synthesized the inhibitors and wrote the synthesis part; J. S., D. M., J. E. L.‐wrote the paper; W‐Y. C., J. E. L., V. P.‐provided critical feedback. All authors have read and approved the final version of the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Authors thank Gleb Bourenkov for the help with data collection at P13 EMBL beamline at PETRA III ring of the DESY synchrotron (access to the DESY synchrotron EMBL beam line P13), supported by iNEXT grant number 653706 (to A. S., E. M., S. G.) funded by the Horizon 2020 program of the European Commission. This research was supported by the grant MSF‐JM‐6 from Vilnius University. A. Za. was supported by the grant S‐SEN‐20‐10 from the Research Council of Lithuania.
J. Smirnovienė, A. Smirnov, A. Zakšauskas, A. Zubrienė, V. Petrauskas, A. Mickevičiūtė, V. Michailovienė, E. Čapkauskaitė, E. Manakova, S. Gražulis, L. Baranauskienė, W.-Y. Chen, J. E. Ladbury, D. Matulis, ChemistryOpen 2021, 10, 567.
References
- 1. Zhao J., Cao Y., Zhang L., Comput. Struct. Biotechnol. J. 2020, 18, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Esbaugh A. J., Tufts B. L., Respir. Physiol. Neurobiol. 2006, 154, 185–198. [DOI] [PubMed] [Google Scholar]
- 3. Lee D., Hong J. H., Int. J. Mol. Sci. 2020, 21, 339. [Google Scholar]
- 4. Sterling D., Reithmeier R. A., Casey J. R., Jop 2001, 2, 165–70. [PubMed] [Google Scholar]
- 5. Widdas W. F., Baker G. F., Baker P., Cytobios 1994, 80, 7–24. [PubMed] [Google Scholar]
- 6. Ingram C. J., Brubaker R. F., Am. J. Ophthalmol. 1999, 128, 292–296. [DOI] [PubMed] [Google Scholar]
- 7. Silver L. H., Am. J. Ophthalmol. 1998, 126, 400–408. [DOI] [PubMed] [Google Scholar]
- 8. Wile D., Ann. Clin. Biochem. 2012, 49, 419–431. [DOI] [PubMed] [Google Scholar]
- 9. Brigo F., Lattanzi S., Igwe S. C., Behzadifar M., Bragazzi N. L., Cochrane Database Syst. Rev. 2018, 10, CD001416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kothare S. V., Kaleyias J., Expert Opin. Drug Metab. Toxicol. 2008, 4, 493–506. [DOI] [PubMed] [Google Scholar]
- 11. Leniger T., Thöne J., Wiemann M., Br. J. Pharmacol. 2004, 142, 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perucca E., Pharmacol. Res. 1997, 35, 241–256. [DOI] [PubMed] [Google Scholar]
- 13. Mboge M. Y., McKenna R., Frost S. C., in Top. Anti-Cancer Res. (Eds.: K. Zaman Atta-ur-Rahman,), Bentham Science Publishers, 2016, pp. 3–42. [Google Scholar]
- 14. Barnett D. H., Sheng S., Charn T. H., Waheed A., Sly W. S., Lin C.-Y., Liu E. T., Katzenellenbogen B. S., Cancer Res. 2008, 68, 3505–3515. [DOI] [PubMed] [Google Scholar]
- 15. Watson P. H., Chia S. K., Wykoff C. C., Han C., Leek R. D., Sly W. S., Gatter K. C., Ratcliffe P., Harris A. L., Br. J. Cancer 2003, 88, 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ilie M. I., Hofman V., Ortholan C., Ammadi R. E., Bonnetaud C., Havet K., Venissac N., Mouroux J., Mazure N. M., Pouyssegur J., Hofman P., Int. J. Cancer 2011, 128, 1614–1623. [DOI] [PubMed] [Google Scholar]
- 17. Viikilä P., Kivelä A. J., Mustonen H., Koskensalo S., Waheed A., Sly W. S., Doisy E. A., Pastorek J., Pastorekova S., Parkkila S., Haglund C., World J. Gastroenterol. 2016, 22, 8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gu X.-F., Shi C.-B., Zhao W., Int. J. Clin. Exp. Pathol. 2019, 12, 2173–2183. [PMC free article] [PubMed] [Google Scholar]
- 19. Haapasalo J., Hilvo M., Nordfors K., Haapasalo H., Parkkila S., Hyrskyluoto A., Rantala I., Waheed A., Sly W. S., Pastorekova S., Pastorek J., Parkkila A.-K., Neurooncology 2008, 10, 131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kopecka J., Campia I., Jacobs A., Frei A. P., Ghigo D., Wollscheid B., Riganti C., Oncotarget 2015, 6(9):6776-6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liao S.-Y., J. Med. Genet. 2003, 40, 257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kivelä A., Parkkila S., Saarnio J., Karttunen T. J., Kivelä J., Parkkila A.-K., Waheed A., Sly W. S., Grubb J. H., Shah G., Türeci Ö., Rajaniemi H., Am. J. Pathol. 2000, 156, 577–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parkkila S., Parkkila A.-K., Saarnio J., Kivelä J., Karttunen T. J., Kaunisto K., Waheed A., Sly W. S., Türeci Ö., Virtanen I., Rajaniemi H., J. Histochem. Cytochem. 2000, 48(12):1601-1608. [DOI] [PubMed] [Google Scholar]
- 24. Kivelä A. J., Parkkila S., Saarnio J., Karttunen T. J., Kivelä J., Parkkila A.-K., Pastoreková S., Pastorek J., Waheed A., Sly W. S., Rajaniemi H., Histochem. Cell Biol. 2000, 114, 197–204. [DOI] [PubMed] [Google Scholar]
- 25. Leppilampi M., Saarnio J., Karttunen T. J., Kivela J., Pastorekova S., Pastorek J., Waheed A., Sly W. S., Parkkila S., World J. Gastroenterol. 2003, 9, 1398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Syrjänen L., Luukkaala T., Leppilampi M., Kallioinen M., Pastorekova S., Pastorek J., Waheed A., Sly W. S., Parkkila S., Karttunen T., APMIS Acta Pathol. Microbiol. Immunol. Scand. 2014, 122, 880–889. [DOI] [PubMed] [Google Scholar]
- 27. Karhumaa P., Mol. Hum. Reprod. 2000, 6, 68–74. [DOI] [PubMed] [Google Scholar]
- 28. Karhumaa P., Kaunisto K., Parkkila S., Waheed A., Pastorekov́ S., Pastorek J., Sly W. S., Rajaniemi H., Mol. Hum. Reprod. 2001, 7, 611–616. [DOI] [PubMed] [Google Scholar]
- 29. Muhammad E., Leventhal N., Parvari G., Hanukoglu A., Hanukoglu I., Chalifa-Caspi V., Feinstein Y., Weinbrand J., Jacoby H., Manor E., Nagar T., Beck J. C., Sheffield V. C., Hershkovitz E., Parvari R., Hum. Genet. 2011, 129, 397–405. [DOI] [PubMed] [Google Scholar]
- 30. Power K. A., Grad S., Rutges J. P. H. J., Creemers L. B., van Rijen M. H. P., O'Gaora P., Wall J. G., Alini M., Pandit A., Gallagher W. M., Arthritis Rheum. 2011, 63, 3876–3886. [DOI] [PubMed] [Google Scholar]
- 31. Waheed A., Sly W. S., Gene 2017, 623, 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Whittington D. A., Waheed A., Ulmasov B., Shah G. N., Grubb J. H., Sly W. S., Christianson D. W., Proc. Natl. Acad. Sci. USA 2001, 98, 9545–9550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jogaitė V., Zubrienė A., Michailovienė V., Gylytė J., Morkūnaitė V., Matulis D., Bioorg. Med. Chem. 2013, 21, 1431–1436. [DOI] [PubMed] [Google Scholar]
- 34. Ulmasov B., Waheed A., Shah G. N., Grubb J. H., Sly W. S., Tu C., Silverman D. N., Proc. Natl. Acad. Sci. USA 2000, 97, 14212–14217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zakšauskas A., Čapkauskaitė E., Jezepčikas L., Linkuvienė V., Kišonaitė M., Smirnov A., Manakova E., Gražulis S., Matulis D., Eur. J. Med. Chem. 2018, 156, 61–78. [DOI] [PubMed] [Google Scholar]
- 36. Zakšauskas A., Čapkauskaitė E., Jezepčikas L., Linkuvienė V., Paketurytė V., Smirnov A., Leitans J., Kazaks A., Dvinskis E., Manakova E., Gražulis S., Tars K., Matulis D., Eur. J. Med. Chem. 2020, 185, 111825. [DOI] [PubMed] [Google Scholar]
- 37. Smirnov A., Zubrienė A., Manakova E., Gražulis S., Matulis D., PeerJ 2018, 6, e4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Čapkauskaitė E., Zubrienė A., Smirnov A., Torresan J., Kišonaitė M., Kazokaitė J., Gylytė J., Michailovienė V., Jogaitė V., Manakova E., Gražulis S., Tumkevičius S., Matulis D., Bioorg. Med. Chem. 2013, 21, 6937–6947. [DOI] [PubMed] [Google Scholar]
- 39. Dudutienė V., Matulienė J., Smirnov A., Timm D. D., Zubrienė A., Baranauskienė L., Morkūnaite V., Smirnovienė J., Michailovienė V., Juozapaitienė V., Mickevičiūtė A., Kazokaitė J., Bakšytė S., Kasiliauskaitė A., Jachno J., Revuckienė J., Kišonaitė M., Pilipuitytė V., Ivanauskaitė E., Milinavičiūtė G., Smirnovas V., Petrikaitė V., Kairys V., Petrauskas V., Norvaišas P., Lingė D., Gibieža P., Capkauskaitė E., Zakšauskas A., Kazlauskas E., Manakova E., Gražulis S., Ladbury J. E., Matulis D., J. Med. Chem. 2014, 57, 9435–9446. [DOI] [PubMed] [Google Scholar]
- 40. Dudutienė V., Zubrienė A., Smirnov A., Timm D. D., Smirnovienė J., Kazokaitė J., Michailovienė V., Zakšauskas A., Manakova E., Gražulis S., Matulis D., ChemMedChem 2015, 10, 662–687. [DOI] [PubMed] [Google Scholar]
- 41. Zubrienė A., Smirnovienė J., Smirnov A., Morkūnaitė V., Michailovienė V., Jachno J., Juozapaitienė V., Norvaišas P., Manakova E., Gražulis S., Matulis D., Biophys. Chem. 2015, 205, 51–65. [DOI] [PubMed] [Google Scholar]
- 42. Dudutienė V., Zubrienė A., Smirnov A., Gylytė J., Timm D., Manakova E., Gražulis S., Matulis D., Bioorg. Med. Chem. 2013, 21, 2093–2106. [DOI] [PubMed] [Google Scholar]
- 43. Linkuvienė V., Zubrienė A., Manakova E., Petrauskas V., Baranauskienė L., Zakšauskas A., Smirnov A., Gražulis S., Ladbury J. E., Matulis D., Q. Rev. Biophys. 2018, 51, DOI 10.1017/S0033583518000082. [DOI] [PubMed] [Google Scholar]
- 44. Pinard M. A., Mahon B., McKenna R., BioMed Res. Int. 2015, 2015, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bhatt A., Mahon B. P., Cruzeiro V. W. D., Cornelio B., Laronze-Cochard M., Ceruso M., Sapi J., Rance G. A., Khlobystov A. N., Fontana A., Roitberg A., Supuran C. T., McKenna R., ChemBioChem 2017, 18(2):213-222 . [DOI] [PubMed] [Google Scholar]
- 46. Smirnovienė J., Smirnovas V., Matulis D., Anal. Biochem. 2017, 522, 61–72. [DOI] [PubMed] [Google Scholar]
- 47. Zubrienė A., Matulis D., in Carbonic Anhydrase Drug Target Thermodyn. Struct. Inhib. Bind. (Ed.: Matulis D.), Springer International Publishing, Cham, 2019, pp. 51–59. [Google Scholar]
- 48. Balandis B., Ivanauskaitė G., Smirnovienė J., Kantminienė K., Matulis D., Mickevičius V., Zubrienė A., Bioorg. Chem. 2020, 97, 103658. [DOI] [PubMed] [Google Scholar]
- 49. Pacchiano F., Carta F., McDonald P. C., Lou Y., Vullo D., Scozzafava A., Dedhar S., Supuran C. T., J. Med. Chem. 2011, 54, 1896–1902. [DOI] [PubMed] [Google Scholar]
- 50. Kišonaitė M., Zubrienė A., Čapkauskaitė E., Smirnov A., Smirnovienė J., Kairys V., Michailovienė V., Manakova E., Gražulis S., Matulis D., PLoS One 2014, 9, e114106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Singh H., Das C. K., Vasa S. K., Grohe K., Schäfer L. V., Linser R., Angew. Chem. Int. Ed. 2020, 59(51):22916-22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dudutienė V., Zubrienė A., Kairys V., Smirnov A., Smirnovienė J., Leitans J., Kazaks A., Tars K., Manakova L., Gražulis S., Matulis D., Biophys. J. 2020, 119(8):1513-1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lomelino C. L., Andring J. T., McKenna R., Int J Med Chem 2018, 2018, 9419521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Behravan G., Jonasson P., Jonsson B.-H., Lindskog S., Eur. J. Biochem. 1991, 198, 589–592. [DOI] [PubMed] [Google Scholar]
- 55. Ren X., Jonsson B.-H., Lindskog S., Eur. J. Biochem. 1991, 201, 417–420. [DOI] [PubMed] [Google Scholar]
- 56. Avvaru B. S., Busby S. A., Chalmers M. J., Griffin P. R., Venkatakrishnan B., Agbandje-McKenna M., Silverman D. N., McKenna R., Biochemistry 2009, 48, 7365–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fisher Z., Boone C. D., Biswas S. M., Venkatakrishnan B., Aggarwal M., Tu C., Agbandje-McKenna M., Silverman D., McKenna R., Protein Eng. Des. Sel. 2012, 25, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Henry R. P., Am. Zool. 1984, 24, 241–251. [Google Scholar]
- 59. Kazokaitė J., Kairys V., Smirnovienė J., Smirnov A., Manakova E., Tolvanen M., Parkkila S., Matulis D., Sci. Rep. 2019, 9, 12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Morkūnaitė V., Gylytė J., Zubrienė A., Baranauskienė L., Kišonaitė M., Michailovienė V., Juozapaitienė V., Todd M. J., Matulis D., J. Enzyme Inhib. Med. Chem. 2015, 30, 204–211. [DOI] [PubMed] [Google Scholar]
- 61. Krishnamurthy V. M., Kaufman G. K., Urbach A. R., Gitlin I., Gudiksen K. L., Weibel D. B., Whitesides G. M., Chem. Rev. 2008, 108, 946–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Baranauskienė L., Matulis D., BMC Biophys. 2012, 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kazokaitė J., Milinavičiūtė G., Smirnovienė J., Matulienė J., Matulis D., FEBS J. 2015, 282, 972–983. [DOI] [PubMed] [Google Scholar]
- 64. Linkuvienė V., Matulienė J., Juozapaitienė V., Michailovienė V., Jachno J., Matulis D., Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 708–718. [DOI] [PubMed] [Google Scholar]
- 65. Zubrienė A., Matulis D., in Carbonic Anhydrase Drug Target Thermodyn. Struct. Inhib. Bind. (Ed.: Matulis D.), Springer International Publishing, Cham, 2019, pp. 107–123. [Google Scholar]
- 66. Zubrienė A., Smirnov A., Dudutienė V., Timm D. D., Matulienė J., Michailovienė V., Zakšauskas A., Manakova E., Gražulis S., Matulis D., ChemMedChem 2017, 12, 161–176. [DOI] [PubMed] [Google Scholar]
- 67. Čapkauskaitė E., Zubrienė A., Paketurytė V., Timm D. D., Tumkevičius S., Matulis D., Bioorg. Chem. 2018, 77, 534–541. [DOI] [PubMed] [Google Scholar]
- 68. Rutkauskas K., Zubrienė A., Tumosienė I., Kantminienė K., Kažemėkaitė M., Smirnov A., Kazokaitė J., Morkūnaitė V., Čapkauskaitė E., Manakova E., Gražulis S., Beresnevičius Z., Matulis D., Molecules 2014, 19, 17356–17380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rutkauskas K., Zubrienė A., Tumosienė I., Kantminienė K., Mickevičius V., Matulis D., Med. Chem. Res. 2017, 26, 235–246. [Google Scholar]
- 70. McDonald P. C., Chia S., Bedard P. L., Chu Q., Lyle M., Tang L., Singh M., Zhang Z., Supuran C. T., Renouf D. J., Dedhar S., Am. J. Clin. Oncol. 2020, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jonsson B.-H., Liljas A., Biophys. J. 2020, 119, 1275–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Čapkauskaitė E., Zubrienė A., Baranauskienė L., Tamulaitienė G., Manakova E., Kairys V., Gražulis S., Tumkevičius S., Matulis D., Eur. J. Med. Chem. 2012, 51, 259–270. [DOI] [PubMed] [Google Scholar]
- 73. Kazokaitė J., Niemans R., Dudutienė V., Becker H. M., Leitāns J., Zubrienė A., Baranauskienė L., Gondi G., Zeidler R., Matulienė J., Tārs K., Yaromina A., Lambin P., Dubois L. J., Matulis D., Oncotarget 2018, 9, DOI 10.18632/oncotarget.25508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zubrienė A., Čapkauskaitė E., Gylytė J., Kišonaitė M., Tumkevičius S., Matulis D., J. Enzyme Inhib. Med. Chem. 2014, 29, 124–131. [DOI] [PubMed] [Google Scholar]
- 75. Kabsch W., Acta Crystallogr. Sect. D 2010, D66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Evans P., Acta Crystallogr. Sect. D. 2006, 62, 72–82. [DOI] [PubMed] [Google Scholar]
- 77. Vagin A., Teplyakov A., Acta Crystallogr. Sect. D 2010, 66, 22–25. [DOI] [PubMed] [Google Scholar]
- 78. Hanwell M. D., Curtis D. E., Lonie D. C., Vandermeersch T., Zurek E., Hutchison G. R., J. Cheminf. 2012, 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., Murshudov G. N., Acta Crystallogr. 2004, 60, 2184–95. [DOI] [PubMed] [Google Scholar]
- 80. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Acta Crystallogr. Sect. D 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Emsley P., Lohkamp B., Scott W. G., Cowtan K., Acta Crystallogr. Sect. D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Murshudov G. N., Vagin A. A., Dodson E. J., Acta Crystallogr. 1997, 53, 240–55. [DOI] [PubMed] [Google Scholar]
- 83. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A., Acta Crystallogr. Sect. D 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Matulis D., Kranz J. K., Salemme F. R., Todd M. J., Biochemistry 2005, 44, 5258–5266. [DOI] [PubMed] [Google Scholar]
- 85. Petrauskas V., Zubrienė A., Todd M. J., Matulis D., in Carbonic Anhydrase Drug Target Thermodyn. Struct. Inhib. Bind. (Ed.: Matulis D.), Springer International Publishing, Cham, 2019, pp. 63–78. [Google Scholar]
- 86. Cimmperman P., Baranauskienė L., Jachimovičiūtė S., Jachno J., Torresan J., Michailovienė V., Matulienė J., Sereikaitė J., Bumelis V., Matulis D., Biophys. J. 2008, 95, 3222–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Keller S., Vargas C., Zhao H., Piszczek G., Brautigam C. A., Schuck P., Anal. Chem. 2012, 84, 5066–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhao H., Piszczek G., Schuck P., Methods 2015, 76, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Morrison J. F., Biochim. Biophys. Acta Enzymol. 1969, 185, 269–286. [DOI] [PubMed] [Google Scholar]
- 90. Williams J. W., Morrison J. F., in Methods Enzymol., Academic Press, 1979, pp. 437–467. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary